
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling Pt nanoparticle sintering by sub-monolayer MgO ALD thin films†
Zhiwei
Zhang
 a,
Matthias
Filez
ab,
Eduardo
Solano
c,
Nithin
Poonkottil
a,
Jin
Li
a,
Matthias M.
Minjauw
a,
Hilde
Poelman
d,
Martin
Rosenthal
e,
Philipp
Brüner
f,
Vladimir V.
Galvita
d,
Christophe
Detavernier
a and
Jolien
Dendooven
*a
a,
Matthias
Filez
ab,
Eduardo
Solano
c,
Nithin
Poonkottil
a,
Jin
Li
a,
Matthias M.
Minjauw
a,
Hilde
Poelman
d,
Martin
Rosenthal
e,
Philipp
Brüner
f,
Vladimir V.
Galvita
d,
Christophe
Detavernier
a and
Jolien
Dendooven
*a
aConformal Coating of Nanomaterials (CoCooN), Department of Solid State Sciences, Ghent University, Krijgslaan 281/S1, 9000 Ghent, Belgium. E-mail: Jolien.Dendooven@UGent.be
bCentre for Membrane Separations Adsorption Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
cNCD-SWEET beamline, ALBA synchrotron light source, Carrer de la Llum 2-26, 08290, Cerdanyola del Vallès, Spain
dLaboratory for Chemical Technology, Ghent University, Technologiepark 125, 9052 Ghent, Belgium
eDUBBLE beamline, ESRF - The European Synchrotron, 71 Avenue des Martyrs, 38000 Grenoble, France
fIONTOF Technologies GmbH, Heisenbergstr. 15, 48149 Muenster, Germany
First published on 15th February 2024
Abstract
Metal nanoparticle (NP) sintering is a major cause of catalyst deactivation, as NP growth reduces the surface area available for reaction. A promising route to halt sintering is to deposit a protective overcoat on the catalyst surface, followed by annealing to generate overlayer porosity for gas transport to the NPs. Yet, such a combined deposition-annealing approach lacks structural control over the cracked protection layer and the number of NP surface atoms available for reaction. Herein, we exploit the tailoring capabilities of atomic layer deposition (ALD) to deposit MgO overcoats on archetypal Pt NP catalysts with thicknesses ranging from sub-monolayers to nm-range thin films. Two different ALD processes are studied for the growth of MgO overcoats on Pt NPs anchored on a SiO2 support, using Mg(EtCp)2 and H2O, and Mg(TMHD)2 and O3, respectively. Spectroscopic ellipsometry and X-ray photoelectron spectroscopy measurements reveal significant growth on both SiO2 and Pt for the former process, while the latter exhibits a drastically lower growth per cycle with an initial chemical selectivity towards Pt. These differences in MgO growth characteristics have implications for the availability of uncoated Pt surface atoms at different stages of the ALD process, as probed by low energy ion scattering, and for the sintering behavior during O2 annealing, as monitored in situ with grazing incidence small angle X-ray scattering (in situ GISAXS). The Mg(TMHD)2–O3 ALD process enables exquisite coverage control allowing a balance between physically blocking the Pt surface to prevent sintering and keeping Pt surface atoms free for reaction. This approach avoids the need for post-annealing, hence also safeguarding the structural integrity of the as-deposited overcoat.
Introduction
Metal nanoparticle (NP) catalysts are an iconic branch of catalysts which are used in a variety of chemical industrial processes, such as ammonia, methanol or Fischer–Tropsch synthesis, and light alkane dehydrogenation.1 In the coming decades, these nanomaterials will form the key enabling technology to achieving a circular, carbon-neutral economy, e.g. by converting CO2 into renewable fuels or by producing H2 during water electrolysis.2–6 Yet, the economic viability of metal NP catalysts is greatly hindered by their long-term degradation.7,8 The main phenomena leading to catalyst deactivation include fouling, poisoning, vapor formation, reactions between different solids, sintering, and mechanical wear such as attrition or crushing.9 While some deactivation processes can be reversed, others, like sintering, are mainly irreversible.10 During sintering, the NP surface energy is reduced by NP growth, thus decreasing the NPs’ surface-to-volume ratio.11 The inherent drive of NPs to grow is therefore a continuous challenge to be tackled in order to safeguard the long-term stability of NP catalysts.10–13In order to delay or prevent sintering, a gamut of strategies have been developed: synthesizing uniformly-sized NPs,14,15 solid NP anchoring on strongly interacting support oxides,16,17 introducing kinetic barriers to prevent transport on the support,18,19 and physically confining NPs.20–24 With respect to the latter strategy, NPs can be introduced in high surface-area supports, such as hydrotalcites consisting of 2D stacked sheets,20 3D mesoporous matrixes or zeolites.20 An alternative, more tailored approach is to confine NPs in a porous core–shell-type structure, akin to ship-in-a-bottle, or by overcoating NPs.22–24
A remarkable technology to deposit conformal overcoats on NPs with sub-monolayer thickness control is atomic layer deposition (ALD).25–29 ALD uses sequential self-saturating reactions between gaseous molecules and a solid surface to grow thin films in a layered fashion.30,31 A landmark study which exploits the conformality of ALD to deposit overcoats on NP catalysts is reported by Stair et al.32 This study shows that Al2O3 ALD overcoats on Pd/Al2O3 catalysts followed by high-temperature annealing leads to significantly improved stability of the NP population due to their physical confinement and results in an enhanced ethane dehydrogenation performance.32 The high-temperature annealing step is necessary to crack the Al2O3 overlayer and introduce mesoporosity for gas transport to the NPs. However, this approach offers a limited level of control over the nature and number of NP surface atoms available for reaction.
More recently, Chen et al. introduced the concept of site- and facet-selective ALD.33 This would enable the selective coating (or blocking) of specific NP edges and facets which display low selectivity and hence degrade the catalyst performance. While they suggested that selective ALD coating of the edges of NPs could hinder growth and sintering, to date no reports provide rich evidence of this intriguing hypothesis.
In this work, we bridge the gap between both discussed ALD approaches of (i) site-selective, sub-monolayer ALD on NP edges and facets on the one hand (Fig. 1a), and (ii) conformal, multi-layer ALD on the entire NP catalyst, followed by annealing and layer cracking on the other hand (Fig. 1b). In particular, through cycle-by-cycle deposition of MgO, we navigate from bare over partially to fully covered NP surfaces, eventually evolving to the thin film limit of nm-thick overlayers (Fig. 1c). To have structural control over the deposited overcoat, and hence also the NP surface atoms available for reaction, no post-annealing treatment is implemented as part of the fabrication process. Two MgO ALD processes are exploited to overcoat archetypal Pt NPs anchored on a SiO2 support with (sub-)monolayer thickness control. The MgO growth characteristics are interrogated by complementary ellipsometry and X-ray photo-electron spectroscopy (XPS). In addition, low energy ion scattering (LEIS) is applied to probe the availability of uncoated Pt surface atoms at different stages of MgO deposition. Secondly, the sintering behavior of the MgO overcoated Pt NPs is tested during (i) annealing in O2 to 800 °C and (ii) cycles of propane dehydrogenation reaction (C3H8 = C3H6 + H2, endothermic: ΔH298 = +124 kJ mol−1) and catalyst regeneration in O2 at 600 °C. Monitoring the NP spacing and size in real-time by in situ grazing-incidence small-angle X-ray scattering (GISAXS) provides first-hand insights in the impact of the MgO overcoat on the achieved sintering delay.
