
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and properties of spindle-shaped aragonite mesocrystals from Mg-bearing solutions†
Zsombor
Molnár
 *ab,
Péter
Pekker
a,
Aleksander
Rečnik
c and
Mihály
Pósfai
ab
*ab,
Péter
Pekker
a,
Aleksander
Rečnik
c and
Mihály
Pósfai
ab
aUniversity of Pannonia, Research Institute of Biomolecular and Chemical Engineering, Nanolab, Egyetem st. 10, 8200, Veszprém, Hungary. E-mail: molnar.zsombor@mk.uni-pannon.hu
bHUN-REN-PE Environmental Mineralogy Research Group, Egyetem st. 10, 8200, Veszprém, Hungary
cJožef Stefan Institute, Department of Nanostructured Materials, Jamova cesta 39, 1000, Ljubljana, Slovenia
First published on 9th January 2024
Abstract
The formation of aragonite under ambient conditions is typically linked to Mg-rich aqueous environments. The grains that form in such environments show peculiar properties such as aggregate-like appearance and mesocrystalline character. We tested the effect of dissolved Mg2+ ions on the formation of aragonite mesocrystals by synthesizing aragonite with an automatic titrator at constant pH and at different dissolved Mg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ca ratios, and by studying the properties of the precipitated material with various scanning transmission electron microscopy (STEM) techniques. At all studied Mg:Ca ratios the firstly condensed carbonate phase was Mg-bearing amorphous calcium carbonate (Mg-ACC) that transformed into aragonite during the synthesis experiments. The aragonite grains had typically aggregate-like appearance and spindle shapes, with the external morphologies of the spindles unaffected by variation in solution chemistry. The alignment of the nanocrystals within the aggregates was crystallographically highly coherent, the [001] directions of nanocrystals showing only a small misorientation with respect to one another; however, both parallel and twin assembly of neighbouring crystals occurred. An increase in the dissolved Mg concentration decreased the crystallographic coherence between the aragonite nanocrystals, suggesting an important role of Mg2+ ions in the assembly of aragonite spindles. Whereas the mesoscale-ordered arrangement of nanocrystals implies a particle-mediated assembly, the observed differences in particle size and composition between the amorphous precursor and the crystalline end-product suggest that the crystallization includes at least partial dissolution and re-precipitation. These findings provide insight into the formation of aragonite and could contribute to the understanding of important aspects of the formation of mesocrystals and hierarchically structured biogenic minerals.
:Ca ratios, and by studying the properties of the precipitated material with various scanning transmission electron microscopy (STEM) techniques. At all studied Mg:Ca ratios the firstly condensed carbonate phase was Mg-bearing amorphous calcium carbonate (Mg-ACC) that transformed into aragonite during the synthesis experiments. The aragonite grains had typically aggregate-like appearance and spindle shapes, with the external morphologies of the spindles unaffected by variation in solution chemistry. The alignment of the nanocrystals within the aggregates was crystallographically highly coherent, the [001] directions of nanocrystals showing only a small misorientation with respect to one another; however, both parallel and twin assembly of neighbouring crystals occurred. An increase in the dissolved Mg concentration decreased the crystallographic coherence between the aragonite nanocrystals, suggesting an important role of Mg2+ ions in the assembly of aragonite spindles. Whereas the mesoscale-ordered arrangement of nanocrystals implies a particle-mediated assembly, the observed differences in particle size and composition between the amorphous precursor and the crystalline end-product suggest that the crystallization includes at least partial dissolution and re-precipitation. These findings provide insight into the formation of aragonite and could contribute to the understanding of important aspects of the formation of mesocrystals and hierarchically structured biogenic minerals.
Introduction
Among the three anhydrous CaCO3 polymorphs, orthorhombic aragonite is typically described as the most stable phase at high pressure.1 Nevertheless, it forms abundantly under surface conditions, typically in aqueous environments characterized by either high dissolved Mg2+ concentrations or elevated temperatures.2–5 Despite its abundance and geochemical importance,6,7 the abiotic formation of aragonite is less studied than that of calcite.8Aragonite particles that form in Mg-rich aqueous media have a peculiar spindle-like appearance. They are composed of a large number of nanosized crystals that collectively produce single-crystalline electron diffraction patterns, implying a mesocrystalline character.9–12 These properties have been observed in both natural13 and synthetic samples, the latter including both abiotically6,9,11,14–23 and biomimetically synthesized aragonite.12,24–27 Whether they formed homogeneously10 or in the presence of additives (such as polyaspartic and amino acids,28 solely amino acids,26 polyamides,12 or swelling clay minerals11), the mesocrystalline characters of aragonite aggregates were similar. Despite their widespread occurrence, the formation mechanism of aragonite spindles is not clearly understood, even though such an understanding could shed light on important aspects of the formation of mesocrystals.
The formation of mesoscopically structured crystals (mesocrystals), especially the cohesion between their fundamental units is a widely studied yet puzzling phenomenon.29 According to the original definition,30 mesocrystals form by the attachment of nanocrystals, a process regulated by the interactions of their different facets, and potentially mediated by organic polymers or gels.31–33 However, Zhu et al.34 suggested that mesocrystals could alternatively be produced via conversion of amorphous nanoparticles into nanocrystals near mineral surfaces, in a process that involves both dissolution–re-precipitation and oriented attachment. Regardless of their assumed formation mechanism, the properties of mesocrystals differ from those of bulk single crystals,35e.g., have larger surface area and porosity, making them effective substrates for adsorption.36
With respect to the possible role of an amorphous precursor in the formation of aragonite mesocrystals, Walker et al.10 performed cryo-TEM analysis on freshly precipitated aragonite, synthesized in ethanol-rich aqueous media. They showed the formation of spindle-shaped aragonite mesocrystals in co-occurrence with ACC. The authors suggested that the formation of the spindles was initiated by the aggregation of ACC nanoparticles, and the crystallization of aragonite started within the amorphous aggregate from independent centres, with the amorphous to crystalline transformation taking place in solid state. Huang et al.26 and Nahi et al.12 studied the effect of amino acids and the combined effects of polyamines and dissolved Mg2+ ions on the formation of aragonite. Both studies reported similar aggregates of aragonite nanocrystals with a typical spindle-shaped external morphology, and observed the presence of small ACC globules on the surfaces or even within the cavities of aragonite aggregates. Based on the crystallographically highly oriented assembly of aragonite nanocrystals, the authors of both studies concluded that the spindle-shaped aragonite aggregates formed via the oriented attachment of nanocrystals. Nevertheless, they assumed that ACC was a precursor of aragonite.
While there are several reports in the literature on the formation of spindle-shaped aragonite aggregates, only a few studies addressed the specific role of Mg2+ ions.6 Since Mg2+ is present (in highly variable concentrations) in almost all types of natural waters, it probably affects contemporary aragonite formation in various environments. In addition, the variation of Mg:Ca ratio over the history of Earth in both marine37 and freshwater systems38 serves as a proxy for environmental changes; thus, knowing the roles of Mg in aragonite formation has importance for paleoenvironmental reconstructions as well.
In this work, laboratory studies were conducted under environmentally relevant conditions to examine the mechanism of aragonite formation in alkaline (pH 9.00), low-ionic-strength (0.02–0.08 M), Mg-rich aqueous media at ambient temperature (20 °C), with a focus on the transition of amorphous precursors into crystalline phases and the roles of dissolved Mg2+ ions. The experiments were conducted with various dissolved Mg:Ca ratios and the products were studied using transmission electron microscopy (TEM) techniques. We demonstrate that the firstly precipitated Mg-bearing ACC precursor transforms rapidly into aragonite, and aragonite mesocrystals form by attachment and subsequent crystallization of amorphous particles on the surfaces of crystalline nuclei. Further, we show that an increase of the Mg:Ca ratio slows the formation of aragonite and affects the size and orientation of the nanocrystalline building blocks within mesocrystals.
Materials and methods
Synthesis of aragonite
Aragonite mesocrystals were synthesized from aqueous solutions by titrating a carbonate buffer solution with mixtures of Mg and Ca chloride solutions under ambient conditions and constant pH. Firstly, 20 mL, 0.01 M Na2CO3 buffer solution was prepared, and the pH set to 9.00 by adding a sufficient amount of 1 M HCl. In order to study the effect of Mg2+ ions, we prepared the Mg-rich titrant solutions as mixtures of aqueous MgCl2 and CaCl2 solutions with 0.01:0.01, 0.04:0.01, 0.06:0.01 and 0.08 M:0.01 M dissolved Mg:Ca ratios, respectively. For the preparation of the solutions, we used analytical graded reagents and double distilled ultra-pure water with 18.2 MΩ resistivity as a solvent.
The titrants were dosed into the buffer solutions by a Metrohm 805 Dosimat automatic titrator. The dosing unit was 5 μL and the dosing speed 0.05 mL min−1. The pH of the solution was kept constant at 9.00 (±0.02) by the dynamic titration of 0.1 M NaOH with a Metrohm 888 Titrando instrument, and measured using a combined, glass pH electrode, with a measuring interval of 2 s. The reactions were monitored by the amount of the dosed NaOH solution. The point of nucleation of solid phases was defined by the concentration where the shape of the titrant curve of the NaOH solution changed from linear to exponential. Each experiment was repeated five times, and the results we present are the mean values for each dissolved Mg:Ca ratio.
The saturation indices (SI) of calcite, aragonite, ordered dolomite and Mg-bearing ACC (Mg-ACC) phases were calculated using the PHREEQC software.39 The known ionic compositions of the solutions at the point of nucleation, and the previously reported log(Ksp) values of calcite, aragonite, dolomite, pure-ACC and Mg-ACC were used for the calculations. References for the applied log(Ksp) values are included in Fig. S1.†
In parallel experiments, we sampled the precipitated materials first at the point of nucleation, then at the point where the volume ratio of the titrant and buffer solutions reached 1, and finally after aging for 2 months. Sampling was done by filtering the whole solution through a 0.45 μm MCE polymer membrane filter with a vacuum pump, then washing the filtered solid materials with 20 mL isopropanol in order to avoid their crystallization upon drying. Four specimens were prepared and analysed for each used Mg:Ca ratio, in order to make sure that incidental drying artefacts (such as individual NaCl or MgCl2 crystals) are recognized. We did not observe the occurrence of such salt phases on the surfaces or in the pores of aragonite crystals.
Material characterization
Samples for transmission electron microscopy (TEM) analyses were prepared by gently touching the surface of the wet filter paper with a carbon-coated lacey Cu grid immediately after the filtering procedure, and drying the grid at room temperature. For TEM analysis we used a Thermo Fisher Talos F200X G2 scanning transmission electron microscope (STEM) that could be operated in both conventional and scanning TEM mode, at 200 kV accelerating voltage. We studied the morphologies of solid particles in bright-field (BF) images, selected-area electron diffraction (SAED) patterns and high-resolution TEM (HRTEM) images for structural characterization of the grains. The beam current was around 80 pA in TEM mode. Elemental maps were obtained in STEM spectrum imaging mode by collecting energy-dispersive X-ray (EDX) spectra in each pixel of the image, using a four-detector SuperX Si drift detector system. In STEM mode the beam current was around 200 pA. The obtained data were processed and evaluated using Velox 2.14 software.Electron tomography measurements were carried out in STEM mode using the high-angle annular dark-field (HAADF) detector for imaging. The chosen grain was tilted in 1° steps in a tilt series between −62° to 60°. We used Inspect3D software for 3D reconstruction and the Avizo software for the visualization of particle morphologies.
Results
In our experiments designed to precipitate aragonite a carbonate buffer was titrated with mixtures of Mg and Ca chloride solutions, while the pH was kept constant by the dosing of NaOH. The measured curves of the dosed amount of NaOH solution showed a typical sigmoidal shape in each experiment, indicating a pH change within the solution that was balanced by the addition of the NaOH titrant (Fig. 1A). The general equation of solid CaCO3 formation is expressed by eqn (1).| Ca2+ + HCO3− ⇄ Ca2+ + CO32− + H+ → CaCO3 + H+ | (1) |
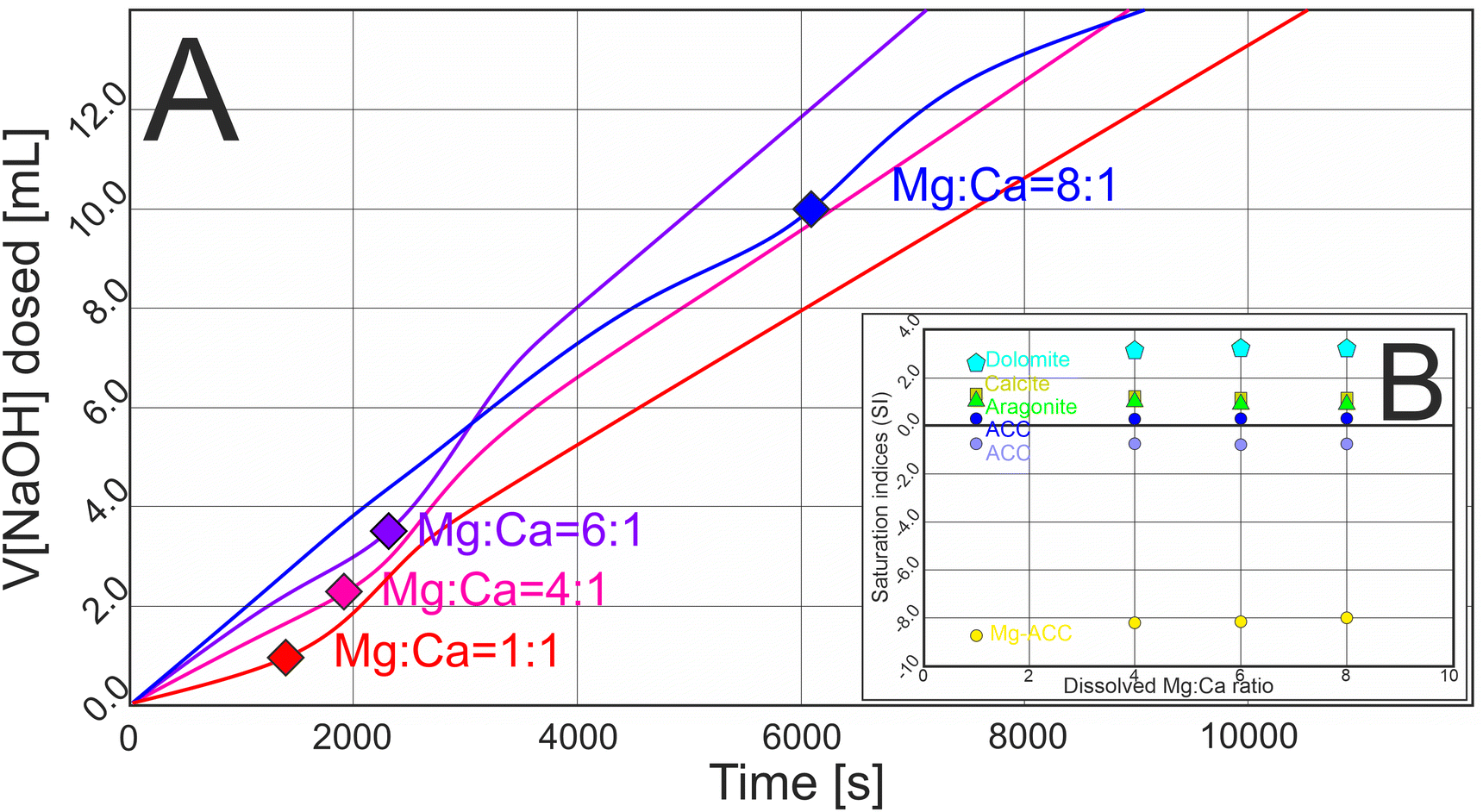 | ||
| Fig. 1 (A) Measured curves of the amount of dosed NaOH solution for four carbonate precipitation experiments with different dissolved Mg:Ca ratios. On each curve a rectangle marks the point where the increment of the dosed solution started to increase, indicating the starting point of the nucleation of carbonate solids (for details see Fig. S1†). (B) Calculated saturation indices (SI) of calcite, aragonite, dolomite, ACC (using two different Ksp values) and Mg-ACC (50 mol% Mg content) at the point of nucleation. The SIs indicate that the solution was undersaturated with respect to Mg-ACC and around the saturation state with respect to ACC. | ||
According to the law of mass action and the principle of mass preservation, HCO3− is the dominant form of carbonate at pH 9.00 (92 mol%), and CO32− occurs as a minor species (5 mol%). When HCO3− dissociates into CO32− and solid CaCO3 forms, H+ ions are liberated that decrease the pH of the solution. Adding NaOH maintained the initial pH 9.00, and the amount of added NaOH solution provided a proxy for identifying the nucleation of CaCO3. We refer to the point where the increment of the dosed NaOH solution started to increase as the point of nucleation.
Increasing the dissolved Mg:Ca ratio shifted the point of nucleation slightly, lengthening the pre-nucleation limb of the titration curves (Fig. 1A); thus, the solid phases formed at larger ionic strength (at 0.024, 0.031, 0.037 and 0.071 M for the 1:1, 4:1, 6:1 and 8:1 dissolved Mg:Ca ratios, respectively; see Fig. S1†). We used the ionic compositions of the solutions at the point of nucleation for calculating the saturation indices (SI) of possible solid phases (Fig. 1B). The SIs suggested that the solutions were supersaturated with respect to calcite, aragonite and dolomite, slightly supersaturated or undersaturated with respect to pure ACC, depending on the used log(Ksp) value (not shown in Fig. 1B), but undersaturated with respect to Mg-bearing ACC in every case (assuming 50 mol% Mg content in the amorphous phase and using the Ksp value of Purgstaller et al.40). The calculated SIs for calcite and aragonite were essentially unaffected by the Mg:Ca ratio (1.2 and 1.1, respectively) (Fig. 1B and S1†).
TEM study of the samples that were taken from the solutions immediately after the point of nucleation revealed co-existing Mg-ACC and aragonite particles. Mg-ACC particles were a few tens of nm large, their shapes spherical or elongated-globular; some individual globules also occurred, but they typically formed small groups in intimate association with aggregates of aragonite nanocrystals at every used Mg:Ca ratio (Fig. 2A). As expected, the groups of Mg-ACC showed amorphous character in both SAED patterns and fast Fourier transforms (FFTs) obtained from HRTEM images, producing diffuse intensity maxima (see the FFT of area 1 in Fig. 2A; Fig. S2 and S3†) with d-values corresponding to those reported by Molnár et al.41 Mg-ACC particles contained a significant amount of Mg (Fig. 2B–C and Fig. S3†) but their Mg:Ca ratio was lower than that of the solution (i.e., 0.45–0.55 vs. 4, respectively). On the other hand, no Mg could be detected in the aragonite aggregates, suggesting that Mg is not incorporated into aragonite even at an initial stage of formation (Fig. 2C). The smallest, presumably incipient aragonite particles were already aggregates that consisted of <10 nm nanocrystals; the entire aggregate produced SAED and FFT patterns reminiscent of single-crystal diffraction (see FFT of area 2 in Fig. 2A). Images such as in Fig. 2A and 3 suggested that aragonite aggregates grew by the attachment of Mg-ACC. Even though we sampled the solution at the point of nucleation, irregular-shaped aggregates (as in Fig. 2A) were rare (we observed about ten particles); instead, the typical aragonite aggregates resembled spindles (Fig. 3A, B and 4A), with specific morphologies and structures.
 | ||
| Fig. 2 Globular Mg-ACC particles with aragonite crystals. (A) HRTEM image with the corresponding FFTs of Mg-ACC and aragonite particles in the inserts, from the areas marked in the main panel. (B) STEM EDX elemental map and (C) corresponding EDX spectra extracted from the marked areas, showing significant Mg in Mg-ACC and almost none in aragonite. The small Mo Kα peak arises from stray radiation from the sample holder. | ||
 | ||
| Fig. 3 Globular Mg-ACC particles on spindle-shaped aragonite aggregates. (A) BF image and (B) STEM EDX elemental map from the same area of the same particle, suggesting that Mg-ACC globules (marked by white arrows) appear only on the surface of the aragonite spindle. | ||
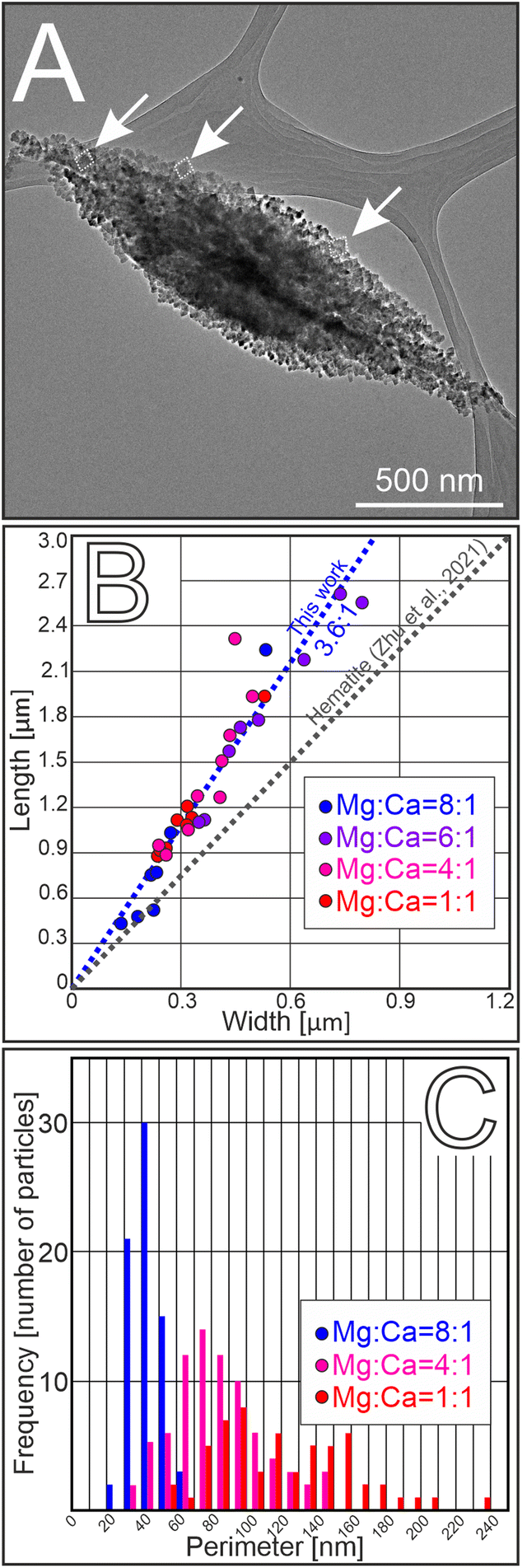 | ||
| Fig. 4 Morphology of spindle-shaped aragonite. (A) BF TEM image showing that the particles have aggregate-like appearance, consist of a large number of nanocrystals and have typical spindle-like morphology. Individual aragonite nanocrystals are framed by white dashed lines and marked by white arrows. (B) The external morphology of the spindle-like aggregates was uniform at every used dissolved Mg:Ca ratio, with the aspect ratio (length-to-width) of the aggregates around 3.6:1 (15 particles were measured for each dissolved Mg:Ca ratio). (C) The sizes of the individual nanocrystals (characterized by their perimeter) that build up the aggregates decreased with increasing dissolved Mg:Ca ratio (80 particles were measured for each dissolved Mg:Ca ratio). | ||
Aragonite spindles had uniform aspect ratios (length to width, l/w = 3.6), independent of the Mg:Ca ratio of the solution (Fig. 4A and B; 15 particles were measured for each dissolved Mg:Ca ratio). The combination of granular texture and single-crystal-like diffraction displayed by the spindles implied a mesocrystalline character. High-resolution study of spindle nanotexture showed that the spindles were composed of few tens-of-nm-large nanocrystals that maintained their mutual orientation relationship with respect to one another, with their c-axis approximately aligned in the direction of the long axis of the spindle (Fig. 4A and 5A–B). The interfaces between the individual nanocrystals were indistinct, with either amorphous material or pores separating the crystalline units, as is characteristic for mesocrystal assemblies.
 | ||
| Fig. 5 Structure analysis of spindle-shaped aragonite aggregates. (A) BF image of a spindle with (B) corresponding SAED pattern that shows elongated, arc-shaped intensity maxima. (C) The arc length of the 002 refection increased with increasing dissolved Mg:Ca ratio (10 particles were measured for each dissolved Mg:Ca ratio). (D) BF image of a spindle with (E) and (F) corresponding FFTs (the marked area shows the places of the two HRTEM images from where the FFTs were calculated, for the original HRTEM images see Fig. S3†), displaying small misorientations between the constituent nanocrystals and the appearance of extra periodicity (marked by yellow arrows) that is inconsistent with the structure of aragonite. | ||
As is also typical for mesocrystals, the alignment of the constituting nanocrystals is imperfect. Typical SAED patterns suggest angular dispersions of nanocrystals, manifested in the intensity maxima appearing as short arcs (Fig. 5A and B). The lengths of the arcs belonging to particular intensity maxima, e.g., to the 002 reflection, increase with the Mg:Ca ratio of the solution (with the arc angle changing from 5 ± 0.5° to 8 ± 0.3° as the Mg:Ca ratio increases from 1:1 to 8:1) (Fig. 5C; 10 particles were measured for each dissolved Mg:Ca ratio). The trend of increasing misalignment of nanocrystals is probably related to the decrease of particle size with increasing Mg:Ca ratio (Fig. 4C). We measured the perimeters of the nanocrystals in random positions within the aggregates. The average perimeters of the nanocrystals were 122, 84 and 44 nm in the spindles precipitated from solutions with 1:1; 4:1 and 8:1 dissolved Mg:Ca ratios, respectively (Fig. 4C; 80 particles were measured for each dissolved Mg:Ca ratio).
SAED patterns and FFTs of HRTEM images obtained from aragonite spindles often show double periodicity along the [110]* directions (Fig. 5D–F; Fig. S4†). Reflections appear that are incompatible with the conventional Pmcn space group of aragonite, since they would have to be assigned non-integer indices (e.g., ½,½,0). Similar reflections have been observed in synthetic aragonite, and interpreted as superstructure reflections.42 In places HRTEM images show neighbouring nanocrystals in [100] and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] orientations (Fig. 6), with their [001] directions almost parallel, suggesting a twin relationship between the two crystals. Based on our observations, two self-assembly modes, (i) lateral (110)[001]||(110)[001] and (ii) twin (110)[001]||(110)[00] are identified. Similar observations were reported for other minerals, such as magnetite43 and rutile.32 We suspect that the above two structural features, i.e., the appearance of half-integer reflections and the presence of twinning, might be related; however, conclusive evidence for this is still lacking.
0] orientations (Fig. 6), with their [001] directions almost parallel, suggesting a twin relationship between the two crystals. Based on our observations, two self-assembly modes, (i) lateral (110)[001]||(110)[001] and (ii) twin (110)[001]||(110)[00] are identified. Similar observations were reported for other minerals, such as magnetite43 and rutile.32 We suspect that the above two structural features, i.e., the appearance of half-integer reflections and the presence of twinning, might be related; however, conclusive evidence for this is still lacking.
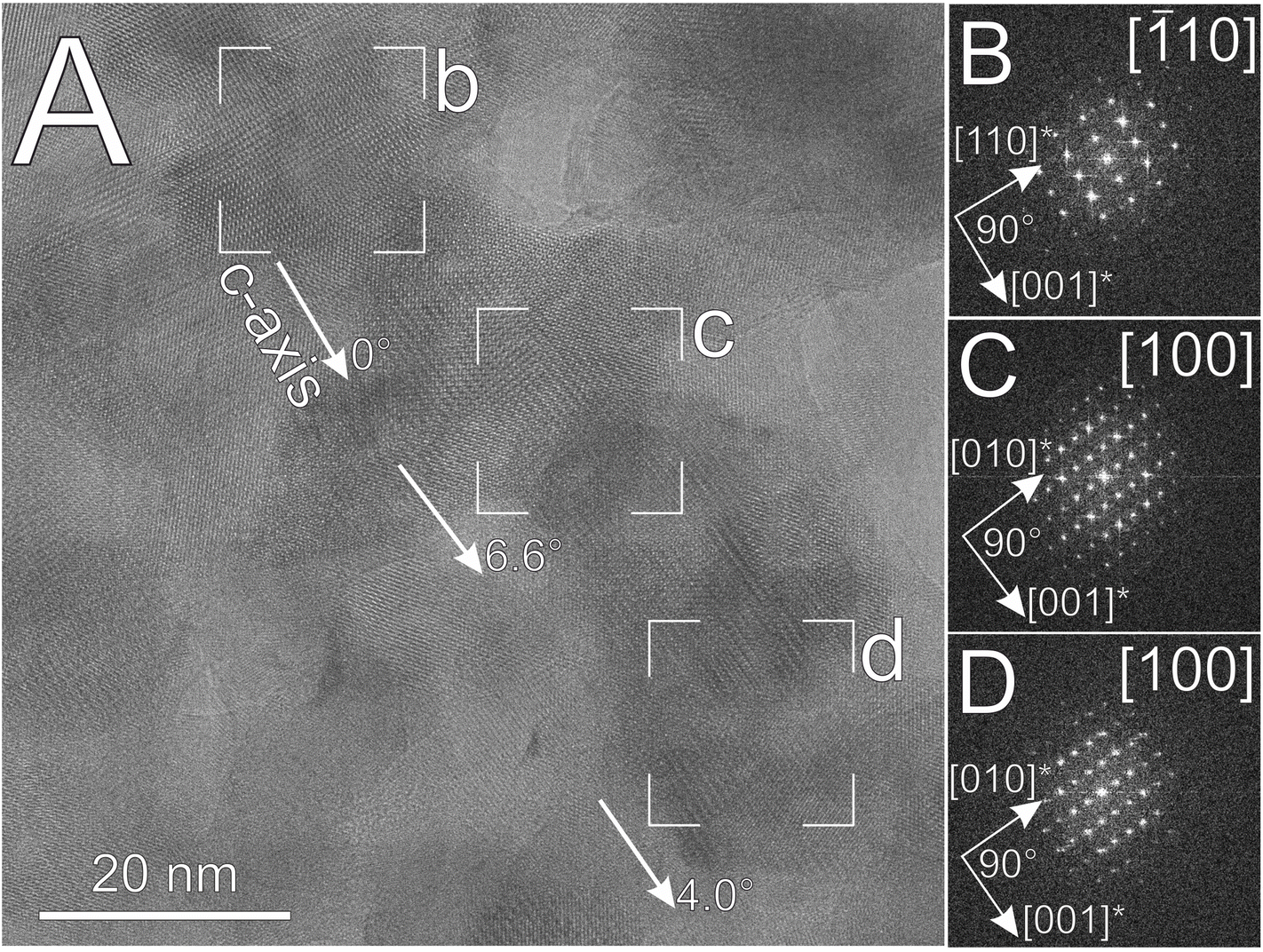 | ||
| Fig. 6 (A) HRTEM image with (B), (C) and (D) corresponding FFTs, obtained from the external part of an aragonite spindle. Two main projections, [10] and [100] were observable for the nanocrystals but their [001]* directions were almost perfectly aligned. | ||
The typical spindle-like morphology of the aragonite aggregates is preserved up to an equal titrant to buffer volume ratio (approximately 3 hours after the nucleation). However, intergrown spindles and more complex, star-like aggregates become more abundant with processing time (Fig. 7A). STEM tomography shows a lamellar morphology of the intergrown spindles (Fig. 7B and Fig. S5 and S6 movies 1–2†). The angles between the long axes of intergrown spindles seem to be specific at first glance, potentially suggesting a twin relationship; however, when measured from the STEM tomographic reconstruction the angles appear to be incidental (Fig. S5†). The tomographic measurements also confirm the presence of holes of various sizes (up to tens of nm) within the aragonite aggregates, located at random positions (Fig. 7B and S6 movie 3†).
 | ||
| Fig. 7 Size and morphology changes during the aging of spindle-shaped aragonite aggregates. (A) TEM BF image and (B) tomographic reconstruction of freshly precipitated aragonite particles consisting of two or more intergrown spindles. (C) TEM BF image of aragonite crystals aged for two months; the mesocrystal-like appearance of the firstly formed aragonite particles vanished and perfect single crystals appeared. | ||
After two months aging, the spindles transformed into assemblies of several-μm-long, well-facetted, lath-shaped aragonite crystals (Fig. 7C) that typically showed the half-integer reflections when viewed along [10] projection (Fig. S7†). These aggregates were loosely bound, and after 120 s ultrasonication disaggregated into individual aragonite crystals.
Discussion
Our study confirmed the results of earlier research regarding two important aspects of aragonite formation, namely that (i) increasing the Mg:Ca ratio of the solution delays the formation of the first solid, and (ii) Mg-ACC is a precursor of aragonite. In our experiments nucleation started at increasing ionic strength as the dissolved Mg:Ca ratio increased (Fig. 1A and S1†), as was also observed earlier,23,44 and the firstly formed phase was Mg-ACC that later transformed into aragonite (Fig. 2). In general, the formation of crystalline CaCO3 is a multistep process involving both liquid and solid precursors. The first solid is typically ACC that transforms into less soluble, crystalline phases via either solid-state transformation or dissolution and re-precipitation processes (DRP), or a combination of both.45 Both in situ TEM46 and SAXS/WAXS45 experiments showed that in the absence of Mg2+ ions ACC particles could transform into vaterite and then to calcite, in agreement with the Ostwald-step rule and as the solubility of the given CaCO3 phases would suggest. As discussed above, in our experiments the solid-state transformation of Mg-ACC can be excluded. On the other hand, a scenario involving its dissolution offers an explanation for the polymorph selection. Once the dissolution of Mg-ACC has started and the dissolved Ca2+ concentration reaches supersaturation with respect to both calcite and aragonite, a new CaCO3 phase is formed. However, the dissolved Mg2+ ions kinetically hinder or decelerate the formation of Mg-bearing calcite crystals via poisoning the growth of calcite nuclei,2,47 thereby enhancing the formation of aragonite,2 the second-least-soluble form of CaCO3.
Morphological features of aragonite spindles (Fig. 4) and the specific crystallographic orientation relationship between their component nanocrystals (Fig. 5 and 6) imply that the spindles formed by particle attachment, i.e., in a non-classical crystallization process, as was also suggested by previous authors.6,12,15,26,35 The formation of mesocrystals via the attachment of crystalline nanoparticles in a crystallographically co-aligned fashion was widely described, e.g., in titanium oxides,32,48 iron oxides,34,49,50 and calcium sulphates.51,51,52 However, the rapid oriented attachment of aragonite nanocrystals is unlikely after their burst nucleation at room temperature, in a weak electrolyte solution, at slightly alkaline pH; moreover, we never observed individual aragonite nanocrystals in our samples. On the other hand, at the initial stages of spindle formation the presence of Mg-bearing ACC on the surfaces of aragonite spindles appeared to be common (Fig. 2 and 3), whereas this phenomenon was absent with increasing reaction time. These observations indirectly suggest that the aragonite spindles grew by the attachment of Mg-ACC particles.
For the growth of aragonite spindles previous studies proposed three models, all of which assumed a particle-mediated growth processes, with their main differences being whether amorphous or crystalline particles are assembled, and where the amorphous to crystalline transformation happens relative to the growing spindles. Walker et al.10 suggested that assembly of amorphous particles is followed by their transformation into aragonite in solid state. Others12,15,26,36 claimed that the growth of the spindles is dominated by the attachment of ACC onto the surfaces of pristine aragonite nanocrystals, with the ACC transforming in solid state and inheriting the structure and orientation of the substrate aragonite nanocrystal. A third model16 suggested that crystalline aragonite nanoparticles attached to one another in a crystallographically well-oriented manner; nevertheless, even in this case the ACC was considered a precursor phase. Our observations support the model by Zhu et al.34 concerning the attachment of Mg-ACC to pre-existing aragonite; however, we assume the amorphous to crystalline transition takes places via a dissolution–reprecipitation process, as discussed above. While the attachment of Mg-ACC to preformed aragonite aggregates is well documented by our TEM images, much less is known about the nucleation of the first nanocrystals. Images obtained from the smallest, presumably incipient aragonite aggregates (Fig. 2A) suggest that once nanocrystals form, they align along the c-axis (Fig. 2) according to an oriented self-assembly mechanism.32,34,53
Compared to the oriented attachment of nanocrystals that are homogeneously precipitated from supersaturated solutions,48,53 the driving forces behind interface-driven nucleation34 are less understood. According to our observations, Mg-ACC is continuously dissolving, thus losing its initial Mg content. Along with this process, new aragonite nanocrystals form in the vicinity of spindles. It remains to be understood why the nanocrystals form in the vicinity of mineral surfaces (i.e., not in direct contact with them), and why they are assembled into mesocrystals instead of growing into single crystals via classical crystal growth.54 In fact, the observed mechanism is not too different from conventional mesocrystal assembly, where nanocrystals are attracted and aligned via van der Waals (vdW) forces, and surface interactions control their oriented attachment.33 However, in the case of aragonite spindles these vdW forces may be also involved in the oriented near-surface crystallization of the nanoparticles, when the amorphous precursor reaches the immediate vicinity of the mineral surface.34 At the same time, surface ligands hold the new nanocrystals in a stable separation from the existing crystalline surface, preventing their merging via classical pathways.33 According to the Lifshitz formulation of vdW interactions,54 orientational dependency originates from the anisotropy of bulk dielectric properties defined by structure-dependent dielectric tensors of interacting materials, which conduct force fields that convey structural information beyond the phase boundary, thus aligning the interacting nanocrystals.32 Such forces may be responsible for the oriented, near-surface amorphous-to-crystalline transformation, as was also observed for iron oxides.34 The misalignment of nanocrystals and the mesocrystalline structure (Fig. 5 and 6) of the aragonite spindles support the mechanism of oriented near-surface nucleation.
The highly anisotropic shape of the spindles suggests that the rate of particle attachment is higher along the [001] direction than perpendicular to it. Some authors observed that the amorphous particles prefer a distinct direction with respect to their attachment;55 indeed, newly formed crystalline particles were shown to preferably attach to the tips of the spindles.34 This could be explained by the tendency of the material to maintain its equilibrium crystal shape, as has also been observed in other mesocrystals.56 The measured uniform length-to-width ratio of the spindles (3.6:1) suggests that this morphology is typical for the early stages of aragonite formation, and the dissolved Mg content does not affect the external shape of the aggregates (Fig. 4B).
The common co-occurrence of the [100] and [10] orientations (Fig. 6), accompanied by the appearance of half-integer ½½0-type reflections suggest the presence of superperiodicity along [110] (Fig. 5E, S4 and S7†). Our findings indicate that double periodicity along the [110]* direction could appear already in the nucleation stage (Fig. S3†). Some analogies can be drawn between aragonite and oxides: in situ TEM studies of iron oxides showed that either the same or twin crystallographic orientations are favoured during the particle attachment process.43 Similarly, both parallel and twin assembly of nanoparticles have been reported for rutile.32 In the case of aragonite, the Mg content of the solution appears to affect the particle attachment process, since Mg was suggested to be responsible for either the formation57 or the enhancement of twin boundaries.21 Thus, the non-integer reflections could possibly arise from nanoscale twinning or polytypism; however, neither could be confirmed from our HRTEM images in the studied projections.
Another feature observed in our products is the angular dispersion of reflections in SAED patterns, associated with mesocrystal assembly. The increasing angular length of 002 with increasing dissolved Mg:Ca ratio of the solution (Fig. 5A–C) possibly originates from the size reduction of the aragonite nanocrystals with the increasing Mg content (Fig. 4C). Interestingly, previous studies on biogenic aragonite (nacre) also reported that small misorientations of the [001] zone axis occurred,58 suggesting that growth by particle attachment could be a prevalent strategy in biomineralization.59,60 Moreover, Lew et al.61 showed that the orientational differences between CaCO3 nanocrystals could be essential for increasing the fracture toughness of the mineralized frameworks of corals, seashells and various other organisms. Our observations suggest that even without biological control or organic additives, simply the Mg content of the solution could affect the crystallographic texture of the product carbonate, in addition to controlling the polymorph selection. Thus, while our observations provide important details for a better understanding of the formation of mesoscopically ordered materials in abiotic systems, they also have implications for biomineralization processes.
Conclusions
Aragonite formation from Mg-rich aqueous solutions involves the precipitation of a transient form with a peculiar, spindle-shaped, aggregate-like appearance and mesocrystalline attributes. While our study focused on the properties and assembly of these spindles, our results also revealed the complexity of aragonite formation, and identified several stages in the process. We found that Mg-rich amorphous calcium carbonate (Mg-ACC) globules precipitate first from the solution. In the next stage, elongated, few tens-of-nm-large aggregates of aragonite nanocrystals appear; we assume they form from Mg-ACC, and immediately merge and align along their c-axis, forming the nuclei of spindles. Then, further Mg-ACC particles are attached to the incipient spindles. Once the Mg-ACC particles reach the aragonite spindle, they transform into aragonite nanocrystals, with this transition accompanied by the loss of Mg, suggestive of a dissolution–reprecipitation process. As the process of attachment and conversion of Mg-ACC particles continues, the individual spindles develop into complex aggregates, consisting of two or more spindles, apparently intergrown in a crystallographically unspecified manner. In the last stage of aragonite formation, the morphology of the spindles is reshaped by Ostwald ripening, resulting in lath-shaped single crystals approaching the equilibrium crystal morphology of aragonite.Aragonite spindles are also observed in biologically controlled mineralization, and their formation is typically attributed to the controlling effects of organic compounds. Our results show that spindle-shaped mesocrystal assemblies of aragonite can form in weak electrolyte solutions without organic additives. Instead of organics, the properties of aragonite mesocrystals, such as the development of the typical spindle morphology and the crystallographic coherence between their component nanocrystals, are affected by the presence and concentration of dissolved Mg2+ ions, respectively. These results imply a possible control mechanism by which living organisms could regulate the properties of the forming mesocrystals, bringing us a step closer to understanding aragonite biomineralization.
Author contributions
The manuscript was written by contributions from all authors. ZM initiated and planned the study, performed the precipitation experiments, characterized the products by TEM, interpreted the results and wrote the ms. PP performed electron tomography and processed its results. MP and AR were consulted during the entire study, contributed to the writing of the ms and secured funding. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was part of the project “Aragonite: structure and formation”, supported by a Hungarian-Slovenian bilateral fund (Grant No. SNN-139585 at the National Office of Research, Development and Innovation (NKFIH) of Hungary and N1-0230 at the Slovenian Research and Innovation Agency (ARIS)) and by Grant No. RRF-2.3.1-21-2022-00014 from the NKFIH. The authors are grateful to all participants of the Hungarian-Slovenian bilateral project for helpful discussions, including Péter Németh, Pavel Gavryuskhin, Vesna Ribić and István Dódony.References
- P. N. Gavryushkin, A. B. Belonshko, N. Sagatov, D. Sagatova, E. Zhitova, G. M. Krzhizhanovskaya, A. Rečnik, E. V. Alexandrov, I. V. Medrish, Z. I. Popov and K. D. Litasov, Cryst. Growth Des., 2020, 21(1), 65–74 CrossRef.
- W. Sun, S. Jayaraman, W. Chen, K. A. Persson and G. Ceder, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(11), 3199–3204 CrossRef CAS PubMed.
- B. Jones and X. Peng, Sediment. Geol., 2016, 345, 103–125 CrossRef CAS.
- R. Boch, X. Wang, T. Kluge, A. Leis, K. Lin, H. Pluch, F. Mittermayr, A. Baldermann, E. M. Böttcher and M. Dietzel, Sedimentology, 2018, 66(2), 604–635 CrossRef.
- Y. Fang, F. Hobbs, Y. Yang and H. Xu, Sedimentology, 2023, 70(4), 1328–1347 CrossRef.
- M. Boon, W. D. A. Rickard, A. L. Rohl and F. Jones, Cryst. Growth Des., 2020, 8, 5006–5017 CrossRef.
- V. Mavromatis, J.-M. Brazier and K. Goetschl, Geochim. Cosmochim. Acta, 2021, 317(15), 53–64 Search PubMed.
- A. J. Guiffre, C. A. D. Gagnon, J. J. De Yoreo and P. M. Dove, Geochim. Cosmochim. Acta, 2015, 165, 407–417 CrossRef.
- Z. Zhang, Y. Xie, X. Xu, H. Pan and R. Tang, J. Cryst. Growth, 2012, 343, 62–67 CrossRef CAS.
- J. M. Walker, B. Marzec and F. Nudelman, Angew. Chem., Int. Ed., 2012, 56(39), 11740–11743 CrossRef PubMed.
- Z. Molnár, P. Pekker, I. Dódony and M. Pósfai, Earth Planet. Sci. Lett., 2021, 567, 116971 CrossRef.
- O. Nahi, N. Kulak, S. Zhang, X. He, Z. Aslam, M. A. Ilet, I. J. Ford, R. Darkins and F. C. Meldrum, Adv. Sci., 2023, 10(1), 2203759 CrossRef CAS.
- B. Jones, Sediment. Geol., 2017, 353, 64–75 CrossRef CAS.
- I. Lee, S. W. Han, H. J. Chou and K. Kim, Adv. Mater., 2001, 13(21), 1617–1620 CrossRef CAS.
- G.-T. Zhou, Q.-Z. Yao, J. Ni and G. Jin, Am. Mineral., 2009, 94, 293–302 CrossRef CAS.
- S. Shi, Z. Su, H. Wei and X. Chen, J. Appl. Polym. Sci., 2010, 117, 3308–3314 CrossRef CAS.
- K. K. Sand, J.-D. Rodriguez-Blanco, E. Makovicky, L. G. Benning and S. L. S. Stipp, Cryst. Growth Des., 2011, 12, 842–853 CrossRef.
- L. Liu, J. Jiang and S.-H. Yu, Cryst. Growth Des., 2014, 14(11), 6048–6056 CrossRef CAS.
- M.-G. Willinger, J. Polleux, M. Antonietti, H. Cölfen, N. Pinna and N. Nassif, CrystEngComm, 2015, 17, 3927 RSC.
- I. B. Meić, J. Kontrec, D. D. Jurašin, B. N. Džakula, L. Štanjer, D. Lyons, M. D. Sikirić and D. Kralj, Cryst. Growth Des., 2016, 17, 1103–1117 CrossRef.
- S. Fermani, N. B. Džakula, M. Reggi, G. Falini and D. Kralj, CrystEngComm, 2017, 19, 2451–2455 RSC.
- S.-H. Yu, H. Li, Q.-Z. Yao, S.-Q. Fu and G.-T. Zhou, J. Environ. Chem. Eng., 2017, 5(1), 893–905 CrossRef CAS.
- F. Konrad, B. Purgstaller, F. Gallien, V. Mavromatis, P. Gane and M. Dietzel, J. Cryst. Growth, 2018, 498, 381–390 CrossRef CAS.
- B. R. Heywood and S. Mann, Chem. Mater., 1994, 6, 311–318 CrossRef CAS.
- K. W. Park, S.-J. Ko, S. W. Lee, K.-H. Cho, J.-W. Ahn and C. Han, J. Cryst. Growth, 2008, 310, 2593–2601 CrossRef.
- Z.-Q. Huang, G.-S. Zhang and Y. Tan, J. Nanomater., 2019, 7328478 CAS.
- Z. Han, R. Meng, H. Zhao, X. Gao, Y. Zhao, Y. Han, F. Liu, M. E. Tucker, J. Deng and H. Yan, Front. Microbiol., 2023, 14, 1078430 CrossRef PubMed.
- A. Sugawara and T. Kato, Chem. Commun., 2000, 6, 487–488 RSC.
- M. Niederberger and H. Cölfen, Chem. Rev., 2008, 108(11), 4332–4432 CrossRef PubMed.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44(35), 5576–5591 CrossRef PubMed.
- Y.-Y. Kim, A. S. Schenk, J. Ihli, A. N. Kulak, N. B. J. Hetherington, C. C. Tang, W. W. Schmahl, E. Griesshaber, G. Hyett and F. C. A. Meldrum, Nat. Commun., 2014, 5, 1–14 Search PubMed.
- V. Jordan, U. Javornik, J. Plavec, A. Podgornik and A. Rečnik, Sci. Rep., 2016, 6, 24216 CrossRef CAS PubMed.
- M. Jehannin, A. Rao and H. Cölfen, J. Am. Chem. Soc., 2019, 141, 10120–10136 CrossRef CAS PubMed.
- G. Zhu, M. L. Sushko, J. S. Loring, B. A. Legg, M. Song, J. A. Soltis, X. Huang, K. M. Rosso and J. J. De Yoreo, Nature, 2021, 590, 416–422 CrossRef CAS PubMed.
- T. M. Stawski, G. J. Smales, E. Scoppola, D. Jha, L. F. G. Morales, A. Moya, R. Wirth, B. R. Pauw, F. Emmerling and A. E. S. Van Driessche, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(48), e2111213118 CrossRef CAS PubMed.
- S. Yu, H. Li, Q.-Z. Yao, S.-Q. Fu and G.-T. Zhou, J. Environ. Chem. Eng., 2017, 5, 839–905 Search PubMed.
- A. Knoll, Rev. Mineral. Geochem., 2003, 54(1), 329–356 CrossRef CAS.
- G. Müller, G. Irion and U. Förstner, Naturwissenschaften, 1972, 59, 158–164 CrossRef.
- D. L. Parkhurst and C. A. J. Appelo, Book chapter in: U.S. Geological Survey Techniques and Methods, 2013, book 6, ch. A43, p. 497 Search PubMed.
- B. Purgstaller, K. E. Goetschl, V. Mavromatis and M. Dietzel, CrystEngComm, 2019, 21, 155–164 RSC.
- Z. Molnár, I. Dódony and M. Pósfai, Geochim. Cosmochim. Acta, 2023, 345, 90–101 CrossRef.
- Y. Hiragi, S. Kachi, T. Takada and N. Nakanishi, Bull. Chem. Soc. Jpn., 1966, 39(11), 2361–2364 CrossRef CAS.
- Y.-S. Li, J. S. Church and A. L. Woodhead, J. Magn. Magn. Mater., 2012, 324(8), 1543–1550 CrossRef CAS.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, J. Cryst. Growth, 2003, 254(1–2), 206–218 CrossRef CAS.
- P. Bots, L. G. Benning, J.-D. Rodriguez-Blanco, T. Roncal-Herrero and S. Shaw, Cryst. Growth Des., 2012, 12(7), 3806–3814 CrossRef CAS.
- D. Li, M. H. Nielsen, J. R. I. Lee, C. Frandsen, J. Banfield and J. J. De Yoreo, Science, 2012, 336, 1014–1018 CrossRef CAS PubMed.
- K. J. Davis, P. M. Dove and J. De Yoreo, Science, 2000, 290(5494), 1134–1137 CrossRef CAS PubMed.
- R. L. Penn and J. F. Banfield, Science, 1998, 281(5379), 969–971 CrossRef CAS PubMed.
- J. Baumgartner, A. Dey, P. H. H. Bomans, C. Le Coadou, P. Fratzl, N. A. J. M. Sommerdijk and D. Faivre, Nat. Mater., 2013, 12, 310–314 CrossRef CAS PubMed.
- L. Kuhrts, S. Prévost, D. M. Chevrier, P. Pekker, O. Spaeker, M. Egglseder, J. Baumgartner, M. Pósfai and D. Faivre, J. Am. Chem. Soc., 2021, 143(29), 10963–10969 CrossRef CAS PubMed.
- T. M. Stawski, A. Van Driessche, M. Ossiro, J.-D. Rodriguez-Blanco, R. Besselink and L. G. Benning, Nat. Commun., 2016, 7(1), 11177 CrossRef CAS PubMed.
- T. M. Stawski, H. M. Freeman, A. Van Driessche, J. Hövelmann, R. Besselink, R. Wirth and L. G. Benning, Cryst. Growth Des., 2019, 19(7), 3714–3721 CrossRef CAS.
- J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Science, 2015, 349(6247), aaa6760 CrossRef PubMed.
- J. C. Hopkins, R. Podgornik, W.-Y. Ching, R. H. French and V. A. Parsegian, J. Phys. Chem. C, 2015, 119, 19083–19094 CrossRef CAS.
- Q. Wang, B. Yuan, W. Huang, H. Ping, J. Xie, K. Wang, W. Wang, Z. Zou and Z. Fu, Natl. Sci. Rev., 2023, 10(4), nwad014 CrossRef PubMed.
- V. Jordan, V. D. B. C. Dasireddy, B. Likozar, A. Podgornik and A. Rečnik, Cryst. Growth Des., 2018, 28(8), 4484–4494 CrossRef.
- P. Németh, E. Mugnaioli, M. Gemmi, G. Czuppon, A. Demény and C. Spötl, Sci. Adv., 2018, 4, eaau6178 CrossRef PubMed.
- X. San, M. Gong, J. Wang, X. Ma, R. dos Reis, P. J. M. Smeets, V. P. Dravid and X. Hu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119(14), e2122218119 CrossRef CAS PubMed.
- K. Benzerara, N. Menguy, N. M. Obst, J. Stolarski, M. Mazur, T. Tylisczak, G. E. Brown and A. Meibom, Ultramicroscopy, 2011, 111, 1268–1275 CrossRef CAS PubMed.
- I. Polishchuk, A. A. Bracha, L. Bloch, D. Levy, S. Kozachkevich, Y. Etinger-Geller, Y. Kauffmann, M. Burghammer, C. Giacobbe, J. Villanova, G. Hendler, C.-Y. Sun, A. J. Giuffre, M. A. Marcus, L. Kundanati, P. Zaslansky, N. M. Pugno, P. U. P. A. Gilbert, A. Katsman and B. Pokroy, Science, 2017, 358(6368), 1294–1298 CrossRef CAS PubMed.
- A. J. Lew, A. C. Stifler, A. Tits, C. A. Schmidt, A. Scholl, A. Cantamessa, L. Müller, Y. Delaunois, P. Compére, D. Ruffoni, M. J. Buehler and P. U. P. A. Gilbert, Adv. Mater., 2023, 35, 2300373 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Uploaded document and movies. See DOI: https://doi.org/10.1039/d3nr04672a |
| This journal is © The Royal Society of Chemistry 2024 |