
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of water on gold supported chiral graphene nanoribbons: rupture of conjugation by an alternating hydrogenation pattern†
Alejandro
Berdonces-Layunta‡
*ab,
Adam
Matěj‡
 *cde,
Alejandro
Jiménez-Martín
cdf,
James
Lawrence
ab,
Mohammed S. G.
Mohammed
ab,
Tao
Wang
ab,
Benjamin
Mallada
cde,
Bruno
de la Torre
d,
Adrián
Martínez
g,
Manuel
Vilas-Varela
g,
Reed
Nieman
h,
Hans
Lischka
h,
Dana
Nachtigallová
ik,
Diego
Peña
g,
Pavel
Jelínek
*cd and
Dimas G.
de Oteyza
*abj
*cde,
Alejandro
Jiménez-Martín
cdf,
James
Lawrence
ab,
Mohammed S. G.
Mohammed
ab,
Tao
Wang
ab,
Benjamin
Mallada
cde,
Bruno
de la Torre
d,
Adrián
Martínez
g,
Manuel
Vilas-Varela
g,
Reed
Nieman
h,
Hans
Lischka
h,
Dana
Nachtigallová
ik,
Diego
Peña
g,
Pavel
Jelínek
*cd and
Dimas G.
de Oteyza
*abj
aDonostia International Physics Center, 20018 San Sebastian, Spain. E-mail: ABerdonces95@gmail.com
bCentro de Fisica de Materiales, 20018 San Sebastian, Spain
cInstitute of Physics, Czech Academy of Sciences, 16200 Prague, Czech Republic. E-mail: mateja@fzu.cz
dRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacky University, 783 71 Olomouc, Czech Republic. E-mail: jelinekp@fzu.cz
eDepartment of Physical Chemistry, Faculty of Science, Palacky University, 779 00 Olomouc, Czech Republic
fFaculty of Nuclear Sciences and Physical Engineering, Czech Technical University in Prague, Brehova 7, Prague 1 115 19, Czech Republic
gCentro Singular de Investigacion en Quimica Bioloxica e Materiais Moleculares (CiQUS), and Departamento de Quimica Organica, Universidade de Santiago de Compostela, 15705 Santiago de Compostela, Spain
hDepartment of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA
iInstitute of Organic Chemistry and Biochemistry, Czech Academy of Sciences, 16000 Prague, Czech Republic
jNanomaterials and Nanotechnology Research Center (CINN), CSIC-UNIOVI-PA, 33940 El Entrego, Spain. E-mail: d.g.oteyza@cinn.es
kIT4Innovations, VSB-Technical University of Ostrava, 17. listopadu 2172/15, Ostrava-Poruba 70800, Czech Republic
First published on 17th November 2023
Abstract
In the last few years we have observed a breakpoint in the development of graphene-derived technologies, such as liquid phase filtering and their application to electronics. In most of these cases, they imply exposure of the material to solvents and ambient moisture, either in the fabrication of the material or the final device. The present study demonstrates the sensitivity of graphene nanoribbon (GNR) zigzag edges to water, even in extremely low concentrations. We have addressed the unique reactivity of (3,1)-chiral GNR with moisture on Au(111). Water shows a reductive behaviour, hydrogenating the central carbon of the zigzag segments. By combining scanning tunnelling microscopy (STM) with simulations, we demonstrate how their reactivity reaches a thermodynamic limit when half of the unit cells are reduced, resulting in an alternating pattern of hydrogenated and pristine unit cells starting from the terminal segments. Once a quasi-perfect alternation is reached, the reaction stops regardless of the water concentration. The hydrogenated segments limit the electronic conjugation of the GNR, but the reduction can be reversed both by tip manipulation and annealing. Selective tip-induced dehydrogenation allowed the stabilization of radical states at the edges of the ribbons, while the annealing of the sample completely recovered the original, pristine GNR.
Introduction
Since the discovery of graphene, many studies put their focus on the interaction between graphene nanostructures and water.1–4 Some of graphene's target applications, such as filtration5–8 or inverse osmosis,9 require direct contact with the solvent. However, even for applications in dry media, the ability to withstand an exposure to ambient moisture is required for their scalability and industrial feasibility.10,11 The interaction between water and nanographenes, including GNR and polycyclic aromatic hydrocarbons (PAHs), is generally assumed to be weak3,12–14 and dominated by London dispersion forces.1,3 For this reason, bigger, more polarizable nanographenes have a stronger interaction.1,3,4 The utmost example of a conjugated carbon lattice, graphene, appears to be immune to water's chemical attack. However, a detail slipped out of focus: most of the studies include the bulk (the graphene surface itself) and ignore the edges15 which, in some cases, largely determine the properties of the carbon-based materials, as observed e.g. for porous graphene16,17 or graphene nanoribbons (GNRs).18–20 For this reason, we decided to test experimentally how GNRs interact with water.GNRs display a great edge to surface ratio and optoelectronic properties with a remarkable dependence on their width and edge shape. For example, whereas armchair-edged ribbons are characterized by a semiconducting band structure, zigzag GNRs are metallic.20 In order to experiment with both types of edges, we chose chiral (ch-)GNRs, characterized by alternating zigzag and armchair edge segments.21,22 Note that the narrow width of the utilized chGNRs renders them semiconducting22,23 and with only a low radical character, as corroborated with calculations.24 For this reason, we consider them to be at the low end with regard to the reactivity of common zigzag edges in PAHs.25–27 In previous experiments, we showed how these chGNRs react with molecular oxygen.28,29 O2 preferentially attacks the zigzag segments to the point of breaking the carbon backbone. However, it remains unknown how the chGNRs react with H2O, as well as how the aromaticity of the GNR relates to their reactivity. Although the interaction between molecules and water on a metallic surface30,31 as well as some inorganic reactions32,33 have been already studied, to the best of our knowledge this is the first report of an organic reaction involving water directly visualized by scanning tunneling microscopy (STM).
