
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amphiphilic titania Janus nanoparticles containing ionic groups prepared in oil–water Pickering emulsion†
Lucas
Niedner
 a and
Guido
Kickelbick
*ab
a and
Guido
Kickelbick
*ab
aSaarland University, Inorganic Solid State Chemistry, Campus, Building C4 1, 66123 Saarbrücken, Germany. E-mail: guido.kickelbick@uni-saarland.de
bSaarene – Saarland Center for Energy Materials and Sustainability, Campus C4 2, 66123 Saarbrücken, Germany
First published on 27th February 2024
Abstract
Titania nanoparticles with a diameter of 8 nm underwent an anisotropic modification using apolar 6-bromohexylphosphonic acid and cationic polar N,N,N-trimethyl-6-phosphonohexan-1-aminium bromide. The Janus modification was achieved through a straightforward one-step Pickering emulsion approach using toluene–water mixtures. The resulting Janus particles were compared with isotropically and statistically modified titania particles, where either a single coupling agent is attached to the surface or both coupling agents are assembled over the surface randomly, respectively. The covalent binding of the phosphonic acids to the titania surface was confirmed by FTIR and 31P solid-state CP-MAS NMR analyses. The grafting density was assessed using TGA, elemental analysis, and ICP-MS, revealing grafting densities of 0.1 mmol g−1 to 0.5 mmol g−1 for the cationic coupling agent and 1.2 mmol g−1 to 1.5 mmol g−1 for the apolar coupling agent, respectively. ζ-Potential titration measurements of both pristine and modified particles revealed isoelectric points at pH 4.5 to 9.3, depending on the type of modification. The ability of the particles to stabilize Pickering emulsions was tested under various conditions, with statistically and Janus-modified particles demonstrating a significant increase in stabilization compared to their isotropically modified counterparts. Furthermore, Janus particles were deposited onto glass substrates by a simple layer-by-layer approach. Through the self-assembly of these Janus particles, the glass substrate's properties could be tailored from hydrophilic to hydrophobic to hydrophilic, depending on the dipping cycle.
Introduction
The scientific interest in Janus particles has been greatly piqued due to their ability to exhibit a wide range of properties and their tendency to self-assemble at interfaces.1,2 By tailoring their polarity, surface wettability or electrical, optical and magnetic properties they can be used in a variety of areas, such as medicine, catalysis, waste water treatment, optics or electronic devices.3–7 Such anisotropic systems are either a combination of two different particles with different chemical compositions,8,9e.g. Au-TiO2 Janus particles,10 or a core particle whose surface is anisotropically modified with different surfactants or molecules.11–14 The latter in particular show unique self-assembly and a high interfacial affinity.13,15–17 Therefore these systems are suitable to form surfactant-free emulsions and functional coatings.18–20 To synthesize these anisotropically modified particles it is necessary to synthetically discriminate between two hemispheres of the particle and to address the separated surfaces by different reactions. For this purpose, the particle is usually immobilised at the liquid–liquid, solid–liquid or solid–gas interface to modify the free-standing hemisphere.21 A commonly applied synthetic approach for higher yields is the use of Pickering emulsions in which the emulsified droplets are stabilised by particles at the liquid–liquid interface. By using molten wax as dispersed oil the particles are immobilised at the wax surface after cooling the wax below its melting point. Thereby, one hemisphere is masked whereby the other one can be modified.22,23 After the first modification step the wax is dissolved and the second hemisphere can now be modified by a second surfactant. The solid wax approach can be substituted by a typical liquid–liquid emulsion if the particle surface can be modified, at least on one side, with molecules that attach very rapidly and have the polarity of one of the two phases (hydrophobic or hydrophilic) (Scheme 1). If the attachment of the molecules was successful particle rotation or phase transfer of the particle from one phase into the other is prevented and after breaking the Pickering emulsion Janus particles are obtained. | ||
| Scheme 1 Amphiphilic Janus modification in a Pickering emulsion with dispersion medium (blue), dispersed phase (red), and solid micro-/nanoparticles (grey). R and R′ are polar and apolar groups depending on the polarity of the two liquids. | ||
In general, Pickering emulsions are better stabilized by larger particles, making amphiphilic functionalization of nanoparticles more difficult.24,25 Therefore, the rapid attachment of the linkers to the particles’ surface is particularly important in the anisotropic functionalization in Pickering emulsions to ensure that the particle is trapped at the interface. It is known that phosphonic acids bind very fast onto transition metal oxide surfaces and build strong covalent bonds, e.g. on Al2O3, ZrO2, Fe3O4 and TiO2 surfaces.11,26–28 In previous studies, we already took advantage of the fast coupling reaction to amphiphilically modify titania particles in toluene–water emulsions with hydrophilic and hydrophobic molecules containing phosphonic acids as linker, e.g. (poly)ethylene glycol, polydimethylsiloxane and alkyl phosphonic acids.11,12 In addition to the synthesis, the characterization of Janus nanoparticles is a particular challenge. Due to the small size of the particles and molecules, the anisotropy of the particles can rarely be detected by optical or electron microscopy. Therefore, advancing methods and indirect methods are required to reveal their Janus character. One approach is to shield one side of the particle to study the free-standing side spectroscopically. Bourone et al. investigated the Janus character of amphiphilic gold nanoparticles functionalized with 1-octanethiol and 8-mercaptooctanoic acid by assembling the particles on hydrophilic and hydrophobic substrates to investigate the free-standing particle hemisphere by infrared reflection adsorption spectroscopy.15 Another approach takes advantage of the unique interfacial properties created by Janus functionalization. Because of their amphiphilic nature, Janus particles should stabilise Pickering emulsions much better than uniformly modified particles in which the different surface molecules are distributed statistically over the particles surface. Binks et al. calculated up to a threefold increase in the energy required to desorb Janus particles from a liquid–liquid interface compared to homogeneously modified particles.29
Regarding the type of surface modification, particles with a charged surface are of particular interest. Nanoparticles with a surface charge offer the possibility of an electrostatically driven self-assembly, e.g., in the layer-by-layer (LbL) approach to obtain various kinds of coatings.28,30–32 A simple approach to induce a surface charge on a metal oxide surface is the protonation and deprotonation of hydroxide groups on its surface. Drawback of this method is the sensitivity of the induced charge to pH, solvents, and other environmental conditions. To generate more stable charges and to adjust the ζ-potential of the particles molecules and macromolecules carrying functional groups are attached to the surface via suitable linkers. In order to obtain Janus particles with one positively charged hemisphere that is stable over a wide pH range mainly polyelectrolytes with primary or ternary ammonium groups, such as poly[2-aminoethyl methacrylate] and poly[2-(dimethylamino)ethyl methacrylate] are grafted to the particles surface via surface-initiated atom transfer radical polymerization (SI-ATRP).17,33,34 A less complex way, which has rarely been reported in the literature so far is the anisotropic attachment of an already charged molecular surfactant carrying an ammonium group. By using these molecular surfactants, the particles surface stays positively charged as well although lower charge densities are obtained compared to polyelectrolytes.28,35 By using an quaternary ammonium salt instead of an amine the charge of the surfactant is independent of the pH.
Regarding the type of particles, titania particles are widely studied and often used for stabilizing Pickering emulsions due to their surface properties, photocatalytic effect and high chemical and mechanical robustness.12,36–39 The titania particles are therefore either combined with another inorganic particle, like Au or SiO2 or their surface is amphiphilically modified. However, to the best of our knowledge, titania particles have not yet been anisotropically modified with pH independent ionic groups on one hemisphere to make use of an electrostatically driven self-assembly.
In this study, we report a simple and fast preparation of amphiphilic titania Janus particles (J-Br/NMe3+@TiO2) carrying a stable cationic charge at one hemisphere and an apolar bromohexyl group on the opposing hemisphere. In detail we modified titania particles in the size range of 8 nm with 6-bromohexylphosphonic acid (BrC6PA) and N,N,N-trimethyl-6-phosphonohexane-1-ammonium bromide (NMe3+C6PA) in a one-step Pickering emulsion approach. To investigate the Janus character the Janus particles are compared with hydrophilic particles (NMe3+@TiO2) isotropically modified with NMe3+C6PA, hydrophobic particles (Br@TiO2) isotropically modified with BrC6PA, and statistically modified particles (s-Br/NMe3+@TiO2) at which the two surfactants are distributed randomly over the surface. The obtained particles are afterwards used to stabilise oil–water emulsions. Furthermore, the Janus particles J-Br/NMe3+@TiO2 are assembled at a glass substrate in a layer-by-layer approach without using an additional particle type or polyelectrolyte. This enabled us to tailor the wettability of the glass substrate from hydrophobic to highly hydrophilic depending on the dipping cycle.
Experimental section
Materials
Titanium(IV) isopropoxide was purchased from abcr GmbH (Karlsruhe, Germany). 1,6-Dibromohexane, triethyl phosphite, trimethyl amine, tetramethylammonium chloride, and bromo trimethylsilane were purchased from Sigma-Aldrich (St. Louis, USA). Dry dichloromethane was purchased from Acros Organics (Geel, Belgium). Methanol, ethanol, and 2-propanol were purchased from Biesterfeld Spezialchemie (Hamburg, Germany). Sodium chloride was purchased from BCD Chemie (Hamburg, Germany). All chemicals were used as received without any further purification. Glass slides were purchased from VWR International (Radnor, USA).Characterisation
Powder X-ray diffraction (PXRD) patterns were recorded on a D8-A25-Advance diffractometer (Bruker AXS, Karlsruhe, Germany) in Bragg-Brentano θ-θ-geometry (goniometer radius 280 mm) with Cu Kα-radiation (λ = 154.0596 pm). A 12 μm Ni foil working as Kβ filter and a variable divergence slit were mounted at the primary beam side. A LYNXEYE 1D detector with 192 channels was used at the secondary beam side. Fluorescence induced background was reduced by detector discrimination. Data acquisition was carried out in a 2θ range from 7 to 120° with a step size of 0.013° and a total scan time of 2 h. TOPAS 5.0 was used for the Rietveld refinements.Solution NMR spectra were recorded on a Avance III HD 400 MHz spectrometer (Bruker, Billerica, USA) with 400.13 MHz for 1H NMR spectra, 100.61 MHz for 13C NMR spectra, and 161.97 MHz for 31P NMR spectra. NMR samples were prepared in chloroform-d (CDCl3) or deuterium oxide (D2O).
Solid-state CP-MAS NMR spectra were recorded on an Avance III HD-Ascend 400WB spectrometer (Bruker, Billerica, USA) at 295 K using 4 mm inner diameter ZrO2 rotors with a 13 kHz rotation frequency and a relaxation delay of 3 s. The resonance frequencies were 100.65 MHz for 13C and 162.04 MHz for 31P NMR spectra.
FTIR spectra were recorded on attenuated total reflectance (ATR) using a Vertex 70 spectrometer (Bruker Optics, Ettlingen, Germany). The spectra were obtained as the average of 32 scans in the wavelength range from 400 cm−1 to 4500 cm−1 at a resolution of 4 cm−1. For evaluation, the spectra were normalized to their intensity maximum.
Inductively coupled plasma mass spectrometry (ICP-MS) was conducted using a commercially available ICP-MS system (8900 Triple Quad and SPS4 autosampler, Agilent, Santa Clara, USA). Stock solutions of single element ICP-MS standards of Br (Merck Millipore, Darmstadt, Germany), Ti (Radnor, USA) and P (AccuStandard, New Heaven, USA) were used. The detector dwell time was 100 μs, the experiments were repeated three times, and the measured isotopes were 31P via mass pair (Q1, Q2) = (31, 47) using O2 as reaction gas, 79Br and 47Ti using He as collision gas as well as 45Sc and 165Ho (all used modes) as the internal standard. For the ICP-MS measurements, the particles were dissolved for 1 h at 160 °C in hot sulphiric acid. A colorless solution was obtained for pure titanium dioxide, while the solution for modified particles turned slightly yellowish.
Dynamic light scattering (DLS) on the particle suspensions was recorded using an ALV/CGS-3 compact goniometer system from ALV (Langen, Germany) with an ALV/LSE-5003 correlator at a wavelength of 632.8 nm (HeNe laser) and a measurement angle of 90°. The evaluation was carried out using autocorrelation according to the g2(t) function. The DLS size distribution curves presented in the paper are number weighted. For each measurement 10 runs for 10 s were performed.
Three-phase contact angle measurements were performed using a home-built system equipped with an adjustable sample stage, back illumination of the sample, as well as a microliter glass syringe and a digital microscope camera from Dino-Lite (Stuttgart, Germany). The evaluation was performed with a contact angle plugin for the software ImageJ. For each dipping step the contact angle was measured three times.
pH measurements were performed with a seven multi pH meter from Mettler Toledo (Columbus, USA) equipped with a Toledo inLab Pro electrode.
UV-Vis spectroscopy was performed on a Lambda 750 spectrometer (PerkinElmer, Waltham, USA) equipped with a 100 mm integration sphere. Measurements were performed in a wavelength range from 800 nm to 200 nm with an increment of 4 nm and an integration time of 0.2 s. PerkinElmer quartz cuvettes were used to measure the particle suspensions, with the sample placed in front of the integration sphere and the reference cuvette with suspending agent placed behind it. For the measurement of the coated glass slides, these were clamped in front of the integration sphere.
Thermogravimetric analysis (TGA) was performed using an Iris TG 209 C instrument (Netzsch, Selb, Germany). Samples were first heated to 100 °C in alumina crucibles at a heating rate of 20 K min−1 and held at 100 °C for 20 min to remove absorbed water and solvent. Subsequently, the samples were then brought to 700 °C under nitrogen atmosphere (40 mL min−1) at 20 K min−1 and heated to 1000 °C under a mixture of oxygen and nitrogen (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) (20 mL min−1) in a final step. The grafting density using TGA results was calculated as follows: Assuming that the total mass loss from 100 °C to 1000 °C is caused only by the decomposition of the phosphonic acid, the molar amount of the phosphonic acid nPA for the specific sample mass is:
:9) (20 mL min−1) in a final step. The grafting density using TGA results was calculated as follows: Assuming that the total mass loss from 100 °C to 1000 °C is caused only by the decomposition of the phosphonic acid, the molar amount of the phosphonic acid nPA for the specific sample mass is:
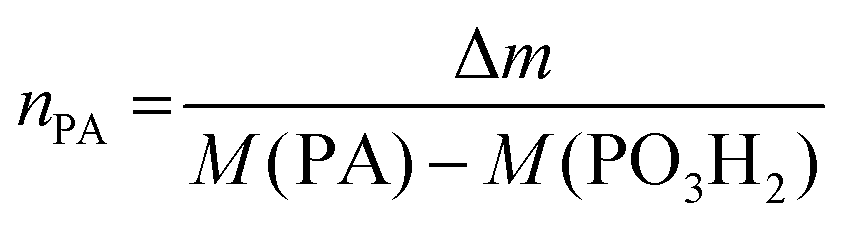 | (1) |
 in mmol g−1 is given by eqn (2):
in mmol g−1 is given by eqn (2): | (2) |
In order not to overestimate the remaining particle mass, the mass of the remaining PO3H2 linker needs to be subtracted from m1000 °C.
Transmission electron microscopy (TEM) was recorded using a JEM-2010 electron microscope (JEOL, Akishima, Japan). 30 μL of the particle suspension in water or methanol was dropped onto a Plano S160-3 copper mesh coated with a carbon film, and the suspending agent was evaporated under normal conditions. The TEM images were analysed with the program ImageJ.
Elemental analysis (EA) was performed on a Vario Micro Cube (Elementar, Langenselbold, Germany). The surface coverage of the modified particles using the combination of EA and TGA is calculated by eqn (3):
 | (3) |
ζ-Potential titrations were performed with a Malvern Zetasizer Ultra equipped with Malvern MPT-2 titrator using a DTS1070 flow cell (Malvern Panalytical, Worcestershire, UK). Aq. HCl and aq. NaOH solutions were used as titrants. Because of the poor dispersability of the Br@TiO2, s-Br/NMe3@TiO2 and J-Br/NMe3@TiO2 particles, all measurements were performed in ethanol–water mixtures (1:1; v:v).
Pickering emulsions were visualized with an Axioskop 50 transmitted light/fluorescence microscope with an AxioCam MRc (1388 × 1040 pixel).
Titanium dioxide nanoparticles
Titanium(IV) oxide particles were synthesised according to previous literature by an aqueous sol–gel reaction.11 Briefly, a solution of 7.5 mL (25 mmol) freshly distilled titanium isopropoxide in 25 mL dry ethanol under argon atmosphere was added dropwise to an aq. HCl solution (4 °C) at pH 1.5 under vigorous stirring. A milky white suspension formed. Subsequently the reaction was stirred at room temperature for several days till the suspension turned transparent. The solvent was removed under reduced pressure, the remaining yellow solid was dispersed in water again and stored under ambient conditions.Phosphonic acids
Organophosphorus compounds 6-bromohexylphosphonic acid (BrC6PA) and N,N,N-trimethyl-6-phosphonohexan-1-aminium bromide (NMe3+C6PA) were synthesised following previous literature.28Isotropic surface modification with NMe3+C6PA
TiO2 particles were dispersed in an aqueous HCl solution (pH = 3.0) to prepare a 0.8 wt% particle suspension. An aqueous stock solution of NMe3+C6PA was added dropwise to this suspension under vigorous stirring so that a concentration of 5 mmol g−1 phosphonic acid per TiO2 particle mass was achieved. After the reaction was stirred for 24 h or 20 min at room temperature, the particles were collected by centrifugation (8000 rpm, 15 min, 21 °C) and washed three times with water. The particles were subsequently stored dispersed in water. The particle content of the dispersion was determined gravimetrically. For PXRD, FTIR, TGA and CHN analysis small amounts of the particles were dried under reduced pressure (15 mbar) for 20 h to obtain a white powder.Isotropic surface modification with BrC6PA
TiO2 particles were dispersed in an aqueous HCl solution (pH = 3.0) to prepare a 0.8 wt% particle suspension. A solution of BrC6PA in ethanol was added dropwise under rigorous stirring to achieve a concentration of 5.0 mmol g−1 phosphonic acid per TiO2 particle mass. During the addition, the transparent suspension turned opaque milky-white. After 20 min of stirring, the particles were separated by centrifugation and washed several times with ethanol. The particles were redispersed in ethanol and the ethanolic suspension was stored at ambient conditions.Statistic surface modification with BrC6PA and NMe3+C6PA
TiO2 particles were dispersed in a mixture of aq. HCl solution (pH = 3.0) and ethanol (1:1; v:v) to prepare a 0.8 wt% particle suspension. To this suspension, stock solutions of BrC6PA in ethanol and NMe3+C6PA in water were added simultaneously so that a concentration of 2.5 mmol g−1 of each phosphonic acids per mass of TiO2 particles was obtained. After the reaction was stirred for 20 min at room temperature, the particles were collected by centrifugation (8 krpm, 15 min, 21 °C) and washed three times with ethanol and water. The particles were stored as aqueous suspensions. The particle content of the dispersion was determined gravimetrically. For PXRD, FTIR, TGA and CHN analysis small amounts of the particles were dried under reduced pressure (15 mbar) for 20 h to obtain a white powder.
Anisotropic surface modification with BrC6PA and NMe3+C6PA in Pickering emulsion
TiO2 particles were dispersed in an aqueous HCl solution (pH = 3.0) to prepare a 0.8 wt% particle suspension. 23 mL toluene were added to 92 mL of the particle suspension, and the mixture was stirred with Heidolph Silent Crusher M at 13000 rpm to from a Pickering emulsion. Stock solutions of BrC6PA dissolved in ethanol and NMe3+C6PA dissolved in distilled water were simultaneously added to this emulsion with constant stirring. The concentrations were adjusted to 2.5 mmol g−1 of each phosphonic acid per mass of particles. After stirring the reaction at 13000 rpm for 20 min, the emulsion was centrifugated at 8000 rpm for 15 min. The emulsion broke and the Janus particles assembled at the toluene–water interface. The Janus particles were carefully removed. The particles remaining in the water phase were discarded. The collected Janus particles were washed with ethanol and water several times by centrifugation and redispersion. Subsequently, the particles were stored dispersed in water. The particle content of the dispersion was determined gravimetrically. For PXRD, FTIR, TGA and CHN analysis small amounts of the particles were dried under reduced pressure (15 mbar) for 20 h to obtain a white powder.
Particle deposition on glass slides
Microscopy glass slides were pre-cleaned in water, ethanol, and acetone baths with ultrasonic treatment for 10 min each, followed by argon plasma treatment for 5 min. The cleaned substrates were immersed in a 0.1 wt% particle suspension of the modified titania dispersed in ethanol or water for 10 min. Afterwards the substrates were removed with a constant speed of 60 mm min−1. To remove poorly attached particles, the slides were immersed in distilled water for 10 min in between each deposition step. Before further characterisation the slides were dried under ambident conditions.Preparation of dyed Pickering emulsions
Dyed Pickering emulsions were prepared by mixing an aqueous suspensions containing 0.8 wt% of the respective particles with the organic solvent containing 0.01 wt% Lumogen red 305 as dye. The mixture was then shaken vigorously by hand for 30 seconds.Results and discussion
Titania particles
TiO2 nanoparticles were synthesised by a sol–gel method according to literature.40,41 Ti(OiPr)4 was reacted with aq. HCl instead of the commonly used aq. HNO3 to prevent interfering nitrate signals in the FTIR spectra. This change has no effect on the phase composition, crystallinity, or particle size. DLS, FTIR and XRD measurements of differently synthesized particles are compared in the ESI (Fig. S1–S3†). Titania particles with a hydrodynamic radius of 4.3 ± 0.7 nm and good dispersibility in water were obtained (Fig. 1). The counting of 200 particles in the TEM resulted in an average diameter of 8.2 ± 2.2 nm. The rietveld refinement of the powder X-ray diffraction (PXRD; Fig. 1) indicated that the particles consist of 64% anatase and 36% brookite with crystallite sizes of 5.1 ± 0.8 nm. FTIR spectra of the pristine titania revealed the TiO2 rocking vibrations at 400 nm and some residual water that is adsorbed on the particles surface (Fig. 2). | ||
| Fig. 1 (a) TEM image, (b) DLS and (c) PXRD pattern (black) with Rietveld refinement (red), difference (grey) and hkl values for anatase (green) and brookite (blue) of pristine TiO2 particles. | ||
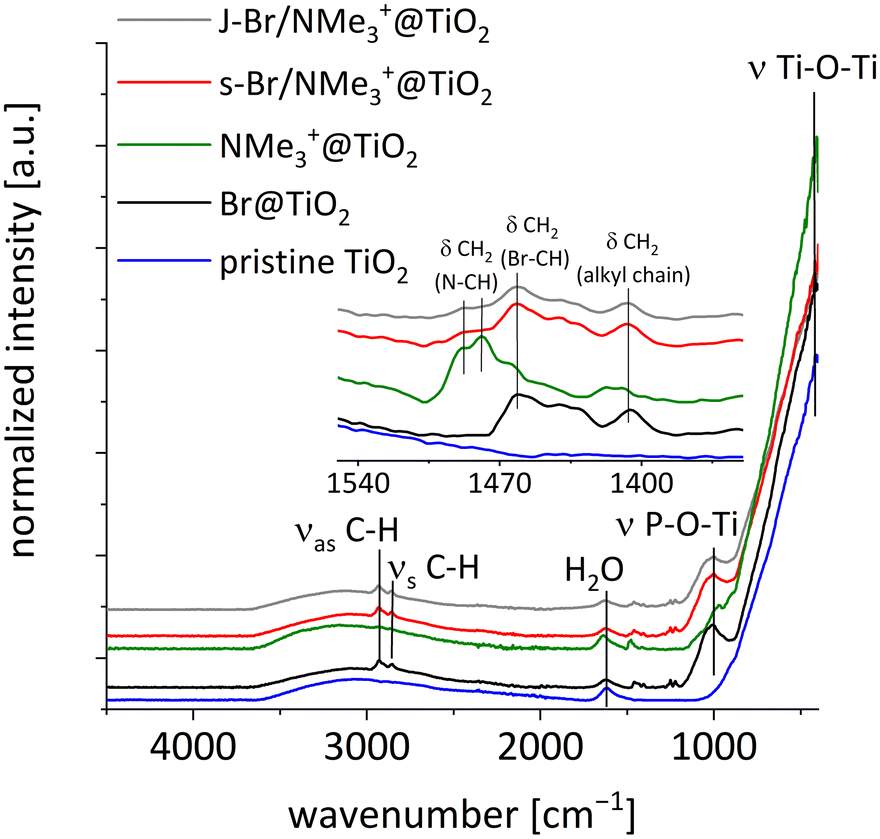 | ||
| Fig. 2 FTIR spectra of titania particles: pristine TiO2 (blue), hydrophobically modified Br@TiO2 (black), cationically modified NMe3+@TiO2 (green), statistically modified s-Br/NMe3+@TiO2 (red) as well as Janus-modified J-Br/NMe3+@TiO2 (grey). | ||
Organophosphonic acids as coupling molecules
BrC6PA and NMe3+C6PA were synthesised as surface modifiers of the titania particles. Both phosphonic acids were prepared according to previously published literature (Scheme 2).28 Starting from 1,6-dibromohexane, diethyl-(6-bromohexyl)phosphonate was prepared via Michaelis–Arbuzov reaction. Subsequent hydrolysation led to BrC6PA. NMe3+C6PA was obtained by reacting diethyl-(6-bromohexyl)phosphonate with trimethylamine, followed by hydrolysation. 1H, 31P, 13C NMR and FTIR spectra of the molecules are shown in the ESI (Fig. S5–S11†). | ||
| Scheme 2 Synthesis of the two phosphonic acids BrC6PA and NMe3+C6PA. | ||
Isotropic, statistic and anisotropic surface functionalization
Amphiphilic titania Janus particles (J-Br/NMe3+@TiO2) were prepared in a simple one-step Pickering emulsion approach. For this purpose, a 0.8 wt% aqueous titania particle suspension (pH = 3.0) and toluene were mixed in a 4:1 volume ratio and stirred at 13000 rpm with a Ultra-Turrax dispersing device to obtain a toluene-in-water Pickering emulsion. To this Pickering emulsion, an aqueous solution of NMe3+C6PA and a solution of BrC6PA in toluene were added simultaneously to anisotropically modify the titania particles located at the liquid–liquid interface. After continuous stirring for 20 min the emulsion was broken by centrifugation and the Janus particles assembled at the toluene–water interface. The Janus particles were carefully collected from the interface and washed several times with ethanol, whereas the particles remaining in the aqueous phase were discarded. The obtained J-Br/NMe3+@TiO2 were compared with isotropically and statistically modified particles Br@TiO2, NMe3+@TiO2 and s-Br/NMe3+@TiO2 to fully evaluate the Janus character (Scheme 3). For isotropic modification the titania particle suspension (pH = 3.0) was stirred with either an aqueous solution of NMe3+C6PA or an ethanolic solution of BrC6PA to obtain hydrophilic NMe3+@TiO2 or hydrophobic Br@TiO2, respectively. Statistically modified particles s-Br/NMe3+@TiO2 on which the two surfactants are distributed randomly were prepared by simultaneously adding an aqueous solution of NMe3+C6PA and an ethanolic solution of BrC6PA to the aqueous titania particle suspension (pH = 3.0) and stirring the mixture for 20 min. To get rid of any unreacted phosphonic acid all particles were washed with ethanol or water several times. The particles were then stored in suspension. For characterization purposes, parts of the suspensions were dried under reduced pressure to obtain white-yellowish powders. PXRD results (Fig. S12†) of the modified particles show no changes in phase composition or crystallite size.
 | ||
| Scheme 3 Schematic illustration of hydrophobic modified Br@TiO2, hydrophilic modified NMe3+@TiO2, statistically modified s-Br/NMe3+@TiO2 and Janus titania particles J-Br/NMe3+@TiO2. | ||
FTIR
The successful modification of titania particles with BrC6PA and NMe3+C6PA was demonstrated by FTIR spectroscopy (Fig. 2). All spectra show the typical Ti–O–Ti skeleton vibration at 400 cm−1. A broad band in the region from 2600 cm−1 to 3600 cm−1 indicates the presence of water and OH groups on the particle surface. Asymmetric and symmetric C–H stretching vibrations (νas(CH), νs(CH)) appear at 2935 cm−1 and 2858 cm−1, which in case of NMe3+C6PA are caused by the alkyl chain spacer and the methyl groups. The broad band from 1180 cm−1 to 900 cm−1 can be assigned to P–O–Ti stretching vibrations (ν(P–O)) and therefore proves the successful bonding of the phosphonate groups to the particle surface. The two phosphonic acids can be distinguished by the C–H deformation vibration of the CH group next to the bromine (δ(Br-CH2) = 1462 cm−1) and the ammonium bromide group (δ(N-CH2) = 1479 cm−1, δ(N-CH3) = 1488 cm−1). Furthermore, the C–H deformation vibrations of the alkyl chains are slightly shifted (δ((CH3)3NCH2-CH2) = 1407 cm−1, δ(BrCH2-CH2) = 1405 cm−1). This shift also occurs in the spectra of the phosphonic acids NMe3+C6PA and BrC6PA (Fig. S5†). Comparing the intensities of the ν(P–O), ν(C–H) and δ(C–H) relative to the Ti–O–Ti bands, it becomes clear that the grafting density must be higher in case of Br@TiO2. Due to the electrostatic repulsion of the cationic ammonium groups, the grafting density is lower in case of NMe3+C6PA. In the spectra of the statistically (s-Br/NMe3+@TiO2) and Janus-modified particles (J-Br/NMe3+@TiO2) deformation vibrations δ(N-CH) and δ(Br-CH2) indicate the presence of BrC6PA as well as NMe3+C6PA on the particle surfaces. Because of the low grafting density with the cationic surfactant NMe3+C6PA the deformation vibration δ(N-CH2) only appears as slight shoulder of the neighbouring deformation vibration δ(Br-CH2).Solid-state NMR spectroscopy
Besides FTIR spectroscopy the modified particles were analysed by solid-state 13C and 31P CP-MAS NMR spectroscopy. Especially solid-state 31P CP-MAS NMR is a powerful tool to investigate the binding of phosphorous compounds onto metal oxide surfaces.28,42–46 Generally, phosphonic acids can bind coordinatively or covalently to the metal oxide surface or form bulk metal phosphonates depending on the type of metal oxide, the phosphonic acid and the reaction conditions.28,43,46 The solid-state 31P CP-MAS NMR spectra of the phosphonic acids and the modified titania particles are shown in Fig. 3. Chemical shifts are summarized in Table S1 in the ESI.† NMe3+C6PA and BrC6PA show narrow peaks at 32.1 ppm and 32.5 ppm, respectively. In comparison there is an upfield shift in the 31P signal after the modification step for modified particles, indicating covalent attachment of the phosphonic acid onto the titania surface.43,46,47 Furthermore, a significant peak broadening occurs, which implies the presence of several binding modes i.e., mono-, bi- and tridentate covalent attachment.42,43,46,47 Fitting the spectra with Gaussian functions shows at least three types of bindings. It is difficult to quantitatively distinguish between the different modes because cross-polarization used in the NMR experiments are known to distort the signal intensities.47 The absence of any peak in the range from −4 ppm to 7 ppm suggests that no bulk titanium phosphonate phase is formed and that under the given conditions phosphonic acids only bind to the titania surface. This observation is in good agreement with literature which states that for titania significant formation of bulk phosphonates occurs only at elevated temperatures.42,43,46 Furthermore, solid-state 13C CP-MAS NMR show the typical signals of the phosphonates (Fig. S13†). | ||
| Fig. 3 31P solid-state CP-MAS NMR spectra of phosphonic acids and modified titania. Schematical illustration of (a) mono-, (b) bi- and (c) tridentate bonding modes of phosphonic acids. | ||
Surface coverage
The detection of the surface coverage is an important parameter for the successful modification of the particles. It was investigated by thermogravimetric analysis (TGA), elemental analysis (EA) and inductively coupled plasma mass spectroscopy (ICP-MS). In a typical TGA measurement the samples were heated to 100 °C for 10 min under nitrogen flow to evaporate adsorbed water and organic solvents. Afterwards the samples were heated to 700 °C under nitrogen flow and further to 1000 °C under a mixture of nitrogen and oxygen (1:9) (Fig. 4). For better comparison the initial weight loss up to 100 °C caused by evaporation of residual water and solvent is not displayed here. The complete TGA curves from 30 °C to 1000 °C can be found in the ESI (Fig. S4†). For pristine titania particles 9.5 wt% mass loss were detected between 130 °C to 377 °C caused by desorption of water that evolves due to further condensation of the OH groups at the particles surface. Two main mass losses were detected for all modified particles before and after the use of oxygen as purging gas at 700 °C. The first mass loss consists of several individual steps that cannot be separated from each other. To compare and calculate the surface coverage the total mass loss from 100 °C to 1000 °C was considered. Isotropically modified particles NMe3+@TiO2 and Br@TiO2 showed total mass losses of 10.2 wt% and 21.2 wt%, respectively. For statistically modified particles s-Br/NMe3+@TiO2 and Janus particles J-Br/NMe3+@TiO2 mass losses of 20.0 wt% and 18.8 wt% were detected, respectively. In case of isotropically modified particles, the molar amount of phosphonic acid per gram particle can be calculated directly from the total mass loss, as stated in previous literature.28 The calculation must take into account that the phoshonate group remains attached to the particles surface even after the heat treatment up to 1000 °C due to the strong P–O–Ti bonding. This effect was also previously observed for ZrO2 and demonstrated by FTIR measurements after TGA, where the P–O–Ti vibration peak is still visible (Fig. S14†).28 The equations used for the calculation are given in the Experimental section (eqn (1) and (2)). Because the decomposition of the two phosphonic acids cannot be distinguished in the TGA, this calculation is impractical for determining the amount of NMe3+C6PA and BrC6PA in s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2. CHN analysis allows the determination of the amount of NMe3+C6PA from the nitrogen content using eqn (3). By subtracting the theoretical carbon content caused by NMe3+C6PA from the total C content, the amount of BrC6PA can then be calculated from the residual carbon content. All results of the elemental analysis and the calculated surface coverage are listed in Table 1. Comparing TGA and elemental analysis, the calculated surface coverages from mass loss, C content and N content match well. The surface coverage for Br@TiO2 and NMe3+@TiO2 is 1.5 mmol g−1 to 1.7 mmol g−1 and 0.5 mmol g−1, respectively. The lower surface coverage in case of NMe3+C6PA is caused by the electrostatic repulsion of the ammonium ion.28 Surprisingly this effect is even more pronounced in case of statistically and Janus-modified particles in which 0.1 mmol g−1 NMe3+C6PA and 1.2 mmol g−1 to 1.3 mmol g−1 BrC6PA are found at the surface. This suggest that the binding of BrC6PA to the titania surface is also kinetically favoured over NMe3+C6PA.
 | ||
| Fig. 4 TGA of titania particles: pristine TiO2 (blue), hydrophobically modified Br@TiO2 (black), cationically modified NMe3+@TiO2 (green), statistically modified s-Br/NMe3+@TiO2 (red) as well as Janus-modified J-Br/NMe3+@TiO2 (grey). | ||
| Sample | Elemental analysis [%] | Surface coverage [mmol g−1] calculated from | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C | H | N | TGA | C | H | N | ICP-MS (P:Ti) |
ICP-MS (Br:Ti) |
|
| Pristine TiO2 | 0.73 | 1.10 | 0 | — | — | — | — | — | — |
| Br@TiO2 | 8.45 | 1.89 | 0 | 1.7 | 1.5 | 1.8 | — | 1.42 | 1.18 |
| NMe3+@TiO2 | 4.72 | 1.75 | 0.51 | 0.5 | 0.5 | 1.1 | 0.4 | 0.46 | 0.06 |
| s-Br/NMe3+@TiO2 | 8.20 | 1.91 | 0.12 | — | 1.3 (BrC6PA) | — | 0.1 (NMe3+C6PA) | 1.54 (total) | 0.69 (total) |
| J-Br/NMe3+@TiO2 | 7.62 | 1.84 | 0.1 | — | 1.2 (BrC6PA) | — | 0.1 (NMe3+C6PA) | 1.30 (total) | 0.94 (total) |
Besides TGA and elemental analysis, induced coupled plasma mass spectroscopy (ICP-MS) was used to calculate the surface coverage of the particles. Therefore, all particle samples were dissolved in hot sulfuric acid and the Ti, P and Br amount were measured. The surface coverage can be calculated using the P:Ti and the Br:Ti ratio (Table 1). The coverage calculated by P:Ti and carbon content from elemental analysis are in good agreement for Br@TiO2 and NMe3+@TiO2. For s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2 it is not possible to distinguish between the two phosphonic acids. Therefore, only the total amount of phosphonic acid at the surface can be calculated. In both cases, however, the sum of BrC6PA amount and NMe3+C6PA amount from elemental analysis fits the total amount calculated from the P:Ti ratio. This indicates the correctness of the values calculated from elemental analysis. In contrast, significantly lower surface coverages are obtained based on Br:Ti ratios. Especially for NMe3+@TiO2 almost no Br could be detected at the particles. To check the purity of the phosphonic acids, their Br and P content were also determined in ICP-MS measurements. Br:P ratios of 1.03 and 0.98 were obtained for BrC6PA and NMe3+C6PA, respectively which fits the expected value of 1.00. The Br content thus changes during the modification step. Since the modification step is carried out in aq. HCl solutions the bromide is exchanged for chloride as counterion. Bromide ions are subsequently removed during the washing process. In case of BrC6PA the bromine is partially substituted by chloride. Therefore, lower Br:Ti ratios are achieved in all cases compared to P:Ti ratios. In addition to exchange of bromide with chloride, bromide can also be removed during the dissolution in hot sulfuric acid by evaporation of HBr. Therefore, the total surface coverage from ICP-MS cannot be determined by the Br:Ti but only by the P:Ti ratio.
ζ-Potential titrations
The surface charge of the modified titania partuiles was investigated by ζ-potential titrations. The effect of quaternary ammonium groups on the ζ-potential has been studied for example by Radchanka et al. who showed the pH independency of the ζ-potential of quantum dots modified with trimethyl ammonium groups.35 In a previously published study we were able to tune the isoelectric point (IEP) of ZrO2 nanoparticles from 5.4 up to 8.7 by surface modifications with varying ratios of ammonium and methyl groups using NMe3+C6PA and methylphosphonic acid as surface modifiers.28 Furthermore NMe3+C6PA was used to switch the IEP of magnetite nanoparticles from 7.3 to 10.6 to embed the particles in an anionic polymer matrix.48 Here, the IEPs of titania particles modified with BrC6PA and NMe3+C6PA were compared with pristine titania obtained from a sol–gel synthesis. Due to the poor dispersibility of Br@TiO2, s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2 in water all measurements were conducted in an ethanol–water mixture (1:1; v:v). The titration curves and IEP are shown in Fig. 5 and Table 2, respectively. In the ethanol–water mixture, the pristine particles and the cationic NMe3+@TiO2 particles exhibit the highest ζ-potential of 10.2 mV at a pH of 2.83 and 4.89. In the case of pristine particles, this is caused by the protonation of OH groups on the surface. In case of NMe3+@TiO2, the high initial value is due to the cationic charge of the ammonium group. For Br@TiO2, s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2, all graphs start at lower ζ-potentials at around 6 mV. Modification with BrC6PA reduces the number of OH groups on the surface due to condensation with the phosphonic acid linker. The bromohexyl group itself cannot be protonated or deprotonated under these conditions and does not carry any ionic group. Therefore, a reduction in the amount of OH groups leads to a decrease in the ζ-potential. The low ζ-potential for s-Br/NM3+@TiO2 and J-Br/NMe3+@TiO2 is caused by the low amounts of NMe+C6PA on the surface compared to the amount of BrC6PA. For these particles the sum of positively charged groups on the surface (–H3O+, –NMe3+) is lower compared to pristine TiO2 which leads to a lower ζ-potential. This effect was previously observed for zirconia particles modified with methylphosphonic acid and NMe3+C6PA. For the modification with ten times the amount of methylphosphonic acid compared to NMe3+C6PA the ζ-potential decreased significantly compared to pristine zirconia.28 Untreated titania has an almost neutral IEP at 6.98 whereas the IEP of Br@TiO2 is in the acidic range at 4.13. Cationic NMe3+@TiO2 particles are positively charged over a larger pH range but still show an IEP at 9.33 although a pH independent cationic charge might be expected. Due to the low grafting density of the NMe3+C6PA, unreacted OH groups remain at the titania surface, which are deprotonated under basic conditions and therefore reduce the ζ-potential at higher pH. This effect is also observed for ammonium modified zirconia particles.28 Furthermore, the saturation of negatively charged OH− counterions in the solution can lead to an decrease of the ζ-potential.34 The IEP of s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2 only differs slightly from the IEP of Br@TiO2 due to the very low grafting density with NMe3+C6PA. In both cases only 0.1 mmol g−1 bind to the titania surface. Comparing s-Br/NM3+@TiO2 and J-Br/NMe3+@TiO2 the IEP of amphiphilic modified particles is slightly higher compared to statistically modified particles. Based on the total grafting densities (Table 1), it can be assumed that there are more unreacted OH-groups on the surface of J-Br/NMe3+@TiO2 compared to s-Br/NMe3+@TiO2. This is caused by an uncomplete modification of the cationic modified hemisphere in the Pickering emulsion modification step. During this process, unreacted OH-groups can not react with BrC6PA due to the insolubility of BrC6PA in water. In case of s-Br/NMe3+@TiO2, OH-groups between two NM3+-groups on the surface can react with BrC6PA, resulting in an overall higher grafting density and a lower number of unreacted OH-groups. This difference in surface hydroxy groups influences the ζ-potential, so that the IEP of J-Br/NMe3+@TiO2 is shifted to slightly higher values compared to s-Br/NMe3+@TiO2.
 | ||
| Fig. 5 ζ-Potential titration of modified titania particles dispersed in ethanol–water mixture (1:1; v:v). | ||
| Sample | IEP |
|---|---|
| TiO2 | 6.98 |
| Br@TiO2 | 4.13 |
| NMe3+@TiO2 | 9.33 |
| s-Br/NMe3+@TiO2 | 4.21 |
| J-Br/NMe3+@TiO2 | 4.53 |
Emulsion stabilization
Due to their better wettability with water and oil, amphiphilic particles should stabilise oil–water emulsions better than completely hydrophobic or hydrophilic modified particles. Binks calculated a threefold increase in the stabilization of Pickering emulsions using Janus particles compared to homogeneously modified particles.29 This theoretical assumption was later verified experimentally by Xue et al.16 Here, pristine and modified titania were compared in their behaviour to stabilize chloroform–water emulsions. For better visualization of the interface, red dye (Lumogen Red 305) has been added to the oil phase. The emulsions were prepared by shaking mixtures of the respective aqueous particle suspensions with equal volumes of the coloured chloroform solution for 30 seconds. Afterwards the emulsions were stored at room temperature. Pickering emulsions are obtained in all cases, except for NMe3+@TiO2 (Fig. 6). On one hand, this is caused by the hydrophilic character of the particles and the resulting poor wettability of the particles with the chloroform phase. On the other hand, the ionic groups cause a strong electrostatic repulsion, which prevents the particles from assembling in a dense layer at the liquid–liquid interface. To investigate the type of emulsion, fluorescence microscopy images were taken from the coloured emulsions (Fig. 7). Pristine TiO2 forms oil-in-water emulsions (O/W) whereas Br@TiO2 forms water-in-oil emulsions (W/O). According to Bancroft's rule, O/W are formed by hydrophilic particles and W/O are formed by hydrophobic particles if equal volume fractions of oil and water are used.24 Surprisingly, s-Br/NMe3+@TiO2 as well as J-Br/NMe3+@TiO2 stabilise O/W emulsions although the amount of BrC6PA at the particle surface is much higher compared to NMe3+C6PA and the particles tend to be more hydrophobic. This anti-Bancroft behaviour was previously observed for other systems, like partially hydrophobic silica particles for O/W emulsions and charged latex particles for W/O emulsions.49–51 The formation of such Pickering emulsions depends on the oil to water volume ratio, as well as on the particles wettability and the phase the particles were initially dispersed in ref. 49, 52 and 53. Because the particles were first dispersed in water, the emulsification process leads to O/W emulsions instead of W/O emulsions. The obtained Pickering emulsions were observed over several days to investigate the demulsification process (Fig. 6). The W/O emulsion stabilized by Br@TiO2 (vial B in Fig. 6) starts to break after several minutes. After 23 minutes already half of the volume is demixed. After three days the two phases separated completely and the Br@TiO2 particles settle to the bottom of the vial. Emulsions stabilized by s-Br/NMe3+@TiO2 starts to demulsify after 20 minutes. Hereby, the particles agglomerate and form a gel-like structure in the aqueous phase (vial D in Fig. 6). For O/W emulsions stabilized by pristine TiO2 and J-Br/NMe3+@TiO2 a rapid sedimentation of the emulsified oil droplets is observed due to the higher density of the chloroform compared to water. To compare the emulsions after three days, the height of the emulsified portion is set in relation to the total height. The ratio is given in percentage. For s-Br/NMe3+@TiO2 only 17% remain as O/W emulsion. For TiO2 and J-Br/NMe3+@TiO2, the emulsions phase accounts for 55% and 57% of the total volume after sedimentation. The increased emulsion volume that can be stabilization by J-Br/NMe3+@TiO2 compared to s-Br/NMe3+@TiO2 already shows the amphiphilic behaviour of the Janus particles. To further compare the stabilization behaviour of s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2, the particle content, solvent and pH were varied (Fig. S16–S18†). Generally, an increase in particle concentration leads to an increase in emulsion stability and a decrease in droplet size, because a larger interfacial area can be covered by the particles.53 This effect is most pronounced at lower concentrations.54 For all tested particle concentrations (0.8 wt%, 1.0 wt%, 2.0 wt%, 4.0 wt%) O/W emulsions were obtained, and no significant change in their stability or droplet size is observed (Fig. S16†). Probably the concentrations are already high enough, so no significant changes can be observed. Besides the particle concentration, the wettability of the particles and the interfacial tension of the oil–water interface has a crucial influence on the emulsion stability. In order to stabilize emulsions, the particles need to be wettened partially by both liquids. Stabilization therefore depends on the oil phase used.53 We therefore used solvents with different polarities to prepare emulsions. Besides chloroform, s-Br/NMe3+@TiO2 and J-Br/NMe3+@TiO2 can stabilize emulsions with toluene, cyclohexane, and ethylene acetate (Fig. S17†). With ethylene acetate, smaller oil droplets are observed, which could be due to the higher polarity compared to the other solvents. When switching the pH to basic conditions (pH = 9) no emulsion is formed with statistically modified s-Br/NMe3+@TiO2, whereas with J-Br/NMe3+@TiO2 a small portion of the oil phase is dispersed in water, forming large chloroform droplets in the water phase (Fig. S18†). The increase in droplet size indicates coalescence of the emulsion and thus a less stable emulsion. Compared to s-Br/NMe3@TiO2, however, an emulsion is formed with J-Br/NMe3+@TiO2. | ||
| Fig. 6 Chloroform–water mixture stabilised with pristine TiO2 (a), Br@TiO2 (b), NMe3+@TiO2 (c), s-Br/NMe3+@TiO2 (d), J-Br/NMe3+@TiO2 (e) after 0 min, 23 min and 3 d. Chloroform phase is coloured with Lumogen Red 305. | ||
 | ||
| Fig. 7 Fluorescence microscopy images of chloroform–water Pickering emulsions immediately after preparation stabilised with TiO2 (a), Br@TiO2 (b), s-Br/NMe3+@TiO2 (c) and J-Br/NMe3+@TiO2 (d). Chloroform phase is coloured with Lumogen Red 305. | ||
Layer-by-layer deposition
To investigate layer formation and their Janus character, the modified particles J-Br/NMe3+@TiO2 were deposited on glass slides in a layer-by-layer approach. In previous studies, we used metal oxide particles modified with NMe3+C6PA to grow stable layers on glass slides by a simple dip coating process.28 In this process, the layer thickness grows linearly with the dipping cycle. In the present study, we deposit the Janus particles in a similarly using 0.1 wt% ethanolic particle dispersions. UV-Vis spectroscopy and three-phase contact angle (CA) measurements were subsequently performed to verify the deposition of the particles. All UV-Vis measurements are shown in the ESI (Fig. S15†). Since the glass slides used show total absorption at 300 nm the transmission of the samples was compared at 350 nm. The transmission values are plotted against the dipping cycles (Fig. 8). The decrease in transmission shows the adsorption of the particles at the glass surface. Besides the UV-Vis transmission, the three-phase contact angle of water on the obtained particle layer was also investigated (Fig. 9). On uncoated glass slides water forms a CA of 49°. After three dipping cycles the CA increases up to 88°. This value agrees well with the CA of 98° for Br@TiO2, which was obtained by drop casting Br@TiO2 particles on glass slides and measuring the three-phase contact angle. The CA remains almost constant from 6 to 18 dipping cycles but decreases to 34° after 42 cycles. The curve of the CA change follows the curve of the change in transmission (Fig. 8). This clearly indicates that no partial delamination takes place after several dipping cycles, but with each dipping cycle new particles adsorb to the substrate. To compare the CA values, suspensions of pristine and isotropically modified particles were drop casted on glass slides to get a dense layer of the respective particle. Subsequently, the three-phase CA was measured. 13°, 11° and 98° were obtained for pristine titania, NMe3+@TiO2 and Br@TiO2, respectively. The CA for J-Br/NMe3+@TiO2 after 18 dipping cycles is in good agreement with the measured values for Br@TiO2. In the first dipping cycles, the hemisphere of the cationically modified particles will align towards the glass, while the hydrophobic part will face outwards. This leads to a poor wettability of the obtained layer. With each dipping cycle, the particles adsorb to the glass until a dense layer has formed and there is no more free space on the hydrophilic glass surface. At this stage, further particles can only adsorb via hydrophobic van der Waals interactions. The hemisphere of the hydrophilic cationic particles is therefore directed outwards and wettability increases again. This causes the decrease in CA from 32 to 42 dipping cycles. The difference between the CA of NMe3+@TiO2 compared to Janus particles after 42 dipping cycles is caused by the low amount of NMe3+C6PA and a comparatively high amount of BrC6PA on the surface of J-Br/NMe3+@TiO2. Still, the presented Janus particles J-Br/NMe3+@TiO2 are suitable for tailoring wettability of plane substrates by an easy feasible dip coating process. | ||
| Fig. 8 UV-Vis transmission at 350 nm wavelength of glass substrates coated with J-Br/NMe3+@TiO2. | ||
 | ||
| Fig. 9 Mean three-phase contact angle on glass substrates after dip coating with J-Br/NMe3+@TiO2 and images after 0 (a), 3 (b), 18 (c) and 39 (d) dipping cycles. Standard deviations from three measurements are given as bars. | ||
Conclusion
In this work, we have presented a straightforward method for the preparation of amphiphilic titania Janus nanoparticles, carrying an apolar bromine functionality on one hemisphere and a cationic quaternary ammonium group on the opposite hemisphere, using organophosphorus molecules as coupling agents. This Janus modification was achieved in a toluene–water Pickering emulsion. For comparison, titania particles were also isotropically modified to obtain particles covered solely with either the bromine or quaternary ammonium functionality, and statistically modified to achieve particles where both coupling molecules cover the surface in a statistically ordered manner. FTIR and 31P solid-state CP MAS NMR confirmed the covalent attachment of the phosphonic acids onto the surface of the titania particles whereas no bulk titania phosphonates could be observed. The surface coverage of modified particles was investigated through a combination of elemental analysis, TGA, and ICP-MS. For particles modified with bromohexylphosphonic acid grafting densities of 1.5 mmol g−1, 1.3 mmol g−1 and 1.2 mmol g−1 are obtained for isotropic, statistical and Janus modification, respectively. Functionalization with the cationic coupling agent bearing the quaternary ammonium group resulted in grafting densities of 0.5 mmol g−1, 0.1 mmol g−1 and 0.1 mmol g−1 for isotropic, statistic and Janus modification, respectively. Both statistically modified and Janus-modified particles were found to stabilise O/W Pickering emulsions with different solvents and in a wide pH range. Notably, Janus particles exhibited exceptional stability, successfully stabilizing chloroform-in-water emulsions for over three days, attributable to their amphiphilic nature. Furthermore, the Janus particles were employed to manipulate the wettability of a glass slide through a dip coating process, leading to a transition from hydrophilic to hydrophobic to hydrophilic surfaces depending on the dipping cycle. This demonstrates the versatility and potential applications of Janus nanoparticles in controlling interfacial properties for various technological and industrial purposes.Author contributions
L. N. conceptualization, investigation, formal analysis, methodology, writing original draft, visualization; G. K.: conceptualization, funding acquisition, supervision, project administration, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Instrumentation and technical assistance for this work were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and German Science Foundation (project number INST 256/349-1). ICP-MS instrumentation for this work was provided by the Elemental analysis group, with financial support from Saarland University and German Science Foundation (project number INST 256/553-1). We thank Susanne Harling for the elemental analysis, Prof. Dr Marc Schneider and Armin Novak from Saarland University (Germany) for provision of the zeta sizer and Elias Gießelmann for solid-state CP-MAS NMR measurements.References
- A. Walther and A. H. E. Müller, Chem. Rev., 2013, 113, 5194–5261 CrossRef CAS PubMed.
- J. Hu, S. Zhou, Y. Sun, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 4356–4378 RSC.
- H. Wang, S. Yang, S. N. Yin, L. Chen and S. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 8827–8833 CrossRef CAS PubMed.
- L. Kong, C. C. Mayorga-Martinez, J. Guan and M. Pumera, ACS Appl. Mater. Interfaces, 2018, 10, 22427–22434 CrossRef CAS PubMed.
- N. Babajani, C. Kaulen, M. Homberger, M. Mennicken, R. Waser, U. Simon and S. Karthäuser, J. Phys. Chem. C, 2014, 118, 27142–27149 CrossRef CAS.
- C. Xu, J. Xie, D. Ho, C. Wang, N. Kohler, E. G. Walsh, J. R. Morgan, Y. E. Chin and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 173–176 CrossRef CAS PubMed.
- K. Panwar, M. Jassal and A. K. Agrawal, Cellulose, 2018, 25, 2711–2720 CrossRef CAS.
- F. Liu, S. Goyal, M. Forrester, T. Ma, K. Miller, Y. Mansoorieh, J. Henjum, L. Zhou, E. Cochran and S. Jiang, Nano Lett., 2019, 19, 1587–1594 CrossRef CAS PubMed.
- D. Jishkariani, Y. Wu, D. Wang, Y. Liu, A. van Blaaderen and C. B. Murray, ACS Nano, 2017, 11, 7958–7966 CrossRef CAS PubMed.
- Z. W. Seh, S. Liu, M. Low, S. Y. Zhang, Z. Liu, A. Mlayah and M. Y. Han, Adv. Mater., 2012, 24, 2310–2314 CrossRef CAS PubMed.
- A. Bachinger, S. Ivanovici and G. Kickelbick, J. Nanosci. Nanotechnol., 2011, 11, 8599–8608 CrossRef CAS PubMed.
- N. Zahn and G. Kickelbick, Colloids Surf., 2014, 461, 142–150 CrossRef CAS.
- J. Song, B. Wu, Z. Zhou, G. Zhu, Y. Liu, Z. Yang, L. Lin, G. Yu, F. Zhang, G. Zhang, H. Duan, G. D. Stucky and X. Chen, Angew. Chem., Int. Ed., 2017, 56, 8110–8114 CrossRef CAS PubMed.
- Y. Guo, A. U. Khan, K. Cao and G. Liu, ACS Appl. Nano Mater., 2018, 1, 5377–5381 CrossRef CAS.
- S. D. Bourone, C. Kaulen, M. Homberger and U. Simon, Langmuir, 2016, 32, 954–962 CrossRef CAS PubMed.
- W. Xue, H. Yang and Z. Du, Langmuir, 2017, 33, 10283–10290 CrossRef CAS PubMed.
- J. Luo, M. Zeng, B. Peng, Y. Tang, L. Zhang, P. Wang, L. He, D. Huang, L. Wang, X. Wang, M. Chen, S. Lei, P. Lin, Y. Chen and Z. Cheng, Angew. Chem., Int. Ed., 2018, 57, 11752–11757 CrossRef CAS PubMed.
- A. Kirillova, C. Marschelke and A. Synytska, ACS Appl. Mater. Interfaces, 2019, 11, 9643–9671 CrossRef CAS PubMed.
- S. Berger, L. Ionov and A. Synytska, Adv. Funct. Mater., 2011, 21, 2338–2344 CrossRef CAS.
- F. Liang, B. Liu, Z. Cao and Z. Yang, Langmuir, 2018, 34, 4123–4131 CrossRef CAS PubMed.
- A. Perro, S. Reculusa, S. Ravaine, E. Bourgeat-Lami and E. Duguet, J. Mater. Chem., 2005, 15, 3745 RSC.
- L. Hong, S. Jiang and S. Granick, Langmuir, 2006, 22, 9495–9499 CrossRef CAS PubMed.
- A. Perro, F. Meunier, V. Schmitt and S. Ravaine, Colloids Surf., 2009, 332, 57–62 CrossRef CAS.
- B. P. Binks and S. O. Lumsdon, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- E. Hoque, J. A. DeRose, G. Kulik, P. Hoffmann, H. J. Mathieu and B. Bhushan, J. Phys. Chem. B, 2006, 110, 10855–10861 CrossRef CAS PubMed.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS PubMed.
- C. Heinrich, L. Niedner, B. Oberhausen and G. Kickelbick, Langmuir, 2019, 35, 11369–11379 CrossRef CAS PubMed.
- B. P. Binks and P. D. I. Fletcher, Langmuir, 2001, 17, 4708–4710 CrossRef CAS.
- D. Lee, M. F. Rubner and R. E. Cohen, Nano Lett., 2006, 6, 2305–2312 CrossRef CAS PubMed.
- R. K. Iler, J. Colloid Interface Sci., 1966, 21, 569–594 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- J. Y. Lee, K. H. Choi, J. Hwang, M. Sung, J. E. Kim, B. J. Park and J. W. Kim, Chem. Commun., 2020, 56, 6031–6034 RSC.
- A. Synytska, M. S. A. Kirillova and L. Isa, ChemPlusChem, 2014, 79, 656–661 CrossRef CAS.
- A. Radchanka, V. Hrybouskaya, A. Antanovich and M. Artemyev, ChemNanoMat, 2022, 8, e202100538 CrossRef CAS.
- C. Marschelke, A. Fery and A. Synytska, Colloid Polym. Sci., 2020, 298, 841–865 CrossRef CAS.
- A. Bachinger and G. Kickelbick, Monatsh. Chem., 2010, 141, 685–690 CrossRef CAS.
- Y. Zhou, F. Shen, S. Zhang, Q. Zhao, Z. Xu and H. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 29876–29882 CAS.
- M. Rehosek, M. Laupheimer and F. Marlow, Colloid Polym. Sci., 2024, 302, 253–260 CrossRef CAS.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12, 6429–6435 CrossRef CAS.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS.
- P. H. Mutin, G. Guerrero and A. Vioux, J. Mater. Chem., 2005, 15, 3761 RSC.
- G. P. Holland, R. Sharma, J. O. Agola, S. Amin, V. C. Solomon, P. Singh, D. A. Buttry and J. L. Yarger, Chem. Mater., 2007, 19, 2519–2526 CrossRef CAS.
- M. Tassi, G. Reekmans, R. Carleer and P. Adriaensens, Solid State Nucl. Magn. Reson., 2016, 78, 50–55 CrossRef CAS PubMed.
- H. Souma, R. Chiba and S. Hayashi, Bull. Chem. Soc. Jpn., 2011, 84, 1267–1275 CrossRef CAS.
- B. Oberhausen and G. Kickelbick, Nanoscale Adv., 2021, 3, 5589–5604 RSC.
- D. M. Ramos, V. Sadtler, P. Marchal, C. Lemaitre, F. Niepceron, L. Benyahia and T. Roques-Carmes, Nanomaterials, 2023, 13, 371 CrossRef CAS PubMed.
- D. M. Ramos, V. Sadtler, P. Marchal, C. Lemaitre, L. Benyahia and T. Roques-Carmes, Colloids Surf., 2023, 673, 131782 CrossRef CAS.
- T. Nallamilli, B. P. Binks, E. Mani and M. G. Basavaraj, Langmuir, 2015, 31, 11200–11208 CrossRef CAS PubMed.
- N. Yan, M. R. Gray and J. H. Masliyah, Colloids Surf., 2001, 193, 97–107 CrossRef CAS.
- D. Gonzalez Ortiz, C. Pochat-Bohatier, J. Cambedouzou, M. Bechelany and P. Miele, Engineering, 2020, 6, 468–482 CrossRef.
- J. Frelichowska, M.-A. Bolzinger and Y. Chevalier, J. Colloid Interface Sci., 2010, 351, 348–356 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TGA, FTIR, liquid 1H, 13C, 31P NMR, solid-state 13C CP-MAS NMR, UV-Vis spectra and photographs of Pickering emulsions. See DOI: https://doi.org/10.1039/d3nr04907h |
| This journal is © The Royal Society of Chemistry 2024 |