
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in biomaterial designs for assisting CAR-T cell therapy towards potential solid tumor treatment
Yuting
Lin
a,
Ying
Chen
a,
Zheng
Luo
ab and
Yun-Long
Wu
 *a
*a
aFujian Provincial Key Laboratory of Innovative Drug Target Research and State Key Laboratory of Cellular Stress Biology, School of Pharmaceutical Sciences, Xiamen University, Xiamen 361102, China. E-mail: wuyl@xmu.edu.cn
bInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), Singapore 138634, Singapore
First published on 11th January 2024
Abstract
Chimeric antigen receptor T (CAR-T) cells have shown promising outcomes in the treatment of hematologic malignancies. However, CAR-T cell therapy in solid tumor treatment has been significantly hindered, due to the complex manufacturing process, difficulties in proliferation and infiltration, lack of precision, or poor visualization ability. Fortunately, recent reports have shown that functional biomaterial designs such as nanoparticles, polymers, hydrogels, or implantable scaffolds might have potential to address the above challenges. In this review, we aim to summarize the recent advances in the designs of functional biomaterials for assisting CAR-T cell therapy for potential solid tumor treatments. Firstly, by enabling efficient CAR gene delivery in vivo and in vitro, functional biomaterials can streamline the difficult process of CAR-T cell therapy manufacturing. Secondly, they might also serve as carriers for drugs and bioactive molecules, promoting the proliferation and infiltration of CAR-T cells. Furthermore, a number of functional biomaterial designs with immunomodulatory properties might modulate the tumor microenvironment, which could provide a platform for combination therapies or improve the efficacy of CAR-T cell therapy through synergistic therapeutic effects. Last but not least, the current challenges with biomaterials-based CAR-T therapies will also be discussed, which might be helpful for the future design of CAR-T therapy in solid tumor treatment.
 Yuting Lin | Yuting Lin is a graduate student at the School of Pharmaceutical Sciences, Xiamen University, Xiamen (China). She received her B. E. degree in pharmaceutical engineering from Fuzhou University (China) in 2023. Currently, she is conducting research within the pharmacy related field of drug/gene vectors. |
 Ying Chen | Ying Chen is a graduate student at the School of Pharmaceutical Sciences, Xiamen University, Xiamen (China). She received her B.S. degree from the School of Pharmaceutical Sciences, Xiamen University, Xiamen (China) in 2021. Currently, her main research focus is on the application of composite enzyme nanogels. |
 Zheng Luo | Zheng Luo is a Ph.D. student at the School of Pharmaceutical Sciences, Xiamen University. He received his master's degree from Xiamen University (China) in 2020. Currently, his main research interests are in the design of biomaterials and their biological applications. |
1. Introduction
In recent decades, immunotherapy, which relies on the body's immune system to target and destroy cancer cells by activating the immune response, has been successful in increasing cure rates and reducing side effects.1 As an important approach of immunotherapy, chimeric antigen receptor T (CAR-T) cell therapy involves collecting, modifying, and re-injecting a patient's T-cells to enhance the immune system's ability to attack cancer cells. Although CAR-T cell therapy has achieved remarkable efficacy in the treatment of hematological tumors,2–5 there are still limitations for solid tumor treatments, such as high cost, uncontrolled safety, lack of precision, and poor infiltration or visualization ability.6 In particular, the complex tumor microenvironment (TME) in solid tumors affects the proliferation and infiltration of CAR-T cells.7 Fortunately, recent reports have shown that biomaterial vectors are widely used in CAR-T therapy to address these challenges. They can enhance the immunomodulatory effects of CAR-T therapy, deliver targeted treatments, and provide early warning of toxic side effects. For CAR gene delivery vectors, non-viral gene delivery vectors such as liposomes and polymers are popular biomaterial designs and have low toxicity or immunogenicity.8 In the case of overcoming the complex TME, biomaterial designs can be loaded with cytokines to induce CAR-T cell proliferation9 and biomaterial scaffolds loaded with cytokines can provide a three-dimensional microenvironment for CAR-T cell proliferation and survival.10 Furthermore, functional biomaterial designs can promote CAR-T cell infiltration by modulating the TME, and provide a platform for CAR-T cell therapy in combination with other therapies such as radiotherapy, chemotherapy, photothermal therapy (PTT), immune checkpoint inhibitor therapy and sonodynamic therapy (SDT).11–14 Last but not least, biomaterials with environmentally responsive design might also improve the precision of delivering CAR-T cells and reduce toxic side effects.15 In short, this review aims to summarize the current challenges of CAR-T cell therapy in solid tumors, with focus on the role of functional biomaterials in addressing these issues, as well as possible strategies to promote the solid tumor treatment efficacy of CAR-T cell therapy (Fig. 1). | ||
| Fig. 1 The application of biomaterial designs (in terms of liposomes, exosomes, nanoparticles, hydrogel, cationic polymers, or implantable scaffold) in assisting CAR-T cell therapy towards solid tumor treatment. | ||
2. Challenges of CAR-T cell therapy in solid tumor treatment
So far, the FDA has approved six CAR-T products, based on the outstanding success of CAR-T cell therapies in patients with hematologic cancers.16–18 However, CAR-T cell therapy still faces several limitations or challenges in solid tumor treatments (Fig. 2). Firstly, CAR-T cell therapy involves four stages: T cell isolation and enrichment, activation, genetic engineering and expansion.19 The complex process and the individualization for patients usually lead to high cost,20 limiting the advancement and accessibility of the product towards clinical trials. Currently, lentiviral vectors (LVs) and retroviral vectors (RVs) are frequently utilized in CAR-T products.17,21–24 But these viral vectors are hindered by immunogenicity, cytotoxic and low capacity issues.25 Furthermore, T cell culture26,27 and virus purification25,28,29 are time-consuming, which contributes to the high cost of CAR-T treatments. Secondly, the complex tumor microenvironment (TME) in a solid tumor provides physical and biological impediments, in cases of abnormal vascular distribution,30 hypoxia,7 or acidic microenvironment31 within tumor tissues.32 It also limits the proliferation and infiltration of CAR-T cells, resulting in poor outcomes in the treatment of solid tumors.9,33 The TME also contains a variety of cells with immunosuppressive capacity, such as regulatory T cells and tumor-associated macrophage (TAM). Their overexpression of cytokines with immunosuppressive capacity such as transforming growth factor β (TGFβ), interleukin-10 (IL-10), interleukin-8 (IL-8)7,34 creates a molecular biology barrier. These factors constitute an immunosuppressive environment that can significantly reduce the efficacy of CAR-T cells.9 Thirdly, CAR-T cell therapy for cancer has concerns on undesired off-target effects.35 Another problem posed by the lack of precision in CAR-T therapy is the cytokine storm. CAR-T cells are transfused back into the patient's body, the body will release a large amount of cytokine, resulting in toxic side effects such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS).36–38 Studies have shown that the critical factors, determining the incidence and severity of CRS or ICANS, are the design of the CAR, the specific target, and the tumor type.39 Last but not least, monitoring the efficacy and safety of CAR-T cell therapy is challenging.40,41 Due to the uncertainty of CAR-T therapy and possible side effects accompanying the entire process, in vivo assays are needed to understand its efficacy and pharmacodynamics.42 Imaging techniques such as computerized tomography (CT) and magnetic resonance imaging (MRI) fail to detect the spatial distribution of CAR-T cells in solid tumors.43 Thus, the incapacity to monitor the accumulation of CAR-T cells in tumors using clinical imaging modalities is a key obstacle in CAR-T cell therapy for solid tumors. In short, the above issues, including high cost connected with the complex manufacturing process, complex tumor microenvironment, lack of precision, and poor visualization ability, are significant endeavors that should be resolved for CAR-T therapies towards potential solid tumor treatments. | ||
| Fig. 2 Schematic of the complex manufacturing process, difficulty in proliferation and infiltration, lack of precision, and poor visualization ability in CAR-T cell therapy towards solid tumor treatment. | ||
Recent studies have shown that combining functional biomaterials with CAR-T cell therapy is a promising strategy.44 In the treatment of solid tumors, functional biomaterials have shown vitality in drug delivery and imaging therapy.45,46 Functional biomaterials allow the development of novel transfection reagents. Biomaterials, in terms of liposomes or cell vesicles, possess low immunogenicity and biocompatible or cost-effective advantages, making them ideal inert carriers for CAR gene transfection.47,48 Furthermore, T-cell transfection in situ is made possible by introducing functional biomaterials. It can streamline the preparation procedure and make CAR-T cell therapy significantly less expensive. There are also functional biomaterials that have undergone modifications to exhibit immunomodulatory characteristics. These materials can respond to and regulate the TME, enhancing the efficacy of CAR-T cell therapy.49 When loaded with cytokines, biomaterials can even further enhance T cell proliferation and differentiation in situ.50 Furthermore, functional biomaterials can be employed as carriers to enclose CAR-T cells and deliver them to the tumor location by controlled release. It can improve the targeting of CAR-T cell therapy and reduce off-target effects. While developing CAR-T therapies, it is also necessary to develop corresponding diagnostic technologies to monitor CAR-T concentration and therapeutic efficacy in real time, as well as to monitor treatment-related side effects. It is possible to reflect T cell activation and functional status by targeting cell surface markers or key substances in metabolic pathways.51,52 Therefore, the application of functional biomaterials in assisting CAR-T cell therapy might be with promising future.