The ribbons are grown on a clean crystal of Au(111) under ultra-high vacuum, which allows us to expose them to high purity gases at low partial pressures. In this case we used water (H2O) and ethanol (EtOH) for control experiments, and studied the result by means of STM. The chGNRs are hydrogenated, suggesting a reducing behaviour of both species on the gold surface. The resulting hydrogenation is regioselective towards the central carbon of the zigzag segments, but restricting adjacent unit cells from reacting at once. Thus, it causes an alternating pattern of hydrogenated and pristine unit cells in the exposed chGNRs, justified thermodynamically by theoretical calculations. The use of CO-functionalized probes allows for bond-resolving (BR-STM) images of the resulting hydrogenation,34,35 whereas scanning tunnelling spectroscopy (STS) allows monitoring their effect on the electronic properties. In addition, we show how we can reverse the hydrogenation either locally with the scanning probe, whereby we can create radicals on the ribbons at will, or globally with annealing treatments, whereby we return the ribbons to their pristine state.
Results
Multiple samples of chiral GNRs were grown following an already published method,21,22,36 the quality of which was checked by means of STM/STS/BR-STM with a CO-functionalized tip (see Fig. 1a–c). A variety of exposure conditions were used: going from pressures around 2 × 10−7 mbar to 8 × 10−7 mbar and exposure times from 1 to 2.5 hours, altogether ranging from ∼540 to ∼5400 Langmuir (see ESI Fig. 1 and 2†). The sample was held at room temperature during the exposure. Control experiments with all filaments within the vacuum chamber turned off precluded their influence on the results described henceforth. In a first check it is hard to appreciate the effect of water, as the ribbons retain their mesoscopic structure (see Fig. 1e). A closer look at the topography image shows a series of transversal protrusions in the central part of some unit cells (circled in green in Fig. 1e), which when observed with a CO-functionalized tip appear as a wider, darker central ring of the zigzag segment (marked with yellow arrows in Fig. 1g). Their appearance resembles our previous observations of hydrogenated C sp3 atoms in PAH.28,37,38 The same conclusion is reached with nc-AFM measurements (Fig. 1h). However, we could not rule out a C sp3 rehybridization caused by a hydroxyl functionalization either, with both cases requiring prior or concerted water dissociation.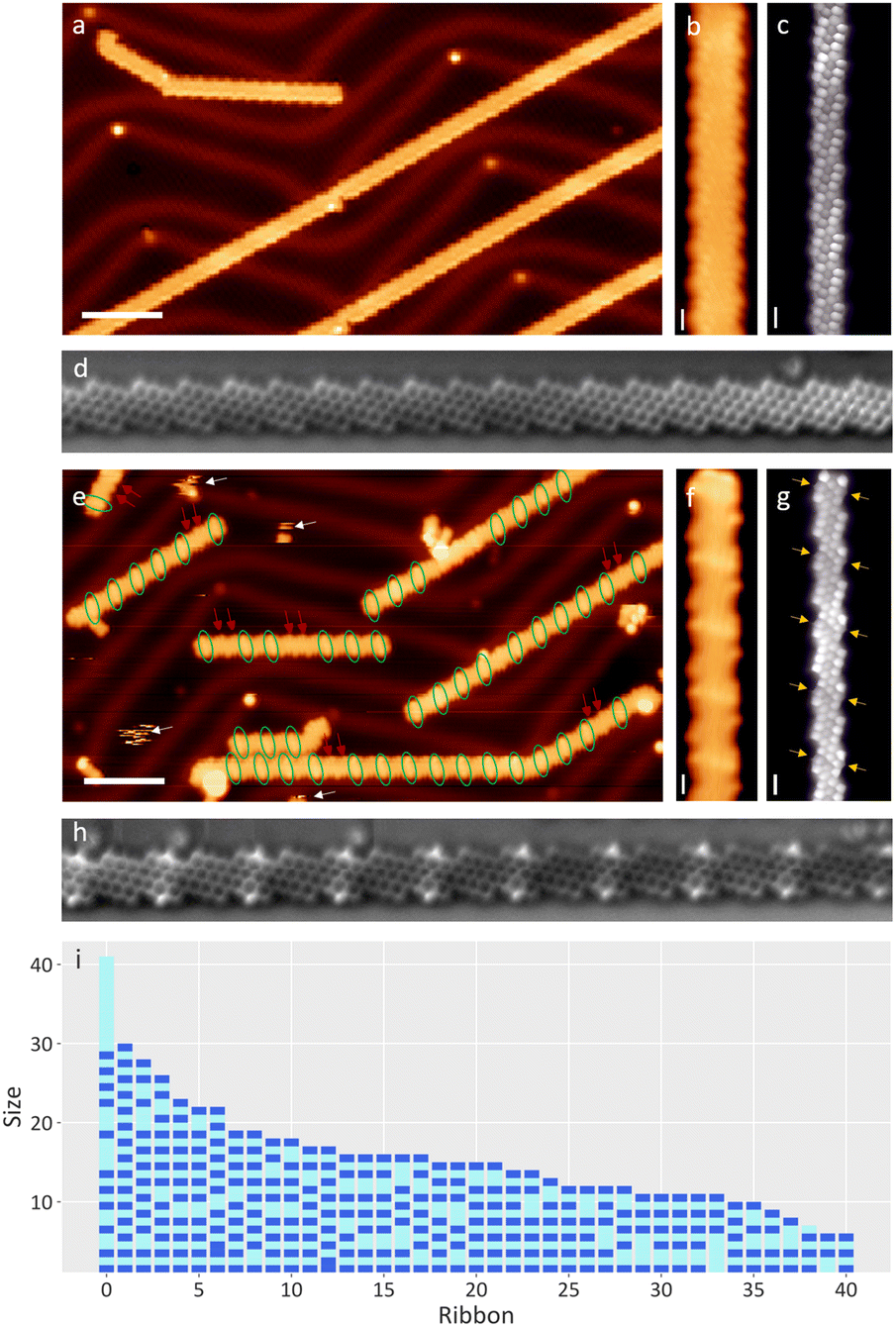 | ||