 | ||
| Fig. 1 Schematic illustration of state-of-the-art ALD approaches to overcoat NPs: (a) site-selective sub-monolayer ALD on NP edges, (b) multilayer overcoats followed by annealing to produce cracks in the layer. (c) Here, the full thickness range, from (sub-)monolayer to nm-thick thin films, is explored to find the optimal balance between physically blocking the Pt surface to prevent sintering and keeping Pt surface atoms free for reaction. No post-annealing treatment is implemented to guarantee the structural integrity of the as-deposited overcoat. | ||
Experimental methods
Chemicals
Pt is deposited using alternate exposures of (methylcyclopentadienyl)-trimethylplatinum (MeCpPtMe3, 99%, Strem Chemicals) and O2. MgO is deposited using bis(ethylcyclopentadienyl)magnesium (Mg(EtCp)2, >98%, Strem Chemicals) and deionized water, or bis(2,2,6,6-tetramethyl-3,5-heptanedionato)magnesium (Mg(TMHD)2, anhydrous, >98%, Strem Chemicals) and O3. Ar with 99.999% purity (Air Liquide) is used as a carrier gas for all precursors. O3 is produced from a 99% O2/N2 mixture with an AC-2025 (IN USA Inc.) generator, resulting in an O3 concentration of 200 μg ml−1.Preparation of supported Pt nanoparticles
ALD of Pt metal NPs is performed in a custom-built vacuum system with a base pressure of 10−6 mbar. Coupons of Si wafer with native oxide are used as a substrate. Prior to Pt ALD, the surface is pre-cleaned and oxidized in situ by exposing it for 30 s to O2 plasma, using a remote inductively coupled plasma source with a 13.56 MHz radio frequency (RF) Advanced Energy power supply and matching network operating at 200 W. MeCpPtMe3 is contained in a stainless-steel bottle heated to 35 °C. The ALD cycle consists of 8 s precursor exposure, 35 s of pumping, 6 s O2 exposure and 30 s of pumping. 25 ALD cycles are applied at a deposition temperature of 300 °C. A static mode is used where the valves to the pumping system are closed during the precursor and O2 exposure.34 The pressure in the chamber reaches ca. 1 mbar during both exposures.ALD of MgO
Mg(EtCp)2 and deionized water are contained in stainless bottles. The water bottle is kept at room temperature while the delivery line is heated to 70 °C. The Mg(EtCp)2 bottle and delivery line are heated to 40 and 50 °C, respectively. The temperature of the sample stage is set to 150 or 200 °C. The Mg(EtCp)2 and water process is applied in dynamic mode, which means that the precursors are introduced while the chamber is continuously pumped by the turbomolecular pump. A 10 s exposure time is used for both Mg(EtCp)2 and water, and the pressure in the chamber reaches 10−3 mbar for each pulse. The pumping times after Mg(EtCp)2 and H2O exposure are set to 60 s. Mg(TMHD)2 is stored in a stainless steel container at 120 °C and O3 is used as the reactant. The pulse time of both reagents is 10 s, while the pumping times after the Mg(TMHD)2 and O3 exposure are 45 s and 60 s, respectively. A static mode is employed for the Mg(TMHD)2 precursor, and the pressure in the chamber reaches ∼1 mbar during the precursor exposure. During the O3 pulse, a pressure of 10−3 mbar is obtained. The deposition temperature is 250 °C. The deposition of MgO is studied on coupons of Si wafer with native oxide, coupons of Si wafer coated with 70 nm of Pt by magnetron sputtering, and on Pt NPs deposited by ALD on surface oxidized Si, hereafter noted as Pt/SiO2.Thin film and surface characterization methods
In situ spectroscopic ellipsometry (SE) is performed during ALD of MgO on Si with native oxide and on Si with sputtered Pt using a portable spectrometer (M-2000, J.A. Woollam) fitted directly onto the ALD reactor. The growing MgO layer is fitted with a Cauchy model. It should be noted that the same optical model is used to fit all acquired SE spectra, even if the MgO thin film is likely non-continuous at small thicknesses. For the thin films deposited on native SiO2, the final thickness is corroborated by ex situ X-ray reflectivity measurements using a Bruker diffractometer with Cu Kα radiation. A Bruker Artax X-ray fluorescence (XRF) system with Mo X-ray source and XFlash Si drift detector is used to determine the Pt loading of the Pt/SiO2 samples. X-ray photoelectron spectra (XPS) are collected on a Thermo Scientific Theta Probe system operating at a pass energy of 200 eV. Al Kα (λ = 0.834 nm) X-rays are generated at 15 kV/70 W and focused to a spot size of 0.3 mm by an MXR1 monochromator. XPS data analysis was performed using the CasaXPS software package.35 Scanning electron microscopy (SEM) images are acquired using a FEI Quanta 200F instrument. Low energy ion scattering (LEIS) measurements are performed on a double toroidal energy analyzer (Qtac100, IONTOF GmbH) with co-axially integrated ion gun and integrated charge compensation system. The thickness of the MgO coating on the Pt nanoparticles is extracted by measuring the energy loss of 3 keV He+ ions scattered in the bulk of the nanoparticles. The position of the edge of the sub-surface Pt signal on the energy scale, relative to the Pt surface peak, is converted to the MgO average layer thickness using a stopping power of 197 eV nm−1 for 3 keV He+ ions in MgO, calculated by SRIM, a software package concerning the Stopping and Range of Ions in Matter.36 This analysis is only done for fully closed MgO layers, as seen from the absence of the Pt surface peak in the spectra. For samples with Pt NPs that are only partially covered with MgO, the fraction of uncoated Pt surface atoms is obtained from LEIS spectra measured with 5 keV Ne+ ions by comparing the area of the peak originating from Pt surface atoms to the area of the same peak measured on an uncoated Pt NP sample. Variations in the latter for different bare Pt/SiO2 samples lead to a percent error of 12% on the extracted fractions.In situ GISAXS measurements
GISAXS experiments are performed at the DUBBLE BM26B beamline of ESRF (Grenoble, France) and NCD-SWEET beamline of ALBA Synchrotron Light Source (Cerdanyola del Vallès, Spain) using a beam energy of 12 and 12.4 keV, respectively. A beam size of 600 × 300 μm2 [H × V] is used at BM26B, while the size is 150 × 70 μm2 [H × V] at NCD-SWEET. All measurements are performed with an incidence angle of 0.4°. The scattering patterns are acquired with a Dectris® Pilatus 3S 1 M detector positioned at a distance of ∼4.5 m from the sample, using a 30 s (ESRF) or 10 s (ALBA) acquisition time. GISAXS patterns are continuously recorded during ramp anneals in O2 and during propane dehydrogenation–oxidation cycling. Samples are annealed to 800 °C in a custom-built vacuum chamber with a dedicated high-temperature heating stage.37,38 The heater is controlled with a Eurotherm PID regulator. The chamber is evacuated to 10−2 mbar and then filled to 1 bar with a mixture of 20% O2 (99.999%) in He (99.999%) using a continuous flow of 500 sccm set using a mass flow controller system (MKS instruments). Propane dehydrogenation–oxidation looping at 600 °C is done at the NCD-SWEET beamline of ALBA, using a high-temperature annealing chamber.39 Gas mixtures of 5% propane in He and 20% O2 in He are connected to the system, together with high purity He (99.9999%) that is used to purge the chamber. During gas looping, a sequence of O2 (240 s)–He (60 s)–C3H8 (240 s)–He (60 s) is repeated five times.GISAXS data analysis
The analysis of the recorded GISAXS scattering patterns is performed in a similar way as in our previous work.37,39,40 A computing routine in Python is applied to each of the recorded GISAXS images. The first step consists of extracting a 1D horizontal profile, i.e. intensity as a function of qy, I(qy), from the 2D images at the Si Yoneda peak along qz.41 The qz value that corresponds to the critical angle of Si is calculated and the horizontal profile I(qy) is obtained by integrating the intensity over 5 neighboring pixels in the qz direction. The main scattering feature in the 2D GISAXS patterns gives rise to a peak in the 1D horizontal profiles. The second step is to fit a Gaussian function to this peak and extract the qy value of the peak maximum with a confidence interval of 95%. This yields the qy,max value that is followed throughout this work to monitor nanoparticle sintering.Results
MgO ALD on planar reference substrates
Two MgO ALD processes are explored and compared with the aim to coat Pt NPs in a later stage. The Mg(EtCp)2–H2O process has been widely used for MgO deposition under mild conditions (<250 °C) with a moderate growth per cycle (GPC) of around 0.15 nm per cycle.42 To study the effect of deposition temperature on the MgO growth behavior, the Mg(EtCp)2–H2O process is investigated at 150 and 200 °C. As a second process, the Mg(TMHD)2–O3 combination has been reported with a low GPC (∼0.025 nm per cycle) within a relatively high temperature window (225–250 °C), also requiring a high heating temperature of the Mg precursor (130 °C).43 Therefore, the deposition temperature of the Mg(TMHD)2–O3 process is chosen at 250 °C. Fig. 2a and b display the chemical structures of the Mg metalorganic precursors as well as the two MgO ALD processes studied. | ||
| Fig. 2 Chemical structures of the two metalorganic precursors used in this work: (a) Mg(EtCp)2 and (b) Mg(TMHD)2. The co-reactants and deposition temperatures are also indicated. (c–e) Film thickness as a function of the number of ALD cycles, as obtained from in situ spectroscopic ellipsometry during 30 cycles of the Mg(EtCp)2–H2O process at (c) 150 °C and (d) 200 °C, and during 100 cycles of the Mg(TMHD)2–O3 process at (e) 250 °C, each time on Pt (sputtered Pt thin film on Si wafer) and on SiO2 (native oxide on Si wafer). | ||
The model Pt/SiO2 catalyst, onto which MgO deposition is aimed at, consists of Pt NPs deposited on a planar Si wafer with native SiO2 surface oxide. MgO ALD growth can be more selective on either Pt or SiO2 due to their different surface reactivity with the Mg precursor. To substantiate MgO growth selectivity, sputtered Pt thin film (ca. 70 nm) and native SiO2 (ca. 3 nm) samples are first adopted as reference substrates to characterize the growth of MgO on these surfaces separately. In particular, 30 cycles of Mg(EtCp)2–H2O ALD and 100 cycles of Mg(TMHD)2–O3 ALD are applied on both substrates. The MgO film thickness is monitored by in situ spectroscopic ellipsometry as a function of the number of ALD cycles on sputtered Pt and native SiO2 model substrates (Fig. 2c–e).
For both deposition temperatures, linear growth curves are observed during Mg(EtCp)2–H2O ALD on SiO2 without nucleation delay or growth enhancement in the initial stages of the process. On Pt, the growth slows down after the first ALD cycle and accelerates again after around 5 ALD cycles. A ∼4.8 nm film is obtained after 30 ALD cycles at 150 °C on both Pt and SiO2, while ∼4.0 nm is deposited at 200 °C, hence a lower MgO film thickness than at 150 °C. More importantly, in the initial 3–20 ALD cycles, this MgO ALD process exhibits a minor deposition preference on the native SiO2 substrate. Acharya et al. also reported the slower nucleation on metal surfaces compared to oxides for ultrathin MgO films due to the lower surface density of hydroxyl reaction sites.44 After 15 cycles, the film thicknesses are nearly identical on both substrates. Based on these results, it can be concluded that Mg(EtCp)2–H2O allows for simultaneous, rather non-selective deposition on Pt and native SiO2 substrates.
The growth of MgO on sputtered Pt and native SiO2 substrates is also investigated by recording ex situ Mg 1s, Pt 4f and Si 2p X-ray photoelectron spectra (XPS) after 1, 5, 10, 15 and 30 ALD cycles (Fig. S1 and S2†). In accordance with the ellipsometry results, a swift and steady deposition with the Mg(EtCp)2–H2O processes at 150 and 200 °C is confirmed on both substrates, though the higher GPC at 150 °C is less pronounced in the XPS data.
For the Mg(TMHD)2–O3 ALD process, the amount of deposited MgO is comparatively low, only leading to a MgO film thickness of ∼1.0 nm on Pt and ∼0.3 nm on SiO2 after 100 ALD cycles (Fig. 2e). During the first 40 cycles, a linear growth regime is observed with a larger GPC on Pt than on SiO2. Therefore, in the initial stages of the process, the Mg(TMHD)2–O3 process is more selective towards growth on Pt. After 40 ALD cycles, the deposition slows down considerably on both Pt and SiO2 substrates, likely owing to the identical MgO-termination of the surface. Beyond 100 ALD cycles, the deposition of MgO slowly continues in a similar fashion for both Pt and SiO2 type samples, as shown by XPS (Fig. S3c and S4c†).
MgO ALD on supported Pt nanoparticles
Prior to MgO deposition, Pt NPs are first deposited by applying 25 ALD cycles of the MeCpPtMe3–O2 process (300 °C) on native SiO2, grown on top of a surface oxidized Si wafer.34 The size of the as-deposited NPs is Gauss-distributed, showing an average of ∼8 nm, and can be clearly observed with scanning electron microscopy (SEM) (Fig. S5†). After Pt NP deposition, samples with different amounts of MgO are prepared by varying the MgO ALD process and number of ALD cycles. Particularly, 1, 3, 5, 10, 15 and 30 cycles are applied for the Mg(EtCp)2–H2O process (150 °C, 200 °C), and 10, 30, 50, 100, 300 and 500 cycles for the Mg(TMHD)2–O3 process (250 °C).The intensity evolution of the Mg 1s XPS peak versus the number of Mg(EtCp)2–H2O ALD cycles is shown in Fig. 3a, both for depositions at 150 and 200 °C (extracted from Fig. S6a and S6d†). The Mg 1s peak area increases significantly upon applying the first ALD cycle(s) and then increases more mildly. Correspondingly, as shown in Fig. 3b (extracted from Fig. S6b and S6e†) and Fig. 3c (extracted from Fig. S6c and S6f†), the intensities of the Pt 4f and Si 2p peaks decrease gradually with an increasing number of MgO ALD cycles, and evolve to very low intensities. The consistent decline of both the Pt 4f and Si 2p peak intensities during Mg(EtCp)2–H2O ALD demonstrates that MgO grows on the Pt NPs and the SiO2 support indiscriminately, progressively covering the entire sample surface. It should be noted that the Pt 4f and Si 2p XPS signals are lost after ∼15 ALD cycles (Fig. 3b and c), while the Mg 1s intensity seems to saturate (Fig. 3a). This indicates that a MgO film of around 5 nm thick (i.e. the information depth of XPS) is deposited on top of the Pt/SiO2 sample, in accordance to the ellipsometry data (Fig. 2c and d) which shows significant MgO growth both on Pt and SiO2.
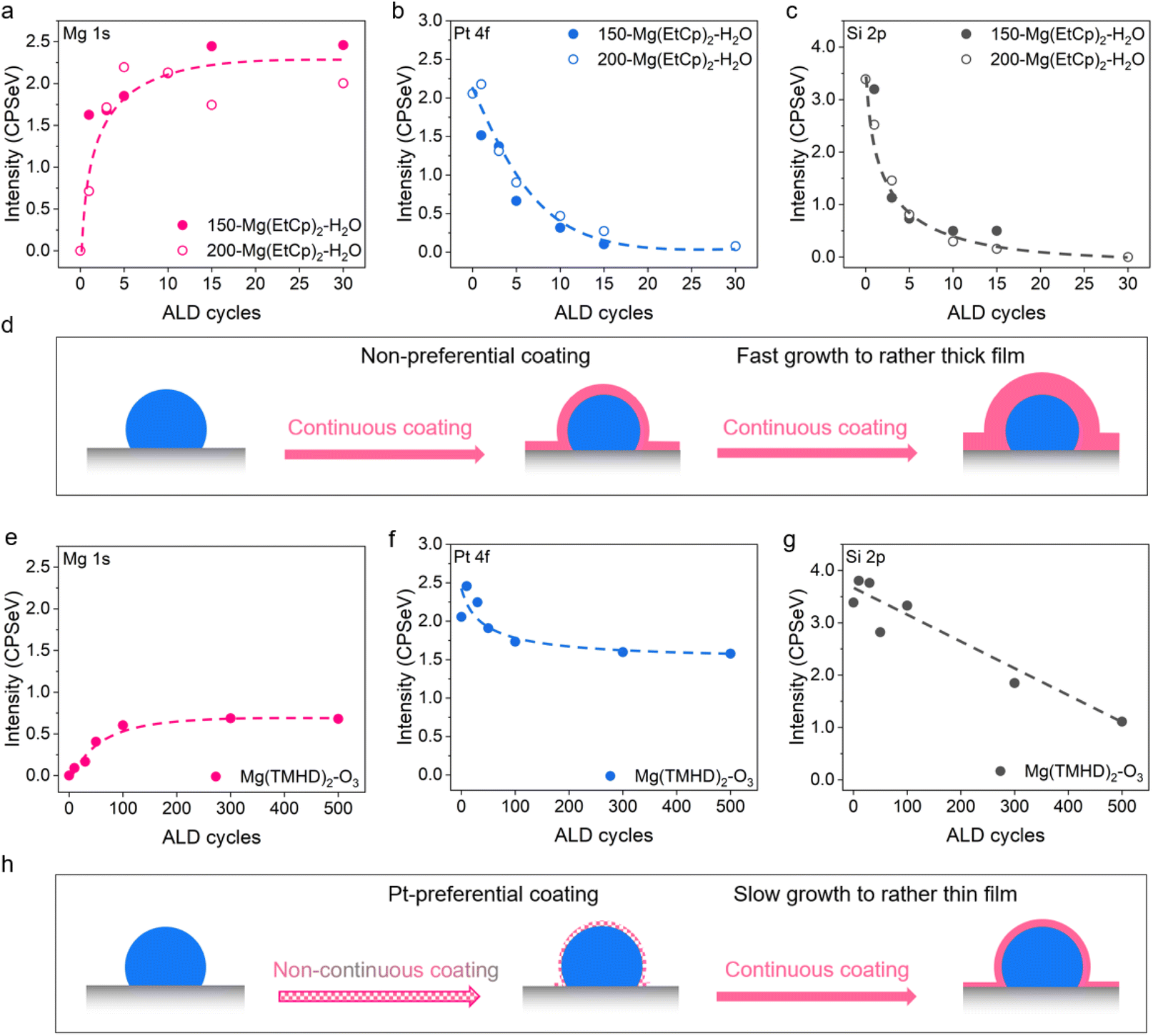 | ||
| Fig. 3 Integrated and background-subtracted peak area of the (a) Mg 1s, (b) Pt 4f and (c) Si 2p XPS signals as a function of the number of Mg(EtCp)2–H2O ALD cycles at 150 and 200 °C on Pt/SiO2. The dashed lines serve as guides to the eye. (d) Schematic overview of MgO thin film deposition with the Mg(EtCp)2–H2O ALD process on Pt/SiO2. (e–g) Integrated and background-subtracted peak area of the (e) Mg 1s, (f) Pt 4f and (g) Si 2p XPS signals as a function of the number of Mg(TMHD)2–O3 ALD cycles at 250 °C on Pt/SiO2. The dashed lines serve as guides to the eye. (h) Schematic overview of MgO thin film deposition with the Mg(TMHD)2–O3 ALD process on Pt/SiO2. | ||
The Mg atomic fraction is calculated by dividing the peak area of the Mg 1s XPS signal by the summed areas of the Mg 1s, Pt 4f and Si 2p peaks, after subtracting backgrounds and dividing by the relative sensitivity factors for each term (Fig. S6g and S6h†). For both the depositions at 150 and 200 °C, a steady increase with the number of cycles is observed up to a Mg atomic fraction of 70%, in line with the gradual, quasi-linear growth observed on the reference substrates (Fig. 2c and d). For higher loadings, the Mg atomic fraction progresses more slowly towards 100%, an effect that can be linked to the MgO film thickness becoming comparable to the XPS information depth. Similar Mg atomic fractions are obtained for the depositions performed at 150 versus 200 °C, despite the difference in GPC observed with ellipsometry on the planar reference substrates (Fig. 2c and d).
The surface morphology of the MgO-coated Pt/SiO2 samples is characterized as a function of the number of Mg(EtCp)2–H2O ALD cycles by SEM (Fig. S7 and S8†). Both at 150 and 200 °C, the initially clearly defined NP features become less observable and evolve into a rather rough surface with an increasing number of MgO ALD cycles.
Combining the XPS and SEM results, a schematic illustration of the MgO thin film growth with the Mg(EtCp)2–H2O ALD process on Pt/SiO2 is depicted in Fig. 3d: the Pt NPs and SiO2 support are coated simultaneously by MgO in the initial stages of the process, and continued substantial MgO growth is observed afterwards, eventually yielding a conformal and relatively thick film.
For the Mg(TMHD)2–O3 process, the intensity evolution of the Mg 1s XPS peak is shown as a function of the number of ALD cycles in Fig. 3e (integrated from Fig. S9a†). The Mg 1s intensity shows a clear and gradual increase from 0 to 100 cycles and levels off afterwards. The intensity is, however, much lower than what is observed for the Mg(EtCp)2–H2O process (Fig. 3a), in line with the significantly lower GPC for the Mg(TMHD)2–O3 process on the reference substrates (Fig. 2c–e). The saturation of the Mg 1s intensity resembles the growth curves on bare Pt and SiO2 (Fig. 2e) with a slowdown in growth after ∼40 ALD cycles. The Mg atomic fraction (Fig. S9d†) evolves slowly to ∼20% after 500 ALD cycles, confirming the low growth for the Mg(TMHD)2–O3 process.
Given the preferred MgO deposition on Pt (Fig. 2e), the intensity evolution of the Pt 4f peaks (Fig. 3f) shows a logic opposite trend to the one of Mg 1s (Fig. 3e). The intensity evolution of the Si 2p XPS signal, on the other hand, decreases more linearly throughout the process (Fig. 3g), which indicates a more gradual MgO deposition on SiO2. Based on these results, it can be inferred that more MgO is deposited on Pt in the initial 100 cycles compared to SiO2, after which SiO2 becomes the most favorable location of MgO deposition as Pt is already fully covered by MgO.
SEM images show that the NP features remain generally visible during the full duration of the Mg(TMHD)2–O3 ALD process (Fig. S10†). This can be clarified by the low deposition rate of the process. Based on the above results, the evolution in MgO thin film growth with the Mg(TMHD)2–O3 process is schematized in Fig. 3h. While MgO deposition takes place on both SiO2 and Pt initially (<100 ALD cycles), Pt is more preferentially coated. Nevertheless, not all Pt surface atoms are covered at this stage (vide infra), leading to a non-continuous coating. After 100 cycles, MgO deposition slows down and the growth is mostly manifested on SiO2, ultimately forming a thin continuous film with thickness lower than 1.5 nm.
The combined ellipsometry-XPS characterization approach provides insights in the MgO thin film formation on Pt/SiO2 substrates. However, in view of catalytic applications, the MgO coverage on the Pt surfaces should be in balance with the availability of the active sites. Particularly, low (high) MgO coverages will lead to a high (low) number of NP surface sites available for catalytical reactions. Characterizing the MgO thickness and selectivity of deposition is therefore not sufficient to assess the ability of the catalyst surface to accommodate reaction. Therefore, low energy ion scattering (LEIS) is applied to detect which fraction of the Pt NP surface is either MgO-covered or MgO-free.45 LEIS is an ion scattering technique which is selective to the atoms in the outermost surface layer, which would also be accessible to molecules during a catalytic process.
For each of the three studied ALD processes, 2–3 samples with low MgO loading are selected based on the XPS characterization. LEIS measurements are first performed using a 3 keV He+ beam to probe the composition of the top surface layer. Prior to analysis, each sample is exposed to atomic oxygen to remove adventitious carbon, which results in a fully oxidized surface. The bare Pt/SiO2 surface therefore shows clear signals of Pt (at ∼2750 eV), Si (at ∼1750 eV) and O (at ∼1130 eV) (Fig. S11†). In addition to the Pt signal from the surface atoms, a broad shoulder at lower energy is visible that corresponds to signal from sub-surface Pt atoms. It originates from neutralized He ions that penetrate sub-surface, undergo backscattering towards the surface and reionize at the surface.45 For MgO-coated samples, an additional prominent peak corresponding to Mg (at ∼1580 eV) is detected. Some samples present signals from F and P contamination corresponding to a few percent. Whether these originate from the synthesis or the packaging for transport is unknown.
More importantly, the presence of an MgO overcoat changes the Pt signature in the spectra with respect to the bare Pt/SiO2 reference (Fig. 4a–c). For the Mg(EtCp)2–H2O process, the intensity of the Pt surface peak (at ∼2750 eV) decreases after 1 MgO ALD cycle, and is completely absent after 3 cycles of the 150 °C process (Fig. 4a) or 5 cycles of the 200 °C process (Fig. 4b). The latter means that no Pt atoms are present on the surface, hence a continuous MgO coating is formed on top of the Pt NPs. For a fully closed MgO layer, its thickness can be extracted from the position of the edge of the sub-surface Pt signal (dashed lines in lower panels of Fig. 4a–c) with respect to the position of the Pt surface peak (at ∼2750 eV) on the energy scale.45,46 MgO layer thicknesses of ∼0.4 nm and ∼0.5 nm are extracted after three cycles of the 150 °C process (Fig. 4a), respectively five cycles of the 200 °C process (Fig. 4b), based on the energy differences. For the Mg(TMHD)2–O3 process (Fig. 4c), a reduced Pt surface peak is visible after 30 ALD cycles and some intensity at the characteristic energy (at ∼2750 eV) is still recorded after 100 cycles. After 500 cycles, a fully closed layer of MgO is formed, with a thickness of ∼0.4 nm.
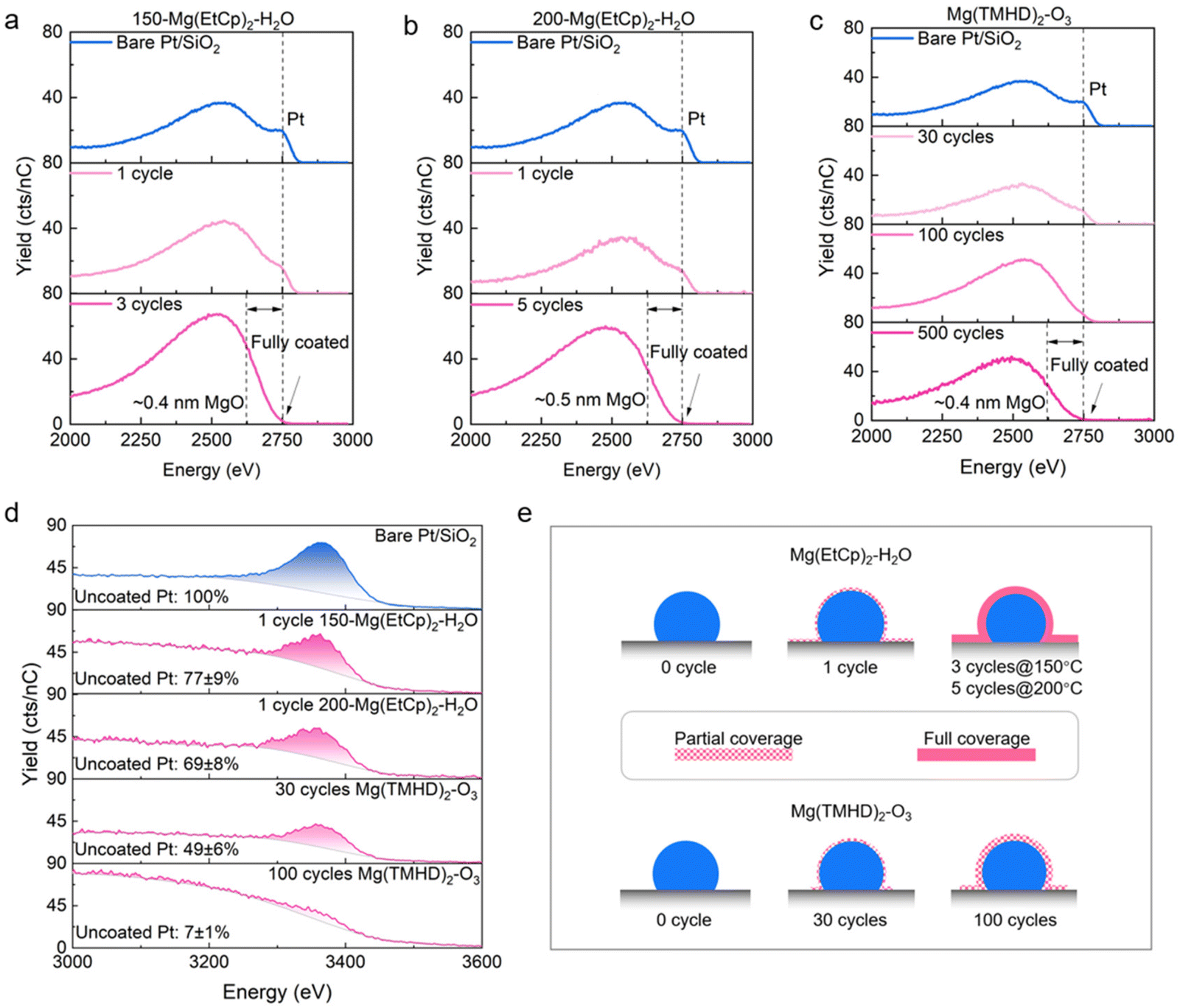 | ||
| Fig. 4 (a–c) 3 keV He+ LEIS spectra of Pt/SiO2 without and with MgO overcoat, deposited with the Mg(EtCp)2–H2O ALD process at (a) 150 °C and (b) 200 °C, and (c) with the Mg(TMHD)2–O3 process. Dashed lines indicate the Pt surface peak (∼2750 eV) and the edge of the Pt sub-surface peak (only in the absence of a Pt surface peak), respectively. (d) 5 keV Ne+ LEIS spectra for bare Pt/SiO2 and for selected samples with MgO overcoat. (e) Schematic representation of the partial MgO coverage on the Pt NPs, as deduced from the LEIS spectra in (d). | ||
To quantify the relative amount of Pt surface atoms for the partially covered NPs, LEIS spectra are recorded with a 5 keV Ne+ beam which yields a higher peak-to-shoulder ratio (Fig. 4d). This allows to integrate the surface peak and to compare its area with the one obtained for the bare Pt/SiO2 sample. After 1 ALD cycle of both Mg(EtCp)2–H2O processes, the fraction of uncoated Pt surface atoms amounts to ∼70–80%, while this is ∼50% after 30 cycles of the Mg(TMHD)2–O3 process. Nearly full coverage is achieved after 100 Mg(TMHD)2–O3 cycles, where only ∼7% of the Pt surface atoms remains uncovered. On the other hand, the XPS results show a roughly 3 to 5 times higher MgO loading after 1 Mg(EtCp)2–H2O cycle than after 30 Mg(TMHD)2–O3 cycles (Fig. 3a and e). Even after 100 Mg(TMHD)2–O3 cycles, the MgO loading still remains relatively lower, confirming the initial selectivity of the latter process towards the Pt NP surface.
In summary, initially, both precursor chemistries partially cover the Pt NPs (Fig. 4e). The Mg(TMHD)2–O3 process deposits MgO preferentially on the Pt NPs, yet, in spite of this, the very low GPC allows for a partial coverage that very gradually transitions to full coverage with increase in ALD cycle number. In contrast, the high GPC of the Mg(EtCp)2–H2O process results in a fast transition to full coverage. These results confirm that ALD, and in particular the Mg(TMHD)2–O3 process, holds a means to control the MgO coverage on the Pt NP surface.
Sintering behavior of MgO-coated Pt nanoparticles
The sintering behavior of bare Pt NPs anchored on a SiO2 support has been studied previously during ramp anneals in O2 environment.37,38 A thermal activation is required to induce sintering and an onset temperature can be defined above which the average size of the Pt NPs starts to increase with annealing temperature. Because multiple smaller NPs merge into a larger one, the areal density of NPs on the support, i.e. the number of NPs per μm2, decreases with temperature, and as a result the average center-to-center distance between the Pt NPs increases.To quantify the delay in Pt NP sintering for the different types of MgO coatings synthesized in this work, from thin overlayers partially or fully covering the NP surface to nm-thick overlayers, and to explore the delicate balance between (i) physically blocking the Pt surface to prevent sintering and (ii) keeping Pt surface atoms free for reaction. To this end, the sintering behavior is investigated for the uncoated Pt/SiO2 sample and the three series of samples with MgO overcoats generated with 1, 3, 5, 10 and 15 cycles of the Mg(EtCp)2–H2O processes (150 and 200 °C), and 10, 30, 50, 100, 300 and 500 cycles of the Mg(TMHD)2–O3 process (250 °C), respectively.
In a first experiment, all samples are annealed in 20% O2 in He from room temperature to 800 °C at a rate of 12 °C min−1 (SEM images in Fig. S12†). GISAXS patterns are recorded during the ramp anneal, offering a means to monitor the growth of the Pt NPs in real time, with a 30 s time resolution, corresponding to a temperature resolution of ∼6 °C. The main feature in the GISAXS patterns originates from scattering caused by the Pt NPs, and the position of its maximum along the qy direction in reciprocal space, qy,max, is inversely proportional to the average center-to-center distance between the Pt NPs (D = 2π/qy,max).36,40,41 Therefore, a shift of the main scattering feature to lower qy values points at an increase in NP spacing and is indicative of NP sintering.36,38
The method used to extract the qy,max value of the 2D GISAXS patterns is detailed in Fig. S12,† and the results are depicted in Fig. 5a–c (with representative 2D patterns included in Fig. S13–S15†). The grey curve in these figures corresponds to the bare Pt/SiO2 NPs and shows a stable qy,max value up to ∼550 °C, followed by a gradual decrease and then a steep decline of qy,max in the range 620–700 °C, indicative of severe NP sintering at these temperatures. The center-to-center distance evolves from ∼10 nm to more than 30 nm (Fig. S16†). The onset temperature for NP sintering, ∼680 °C, is extracted by determining the minimum in the derivative of this curve (Fig. S17†).37,39
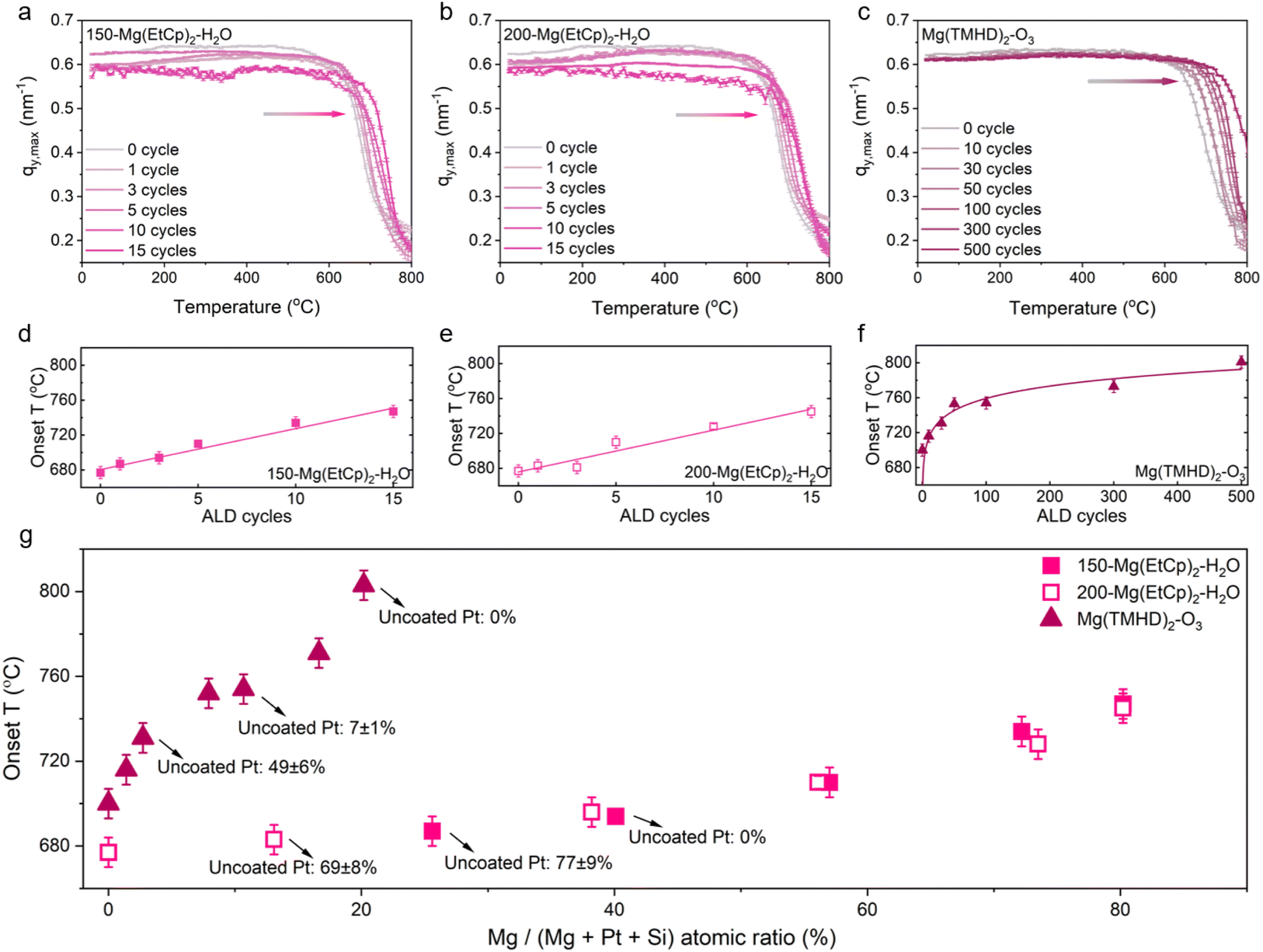 | ||
| Fig. 5 In situ GISAXS data recorded during a ramp anneal (12 °C min−1) in O2 (20% O2 in He), showing the evolution in the qy,max position with temperature for three series of MgO-coated Pt/SiO2, synthesized with the Mg(EtCp)2–H2O process at (a) 150 °C, (b) 200 °C and (c) the Mg(TMHD)2–O3 process at 250 °C. (d–f) Sintering onset temperature as a function of the number of ALD cycles, extracted from the data in (a–c). The error bar (± 6 °C) indicates the temperature resolution obtained in the measurement. (g) Sintering onset temperatures of the three sample series plotted against the Mg atomic fraction, obtained from XPS. | ||
For the three investigated ALD processes, the steep decline in qy,max occurs at progressively higher temperatures as the number of MgO ALD cycles is increased. The extracted onset temperatures are shown in Fig. 5d–f as a function of the number of ALD cycles. For the Mg(EtCp)2–H2O processes, the onset temperature increases gradually with the number of cycles, reaching ∼745 °C after 15 ALD cycles (Fig. 5d and e). For the Mg(TMHD)2–O3 process, the onset temperature increases to ∼755 °C after 50 ALD cycles, followed by a relatively slower increase with the number of ALD cycles (Fig. 5f). The largest delay in sintering is observed for the Pt/SiO2 sample covered with 500 cycles of the Mg(TMHD)2–O3 process, marked by an onset temperature of ∼800 °C.
In Fig. 5g, the onset temperatures for Pt NP sintering are plotted against the Mg atomic fraction, as derived from XPS. This allows for a comparison of the effect of the different types of MgO overlayers on the sintering behavior. Not surprisingly, the data points for the Mg(EtCp)2–H2O processes at 150 and 200 °C follow exactly the same trend, pointing to a negligible impact of the deposition temperature for this process. Clearly, the Mg(TMHD)2–O3 process enables a higher onset temperature with a lower loading of MgO. For instance, an onset temperature of ∼750 °C is achieved for a Mg atomic fraction of 10%, while the Mg(EtCp)2–H2O processes require a loading corresponding to a Mg atomic fraction of 80% to achieve the same sintering delay. The fraction of uncoated Pt surface atoms is also indicated in Fig. 5g, for those samples that are analyzed with LEIS. From Fig. 5g it follows that only the Mg(TMHD)2–O3 process enables a delay in sintering while keeping a fraction of the Pt surface atoms available for reaction. The two characteristics of the Mg(TMHD)2–O3 ALD process on Pt/SiO2 systems, i.e. (i) the initial selectivity towards Pt over SiO2, and (ii) the very low GPC, turn out to be beneficial to increase the thermal stability of the Pt NPs without fully covering them.
In our previous work,37 we argued that NP sintering during annealing in O2 presents two regimes with temperature. When the onset temperature for sintering is surpassed, thermal decomposition of PtO2 formed on the NP surface leads to unstable and mobile PtOx species that cause NP growth via Ostwald ripening. At higher temperatures, PtOx desorbs from the surface inducing more severe sintering, while sintering via particle migration cannot be excluded. The non-continuous MgO coating applied in this work is believed to physically hinder the migration and desorption of mobile PtOx species and/or small particles on the surface, explaining the reduced sintering.
In a second experiment, Pt NPs partially covered with ultrathin MgO overlayers are exposed to five cycles of propane dehydrogenation reaction and O2 regeneration at 600 °C, namely 5 × [C3H8/He/O2/He]. During propane dehydrogenation reaction (C3H8 = C3H6 + H2), undesired carbon-rich species poison the Pt NP surface and inhibit further reaction.47 Therefore, industrially, O2 regeneration pulses are applied frequently to burn these carbon deposits (termed ‘coke’) and rejuvenate the catalyst. However, during such reaction–regeneration cycling, NP sintering takes place, leading to irreversible catalyst deactivation. In this perspective, MgO overlayers on Pt could form a viable strategy to prevent rapid NP sintering in this industrial reaction, prolonging the process lifespan.
To study the sintering of MgO-overcoated Pt NPs during reaction–regeneration cycling, the samples with the lowest MgO loading and largest fraction of uncoated Pt surface atoms are selected for this experiment, i.e. Pt/SiO2 samples coated with 1 Mg(EtCp)2–H2O ALD cycle at 200 °C and with 10 Mg(TMHD)2–O3 ALD cycles. Bare Pt/SiO2 is included as a reference. The samples are first heated from room temperature to 600 °C at 12 °C min−1 in 20% O2 in He. Remark that the reaction temperature of 600 °C lies below the onset temperature of significant NP sintering (Fig. 5). Then, the gas environment is periodically alternated between O2 (i.e. 20% O2 in He) and propane (i.e. 5% propane in He). In between each gas exposure of 240 s, the heated reactor is purged for 60 s with pure He in order to prevent direct mixing of O2 and propane. This 10 min reaction–regeneration cycle is repeated 5 times, during which in situ GISAXS data are recorded (Fig. S18†). The extracted qy,max values as a function of reaction time are shown in Fig. 6. During the initial O2 exposure, the qy,max values remain relatively constant for the three investigated samples and no significant sintering occurs. When propane is introduced, immediate NP sintering is observed only for the bare Pt/SiO2 NPs and for the NPs coated with 1 cycle of Mg(EtCp)2–H2O ALD, as seen from the sudden drop in qy,max at the start of the propane exposure. This is followed by more gradual sintering throughout the further propane exposure for all three samples. When O2 is re-injected in the second reaction cycle, the uncoated Pt NPs again undergo abrupt sintering, while the MgO-coated NPs remain stable. In the subsequent reaction cycles, qy,max decreases at a particularly slow rate for all three samples. Throughout the experiment, the center-to-center NP distance increases from ∼10 nm to ∼18 nm, ∼15.5 nm and ∼14 nm for the uncoated Pt NPs, the MgO-coated sample prepared with 1 Mg(EtCp)2–H2O ALD cycle and the MgO-coated sample prepared with 10 Mg(TMHD)2–O3 ALD cycles, respectively (Fig. S19†).
 | ||
| Fig. 6 In situ GISAXS data recorded during five cycles of propane dehydrogenation reaction (5% C3H8 in He) and regeneration in O2 (20% O2 in He) at 600 °C, showing the evolution in the qy,max position with time for three samples: bare Pt/SiO2 and two MgO-coated Pt/SiO2, one synthesized with 1 Mg(EtCp)2–H2O ALD cycle at 200 °C and one synthesized with 10 Mg(TMHD)2–O3 ALD cycles at 250 °C. | ||
The sintering of bare Pt NPs during the first propane exposure is most likely caused by the exothermic reaction of propane with residual oxygen, e.g. on the Pt NP surface. This might lead to heat generation during the (partial) oxidation of propane, hence triggering significant NP sintering. Upon further propane exposure, NP sintering is continued presumably as a result of increased mobility of CxHy-adsorbed Pt species. In the following O2 step, coke deposits on Pt burn into CO2 and H2O, again producing heat due to the exothermic nature of the reaction which might trigger NP sintering.
By partially covering the Pt surface with MgO via ALD, sintering during the propane step can be reduced to some extent, while the abrupt sintering upon exposure to O2 is prevented. Potentially the MgO decorated Pt surface is less prone to carbon depositions during propane dehydrogenation, resulting in less carbon oxidation and hence less sintering during the subsequent O2 step. Alternatively, the partial MgO overcoat may form a protection barrier against sintering upon exposure to O2.
Conclusion
In this work, two ALD precursor chemistries for the deposition of MgO on Pt NPs anchored on a SiO2 support are investigated to understand the opportunities for delaying NP sintering while keeping Pt surface atoms available for reaction. The cycle-by-cycle deposition nature of ALD allows to achieve overcoats, from (sub)monolayers to thin films of a few nm's, as confirmed by a combination of ellipsometry, XPS and LEIS characterizations.The Mg(TMHD)2–O3 ALD process at 250 °C initially deposits MgO preferentially on the Pt NPs, with less growth on the SiO2 support. Moreover, the process is characterized by a very low GPC and the growth slows further down when the Pt or SiO2 surface turn into a MgO-terminated surface. This leads to low MgO loadings and film thicknesses in the Å-range, even for hundreds of ALD cycles. As a result, a tunable partial coverage of MgO on the Pt surface can be achieved with this process, in spite of the preferential growth on Pt.
In contrast, the Mg(EtCp)2–H2O process is characterized by a GPC exceeding 1 Å per cycle, at both investigated deposition temperatures of 150 and 200 °C, and no pronounced surface selectivity is observed for growth on Pt versus SiO2. This results in higher MgO loadings, and a fast transition from an initial partial coverage (for 1 ALD cycle) to a full coverage of the Pt surface (for 3–5 ALD cycles).
The sintering behavior of the MgO-coated Pt NPs is studied via in situ GISAXS. Higher onset temperatures for sintering point to a successful strategy to stabilize the Pt NPs and delay sintering. With increasing number of ALD cycles, both ALD processes enable an increase in the onset temperature for sintering. However, in case of the Mg(EtCp)2–H2O process, rather thick MgO films that fully cover the Pt surface are required, while the Mg(TMHD)2–O3 ALD process achieves larger delays in NP sintering for MgO loadings that only partially cover the Pt surface.
In summary, different ALD chemistries allow for a different structural control over the deposited MgO overcoat, and hence also the fraction of uncovered Pt surface atoms available for reaction. The Mg(TMHD)2–H2O process can yield sub-monolayer coverages of MgO on the Pt NPs, which can significantly delay NP sintering whilst still exposing a major fraction of NP surface atoms for reaction. This avoids the need for post-annealing to create porosity in the overcoat, guaranteeing the structural integrity of the as-deposited MgO.
Author contributions
C. D. and J. D. conceived the project in collaboration with H. P. and V. V. G.; M. F. and J. D. supervised the research; Z. Z. prepared the samples, performed the experiments and analyzed the data; Z. Z., M. F., E. S., N. P., M. R., C. D. and J. D. performed the in situ GISAXS measurements; J. L. and M. M. M. performed the XPS measurements; P. B. performed the LEIS measurements; Z. Z. prepared the manuscript (first draft); M. F. and J. D. revised the manuscript with contribution from all co-authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the special research fund BOF of Ghent University (BOF-GOA 01G01019, BOF-GOA 01G02124, Starting Grant 01N14319) and the Research Foundation – Flanders (FWO projects 3G021220, G093823N, G0A5923N). N. P. acknowledges funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no. 765378. M. F. acknowledges the FWO for a senior postdoctoral research fellowship (1280621N). E. S. acknowledges the Spanish MICINN/FEDER project PID2021-124572OB-C33. GISAXS experiments were performed at the NCD-SWEET beamline at ALBA Synchrotron with the collaboration of ALBA staff. We acknowledge the European Synchrotron Radiation Facility (ESRF) for provision of synchrotron radiation facilities and we would like to thank the DUBBLE staff for assistance and support in using beamline BM26B. The authors acknowledge Olivier Janssens for his technical support.References
- A. Motagamwala, R. Almallahi, J. Wortman, V. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- S. Mitchell, R. Qin, N. Zheng and J. Pérez-Ramírez, Nat. Nanotechnol., 2021, 16, 129–139 CrossRef CAS PubMed.
- S. Yu, A. Wilson, J. Heo and P. Jain, Nano Lett., 2018, 18, 2189–2194 CrossRef CAS PubMed.
- G. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. Prince, V. Matolın, L. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- L. Shi, G. Deng, W. Li, S. Miao, Q. Wang, W. Zhang and A. Lu, Angew. Chem., Int. Ed., 2015, 54, 13994–13998 CrossRef CAS PubMed.
- Z. Zhang, L. Deng, Z. Zhao, Y. Zhao, J. Yang, J. Jiang, G. Huang and Y. Mei, J. Mater. Chem. A, 2020, 8, 3499–3508 RSC.
- Y. Nagai, K. Dohmae, Y. Ikeda, N. Takagi, T. Tanebe, N. Hara, G. Guilera, S. Pascarelli, M. Newton, O. Kuno, H. Jiang, H. Shinjoh and S. Matsumoto, Angew. Chem., Int. Ed., 2008, 47, 9303–9306 CrossRef CAS PubMed.
- G. Prieto, J. Zečević, H. Friedrich, K. de Jong and K. de Jongh, Nat. Mater., 2013, 12, 34–39 CrossRef CAS PubMed.
- A. Martín, S. Mitchell, C. Mondelli, S. Jaydev and J. Perez-Ramirez, Nat. Catal., 2022, 5, 854–866 CrossRef.
- Y. Dai, P. Lu, Z. Cao, C. Campbell and Y. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- T. Hansen, A. DelaRiva, S. Challa and A. Datya, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- L. Wang, L. Wang, X. Meng and F. Xiao, Adv. Mater., 2019, 31, 1901905 CrossRef CAS PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2021, 121, 834–881 CrossRef CAS PubMed.
- J. Sun, D. Ma, H. Zhang, X. Liu, X. Han, X. Bao, G. Weinberg, N. Pfander and D. Su, J. Am. Chem. Soc., 2006, 128, 15756–15764 CrossRef CAS PubMed.
- S. Zhang, S. Muratsugu, N. Ishiguro and M. Tada, ACS Catal., 2013, 3, 1855–1864 CrossRef CAS.
- P. Lu, C. Campbell and Y. Xia, Nano Lett., 2013, 13, 4957–4962 CrossRef CAS PubMed.
- X. Liu, M. Liu, Y. Luo, C. Mou, S. Lin, H. Cheng, J. Chen, J. Lee and T. Lin, J. Am. Chem. Soc., 2012, 134, 10251–10258 CrossRef CAS PubMed.
- C. Han, P. Majumdar, E. Marinero, A. Tapia, R. Zanella, J. Greeley and V. Ortalan, Nano Lett., 2015, 15, 8141–8147 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. DeLaRiva, E. Peterson, H. Pham, S. Challa, G. Qi, S. Oh, M. Wiebenga, X. Hernandez, Y. Wang and A. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- T. Onn, M. Monai, S. Dai, E. Fonda, T. Montini, X. Pan, G. Graham, P. Fornasiero and R. Gorte, J. Am. Chem. Soc., 2018, 140, 4841–4848 CrossRef CAS PubMed.
- L. Shang, T. Bian, B. Zhang, D. Zhang, L. Wu, C. Tung, Y. Yin and T. Zhang, Angew. Chem., Int. Ed., 2014, 53, 250–254 CrossRef CAS PubMed.
- C. Lin, J. Jang, L. Zhang, E. Stach and R. Gorte, ACS Catal., 2018, 8, 7679–7687 CrossRef CAS.
- N. Srinath, A. Longo, H. Poelman, R. Ramachandran, J. Feng, J. Dendooven, M. Reyniers and V. Galvita, ACS Catal., 2021, 11, 11320–11335 CrossRef CAS.
- A. Aitbekova, C. Zhou, M. Stone, J. Pacheco, A. Yang, A. Hoffman, E. Goodman, P. Huber, J. Stebbins, K. Bustillo, P. Ercius, J. Ciston, S. Bare, P. Plessow and M. Cargnello, Nat. Mater., 2022, 21, 1290–1297 CrossRef CAS PubMed.
- W. McNeary, S. Tacey, G. Lahti, D. Conklin, K. Unocic, E. Tan, E. Wegener, T. Erden, S. Moulton, C. Gump, J. Burger, M. Griffin, C. Rarberow, M. Watson, L. Tuxworth, K. Allsburg, A. Dameron, K. Buechler and D. Vardon, ACS Catal., 2021, 11, 8538–8549 CrossRef CAS.
- H. Yan, K. He, I. Samek, D. Jing, M. Nanda, P. Stair and J. Notestein, Science, 2021, 371, 1257–1260 CrossRef CAS PubMed.
- E. Sarnello, Z. Lu, S. Seifert, R. Winans and T. Li, ACS Catal., 2021, 11, 2605–2619 CrossRef CAS.
- V. Pramhaas, M. Roiaz, N. Bosio, M. Corva, C. Rameshan, E. Vesselli, H. Gronbeck and G. Rupprechter, ACS Catal., 2021, 11, 208–214 CrossRef CAS PubMed.
- Z. Li, R. Li, H. Jing, J. Xiao, H. Xie, F. Hong, N. Ta, X. Zhang, J. Zhu and C. Li, Nat. Catal., 2023, 6, 80–88 CrossRef CAS.
- C. Detavernier, J. Dendooven, S. Sree, K. Ludwig and J. Martens, Chem. Soc. Rev., 2011, 40, 5242–5253 RSC.
- Z. Zhang, Y. Zhao, Z. Zhao, G. Huang and Y. Mei, Chem. Mater., 2020, 32, 9056–9077 CrossRef CAS.
- J. Lu, B. Fu, M. Kung, G. Xiao, J. Elam, H. Kung and P. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- J. Cai, J. Zhang, K. Cao, M. Gong, Y. Lang, X. Liu, S. Chu, B. Shan and R. Chen, ACS Appl. Nano Mater., 2018, 1, 522–530 CrossRef CAS.
- J. Dendooven, R. Ramachandran, E. Solano, M. Kurttepeli, L. Geerts, G. Heremans, J. Ronge, M. Minjauw, T. Dobbelaeere, K. Casier, J. Martens, A. Vantomme, S. Bals, G. Portale, A. Coati and C. Detavernier, Nat. Commun., 2017, 8, 1074 CrossRef PubMed.
- N. Fairly, V. Fernandez, M. R. Plouet, C. G. Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci. Adv., 2021, 5, 100112 CrossRef.
- J. F. Ziegler, M. D. Ziegler and J. P. Biersack, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1818–1823 CrossRef CAS.
- E. Solano, J. Dendooven, R. K. Ramachandran, K. Kerckhove, T. Dobbelaere, D. Merino and C. Detavernier, Nanoscale, 2017, 9, 13159–13170 RSC.
- E. Solano, J. Dendooven, D. Deduytsche, N. Poonkottil, J.-Y. Feng, M. B. J. Roeffaers, C. Detavernier and M. Filez, Small, 2023, 19, 2205217 CrossRef CAS PubMed.
- E. Solano, J. Dendooven, J. Feng, P. Bruner, M. Minjauw, R. Ramachandran, M. Daele, D. Merino and C. Detavernier, Nanoscale, 2020, 12, 11684–11693 RSC.
- J. Dendooven, M. Daele, E. Solano, R. K. Ramachandran, M. Minjauw, A. Resta, A. Vlad, Y. Garreau, A. Coati, G. Portale and C. Detavernier, Phys. Chem. Chem. Phys., 2020, 22, 24917–24933 RSC.
- Y. Yoneda, Phys. Rev., 1963, 131, 2010–2013 CrossRef.
- B. Burton, D. Goldstein and S. George, J. Phys. Chem. C, 2009, 113, 1939–1946 CrossRef CAS.
- M. Putkonen, L. Johansson, E. Rauhala and L. Niinisto, J. Mater. Chem. C, 1999, 9, 2449–2452 RSC.
- J. Acharya, R. Goul, D. Romine, R. Sakidja and J. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 30368–30375 CrossRef CAS PubMed.
- C. Cushman, P. Brüner, J. Zakel, G. Major, B. Lunt, T. Grehl, N. Smith and M. Linford, Anal. Methods, 2016, 8, 3419–3439 RSC.
- H. Brongersma, T. Grehl, R. Schofeild, P. Smith and J. Veen, Platinum Metals Rev., 2010, 54, 81–87 CrossRef CAS.
- J. Sattler, J. Martinez, E. Jimenez and B. Weckhuysen, Chem. Rev., 2014, 114, 10613–10653 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XPS spectra for MgO coatings on sputtered Pt film, native SiO2 substrate, and Pt/SiO2 NP systems, SEM images of bare and MgO coated Pt NPs, LEIS spectra, schematic of the GISAXS analysis strategy, selection of 2D GISAXS patterns recorded during O2 anneals, evolution in center-to-center NP distance with temperature, derivative of the qy,maxversus temperature curves, selection of 2D GISAXS patterns recorded during propane dehydrogenation-oxidation cycling and the evolution of interparticle distance during five cycles of propane dehydrogenation-oxidation cycling. See DOI: https://doi.org/10.1039/d3nr05884k |
| This journal is © The Royal Society of Chemistry 2024 |