3. Functional biomaterials enhance the efficacy of CAR-T cell immunotherapy
The process of CAR-T therapy for treating solid tumors involves the use of functional biomaterials such as nanoparticles, polymers, liposomes, and exosomes53 (Table 1). Firstly, functional biomaterials can improve the manufacturing of CAR-T cells. Biomaterial-based non-viral genetic vectors with low cytotoxicity, low immunogenicity and high safety provide new ideas for the transfection of CAR-T cells. Gene delivery vectors based on functional biomaterials are safer and with less cytotoxic effects, in comparison with traditional viral vectors. It can improve the manufacturing process of CAR-T cells in a more cost-effective mass production manner. Secondly, the application of various functional biomaterials can remodel TME to promote the proliferation and infiltration of CAR-T cells, and improve the effect of tumor immunotherapy. In addition, functional biomaterials can provide a combination therapy platform for CAR-T cell therapy, which can also improve the targeting of CAR-plasmids and CAR-T cells, and mitigate the off-target effect of solid tumors. Moreover, it enables the integration of diagnosis and treatment to avoid the toxic side effects of CAR-T therapy. The following section will explore the application of functional biomaterials in CAR cell therapy from the above perspective.| Types | Strategies | Functions | Ref. | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Liposomes | Engineering lipid nanoparticles of CD19 CAR | Non-viral gene vectors | 54 | Delivery of drugs or genes through fusion with cell membranes, good biocompatibility, low cytotoxicity, high drug stability | Low encapsulation rate and prone to leakage |
| To add SCH-58261 to lipids, encapsulate the A2aR antagonist SCH-58261 into lipid nanoparticles, and embellish to CAR-T cells | To promote infiltration of CAR-T cells | 49 | |||
| Polymers | mRNA core coalesced with poly(β-amino) ester with anti-CD3/CD8-modified poly (glutamic acid) as shell | Non-viral gene vectors | 55 | ||
| Preparation of hyaluronic acid-based hydrogels to load CAR-T cells targeting human chondroitin sulfate proteoglycan 4 | Implantable stents | 56 | |||
| Mixing CAR-T cells, cytokines and light curable gelatin methacrylate hydrogel | Partial delivery of CAR-T cells | 10 | |||
| PMN-mediated CAR-T cells | To promote infiltration of CAR-T cells | 57 | Higher stability and drug retention efficiency with different polymers for multiple drug delivery modes | Controllability and biocompatibility of drug release kinetics in polymer delivery systems need to be improved | |
| Poly(lactic-glycolic) acid (PLGA) nanoparticle loaded with indocyanine green | Photothermal therapy | 58 | |||
| Interleukin-15 and an injectable hydrogel | To improve targeting | 59 | |||
| IL-6-specific antibody conjugated to a thermo-sensitive hydrogel poly(N-isopropylacrylamide-co-methacrylic acid) | Suppress CAR-T-cell-induced cytokine storm | 60 | |||
| Exosomes | Loading specific mRNA into exosomes using phage shell protein MS2 | Non-viral gene vectors | 61 | Good stability in blood, natural targeting ability, low immunogenicity | Difficulty in purifying exosomes and obtaining high yields of pure exosomes |
| Silicon nanotubes | Transfection of CARs into primary T cells via vertically aligned silicon nanotube arrays | Non-viral gene vectors | 62 | Good biocompatibility and tunability, low toxicity, self-degradable | Preparation of stable structures is more difficult to achieve |
| Nanoparticles | Loading of IL 12 in human serum albumin nanoparticles and incorporation into CAR-T cells to prepare redox-responsive protein nanopreparations | Partial delivery of CAR-T cells | 63 | High stability, low cost, wide range of applications | Complex preparation process and poor batch-to-batch reproducibility |
| Nanoparticles loaded with antigen | To improve targeting | 64 | |||
| PNP-nanoparticle-based hydrogels | Enhanced expansion of CAR-T cells | 65 | |||
| Lipid nanoparticle system with a surface modified by CD3 antibody was constructed containing IL-6 shRNA and CD19-CAR | Transfection of CAR-T cells | 66 |
3.1. Transfection of CAR-T cells
Gene delivery vectors can be optimized for different types of T cells to achieve higher efficiency and selectivity, an important aspect of CAR-T cell manufacturing. Currently, most CAR-T therapies use LVs or RVs to introduce genes into the precursor or stem cell genomes in vitro.67–71 Although viral vectors have high gene delivery efficiency, they can cause immune responses and uncertainty in gene insertion.72 Yu et al. constructed epidermal growth factor receptor variant III CAR expression plasmid vectors for third-generation intracellular expression of CAR-targeting EGFR variant III. It was found that self-assembled nanoparticles (pEGFRvIII-CAR@SNPs) could exhibit high efficiency and reduced cytotoxicity in gene transfection of Jurkat cells, which has considerable potential for T cell transient CAR modification.73 With the wide use of mRNA coronavirus vaccines, non-viral vectors have attracted more and more attention.74 Compared to viral vectors, non-viral vectors are less cytotoxic, less immunogenic and less mutagenic, and the target gene is not integrated into the host genome after transfection using non-viral vectors.75 Chen et al. successfully transfected CAR into primary T cells with the help of vertically aligned silicon nanotubes (Fig. 3A). It has higher delivery efficiency and CAR expression efficiency than electroporation.62 Inoo et al. found that electroporating mRNA into T cells to create anti-VEGFR2 CAR-T cells has similar anti-tumor effects as single retroviral vector-modified CAR-T cells.76 In addition, non-viral vectors for CAR-T cell therapy products are simpler to manufacture, leading directly to lower costs and easier quality control. Bozza et al. developed non-integrated DNA nanocarrier nano-S/MARt vectors that can generate active CAR-T cells. This platform is advantageous because it does not contain viral components, making it non-immunogenic, simple, and versatile. It is capable of extrachromosomal replication in the nucleus of dividing cells and provides sustained transgene expression in human T cells without compromising their behavioral and molecular integrity.77 Billingsley et al. screened a sequential library of ionizable lipid nanoparticle (LNP) formulations with different excipient components, and the prepared B10 LNPs of formulation increased mRNA delivery by 3-fold. It also reduced cytotoxicity and showed potent cancer cell killing power, and the prepared B10 LNPs provided new ideas for an effective mRNA delivery platform for iMPIT cell engineering.78 But both transduction in vitro and electroporation processes carry significant costs and risks.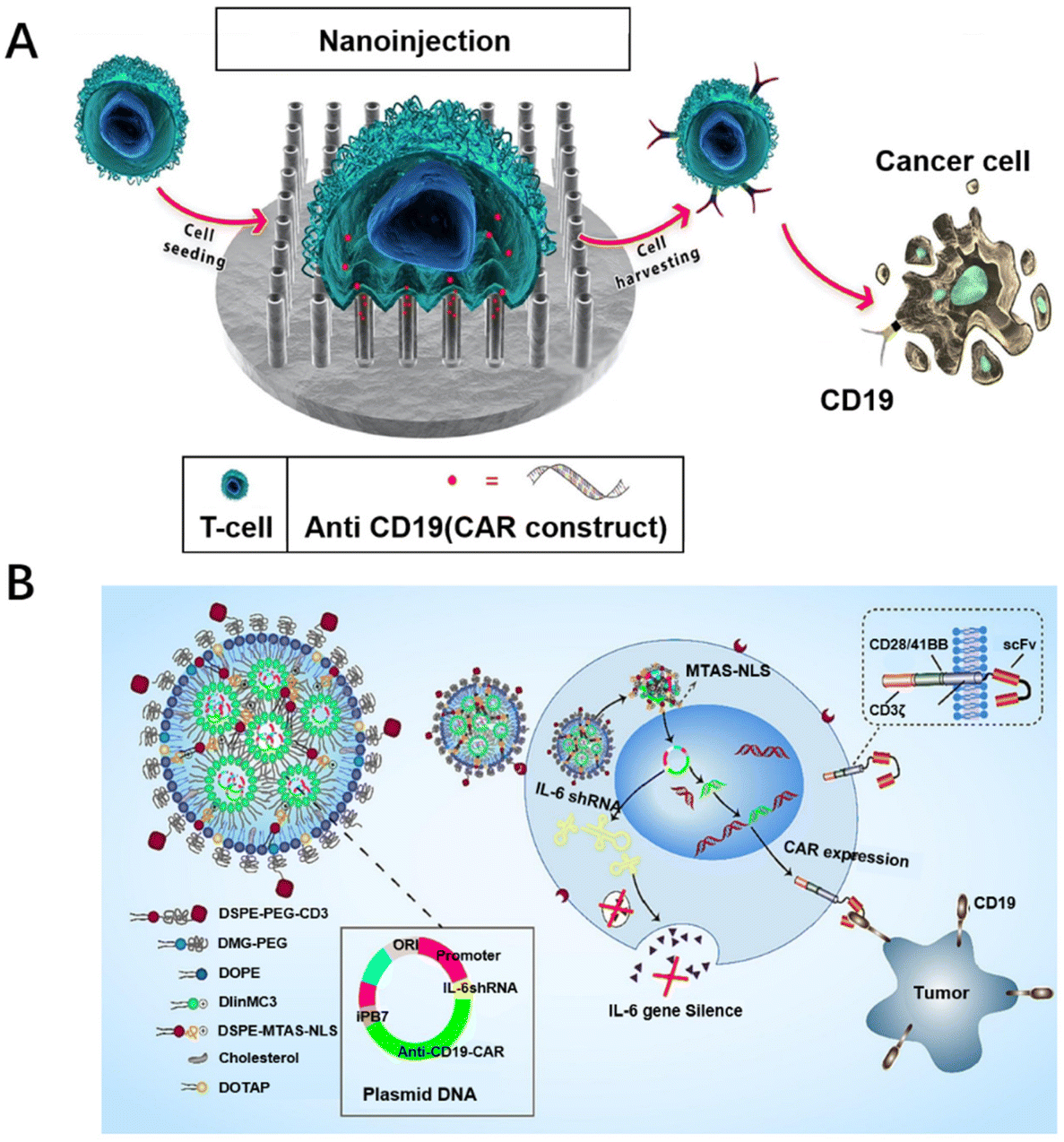 | ||
| Fig. 3 Biomaterial designs for promoting the transfection of CAR-T cells. (A) Vertically aligned silicon nanotube arrays successfully transfect CAR into primary T cells. Reproduced with permission.62 Copyright 2023, Elsevier. (B) A lipid nanoparticle system with a surface modified with CD3 antibodies was constructed containing plasmids of interleukin 6 short hairpin RNA (IL-6 shRNA) and CD19-CAR (antiCD3-LNP/CAR19+ shIL6) combination genes to stably transfect T cells. Reproduced with permission.66 Copyright 2022, Elsevier. | ||
Therefore, researchers have turned their attention to generating CAR-T cells in vivo. Since the process of producing CAR-T cells in vitro is not required, the cost of T-cell culture can be reduced, resulting in a substantial reduction in the price of CAR-T cell therapy with this strategy. Furthermore, it does not require the use of initial cells and there are no immune problems. Compared with in vitro CAR-T cells, in vivo CAR-T cells have better performance and self-renewal ability. Agarwal et al. used LV targeting CD8+ cells to induce CAR-T cells in situ in immune-deficient NOD-scid-IL2Rc null mice.79 However, in their study, CAR-positive NK and NKT cells would also be detected, which might be due to the non-specificity of the lentiviral vector. To improve the non-specificity of lentiviral vectors, non-viral vectors modified with biomaterials have been reported. As a typical example, Zhou et al. constructed a CD3 antibody-modified LNP (antiCD3-LNP/CAR19+ sh-IL6) wrapped in a plasmid system, mediated by CD3 antibodies to target T cells, stably transfecting T cells in vivo with the nuclear localization peptide MTAS-NLS, which produced IL-6 hypersecretory CAR-T cells directly in vivo to kill CD19-high expressing leukemia tumor cells (Fig. 3B).66 Recently, Zhu et al. achieved in situ manufacture of CAR-T cells in mice by self-assembling mPEG-PCL-PEI with T-cell-targeted anti-CD3e f(ab′)2 fragment and α-cyclodextrin, loading plasmid CAR, and realizing the effective aggregation of CAR-T cells at tumor sites.80 Biomaterial designs can be used to deliver CAR-expressing genes directly to circulating T cells in vivo, enabling the in situ generation of CAR-T cells, simplifying the process of CAR-T cell preparation, making it simple and economical to prepare, and providing a new idea for CAR-T cell therapy.
In short, functional biomaterial designs have become a popular choice for gene delivery due to their low cost, good biocompatibility, low toxicity and low immunogenicity. Different biomaterials such as liposomes,81 polymers82 and exosomes83,84 as vectors can overcome the insecurity of viral vectors and electroporation methods, providing new opportunities for the widespread use of CAR-T therapies for solid tumors (Table 2). CAR-T cells can be generated in vivo by using different biomaterials as gene delivery vectors, simplifying the CAR-T cell preparation process and reducing costs, with a similar therapeutic effect.
| Gene vectors | Types | Genes | Models | In vivo/vitro | Ref. |
|---|---|---|---|---|---|
| Viral vectors | Lentiviral vectors | ||||
| SINV-LV | — | B Cellular tumors | In vivo | 85 | |
| Retroviral vectors | |||||
| Bicistronic retroviral vector encoding | — | Cell ablation | In vivo | 86 | |
| Non-viral vectors | Lipid nanoparticles | ||||
| AntiCD3-LNP/CAR19+ shIL6 | shRNA | Leukemia | In vivo | 66 | |
| CAR mRNA/9322-O16B LNP | mRNA | Leukaemia | In vitro | 87 | |
| Cationic polymers | |||||
| Polymeric nanomicelles carrier for piggyBac | DNA | T lymphocytes | In vitro | 88 | |
| pDNA@SNPsx/y | pDNA | Jurkat T cells | in vitro | 73 | |
| PBAE 447 polymer-microtubule-associated nuclear localization peptide | DNA | Leukaemia | In vivo | 89 | |
| CD3-targeted polymeric nanoparticles carrying Cy5-labeled mRNA | mRNA | T lymphocytes | In vitro | 55 | |
| Exosomes | |||||
| Exosome delivery platform | mRNA | T lymphocytes | In vitro | 61 | |
3.2. Promotion of the proliferation of CAR-T cells
Providing appropriate growth factors and cell culture conditions can promote the proliferation of CAR-T cells and afford sufficient numbers of CAR-T cells. Consequently, cytokines loaded on biomaterials can induce T cell proliferation and differentiation in situ. This approach has been shown to enhance the efficacy and safety of preclinical models.44 For example, Liu et al. used integrated mesoporous silica nanoparticles as carriers loaded with IL-2 and transforming growth factor-β (TGF-β) to achieve the combined release of IL-2/TGF-β, promoting the proliferation and differentiation of terminal effector T cells through the up-regulation of perforin, granzyme B and interferon-γ (Fig. 4A).90 By regulating the immune microenvironment, inducing vascular normalization, recruiting cytotoxic T cells and inducing T cell proliferation and differentiation, it can effectively improve the efficacy of CAR-T cell therapy.91 Meanwhile, the implantable scaffold provides a platform technology for the programming of CAR-T cells that may stimulate novel therapeutic approaches. Implantable scaffolds are scaffold structures that can be implanted in the body and are usually made of biodegradable materials. They provide a three-dimensional microenvironment in which CAR-T cells can grow and differentiate, thereby enhancing their viability and aggressiveness.92 Agarwalla et al. have developed a multifunctional alginate scaffold (MASTER) for T cell programming and release, which is modified with antibodies and encapsulates cytokines to promote cell proliferation. Mice treated with MASTER CAR-T cells showed better anti-tumor effects and greater persistence (Fig. 4B).50 Therefore, specific cytokines can be delivered by using biomaterial designs to promote CAR-T cell proliferation.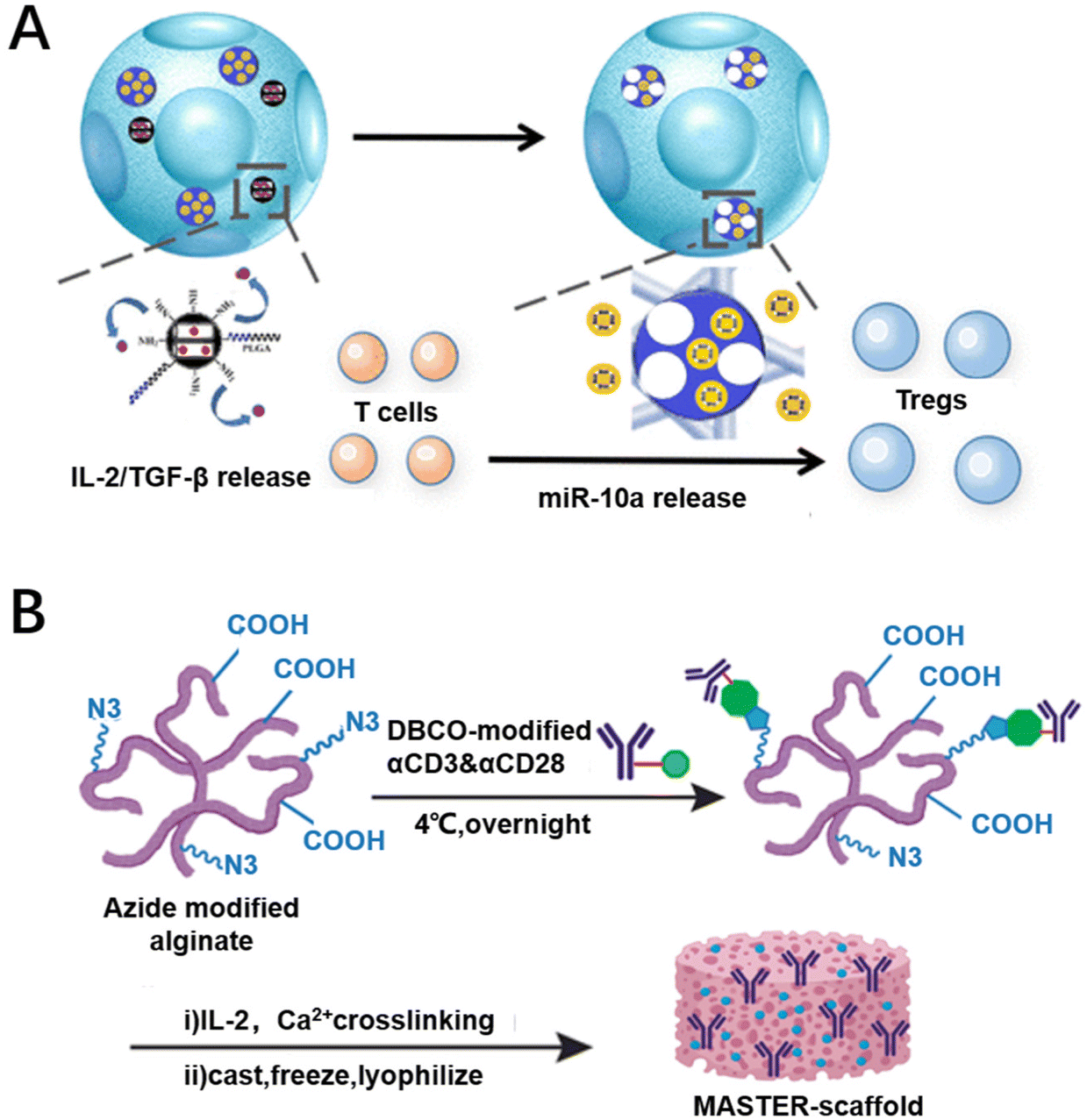 | ||
| Fig. 4 Biomaterial designs for promoting the proliferation of CAR-T cells. (A) Mesoporous silica nanoparticles (MSNs) loaded with IL-2 and TGF-β release IL-2 and TGF-β to promote CAR-T cell proliferation. Reproduced with permission.90 Copyright 2018, American Chemical Society. (B) Multifunctional alginate scaffold (MASTER) encapsulating cytokines that promote CAR-T cell proliferation. Reproduced with permission.50 Copyright 2022, Springer Nature. | ||
3.3. Promotion of the infiltration of CAR-T cells
Biomaterial-based delivery systems have been developed to modify and improve TME by addressing abnormal vascular distribution, hypoxia, acidity, and heterogeneity. They also promote the infiltration of various immune cells and enhance the activity of anti-tumor immune cells, resulting in improved efficacy of immunotherapies.93 For example, Deng et al. have developed a nanomedicine called F3-low molecular weight heparin–hydrazone–gambogic acid (FLG) that can repair abnormal tumor vasculature by inhibiting the VEGF/VEGFR2 pathway. They also designed another nanomedicine, MAR/MPA, by loading maraviroc (MAR) loaded with methoxypoly(ethylene glycol)–poly(lactic acid) (mPEG–PLA, MPP) nanoparticles modified with benzamide lipid derivatives (MPA), which can selectively inhibit the C–C motif chemokine ligand 5 (CCL5)/C–C chemokine receptor type 5 (CCR5)-related pathway in the tumor tissues. The nanosystem with the combination of FLG and MAR/MPA directly regulates vascular endothelial cells, induces vascular normalization, promotes recovery of the vascular system, remodels the TME, and improves therapeutic efficacy.94 Luo et al. used alginate microspheres loaded with interleukin-15 (IL-15) to fabricate an injectable immuno-microchip (i-G/MC) system. They also used marine extracellular hemoglobin as an oxygen carrier, which continuously released oxygen after injection to remodel the TME, contributing to the infiltration of CAR-T cells.95 For example, Yang et al. modified a biodegradable hollow manganese dioxide (H–MnO2) nanoplatform with poly(ethylene glycol) (PEG). The photosensitizer chlorine e6 (Ce6) and the anticancer drug doxorubicin can be co-loaded in this hollow H–MnO2–PEG nanoplatform with high loading capacity (HMnO2–PEG/C&D). The obtained H–MnO2–PEG/C&D dissociates at acidic pH to release the loaded drug while triggering the decomposition of the tumor's endogenous H2O2 to produce oxygen, thus alleviating hypoxia.96 Adequate oxygen in the TME can downregulate the expression of hypoxia inducible factor 1α (HIF-1α) and facilitate the infiltration of CAR-T cells. In another case, Jie et al. designed a self-assembling peptide-based scaffold for mimicking cellular matrices, and local delivery of CAR-T cells by the scaffold resulted in long-term retention and increased CAR-T cell infiltration.97 More interestingly, Li et al. prepared porous microneedle (PMN) patches made of poly(lactic-glycolic) acid (PLGA). PMN patch-assisted delivery enables dispersed inoculation of CAR-T cells in solid tumors. This patch disrupts the physical barrier formed by the extracellular matrix and facilitates the infiltration of CAR-T cells (Fig. 5A).98 Furthermore, Li et al. developed a genetically programmable cell vesicle (D@aPD-L1 NVs) containing an anti-programmed death ligand 1 single-chain variable fragment (anti-PD-L1 scFv) and a glutamine antagonist. Using D@aPD-L1 NVs, it is possible to distribute glutamine antagonists specifically to the tumor location, lower the proportion of immunosuppressive cells, encourage the recruitment of inflammatory cells and the release of inflammatory cytokines in tumor tissues, and enhance the ability of CAR-T cells to infiltrate the tumor (Fig. 5B).99 Therefore, in order to facilitate the entry of CAR-T cells into solid tumors, unique biomaterials can be designed to improve the therapeutic effect of CAR-T cells. These biomaterial designs have shown promising applications in improving the therapeutic effect of CAR-T cells by overcoming the TME and might be expected to be an important adjunct to CAR-T cell therapy in future. | ||
| Fig. 5 Biomaterial designs to promote CAR-T cell infiltration. (A) The delivery of CAR-T cells by porous microneedles (PMN) patches made of PLGA can evenly disperse CAR-T cells into solid tumors and promote their proliferation and infiltration. Reproduced with permission.98 Copyright 2022, Oxford University Press. (B) The cellular nanovesicles (NVs) that express high-affinity anti-murine PD-L1 single chain variable fragment (aPD-L1 scFv) are loaded with glutamine antagonists (D@aPD-L1 NVs) to improve the infiltration of CAR-T cells. Reproduced with permission.99 Copyright 2023, John Wiley and Sons. | ||
3.4. Providing a combination therapy platform for CAR-T cell therapy
In short, previous studies have shown that CAR-T cell therapy alone is not effective at treating solid tumors. In recent years, it has been found that combining CAR-T cell therapy with chemotherapy, radiotherapy, photothermal and other therapies through functional biomaterials might be a good option to improve the efficacy of CAR-T therapy and to reduce side effects. Furthermore, the unique properties of biomaterial designs (e.g., liposomes or polymers) might further enhance the effect of combination therapy and might be beneficial for improving the safety or efficacy of immunotherapy.3.5. Improvement of the safety of CAR-T therapy
When using CAR-T therapies for solid tumors, there are concerns about their safety due to off-target effects and cytokine storms. Currently, researchers are addressing the safety of CAR-T therapy by improving targeting and diagnostic techniques. Targeting systems by using functional biomaterial designs, such as nanoparticles or peptides, have been carried out to enhance the targeting of CAR-plasmids or CAR-T cells and reduce off-target effects. Furthermore, a platform has been created based on these materials to monitor the effectiveness of CAR-T cell treatment in vivo and to detect possible toxic side effects, which might be beneficial for the application of CAR-T therapy in solid tumors.As one strategy for enhancing the precision of CAR-plasmids transported to T cells, Rurik et al. developed a therapeutic approach to generate transient CAR-T cells. The designed CAR-plasmid was packaged in CD5-targeted liposome nanoparticles to transduce mRNA into T cells in vivo. The lipid nanoparticles can specifically bind T cells and enter inside them through cytocytosis to produce transient, effective CAR-T cells in vivo.124 In another case, Siriwon et al. cross-linked CAR-T with multilayer liposome vesicles (cMLVs) containing the small molecule antagonist SCH-5826. The cMLV nanoparticles could be anchored to the surface of CAR-T cells. And cMLV-anchored CAR-T cells were able to target the spleen, lymph nodes, and lungs, more effectively lymphoid organs and tumors.49 Wang's team divided the intracellular activation domain of CAR into two parts, inserted a heterodimer between the two parts, and loaded the switchable molecule with gelatinase-targeted nanoparticles. The switchable molecule is specifically delivered to the tumor to achieve CAR-T localization-specific activation in the tumor. The switchable CAR-T delivery composite system was found to improve the targeting of CAR-T cells and achieve tumor-specific killing, effectively solving the problem of the off-target effect of CAR-T cell therapy for solid tumors (Fig. 6A).125 Sun et al. found that it is possible to load exogenous antigens onto nanoparticles (F-AgNPs). The F-AgNPs modify exogenous antigens to tumor cell membranes via membrane fusion. The modification of the tumor cell membrane in situ provides sufficient targets for subsequent CAR-T cell recognition, allowing CAR-T cells to redirect to tumors without corresponding targets (Fig. 6B).64
 | ||
| Fig. 6 Biomaterial designs for improved targeting. (A) Primitive gelatinase-responsive nanoparticles were used to selectively deliver heterodimerization switches that CAR-T cells consistently and precisely activate only on tumors. Reproduced with permission.125 Copyright 2023, John Wiley and Sons. (B) The F-AgNP modified exogenous antigens onto the tumor cell membranes, which provided sufficient targets for CAR-T cell recognition, enabling CAR-T cell to exert antitumor effects independent of tumor inherent antigen profiles. Reproduced with permission.64 Copyright 2022, Springer Nature. | ||
Furthermore, the targeting ability of CAR-T cells can be improved by the assistance of the unique responsiveness of biomaterials. Miller et al. added a heat shock protein transcription start sequence upstream of the CAR structural gene to enable engineered T cells to express CAR only in high-temperature environments. Upon irradiation with near-infrared light, polyethylene glycol (PEG)-coated plasmonic gold nanorods (AuNRs) aggregated in tumor tissues in vivo generating heat, which promotes the expression of engineered T-cells in the vicinity of the tumor tissues to be converted into CAR-T and thus enhances the targeting of CAR-T.126 Nguyen et al. separated the functional structural domains of the CAR structure and added a light-responsive module to the separated functional structural domains, which could re-fuse the separated functional structural domains of the CAR under the action of specific wavelengths emitted by the converted nanomaterials, thus enabling the targeted generation of CAR-T within tumor tissues.127
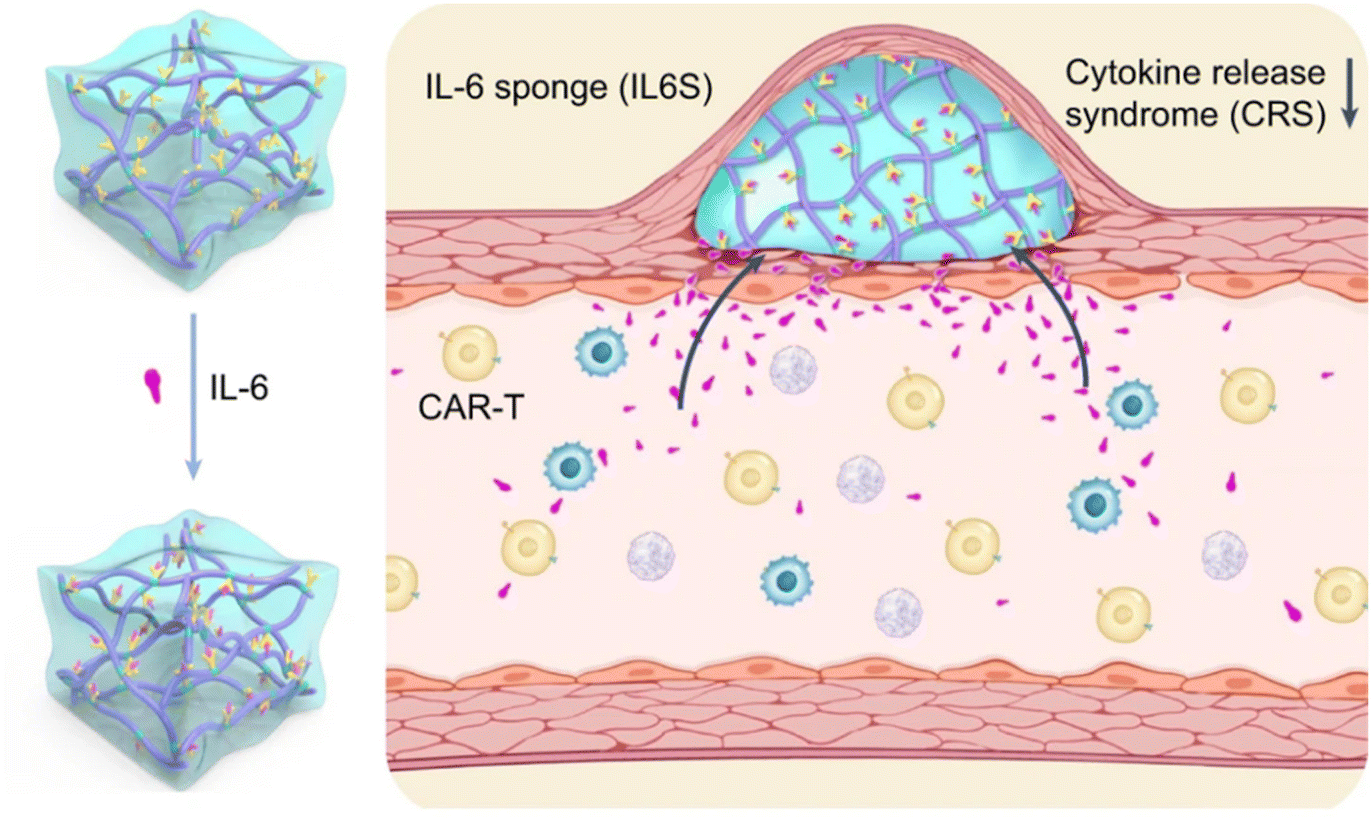 | ||
| Fig. 7 Biomaterial designs inhibit cytokine release syndrome. A temperature-sensitive hydrogel functionalized with antibodies targeting the pro-inflammatory cytokine interleukin-6 (IL-6) in mitigating cytokine release syndrome during CAR-T-cell therapy. Reproduced with permission.60 Copyright 2023, Springer Nature. | ||
4. Conclusion and perspectives
In summary, CAR-T cell therapy has shown great success in the treatment of hematological tumors, and researchers are now focusing on improving its efficacy in the treatment of solid tumors. The biomaterial-based strategy promises to change the manufacturing methods of CAR-T cells, regulate the TME, improve the safety and enhance the efficacy of CAR-T cells for the treatment of solid tumors. However, there are several challenges that need to be addressed to achieve this goal. Firstly, non-viral gene vectors based on biomaterial designs suffer from low transfection efficiency compared to viral vectors. It requires the extraction of more cells from patients and a longer time for expanding the CAR-T cells. Therefore, future research requires a more in-depth study of non-viral gene vectors, developing novel gene delivery vectors in gene loading and T-cell gene modification. In addition, generating CAR-T cells in vivo simplifies the manufacturing process of CAR-T cells and reduces costs. But it may be transduced in other cells, leading to off-target transduction.130 Fortunately, recent reports have shown that using a switching technique enhances the safety and effectiveness of CAR-T cells. Based on functional biomaterial designs, the method has various designs and is expected to achieve the distinction between T cells and normal cells. Furthermore, recent reports have shown that biomaterial designs can also provide a platform for CAR-T cell therapy as a combination therapy. However, clinical data on combination therapies are limited, and the optimal dosing regimen for combination therapy still needs to be explored. Last but not least, biomaterial designs are often used in therapeutic applications in the form of nanomaterials,131 which are difficult to industrialize and have poor inter-batch reproducibility due to the complexity of the nanomaterial preparation process.132,133 Simplicity should be considered when designing biomaterials to simplify the manufacturing process of CAR-T cells for industrial production. It is also important to emphasize that the long-term compatibility, degradation characteristics and safety of implantable scaffolds still need to be explored. Polymeric materials implanted in the human body may release degradation products that may be toxic to humans.134–136 It needs optimization of the design of biomaterial systems based on their own physicochemical properties, biocompatibility, targeting and other characteristics to achieve effective modulation of the TME and sustained intra-tumor immune activation. Gong et al. used materials alone to modulate cell–cell interactions for disease treatment and reduce CRS and neurotoxicity by constructing in situ PEGylation of CAR-T cells.137 It provides a safer strategy and direction for biomaterial-based CAR-T cell therapy. There remains a significant market opportunity for CAR-T clinical therapy, particularly in the treatment of solid tumors. Allogeneic CAR-T cells are one of the future directions, but they can produce host-versus-graft reactions and graft-versus-host disease problems.138,139 It can be designed together with special functional biomaterials to improve the biocompatibility and durability of allogeneic CAR-T cells, paving the way for breakthroughs in the treatment of solid tumors. In short, these insights will provide important guidelines for the development of safe and inexpensive biomaterial designs to enhance the efficacy of CAR-T cell therapy. As functional biomaterials continue to advance, this strategy might show great potential for creating a promising therapeutic approach towards solid tumor treatment.Author contributions
Literature review and writing—original draft preparation, Y. L. and writing—review and editing and supervision, Y. C., Z. L. and Y.-L. W. All authors have read and agreed to the current version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Natural Science Foundation of China grant 81971724, 82173750, and 82372119 and the National Key R&D Program of China grant 2020YFA0908100.References
- J. C. Del Paggio, Nat. Rev. Clin Oncol., 2018, 15, 268–270 CrossRef PubMed.
- N. Gagelmann, K. Riecken, C. Wolschke, C. Berger, F. A. Ayuk, B. Fehse and N. Kröger, Leukemia, 2020, 34, 2317–2332 CrossRef PubMed.
- Z. Yin, Y. Zhang and X. Wang, Biomarker Res., 2021, 9, 58 CrossRef PubMed.
- J. Gideon, Clin. J. Oncol. Nurs., 2022, 26, 597–601 Search PubMed.
- C. H. June, R. S. O'Connor, O. U. Kawalekar, S. Ghassemi and M. C. Milone, Science, 2018, 359, 1361–1365 CrossRef PubMed.
- M. C. Milone and U. O'Doherty, Leukemia, 2018, 32, 1529–1541 CrossRef PubMed.
- G. Liu, W. Rui, X. Zhao and X. Lin, Cell. Mol. Immunol., 2021, 18, 1085–1095 CrossRef PubMed.
- M. P. Lokugamage, C. D. Sago, Z. B. Can, B. R. Krupczak and J. E. Dahlman, Adv. Mater., 2019, 31, e1902251 CrossRef PubMed.
- S. Zuo, J. Song, J. Zhang, Z. He, B. Sun and J. Sun, Theranostics, 2021, 11, 7471–7487 CrossRef CAS PubMed.
- W. Zhou, S. Lei, M. Liu, D. Li, Y. Huang, X. Hu, J. Yang, J. Li, M. Fu, M. Zhang, F. Wang, J. Li, K. Men and W. Wang, Biomaterials, 2022, 291, 121872 CrossRef CAS PubMed.
- Q. Chen, M. C. Chen and Z. Liu, Chem. Soc. Rev., 2019, 48, 5506–5526 RSC.
- S. Y. Shen, X. Xu, S. Q. Lin, Y. Zhang, H. Y. Liu, C. Zhang and R. Mo, Nat. Nanotechnol., 2021, 16, 104–113 CrossRef CAS PubMed.
- K. Perica, A. Tu, A. Richter, J. G. Bieler, M. Edidin and J. P. Schneck, ACS Nano, 2014, 8, 2252–2260 CrossRef CAS PubMed.
- A. K. Kosmides, K. Necochea, J. W. Hickey and J. P. Schneck, Nano Lett., 2018, 18, 1916–1924 CrossRef CAS PubMed.
- M. Zhan, J. Qiu, Y. Fan, L. Chen, Y. Guo, Z. Wang, J. Li, J.-P. Majoral and X. Shi, Adv. Mater., 2022, 35, e2208277 CrossRef PubMed.
- V. Prasad, Nat. Rev. Clin Oncol., 2018, 15, 11–12 CrossRef PubMed.
- M. B. Leick, M. V. Maus and M. J. Frigault, Mol. Ther., 2021, 29, 433–441 CrossRef CAS PubMed.
- F. Liu, H. Zhang, X. Wang, Y. Xiong, Y. Cao, Y. Su, H. Yi, J. Feng, W. Zhang, Y. Ma, M. Wada and Y. Ma, Blood, 2019, 134, 38 CrossRef.
- P. Vormittag, R. Gunn, S. Ghorashian and F. S. Veraitch, Curr. Opin. Biotechnol, 2018, 53, 164–181 CrossRef CAS PubMed.
- I. Hernandez, V. Prasad and W. F. Gellad, JAMA Oncol., 2018, 4, 994–996 CrossRef.
- A. Mullard, Nat. Rev. Drug Discovery, 2022, 21, 249 Search PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 166 Search PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2021, 20, 332 Search PubMed.
- R. Voelker, J. Am. Med. Assoc., 2020, 324, 832 Search PubMed.
- A. S. Moreira, T. Q. Faria, J. G. Oliveira, A. Kavara, M. Schofield, T. Sanderson, M. Collins, R. Gantier, P. M. Alves, M. J. T. Carrondo and C. Peixoto, Sep. Purif. Technol., 2021, 274, 118598 CrossRef.
- A. A. Leech and S. B. Dusetzina, J. Natl. Cancer Inst., 2018, 111, 644–645 CrossRef.
- S. Jagannath, N. Joseph, C. Crivera, C. C. Jackson, S. Valluri, P. Cost, H. Phelps, R. Slowik, T. Klein, X. Yu, L. Smolen and A. D. Cohen, Blood, 2021, 138, 4964 CrossRef.
- S. Leung, A. T. McLeod, Z. Sheikholeslami, S. Shoaebargh, T. Ho, R. Ramanuj, P. Beaudette, N. Bakhtyar and A. Ghorbani, Cytotherapy, 2020, 22, S184–S185 CrossRef.
- M. Potter, B. Lins, M. Mietzsch, R. Heilbronn, K. Van Vliet, P. Chipman, M. Agbandje-McKenna, B. D. Cleaver, N. Clément, B. J. Byrne and S. Zolotukhin, Mol. Ther.–Methods Clin. Dev., 2016, 1, 14034 CrossRef PubMed.
- R. K. Jain, Nat. Med., 2003, 9, 685–693 CrossRef PubMed.
- Y. Yang, D. Fan, B. H. Zheng and S. T. Zhou, Sichuan Daxue Xuebao, Yixueban, 2023, 54, 497–504 Search PubMed.
- A. Maali, M. Gholizadeh, S. Feghhi-Najafabadi, A. Noei, S. S. Seyed-Motahari, S. Mansoori and Z. Sharifzadeh, Front. Immunol., 2023, 14, 1012841 CrossRef PubMed.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef PubMed.
- Z. Luo, X. Yao, M. Li, D. Fang, Y. Fei, Z. Cheng, Y. Xu and B. Zhu, Adv. Drug Delivery Rev., 2022, 185, 114301 CrossRef CAS PubMed.
- P. Yang, Y. Wang, Z. Yao, X. Gao, C. Liu, X. Wang, H. Wu, X. Ding, J. Hu, B. Lin, Q. Li, M. Li, X. Li, X. Chen, W. Qi, W. Li, J. Xue and H. Xu, J. Am. Chem. Soc., 2020, 23, 14954 Search PubMed.
- S. Rafiq, C. S. Hackett and R. J. Brentjens, Nat. Rev. Clin Oncol., 2020, 17, 147–167 CrossRef PubMed.
- S. S. Neelapu, S. Tummala, P. Kebriaei, W. Wierda, C. Gutierrez, F. L. Locke, K. V. Komanduri, Y. Lin, N. Jain, N. Daver, J. Westin, A. M. Gulbis, M. E. Loghin, J. F. de Groot, S. Adkins, S. E. Davis, K. Rezvani, P. Hwu and E. J. Shpall, Nat. Rev. Clin Oncol., 2018, 15, 47–62 CrossRef CAS.
- H. J. Hawkins, T. A. Gooley and M. Tees, Blood, 2021, 22, 1851–1860 Search PubMed.
- J. Gauthier, A. Cearley, P. Perkins, A. Kirk, M. Shadman, S. Williamson, J. Myers, A. I. Chen, S. Nagle, B. M. Hayes-Lattin, L. G. Schachter, D. G. Maloney, C. J. Turtle, M. L. Sorror and R. T. Maziarz, J. Clin. Oncol., 2021, 39, 7532 Search PubMed.
- J. Paludo, R. Bansal, A. T. Holland, K. L. Haugen, M. T. Spychalla, A. L. McClanahan, T. A. Truong, M. A. Hathcock, A. Khurana, H. B. Alkhateeb, D. Dingli, S. R. Hayman, P. Kapoor, M. A. Gertz, Y. Wang, S. S. Kenderian, T. Kourelis, S. Kumar, M. V. Shah, M. A. Siddiqui, R. M. Warsame, J. C. Villasboas, N. Bennani, P. B. Johnston, S. M. Ansell, T. C. Haddad and Y. Lin, Blood, 2021, 138, 568 CrossRef.
- A. M. Goodman, K. A. Holden, A.-R. Jeong, L. Kim, K. D. Fitzgerald, E. Almasri, G. McLennan, M. Eisenberg, A. H. Jahromi, C. Hoh, M. Hurley, C. Mulroney, D. Tzachanis, E. D. Ball, T. J. Jensen and R. Kurzrock, Blood, 2020, 136, 17 CrossRef.
- C. B. García-Calderón, B. Sierro-Martínez, E. García-Guerrero, L. Sanoja-Flores, R. Muñoz-García, V. Ruiz-Maldonado, M. R. Jimenez-Leon, J. Delgado-Serrano, Á. Molinos-Quintana, B. Guijarro-Albaladejo, I. Carrasco-Brocal, J.-M. Lucena, J.-R. García-Lozano, C. Blázquez-Goñi, J. L. Reguera-Ortega, M.-F. González-Escribano, M. Reinoso-Segura, J. Briones, J. A. Pérez-Simón and T. Caballero-Velázquez, Front. Immunol., 2023, 14, 1152498 CrossRef PubMed.
- K. Feng, Y. Guo, H. Dai, Y. Wang, X. Li, H. Jia and W. Han, Sci. China: Life Sci., 2016, 59, 468–479 CrossRef CAS PubMed.
- Z. Liao, W. Zhang, H. Zheng, Y. Wang, J. Yu, H. Li and Z. Gu, J. Controlled Release, 2022, 344, 272–288 CrossRef CAS.
- D. Shan, C. Ma and J. Yang, Adv. Drug Delivery Rev., 2019, 148, 219–238 CrossRef CAS PubMed.
- M. Kanamala, W. R. Wilson, M. Yang, B. D. Palmer and Z. Wu, Biomaterials, 2016, 85, 152–167 CrossRef CAS PubMed.
- M. Dymek and E. Sikora, Adv. Colloid Interface Sci., 2022, 309, 102757 CrossRef CAS.
- M. Zabara, B. Senturk, M. Gontsarik, Q. Ren, M. Rottmar, K. Maniura-Weber, R. Mezzenga, S. Bolisetty and S. Salentinig, Adv. Colloid Interface Sci., 2019, 29, 1904007 Search PubMed.
- N. Siriwon, Y. J. Kim, E. Siegler, X. H. Chen, J. A. Rohrs, Y. R. Liu and P. Wang, Cancer Immunol. Res., 2018, 6, 812–824 CrossRef.
- P. Agarwalla, E. A. Ogunnaike, S. Ahn, K. A. Froehlich, A. Jansson, F. S. Ligler, G. Dotti and Y. Brudno, Nat. Biotechnol., 2022, 40, 1250–1258 CrossRef PubMed.
- J. A. Ronald, B. S. Kim, G. Gowrishankar, M. Namavari, I. S. Alam, A. D'Souza, H. Nishikii, H. Y. Chuang, O. Ilovich, C. F. Lin, R. Reeves, A. Shuhendler, A. Hoehne, C. T. Chan, J. Baker, S. S. Yaghoubi, H. F. VanBrocklin, R. Hawkins, B. L. Franc, S. Jivan, J. B. Slater, E. F. Verdin, K. T. Gao, J. Benjamin, R. Negrin and S. S. Gambhir, Cancer Res., 2017, 77, 2893–2902 CrossRef.
- M. Krekorian, G. O. Fruhwirth, M. Srinivas, C. G. Figdor, S. Heskamp, T. H. Witney and E. H. J. G. Aarntzen, Theranostics, 2019, 9, 7924–7947 CrossRef.
- Y. E. Xue, J. Y. Che, X. M. Ji, Y. N. Li, J. B. Xie and X. Y. Chen, Chem. Soc. Rev., 2022, 51, 1766–1794 RSC.
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Nano Lett., 2020, 20, 1578–1589 CrossRef PubMed.
- H. F. Moffett, M. E. Coon, S. Radtke, S. B. Stephan, L. McKnight, A. Lambert, B. L. Stoddard, H. P. Kiem and M. T. Stephan, Nat. Commun., 2017, 8, 389 CrossRef.
- Q. Y. Hu, H. J. Li, E. Archibong, Q. Chen, H. T. Ruan, S. Ahn, E. Dukhovlinova, Y. Kang, D. Wen, G. Dotti and Z. Gu, Nat. Biomed. Eng., 2021, 5, 1038–1047 CrossRef.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M. G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef.
- Z. Chen, H. Pan, Y. Luo, T. Yin, B. Zhang, J. Liao, M. Wang, X. Tang, G. Huang, G. Deng, M. Zheng and L. Cai, Small, 2021, 17, e2007494 CrossRef.
- K. Wang, Y. H. Chen, S. Ahn, M. Zheng, E. Landoni, G. Dotti, B. Savoldo and Z. C. Han, Nat. Cancer, 2020, 1, 990–997 CrossRef.
- L. Xianlei, G. Ningqiang, T. Falin, Z. Shangkun, Z. Yuxuan, W. Yufei, Q. Guangchao, W. Yongchao, L. Fangzhou, X. Yihui, Z. Linlin, W. Jinjin, N. Qiankun, G. Yaling, G. Chaojiang, J. Huaidong, H. Xingxu, S. Xinghua, Z. Tongcun, W. Yan and L. Xing-Jie, Nat. Biomed. Eng., 2023, 7, 1129–1141 CrossRef PubMed.
- K. Si, Z. Dai, Z. Li, Z. Ye, B. Ding, S. Feng, B. Sun, Y. Shen and Z. Xiao, Cytotherapy, 2023, 25, 615–624 CrossRef PubMed.
- C. Yaping, M. Melanie, S. Ali-Reza, Y. Hao Zhe, C. B. David, M. Takahide, S. Koukou, M. Yasuhiro, C. B. Simon, M. Kenneth, E. Roey and H. V. Nicolas, Mater. Today, 2023, 63, 8–17 CrossRef.
- Y. M. Luo, Z. Chen, M. J. Sun, B. H. Li, F. Pan, A. Q. Ma, J. H. Liao, T. Yin, X. F. Tang, G. J. Huang, B. Z. Zhang, H. Pan, M. B. Zheng and L. T. Cai, Biomaterials, 2022, 281, 121341 CrossRef.
- Z. Sun, R. Li, Y. Shen, S. Tan, N. Ding, R. Xu, X. Wang, J. Wei, B. Liu and F. Meng, J. Hematol. Oncol., 2022, 15, 29 CrossRef.
- A. K. Grosskopf, L. Labanieh, D. D. Klysz, G. A. Roth, P. Xu, O. Adebowale, E. C. Gale, C. K. Jons, J. H. Klich, J. Yan, C. L. Maikawa, S. Correa, B. Ou, A. I. d'Aquino, J. R. Cochran, O. Chaudhuri, C. L. Mackall and E. A. Appel, Sci. Adv., 2022, 8, eabn8264 CrossRef.
- J. E. Zhou, L. Sun, Y. Jia, Z. Wang, T. Luo, J. Tan, X. Fang, H. Zhu, J. Wang, L. Yu and Z. Yan, J. Controlled Release, 2022, 350, 298–307 CrossRef PubMed.
- A. Borogovac, A. Keruakous, M. Bycko, J. Holter Chakrabarty, S. Ibrahimi, M. Khawandanah, G. B. Selby, C. Yuen, S. Schmidt, M. T. Autry, T. Al-Juhaishi, M. J. Wieduwilt and A. S. Asch, Bone Marrow Transplant., 2022, 57, 1025–1027 CrossRef PubMed.
- M. Sadeqi Nezhad, M. Yazdanifar, M. Abdollahpour-Alitappeh, A. Sattari, A. Seifalian and N. Bagheri, Biotechnol. Bioeng., 2021, 118, 3691–3705 CrossRef.
- R. Mohty and J. Gauthier, Bone Marrow Transplant., 2021, 56, 2630–2636 CrossRef.
- N. Albinger, J. Hartmann and E. Ullrich, Gene Ther., 2021, 28, 513–527 CrossRef PubMed.
- P. P. Zheng, J. M. Kros and J. Li, Drug Discovery Today, 2018, 23, 1175–1182 CrossRef PubMed.
- D. A. Savenkova, A. A. Makarova, I. K. Shalik and D. V. Yudkin, Int. J. Mol. Sci., 2022, 23, 14954 CrossRef.
- Q. Yu, M. Zhang, Y. Chen, X. Chen, S. Shi, K. Sun, R. Ye, Y. Zheng, Y. Chen, Y. Xu and J. Peng, Int. J. Nanomed., 2020, 15, 483–495 CrossRef.
- V. Gote, P. K. Bolla, N. Kommineni, A. Butreddy, P. K. Nukala, S. S. Palakurthi and W. Khan, Int. J. Mol. Sci., 2023, 24, 2700 CrossRef CAS.
- X. Han, A. Alu, H. Liu, Y. Shi, X. Wei, L. Cai and Y. Wei, Bioact. Mater., 2022, 17, 29–48 CAS.
- K. Inoo, R. Inagaki, K. Fujiwara, S. Sasawatari, T. Kamigaki, S. Nakagawa and N. Okada, Mol. Ther.–Oncolytics, 2016, 3, 16024 CrossRef PubMed.
- M. Bozza, A. De Roia, M. P. Correia, A. Berger, A. Tuch, A. Schmidt, I. Zornig, D. Jager, P. Schmidt and R. P. Harbottle, Sci. Adv., 2021, 7, eabf1333 CrossRef.
- M. M. Billingsley, A. G. Hamilton, D. Mai, S. K. Patel, K. L. Swingle, N. C. Sheppard, C. H. June and M. J. Mitchell, Nano Lett., 2022, 22, 533–542 CrossRef.
- S. Agarwal, T. Weidner, F. B. Thalheimer and C. J. Buchholz, OncoImmunology, 2019, 8, e1671761 CrossRef PubMed.
- C. Zhu, L. Ke, X. Ao, Y. Chen, H. Cheng, H. Xin, X. Xu, X. J. Loh, Z. Li, H. Lyu, Q. Wang, D. Zhang, Y. Ping, C. Wu and Y. L. Wu, Adv. Mater., 2023, e2310078 CrossRef PubMed.
- H. Li, M. Somiya and S. Kuroda, Biomaterials, 2021, 268, 120601 CrossRef.
- H. Zu and D. Gao, AAPS J., 2021, 23, 78 CrossRef.
- Y. Lu, W. Huang, M. Li and A. Zheng, Pharmaceutics, 2023, 15, 598 CrossRef CAS PubMed.
- A. Amiri, R. Bagherifar, E. Ansari Dezfouli, S. H. Kiaie, R. Jafari and R. Ramezani, J. Transl. Med., 2022, 20, 125 CrossRef CAS.
- J. T. Huckaby, E. Landoni, T. M. Jacobs, B. Savoldo, G. Dotti and S. K. Lai, J. Immunother. Cancer, 2021, 9, e002737 CrossRef PubMed.
- E. Lanitis, G. Rota, P. Kosti, C. Ronet, A. Spill, B. Seijo, P. Romero, D. Dangaj, G. Coukos and M. Irving, J. Exp. Med., 2021, 218, e20192203 CrossRef.
- Z. Ye, J. Chen, X. Zhao, Y. Li, J. Harmon, C. Huang, J. Chen and Q. Xu, ACS Biomater. Sci. Eng., 2022, 8, 722–733 CrossRef.
- Y. Zheng, Z. R. Li, R. Yue, Y. L. Fu, Z. Y. Liu, H. Y. Feng, J. G. Li and S. Y. Han, Am. J. Transl. Res., 2019, 11, 7126–7136 Search PubMed.
- T. T. Smith, S. B. Stephan, H. F. Moffett, L. E. McKnight, W. H. Ji, D. Reiman, E. Bonagofski, M. E. Wohlfahrt, S. P. S. Pillai and M. T. Stephan, Nat. Nanotechnol., 2017, 12, 813–820 CrossRef.
- Z. N. Liu, X. Chen, Z. P. Zhang, X. J. Zhang, L. Saunders, Y. S. Zhou and P. X. Ma, ACS Nano, 2018, 12, 9785–9799 CrossRef PubMed.
- S. H. Ross and D. A. Cantrell, Annu. Rev. Immunol., 2018, 36, 411–433 CrossRef CAS PubMed.
- D. Karami, N. Richbourg and V. Sikavitsas, Cancer Lett., 2019, 449, 178–185 CrossRef CAS PubMed.
- Y. Chao and Z. Liu, Nat. Rev. Bioeng., 2023, 1, 125–138 CrossRef.
- Y. Deng, Z. Jiang, Y. Jin, J. Qiao, S. Yang, H. Xiong and J. Yao, J. Controlled Release, 2021, 340, 87–101 CrossRef.
- Z. Luo, Z. Liu, Z. Liang, J. Pan, J. Xu, J. Dong, Y. Bai, H. Deng and S. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 56712–56722 CrossRef.
- G. B. Yang, Nat. Commun., 2017, 8, 902 CrossRef.
- J. Jie, D. Mao, J. Cao, P. Feng and P. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 37514–37527 CrossRef.
- H. J. Li, Z. J. Wang, E. Archibong, Q. Wu, G. J. Chen, Q. Y. Hu, T. Y. Ci, Z. W. Chen, J. Q. Wang, D. Wen, H. W. Du, J. Jiang, J. Sun, X. C. Zhang, G. Dotti and Z. Gu, Natl. Sci. Rev., 2022, 9, nwab172 CrossRef PubMed.
- X. Li, T. Zhu, R. Wang, J. Chen, L. Tang, W. Huo, X. Huang and Q. Cao, Adv. Mater., 2023, 35, e2211138 CrossRef.
- J. P. Murad, D. Tilakawardane, A. K. Park, L. S. Lopez, C. A. Young, J. Gibson, Y. Yamaguchi, H. J. Lee, K. T. Kennewick, B. J. Gittins, W. C. Chang, C. P. Tran, C. Martinez, A. M. Wu, R. E. Reiter, T. B. Dorff, S. J. Forman and S. J. Priceman, Mol. Ther., 2021, 29, 2335–2349 CrossRef PubMed.
- M. E. Lutsiak, R. T. Semnani, R. De Pascalis, S. V. Kashmiri, J. Schlom and H. Sabzevari, Blood, 2005, 105, 2862–2868 CrossRef PubMed.
- R. Ramakrishnan, C. Huang, H. I. Cho, M. Lloyd, J. Johnson, X. Ren, S. Altiok, D. Sullivan, J. Weber, E. Celis and D. I. Gabrilovich, Cancer Res., 2012, 72, 5483–5493 CrossRef PubMed.
- Y. Chao, T. Wei, Q. Li, B. Liu, Y. Hao, M. Chen, Y. Wu, F. Song, Q. Chen and Z. Liu, Biomaterials, 2023, 295, 122052 CrossRef PubMed.
- D. Zhang, Q. Li, X. Chen, X. Nie, F. Xue, W. Xu and Y. Luan, Small, 2022, 18, e2202663 CrossRef.
- J. Krombach, R. Hennel, N. Brix, M. Orth, U. Schoetz, A. Ernst, J. Schuster, G. Zuchtriegel, C. A. Reichel, S. Bierschenk, M. Sperandio, T. Vogl, S. Unkel, C. Belka and K. Lauber, OncoImmunology, 2019, 8, e1523097 CrossRef PubMed.
- F. Park, Physiol. Genomics, 2007, 31, 159–173 CrossRef.
- E. A. Reits, J. W. Hodge, C. A. Herberts, T. A. Groothuis, M. Chakraborty, E. K. Wansley, K. Camphausen, R. M. Luiten, A. H. de Ru, J. Neijssen, A. Griekspoor, E. Mesman, F. A. Verreck, H. Spits, J. Schlom, P. van Veelen and J. J. Neefjes, J. Exp. Med., 2006, 203, 1259–1271 CrossRef PubMed.
- C. Qu, N. Ping, L. Kang, H. Liu, S. Qin, Q. Wu, X. Chen, M. Zhou, F. Xia, A. Ye, D. Kong, C. Li, L. Yu, D. Wu and Z. Jin, J. Immunother., 2020, 43, 32–37 CrossRef.
- Y. Nai, L. Du, M. Shen, T. Li, J. Huang, X. Han, F. Luo, W. Wang, D. Pang and A. Jin, Front. Mol. Biosci., 2021, 8, 756599 CrossRef.
- C. Liang, Y. Chao, X. Yi, J. Xu, L. Feng, Q. Zhao, K. Yang and Z. Liu, Biomaterials, 2019, 197, 368–379 CrossRef.
- Q. Chen, Q. Hu, E. Dukhovlinova, G. Chen, S. Ahn, C. Wang, E. A. Ogunnaike, F. S. Ligler, G. Dotti and Z. Gu, Adv. Mater., 2019, 31, e1900192 CrossRef.
- L. Zhu, J. Liu, G. Zhou, T. M. Liu, Y. Dai, G. Nie and Q. Zhao, Small, 2021, 17, e2102624 CrossRef PubMed.
- W. Ma, D. Zhu, J. Li, X. Chen, W. Xie, X. Jiang, L. Wu, G. Wang, Y. Xiao, Z. Liu, F. Wang, A. Li, D. Shao, W. Dong, W. Liu and Y. Yuan, Theranostics, 2020, 10, 1281–1295 CrossRef PubMed.
- P. Sharma and J. P. Allison, Science, 2015, 348, 56–61 CrossRef PubMed.
- S. Tan, D. Chen, K. Liu, M. He, H. Song, Y. Shi, J. Liu, C. W. Zhang, J. Qi, J. Yan, S. Gao and G. F. Gao, Protein Cell, 2016, 7, 866–877 CrossRef PubMed.
- J. Wang, Q. Deng, Y. Y. Jiang, R. Zhang, H. B. Zhu, J. X. Meng and Y. M. Li, Oncol. Lett., 2019, 18, 4415–4420 Search PubMed.
- P. Zhao, D. Atanackovic, S. Dong, H. Yagita, X. He and M. Chen, Mol. Pharmaceutics, 2017, 14, 1494–1500 CrossRef PubMed.
- Z. Su, S. Dong, Y. Chen, T. Huang, B. Qin, Q. Yang, X. Jiang and C. Zou, Adv. Sci., 2023, 10, e2206213 CrossRef.
- V. G. Deepagan, D. G. You, W. Um, H. Ko, S. Kwon, K. Y. Choi, G.-R. Yi, J. Y. Lee, D. S. Lee, K. Kim, I. C. Kwon and J. H. Park, Nano Lett., 2016, 16, 6257–6264 CrossRef PubMed.
- H. Baek, D. Lockwood, E. J. Mason, E. Obusez, M. Poturalski, R. Rammo, S. J. Nagel and S. E. Jones, Front. Neurol., 2022, 13, 880814 CrossRef.
- Y. Wu, Y. Liu, Z. Huang, X. Wang, Z. Jin, J. Li, P. Limsakul, L. Zhu, M. Allen, Y. Pan, R. Bussell, A. Jacobson, T. Liu, S. Chien and Y. Wang, Nat. Biomed. Eng., 2021, 5, 1336–1347 CrossRef PubMed.
- Z. Yang, Biomaterials, 2022, 280, 121250 CrossRef PubMed.
- D. Wang, M. Zhang, Y. Zhang, G. Qiu, J. Chen, X. Zhu, C. Kong, X. Lu, X. Liang, L. Duan, C. Fang, J. Liu, K. Zhang and T. Luo, Adv. Sci., 2022, 9, e2203106 CrossRef.
- J. G. Rurik, I. Tombacz, A. Yadegari, P. O. Mendez Fernandez, S. V. Shewale, L. Li, T. Kimura, O. Y. Soliman, T. E. Papp, Y. K. Tam, B. L. Mui, S. M. Albelda, E. Pure, C. H. June, H. Aghajanian, D. Weissman, H. Parhiz and J. A. Epstein, Science, 2022, 375, 91–96 CrossRef PubMed.
- X. Wang, F. Meng, X. Li, L. Xue, A. Chen, Y. Qiu, Z. Zhang, L. Li, F. Liu, Y. Li, Z. Sun, Y. Chu, R. Xu, L. Yu, J. Shao, M. Tian, X. Qian, Q. Liu, B. Liu and R. Li, Adv. Sci., 2023, 10, e2205044 CrossRef.
- I. C. Miller, A. Zamat, L. K. Sun, H. Phuengkham, A. M. Harris, L. Gamboa, J. Yang, J. P. Murad, S. J. Priceman and G. A. Kwong, Nat. Biomed. Eng., 2021, 5, 1348–1359 CrossRef PubMed.
- N. T. Nguyen, K. Huang, H. X. Zeng, J. Jing, R. Wang, S. H. Fang, J. Chen, X. Liu, Z. X. Huang, M. J. You, A. Rao, Y. Huang, G. Han and Y. B. Zhou, Nat. Nanotechnol., 2021, 16, 1424–1434 CrossRef.
- L. Kiru, A. Zlitni, A. M. Tousley, G. N. Dalton, W. Wu, F. Lafortune, A. Liu, K. M. Cunanan, H. Nejadnik, T. Sulchek, M. E. Moseley, R. G. Majzner and H. E. Daldrup-Link, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2102363119 CrossRef.
- B. W. Shi, D. Li, W. W. Yao, W. F. Wang, J. Jiang, R. H. Wang, F. H. Yan, H. Liu, H. Zhang and J. Ye, Biomater. Sci., 2022, 10, 2577–2589 RSC.
- A. Raguram, S. Banskota and D. R. Liu, Cell, 2022, 185, 2806–2827 CrossRef PubMed.
- H. Zheng, Q. Wu, H. Li and Z. Gu, Synth. Biol. J., 2022, 3, 279–301 Search PubMed.
- F. Peng, M. I. Setyawati, J. K. Tee, X. Ding, J. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nat. Nanotechnol., 2019, 14, 279–286 CrossRef PubMed.
- J. Zhang, Y. Li, F. F. An, X. Zhang, X. Chen and C. S. Lee, Nano Lett., 2015, 15, 313–318 CrossRef PubMed.
- S. S. Soni and C. B. Rodell, Acta Biomater., 2021, 133, 139–152 CrossRef.
- H. Bai, J. Wang, Z. Li and G. Tang, Int. J. Mol. Sci., 2019, 20, 2097 CrossRef.
- L. Bachelet, I. Bertholon, D. Lavigne, R. Vassy, M. Jandrot-Perrus, F. Chaubet and D. Letourneur, Biochim. Biophys. Acta, 2009, 1790, 141–146 CrossRef PubMed.
- N. Gong, X. Han, L. Xue, R. El-Mayta, A. E. Metzloff, M. M. Billingsley, A. G. Hamilton and M. J. Mitchell, Nat. Mater., 2023, 22, 1571–1580 CrossRef PubMed.
- Y. Zhang, C. Li, M. Du, H. Jiang, W. Luo, L. Tang, Y. Kang, J. Xu, Z. Wu, X. Wang, Z. Huang, Y. Zhang, D. Wu, A. H. Chang, Y. Hu and H. Mei, Blood Cancer J., 2023, 13, 61 CrossRef PubMed.
- D. Vasic, J. B. Lee, Y. Leung, I. Khatri, Y. Na, D. Abate-Daga and L. Zhang, Sci. Immunol., 2022, 7, eabl3642 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |