
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphene oxide films as a novel tool for the modulation of myeloid-derived suppressor cell activity in the context of multiple sclerosis†
Celia
Camacho-Toledano
ab,
Isabel
Machín-Díaz
ab,
Rafael
Lebrón-Galán‡
a,
Ankor
González-Mayorga§
c,
Francisco J.
Palomares
d,
María C.
Serrano
 *d and
Diego
Clemente
*abe
*d and
Diego
Clemente
*abe
aNeuroimmune-Repair Group, Hospital Nacional de Parapléjicos (HNP), SESCAM, Finca La Peraleda s/n, 45071-Toledo, Spain. E-mail: dclemente@sescam.jccm.es
bCentro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Carlos III Health Institute, Av. Monforte de Lemos, 3-5, 28029-Madrid, Spain
cLaboratory of Interfaces for Neural Repair, Hospital Nacional de Parapléjicos, SESCAM, Finca La Peraleda s/n, 45071- Toledo, Spain
dInstituto de Ciencia de Materiales de Madrid (ICMM), Consejo Superior de Investigaciones Científicas (CSIC), Calle Sor Juana Inés de la Cruz 3, 28049-Madrid, Spain. E-mail: mc.terradas@csic.es
eDesign and development of biomaterials for neural regeneration, HNP, Associated Unit to CSIC through ICMM, Finca La Peraleda s/n, 45071-Toledo, Spain
First published on 18th March 2024
Abstract
Despite the pharmacological arsenal approved for Multiple Sclerosis (MS), there are treatment-reluctant patients for whom cell therapy appears as the only therapeutic alternative. Myeloid-derived suppressor cells (MDSCs) are immature cells of the innate immunity able to control the immune response and to promote oligodendroglial differentiation in the MS animal model experimental autoimmune encephalomyelitis (EAE). However, when isolated and cultured for cell therapy purposes, MDSCs lose their beneficial immunomodulatory properties. To prevent this important drawback, culture devices need to be designed so that MDSCs maintain a state of immaturity and immunosuppressive function similar to that exerted in the donor organism. With this aim, we select graphene oxide (GO) as a promising candidate as it has been described as a biocompatible nanomaterial with the capacity to biologically modulate different cell types, yet its immunoactive potential has been poorly explored to date. In this work, we have fabricated GO films with two distintive redox and roughness properties and explore their impact in MDSC culture right after isolation. Our results show that MDSCs isolated from immune organs of EAE mice maintain an immature phenotype and highly immunosuppressive activity on T lymphocytes after being cultured on highly-reduced GO films (rGO200) compared to those grown on conventional glass coverslips. This immunomodulation effect is depleted when MDSCs are exposed to slightly rougher and more oxidized GO substrates (rGO90), in which cells experience a significant reduction in cell size associated with the activation of apoptosis. Taken together, the exposure of MDSCs to GO substrates with different redox state and roughness is presented as a good strategy to control MDSC activity in vitro. The versatility of GO nanomaterials in regards to the impact of their physico-chemical properties in immunomodulation opens the door to their selective therapeutic potential for pathologies where MDSCs need to be enhanced (MS) or inhibited (cancer).
Introduction
Multiple sclerosis (MS) is a chronic neurological disease affecting 2.5 M people worldwide. In MS, the harmony of immune tolerance related to the nervous system is broken. As a consequence, the immune system attacks the brain and spinal cord producing myelin destruction (demyelination) and neuronal death (neurodegeneration), which leads to sensory, motor and cognitive symptoms and a gradual increase in patient disability.1 85% of MS patients experience the relapsing-remitting MS (RRMS), in which periods of symptoms exacerbation are followed by full or partial recoveries.2 Due to an extensive investigation in therapies for MS, over 20 disease modifying treatments are available for RRMS patients to control relapses and slow the progression of disability.3 However, in an important proportion of RRMS patients showing early and highly active MS, the risk of disability accumulation is high and for many of them current therapeutic options provide suboptimal efficacy, with cell therapy being the only alternative.4,5 In this context, cells controlling the immune response (regulatory cells) of the innate and adaptive immunity appear as a suitable therapeutic target to regulate the exacerbated immune system, favour the restoration of immune tolerance, and induce nervous tissue regeneration.6In the last years, the study of myeloid-derived suppressor cells (MDSCs), a highly immature regulatory cell type of the innate immune response, is gaining importance in the context of MS.7,8 MDSCs have been shown as key elements in the control of symptoms recovery in the murine MS model, experimental autoimmune encephalomyelitis (EAE).9 In fact, MDSCs are being identified as a target for new MS interventions due to their multifaceted roles as orchestrators of the control of the adaptive immune response (i.e., T lymphocytes),10–13 together with their involvement in favouring myelin restoration14 and preventing the harmful microbiota disbiosis induced by EAE.7In vivo studies in EAE have shown that the maintenance of the number and immature state of MDSCs is crucial for them to exert their potent control of T cell viability (immunosuppression) and moderate disease severity.9,13 However, once isolated from the donor organism, MDSCs tend to differentiate spontaneously, which greatly impairs their potential usefulness for future cell therapies requiring their isolation and prior expansion on standard cell culture conditions. For this reason, research is needed on new materials for cell growth able to preserve MDSCs as immature and highly immunocompetent cells.
Nanomedicine has emerged as a revolutionary field to provide novel customizable therapies in the nanoscale for a wide plethora of medical applications. Specifically, it makes use of nanosized tools for the diagnosis, prevention and treatment of diseases.15 The interest is such that an increasing number of applications and products containing nanomaterials, or at least with nano-based claims, have been made available to date. Within those, nanosized particle-based platforms for immune-related biomedical applications are a relatively novel field in which both immunosuppression and immune activation are being pursued, including immunotherapy tools, vaccine carriers, adjuvants, and drug delivery systems to target inflammatory cells. Among the most relevant nanomaterials under investigation for biomedical applications, graphene-derived materials (GDMs) are becoming promising candidates for both diagnostic and therapeutic uses. Regarding immune-related applications, the exploration of GDMs is limited and mainly focused on three major topics: (i) their interaction with immune cells for systemic biocompatibility assessment and the induction of specific immune responses, (ii) the development of immune-biosensors and (iii) their use in combination with antibodies for tumor targeting.16 In the first case, although largely unexplored until recently, GDMs are displaying a surprisingly attractive ability to interact with immune system elements, either by stimulating or suppressing specific responses. To this regard, their different physico-chemical properties including purity, shape dimension, redox state, and functionalization are essential drivers of this immune interaction. However, and despite discrete progress in the field, the role that these properties play on the specific interaction of GDMs with immune cells is still poorly understood.
Recent advances on the exploration of GDMs, including graphene oxide (GO), on immune cell interactions have shown important immunosuppressive actions over different myeloid cells,17 being more extensively explored in macrophages.18 Particularly, GO seems to have an outstanding ability to induce macrophage cytotoxicity, but also to alter their phagocytic capacity and, more importantly, polarize them towards either destructive or regenerative phenotypes by manipulating different physico-chemical properties of this nanomaterial such as its oxidation degree.19,20 Alternatively, oxidised multiwall carbon nanotubes, another class of GDMs, can be engulfed by dendritic cells (DCs) without altering the expression of specific markers such as those important for antigen presentation, i.e. major histocompatibility complex class II (MHCII), or the co-stimulatory molecule CD86, and cytokines such as TNF-α, but promote Th1 pro-inflammatory polarization of T lymphocytes by inhibiting the expression of anti-inflammatory Th2 cytokines.21 When further functionalized with a recombinant form of the human surfactant protein D, oxidised and carboxymethyl cellulose-coated carbon nanotubes are phagocytosed by macrophages/monocytic cell lines and increase pro-inflammatory destructive responses.22 Additionally, GDMs are able to modulate mesenchymal and neural stem cell differentiation in vitro,23 thus bringing an alternative therapeutic potential to these nanosized carbon materials for their use in cell-based treatments as culture devices to either favour or hamper cell differentiation.
Herein, we describe the pioneer use of GO-based culture substrates for the modulation of MDSC phenotype, specifically preventing the loss of their highly anti-proliferative activity (immunosuppression) over activated T lymphocytes in vitro in the context of the murine model of MS. Moreover, we identify that the oxidative degree and roughness of GO exerts a pivotal role on cell shape and differentiation, immunosuppressive activity, and viability.
Results
Fabrication and characterization of rGO substrates for MDSC culture
GO substrates for MDSC culture were fabricated by spin coating as thin layers on top of glass coverslips from a commercial GO slurry. GO sheets in the slurry were composed of a few layers of carbon, with lateral sizes at the micron scale as proved by transmission electron microscopy (TEM) (Fig. 1A). After thermal reduction at 200 °C, the resulting films (rGO200) were visualized by scanning electron microscopy (SEM), confirming that rGO sheets compacted and extensively covered the surface of the glass coverslips as a continuous coating with irregular wrinkles of diverse height and length (Fig. 1B). The specific roughness of such rGO coatings were measured by atomic force microscopy (AFM) (Fig. 1C). Specifically, these rGO200 substrates were characterized by the following surface roughness parameters: a root mean square deviation (Rq) value of 38.8 ± 4.26 nm, an arithmetic average roughness (Ra) of 30.4 ± 3.37 nm and a maximum roughness depth (Rmax) of 290.1 ± 38.54 nm (further details on these variables are included in the Experimental section). It is important to note that these rGO films had certain degree of three-dimensionality, as proved by AFM studies. However, the relatively small dimensions of these substrates in the Z-axe (a few nanometers) in comparison to the size of cells and their respective protrusions could make them to be considered as 2D rather than 3D substrates. Nonetheless, this is a relevant feature to take into consideration based on the well-known impact that the acquisition of a third dimension plays in the biological responses to biomaterials.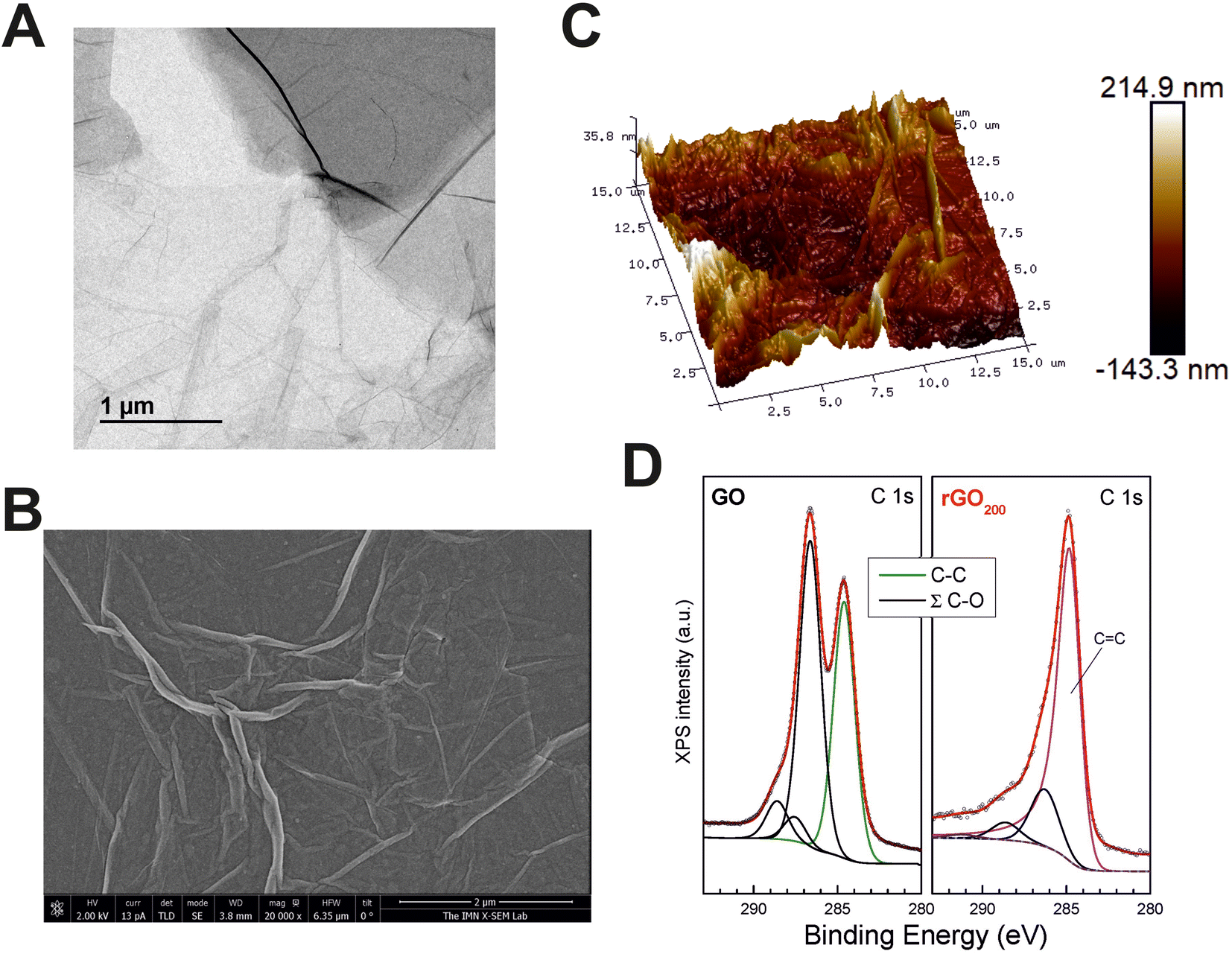 | ||
Fig. 1 Physico-chemical characterization of rGO200 substrates for the modulation of MDSC phenotype in vitro. (A) Representative transmission electron microscopy image of GO nanosheets in the slurry. (B) Representative scanning electron microscopy images illustrating major topographical surface features of the resulting film. (C) 3D surface plots as measured by atomic force microscopy. (D) X-ray photoelectron spectroscopy spectra of the C 1s corresponding to GO slurry (left) and rGO200 (right), together with their corresponding fits composed of chemically shifted components associated to different C chemical bondings. These spectra were normalized to the maximum peak intensity in each case for a better visual inspection and direct comparison among samples in order to highlight line shape differences and contribution for each C chemical environment. Data points in every spectrum are represented as open symbols and Shirley background and component peaks using solid lines. The fitting curve (red line) resulted from the addition of several contributions belonging to: C–C bonding for GO (C–H, C–C defective sp3/sp2 configurations; green), graphitic structures for rGO200 (magenta), oxygen functional groups grouped as ∑(C–O) (C–OH carbonyl, O–C–O carbonyl and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carboxylic groups at the energy shifted values reported in the literature; black) and π–π* transitions coming from graphitic structures for rGO200 at a binding energy of ca. 291.0 eV (magenta). O carboxylic groups at the energy shifted values reported in the literature; black) and π–π* transitions coming from graphitic structures for rGO200 at a binding energy of ca. 291.0 eV (magenta). | ||
Next, a careful chemical characterization was performed by X-ray photoelectron spectroscopy (XPS). Fig. 1D displays the comparison between X-ray XPS spectra of the C 1s core level of the GO slurry and rGO200. Spectra were normalized to the maximum intensity to highlight line shape differences, which provides direct valuable insights regarding the chemical environment of C atoms. The elemental content of C and O for the different samples is provided in Table 1, as well as the relative percentage of the different C–O functional groups. From the quantitative analysis of XPS peaks upon sample annealing, it is observed that the O versus C (O/C) atomic content ratio was dramatically reduced from 0.86 (GO slurry) to 0.10 (rGO200). This fact is also in line with the signal in the energy region of C 1s where the photoelectrons from C–OH and O–C–O groups are emitted, whose intensity also decreased with the O 1s signal (see below). When analyzed in detail, XPS spectra of GO exhibit its characteristic photoelectron emission comprised of two wide and intense peaks in the binding energy range from 282 to 290 eV. Detailed peak shape analysis was performed by the deconvolution of the C 1s spectrum with several Gaussian/Lorentzian symmetric components (ratio of 85/15) using a least-squares fitting routine. The energy position of the peaks and their relative heights were determined to account for the emission ascribed to the different chemical environment of carbon atoms according to the values reported in previous work.24,25 The result of this fit provides a single symmetric peak from C–C emission representative of the oxide nature of GO and several components attributed to the presence of various oxygen functional groups denoted as ∑(C–O) in Table 1. On the contrary, C 1s emission from rGO200 displays a very different lineshape dominated by an intense and asymmetric peak centered close to 284.5 eV. In addition, the long tail on the high energy side of the main peak usually reveals the metallic character of the sample, which certainly supports its graphitic-like nature and ascribes to sp2 bonding. Then, core level fitting of the spectrum is done by the deconvolution of an asymmetric component characteristic of C sp2 bonding (whose parameter values – peak position and full width half maximum – were obtained by fitting the C 1s signal from a HOPG reference sample) besides the symmetric components from the functional groups ∑(C–O). In this case, curve fitting of the whole spectrum makes it hard to clearly identify the sole emission from C–O and CO chemical states, so their full contribution is added in Table 1. Moreover, a weak component appears, broader that any of the previous ones, shifted at a higher binding energy value of ca. 6.5 eV from the main C 1s peak, which is clearly linked to the characteristic π–π* shake-up transition consistent with the majority existence of sp2 bonding in the sample. Note that a small amount of disordered regions where C atoms with defect-like sp2/sp3 bonding nature might also exist and those are included in the C–C component, but with no significant weight due to the low chemical shift and their minor contribution.
| Sample | O/C (at) | C–C | Σ(C–O) | C sp2 | C–C | C–O | CO |
O–CO |
π–π* |
|---|---|---|---|---|---|---|---|---|---|
| GO | 0.86 | 42.4 | 57.6 | — | 42.4 | 47.9 | 3.8 | 5.9 | — |
| rGO90 | 0.48 | 49.6 | 50.4 | — | 49.6 | 43.0 | 2.0 | 5.4 | — |
| rGO200 | 0.10 | 76.5 | 23.5 | 75.3 | — | 18.0 | 5.5 | 1.2 |
In order to confirm the reproducibility of substrate preparation, we repeated AFM and XPS measurements in a representative sample of each batch prepared. Surface roughness was comparable in all batches tested for each particular substrate (Fig. S1†). Specifically, rGO200 samples turned out to be very similar intra- and inter-batches (e.g. Rq values differed less than 2.5% within each particular batch and less than 17% between batches). Statistical comparisons corroborated the absence of significant differences for samples from different batches (p = 0.458 for Rq, p = 0.450 for Ra and p = 0.384 for Rmax) and samples from the same batch (p = 0.876 for Rq, p = 0.791 for Ra and p = 0.694 for Rmax). XPS spectra analyses also confirmed reproducibility among batches with respect to chemical composition (Fig. S2†).
rGO200 prevented spontaneous differentiation of MDSCs towards pro-inflammatory phenotypes and increased their immunosuppressive capacity on stimulated T lymphocytes
It has been already proved that splenic MDSCs from EAE mice differentiate towards mature myeloid cell subsets (macrophages, DCs, granulocytes) in culture.9 This causes them to lose their immunosuppressive activity, which can be prevented, for instance, if the cells are exposed to interferon-β.13 However, there are numerous MS patients who are non-responders to this therapy26 and current drugs fail therapeutically in patients with highly active MS.Encouraged by the reported implications of GDMs on cell differentiation,23 rGO200 films spin-coated on top of conventional glass coverslips were used to modulate the phenotype of splenic MDSCs in vitro (Fig. 2A). First, we confirmed that glass did not exert a noticeable alteration of MDSC differentiation after 24 h in culture compared to those grown on tissue culture plastic (treated polystyrene) except for an increase in the macrophage marker F4/80 (epidermal growth factor-like module-containing mucin-like hormone receptor-like 1) in the absence of the differentiation markers of DCs (the integrin alpha X, known as CD11c, their most widely used defining marker) and antigen presenting cells (APCs; marked with MHC-II, only found on cells with the ability of presenting antigens to T lymphocytes) (Fig. S3†). Mean fluorescence intensity (MFI) is a valuable technical approximation used in flow cytometry to define the extent of immunostaining and, in our particular case, cell differentiation, i.e. the higher the MFI of phenotype-defining markers, the more differentiated state of the immune cells tested. The absence of an increase of the MFI values for Ly-6G (typical marker of granulocytes) and CD11c (DCs) after culture on glass ruled out the possibility of a spontaneous differentiation of our MDSCs towards those two immune cell types in vitro.9 However, these same MDSCs on glass showed a significant increase of the macrophage marker (F4/80), which is also a specific marker of monocytic MDSCs.27 One of the main characteristics of MDSCs is their absence of antigen-presenting capacity measured by MHC-II expression (which differentiates them from pro-regenerative macrophages that present a mild expression of MHC-II28). Therefore, the negligible MHC-II immunolabelling found in these cells indicates that the increase in F4/80 found is not indicative of their maturation towards either pro-inflammatory (high expression of MHC-II) or pro-regenerative (low/intermediate expression of MHC-II) macrophages. Finally, CD11b (a panmarker of myeloid cells) and Ly-6C (marker for monocytic MDSCs), which have been both endorsed as markers for elevated immunosuppressive activity in MDSCs,13,29 did not show any significant alteration. Based on this, these data indicate that culture on glass does not induce a remarkable modification of the cell phenotype of these MDSCs compared to tissue culture plastic in terms of maturation and activation.
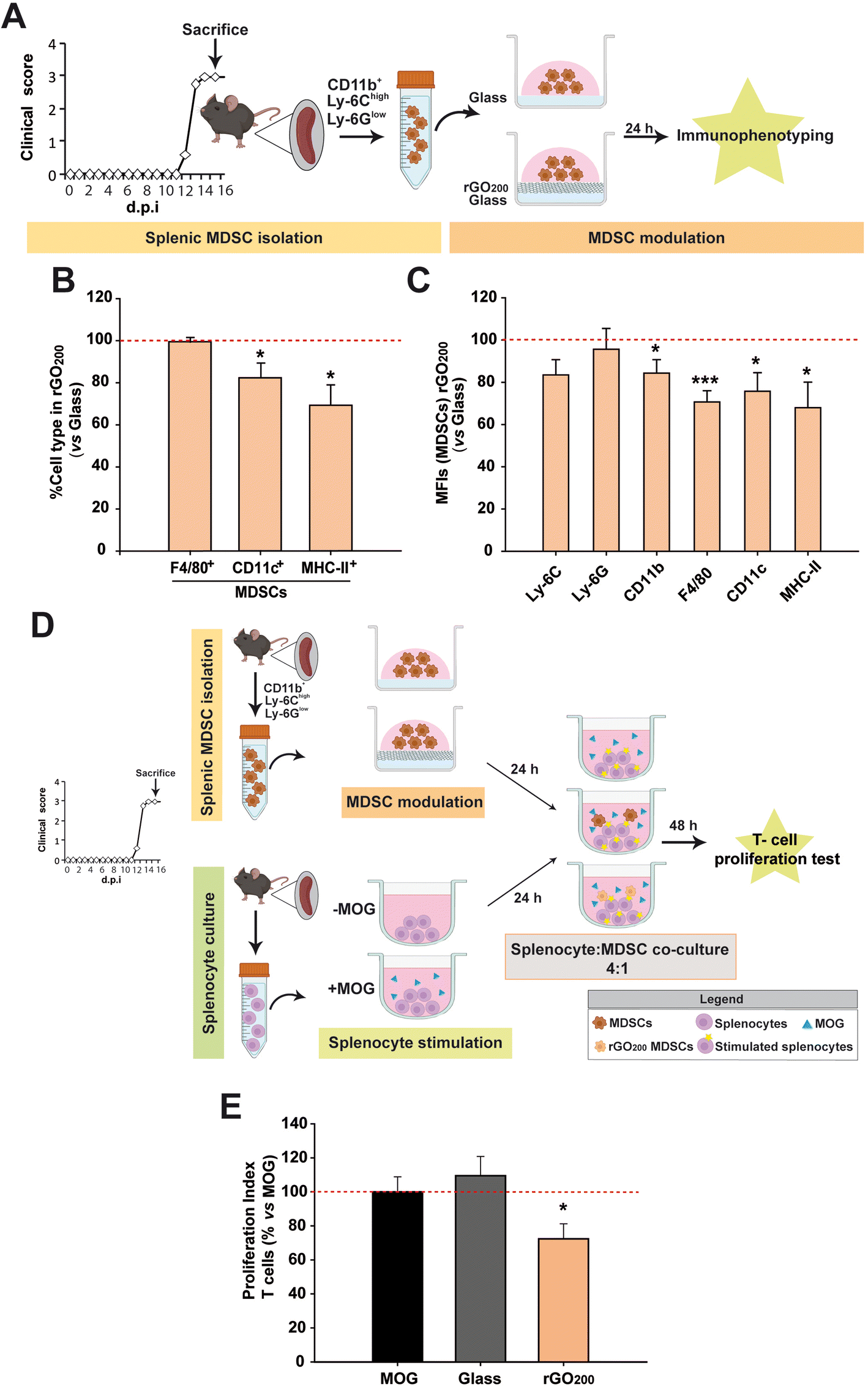 | ||
| Fig. 2 Preservation of the undifferentiated state of splenic MDSCs and enhancement of their anti-proliferative capacity over activated T cells after culture on rGO200 substrates. (A) Schematic representation of the experimental procedure followed for phenotype characterization of these cells. (B and C) rGO200 blocked the cell percentage (B) and expression (measured as the mean fluorescence intensity, MFI; (C) of maturation markers of splenic MDSCs compared to glass coverslips (red dashed line). (D) Schematic of the experimental procedure followed to test the effect of rGO200 on MDSC immunosuppression activity over splenocytes from EAE mice stimulated with MOG, the immunogen used for this animal model induction (positive control). (E) Results on the immunosuppression activity of splenic MDSCs found (red dashed line represents the positive control). Data are shown as the mean ± standard error of the mean of N = 3 independent experiments in all cases. | ||
On the contrary, rGO200 substrates were able to maintain splenic MDSCs in a less matured state than those cultured in conventional glass coverslips at the same time point (Fig. 2B and C). Specifically, rGO200 substrates significantly prevented splenic MDSC differentiation towards mature macrophages (F4/80 in MFI), DCs (CD11c+; both in percentage of cells and MFI) and APCs (MHC-II+; both in percentage of cells and MFI).
In order to determine whether maintaining the undifferentiated state of splenic MDSCs after culture on rGO200 could have any impact on their immunosuppressive capacity, these cells were co-cultured (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4) with splenocytes obtained from EAE mice at the peak of their clinical course and previously stimulated with the myelin oligodendrocyte glycoprotein peptide (MOG35–55), the same molecule used for the immunization (induction) of the EAE model, (Fig. 2D). As previously showed for tissue culture plastic,13 splenic MDSCs cultured for 24 h on glass coverslips did not exert any immunosuppression on MOG-stimulated T lymphocytes, i.e. T cells that respond to the immunogenic stimulus used for active immunization (proliferation index (PI) vs. non-stimulated splenocytes, PI: MOG = 5.7 ± 0.8 and glass coverslip = 6.4 ± 0.8%; p = 0.516). Remarkably, splenic MDSCs exposed to rGO200 for 24 h were able to significantly suppress T cell proliferation (PI: rGO200 = 4.1 ± 0.5%, p < 0.05 vs. MOG; Fig. 2E). These data point to rGO200 films as substrates to potently preserve the immunosuppressive capacity of splenic MDSCs by keeping them in an undifferentiated state in culture.
:4) with splenocytes obtained from EAE mice at the peak of their clinical course and previously stimulated with the myelin oligodendrocyte glycoprotein peptide (MOG35–55), the same molecule used for the immunization (induction) of the EAE model, (Fig. 2D). As previously showed for tissue culture plastic,13 splenic MDSCs cultured for 24 h on glass coverslips did not exert any immunosuppression on MOG-stimulated T lymphocytes, i.e. T cells that respond to the immunogenic stimulus used for active immunization (proliferation index (PI) vs. non-stimulated splenocytes, PI: MOG = 5.7 ± 0.8 and glass coverslip = 6.4 ± 0.8%; p = 0.516). Remarkably, splenic MDSCs exposed to rGO200 for 24 h were able to significantly suppress T cell proliferation (PI: rGO200 = 4.1 ± 0.5%, p < 0.05 vs. MOG; Fig. 2E). These data point to rGO200 films as substrates to potently preserve the immunosuppressive capacity of splenic MDSCs by keeping them in an undifferentiated state in culture.
In an attempt to discard that rGO200 nanosheets were being released from the culture substrates and exert a direct effect on T cells in suspension, culture media from the different conditions under investigation (but in absence of cells) were analysed by dynamic light scattering (DLS). We identified a limited population of particles smaller than 100 nm in Z average (Polydispersity Index, PDI = 0.2 and mean size = 10 nm). Comparatively, the hydrodynamic size of the original GO slurry in suspension was much higher (Zaverage = 5400 nm, PDI = 0.2 and mean size = 1350 nm) (Fig. S4†). Therefore, the observed decrease in T-cell proliferation after exposure of MDSCs to rGO200 should be attributed to a modification of cell activity by direct contact with the film rather than to the presence in suspension of rGO nanosheets.
The activity state of MDSCs was substantially affected by the physico-chemical properties of rGO films
We next investigated the role that the specific physico-chemical properties of these rGO200 films had on the immunosuppression-enhancing effect found in MDSCs. For this purpose, we fabricated a more oxidized rGO substrate, i.e. rGO90, by means of a milder annealing process of the GO-coated glass coverslips (90 °C for 15 min). Morphological analysis by SEM revealed comparable features to those identified in rGO200 (Fig. S5A†). Regarding roughness, rGO90 films were characterized by Rq values of 63.6 ± 7.45 nm, Ra of 49.8 ± 5.60 nm and Rmax of 435.2 ± 55.25 nm (Fig. S5B†). Statistical analyses confirmed slight but significant differences for both Rq (p = 0.017) and Ra (p = 0.014) values between rGO90 and rGO200, but not for Rmax (p = 0.062). More importantly, we corroborated a differential reduction degree (O/C = 0.48) with respect to rGO200 (Fig. S5C† and Table 1). Lineshape analysis of rGO90 XPS spectra revealed no significant differences with that of GO slurry, where both main peaks remained with a change in their relative intensity. This result is consistent with the lower O/C atomic ratio obtained and the slightly higher C–C bonding content. On the contrary, as previously outlined, C 1s emission from rGO200 displayed a very different lineshape dominated by an intense and asymmetric peak centered close to 284.5 eV. Taken together, these results indicate a lighter reduction of rGO90 than rGO200, supported by a significantly higher presence of oxygen-containing groups in the former substrates, which remained chemically closer to the original GO slurry (Fig. 1D). Importantly, just by changing the annealing temperature, we were able to prepare comparable substrates with slight but significant differences in surface roughness and oxygen content. It is worth to note that GO films (without any thermal treatment) could not be used for cell culture due to their physical instability and massive sheets detachment under aqueous conditions, even at short time points of incubation.After substrates characterization, the immunophenotype and immunosuppressive activity of MDSCs were addressed after cultured on rGO90versus rGO200. The ambitioned use of MDSCs as a cell-based therapy in autogenic transplants forced the use of the bone marrow instead of the spleen as the main tissue source for this regulatory cell type. For this reason, we next used bone marrow MDSCs instead of splenic MDSCs to facilitate the future translation of our findings. The exposition to either rGO90 or rGO200 substrates reduced both the cell percentage and MFI of CD11c+ (DCs) and MHC-II+ cells (APCs) in bone marrow MDSCs (Fig. 3A–D), similarly to results described for splenic MDSCs. Interestingly, rGO90 exposition dramatically diminished the percentage, not only the MFI value, of bone marrow MDSCs expressing the macrophage marker F4/80, not observed before for rGO200. Comparing both types of rGO substrates, rGO90 induced a more noticeable effect than rGO200 on the cell percentage and MFI value for both F4/80 and MHC-II markers (Fig. 3E and F).
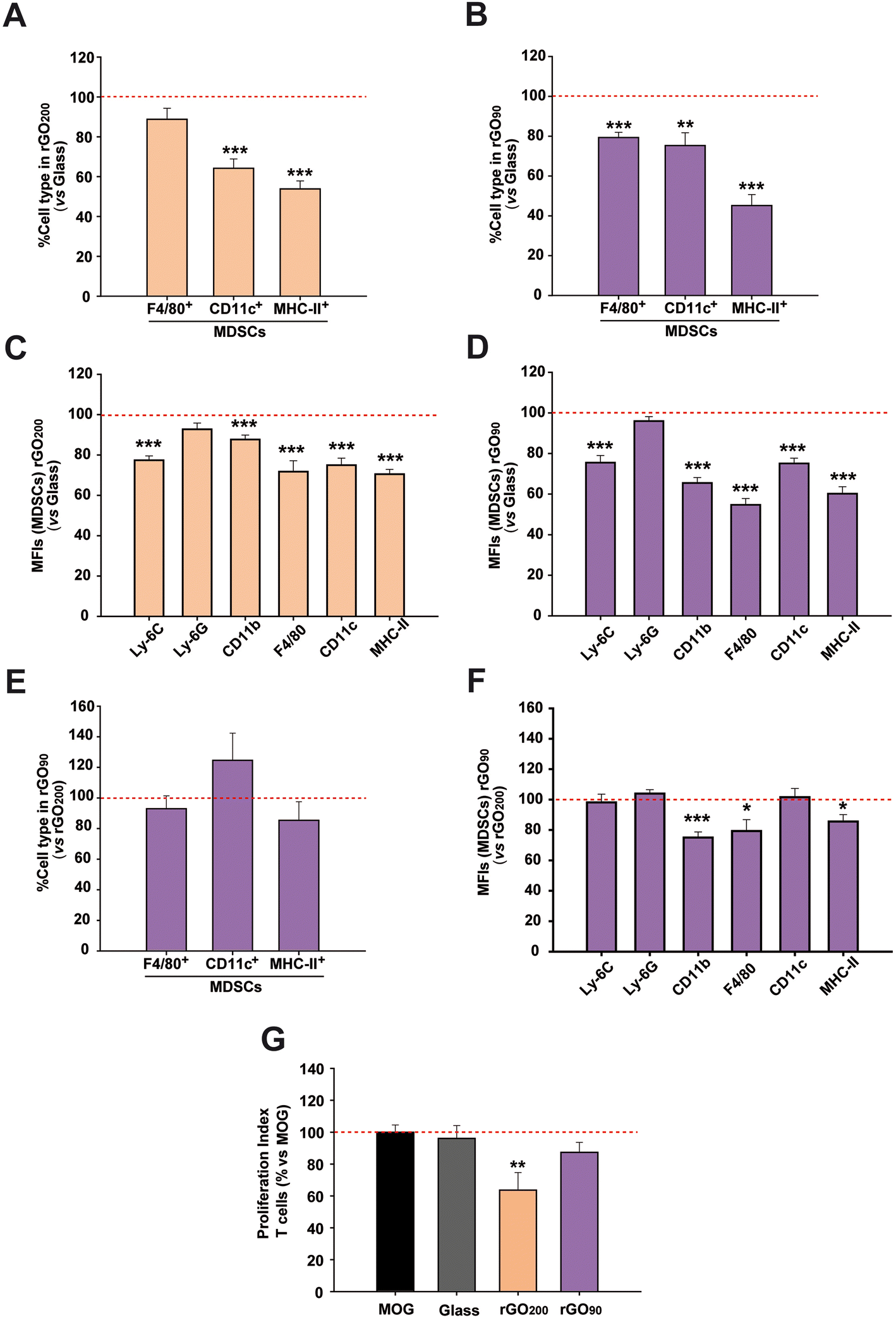 | ||
| Fig. 3 The physico-chemical properties of rGO culture substrates seem pivotal to modulate the immunosuppressive activity of bone marrow MDSCs. (A–D) Both rGO200 and rGO90 induced a marked reduction in the cell percentage (A and B) and expression (mean fluorescence intensity, MFI; (C and D) values of cells presenting the selected maturation markers compared to those cells cultured on glass coverslips (red dashed lines). (E and F) This effect was more prominent when exposed to rGO90, as expressed both in cell percentage and MFI, respectively. (G) The exposition to rGO200, but not to glass or rGO90, more effectively preserved their immunosuppressive activity over MOG-stimulated T cells [red dashed lines represent the proliferative activity of T cells stimulated by MOG, the immunogen used for the induction of this animal model (positive control), – in the absence of bone marrow MDSCs]. Data are shown as the mean ± standard error of the mean of N = 3 independent experiments for (A–F) and N = 5 independent experiments for (G). | ||
To investigate how the culture on rGO200 and rGO90 films could impact the immunosuppressive activity of bone marrow MDSCs, cells were co-cultured with MOG-stimulated splenocytes isolated from EAE mice sacrificed at the peak of the clinical course. As for splenic MDSCs, these bone marrow MDSCs lost their immunosuppressive activity over antigen specific T cells when cultured on glass coverslips (PI vs. non-stimulated splenocytes: MOG-stimulated splenocytes = 8.6 ± 1.2; glass coverslip = 7.4 ± 1.2; p = 0.649). Contrarily, this immunosuppression was preserved when previously exposed to rGO200 films (PI: GO200 = 4.4 ± 1.0; p < 0.05 vs. MOG), but not when exposed to rGO90 (PI: rGO90 = 8.4 ± 2.0; p = 0.141 vs. MOG; Fig. 3G), despite the reduction in bone marrow MDSC differentiation markers also found for rGO90. These data indicate that bone marrow MDSCs cultured on rGO200 films, but not those on rGO90, retain the physiological ability to prevent the proliferation of T lymphocytes responsive to the immunogen causing the development of the EAE MS model.
The exposure to rGO substrates prevented the increase in cell size and morphology of bone marrow MDSCs in culture, being more pronounced for rGO90
In our study, although both types of rGO induced a marked reduction on MDSC differentiation markers, the more accentuated modulation after culture on rGO90 substrates, together with the loss of immunosuppression on those cells, should involve other cellular events rather than the maintenance of immaturity. Since alterations in the morphology of cultured peripheral myeloid cells have been associated with changes in cell functionality,30 a morphological analysis of bone marrow MDSCs was next carried out by SEM (Fig. S6†). Consistent with the data described earlier for immunophenotyping, culture on rGO prevented bone marrow MDSCs from acquiring morphological characteristics of myeloid cells in the process of spontaneous maturation on glass coverslips (i.e. an expanded cytoplasm and presence of protrusions; Fig. 4A). Specifically, bone marrow MDSCs on both types of rGO films showed a significantly lower total cell and protrusion area (Fig. 4B and C, respectively). In fact, the exposition to rGO200 and rGO90 induced a respective reduction of 30.51 ± 3.36% and 41.03 ± 1.71% in total cell area, together with a 60.44 ± 5.71% and 71.04 ± 3.96% reduction in the area of the protrusions vs. glass, respectively. When expressed as a percentage of the total cell area, protrusions also significantly diminished in cells cultured on rGO substrates (Fig. 4D; 38.66 ± 6.38% and 40.72 ± 6.66% of reduction vs. glass for rGO200 and rGO90, respectively). Similarly, the main cytoplasm area markedly decreased in comparison to glass (Fig. 4E), showing a 12.89 ± 2.86% and 23.37 ± 1.74% of reduction for rGO200 and rGO90, respectively. Indeed, culture on rGO90 induced a more pronounced shrinkage on bone marrow MDSCs than rGO200, (percentage of reduction of cells grown on rGO90vs. rGO200: total cell area: 15.14 ± 2.47%; area of the protrusions: 26.79 ± 9.99%; percentage of protrusions area: 3.36 ± 10.86%), although only statistically significant for the main cytoplasm area reduction in rGO90vs. rGO200: 12.03 ± 1.99%. All these data indicate that rGO films maintain bone marrow MDSCs in a less expanded morphology, such effect being significantly more pronounced for the more oxidized substrate (rGO90). In agreement with differentiation findings presented above, the more dramatic changes induced by rGO90 may indicate that the slight but significant differences in the physico-chemical properties of these two rGO substrates (i.e. roughness and redox state) markedly and dissimilarly impact bone marrow MDSC behaviour.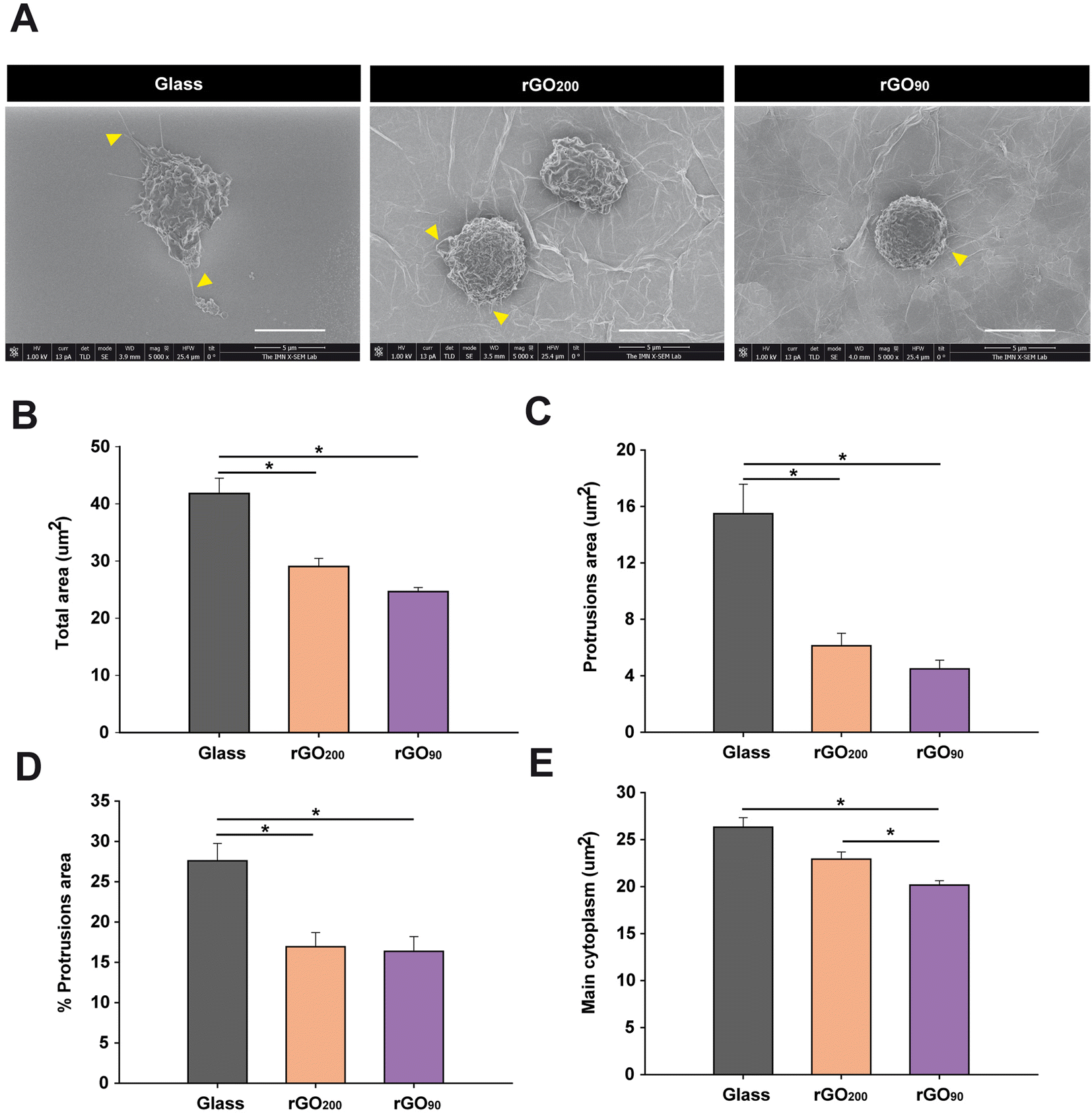 | ||
| Fig. 4 rGO substrates significantly affect bone marrow MDSC size and morphology. (A) Representative SEM images of MDSCs after 24 h culture on glass coverslips, rGO200 and rGO90. Yellow arrowheads indicate examples of cell protrusions. Scale bars represent 5 μm. (B–E) Both types of rGO films impeded cell size increase and protrusion expansion compared to cells grown on glass coverslips. Statistics: ANOVA on RANKS; p < 0.001 in all cases; differences between each group and MOG-stimulated are represented by *p < 0.05 after a Dunn's post hoc test. Data are shown as the mean ± standard error of the mean of n = 85 (glass coverslip), n = 57 (rGO90) and n = 66 (rGO200) cells from N = 2 independent experiments. | ||
The physico-chemical differences of rGO substrates dissimilarly target bone marrow MDSC viability
The differentiation impairment in bone marrow MDSCs seems to be related to their maintenance in a highly immature state. However, the more exacerbated response in terms of reduction of phenotypic markers and smaller sizes found in cells cultured on rGO90 films could be also explained as a result of their entry into programmed cell death (apoptotic) pathways. In order to verify this hypothesis, an annexin V (AV)/propidium iodide (PrI) assay was carried out (Fig. 5A). We first found that the percentage of viable bone marrow MDSCs (AV−PrI−) within the total cells cultured on rGO200 was similar to that found on glass coverslips (Fig. 5B). However, rGO90 dramatically reduced the percentage of viable cells after 24 h in culture. Importantly, both types of rGO substrates reduced the percentage of cells in the first stages of apoptosis (early apoptosis; AV+PrI−) compared to glass coverslips, being even lower in the case of rGO90 (Fig. 5C). Finally, whereas neither rGO200 nor rGO90 altered the percentage of unviable cells (late apoptosis/death; AV+PrI+) within the whole cultured cells (Fig. 5D), rGO90 promoted a remarkable increase in the proportion of dead cells compared to both glass and rGO200 (Fig. 5E). Then, these results corroborated a significant impact of rGO films on bone marrow MDSCs viability, markedly modulated again by the slight but different physico-chemical properties of these two types of rGO films.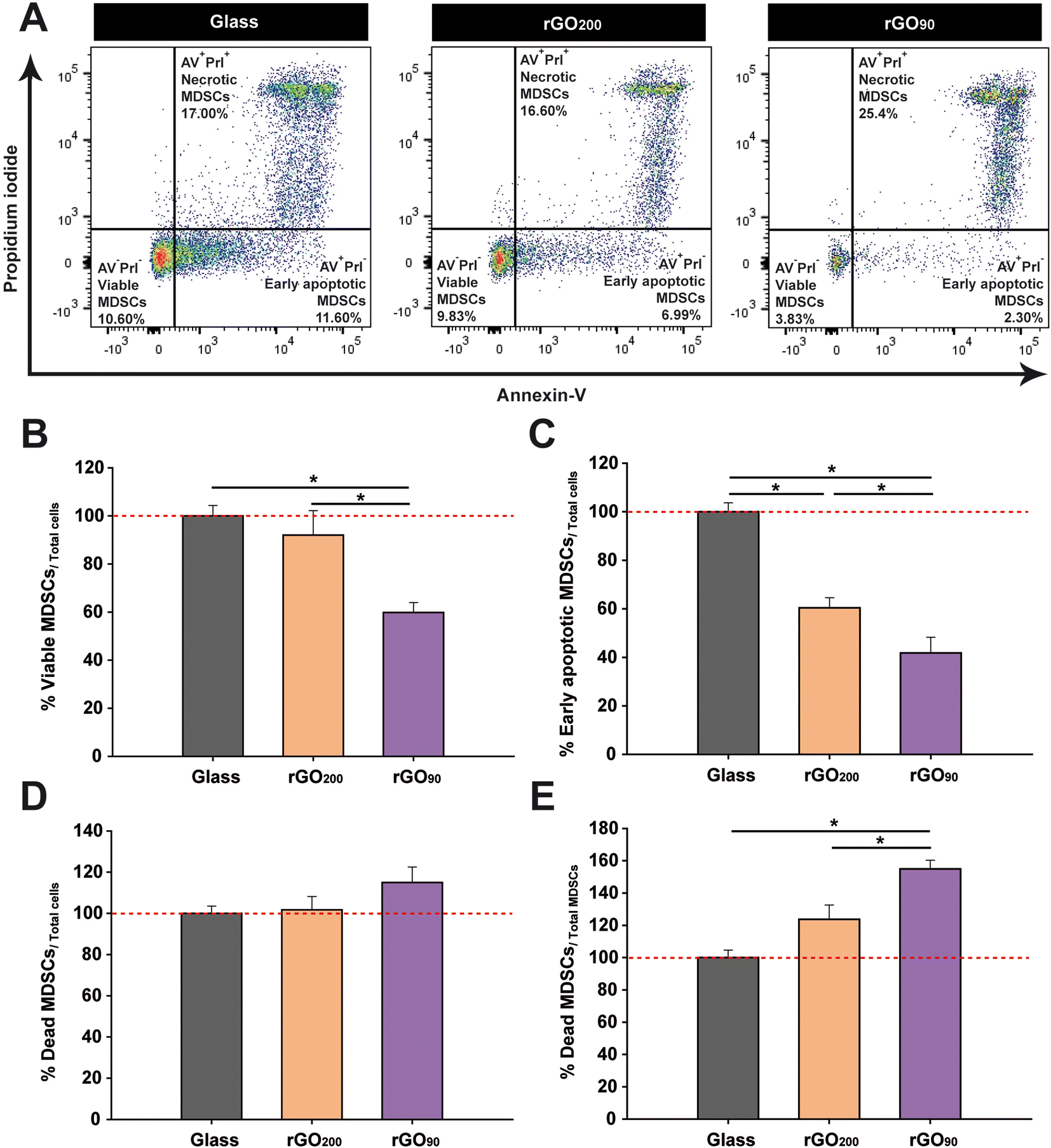 | ||
| Fig. 5 rGO substrates significantly impact viability and cell death (apoptosis) of bone marrow MDSCs depending on their physico-chemical properties. (A) Representative dot plots of a cell viability assay with annexin V (AV) and propidium iodide (PrI) on glass coverslips, rGO200 and rGO90. Viable (AV−PrI−), early apoptotic (AV+PrI−) and late apoptotic/dead (AV+PrI+) cells were defined. (B) rGO90 dramatically decreased MDSC viability compared to both glass and rGO200 substrates. (C) Both rGO substrates reduced the presence of early apoptotic MDSCs, being more remarkable in the case of rGO90. (D) rGO substrates had no impact on the percentage of late apoptotic MDSCs with respect to total cultured cells. (E) rGO90 induced a statistically significant enrichment in late apoptotic cells within the population of MDSCs with respect to glass and rGO200. Statistics: ANOVA (C–E) or ANOVA on RANKS (B) was carried out (**p < 0.01 in B, ***p < 0.001 in C and E, and p = 0.121 in D). Data are shown as the mean ± standard error of the mean of N = 3 independent experiments. | ||
Discussion
To the best of our knowledge, this study is the first highlighting the relevance that the physico-chemical properties of rGO play on the activation and phenotype modulation of MDSCs. More importantly, it is the first to prove the capacity of a nanomaterial-based film (rGO200) to preserve MDSCs in an immature and modulatory phenotype with therapeutic potential for autoimmune diseases.Studies addressing the modulation of splenic MDSCs by the exposure to different biomaterials are very scarce in the literature, only circumscribed to cancer research and without analyzing each MDSC subset (polymorphonuclear or monocytic) separately. In this sense, immunosuppressive activity of MDSCs has been downregulated through their exposure to the DNA demethylating agent hydralazine (together with mitoxantrone as a chemotherapeutic agent) by means of a multifunctional nanoplatform.31 Moreover, repolarization of MDSCs toward a more pro-inflammatory phenotype has been also proved by using two cationic polymers: cationic dextran and polyethylenimine.32 In this context, our study pioneers to prove specific immunomodulation on the subset of monocytic MDSCs, when isolated from both the spleen and the bone marrow of mice with EAE.
Importantly, most of the studies carried out to date with GDMs and immune cells have been performed with nanomaterials in suspension, likely maximizing their impact on cell responses. Indeed, the exposure to nanomaterials in solution is expected to have a larger biological impact as the contact surface for interaction for both cells and nanomaterials is maximized, so internalization might be boosted. Our particular approach deals with rGO substrates in which rGO nanosheets are integrated in stable films attached to the underlying glass coverslips. In fact, our DLS data proved that drifting rGO nanosheets were almost absent in all culture conditions, thus ruling out any major biological responses mediated by rGO nanosheets in suspension. Interestingly, previous studies by others have demonstrated that GO films could induce cell proliferation and apoptosis by cell contact, independently of cell engulfment. For instance, Escudero et al. demonstrated that coating CoCr alloys with electrochemically reduced GO caused lower damage to cell plasma membranes in J774A.1 macrophages than pristine alloys.33 In a different study, the diminished GO internalization observed in bone marrow macrophages with a significant deficiency in phagocytosis did not affect cell necrosis induced by these GO nanosheets.34 In the same sense, previous work has shown that neither pristine nor functionalized graphene flakes induced any direct effect on human T-cell proliferation and suppression.35 Interestingly, a former study using an immune array of human peripheral blood mononuclear cells (PBMCs) provided convincing evidence that the adaptive immune response observed after GO exposure was not directly, but indirectly, affected by the primary effect on myeloid cells. Indeed, genes involved in direct T-cell activation, such as IL-2 and IFN-γ, were not affected, which correlated with the absence of CD69 and CD25 expression in these cells after GO exposure.36
In our study, GO films were early discarded as culture substrates because GO nanosheets without any thermal treatment got rapidly released from the films, thus causing a major biological impact of nanomaterials mediated by interactions in suspension rather than from the culture substrate. Thermal conditions for rGO90 were chosen as the ones sufficient both to significantly reduce GO nanosheets (O/C ratio from 0.86 to 0.48) and to preserve the integrity of the film without nanosheets release. Finally, those conditions to obtain rGO200 were designated again to produce a further significant reduction of rGO (O/C ratio of 0.10). Based on previous work by others, the oxygen-containing groups remaining in our rGO200 films would need much higher temperatures (>600 °C) to be effectively removed from the films.37 Indeed, our observations clearly indicate that annealing at 200 °C is an optimal option to maintain MDSCs in a highly active anti-inflammatory state. This observation is in agreement with the decreased oxidative stress and pro-inflammatory cytokine secretion observed in RAW-264.7 macrophages after exposure to suspensions of rGO nanostructures also annealed at 200 °C for 30 min.20 In both cases, the thermal reduction of GO at 200 °C improved the biocompatibility of this nanomaterial. Therefore, our data provide further insights into the possibility of triggering an anti-inflammatory response by GDMs, such as the site-specific Th2 response induction (i.e. a significant increase in IL-5, IL-3, IL-33, and their soluble receptor, sST2), as previously postulated by Wang et al. for T cells exposed to graphene nanosheets in suspension.38 Our data also indicate that, by modulating the physico-chemical characteristics of rGO culture substrates, we might exert either pro- or anti-inflammatory effects on bone marrow MDSC activity. These findings are in agreement with previous reports showing the ability of GDMs in suspension to induce changes on immune cell activity. For instance, small GO nanosheets were able to up-regulate critical genes involved in both Th1 and Th2 immune responses such as CSF2, TNF, IL6, IL10, CD80, IL1, IL1R1, TICAM1, IL8, IL23A, NFKB1, TBX21, CD40, CCR6, and IFNAR1 on human PBMCs. Also, this GO nanomaterial stimulated the release of some Th1 but also Th2 cytokines such as IL-1β, IL-1α, TNF-α, IL-10, IL-6, and IL-8 in the same cells.36
Our morphological studies by SEM showed that both rGO200 and rGO90 inhibit the spontaneous increase in cell size and protrusion elongation typical of myeloid cells in culture.30 In agreement with our data, Sasidharan et al. showed that pristine graphene flakes in suspension (fabricated by the arc discharge physical method) impeded myeloid cells such as RAW264.7 macrophages to attain their normal stretched morphology and the formation of filopodial extensions,35 this effect being significantly reverted by carboxyl functionalization of the flakes. In a different work, Hung et al. showed that GO significantly elicited dendritic and/or round-like morphologies in human monocytes as compared with the well-spread morphology of the control group (glass coverslips).39 Although our data point towards the direct physical interaction with rGO films in culture as responsible for this limitation in cell body spreading, we cannot exclude the possibility of an indirect effect of rGO. It has been previously shown that changes in myeloid cell morphology induced by GDMs are mediated in an autocrine manner.40 When cultured in control conditioned media collected from untreated RAW264.7 cells, naïve RAW264.7 cells frequently displayed a multipolar stellate morphology, typical for macrophage polarization. However, when exposed to conditioned media collected from graphene-exposed RAW264.7 cells, naïve RAW264.7 cells showed a polar spherical phenotype, likely mediated by soluble graphene-induced factors. Contrarily, macrophages grown on electrochemically reduced GO presented elongated cell bodies with characteristic protrusions.33 These controversial observations are probably related to the different physico-chemical properties of the GDMs under investigation, the cell source and phenotype, and the specific strategy followed for cell culture.
Our data also suggest that rGO90, but not rGO200, exerts cytotoxic activity on bone marrow MDSCs as its exposure induces both a reduction in the number of viable cells and an increase in those at a late stage of cell apoptosis. To this regard, the toxicity of GDMs on myeloid cells is still a matter of debate.41 Some authors have reported the so-called “mask effect” to explain cell toxicity induced by GDMs.42 Specifically, the close contact between GO nanosheets and cell membranes could drive to GO internalization. Once inside, GO could directly affect cell parameters such as viability, redox state and activation. The normal signalling and communication of cells with their environment could be also significantly altered by the presence of GO nanomaterials in their surroundings, thus influencing cell homeostasis and altering cell death, proliferation and activation cascades. In this sense, the smaller the size of GO sheets, the larger the mask effect expected. For instance, GO nanosheets (lateral dimension of 1–2 μm) in suspension were found to induce a toll-like receptor 4-mediated necrosis in different lines of macrophages, with a clear decrease on cell protrusions.34 When tested in vivo, subdermal inoculation of GO nanosheets in a rat model increased CD163+ macrophages (M2-like phenotype) compared to control animals, whereas no changes in CD86+ macrophages (M1-like) were detected.39 In sum, myeloid cells are able to engulf and process GDMs, markedly depending on physico-chemical properties such as shape, size, and chemical functionalization of the nanomaterials, which might either induce pro/anti-inflammatory responses or lead to the modulation and disruption of the immune activity.43
Other authors have also postulated nanomaterial concentration as an important factor for the control of the activity of MDSCs. In a particular study with MDSCs derived from PBMCs of healthy human donors, low concentrations of GO nanosheets in suspension (2.5 and 5 μg mL−1) induced MDSC proliferation, while a higher concentration (10 μg mL−1) reduced their viability.44 Other than these properties, chemical modifications (i.e. functionalization) of GDMs have also demonstrated to significantly impact their toxicity and ability to modulate myeloid cell interactions. For instance, polyethylene glycol (PEG) reduced the risk of immune responses of nanomaterials by increasing their stability in physiological conditions and minimizing their interaction with biomolecules. Particularly, Feito et al. described how PEGylated GO diminished the proliferation of macrophages in a concentration-dependent fashion.45 In combination with polyethylenimine to functionalize GO and serve as an adjuvant, PEG promoted DC maturation and enhanced cytokine secretion by acting over toll-like receptor-dependent pathways.46 However, no data are available to date about the effect of GO functionalization over MDSCs, a field that seems pivotal for its future therapeutic use in nanomedicine. It is important to note that other GDMs such as carbon nanotubes could also represent attractive immunomodulatory tools for MDSCs, based on their capacity to positively interact with immune cells such as DCs without altering the expression of some of their specific markers and to promote specific actions on the polarization of T lymphocytes.47,48 Moreover, oxidized multiwall carbon nanotubes initiate macrophage phagocytosis and upregulate CD14, CD11b, TLR-4/MD2, and CD206, without altering MHC-II expression. Macrophages activated with these nanomaterials produce angiogenesis-related cytokines such as MMP-9 and VEGF and reduce levels of pro-inflammatory cytokines.49 The understanding and precise control of the physico-chemical properties of these carbon-based nanomaterials is pivotal for exploiting their therapeutic potential.
In our studies, different thermal annealing conditions resulted in rGO films with slightly but significantly different roughness and redox states. These both physico-chemical parameters were closely dependent on each other based on the thermal treatment used, with the latter being expected to have a superior implication given its more substantial difference between rGO90 and rGO200 participating in the opposite modulation of immunological responses from endogenous monocytic MDSCs isolated from the bone marrow in an autoimmune disease. The specific molecular mechanisms behind the different modulation of MDSC behaviour driven by these two physico-chemically dissimilar rGO substrates will be the focus of future work in our laboratory. Nonetheless, we can hypothesize some pivotal actors contributing to these results based on previous findings in comparable systems. Extensive literature has already outlined the pivotal role that the biophysical cues of biomaterials including nanomaterials play in the manipulation of (stem) cell fates.50 Based on previous work by others, we theorize that the MDSC affectation mediated by rGO90 is likely related to its higher oxygen content in comparison to rGO200. Although still not investigated for MDSCs, published data on neural cells consistently indicate induction of reactive oxygen species (ROS) production, primarily through activation of mitochondria and NADPH oxidases located in the cytoplasm and plasma membrane. These events are followed by lipid peroxidation, cell membrane damage and ROS-disrupted mitochondrial homeostasis associated to the exposure to more oxidized forms of GO nanomaterials in suspension.51 In these studies, the autophagy-lysosomal network was proved to be initiated as a defensive reaction to eradicate oxidative damage to mitochondria, whose failure due to lysosomal dysfunction exacerbated mitochondrial stress and led to cell apoptosis/necrosis. In a different work with a human monocyte cell line (THP-1 cells), a reduction in GO was directly associated with a lower cytotoxicity; while more oxidized GO forms led to increased lipid peroxidation, membrane leakage and cell death.52 In agreement with this, our rGO90 films (with a higher content in oxygen-containing groups as proved by XPS studies) promoted lower viability and higher late apoptotic percentages in comparison to glass and rGO200 substrates.
In our studies, different thermal annealing conditions of GO coatings resulted in rGO films with slightly but significantly different roughness and redox states. These both physico-chemical parameters are therefore closely dependent on each other in this model, being difficult to separate the respective contribution of each one on MDSC responses. Nonetheless, given the more dramatic changes in O/C ratios in comparison to roughness values, we hypothesize the former as a more relevant parameter for the effects found on MDSCs growth, differentiation state and immune functionality.
Thermal treatment of rGO materials typically induces the removal of oxygen-containing groups and the recovery of sp3/sp2 configurations and π–π transitions coming from graphitic structures.37 As a consequence of these chemical changes, there is a certain recovery of the original stacking of the graphene sheets (no longer repulsed from each other by the oxygen-containing groups) that could drive to a more effective disposition of the sheets and, therefore, a lower roughness of the surface of the resulting films. Surface roughness has been also proved to decisively impact cell fate. For instance, scaffold roughness values around 70 nm promoted osteogenic gene expression in mesenchymal stem cells, while those around 15 nm enhanced chondrogenic gene expression of this same cell type.53 Additionally, smooth glass surfaces (Rq = 1 nm) supported rapid proliferation and long-term self-renewal of human embryonic stem cells, while rough glass surfaces at the nanoscale (Rq = 150 nm) induced their spontaneous differentiation.53 Further work has even identified a roughness threshold for controlling pluripotency of embryonic stem cells (Rq < 392 nm for long-term maintenance of pluripotency; Rq > 573 nm for faster unidirectional differentiation).54 Importantly, this type of behaviour has been demonstrated to be cell-type specific, so precise conditions must be defined case by case. Moreover, although extensively explored for a diversity of cells including stem cells, immune cells have been scarcely explored in these scenarios, so conclusions must be taken carefully. For instance, differences between GO and rGO nanosheets have been explored in terms of cell toxicity, morphology and immunomodulation of bone marrow-derived macrophages with rGO being more cytotoxic than GO, with a marked reduction on cell complexity and exhibiting a more pro-inflammatory activity.55 Although these cell-specific effects exerted by rGO were concomitant with a substantial modification of nanosheet morphology (i.e., from a sheet-like structure for GO to a curly polygonal shape for rGO), its significance on cellular activity and the mechanisms underlying this impact on immune cells were not further explored. In a different study, it has been also reported that thermally-reduced GO sheets promoted RAW-264.7 macrophages polarization towards a more anti-inflammatory activity state.38 In such work, although the thermal reduction of GO provoked a more crumpling surface morphology of the tested sheets compared to the flat-like GO, the direct effect of this morphological change on macrophage cell activity was not explored. rGO200 films are herein presented as attractive substrates for the preservation of the immature phenotype of MDSCs in culture, with the consequent potential for their use as a cell therapy for immune diseases. Alternatively, those based on rGO90 films could be of interest for the implementation of novel therapeutics in cancer, including the incorporation of stimuli-responsive nanomedicines with a mediated activation by external stimuli such as light and radiation.56
Conclusions
We have demonstrated that rGO200 films (O/C ratio = 0.10 and Rq = 38.8 nm) keep the immunosuppressive activity of MDSCs in vitro. Our results also prove that, by controlling the particular physico-chemical properties of GDM-based substrates (e.g., surface roughness and redox state), one could minimize toxicity effects and take advantage of the full potential of these nanomaterials in the field of immunotherapeutics. Our findings open new perspectives in the use of rGO-based culture platforms for the expansion of MDSCs ex vivo as an autologous cell therapy for autoimmune pathologies such as MS. To the best of our knowledge, this is the first report of a GO-based film capable of modulating the phenotype of MDSCs, a cell subset with a clear therapeutic potential. They also emphasize the enormous versatility of rGO nanomaterials, which are being discovered as powerful tools for biomedicine. If properly controlled, the immunomodulatory effects of highly reduced GO substrates on MDSCs could be exploited for the treatment of diseases with a clear autoimmune etiology, such as MS. Conversely, a more oxidized form of GO (O/C ratio = 0.48 and Rq = 63.6 nm) would be an excellent nanotechnological tool for cancer, in which the elimination of MDSCs appears to be a successful strategy for the control of the exacerbated immune evasion that is behind the uncontrolled growth of tumour cells.Experimental
Preparation of rGO films
GO slurry (Graphenea, S.A.; Batch #C1250/GOB125/D; 4.6 wt% concentration, >95% monolayer content) was dispersed into distilled water by gentle mixing (∼10 mg mL−1). This GO suspension was used to homogeneously coat circular glass coverslips (10 mm in diameter) by spin-coating at 3600 rpm for 10 s (30 μl per glass coverslip, pre-stabilization of the drop for 1 min at room temperature). The so-obtained GO films were then exposed to two different thermal treatments: (i) 90 °C for 15 min (more oxidized state, rGO90) and (ii) 200 °C for 30 min (more reduced state, rGO200). The rationale behind the selection of these thermal treatments is explained in the Results section.Physico-chemical characterization of GO slurry and rGO films
TEM studies were performed by using a Jeol JEM 1010 microscope (Tokyo, Japan) at 80–100 kV with a coupled digital camera (Gatan SC200, Pleasanton, CA, USA) for image acquisition. Surface topography of rGO films was visualized by using a last generation ultrahigh resolution FEI VERIOS 460 scanning electron microscope. The analysis of the elemental and chemical composition of GO, rGO90 and rGO200 samples was performed by XPS. Experiments were carried out in an UHV chamber with a base pressure of 10−10 mbar equipped with a hemispherical electron energy analyzer (SPECS Phoibos 150 spectrometer) and a 2D delay-line detector, using an X-ray source of Mg-Kα (1253.6 eV). XPS spectra were acquired at normal emission take-off angle, using an energy step of 0.50 and 0.05 eV and a pass-energy of 40 and 20 eV for survey spectra and detailed core level regions, respectively. Surface charging effect built up upon the photoemission experiments, which produces both energy shift and lineshape modifications of the spectra, has been compensated using a low energy electron flood gun. The spectra were analyzed with the CasaXPS program (Casa Software Ltd, Cheshire, UK) using a Shirley method for background subtraction and data processing for quantitative XPS analysis. Spectra are displayed after the subtraction of the contribution of the Mg-Kα satellite emission. The absolute binding energies of the photoelectron spectra were determined by referencing to the sp2 transition of C 1s at 284.6 eV determined from a freshly cleaved HOPG (Highly Oriented Pyrolytic Graphite) sample. The overall sample composition was determined from survey spectra which showed intense signals from C and O, as well as a minor contribution from Na, K and S (only in GO and rGO90 samples). O/C atomic concentrations were determined by measuring the integral areas of O 1s and C 1s spectra, taken at a pass energy of 20 eV, after background subtraction and normalization using the sensitivity factors proportional to the Scofield cross section provided by the electron energy analyzer manufacturer.24AFM studies were carried out by using a Nanoscope V forces microscope (Bruker) in tapping mode with a TESPSS tip (Bruker). Surface roughness parameters (i.e. Rq, Ra and Rmax) were obtained by using the Nanoscope Analysis software v2.0. Rq is the root-mean-square (rms) value of the profile heights over the evaluation length. Ra is defined as the arithmetic average of the profile heights over the evaluation length. Rmax is the largest of the successive values of the vertical distance between the highest and lowest points of the profile calculated over the evaluation length.
Culture media after incubation with both types of rGO culture substrates were analyzed by DLS for the detection of rGO nanosheets released from the substrates. Measurements were carried out in a ZetasizerNano ZS instrument (Malvern). Temperature was set to 25 °C and samples were diluted with MilliQ distilled water. Data analysis was performed considering the Gaussian distributions intensity-weighted and numbered weighted, obtaining the Z average and PDI from the first, and the mean particle size in number from the second one.
EAE induction
Female 6 to 8 weeks-old C57BL/6 mice were purchased from Janvier Labs (France). Chronic progressive EAE model was induced by subcutaneous immunization with 200 μg of myelin oligodendrocyte glycoprotein peptide (MOG35–55, Genscript, New Jersey, USA) emulsified in complete Freund's Adjuvant (CFA) containing 4 mg mL−1 of inactivated particles of Mycobacterium tuberculosis (H37Ra; BD Biosciences, Franklin Lakes, New Jersey, USA) and at a final volume of 200 μL. Each mouse was intravenously administered 250 ng of pertussis toxin (Sigma Aldrich, St Louis, MO, USA) diluted in 100 μL of phosphate buffered saline (PBS, pH 7.4) through the tail vein the day of immunization and 2 days later. Clinical scores were evaluated on a daily basis in a double-blind manner according to the following scale: 0, no detectable signs of EAE; 1, paralyzed tail; 2, weakness or unilateral partial hindlimb paralysis; 3, complete bilateral hindlimb paralysis; 4, total paralysis of forelimbs and hindlimbs; and 5, death. Following ethical standards and regulations, humane endpoint criteria were applied when an animal reached a clinical score ≥ 4, when clinical score ≥ 3 was reached for more than 48 h or whether existed self-mutilation, persistent retention of urine, 35% weight loss and signs of stress or pain for more than 48 h, even if EAE score was <3. Animal manipulations were approved by the institutional ethical committees (Comité Ético de Experimentación Animal del Hospital Nacional de Parapléjicos), and all experiments were performed in compliance with the European guidelines for animal research (European Communities Council Directive 2010/63/EU, 90/219/EEC, Regulation No. 1946/2003) and the Spanish National and Regional Guidelines for Animal Experimentation (RD 53/2013 and 178/2004, Ley 32/2007 and 9/2003, and Decreto 320/2010).Splenic and bone marrow MDSC sorting
For splenic-MDSC isolation, fresh spleens were collected from euthanized mice at the peak of their clinical symptoms defined as the day with a repeated clinical score of 3. The tissue was homogenized to a single cell suspension, passed through a 40 μm filter (BD Biosciences) and washed in Roswell Park Memorial Institute (RPMI) medium (Gibco-Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 2 mM L-glutamine (Thermofisher), 10% heat-inactivated Fetal Bovine Serum (FBS, Capricorn) and 1% penicillin/streptomycin (P/S, Gibco). Erythrocytes were lysed with ACK lysis buffer (8.29 g L−1 NH4Cl, 1 g L−1 KHCO3, 1 mM EDTA in distilled H2O at pH 7.4) and the remaining splenocytes were centrifuged at 437g for 5 min and resuspended in supplemented PBS (sPBS: 10% FBS, 25 mM HEPES and 2% P/S diluted in sterile PBS).To obtain MDSCs from bone marrow, mice were euthanized at the peak of their clinical symptoms defined as for splenic MDSCs. Both femurs and tibiae were dissected out and the bone marrow was flushed with supplemented RPMI media. Bone marrow cells were centrifuged at 437g for 5 min and the obtained pellet was resuspended and red cell removed with ACK lysis buffer. After that, BM-derived cells were centrifuged at 437g for 5 min, resuspended in sPBS, and passed through a 100 μm filter (BD Biosciences).
Splenocytes or bone marrow cells were resuspended in sPBS and the Fc receptors were blocked for 10 min at 4 °C with 10 μg mL−1 anti-CD16/CD32 antibodies (BD Biosciences 553142). After blocking, FITC conjugated rat anti-mouse Ly-6C (AL-21 clone), PE conjugated rat anti-mouse Ly-6G (1A8 clone) and PerCP-Cy5.5 conjugated rat anti-mouse CD11b (M1/70 clone) antibodies were added to the cell suspension and incubated for 30 min at 4 °C in the dark (Table S1†). The samples were then rinsed with sPBS, centrifuged at 371g for 5 min, resuspended again in sPBS, and sorted in a Fluorescence Activated Cell Sorter (FACS) Aria (BD Biosciences) located at the Flow Cytometry Service of the Hospital Nacional de Parapléjicos. Monocytic splenic and bone marrow MDSCs were identified as Ly-6Chigh Ly-6G−/low gated on CD11b+ myeloid cells and recovered at a purity >95%.
MDSC cultures on rGO substrates
Splenic and bone marrow MDSCs were resuspended in Iscove's Modified Dulbecco's Medium (IMDM; Biowest) supplemented with 10% FBS, 1% P/S, 50 μM β-mercaptoethanol (β-ME; Sigma), and 2 mM L-glutamine (Thermofisher). A total of 105 MDSCs were seeded on glass coverslips or reduced GO (rGO)-based substrates. After 24 h, MDSCs were harvested, centrifuged at 371g for 5 min and resuspended in the appropriate medium for further experiments.Splenocyte stimulation and co-culture with MDSCs
Single-cell suspensions from the spleen were prepared as described above and splenocytes were exposed to 2-μM Tag-it Violet™ Proliferation and Cell Tracking Dye (Biolegend) diluted in PBS supplemented with 0.1% bovine serum albumin at 37 °C for 20 min in darkness following manufacturer's protocol. After washing, 2 × 105 Tag-it Violet-labelled splenocytes were cultured in 96-well U-bottom plates (Nunclon Delta Surface; Thermo Scientific, Waltham, MA, USA) at a final volume of 200 μL of supplemented IMDM per well. Splenocytes were stimulated for 72 h with 5 μg mL−1 MOG35–55 (or culture medium alone for the unstimulated controls). To analyze the effect of MDSCs growing on rGO substrates, cells were collected from glass coverslip/rGO90/rGO200 devices and the number of viable cells was quantified using the Trypan Blue viability dye. After that, 5 × 104 live MDSCs were exposed to pre-activated splenocytes during 24 h at a 1:4 ratio (MDSCs:splenocytes). After 48 h, all cells were harvested from the culture plates and the cell Fc receptors were blocked for 10 min at 4 °C with 10 μg mL−1 anti-CD16/CD32 antibodies (BD Biosciences 553142). Then, cells were incubated for 20 min at 4 °C in the dark with the following fluorochrome-conjugated monoclonal antibodies: anti-CD11b PerCP-Cy5.5 (M1/70 clone), anti-CD3e APC (145-2C11 clone), anti-CD4 PE (RM4-5 clone), and anti-CD8a FITC (53-6.7 clone; all from BD Biosciences). Finally, T cell proliferation, indicated by Tag-it Violet dilution, was analyzed by flow cytometry in a fluorescence activated FACS Canto II analyzer (BD Biosciences) located at the Flow Cytometry Service of the Hospital Nacional de Parapléjicos. The data obtained was analyzed using the FlowJo 10.6.2 software (Tree Star Inc.).
Phenotypic and cell viability analysis of MDSCs grown on rGO substrates
After 24 h of culture on glass coverslips or rGO-based films, MDSCs were harvested, centrifuged at 371g for 5 min, and washed in PBS. Cell suspensions were then used for phenotypic and viability analyses. For myeloid markers analysis, Fc receptors from splenic or bone marrow MDSCs were blocked for 10 min at 4 °C with 10 μg mL−1 anti-CD16/CD32 antibodies. Then, cells were labeled for 20 min at 4 °C in the dark with the following fluorochrome-conjugated monoclonal antibodies: anti-Ly-6C FITC (AL-21 clone), anti-Ly-6G PE (1A8 clone) and anti-CD11b PerCP-Cy5.5 (M1/70 clone) all from BD Biosciences; anti-MHC-II PE-Cy7 (M5/114.15.2 clone), anti-CD11c APC (N418 clone), and anti-F4/80 eFluor450 (BM8 clone) from eBioscience-Thermo Fisher Scientific (Table S1†). MDSCs were then rinsed in PBS, recovered by centrifugation at 371g for 5 min, and resuspended in PBS.For cell viability analysis, labelled bone marrow MDSCs were stained with the Annexin V-FITC Apoptosis Detection Kit (Beckman Coulter, IL, Italy) following manufacturer's protocol. Briefly, cells were centrifuged at 93g for 3 min, rinsed in PBS and centrifuged again to remove supernatants. Pellets were resuspended in 100 μl of binding buffer at a concentration of 106 cells per mL and stained with 5 μl of AV-FITC conjugate plus 5 μl of PrI for 15 min at room temperature. The different cell staining patterns were classified as follows: (1) viable cells: double negative for AV and PrI (AV−PrI−); (2) early apoptotic cells: positive for AV and negative for PrI (AV+PrI−), and (3) late apoptotic/dead cells: double positive for AV and PrI (AV+PrI+). In all cases, MDSCs were analyzed in a FACS Canto II cytometer (BD Biosciences). The data obtained were analyzed using the FlowJo 10.6.2 software (Tree Star Inc.).
Morphological characterization of MDSCs on rGO substrates by scanning electron microscopy
Cell culture morphology was studied by SEM. Briefly, culture samples were rinsed in PBS twice and fixed with glutaraldehyde (2.5% in PBS) for 45 min. After gentle washing in distilled water, dehydration was performed by using series of ethanol solutions for 15 min (2 washes) and a final dehydration in absolute ethanol for 30 min. Samples were then dried at room temperature for at least 48 h. After mounting in stubs and coating with a nanometer-thick chromium layer under vacuum, cell culture samples were visualized by using a last generation ultrahigh resolution FEI VERIOS 460 electron microscope. From the images collected (N ≥ 50 per substrate), the impact of rGO substrates on MDSC morphology was characterized by measuring the total cell area, the protrusions area and the main cytoplasm area using the Fiji software (Fig. S6†). To avoid biased effects, measurements were carried out by two independent observers.Statistical analysis
Data are expressed as the mean ± standard error of the mean. Statistical analyses were performed with SigmaPlot v11.0 (Systat Software, San Jose, CA, USA). Student's t-test or Mann–Whitney U test for parametric or non-parametric data, respectively, were used to compare the effect on cell phenotype markers, T cell immunosuppression versus control conditions (glass or MOG-stimulated splenocytes, respectively), and rGO roughness (Rq, Ra and Rmax). An ANOVA test or its corresponding ANOVA on RANKS was performed followed by the Tukey's or Dunn's post hoc tests, respectively, to compare changes in cell morphology and viability. The minimum value of statistical significance considered was p < 0.05 and the results of the analysis were represented as: *p < 0.05; **p < 0.01; and ***p < 0.001.Author contributions
CC-T: investigation, formal analysis, writing – original draft, visualization; IM-D: investigation; RL-G: investigation; AGM: investigation; FJP: investigation, formal analysis, writing – review & editing; MCS: conceptualization, methodology, investigation, writing – original draft, resources, writing – review & editing, visualization, supervision, project administration, funding acquisition; DC: conceptualization, methodology, writing – original draft, resources, writing – review & editing, visualization, supervision, project administration, funding acquisition. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work has been supported by the Instituto de Salud Carlos III (PI15/00963, PI18/00255, PI21/00302, RD16/0015/0019, all co-funded by the European Union, and CB22/05/00016), projects MAT2016-58857-R and PID2020-113480RB-I00 funded by MCIN/AEI/10.13039/501100011033/. CC-T holds a predoctoral fellowship from the Instituto de Salud Carlos III (FI19/00132, co-funded by the European Union). The authors would like to thank Jennifer García and Inmaculada Alonso for their technical assistance. Dr Virginia Vila-del Sol and Ángela Marquina Rodríguez from the Flow Cytometry Core Facility of the Hospital Nacional de Parapléjicos are acknowledged for their technical assistance with flow cytometry studies. We also acknowledge the service from the MiNa Laboratory at IMN and funding from CM (project SpaceTec, S2013/ICE2822), MINECO (project CSIC13-4E-1794) and EU (FEDER, FSE) for SEM studies. FJP acknowledges financial support from Grant PID2021-126169OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”. The ICTS – Centro Nacional de Microscopía Electrónica from the Universidad Complutense de Madrid is acknowledged for assistance with AFM studies.References
- C. Walton, R. King, L. Rechtman, W. Kaye, E. Leray, R. A. Marrie, N. Robertson, N. La Rocca, B. Uitdehaag, I. van der Mei, M. Wallin, A. Helme, C. Angood Napier, N. Rijke and P. Baneke, Mult. Scler. J., 2020, 26, 1816–1821 CrossRef PubMed.
- S. L. Hauser and B. A. C. Cree, Am. J. Med., 2020, 133, 1380–1390 CrossRef CAS PubMed.
- L. Freeman, E. E. Longbrake, P. K. Coyle, B. Hendin and T. Vollmer, CNS Drugs, 2022, 36, 1285–1299 CrossRef CAS PubMed.
- G. Arrambide, E. Iacobaeus, M. P. Amato, T. Derfuss, S. Vukusic, B. Hemmer, L. Brundin, M. Tintore, J. Berger, A. Boyko, V. Brinar, W. Brownlee, O. Ciccarelli, A. Coles, J. Correale, G. Cutter, G. Edan, N. Evangelou, O. Fernandez, J. Frederiksen, R. Gold, Y. Hacohen, H. P. Hartung, K. Hellwig, J. Hillert, J. Imitola, T. Kalincik, L. Kappos, S. Khoury, H. J. Kim, E. K. Havrdová, R. Liblau, J. Lycke, X. Montalban, P. Muraro, S. Reingold, K. Schmierer, F. Sellebjerg, P. S. Sørensen, A. Solari, M. P. Sormani, A. Thompson, B. Trapp, H. Tremlett, M. Trojano, C. Tur, A. Uccelli, V. van Pesch and E. Waubant, Mult. Scler. J., 2020, 26, 1045–1063 CrossRef PubMed.
- A. E. Miller, T. Chitnis, B. A. Cohen, K. Costello, N. L. Sicotte and R. Stacom, JAMA Neurol., 2021, 78, 241–246 CrossRef PubMed.
- L. Calahorra, C. Camacho-Toledano, M. P. Serrano-Regal, M. C. Ortega and D. Clemente, Biomedicines, 2022, 10, 1–26 CrossRef PubMed.
- D. Radojević, M. Bekić, A. Gruden-Movsesijan, N. Ilić, M. Dinić, A. Bisenić, N. Golić, D. Vučević, J. Đokić and S. Tomić, Gut Microbes, 2022, 14, 1–28 CrossRef PubMed.
- C. Camacho-Toledano, I. Machín-Díaz, L. Calahorra, M. Cabañas-Cotillas, D. Otaegui, T. Castillo-Triviño, L. M. Villar, L. Costa-Frossard, M. Comabella, L. Midaglia, J. M. García-Domínguez, J. García-Arocha, M. C. Ortega and D. Clemente, J. Neuroinflammation, 2022, 19, 1–22 CrossRef PubMed.
- V. Moliné-Velázquez, M. C. Ortega, V. Vila-del Sol, C. Melero-Jerez, F. de Castro and D. Clemente, Neurobiol. Dis., 2014, 67, 149–164 CrossRef PubMed.
- B. Zhu, Y. Bando, S. Xiao, K. Yang, A. C. Anderson, V. K. Kuchroo and S. J. Khoury, J. Immunol., 2007, 179, 5228–5237 CrossRef CAS PubMed.
- V. Moliné-Velázquez, H. Cuervo, V. Vila-Del Sol, M. C. Ortega, D. Clemente and F. De Castro, Brain Pathol., 2011, 21, 678–691 CrossRef PubMed.
- B. Knier, M. Hiltensperger, C. Sie, L. Aly, G. Lepennetier, T. Engleitner, G. Garg, A. Muschaweckh, M. Mitsdörffer, U. Koedel, B. Höchst, P. Knolle, M. Gunzer, B. Hemmer, R. Rad, D. Merkler and T. Korn, Nat. Immunol., 2018, 19, 1341–1351 CrossRef CAS PubMed.
- C. Melero-Jerez, M. Suardíaz, R. Lebrón-Galán, C. Marín-Bañasco, B. Oliver-Martos, I. Machín-Díaz, Ó. Fernández, F. de Castro and D. Clemente, Neurobiol. Dis., 2019, 127, 13–31 CrossRef CAS PubMed.
- C. Melero-Jerez, B. Fernández-Gómez, R. Lebrón-Galán, M. C. Ortega, I. Sánchez-de Lara, A. C. Ojalvo, D. Clemente and F. de Castro, Glia, 2021, 69, 905–924 CrossRef CAS PubMed.
- A. Hafner, J. Lovrić, G. P. Lakǒ and I. Pepić, Int. J. Nanomed., 2014, 9, 1005–1023 Search PubMed.
- M. Orecchioni, C. Ménard-Moyon, L. G. Delogu and A. Bianco, Adv. Drug Delivery Rev., 2016, 105, 163–175 CrossRef CAS PubMed.
- V. Palmieri, G. Perini, M. De Spirito and M. Papi, Nanoscale Horiz., 2019, 4, 464–471 RSC.
- H. Lin, Z. Song and A. Bianco, J. Environ. Sci. Health, Part B, 2021, 56, 333–356 CrossRef CAS PubMed.
- J. Yan, L. Chen, C. C. Huang, S. C. C. Lung, L. Yang, W. C. Wang, P. H. Lin, G. Suo and C. H. Lin, Colloids Surf., B, 2017, 153, 300–309 CrossRef CAS PubMed.
- M. Cicuéndez, L. Casarrubios, N. Barroca, D. Silva, M. J. Feito, R. Diez-orejas, P. A. A. P. Marques and M. T. Portolés, Int. J. Mol. Sci., 2021, 22, 6701 CrossRef PubMed.
- Y. Dai, X. Nie, J. Liu, J. Meng and H. Xu, Chin. J. Biomed. Eng., 2017, 36, 464–469 Search PubMed.
- K. Pondman, B. Paudyal, R. Sim, A. Kaur, L. Kouser, A. Tsolaki, L. Jones, C. Salvador-Morales, H. A. Khan, B. Ten Haken, G. Stenbeck and U. Kishore, Nanoscale, 2017, 9, 1097–1109 RSC.
- A. Halim, Q. Luo, Y. Ju and G. Song, Nanomaterials, 2018, 8, 736 CrossRef PubMed.
- F. Della Pelle, R. Di Battista, L. Vázquez, F. J. Palomares, M. Del Carlo, M. Sergi, D. Compagnone and A. Escarpa, Appl. Mater. Today, 2017, 9, 29–36 CrossRef.
- A. Domínguez-Bajo, A. González-Mayorga, C. R. Guerrero, F. J. Palomares, R. García, E. López-Dolado and M. C. Serrano, Biomaterials, 2019, 192, 461–474 CrossRef PubMed.
- P. Aliaga-Gaspar, I. Hurtado-Guerrero, N. L. Ciano-Petersen, P. Urbaneja, I. Brichette-Mieg, V. Reyes, J. L. Rodriguez-Bada, R. Alvarez-Lafuente, R. Arroyo, E. Quintana, L. Ramió-Torrentà, A. Alonso, L. Leyva, O. Fernández and B. Oliver-Martos, Front. Immunol., 2021, 12, 778204 CrossRef CAS PubMed.
- F. Veglia, E. Sanseviero and D. I. Gabrilovich, Nat. Rev. Immunol., 2021, 21, 485–498 CrossRef CAS PubMed.
- C. Melero-Jerez, M. C. Ortega, V. Moliné-Velázquez and D. Clemente, Biochim. Biophys. Acta, Mol. Basis Dis., 2016, 1862, 368–380 CrossRef CAS PubMed.
- B. Zhu, J. K. Kennedy, Y. Wang, C. Sandoval-Garcia, L. Cao, S. Xiao, C. Wu, W. Elyaman and S. J. Khoury, J. Immunol., 2011, 187, 2418–2432 CrossRef CAS PubMed.
- P. M. Kou and J. E. Babensee, J. Biomed. Mater. Res., Part A, 2011, 96, 239–260 CrossRef PubMed.
- S. Zhou, Q. Shang, J. Ji and Y. Luan, ACS Appl. Mater. Interfaces, 2021, 13, 47407–47417 CrossRef CAS PubMed.
- W. He, P. Liang, G. Guo, Z. Huang, Y. Niu, L. Dong, C. Wang and J. Zhang, Sci. Rep., 2016, 6, 24506 CrossRef CAS PubMed.
- M. L. Escudero, I. Llorente, B. T. Pérez-Maceda, S. S. José-Pinilla, L. Sánchez-López, R. M. Lozano, S. Aguado-Henche, C. C. de Arriba, M. A. Alobera-Gracia and M. C. García-Alonso, Mater. Sci. Eng., C, 2020, 109, 1–8 CrossRef PubMed.
- G. Qu, S. Liu, S. Zhang, L. Wang, X. Wang, B. Sun, N. Yin, X. Gao, T. Xia, J. J. Chen and G. B. Jiang, ACS Nano, 2013, 7, 5732–5745 CrossRef CAS PubMed.
- A. Sasidharan, L. S. Panchakarla, A. R. Sadanandan, A. Ashokan, P. Chandran, C. M. Girish, D. Menon, S. V. Nair, C. N. R. Rao and M. Koyakutty, Small, 2012, 8, 1251–1263 CrossRef CAS PubMed.
- M. Orecchioni, D. A. Jasim, M. Pescatori, R. Manetti, C. Fozza, F. Sgarrella, D. Bedognetti, A. Bianco, K. Kostarelos and L. G. Delogu, Adv. Healthcare Mater., 2016, 5, 276–287 CrossRef CAS PubMed.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, M. D. Gutiérrez, F. Rodríguez-Reinoso and R. Menéndez, Carbon, 2013, 52, 476–485 CrossRef CAS.
- X. Wang, R. Podila, J. H. Shannahan, A. M. Rao and J. M. Brown, Int. J. Nanomed., 2013, 8, 1733–1748 Search PubMed.
- H. S. Hung, M. L. Kung, F. C. Chen, Y. C. Ke, C. C. Shen, Y. C. Yang, C. M. Tang, C. A. Yeh, H. H. Hsieh and S. H. Hsu, Nanomaterials, 2021, 11, 2046 CrossRef CAS PubMed.
- H. Zhou, K. Zhao, W. Li, N. Yang, Y. Liu, C. Chen and T. Wei, Biomaterials, 2012, 33, 6933–6942 CrossRef CAS PubMed.
- T. Svadlakova, D. Holmannova, M. Kolackova, A. Malkova, J. Krejsek and Z. Fiala, Int. J. Mol. Sci., 2022, 23, 1–24 Search PubMed.
- J. Russier, E. Treossi, A. Scarsi, F. Perrozzi, H. Dumortier, L. Ottaviano, M. Meneghetti, V. Palermo and A. Bianco, Nanoscale, 2013, 5, 11234–11247 RSC.
- L. Fusco, M. Orecchioni, G. Reina, V. Bordoni, C. Fuoco, C. Gurcan, S. Guo, M. Zoccheddu, F. Collino, B. Zavan, E. Treossi, A. Yilmazer, V. Palermo, A. Bianco and L. G. Delogu, NanoImpact, 2021, 23, 100330 CrossRef CAS PubMed.
- S. A. Zamorina, K. Y. Shardina, V. P. Timganova, M. S. Bochkova, A. I. Nechaev, P. V. Khramtsov and M. B. Raev, Bull. Exp. Biol. Med., 2020, 170, 84–87 CrossRef CAS PubMed.
- M. J. Feito, M. Vila, M. C. Matesanz, J. Linares, G. Gonçalves, P. A. A. P. Marques, M. Vallet-Regí, J. M. Rojo and M. T. Portolés, J. Colloid Interface Sci., 2014, 432, 221–228 CrossRef CAS PubMed.
- L. Xu, J. Xiang, Y. Liu, J. Xu, Y. Luo, L. Feng, Z. Liu and R. Peng, Nanoscale, 2016, 8, 3785–3795 RSC.
- D. Yali, N. Xin, L. Jian, M. Jie and X. Haiyan, Chin. J. Biomed. Eng., 2017, 36, 464–469 Search PubMed.
- K. M. Pondman, B. Paudyal, R. B. Sim, A. Kaur, L. Kouser, A. G. Tsolaki, L. A. Jones, C. Salvador-Morales, H. A. Khan, B. ten Haken, G. Stenbecka and U. Kishore, Nanoscale, 2017, 9, 1097–1109 RSC.
- J. Meng, X. Li, C. Wang, H. Guo, J. Liu and H. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 3180–3188 CrossRef CAS PubMed.
- X. Wan, Z. Liu and L. Li, Adv. Funct. Mater., 2021, 31, 2010626 CrossRef CAS.
- F. Xiaoli, Z. Yaqing, L. Ruhui, L. Xuan, C. Aijie, Z. Yanli, H. Chen, C. Lili and S. Longquan, J. Hazard. Mater., 2021, 416, 126158 CrossRef PubMed.
- R. Li, L. M. Guiney, C. H. Chang, N. D. Mansukhani, Z. Ji, X. Wang, Y.-P. Liao, W. Jiang, B. Sun, M. C. Hersam, A. E. Nel and T. Xia, ACS Nano, 2018, 12, 1390–1402 CrossRef CAS PubMed.
- H. Chen, X. Huang, M. Zhang, F. Damanik, M. B. Baker, A. Leferink, H. Yuan, R. Truckenmüller, C. van Blitterswijk and L. Moroni, Acta Biomater., 2017, 59, 82–93 CrossRef CAS PubMed.
- Z. Lyu, H. Wang, Y. Wang, K. Ding, H. Liu, L. Yuan, X. Shi, M. Wang, Y. Wang and H. Chen, Nanoscale, 2014, 6, 6959–6969 RSC.
- Y. Wu, F. Wang, S. Wang, J. Ma, M. Xu, M. Gao, R. Liu, W. Chen and S. Liu, Nanoscale, 2018, 10, 14637–14650 RSC.
- H. Li, Y. Feng, Q. Luo, Z. Li, X. Li, H. Gan, Z. Gu, Q. Gong and K. Luo, Theranostics, 2023, 13, 5386–5417 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Data about used antibodies for flow cytometry, XPS, AFM and DLS. See DOI: https://doi.org/10.1039/d3nr05351b |
| ‡ Present address: Hospital Universitario de Toledo, Avd. Río Guadiana, s/n 45004. Toledo, Spain. |
| § Present address: Hospital Universitario de Navarra, C/Irunlarrea 3, 31008 Pamplona, Navarra, Spain. |
| This journal is © The Royal Society of Chemistry 2024 |