| Fig. 1 (a–d) Pristine sample of 3,1-chGNRs: (a) survey at −1.5 V, 100 pA, (b) zoomed image of the topography and (c) BR-STM and (d) NC-AFM image with a CO functionalized tip. (e–i) After exposure, the ribbon zigzag edges are doubly hydrogenated in alternating unit cells. (e) Survey at −1.5 V, 100 pA. Green circles mark hydrogenated segments, while red arrows mark consecutive pristine units. (f) Zoomed image of the topography of a hydrogenated ribbon. (g) BR-constant height (C.H.) at 5 mV, 120 pA. (h) NC-AFM image and (i) a diagram of 41 representative water-exposed ribbons from a sample exposed to 540 Langmuir of water, ordered by size, revealing the general hydrogenation pattern (not a histogram). The hydrogenated and pristine monomers are marked in dark and light blue, respectively, exhibiting their alternating pattern, along with very uncommon exceptions of two hydrogenated neighbors and the more frequently observed exceptions made up by neighboring pristine unit cells. Scale bars: a and e: 5 nm. b–d and f–h: 5 Å. | ||
In order to address the nature of the substitution on the zig-zag carbon atoms, we repeated the experiment using ethanol. The addition of an ethoxy substituent would be easily identifiable by STM as ethoxy tails at the edges of the ribbons. A new pristine sample held at room temperature was exposed to 7 × 10−8 mbar of ethanol for 20 minutes (63 Langmuir). However, the resulting ribbons displayed defects with an identical appearance to those obtained with water, proving that H˙ as a reducing agent of the ribbons is more favourable than the ˙OH or CH3CH2O˙ introduced. Due to the lower exposure, this sample has significantly less reacted rings (see ESI Fig. 3†), which accumulate on the terminal units of the GNRs, resembling other low concentration water exposure tests that were performed. Although the water-exposed samples exhibited additional adsorbates, their mobility even at cryogenic temperatures (see white arrows in Fig. 1e), along with the lack of chemical sensitivity of our measurements, prevented their detailed analysis and their assignment to, e.g., the remaining hydroxyl groups. Interestingly, the reduction of a unit cell prevents hydrogenation in their immediate neighbors, revealing long range effects that affect the overall kinetic or thermodynamic viability of the reaction. Upon saturation, this results in ribbons with a dominating pattern made up by hydrogenated ends and an alternation of pristine and hydrogenated unit cells towards the ribbon's interior (see Fig. 1i). Exceptions to this pattern are terminal unit cells that are not hydrogenated (accounting for about 5% of the GNR ends, see ESI Fig. 4†), or two neighboring unit cells that are either both hydrogenated or both pristine. Whereas the former only occurs very rarely, the latter is relatively common (marked with red arrows in Fig. 1e). Representative examples of the hydrogenation pattern of ribbons and the associated exceptions are shown in Fig. 1i.
The random distribution of the defects made up by neighboring pristine unit cells hints at the following reaction dynamics: after the terminal cell reacted, the reactive species attacks randomly any other segment of the GNR that is not next to a hydrogenated unit cell. An exposure to 560 Langmuir of water (see Fig. 1e–i) is close to saturation already. Once there are no more pristine segments left that include three or more unit cells, the hydrogenation stops.
Theoretical insights
We employed quantum mechanical calculations to probe the thermodynamic viability of water dissociation on gold adatoms, as a way to unveil the impact of entrapped water molecules in gating hydrogenation reactions onto GNRs. We analyzed the relative binding energies of hydrogen and hydroxyl radicals (for computational details see Methods in ESI†). The calculated bond dissociation energy (O–H bond) of water molecules adsorbed onto gold adatom accounts for 81 kcal mol−1; thus it decreases significantly compared to the value calculated for free water (121 kcal mol−1) (see ESI Fig. 5†). Moreover, theoretical calculations show that both hydrogen and hydroxyl radicals interact with gold adatoms with comparable binding energy (ΔE of 0.5 kcal mol−1). Nonetheless, the hydrogen radical binds to the zig-zag edge of the GNR by 7 kcal mol−1 stronger energy compared to the hydroxyl radical. The combination of water splitting plus GNR hydrogenation, whether sequential or concerted, reduces the energy balance of the reaction to reasonable levels.The validity of such theoretical description clearly depends on the level of computation and methods used in the study. The ribbons are prone to exhibit a multireference character,23,24 a factor that introduces errors in the DFT calculated energies. Multireference methods, while limited to small size systems, can be used to obtain more accurate results. For this reason, we performed multireference average quadratic coupled cluster (MR-AQCC) calculations on dimers and trimers to corroborate the less computationally demanding DFT analysis. It has been shown in previous calculations, e.g. for oligoacenes39 and diindenoacenes,40 that this method provides a balanced description of the biradicaloid and higher radicaloid character in PAHs. The calculated energy trends for both methods are found to correlate well with each other (see ESI Fig. 6†). One can see that the largest error occurs for the first hydrogenation step. This fact is not surprising as the addition of an odd number of hydrogen radicals results in a radical structure, which, together with the GNR's length, is difficult to be correctly described by DFT. Herein we focus on the thermodynamically stable products instead of the kinetics, presenting only the energetics of fully hydrogenated units. Such structures are described by DFT methods sufficiently well. Therefore, we carried out extensive DFT calculations for the relative energies and aromaticity of longer chains, from the dimer to the heptamer. The energetic trends associated with the hydrogenation reaction steps are found to be similar, irrespective of different chain lengths; the computational results for heptamers are presented herein as the system is capable of describing better the variety of possibilities in hydrogenation. The results obtained from the computed relative energies of partially hydrogenated GNRs are shown in Fig. 2. The most stable structure calculated in the first reaction step contains the terminal unit in a reduced form; here, the central sp2 carbon atoms of the zigzag edge reacted with H atoms, and such a process is highlighted in Fig. 2a by a black arrow. The relative energies of the products calculated after the completion of the first and second (Fig. 2b) steps underline the proclivity towards reduction of terminal units versus the inner carbon domains of the GNR. These findings agree well with the experimentally observed products, where the ending portion of the GNRs are found reduced in most cases. Further hydrogenation exhibits a clear opposition to attack of subsequent units as seen in Fig. 2b–d. We calculated the diradical character of the GNR with increasing length as a possible mechanism for hydrogenation protection. The calculated values are summarised in ESI Table 1† and show a saturation of the diradical character y0 around 18%, making the studied GNR a relatively weak diradicaloid. It can be seen that the diradical character of the GNR can be connected to the measured bandgap of the GNR (see Fig. 3a), i.e., longer GNRs have small bandgaps and higher y0 while a pristine monomer shows the largest bandgap and almost negligible y0. Although the diradical character on its own is not sufficient to explain the observed protection of consecutive units against hydrogenation, it offers an explanation of the stability of the final quasi-perfect alternation pattern. The diradical character drops for a pristine monomer within a GNR to a value of around 4%, which appears to be below the threshold for successful hydrogenation in the presence of water. A deeper study of the reaction mechanism and kinetics is beyond the scope of this work as the number of possible mechanisms renders such a study highly convoluted. However, the results from the relative total energies of hydrogenated products correlate well with the experimentally observed products and thus suggest that the hydrogenation caused by water exposure is thermodynamically driven.
 | ||
| Fig. 2 Relative energies of water splitting plus hydrogenation of GNRs at different positions and level of reduction (see Methods in ESI†). Addition of two H radicals to units at different positions for pristine GNRs (a), GNRs with one terminal unit reduced (b), GNRs with both termini reduced (c) and a heptamer GNR with three reduced units as a possible final product (d). Positions of units correspond to structures below the graphs. NICS and ACID plots for each structure are shown below the corresponding graphs. Blue and red circles display aromatic and antiaromatic rings, respectively. ACID isosurfaces are plotted at an isovalue of 0.05 a.u.; for high-resolution images see the ESI.† | ||
 | ||
| Fig. 3 Analogous images of a ribbon with alternating hydrogenated cells and a pristine dimer with a CO tip. (a) Spectroscopy of the different sections of the ribbon. It shows how the hydrogenation disrupts the conjugation and increases the bandgap. (b) Topography. C.C. −800 mV, 50 pA. (c) BR C.H. 10 mV at ∼260 pA. Hydrogenated carbons are visible in the left edge of this backwards scan. (d) dI/dV at the HOMO of the hydrogenated segments. −1.2 V, C.H. (e) dI/dV at the HOMO of the pristine monomers. −600 mV, C.H. (f) dI/dV at the HOMO of the pristine dimer. −350 mV, C.H. Scale bars: 5 Å. | ||
To further investigate the reason for the observed higher stability of the alternating pattern, we carried out nucleus-independent chemical shift41 (NICS) and anisotropy of induced-current density42,43 (ACID) calculations in order to analyze the electronic differences between various hydrogenated products. While NICS values characterize each ring by the amount of magnetic shielding (deshielding) due to aromaticity (antiaromaticity) of that ring, another method should always be included in order to characterize the global properties of the system. The π-ACID method offers visualization of conjugation in molecules allowing for global aromaticity description. The plots in Fig. 2 show ACID and NICS results on heptamer GNRs in various stages of hydrogenation (for high resolution ACID plots on trimers see ESI Fig. 7–11,† NICS of hexamers in ESI Fig. 12†). The results show a globally conjugated circuit along the periphery of the ribbon exhibiting a diatropic current, proving the global aromatic character of the pristine GNR together with Hückel's 4n + 2 rule along the circuit. At the terminal units, the conjugation forms local antiaromatic bays, which effectively extend the conjugated circuit by four more carbons. Although this increased conjugation path does not affect Hückel's rule, the presence of this antiaromatic bay may be responsible for the higher local reactivity of the terminal units. This effect must thus be compensated by a more stable global structure, hinting at an energetic preference for longer conjugation paths. The antiaromatic (aromatic) rings are illustrated by red (blue) circles in NICS plots with a diameter proportional to the magnetic shielding magnitude. Once a unit is reduced, the conjugation changes dramatically depending on the position of the reduced unit. In all cases, the hydrogenation of a unit breaks the global conjugation through this unit, which leads to fragmentation of the peripheral conjugation path. It can be seen that the pristine units extend their conjugation path towards the reduced unit by including one of its formed Clar's sextets (see ESI Fig. 13†). When the terminus is reduced, there is a globally conjugated periphery of pristine units and three isolated benzene rings in the reduced unit cell. By reducing an internal unit cell, two conjugated paths are formed and only two isolated benzene rings form (see Fig. 2d, ESI Fig. 9 and 12c, e†). By considering the reduction of two consecutive unit cells, four extra isolated benzene rings are formed (ESI Fig. 10, 12d, e and 13†). Both calculations and experiments agree that this scenario, forming less conjugated parts (localized Clar sextets) between two consecutive hydrogenated unit cells, is energetically unfavorable. We thus infer that longer and less fragmented conjugation paths are favored.
Discussion
Hydrogenation of the two edges of a unit cell localizes the aromaticity in the contiguous rings (see Fig. 2 and ESI Fig. 8–12†). The resulting structure thus has a lower conjugation and hence a wider bandgap. We performed a series of STS measurements to characterize the electronic structure of the hydrogenated graphene nanoribbons (see Fig. 3a). From the spectra we can observe how the HOMO of the alternating structure appears at an energy around −0.6 eV. Compared to the −0.25 eV valence band onset of the pristine ribbons,22 this indicates a large increase of the ribbon's bandgap, especially considering that the LUMO also shifts from around 0.5 eV beyond 1.1 eV, which corresponds to the stability threshold of the hydrogenated ribbons (as will be shown below, a higher bias dehydrogenates the ribbons back to their pristine form). The bandgap rapidly decreases if larger pristine segments are present. In the case of pristine dimers, we can observe both in the spectra and in the differential conductance images how the HOMO appears at −0.3 eV, and the LUMO around 0.7 eV, implying a bandgap of around 1 eV. This is much closer to the 0.7 eV bandgap of longer ribbons.22Further efforts of measuring the LUMO of the hydrogenated ribbons resulted in the tip-induced dehydrogenation of the ribbons. Fig. 4a shows an example of how acquiring a spectrum from 0 to +1.5 eV at the position of the lightning icon causes the dehydrogenation of the C sp3 under the tip and its associated sp2 rehybridization. Subsequent bond resolved STM images at a low bias display greatly enhanced signals at those positions (Fig. 4b). Conductance spectra show the appearance of a state around the Fermi level (Fig. 4e) at this position that was previously absent. This new state is univocally identified as an unpaired electronic state, or in other terms, a radical. The remaining sp3 carbon on the opposite side of the unit cell causes an imbalance of pz electrons between the two carbon sublattices, as has been reported previously by our group with oxygen-functionalized ribbons.28,37 However, whereas with oxygen-functionalized ribbons the radical state remained occupied and resulted in a net spin moment, in these pure hydrocarbon ribbons the orbitals lie at higher energies (slightly above the substrate's Fermi level). This leads to the unpaired electron being transferred from the ribbon to the underlying Au and thereby depopulates the radical state. Nevertheless, these radical structures reveal a limited stability, and numerous attempts for getting close-by radicals on these ribbons result in the successive full dehydrogenation of the unit cells, regenerating the pristine ribbon.
 | ||
| Fig. 4 (a) Ramping the bias at the hydrogenated sections can promote the dehydrogenation. (b) What is left is a unpaired state, stabilized by the hydrogen on the other side of the ribbon, which looks like a burst in the current around the Fermi level. (c) Successive bias ramps can cause successive dehydrogenations, where the radical-stabilizing hydrogen is always more likely to be removed. The result is the recovery of the (d) pristine structure. Scale bars: 5 Å. (e) Low bias spectra at (B) the red spot. | ||
The hydrogenation caused by water is thus a mild modification process of the graphene nanoribbons. Annealing the sample to 150 °C results in their complete dehydrogenation, recovering the initial sample. The electronic and molecular integrity of the recovered ribbons was confirmed. The recovery is an analogous process of the original planarization of the GNR, but for two extra factors that help the recovery: first, the ribbons are now fully planar but for the edge carbons, so the steric hindrance favours the dehydrogenation. Second, the π network environment of this edge carbon energetically stabilizes the sp2 carbon of the product.26
We consider that both for the hydrogenation and the regeneration, the catalytic effect of the gold surface was influential in the process. However, most of the device preparation processes involve exposing the metal-supported ribbons to solvents for the transfer process, not to forget that these nanographenes have a low open shell character (from 4% in the monomer to only about 18% in long ribbons). Wider ribbons or longer zigzag segments in GNRs of different chirality20,23,26 will have a higher radical character and reactivity. We consider that the phenomena shown in this work are influential for the future applicability of the GNRs, and different protection strategies have to be considered in order to keep the integrity of the ribbons.
Conclusions
We have proved the sensitivity of (3,1,4)-chGNRs on Au(111) to humidity at room temperature. Low pressure exposure of high purity water readily affected the ribbons. Control experiments with ethanol revealed that both water and ethanol split their O–H bond, presumably promoted by the metallic surface, and hydrogenate the central carbon atom of the zigzag edges. The regioselectivity stems from the increased radical character at those positions. The hydrogenation avoids contiguous units, causing an alternating hydrogenation pattern that typically starts from the terminal units. The double hydrogenation breaks the continuous conjugation of the ribbon and opens the bandgap in the affected sections. However, the hydrogenation process can be easily reverted by annealing treatments or by tip-induced manipulation, which further allows dehydrogenating only one side of a unit cell and thereby creating radical states at predefined positions.Author contributions
Alejandro Berdonces-Layunta – experimental investigation, formal analysis of the experimental data, visualization and writing of the original manuscript. Adam Matěj – theoretical investigation, formal analysis of the theoretical data, visualization and writing of the original manuscript. James Lawrence – experimental investigation, review and editing. Mohammed S. G. Mohammed – experimental investigation. Tao Wang – experimental investigation, review and editing. Alejandro Jiménez-Martín, Benjamin Mallada, and Bruno de la Torre – experimental investigation (NC-AFM images). Adrian Martínez – resources and synthesis of the molecular precursors. Manuel Vilas-Varela – methodology for the precursors employed. Reed Nieman – theoretical investigation. Hans Lischka – methodology and supervision. Dana Nachtigalova – theoretical investigation. Diego Peña – methodology for the precursors and supervision. Pavel Jelínek – resources, supervision, methodology, project administration and review and editing of the article. Dimas G. De Oteyza – resources, conceptualization, supervision, methodology, project administration and review and editing of the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from MCIN/AEI/10.13039/501100011033 (grant no. PID2019-107338RB-C62 and PID2019-107338RB-C63) and the European Union “NextGenerationEU”/PRTR (TED2021-132388B-C42 and TED2021-132388B-C43), from MCIN/AEI/10.13039/501100011033/ERDF/EU (PID2022-140845OB-C62 and PID2022-140845OB-C64), the Xunta de Galicia (Centro Singular de Investigación de Galicia, 2019-2022, grant no. ED431G2019/03), and European Regional Development Fund (ERDF). P. J. and A. M. acknowledge the support from Praemium Academie of the Academy of Science of the Czech Republic and Czech Science Foundation projects no. 20-13692X. We acknowledge the computational resources provided by the project “e-Infrastruktura CZ” (e-INFRA CZ LM2018140) supported by MEYS CR. A. M. is thankful for the support from the Internal Student Grant Agency of the Palacký University in Olomouc, Czech Republic, IGA_PrF_2023_018 and the Endowment Fund of the Palacký University in Olomouc, Czech Republic. H. L. wants to acknowledge the support by the National Science Foundation [grant number 2107923], Division of Chemistry. B. T. acknowledges the financial support from Czech Science Foundation 23-06781M and CzechNanoLab Research Infrastructure supported by MEYS CR (LM2023051). We acknowledge financial support of the European Union under the REFRESH – Research Excellence For REgion Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition.References
- E. M. Cabaleiro-Lago, J. A. Carrazana-García and J. Rodríguez-Otero, J. Chem. Phys., 2009, 130, 234307 CrossRef PubMed
.
- S. Tsuzuki and T. Uchimaru, Curr. Org. Chem., 2006, 10, 745–762 CrossRef CAS
- D. Feller and K. D. Jordan, J. Phys. Chem. A, 2000, 104, 9971–9975 CrossRef CAS
- C. S. Lin, R. Q. Zhang, S. T. Lee, M. Elstner, Th. Frauenheim and L. J. Wan, J. Phys. Chem. B, 2005, 109, 14183–14188 CrossRef CAS PubMed
- S. Blankenburg, M. Bieri, R. Fasel, K. Müllen, C. A. Pignedoli and D. Passerone, Small, 2010, 6, 2266–2271 CrossRef CAS PubMed
- J. Ma, M. Yang, F. Yu and J. Zheng, Sci. Rep., 2015, 5, 13578 CrossRef PubMed
- Y. Yang, X. Yang, L. Liang, Y. Gao, H. Cheng, X. Li, M. Zou, R. Ma, Q. Yuan and X. Duan, Science, 2019, 364, 1057–1062 CrossRef CAS PubMed
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed
- D. Cohen-Tanugi and J. C. Grossman, Nano Lett., 2012, 12, 3602–3608 CrossRef CAS PubMed
- S. Han, D. Wu, S. Li, F. Zhang and X. Feng, Adv. Mater., 2014, 26, 849–864 CrossRef CAS PubMed
- P. R. Kidambi, D. D. Mariappan, N. T. Dee, A. Vyatskikh, S. Zhang, R. Karnik and A. J. Hart, ACS Appl. Mater. Interfaces, 2018, 10, 10369–10378 CrossRef CAS PubMed
- H. Li and X. C. Zeng, ACS Nano, 2012, 6, 2401–2409 CrossRef CAS PubMed
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235440 CrossRef
- G. Hong, Y. Han, T. M. Schutzius, Y. Wang, Y. Pan, M. Hu, J. Jie, C. S. Sharma, U. Müller and D. Poulikakos, Nano Lett., 2016, 16, 4447–4453 CrossRef CAS PubMed
- A. H. Holm, R. Møller, K. H. Vase, M. Dong, K. Norrman, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, New J. Chem., 2005, 29, 659 RSC
- Z. Mutlu, P. H. Jacobse, R. D. McCurdy, J. P. Llinas, Y. Lin, G. C. Veber, F. R. Fischer, M. F. Crommie and J. Bokor, Adv. Funct. Mater., 2021, 31, 2103798 CrossRef CAS
- C. Moreno, M. Vilas-Varela, B. Kretz, A. Garcia-Lekue, M. V. Costache, M. Paradinas, M. Panighel, G. Ceballos, S. O. Valenzuela, D. Peña and A. Mugarza, Science, 2018, 360, 199–203 CrossRef CAS PubMed
- Z. Mutlu, J. P. Llinas, P. H. Jacobse, I. Piskun, R. Blackwell, M. F. Crommie, F. R. Fischer and J. Bokor, ACS Nano, 2021, 15, 2635–2642 CrossRef CAS PubMed
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed
-
M. Corso, E. Carbonell-Sanromà and D. G. de Oteyza, in On-Surface Synthesis II, ed. D. G. de Oteyza and C. Rogero, Springer International Publishing, Cham, 2018, pp. 113–152 Search PubMed
- D. G. de Oteyza, A. García-Lekue, M. Vilas-Varela, N. Merino-Díez, E. Carbonell-Sanromà, M. Corso, G. Vasseur, C. Rogero, E. Guitián, J. I. Pascual, J. E. Ortega, Y. Wakayama and D. Peña, ACS Nano, 2016, 10, 9000–9008 CrossRef CAS PubMed
- N. Merino-Díez, J. Li, A. Garcia-Lekue, G. Vasseur, M. Vilas-Varela, E. Carbonell-Sanromà, M. Corso, J. E. Ortega, D. Peña, J. I. Pascual and D. G. de Oteyza, J. Phys. Chem. Lett., 2018, 9, 25–30 CrossRef PubMed
- J. Li, S. Sanz, N. Merino-Díez, M. Vilas-Varela, A. Garcia-Lekue, M. Corso, D. G. de Oteyza, T. Frederiksen, D. Peña and J. I. Pascual, Nat. Commun., 2021, 12, 5538 CrossRef CAS PubMed
- A. Konishi and T. Kubo, Top. Curr. Chem. (Z), 2017, 375, 83 CrossRef PubMed
- E. F. Sheka, N. A. Popova, V. A. Popova, E. A. Nikitina and L. H. Shaymardanova, J. Exp. Theor. Phys., 2011, 112, 602–611 CrossRef CAS
- D. Jiang, B. G. Sumpter and S. Dai, J. Chem. Phys., 2007, 126, 134701 CrossRef PubMed
- D. G. de Oteyza and T. Frederiksen, J. Phys.: Condens. Matter, 2022, 34, 443001 CrossRef CAS PubMed
- A. Berdonces-Layunta, J. Lawrence, S. Edalatmanesh, J. Castro-Esteban, T. Wang, M. S. G. Mohammed, L. Colazzo, D. Peña, P. Jelínek and D. G. de Oteyza, ACS Nano, 2021, 15, 5610–5617 CrossRef CAS PubMed
- J. Lawrence, A. Berdonces-Layunta, S. Edalatmanesh, J. Castro-Esteban, T. Wang, A. Jimenez-Martin, B. De La Torre, R. Castrillo-Bodero, P. Angulo-Portugal, M. S. G. Mohammed, A. Matěj, M. Vilas-Varela, F. Schiller, M. Corso, P. Jelinek, D. Peña and D. G. De Oteyza, Nat. Chem., 2022, 14, 1451–1458 CrossRef CAS PubMed
- K. Lucht, D. Loose, M. Ruschmeier, V. Strotkötter, G. Dyker and K. Morgenstern, Angew. Chem., Int. Ed., 2018, 57, 1266–1270 CrossRef CAS PubMed
- K. Lucht, I. Trosien, W. Sander and K. Morgenstern, Angew. Chem., Int. Ed., 2018, 57, 16334–16338 CrossRef CAS PubMed
- H. Shi, H. Yuan, Y. Sun, X. Ma, Z. Li, D. Zhou, Z. Li and X. Shao, Nano Lett., 2021, 21, 9567–9572 CrossRef CAS PubMed
- O. Bikondoa, C. L. Pang, R. Ithnin, C. A. Muryn, H. Onishi and G. Thornton, Nat. Mater., 2006, 5, 189–192 CrossRef CAS
- D. G. de Oteyza, P. Gorman, Y.-C. Chen, S. Wickenburg, A. Riss, D. J. Mowbray, G. Etkin, Z. Pedramrazi, H.-Z. Tsai, A. Rubio, M. F. Crommie and F. R. Fischer, Science, 2013, 340, 1434–1437 CrossRef CAS PubMed
- L. Gross, F. Mohn, N. Moll, P. Liljeroth and G. Meyer, Science, 2009, 325, 1110–1114 CrossRef CAS PubMed
- N. Merino-Díez, M. S. G. Mohammed, J. Castro-Esteban, L. Colazzo, A. Berdonces-Layunta, J. Lawrence, J. I. Pascual, D. G. de Oteyza and D. Peña, Chem. Sci., 2020, 11, 5441–5446 RSC
- T. Wang, S. Sanz, J. Castro-Esteban, J. Lawrence, A. Berdonces-Layunta, M. S. G. Mohammed, M. Vilas-Varela, M. Corso, D. Peña, T. Frederiksen and D. G. de Oteyza, Nano Lett., 2022, 22, 164–171 CrossRef CAS PubMed
- J. Holec, B. Cogliati, J. Lawrence, A. Berdonces-Layunta, P. Herrero, Y. Nagata, M. Banasiewicz, B. Kozankiewicz, M. Corso, D. G. De Oteyza, A. Jancarik and A. Gourdon, Angew. Chem., Int. Ed., 2021, 60, 7752–7758 CrossRef CAS PubMed
- F. Plasser, H. Pašalić, M. H. Gerzabek, F. Libisch, R. Reiter, J. Burgdörfer, T. Müller, R. Shepard and H. Lischka, Angew. Chem., Int. Ed., 2013, 52, 2581–2584 CrossRef CAS PubMed
- R. Nieman, N. J. Silva, A. J. A. Aquino, M. M. Haley and H. Lischka, J. Org. Chem., 2020, 85, 3664–3675 CrossRef CAS PubMed
- P. V. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. Van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02933f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |