
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bi- and tricyclic diterpenoids: landmarks from a decade (2013–2023) in search of leads against infectious diseases
Olha
Antoniuk
 abc,
Ana
Maranha
bc,
Jorge A. R.
Salvador
abc,
Nuno
Empadinhas
bc and
Vânia M.
Moreira
*abcd
abc,
Ana
Maranha
bc,
Jorge A. R.
Salvador
abc,
Nuno
Empadinhas
bc and
Vânia M.
Moreira
*abcd
aFaculty of Pharmacy, University of Coimbra, Portugal. E-mail: vmoreira@ff.uc.pt
bCentre for Neuroscience and Cell Biology, University of Coimbra, Portugal
cCentre for Innovative Biomedicine and Biotechnology, University of Coimbra, Portugal
dDrug Research Program, Division of Pharmaceutical Chemistry and Technology, Faculty of Pharmacy, University of Helsinki, 00014 Helsinki, Finland
First published on 7th October 2024
Abstract
Covering: 2013 to 2023
In an era where antimicrobial resistance severely threatens our ability to treat infections, the discovery of new drugs that belong to different chemical classes and/or bear original modes of action is urgently needed. In this case, diterpenoids comprise a productive field with a proven track record in providing new anti-infectives to tackle bacterial infections and malaria. This review highlights the potential of both naturally occurring and semi-synthetic bi- and tricyclic diterpenoids to become leads in search of new drugs to treat infections caused by bacteria, fungi, viruses and protozoan parasites. The literature from the last decade (2013–2023) is covered, focusing on naturally occurring and semi-synthetic bicyclic (labdanes and labdane-type) and tricyclic (all classes) diterpenoids, detailing their relevant biological activities in the context of infection, which are explained through structure–activity relationships.
 Olha Antoniuk | Olha Antoniuk was born in Kyiv, Ukraine. She obtained her Pharmaceutical Sciences Degree in 2005 at the National University of Pharmacy (Ukraine) and her Master's Degree in Medicinal Chemistry and Biopharmaceutics in 2022 from the University of Lisbon, Portugal. She is currently a PhD student in Medicinal Chemistry at the University of Coimbra, and a member of the Centre for Neuroscience and Cell Biology (CIBB consortium). Her work is focused on the synthesis and development of antimicrobial drugs based on natural products (diterpenes). |
 Ana Maranha | Ana Maranha obtained her Biology Degree in 2008, her Masters in Molecular Cell Biology in 2010 and her PhD in Biosciences with a specialty in Microbiology in 2016, from the University of Coimbra (UC). Her PhD studies on the biosynthesis of mycobacterial polymethylated polysaccharides included a period at the University of Guelph, Canada. She is currently a researcher at the Molecular Microbiology and Microbiome Group at the Centre for Neuroscience and Cell Biology (CIBB consortium), and her work is focused on microbiome dysbiosis in chronic diseases, and natural products for antimicrobial drug discovery. |
 Jorge A. R. Salvador | Jorge Salvador has a PhD degree in Pharmacy-Pharmaceutical Chemistry, University of Coimbra (UC) in collaboration with the University of York, U.K. He spent one year as a Postdoctoral Student at the University of Sussex, UK, and has a post-graduation in Cancer Biology & Therapeutics-HICR from Harvard-Medical School, University of Harvard, USA. He has a position as a Full Professor at the Faculty of Pharmacy (UC) and is the group leader of the research group “Medicinal Chemistry & Drug Discovery” at the Centre for Innovative Biomedicine and Biotechnology (Portugal). His extensive work has been focused on studies of new anticancer compounds. |
 Nuno Empadinhas | Nuno Empadinhas holds a degree in Biology and a PhD in Biochemistry (2005) with specialty in Microbiology from the University of Coimbra (UC). He is a Principal Investigator at the Centre for Neuroscience and Cell Biology. He studied the physiology of microbes from extreme environments and elucidated the biogenesis of rare mycobacterial polysaccharides, discovering microbial genes/enzymes that were founding members of 17 new families in the IUBMB database. His transdisciplinary research has attracted ∼2M€ funding and was awarded the Mizutani Glycoscience Grant (2012), Mantero Belard Award (2016), Thomé Villar Award (2017), Seed Project UC Award (2020), and Pfizer Prize for Basic Science (2023). |
 Vânia M. Moreira | Vânia M. Moreira holds a PhD in Pharmacy-Pharmaceutical Chemistry, University of Coimbra (UC), in collaboration with the University of Maryland, USA. She holds the “Title of Docent” (Dosentti) in Medicinal Chemistry, University of Helsinki, Finland (2015), and is a Fellow of the Higher Education Academy (FHEA), UK (2018). She is an Associate Professor at the Faculty of Pharmacy, UC. Her work devoted to exploring the medicinal chemistry of terpenoid-based compounds has attracted funding from a panel of international sources, and she has received several distinctions and awards throughout her career, including a highlight by the EFMC as a “New Talent Europe 2016”. |
1. Background and introduction
Naturally occurring and semi-synthetic bi- and tricyclic diterpenoids have been studied over the past years in the search for new leads to boost drug discovery. However, a comprehensive review of their potential interest in the field of infection has been missing. Among the bicyclic diterpenoids, both clerodanes1 and halimanes2 have been the topic of extensive reviews. However, in the case of labdanes, no literature review has been published concerning their bioactivities since 2004.3 Thus, prior reviews4,5 are outdated, and a work dating 2010 (ref. 6) is solely dedicated to the chemistry of the labdane skeleton. A recently published work7 accounts for naturally occurring antimicrobial diterpenoids but only covers the last 5 years and is devoid of semi-synthetic derivatives. Another review focused on natural diterpenes against tuberculosis,8 and some reports are available on the bioactivities of the aromatic abietane dehydroabietic acid.9,10 Herein, we cover the literature from the last decade (2013–2023) concerning naturally occurring and semi-synthetic bicyclic (labdanes and labdane-type) and tricyclic (all classes) diterpenoids, providing details of their relevant biological activities in the context of infections caused by bacteria, fungi, viruses and protozoan parasites. Furthermore, their anti-infective bioactivities are explained through structure–activity relationships (SARs), and directions for future research in this field are provided.1.1 Infection and antimicrobial resistance
In 2019, infectious diseases were responsible for 24.2% of global mortality, resulting in 13.7 million deaths, with low-income countries bearing a disproportionate burden.11,12 Lower respiratory tract infections and diarrhoeal diseases are ranked as the fourth and eighth leading causes of global mortality, respectively, while malaria, tuberculosis (TB), and HIV/AIDS remain among the top ten causes of mortality in low-income countries.13 Among the deaths attributed to a single causative agent, bacterial infections account for 64.8% of the infectious disease mortality globally, followed by viruses (6.1%), fungi (2.4%), and parasites (1.0%).14The discovery of antibiotics marked a significant milestone in modern medicine, shifting the burden of death from communicable to non-communicable diseases such as cardiovascular diseases, cancer, chronic respiratory diseases, diabetes, and neurological disorders. However, the increasing antimicrobial resistance (AMR) now threatens these advancements. With an estimated 4.95 million AMR-related deaths in 2019 alone, the urgency for new antimicrobials is critical.15 The intrinsic or acquired resistance of certain pathogens, such as carbapenem-resistant Acinetobacter and Enterobacterales (CRE), methicillin-resistant Staphylococcus aureus (MRSA), drug-resistant Mycobacterium tuberculosis (DR-TB), and Candida auris, undermines conventional treatment approaches.15,16 Presently, over 20% of bacterial infections are caused by drug-resistant strains, including pan-drug-resistant Gram-negative bacteria, which are non-susceptible to all agents in all antimicrobial categories and have been reported in over twenty countries worldwide.17 In 2019, the average resistance to twelve priority antibiotic–bacterium combinations reached 30% in G20 countries and 20% in 17 Organization for Economic Co-operation and Development (OECD) countries, marking a 3% increase since 2009. Even with a deceleration trend, resistance to last-line antibiotics such as carbapenems can increase by 3.2 times, by 2035, when compared to 2005 levels.17
In 2019, eight pathogens including Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Mycobacterium tuberculosis (Mtb) and Enterococcus faecium were responsible for 80% of AMR-associated deaths globally. In all cases except Mtb, over 60% of the deaths were linked to the AMR variant of the pathogen.14,15 These organisms are prioritised on the WHO Priority Pathogens List (PPL) to guide the research and development of urgently needed effective drugs.18 Additionally, the WHO has created the Fungal Priority Pathogen List (FPPL) to address the increasing threat of invasive fungal diseases. The critical pathogens in this list include Cryptococcus neoformans, Candida auris, Aspergillus fumigatus, and Candida albicans.19,20 The emergence of drug resistance in neglected tropical diseases, such as human African trypanosomiasis21 and leishmaniasis,22 caused by protozoans also poses a significant global health challenge. These diseases disproportionately affect vulnerable populations, and the limited discovery of new agents exacerbates the problem.23 Moreover, the spread of drug resistance to most of the available antimalarial drugs is also a major concern.24
Many pathogens can form biofilms, which are microbial communities that adhere to surfaces and are encased in a self-generated matrix.25 Biofilms are a major virulence factor in various human infections, especially those linked to medical devices and chronic conditions such as chronic wounds and cystic fibrosis.26 They shield microorganisms from environmental stresses, antimicrobials, and the immune system, making them highly resilient and hard to eliminate. Biofilm-related infections show inherent antibiotic tolerance, leading to additional treatment challenges and therapeutic failures. Their presence also promotes resistance evolution, as observed in pathogens such as Staphylococcus aureus, Pseudomonas aeruginosa, and nontuberculous mycobacteria (NTM), notably Mycobacterium abscessus in cystic fibrosis patients, as well as in Mtb in tuberculosis, where biofilms interfere with the efficacy of antibiotics.27–29 AMR poses a risk not only to infectious disease treatment but also to the safety and efficacy of surgical procedures, immunosuppressive chemotherapy, sustainable food production, and the environment.30 Factors such as antibiotic misuse, globalisation, natural disasters, and geopolitical instability contribute to AMR proliferation.30 However, despite the dynamic preclinical research, the clinical pipeline for novel antimicrobials remains insufficient, primarily comprised of derivatives of existing antibiotic classes, with few new compounds entering the pipeline.31,32 The need to safekeep new antibiotics for use in the case of a major sanitary crisis further detracts large pharmaceutical companies from antibiotic development, leaving academic institutions and smaller companies to bear the R&D burden.33 However, despite new policy initiatives to improve the pipeline via push and pull incentives, overall it is believed that at present there is still insufficient targeted support and coordination for academia and small- and medium-sized companies, with drug discovery activities struggling to supply the necessary discovery and preclinical programmes.33
1.2 The impact of terpenoids in the discovery of novel anti-infective drugs
The historical value of natural products (NPs) and their derivatives as sources of new drugs is indisputable, as documented in the comprehensive work by Newman and Cragg.34 In particular, the area of anti-infectives has remained totally dependent on NPs and their structures. About 48% of the total number of antibacterial drugs approved between 1981 and 2019 was either a NP or a NP derivative, and 22.2% was totally synthetic but based on the well-known quinolone scaffold. The pleuromutilins (1–2) (Fig. 1) and the resin acids (3–5) (Fig. 2) are example diterpenoids used for their antibacterial properties, as discussed in Section 2.4. | ||
| Fig. 1 Terpenoids currently in clinics. | ||
 | ||
| Fig. 2 Main resin acids. | ||
Only two antifungal drugs were approved in the same period, both NP-based, reflecting the dearth of research into this topic over the past few decades. Notably, one of the most recent approvals in this field is the triterpene ibrexafungerp (6) (Fig. 1), for both the treatment and reduction of the incidence of vulvovaginal candidiasis.35 Ibrexafungerp (6) is a glucan synthase inhibitor that is a derivative of the NP echinocandin enfumafungin, with a better bioavailability profile, which makes it suitable for oral administration. This compound interacts with the enzyme at a site that is distinct but partially shared with that of the echinocandins. Notably, ibrexafungerp (6) is active against several Candida strains including the multi-drug resistant Candida auris, which causes severe illness and spreads easily among patients in a nosocomial environment, as well as against Candida glabrata and Aspergillus species.36 Finally, artemisinin (7) (Fig. 1), a sesquiterpene lactone isolated from Artemisia annua, with an unusual endoperoxide bridge, is still an important antimalarial agent to date, especially if used in combination regimens to treat drug-resistant malaria, and for helminth infections.37 Its modes of action have been proposed to involve not only the parasite haemoglobin-digestion processes but also the mitochondria and the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA).
2. The diterpenoids
2.1 Introduction to diterpenoids
Diterpenoids (C20), which are composed of four isoprene units, are members of a large super-family of >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 natural products, originating from (E,E,E)-geranylgeranyl diphosphate (GGPP) (Fig. 3A).6,38 In plant plastids, the methylerythritol phosphate (MEP)-dependent pathway generates GGPP from isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). In contrast, fungal diterpenoids are usually synthesised via the mevalonate (MVA) pathway.39,40 Both plants and bacteria can use either pathway, but in plants, these pathways exist with a clear spatial separation, given that the MVA pathway operates only in the cytosol and peroxisomes.38–41 Although the subcellular compartmentalization of the MVA and MEP pathways allows them to operate independently, metabolic exchange can occur between these two pathways.39,40
000 natural products, originating from (E,E,E)-geranylgeranyl diphosphate (GGPP) (Fig. 3A).6,38 In plant plastids, the methylerythritol phosphate (MEP)-dependent pathway generates GGPP from isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). In contrast, fungal diterpenoids are usually synthesised via the mevalonate (MVA) pathway.39,40 Both plants and bacteria can use either pathway, but in plants, these pathways exist with a clear spatial separation, given that the MVA pathway operates only in the cytosol and peroxisomes.38–41 Although the subcellular compartmentalization of the MVA and MEP pathways allows them to operate independently, metabolic exchange can occur between these two pathways.39,40
 | ||
| Fig. 3 (A) Bicyclisation of GGPP to copalyl diphosphate (CPP) mediated by class II diterpene synthases. This reaction generally precedes that mediated by class I diterpene synthases. OPP = diphosphate group. (B) General structures of the bicyclic (labdane) and tricyclic (abietane, pimarane, and cassane) diterpenoids discussed in this work. | ||
Diterpenoid biosynthesis can be initiated by two different types of reactions, both involving carbocationic cascades but triggered in different ways. The reactive allylic bond in GGPP invariably undergoes lysis/ionisation via a carbocationic cascade of reactions mediated by class I diterpene synthases (EC 4.2.3.x). However, this can be preceded by a protonation-initiated (bi)cyclisation reaction, catalysed by class II diterpene cyclases (EC 5.5.1.x), which leaves the allylic diphosphate ester bond of GGPP intact for the subsequent action of class I diterpene synthases (Fig. 3A).38 The formed labda-13-en-8-yl cation intermediate (I–IV) can lead to four different isomers, with a fixed trans configuration across the decalin ring (Fig. 3A). The isomers are named “normal” or antipodal/enantiomeric “ent”, depending on their absolute configuration compared to the stereochemistry of the analogous A/B rings in cholesterol, namely in copalyl diphosphate (CPP), ent-CPP, syn-CPP and syn-ent-CPP. The most observed isomers are ent-CPP and CPP, whereas the production of syn-ent-CPP has not been observed, with the syn-ent stereochemistry only recognized from plants of the Calceolaria genus.6,42
The most basic structures in the labdane-related diterpenoid super-family are the bicyclic labdane, clerodane and halimadane families formed by relevant class II diterpene cyclases, with relevant class I synthases presumably simply removing the diphosphate without catalysing cyclisation (Fig. 3B). The overwhelming majority of further cyclised labdane-related diterpenoids, including the tricyclic diterpenoids abietanes, and pimaranes, are derived from the action of relevant class I diterpene synthases on the various stereoisomers of CPP (Fig. 3B).6,38
2.2 Structure and occurrence of the labdane-type diterpenoids
The labdane diterpenoids are secondary metabolites widely distributed in different parts of plants including their roots, barks, tubers, seeds and leaves, as well as in tissues of fungi, bacteria, insects and marine organisms.5,43 A plethora of plant families can be listed as sources of labdanes including Asteraceae, Labiatae, Cistaceae, Pinaceae, Cupressaceae, Taxodiaceae, Acanthaceae, Annonaceae, Caprifoliaceae, Solanaceae, Apocynaceae, Verbenaceae and Zingiberaceae. Another important source of labdanes are coniferous plants.5 The scientific literature concerning diterpenoids and their natural terrestrial sources was regularly reviewed by Hanson from 1996 to 2019.† The general skeleton of labdane-type diterpenoids is depicted in Fig. 3B, which is comprised of a decalin core and a side chain at C9, consisting of six carbons and can be open or closed. Another common modification involves a furan ring on the side chain. Labdanes occur in nature in both the normal and antipodal series.52.3 Structure and occurrence of the tricyclic diterpenoids
Conifer resin is an abundant source of abietanes, as known as resin acids, among which the main compounds are abietic (3), dehydroabietic (4) and pimaric (5) acids (Fig. 2).44 Rosin, i.e., the solid portion of resin after the evaporation of volatiles, and its derivatives, have been widely used for industrial purposes in glues, inks, varnishes, adhesive plasters, soldering glues and sealing waxes.44,45 Rosin has also been used as a glazing agent in medicines and chewing gum. Besides conifer resin, both abietanes and other tricyclic diterpenoids can be found in plant families including Lamiaceae, particularly in the genus Salvia, and in fungi, bacteria and marine organisms.10,45 The general skeleton of the different classes of tricyclic diterpenoids is depicted in Fig. 3B. Abietanes bear an isopropyl side chain at C13, whereas that of the pimaranes and cassanes is unsaturated. The location of the ring C methyl substituent varies in the pimarane and cassane series.462.4 Diterpenoids in clinics
Diterpenoids have historically provided important drugs for the treatment of human illnesses. Among them, the diterpene taxane paclitaxel or Taxol® (8) (Fig. 1), which is present in the bark of the Pacific yew, is the best known, having widespread use as a broad-spectrum anticancer drug.47 Its four-membered oxetane ring and complex ester side chain are both essential for its antitumoral activity, which occurs through inhibition of microtubule polymerisation, causing cell cycle arrest at the G2/M phase, and finally cell death.47 Paclitaxel (8) is used to treat a variety of cancers including ovarian, lung, breast, head and neck, and melanoma.47 Ingenol mebutate (Picato™) (9) is another relevant diterpenoid derivative that was approved for the treatment of actinic keratosis (a premalignant skin condition) in 2012, but was later discontinued.48Regarding anti-infectives, the pleuromutilins (1–2) (Fig. 1) and resin acids (3–5) (Fig. 2) are significant examples.49–51 The pleuromutilins have been known since the 1950s, when the compound that names the class, i.e., pleuromutilin, was isolated from the mushroom Pleurotus mutilus.49 The pleuromutilin scaffold is comprised of a unique annelation of five-, six-, and eight-membered rings and eight stable chiral centres, as well as a glycolic ester moiety as a side chain. The pleuromutilins inhibit bacterial protein synthesis by binding to the 50S ribosomal subunit at the peptidyl transferase centre, preventing the correct positioning of transfer ribonucleic acid (tRNA) for peptide transfer and new bond formation.49 Due to this mode of action, they exhibit a broad spectrum of action against Gram-positive, Gram-negative and atypical respiratory pathogens, and more importantly low potential for the development of resistance.49 Topical retapamulin (1) (Fig. 1) was the first to be approved for the treatment of impetigo, infected small lacerations, abrasion or sutured wounds caused by Staphylococcus aureus or Streptococcus pyogenes.49 In 2019, the FDA approved lefamulin (2) (Fig. 1) for the treatment of adults with community-acquired pneumonia.52
The antimicrobial properties of the tricyclic abietane-type diterpenoids known as resin acids have been known for several decades, especially in countries in Northern Europe such as Finland, where home-made spruce resin salve has long been used as traditional folk medicine for wound-healing.51,53 Research has shown that the antimicrobial and wound-healing properties of the resin salve are due to the presence of resin acids, which comprise about 90–95% of its solid portion, with the most significant being abietic (3), dehydroabietic (4) and pimaric (5) acids.44 At present, Norway spruce (Picea abies) resin salve is commercially available as Abilar® for the treatment of a plethora of conditions including wounds, scratches, bruises and abrasions, bite and puncture wounds, paronychia, burn injuries, chicken pox-related skin infections and impetigo (where Staphylococcus aureus is an important pathogen), infected and surgical wounds, and skin cracks.51 Moreover, abietic acid (3) is an important component of dental filling materials such as Nishika Plast Seal Quick®, which is commercially available in Japan.50
3. Anti-infective labdane-type diterpenoids
The labdane-type diterpenoids portrayed in the literature over the past decade with significant anti-infective activity are detailed in the following sections. The structures of both naturally occurring and semi-synthetic compounds are depicted in Fig. 4–11, with that from natural sources categorised according to their side chain at C9. Section 3.6 is devoted to labdane-type diterpenoids produced by biotransformation. The data for all reported sources and biological activities for each compound are summarised in Tables 1 and 2. For consistency, we include values for the different reported biological activities only up to roughly 160–200 μM. At concentrations above these values, the compounds are generally too toxic or poorly soluble.| Entry | Compound | Source | Reported biological activitya | Ref. |
|---|---|---|---|---|
| a Units reported according to the original reference. Conversion into micromolar is shown in brackets (μM). MIC = Minimum inhibitory concentration; MFC = Minimum fungicidal concentration; IC50 = Concentration that inhibits the growth of a species by 50%; SI = Selectivity index; MRSA = Methicillin-resistant Staphylococcus aureus; DENV = Dengue virus; WNV = West Nile virus; H1N1 and H3N2 = Influenza A virus; RSV = Respiratory Syncytial virus; CHIKV = Chikungunya virus; *Gram-positive; **Gram-negative. b Vancomycin-sensitive. c Vancomycin-resistant. d Multidrug-resistant. e Methicillin-resistant. f Several strains. g Parasite residing inside cells. h Amastigotes. i Clinical isolates. j Tachyzoites. k Trypamastigotes. l Procyclic forms. m Promastigotes. n Virus residing inside cells. | ||||
| 1 | Manool (10) | Salvia sclarea | MIC (Enterococcus faecium MB 2b,*, Enterococcus gallinarum*) = 4 μg mL−1 (14 μM) | 54–56 |
| Salvia tingitana | MIC (Streptococcus agalactiae*, Streptococcus dysgalactiae*) = 6.25 μg mL−1 (22 μM) | |||
| MIC (Enterococcus faecium MB 152c,*, Enterococcus avium*) = 8 μg mL−1 (28 μM) | ||||
| MIC (Enterococcus faecalis MB 1c,*, Enterococcus casseliflavus*, Enterococcus durans*) = 16 μg mL−1 (55 μM) | ||||
| MIC (Enterococcus faecalis MB 76d,*) = 32 μg mL−1 (110 μM) | ||||
| 2 | Sclareol (11) | MIC (Staphylococcus aureus MB 18*,e, Staphylococcus aureus MB 188*,f, Staphylococcus saprophyticus MB 41*, Staphylococcus capitis MB 71*, Staphylococcus warneri MB 74e,*, Staphylococcus lugdunensis MB 96*, Staphylococcus hominis MB 124e,*, Enterococcus faecalis MB 1*, Enterococcus faecium MB 2b,*, Enterococcus durans*) = 32 μg mL−1 (104 μM) | 55–57 | |
| MIC (Staphylococcus epidermidis MB 165d,*, Staphylococcus epidermidis MB 169*,d, Staphylococcus simulans MB 94*, Staphylococcus haemolyticus MB 115e,*, Enterococcus faecalis MB 76b,*, Enterococcus faecium MB 152c,*, Enterococcus avium MB 119*, Enterococcus casseliflavus MB 159*, Enterococcus gallinarum MB 111*) = 64 μg mL−1 (208 μM) | ||||
| MIC (Candida albicans, Candida auris, Candida parapsilosis) = 50 μg mL−1 (162 μM) | ||||
| 3 | (12) | Salvia tingitana | MIC (Enterococcus faecalis MB 1*, Enterococcus avium MB 119*, Enterococcus gallinarum MB 111*) = 32 μg mL−1 (99 μM) | 55 |
| MIC (Staphylococcus warneri MB 74e,*, Staphylococcus lugdunensis MB 96*, Enterococcus faecalis MB 76*, Enterococcus faecium MB 2b,*, Enterococcus faecium MB 152c,*, Enterococcus casseliflavus MB 159*, Enterococcus durans MB 113*) = 64 μg mL−1 (197 μM) | ||||
| 4 | Salvic acid (13) | Eupatorium salvia Colla | MIC (Staphylococcus aureus*, Bacillus cereus*) = 50 μg mL−1 (155 μM) | 58 |
| 5 | ent-Copalic acid (14) | Kaempferia pulchra | MIC (Streptococcus agalactiae*, Streptococcus dysgalactiae*) = 1.56 μg mL−1 (5.1 μM) | 54, 59 and 60 |
| MIC (Staphylococcus epidermidis*, Staphylococcus aureus*) = 6.25 μg mL−1 (21 μM) | ||||
| MIC (Trichophyton rubrum, Microsporum gypseum) = 50 μg mL−1 (164 μM) | ||||
| 6 | (15) | Copaifera reticulata | IC50 (Enterococcus faecium*) = 9.3 μg mL−1 (28 μM) | 61 |
| IC50 (MRSA*) = 10.7 μg mL−1 (32 μM) | ||||
| IC50 (Trichophyton mentagrophytes) = 38 μg mL−1 (113 μM) | ||||
| IC50 (Trichophyton rubrum) = 44.7 μg mL−1 (133 μM) | ||||
| 7 | (16) | IC50 (Enterococcus faecium*) = 1.6 μg mL−1 (5.3 μM) | ||
| IC50 (MRSA*) = 2.5 μg mL−1 (8.2 μM) | ||||
| 8 | (17) | Caesalpinia decapetala | MIC (MRSA*) = 12 μg mL−1 (36 μM) | 62 |
| 9 | ent-Agathic acid (18) | Copaifera reticulata | IC50 (Trichophyton mentagrophytes) = 30.2 μg mL−1 (90 μM) | 61 |
| IC50 (Trichophyton rubrum) = 41.7 μg mL−1 (125 μM) | ||||
| 10 | (19) | Copaifera oleoresins | MIC (Candida tropicalis) = 9.3 μM | 63 |
| MIC (Candida albicans) = 74.3 μM | ||||
| 11 | Anticopalic acid (20) | Kaempferia elegans | MIC (Bacillus cereus*) = 3.13 μg mL−1 (10 μM) | 64 and 65 |
| Pinus pumila | MIC (Staphylococcus aureus*, Enterococcus faecalis*) = 12.5 μg mL−1 (41 μM) | |||
| MIC90 (Enterococcus faecalis*) = 50 μM | ||||
| 12 | Anticopalol (21) | Kaempferia elegans | MIC (Enterococcus faecalis*, Bacillus cereus*) = 6.25 μg mL−1 (22 μM) | 64 |
| Kaempferia pulchra | MIC (Staphylococcus aureus*) = 12.5 μg mL−1 (43 μM) | |||
| 13 | (22) | MIC (Bacillus cereus*) = 6.25 μg mL−1 (21 μM) | ||
| 14 | Cuceolatin A (23) | Cunninghamia lanceolata | MIC (Bacillus subtilis*) = 8.7 μM | 66 |
| MIC (Staphylococcus aureus*) = 10.3 μM | ||||
| 15 | Cuceolatin B (24) | MIC (Staphylococcus aureus*) = 11.7 μM | ||
| MIC (Bacillus subtilis*) = 18.6 μM | ||||
| 16 | Cuceolatin C (25) | MIC (Bacillus subtilis*) = 24.6 μM | ||
| MIC (Staphylococcus aureus*) = 24.7 μM | ||||
| 17 | (26) | MIC (Staphylococcus aureus*) = 5.9 μM | ||
| MIC (Bacillus subtilis*) = 12.3 μM | ||||
| 18 | Pahangensin B (27) | Alpinia pahangensis | MIC (Bacillus cereus*) = 52.1 μg mL−1 (157 μM) | 67 |
| 19 | (28) | Elytropappus rhinocerotis | MIC (Brevibacterium agri*) = 58 μg mL−1 (179 μM) | 68 |
| 20 | Vitexolin B (29) | Vitex vestita | MIC (Bacillus cereus*,f) = 25–50 μM | 69 |
| 21 | (30) | Talaromyces scorteus | MIC (Vibrio parahaemolyticus**) = 8 μg mL−1 (23 μM) | 70 |
| 22 | (31) | Leucas stelligera | IC50 (Mycobacterium tuberculosis) = 5.95 μg mL−1 (19 μM) | 71 |
| 23 | (32) | IC50 (Mycobacterium tuberculosis) = 9.8 μg mL−1 (30 μM) | ||
| 24 | (33) | Alpinia nigra | MIC (Bacillus subtilis*) = 4 μg mL−1 (13 μM) | 72 and 73 |
| Etlingera coccinea | MIC (Staphylococcus aureus*,f, Bacillus cereus*) = 4–12.5 μg mL−1 (13–41 μM) | |||
| Etlingera sessilanthera | MIC (Salmonella paratyphi**, Yersinia enterocolitica**) = 12.5 μg mL−1 (41 μM) | |||
| MIC (Listeria monocytogenes *, Escherichia coli**) = 25 μg mL−1 (83 μM) | ||||
| 25 | (34) | Alpinia nigra | MIC (Staphylococcus aureus*, Yersinia enterocolitica**) = 3.38 μg mL−1 (11 μM) | 72 |
| MIC (Bacillus cereus*, Salmonella paratyphi**, Escherichia coli**) = 6.25 μg mL−1 (20 μM) | ||||
| MIC (Listeria monocytogenes*) = 12.5 μg mL−1 (39 μM) | ||||
| 26 | (35) | Salvia leriifolia | MIC (Staphylococcus aureus*) = 157 μM | 74 |
| 27 | (36) | Piliostigma thonningii | IC50 (Trypanosoma bruceih) = 3.84 μM | 75 |
| IC50 (Leishmania donovanig) = 7.82 μM | ||||
| 28 | (37) | IC50 (Trypanosoma bruceih) = 3.42 μM | ||
| 29 | (38) | Psiadia arguta | IC50 (Plasmodium falciparum) = 29.1 μM | 76 |
| 30 | (39) | IC50 (Plasmodium falciparum) = 33.2 μM | ||
| 31 | (40) | IC50 (Plasmodium falciparum) = 36.6 μM | ||
| 32 | (41) | IC50 (Plasmodium falciparum) = 22.2 μM | ||
| 33 | Stachyonic acid A (42) | Basilicum polystachyon | IC50 (WNV) = 1.2 μM | 77 and 78 |
| IC50 (DENV) = 1.4 μM | ||||
| IC50 (H1N1) = 4.1 μM | ||||
| IC50 (H3N2) = 18 μM | ||||
| 34 | Forsypensin A (43) | Forsythia suspensa | EC50 (RSV) = 14.6 μM | 79 |
| IC50 (H1N1) = 21.8 μM | ||||
| 35 | Forsypensin B (44) | EC50 (RSV) = 15.4 μM | ||
| IC50 (H1N1) = 23.2 μM | ||||
| 36 | Forsypensin C (45) | EC50 (RSV) = 13.7 μM | ||
| IC50 (H1N1) = 22.9 μM | ||||
| 37 | Forsypensin D (46) | EC50 (RSV) = 11.8 μM | ||
| IC50 (H1N1) = 27.4 μM | ||||
| 38 | Forsypensin E (47) | EC50 (RSV) = 10.5 μM | ||
| IC50 (H1N1) = 24.6 μM | ||||
| 39 | Forsyshiyanin A (48) | EC50 (RSV) = 10.5 μM | 80 | |
| IC50 (H1N1) = 18.4 μM | ||||
| 40 | Forsyshiyanin B (49) | EC50 (RSV) = 13.2 μM | ||
| IC50 (H1N1) = 26.2 μM | ||||
| 41 | (50) | EC50 (RSV) = 14.4 μM | ||
| IC50 (H1N1) = 19.9 μM | ||||
| 42 | (51) | EC50 (RSV) = 12.7 μM | ||
| IC50 (H1N1) = 25.7 μM | ||||
| 43 | (52) | EC50 (RSV) = 10.5 μM | ||
| IC50 (H1N1) = 24.1 μM | ||||
| 44 | (53) | EC50 (RSV) = 11.8 μM | ||
| IC50 (H1N1) = 24.9 μM | ||||
| 45 | (54) | EC50 (RSV) = 12.0 μM | ||
| IC50 (H1N1) = 23.5 μM | ||||
| 46 | (55) | EC50 (RSV) = 11.1 μM | ||
| IC50 (H1N1) = 18.6 μM | ||||
| 47 | ent-Polyalthic acid (57) | Copaifera reticulata | MIC (Peptostreptococcus microsi,*, Porphyromonas gingivalis** = 6.25 μg mL−1 (20 μM) | 61, 81–83, 84 and 85 |
| Copaifera lucens | MIC (Lacticaseibacillus caseii,*) = 12.5 μg mL−1 (40 μM) | |||
| Copaifera duckei | MIC (Streptococcus sobrinus*, Streptococcus mitis*, Streptococcus mutans*, Streptococcus salivariusi,*, Streptococcus sanguinis*, Enterococcus faecalis*, Aggregatibacter actinomycetemcomitans**, Fusobacterium nucleatum**, Actinomyces naeslundii*) = 25 μg mL−1 (80 μM) | |||
| Copaifera trapezifolia | MIC (Streptococcus salivarius*, Streptococcus sanguinisi,*, Lacticaseibacillus casei*, Porphyromonas gingivalisi,*, Fusobacterium nucleatumi,**, Prevotella nigrescens**, Bacteroides fragilis**) = 50 μg mL−1 (158 μM) | |||
| IC50 (Enterococcus faecium*) = 8.5 μM | ||||
| IC50 (MRSA*) = 8.9 μM | ||||
| IC50 (Trichophyton mentagrophytes) = 4.3 μg mL−1 (14 μM) | ||||
| IC50 (Trichophyton rubrum) = 6.8 μg mL−1 (22 μM) | ||||
| MIC (Candida glabrata) = 12.5 μg mL−1 (40 μM) | ||||
| MFC (Candida glabrata) = 25 μg mL−1 (79 μM) | ||||
| IC50 (Trypanosoma bruceik) = 3.87 μg mL−1 (12 μM) | ||||
| IC50 (Leishmania donovanih) = 8.68 μg mL−1 (28 μM) | ||||
| IC50 (Toxoplasma gondiij) = 64 μg mL−1 (202 μM) | ||||
| 48 | Coronarin E (58) | Hedychium ellipticum | MIC (Mycobacterium tuberculosis) = 12.5 μg mL−1 (44 μM) | 86 |
| 49 | Andrographolide (60) | Andrographis paniculata | IC50 (Trypanosoma bruceil) = 8.3 μM, SI = 8.5 | 77, 87–93 |
| IC50 (Leishmania martiniquensism) = 4.04 μg mL−1 (12 μM) | ||||
| EC50 (DENVn) = 21.3–22.7 μM | ||||
| IC50 (CHIKVn) = 77 μM | ||||
| 50 | Vitexolide A (61) | Vitex vestita | MIC (Bacillus cereus*,i, Staphylococcus haemolyticus*) = 6 μg mL−1 (18 μM) | 69 |
| MIC (Staphylococcus aureusf,*) = 12–24 μg mL−1 (36–72 μM) | ||||
| MIC (Corynebacterium striatum*, Enterococcus durans*, Enterococcus faecalis*, Enterococcus gallinarum*, Staphylococcus lugdunensis*, Staphylococcus saprophyticus*, Enterococcus faecalisi,*, Listeria monocytogenesi,*) = 24 μg mL−1 (72 μM) | ||||
| MIC (Enterococcus avium*, Enterococcus faeciumi,*, Enterococcus gallinarum*, Listeria innocuai,*, Staphylococcus sciurii,*) = 48 μg mL−1 (144 μM) | ||||
| 51 | (62) | MIC (Bacillus cereus*,f, Staphylococcus haemolyticus*, Staphylococcus intermedius*, Streptococcus agalactiae*) = 24 μg mL−1 (72 μM) | ||
| MIC (Staphylococcus aureusf,*, Staphylococcus epidermidis*, Staphylococcus lugdunensis*, Staphylococcus saprophyticus*) = 48 μg mL−1 (144 μM) | ||||
| 52 | Vitexolide D (63) | MIC (Bacillus subtilis*,f) = 25 μg mL−1 (79 μM) | ||
| MIC (Bacillus cereus*,f) = 25–50 μg mL−1 (79–157 μM) | ||||
| MIC (Corynebacterium striatum*, Enterococcus faeciumi,*, Enterococcus gallinarumi,*, Listeria monocytogenesi,*, Staphylococcus aureus*,f, Staphylococcus epidermidis*, Staphylococcus intermedius*, Streptococcus agalactiae*) = 50 μg mL−1 (157 μM) | ||||
| 53 | Vitexolide E (64) | MIC (Enterococcus durans*, Enterococcus faecalis*,f, Enterococcus faecium*,f, Streptococcus agalactiae*) = 50 μg mL−1 (157 μM) | ||
| 54 | (65) | Leucas stelligera | IC50 (Mycobacterium tuberculosis) = 5.02 μg mL−1 (16 μM) | 71 |
| 55 | (66) | Hedychium ellipticum | MIC (Mycobacterium tuberculosis) = 6.25 μg mL−1 (20 μM) | 86 |
| 56 | (67) | Colophospermum mopane | MIC (Klebsiella pneumoniae**, Enterococcus faecalis*) = 62.5 μg mL−1 (185 μM) | 94 |
| 57 | (68) | MIC (Klebsiella pneumoniae**, Enterococcus faecalis*) = 62.5 μg mL−1 (164 μM) | ||
| 58 | (69) | MIC (Klebsiella pneumoniae**) = 62.5 μg mL−1 (193 μM) | ||
| 59 | (70) | MIC (Klebsiella pneumoniae**) = 46.9 μg mL−1 (155 μM) | ||
| MIC (Staphylococcus aureus*) = 62.5 μg mL−1 (194 μM) | ||||
| 60 | Acuminolide (71) | Vitex vestita | MIC (Bacillus cereus*) = 23 μM | 69 |
| 61 | (72) | Leucas stelligera | IC50 (Mycobacterium tuberculosis) = 5.55 μg mL−1 (17 μM) | 71 |
| 62 | Isoambreinolide (73) | Vitex trifolia | MIC (Mycobacterium tuberculosis) = 25 μg mL−1 (95 μM) | 95 |
| 63 | Pahangensin A (74) | Alpinia pahangensis | MIC (Bacillus cereus*, Bacillus subtilis*) = 31.25 μg mL−1 (52 μM) | 67 |
| MIC (Staphylococcus aureus*) = 52.08 μg mL−1 (87 μM) | ||||
| 64 | Forsyqinlingine A (75) | Forsythia suspensa | EC50 (RSV) = 5.0 μM | 96 |
| IC50 (H1N1) = 6.9 μM | ||||
| 65 | Forsyqinlingine B (76) | EC50 (RSV) = 4.8 μM | ||
| IC50 (H1N1) = 7.7 μM | ||||
| Entry | Compound | Microorganism | Reported biological activitya | Ref. |
|---|---|---|---|---|
| a Units reported according to the original reference. Conversion into micromolar is shown in brackets (μM). MIC = Minimum inhibitory concentration. | ||||
| 1 | 77 | Cunninghamella elegans | MIC (Candida albicans) = 1.1 μg mL−1 (3.1 μM) | 63 |
| MIC (Candida tropicalis) = 4.4 μg mL−1 (13 μM) | ||||
| 2 | 78 | MIC (Candida albicans) = 1.1 μg mL−1 (3.1 μM) | ||
| MIC (Candida tropicalis) = 4.4 μg mL−1 (13 μM) | ||||
| 3 | 79 | Aspergillus brasiliensis | MIC (Candida glabrata) = 12.5 μg mL−1 (36 μM) | 84 |
| 4 | 80 | MIC (Candida glabrata) = 12.5 μg mL−1 (34 μM) | ||
| 5 | 81 | MIC (Candida glabrata) = 12.5 μg mL−1 (36 μM) | ||
| 6 | 82 | MIC (Candida glabrata) = 12.5 μg mL−1 (36 μM) | ||
3.1 Naturally occurring labdane-type diterpenoids with an acyclic side chain at C9
The antibacterial activity of the labdane alcohol manool (10) (Fig. 4), which is present in Salvia species, has been extensively documented (Table 1, entry 1). Manool (10) is active against streptococci,54 and notably against enterococci, with minimum inhibitory concentration (MIC) values ranging from 4 to 32 μg mL−1.55 Manool (10) was reported to inhibit both ATP production mediated by ATP synthase and ATP hydrolysis. Its ability to bind to this enzyme was further studied by docking and molecular dynamics simulations, which revealed that 10 forms lipophilic interactions with the binding site residues and its terminal vinyl group participates in a NH–π interaction with the backbone nitrogen of residue A278.55 Although the hydroxy group is the only polar group in 10, the simulation revealed that strong H-bonding to specific amino acid residues significantly contributes to the anchoring of this compound to its binding site in the enzyme, which was confirmed through experimental data.55 | ||
| Fig. 4 Naturally occurring labdane-type diterpenoids with an acyclic side chain at C9. | ||
The diols sclareol (11) and 14α-epoxysclareol (12) (Fig. 4) were overall less active against bacteria than manool (10) (Table 1, entries 2 and 3).55 Sclareol (11) and clindamycin displayed synergistic activity, i.e., their fractional inhibitory concentration index (FIC) was lower than 0.5, against MRSA.56 The antifungal activity of sclareol (11) against Candida spp was investigated.57 This compound inhibited the growth of Candida albicans, Candida auris and Candida parapsilosis with an MIC value of 50 μg mL−1 (Table 1, entry 2), inducing apoptosis-like cell death in Candida albicans, with depolarization of the mitochondrial membrane potential and increase in the reactive oxygen species (ROS) levels. Sclareol (11) was also able to inhibit biofilm formation in Candida albicans in a dose-dependent manner, with a decrease in biofilm-related factors including ZAP1, ADH5, CSH1, TPO4 and CAN2. Hyphal formation was inhibited by more than 50% at the MIC value of sclareol (11), both in yeast extract peptone dextrose (YPD) and spider medium.57 Notably, the activity of sclareol (11) was synergistic with that of miconazole against Candida albicans (FIC value of 0.31), with the co-treatment resulting in a 4-fold increase in potency for miconazole.57
Salvic acid (13) (Fig. 4) isolated from the aerial parts of Eupatorium salvia Colla was only active against Gram-positive bacteria, namely Staphylococcus aureus and Bacillus cereus (Table 1, entry 4).58ent-Copalic acid (14) (Fig. 4), the most abundant diterpene in the oleoresin of Copaifera species, displays potent antimicrobial action against staphylococci and streptococci, including clinical isolates,54 and against anaerobic pathogens associated with dental infections and dental biofilm formation (Table 1, entry 5).59
The antibacterial effects of 14 were potentiated by combination with chlorhexidine, a commonly used disinfectant. Significant reductions in the bacterial burden, i.e., from 3 log units, were observed after the treatment of Peptostreptococcus anaerobius with 6.25 μg mL−1 of (14) alone, after 48 h. More importantly, ent-copalic acid (14) could eradicate (3 log units) pre-formed biofilms of both Peptostreptococcus anaerobius and Actinomyces naeslundii at 62.5 and 1000 μg mL−1, respectively. Another study reported that 14 is also active against dermatophytes including Trichophyton rubrum and Microsporum gypseum (Table 1, entry 5).60 A significant reduction in Trichophyton rubrum hyphal growth was observed by fluorescence microscopy after treatment with ent-copalic acid (14) at sub-inhibitory concentrations. Scanning electron microscopy (SEM) revealed the inhibition of hyphal growth and an irregular growth pattern following treatment with the compound.
Notably, labdane-type di-acid 15 and compound 16 (Fig. 4), also present in Copaifera species, were both active against drug-resistant MRSA (Table 1, entries 6 and 7, respectively), with 15 displaying no cytotoxicity at concentrations of up to 100 μg mL−1, in a panel of cell lines.61 Good antibacterial activity against MRSA was also observed for triene di-acid 17 (Fig. 4), isolated from Caesalpinia decapetala (Table 1, entry 8).62 However, ent-agathic acid (18) (Fig. 4) was not active against bacteria but it displayed antifungal activity against the dermatophytes Trichophyton rubrum and Trichophyton mentagrophytes (Table 1, entry 9).61 Di-acid 19 (Fig. 4) inhibited the growth of Candida tropicalis and Candida albicans with MIC values of 9.3 and 74.3 μM, respectively (Table 1, entry 10).63
The activity of three labdane-type diterpenoids, including anticopalic acid (20), anticopalol (21) and 8(17)-ladben-15-ol (22) (Fig. 4), against the Gram-positive Staphylococcus aureus, Enterococcus faecalis and Bacillus cereus was reported (Table 1, entries 11–13, respectively).64,65 The four labdane-type diterpenoids (23–26) (Fig. 4), isolated from Cunninghamia lanceolata, were active against Staphylococcus aureus and Bacillus subtilis, with IC50 values below 25 μM (Table 1, entries 14–17, respectively).66 Pahangensin B (27) (Fig. 4) was reported to have mild activity against Bacillus cereus (Table 1, entry 18),67 whereas labdane (28) (Fig. 4), isolated from Elytropappus rhinocerotis, was active against Brevibacterium agri (Table 1, entry 19).68
Vitexolin B (29) (Fig. 4), from Vitex vestita, was active against Bacillus cereus standard environmental and clinical isolates, with MIC values ranging from 25 to 50 μM (Table 1, entry 20).69 The 5,9-dihydroxylated derivative (30) of isocupressic acid (Fig. 4) was isolated from the fungus Talaromyces scorteus AS-242 and found to inhibit the activity of the Gram-negative Vibrio parahaemolyticus, which is responsible for gastroenteritis in humans, with an MIC value of 8 μg mL−1 (Table 1, entry 21).70
The sclareol-type labdane (31) and the triol (32) (Fig. 4), isolated from Leucas stelligera, inhibited the growth of Mycobacterium tuberculosis, with IC50 values of 5.95 and 8.94 μg mL−1 (Table 1, entries 22 and 23, respectively), whereas no significant activity was observed against Escherichia coli or Mycobacterium smegmatis.71 Compound (32) was particularly selective given that no significant cytotoxicity was observed in any of the cell lines tested, namely MCF-7, Thp-1 and HepG2, at 100 μg mL−1. The di-aldehyde labdane (33) and its epoxide analogue (34) (Fig. 4) have been isolated from diverse plant sources. Both compounds are active against a panel of bacteria, both Gram-positive and -negative (Table 1, entries 24 and 25, respectively).72,73 The labdane-type diterpenoid (35) (Fig. 4), where lactonisation occurs at C6 and C19, was isolated from Salvia leriifolia and found to bear modest antibacterial activity against Staphylococcus aureus (Table 1, entry 26).74
Labdane 36 and alepterolic acid (37) (Fig. 4), isolated from Piliostigma thonningii, were tested against the amastigotes of Leishmania donovani and Trypanosoma brucei (Table 1, entries 27 and 28, respectively).75 The hydroxyl group at C3 was important for their antiprotozoal activity against Trypanosoma brucei. Several labdanes (38–41) (Fig. 4) were isolated from Psiadia arguta and evaluated for their antimalarial effects, with IC50 values ranging from 22.2 to 36.6 μM against Plasmodium falciparum (Table 1, entries 29–32, respectively).76
On African green monkey kidney Vero cells, stachyonic acid A (42) (Fig. 4) was reported as an antiviral agent against Dengue virus, with an IC50 value of 1.4 μM (Table 1, entry 33).77 Another study reported that this compound is also active against the West Nile virus and human influenza viruses H1N1 and H3N2.78 Forsypensins A–E (43–47), forsyshiyanins A (48) and B (49), and other labdanes (50–55) (Fig. 5), isolated from Forsythia suspensa, were all active against the influenza (H1N1) and respiratory syncytial viruses (RSV) (Table 1, entries 34–46), respectively, but less potent than ribavirin, which was used as a positive control.79,80
 | ||
| Fig. 5 Naturally occurring labdane-type diterpenoids with an acyclic side chain at C9 (Cont.). | ||
Cyslabdane A (56) (Fig. 5), produced by Streptomyces cyslabdanicus K04-0144, although not an antibacterial compound itself, was reported to enhance the activity of β-lactam antibiotics against MRSA by 8–32-fold (penam), 4–32-fold (cephem), and 128 to over 1000-fold (carbapenem).97
3.2 Naturally occurring labdane-type diterpenoids bearing a furan ring on the side chain at C9
The activity of ent-polyalthic acid (57) (Fig. 6), present in the oleoresin of Copaifera species, was studied in a similar fashion to that of ent-copalic acid (14) (Fig. 4) against oral pathogens (Table 1, entry 47).81,82 However, unlike 14, synergy studies with chlorhexidine did not result in an improvement in activity. Compound (57) could inhibit biofilm formation in Porphyromonas gingivalis and in the clinical isolate Peptostreptococcus micros only by 50% at 6.5 μg mL−1, but unable to eradicate established biofilms. ent-Polyalthic acid (57) was not toxic when tested on the Caenorhabditis elegans model, even after 72 h, at a high concentration of 1000 μg mL−1.82 The potential of ent-polyalthic acid (57) to affect the parasite Toxoplasma gondii residing inside BeWo cells and in human villous explants was also studied. This compound could inhibit the proliferation of Toxoplasma gondii tachyzoites at concentrations of 32 and 64 μg mL−1 (BeWo cells) and 64 μg mL−1 (villous explants), respectively.82,83 | ||
| Fig. 6 Naturally occurring labdane-type diterpenoids bearing a furan ring on the side chain at C9. | ||
The authors observed that ent-polyalthic acid (57) could downregulate the levels of IL-6, IL-8 and TNF-α in villous explants regardless of Toxoplasma gondii infection and suggested that this immunomodulation of the placental microenvironment could be relevant for targeting the parasite.83 Finally, ent-polyalthic acid (57) was also found to have a significant antifungal effect against the dermatophytes Trichophyton rubrum and Trichophyton mentagrophytes, with IC50 values of 6.8 and 4.3 μg mL−1, respectively (Table 1, entry 47).61
Coronarin E (58) (Fig. 6), isolated from Hedychium ellipticum Buch.-Ham. ex Sm., exhibits activity against Mycobacterium tuberculosis but with a low selectivity index (SI) (<10), as assessed on a panel of human cell lines (Table 1, entry 48).86 Otostegindiol (59) (Fig. 6), isolated from the leaves of Otostegia integrifolia Benth, a plant used in Ethiopia for the treatment of malaria, could to induce the maximum decrease of 73% in parasite burden in mice infected with Plasmodium berghei at a dose of 100 mg kg−1 per day.98
3.3 Naturally occurring labdane-type diterpenoids bearing a lactone on the side chain at C9
Andrographolide (60) (Fig. 7) is one of the most studied labdane-type diterpenoids. It is naturally occurring in Andrographis paniculata, a herbaceous plant of the Acanthaceae family, native to Asian countries and cultivated in Scandinavia and other parts of Europe.88,89 This plant has traditionally been used for medicinal purposes since ancient times and its reported activities include antibacterial, antipyretic, antiviral and antioxidant. Andragrapholide (60) has been identified as its major component and its antiprotozoal and antiviral properties have been documented.87–92 Andragrapholide (60) dose-dependently inhibited the growth of the procyclic (insect vector) forms of the parasite Trypanosoma brucei, with an IC50 of 8.3 μM (Table 1, entry 49) and SI of 8.5.88 Severe morphological alterations such as extensive swelling and disintegration of the cell membrane were observed after treatment with this compound. SEM showed swollen parasites with the loss of flagella, in comparison to the controls. | ||
| Fig. 7 Naturally occurring labdane-type diterpenoids bearing a lactone on the side chain at C9. | ||
Cell cycle arrest at the sub-Go/G1 stage occurred, with externalisation of phosphatidylserine, conclusive of apoptotic-like cell death. In the parasite-treated cells, apoptotic nuclei were observed, with the accumulation of lipid-storage bodies in the cytoplasm, and oxidative stress was triggered by an increase in intracellular ROS. Further evidence of apoptotic-like cell death following treatment with 60 came from the induction of loss of membrane potential, depletion of the antioxidant thiol levels and increase in lipidic peroxidation. Andragrapholide (60) was also reported to be active against the promastigotes of Leishmania martiniquensis, with an IC50 value of 4.04 μg mL−1 (Table 1, entry 49), but its cytotoxicity was high in the same concentration range.87 This compound was also active against the intracellular forms of the parasite.87
The antiviral effects of 60 were studied against Chikungunya virus (CHIKV), the causative agent of chikungunya fever, prevalent in Africa, India, Southeast Asia, and the Americas (Table 1, entry 49).89 Its action was also studied against the hepatitis C (HCV)90 and the Dengue77,91,93 viruses. A study found that 60 caused a 3 log unit decrease in CHIKV burden within HepG2 cells, with an EC50 of 77 μM (Table 1, entry 49) and without cytotoxicity.89 Andrographolide (60) was suggested to act at the post-virus entry stages, given that the reduction of viral protein expression and virus titer was most significant immediately after the infection period of the cells.89 Andrographolide (60) treatment of both Ava5cells, containing the HCV subgenomic replicon, and HCVcc-infected Huh-7 cells resulted in a decrease in viral protein and RNA, with EC50 values of 6 and 5.1 μM, respectively, and without cytotoxicity.90 Synergistic effects were observed with co-treatment of the infected cells with 60 and IFN-α, the HCV NS3/4A protease inhibitor telaprevir and the NS5B polymerase PSI-7977 inhibitor. Moreover, the antiviral effects of 60 were shown to involve the induction of the p38/MAPK/Nrf2/HO-1 pathway.90 The expression of haem oxygenase-1 (HO-1) was upregulated upon treatment with andrographolide (60), leading to increased levels of biliverdin, which suppressed HCV replication by promoting the antiviral responses mediated by IFN and inhibiting NS3/4A protease activity.90 The antiviral effects of 60 were attenuated upon the use of either a HO-1 inhibitor or HO-1 gene knockdown, which evidenced its role in the mode of action of the compound.90
The phosphorylation of p38 mitogen-activated protein kinase (MAPK) was activated in the presence of (60), which resulted in the stimulation of nuclear factor erythroid-2 (Nrf2)-mediated HO-1 expression.90 The antiviral activity of andrographolide (60) against Dengue virus was somewhat less potent.77,91 This compound inhibited the levels of viral infection in both HepG2 and HeLa cell lines, with EC50 values of 21.3 and 22.7 μM, respectively, and reduction of viral proteins DENV E and NS3 in both cell lines.91 In the case of CHIKV, the time of addition studies also showed that the activity of 60 is confined to the post-infection stage. A proteomic analysis of the anti-Dengue virus activity of 60 on HepG2 cells revealed that this activity can be cell-type dependent to a certain extent.93 Andrographolide (60) treatment of infected cells impacts several processes, ultimately resulting in a reduction in viral replication. The authors proposed that the increase in phosphorylation of eukaryotic initiation factor 2 (eIF2α) in response to andrographolide (60) is indeed a major determinant of its anti-Dengue activity and that it occurs as a consequence of the effects of this compound, either direct or not, in the critical regulator of the unfolded protein response GRP78.93
The labdane-type lactones vitexolide A (61), 12-epivitexolide A (62), vitexolide D (63) and vitexolide E (64) (Fig. 7) were assessed against a panel of 46 Gram-positive bacteria species, including clinical isolates (Table 1, entries 50–53).69 Among them, vitexolide A (61) was the most potent with MIC values ranging from 6 to 96 μM.69
Lactone 65 (Fig. 7) displayed selective activity against pathogenic Mycobacterium tuberculosis, with an IC50 of 5.02 μg mL−1, and no significant activity against non-pathogenic Mycobacterium smegmatis (Table 1, entry 54).71 No significant cytotoxicity was observed for compound 65 against MCF-7, Thp-1 or HepG2 cells at a concentration of 100 μg mL−1.71
Labdane 66 (Fig. 7), isolated from Hedychium ellipticum Buch.-Ham. ex Sm., also displayed antitubercular activity but with a low SI (<10) when assessed on a panel of human cell lines (Table 1, entry 55).86
3.4 Naturally occurring epoxy labdane-type diterpenoids
Labdanes 67–70 (Fig. 8), isolated from Colophospermum mopane, displayed MIC values ranging from 46.9 to 62.5 μg mL−1 against a panel of bacterial strains (Table 1, entries 56–59, respectively).94 Acuminolide (71) (Fig. 8), from Vitex vestita, was active against Bacillus cereus N190 with an MIC value of 23 μM (Table 1, entry 60).69 | ||
| Fig. 8 Naturally occurring epoxy labdane-type diterpenoids. | ||
3.5 Other naturally occurring labdane-type diterpenoids
Spiro-tetrahydrofuran labdane derivative 72 (Fig. 9), isolated from Leucas stelligera, inhibited the growth of Mycobacterium tuberculosis (Table 1, entry 61). This compound also inhibited the growth of MCF-7 cells by roughly 40%, at 100 μg mL−1, displaying moderate cytotoxicity.71 | ||
| Fig. 9 Other naturally occurring labdane-type diterpenoids. | ||
Isoambrenolide (73) (Fig. 9), isolated from Vitex folia, was also active against Mycobacterium tuberculosis H37Rv, with an MIC value of 100 μg mL−1 (Table 1, entry 62).95 The bis-lambda-triene lactone pahangensin A (74) (Fig. 9), isolated from Alpinia pahangensis, was moderately active against Gram-positive bacteria and devoid of activity against Gram-negative bacteria (Table 1, entry 63).67 Two labdane-type alkaloids forsyqinlingines A (75) and B (76) (Fig. 9), from the ripe fruits of Forsythia suspensa, displayed antiviral activities against influenza A (H1N1) and respiratory syncytial viruses (RSV), with IC50 values in the range of 6.9–7.7 μM and 4.8–5.0 μM (Table 1, entries 64 and 65), respectively.96
3.6 Labdane-type diterpenoids produced by biotransformation
Among the labdane-type diterpenoids, 77 and 78 (Fig. 10), produced by biotransformation with Cunninghamella elegans, displayed a significant improvement in antifungal activity against several Candida strains (Table 1, entries 1 and 2, respectively).63 Compounds 77 and 78 were 40- and 2.5-fold more potent than the reference fluconazole against Candida albicans and Candida tropicalis, respectively. The microbial transformation of ent-polyalthic acid (57) (Fig. 6) with Aspergillus brasiliensis afforded compounds 79–82 (Fig. 10), with potent antifungal effects against Candida glabrata (Table 2, entries 3–6), being 4-fold more potent than fluconazole.84 | ||
| Fig. 10 Labdane-type diterpenoids produced by biotransformation. | ||
3.7 Semi-synthetic labdane-type diterpenoids
A panel of salvic acid (13) (Fig. 4) esters was synthesised to probe the effect of increased lipophilicity on their antibacterial activity.5 Similar to salvic acid (13), the presence of carboxylic acid at C15 was crucial for the antibacterial activity. The optimal length of the ester at C7 was achieved in compounds 83–89 (Fig. 11), with an 8- to 16-fold improvement in antibacterial potency, displaying MIC values ranging from 3.13 to 6.25 μg mL−1. However, none of these compounds were active against Gram-negative bacteria.58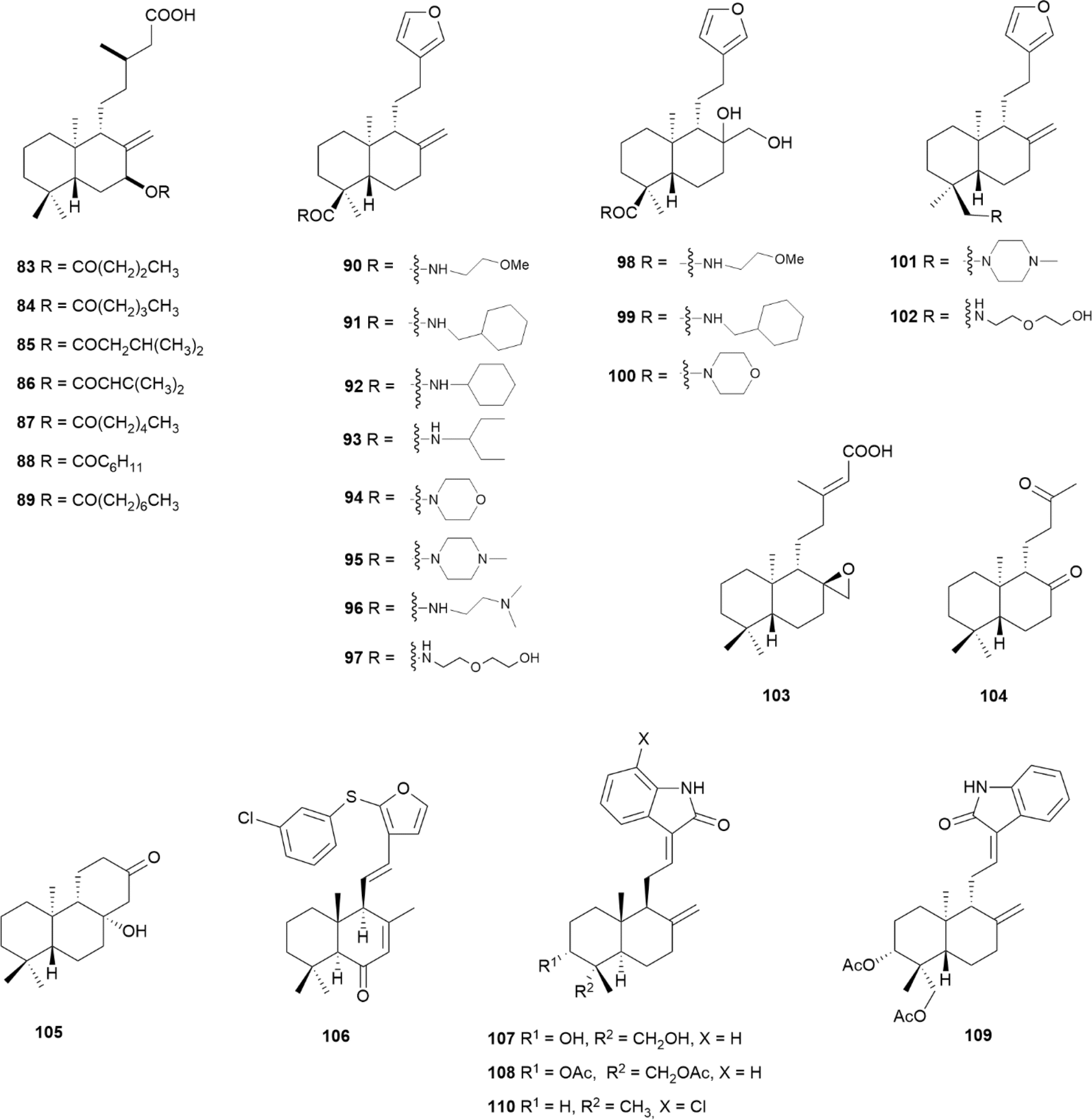 | ||
| Fig. 11 Semi-synthetic labdane-type diterpenoids. | ||
Among a panel of amide derivatives of ent-polyalthic acid (57) (Fig. 6), compounds 90 and 91 (Fig. 11) displayed the best antileishmanial activity against Leishmania donovani axenic amastigotes, with IC50 values of 6.73 and 3.84 μg mL−1, respectively, and compound 90 was also active against Trypanosoma brucei trypomastigotes with an IC50 of 2.54 μg mL−1.85 In the case of antileishmanicidal activity, bulky lipophilic groups were generally preferred as in 91, 92 and 93, with cyclic amides 94 and 95 not showing significant activity. All the amides were active against Trypanosoma brucei, with the exception of 96 and 97, and also diols 98, 99 and 100. The parent ent-polyalthic acid (57) displayed IC50 values of 8.86 and 3.87 μg mL−1 against Leishmania donovani and Trypanosoma brucei, respectively, and neither 57 nor any of the compounds was more potent than pentamidine or amphotericin B, used as positive controls.85
Amides 95 and 97 and amines 101 and 102 (Fig. 11), produced from ent-polyalthic acid 57, were also tested against the Gram-positive bacteria Enterococcus faecalis, Enterococcus faecium, Staphylococcus epidermidis and Staphylococcus aureus, with MIC values ranging from 8 to 32 μg mL−1.99 The inhibition of Staphylococcus epidermidis biofilm formation was achieved in less than 1 log unit (∼97%) with compounds 95 and 101 only, at a high concentration of 512 mg mL−1.99
The derivatives of ent-copalic acid (14) (Fig. 4), compounds 103–105 (Fig. 11), prepared by oxidation and aldol condensation, displayed antitubercular activity with MIC values ranging from 6.25 to 25 μg mL−1 against Mycobacterium tuberculosis H37Rv, and negligible cytoxicity.100 Among a panel of synthesised derivatives of hedychenone, compound 106 (Fig. 11) was the only one showing antibacterial activity against Staphylococcus aureus, as evaluated by the well diffusion assay.101
The synthesis of oxindole derivatives of andrographolide (60) (Fig. 7) led to the discovery of compound 107 (Fig. 11), where the NH-group of the oxindole moiety was crucial for its activity, considering that any derivative devoid of it, lost the antiviral potency against the CHIKV.102 Diacetylated compounds 108 and 109 (Fig. 11) were only slightly less potent, suggesting that the hydroxyl groups on the decalin core were not relevant for the activity. The authors also ruled out that the ent series was not preferred, and that side chain (E) isomers performed better than their (Z) counterparts. Finally, compound 110 was observed to be a potent inhibitor against two isolates from human patients, with minimal cytotoxicity. This compound displayed both prophylactic and therapeutic effects on the host cells, where it was shown to interfere with viral replication.102
4. Anti-infective tricyclic diterpenoids
The tricyclic diterpenoids portrayed in the literature over the past decade, with significant anti-infective activity, are detailed in the following sections. The structures of both naturally occurring and semi-synthetic compounds are depicted in Fig. 12–20, with semi-synthetic compounds grouped according to their parent diterpenoid. The data for all the reported sources and biological activities for each compound are summarised in Tables 3 and 4 and follows the same inclusion criteria as described in Section 3.| Entry | Compound | Source | Reported biological activitya | Ref. |
|---|---|---|---|---|
| a Units reported according to the original reference. Conversion into micromolar is shown in brackets (μM). MIC = Minimum inhibitory concentration; IC50 = Concentration that inhibit the growth of a species by 50%; EC50 = concentration corresponding to 50% growth inhibition of the parasite or cells; SI = Selectivity index; MRSA = Methicillin-resistant Staphylococcus aureus; VRE = Vancomycin-resistant Enterococcus; *Gram-positive; **Gram-negative. b Prior to biofilm establishment. c After biofilms are established. d IC50(post)/IC50(pre). e Promastigotes. f Amastigotes. g Parasite residing inside cells. h Trypomastigotes. i Several strains. j Chloroquine-resistant. k Pentamidine-resistant. l Diminazene-resistant. | ||||
| 1 | Abietic acid (3) | Genus Pinus | MIC (Cutibacterium acnes*) = 4 μg mL−1 (13 μM) | 103–105 and 106 |
| MIC (Staphylococcus epidermidis*) = 8 μg mL−1 (27 μM) | ||||
| MIC (Streptococcus mitis*) = 16 μg mL−1 (53 μM) | ||||
| MIC (Staphylococcus aureus*, Pseudomonas fluorescens**) = 25 μg mL−1 (83 μM) | ||||
| MIC (Salmonella typhimurium**, Rothia mucilaginosa*) = 31 μg mL−1 (103 μM) | ||||
| MIC (Bacillus subtilis*, Escherichia coli**) = 50 μg mL−1 (167 μM) | ||||
| 2 | Dehydroabietic acid (4) | IC50 (preb, Staphylococcus aureus*) = 27.8 μM; IC50 (postc, Staphylococcus aureus*) = 112.8 μM; foldd = 2–4 | 107, 108, 109–111, 106 and 112 | |
| MIC (Staphylococcus aureus*) = 70 μM | ||||
| MIC (Staphylococcus aureus Newman) = 12.5–25 μg mL−1 (41–83 μM) | ||||
| MIC (Bacillus subtilis*, Pseudomonas fluorescens**) = 50 μg mL−1 (166 μM) | ||||
| MIC (Saccharomyces cerevisiae) = 62.5 μg mL−1 (207 μM) | ||||
| 3 | Taxodone (111) | Salvia austriaca | MIC (Staphylococcus aureus*) = 31.25 μg mL−1 (99 μM) | 113 and 114 |
| MIC (Candida albicans) = 62.5 μg mL−1 (198 μM) | ||||
| IC50 (Trypanosoma brucei rhodesiensee) = 1.67 μM, SI = 2.4 | ||||
| IC50 (Plasmodium falciparum) = 3.66 μM, SI = 1.1 | ||||
| IC50 (Trypanosoma cruzif) = 7.63 μM, SI < 1 | ||||
| 4 | Taxodione (112) | Salvia deserta | MIC (Staphylococcus aureus*, MRSA*) = 31.8 μM | 114–117 |
| Salvia austriaca | MIC (Candida glabrata, Cryptococcus neoformans) = 15.9 μM | |||
| Plectranthus barbatus | MIC (Candida krusei) = 31.8 μM | |||
| Taxodium distichum | MIC (Candida albicans) = 63.6 μM | |||
| IC50 (Trypanosoma brucei rhodesiensef) = 0.05 μM, SI = 38 | ||||
| IC50 (Leishmania donovanig) = 1.46 μM | ||||
| IC50 (Plasmodium falciparum) = 1.9 μM, SI = 1 | ||||
| IC50 (Trypanosoma cruzig) = 7.11 μM, SI < 1 | ||||
| IC50 (Trypanosoma bruceie) = 9.8 μM, SI = 2.3 | ||||
| IC50 (Leishmania amazonensisf) = 14.3 μM, SI < 1 | ||||
| IC50 (Leishmania infantumg, Trypanosoma cruzih) = 25.7 μM | ||||
| 5 | (113) | Salvia austriaca | IC50 (Trypanosoma brucei rhodesiensee) = 0.62 μM, SI = 5 | 114 |
| IC50 (Plasmodium falciparum) = 3.37 μM, SI < 1 | ||||
| IC50 (Trypanosoma cruzif) = 7.76 μM, SI < 1 | ||||
| 6 | Horminone (114) | Plectranthus madagascariensis | IC50 (MRSA*) = 29.7 μM | 115 |
| IC50 (Staphylococcus aureus*) = 38.7 μM | ||||
| 7 | Horminone (115) | IC50 (MRSA*) = 6.8 μM | 115 and 118 | |
| IC50 (Staphylococcus aureus*) = 8.3 μM | ||||
| MIC (Mycobacterium tuberculosis) = 11.93–44.19 μM | ||||
| IC50 (Leishmania donovanie) = 29.43 μM | ||||
| 8 | Plectranthroyleanone B (116) | Plectranthus africanus | MIC (Klebsiella pneumoniae**) = 37.5 μg mL−1 (80 μM) | 119 |
| 9 | Plectranthroyleanone C (117) | MIC (Klebsiella pneumoniae**) = 37.5 μg mL−1 (83 μM) | ||
| 10 | (126) | Kaempferia roscoeana | MIC (Staphylococcus aureus*) = 25 μg mL−1 (93 μM) | 120 |
| 11 | (127) | MIC (Staphylococcus epidermidis*, Bacillus cereus*) = 25 μg mL−1 (88 μM) | ||
| 12 | (128) | Plectranthus madagascariensis | MIC (Enterococcus spp.i) = 7.81–15.63 μg mL−1 (20–40 μM) | 118 and 121 |
| Plectranthus grandidentatus | MIC (Mycobacterium tuberculosis) = 39.2–40.08 μM | |||
| 13 | (129) | Plectranthus madagascariensis | MIC (Mycobacterium tuberculosis) = 1.93–15.62 μg mL−1 (5.6–45 μM) | 118 |
| 14 | Torgranol E (130) | Torreya grandis | MIC (Mycobacterium tuberculosis) = 16 μg mL−1 (47 μM) | 122 |
| 15 | (131) | MIC (Mycobacterium tuberculosis) = 16 μg mL−1 (51 μM) | ||
| 16 | (132) | MIC (Mycobacterium tuberculosis, Staphylococcus aureus*) = 16 μg mL−1 (51 μM) | ||
| 17 | Ferruginol (133) | Salvia deserta | MIC (MRSA*) = 17.5 μM | 115, 117, 123, 124 and 125 |
| Salvia hydrangea | MIC (Staphylococcus aureus*) = 34.9 μM | |||
| Taxodium distichum | EC50 (Plasmodium falciparumj) = 0.20 μM | |||
| Salvia sahendica | IC50 (Plasmodium falciparum) = 2.9 μM | |||
| Podocarpus ferruginea | IC50 (Leishmania donovanif) = 5.9 μM | |||
| IC50 (Trypanosoma brucei rhodesienseh) = 16.6 μM | ||||
| IC50 (Leishmania donovanie) = 45.7 μM, SI < 1 | ||||
| IC50 (Leishmania amazonensisf) = 4.4 μg mL−1 (15 μM) | ||||
| IC50 (Leishmania majore) = 12.1 μg mL−1 (42 μM) | ||||
| 18 | (134) | Perovskia abrotanoides | IC50 (Trypanosoma brucei rhodesienseh) = 7.2 μM | 126 and 124 |
| Salvia sahendica | IC50 (Plasmodium falciparum) = 9.6 μM | |||
| IC50 (Leishmania donovanie) = 11.6 μM | ||||
| 19 | Miltiodiol (135) | Perovskia abrotanoides | IC50 (Trypanosoma brucei rhodesienseh) = 0.5 μM | 126 |
| IC50 (Leishmania donovanih) = 17 μM | ||||
| 20 | (136) | IC50 (Trypanosoma brucei rhodesiensef) = 0.8 μM | ||
| IC50 (Leishmania donovanie) = 1.8 μM | ||||
| IC50 (Plasmodium falciparum) = 16.2 μM | ||||
| 21 | (137) | Torreya grandis | MIC (Staphylococcus aureus*) = 4 μg mL−1 (13 μM) | 117 and 122 |
| MIC (Mycobacterium tuberculosis) = 16 μg mL−1 (51 μM) | ||||
| IC50 (Leishmania amazonensisf) = 5.4 μg mL−1 (17 μM) | ||||
| IC50 (Leishmania donovanie) = 7.8 μg mL−1 (25 μM) | ||||
| 22 | (138) | Taxodium distichum | IC50 (Leishmania amazonensisf) = 0.52 μg mL−1 (1.4 μM) | 117 |
| IC50 (Leishmania donovanie) = 2.5 μg mL−1 (6.9 μM) | ||||
| 23 | (139) | Salvia repens | IC50 (Leishmania donovanif) = 0.75 μg mL−1 (2.2 μM), SI = 23.8 | 127 |
| 24 | (143) | Salvia leriifolia | IC50 (Plasmodium falciparum) = 0.4 μM, SI = 84 | 128 |
| IC50 (Trypanosoma brucei rhodesiense) = 19.7 μM | ||||
| IC50 (Trypanosoma cruzih) = 27.6 μM | ||||
| 25 | (144) | IC50 (Trypanosoma brucei rhodesiensef; Leishmania donovanif) = 1.0 μM | ||
| IC50 (Plasmodium falciparum) = 3.6 μM | ||||
| IC50 (Trypanosoma cruzih) = 4.6 μM | ||||
| 26 | Mangiolide (145) | Suregada zanzibariensis | IC50 (MRSA*) = 3.9 μg mL−1 (11 μM) | 129 |
| Suregada zanzibariensis | IC50 (VRE*) = 7.2 μg mL−1 (19 μM) | |||
| IC50 (Cryptococcus neoformans) = 1.2 μg mL−1 (3.2 μM) | ||||
| IC50 (Plasmodium falciparum) = 0.76 μg mL−1 (2 μM), SI < 10 | ||||
| IC50 (Plasmodium falciparumj) = 0.89 μg mL−1 (2.4 μM), SI < 10 | ||||
| 27 | (146) | IC50 (Plasmodium falciparumj) = 1.17 μg mL−1 (3.5 μM), SI < 10 | ||
| IC50 (Plasmodium falciparum) = 1.24 μg mL−1 (3.8 μM), SI < 10 | ||||
| 28 | (147) | Plectranthus barbatus | IC50 (Trypanosoma bruceih) = 1.9 μM, SI = 50.5 | 116 |
| IC50 (Plasmodium falciparum) = 9.2 μM, SI = 10.4 | ||||
| IC50 (Leishmania infantumf, Trypanosoma cruzih) = 25.7 μM, SI = 3.7 | ||||
| 29 | Clinopodiolide A (148) | Salvia clinopodioides | IC50 (Entamoeba histolytica) = 43.0 μM | 130 |
| IC50 (Giardia lamblia) = 67.1 μM | ||||
| 30 | Clinopodiolide B (149) | IC50 (Entamoeba histolytica) = 37.8 μM | ||
| IC50 (Giardia lamblia) = 63 μM | ||||
| 31 | (150) | IC50 (Entamoeba histolytica) = 34.9 μM | ||
| IC50 (Giardia lamblia) = 46.4 μM | ||||
| 32 | Triacetylclinopodiolide B (151) | IC50 (Entamoeba histolytica) = 39.5 μM | ||
| IC50 (Giardia lamblia) = 47.7 μM | ||||
| 33 | Clinopodiolide C (152) | IC50 (Entamoeba histolytica) = 31.3 μM | ||
| IC50 (Giardia lamblia) = 49.0 μM | ||||
| 34 | (153) | Croton cascarilloide | MIC (Corynebacterium spp.*) = 31 μg mL−1 (103 μM) | 131 |
| MIC (Enterococcus faecalis*) = 43 μg mL−1 (142 μM) | ||||
| MIC (Enterococcus spp.*) = 46 μg mL−1 (152 μM) | ||||
| 35 | (154) | MIC (Corynebacterium spp.*) = 40 μg mL−1 (119 μM) | ||
| MIC (Enterococcus faecalis*, Enterococcus spp.*) = 49 μg mL−1 (147 μM) | ||||
| 36 | (155) | MIC (Corynebacterium spp.*) = 35 μg mL−1 (99 μM) | ||
| MIC (Enterococcus faecalis*) = 41 μg mL−1 (116 μM) | ||||
| MIC (Enterococcus spp.*) = 47 μg mL−1 (133 μM) | ||||
| 37 | Eupholide F (156) | Euphorbia fischeriana | MIC (Mycobacterium tuberculosis) = 50 μM | 132 |
| 38 | Eupholide G (157) | MIC (Mycobacterium tuberculosis) = 50 μM | ||
| 39 | Eupholide H (158) | MIC (Mycobacterium tuberculosis) = 50 μM | ||
| 40 | Jolkinolide B (159) | MIC (Mycobacterium smegmatis) = 25 μg mL−1 (77 μM) | 133 | |
| 41 | 17-Hydroxyjolkinolide B (160) | MIC (Mycobacterium smegmatis) = 1.5 μg mL−1 (4.3 μM) | ||
| 42 | (161) | Euphorbia wallichii | MIC (Corynebacterium spp.*) = 35 μg mL−1 (107 μM) | 134 |
| MIC (Enterococcus faecalis*) = 51 μg mL−1 (155 μM) | ||||
| MIC (Enterococcus spp.*) = 59 μg mL−1 (180 μM) | ||||
| 43 | (162) | MIC (Corynebacterium spp.*) = 37 μg mL−1 (108 μM) | ||
| MIC (Enterococcus faecalis*) = 45 μg mL−1 (131 μM) | ||||
| MIC (Enterococcus spp.*) = 56 μg mL−1 (163 μM) | ||||
| 44 | Icacinlactone H (163) | Icacina trichantha | MIC (Helicobacter pyloriI,**) = 8–16 μg mL−1 (22–43 μM) | 135 |
| 45 | Icacinlactone B (164) | MIC (Helicobacter pyloriI,**) = 8–16 μg mL−1 (23–45 μM) | ||
| 46 | Libertellenone A (165) | Eutypella spp. | MIC (Escherichia coli**, Bacillus subtilis*, Vibrio vulnificus**) = 16 μg mL−1 (48 μM) | 136 |
| MIC (Staphylococcus aureus*) = 32 μg mL−1 (96 μM) | ||||
| 47 | Eutypellenoid B (166) | MIC (Escherichia coli**, Staphylococcus aureus*) = 8 μg mL−1 (17 μM) | 137 | |
| MIC (Bacillus subtilis*, Vibrio alginolyticus**, Vibrio vulnificus**, Streptococcus agalactiae*) = 32 μg mL−1 (68 μM) | ||||
| MIC (Candida parapsilosis, Candida albicans) = 8 μg mL−1 (17 μM) | ||||
| MIC (Candida glabrata) = 16 μg mL−1 (34 μM) | ||||
| MIC (Candida tropicalis) = 32 μg mL−1 (68 μM) | ||||
| 48 | Eutypellenoid C (167) | MIC (Escherichia coli**, Staphylococcus aureus*, Bacillus subtilis *) = 32 μg mL−1 (68 μM) | ||
| 49 | Eutypenoid C (168) | MIC (Staphylococcus aureus*) = 32 μg mL−1 (67 μM) | ||
| 50 | (169) | Azadirachta indica | MIC (Pleomorphomonas oryzae**) = 32 μg mL−1 (86 μM) | 138 |
| MIC (Candida albicans) = 64 μg mL−1 (172 μM) | ||||
| 51 | (170) | MIC (Pleomorphomonas oryzae**) = 16 μg mL−1 (40 μM) | ||
| MIC (Candida albicans) = 16 μg mL−1 (40 μM) | ||||
| MIC (Aspergillus niger) = 32 μg mL−1 (80 μM) | ||||
| 52 | (171) | Aldama discolor | IC50 (Plasmodium falciparum) = 3.8 μM, SI = 13 | 139 |
| IC50 (Trypanosoma cruzif) = 15.4 μM, SI < 10 | ||||
| IC50 (Leishmania donovanif) = 18.2 μM, SI < 10 | ||||
| IC50 (Trypanosoma brucei rhodesienseh) = 24.3 μM, SI < 10 | ||||
| 53 | (172) | Aspergillus ochraceus | MIC (Staphylococcus aureusi,*) = 8–10 μg mL−1 (26–33 μM) | 140 |
| MIC (Staphylococcus capitis*, Staphylococcus haemolyticus*, Streptococcus pneumoniae*) = 9 μg mL−1 (29 μM) | ||||
| MIC (Staphylococcus epidermidis *) = 12 μg mL−1 (39 μM) | ||||
| MIC (Enterococcus faecalis *) = 25 μg mL−1 (82 μM) | ||||
| 54 | Talascortene C (173) | Talaromyces scorteus | MIC (Escherichia coli**) = 8 μg mL−1 (23 μM) | 70 |
| 55 | Talascortene D (174) | MIC (Escherichia coli**) = 16 μg mL−1 (48 μM) | ||
| 56 | Talascortene E (175) | MIC (Escherichia coli**) = 1 μg mL−1 (2.9 μM) | ||
| MIC (Micrococcus luteus*) = 8 μg mL−1 (23 μM) | ||||
| 57 | Talascortene F (176) | MIC (Escherichia coli**) = 8 μg mL−1 (23 μM) | ||
| 58 | Talascortene G (177) | MIC (Vibrio parahaemolyticus**) = 8 μg mL−1 (22 μM) | ||
| MIC (Pseudomonas aeruginosa**) = 32 μg mL−1 (88 μM) | ||||
| 59 | (178) | Perovskia abrotanoides | IC50 (Plasmodium falciparum) = 14.8 μM, SI = 5.2 | 124 |
| IC50 (Leishmania donovanif) = 27.7 μM, SI = 2.8 | ||||
| 60 | (179) | Aldama discolor | IC50 (Leishmania donovanif) = 13.8 μM, SI < 10 | 139 |
| IC50 (Plasmodium falciparum) = 16.5 μM | ||||
| IC50 (Trypanosoma cruzif) = 19.4 μM, SI < 10 | ||||
| 61 | (180) | IC50 (Plasmodium falciparum) = 16.1 μM | ||
| IC50 (Leishmania donovanif) = 21.9 μM, SI < 10 | ||||
| 62 | (181) | Swartzia simplex | MIC (Candida albicans) = 32 μg mL−1 (89 μM) | 141 |
| 63 | Simplexene D (182) | MIC (Candida albicans) = 32 μg mL−1 (92 μM) | ||
| 64 | Bokkosin (183) | Calliandra portoricensis | EC50 (Trypanosoma bruceik,e) = 0.33 μg mL−1 (0.5 μM) | 142 |
| EC50 (Trypanosoma bruceie) = 0.69 μg mL−1 (1.1 μM), SI > 200 | ||||
| EC50 (Leishmania mexicanae) = 5.8–9.2 μg mL−1 (9.2–15 μM) | ||||
| EC50 (Trypanosoma congolensee,l) = 17.5 μg mL−1 (28 μM) | ||||
| EC50 (Trypanosoma congolensee) = 21.6 μg mL−1 (34 μM) | ||||
| 65 | Caesalsappanin A (184) | Caesalpinia sappan | IC50 (Plasmodium falciparum) = 7.4 μM, SI < 10 | 143 |
| 66 | Caesalsappanin E (185) | IC50 (Plasmodium falciparum) = 15.7 μM, SI > 10 | ||
| 67 | Caesalsappanin G (186) | IC50 (Plasmodium falciparum) = 0.78 μM, SI > 10 | ||
| 68 | Caesalsappanin H (187) | IC50 (Plasmodium falciparum) = 0.52 μM, SI > 10 | ||
| 69 | Caesalsappanin I (188) | IC50 (Plasmodium falciparum) = 2.5 μM, SI > 10 | ||
| 70 | (189) | Caesalpinia pulcherrima | IC50 (Leishmania majore) = 30 μg mL−1 (65 μM) | 144 |
| Entry | Compound | Reported biological activityb | Ref. |
|---|---|---|---|
| a Units reported according to the original reference. Conversion into micromolar is shown in brackets (μM). MIC = Minimum inhibitory concentration; IC50 = Concentration that inhibit the growth of a species by 50%; EC50 = concentration corresponding to 50% growth inhibition of the parasite or cells; SI = Selectivity index; MRSA = Methicillin-resistant Staphylococcus aureus; DENV = Dengue virus; ZIKV = Colombina Zika virus; CHIKV = Chikungunya virus; HHV = Herpes virus; H1N1 = Influenza A virus; *Gram-positive; **Gram-negative. b Several strains. c Prior to biofilm establishment. d After biofilms are established. e IC50(post)/IC50(pre). f Parasites residing inside cells. g Promastigotes. h Amastigotes. i Post-infection stage. j Chloroquine-resistant. | |||
| 1 | 190 | MIC (Staphylococcus epidermidis*) = 16 μg mL−1 (40 μM) | 104 |
| MIC (Rothia mucilaginosa*) = 31 μg mL−1 (77 μM) | |||
| 2 | 191 | MIC (Staphylococcus aureus*) = 16 μg mL−1 (35 μM) | 145 |
| MIC (Cryptococcus neoformans var. grubii) = 4 μg mL−1 (8.8 μM) | |||
| MIC (Candida albicans) = 8 μg mL−1 (18 μM) | |||
| 3 | 192 | MIC (Candida albicans) = 15.62 μg mL−1 (23 μM) | 146 |
| 4 | 193 | MIC (Staphylococcus aureusb,*) = 1.56–3.13 μg mL−1 (2.5–5 μM) | 110 |
| 5 | 194 | MIC (Staphylococcus aureusb,*) = 1.25–3.13 μg mL−1 (2–5.1 μM) | |
| 6 | 195 | MIC (Staphylococcus aureusb,*) = 1.56–3.13 μg mL−1 (2.9–5.8 μM) | |
| 7 | 196 | MIC (Staphylococcus aureusb,*) = 0.39–6.25 μg mL−1 (0.8–13 μM) | 111 |
| 8 | 197 | MIC (Staphylococcus aureusb,*) = 1.25–3.13 μg mL−1 (2.7–6.8 μM) | |
| 9 | 198 | MIC (Staphylococcus aureusb,*) = 1.56–3.13 μg mL−1 (3.2–6.4 μM) | |
| 10 | 199 | MIC (Staphylococcus aureusb,*) = 1.56–6.25 μg mL−1 (2.7–10 μM) | 147 |
| 11 | 200 | MIC (MRSA*) = 7.8–31.2 μg mL−1 (24–94 μM) | 153 |
| MIC (Staphylococcus aureus*) = 15.6–31.2 μg mL−1 (47–94 μM) | |||
| 12 | 201 | MIC (MRSA*) = 3.9–7.8 μg mL−1 (11–23 μM) | |
| MIC (Staphylococcus aureus*) = 7.8–15.6 μg mL−1 (23–45 μM) | |||
| 13 | 202 | MIC (MRSA*) = 15.6–31.2 μg mL−1 (43–87 μM) | |
| MIC (Staphylococcus aureus*) = 31.2 μg mL−1 (87 μM) | |||
| 14 | 203 | MIC (MRSA*) = 31.2–62.5 μg mL−1 (83–167 μM) | |
| MIC (Staphylococcus aureus*) = 62.5 μg mL−1 (167 μM) | |||
| 15 | 204 | MIC (Staphylococcus aureus*, Escherichia coli**) = 3.1 μg mL−1 (6 μM) | 106 |
| MIC (Pseudomonas fluorescens**) = 6.3 μg mL−1 (12 μM) | |||
| MIC (Bacillus subtilis*) = 12.5 μg mL−1 (24 μM) | |||
| 16 | 205 | MIC (Staphylococcus aureus*, Escherichia coli**, Pseudomonas fluorescens**) = 1.6 μg mL−1 (3.1 μM) | |
| MIC (Bacillus subtilis*) = 3.1 μg mL−1 (6 μM) | |||
| 17 | 206 | MIC (Bacillus subtilis*) = 1.9 μg mL−1 (4.1 μM) | 148 |
| MIC (Escherichia coli**) = 3.9 μg mL−1 (8.4 μM) | |||
| MIC (Staphylococcus aureus*) = 7.8 μg mL−1 (17 μM) | |||
| MIC (Pseudomonas fluorescens**) = 15.6 μg mL−1 (34 μM) | |||
| MIC (Candida albicans) = 31.2 μg mL−1 (67 μM) | |||
| 18 | 207 | MIC (Escherichia coli**, Pseudomonas fluorescens**) = 7.8 μg mL−1 (17 μM) | |
| MIC (Bacillus subtilis*, Staphylococcus aureus*) = 15.6 μg mL−1 (34 μM) | |||
| MIC (Candida albicans, Candida tropicalis) = 31.2 μg mL−1 (69 μM) | |||
| 19 | 208 | MIC (MRSA*) = 31.2 μg mL−1 (74 μM) | 149 |
| 20 | 209 | MIC (Staphylococcus aureus*) = 1.9 μg mL−1 (3.6 μM) | 150 |
| MIC (Bacillus subtilis*) = 3.9 μg mL−1 (7.4 μM) | |||
| MIC (Pseudomonas fluorescens**) = 7.8 μg mL−1 (15 μM) | |||
| MIC (Escherichia coli**) = 15.6 μg mL−1 (30 μM) | |||
| MIC (Candida albicans, Candida tropicalis, Aspergillus niger) = 7.8 μg mL−1 (15 μM) | |||
| 21 | 210 | MIC (Bacillus subtilis*) = 0.9 μg mL−1 (1.6 μM) | |
| MIC (Staphylococcus aureus*) = 1.9 μg mL−1 (3.4 μM) | |||
| MIC (Escherichia coli**, Pseudomonas fluorescens**) = 7.8 μg mL−1 (14 μM) | |||
| MIC (Candida albicans, Candida tropicalis) = 31.2 μg mL−1 (56 μM) | |||
| 22 | 211 | IC50 (prec, Staphylococcus aureus*) = 33.2 μM; IC50 (postd, Staphylococcus aureus*) = 86.1 μM; folde = 3 | 108 and 112 |
| MIC (Staphylococcus aureus*) = 60 μM | |||
| 23 | 212 | IC50 (prec, Staphylococcus aureus*) = 9.4 μM; IC50 (postd, Staphylococcus aureus*) = 27.9 μM; folde = 3 | |
| MIC (Staphylococcus aureus*) = 15 μM | |||
| IC50 (Leishmania donovanif) = 5 μM, SI = 24 | |||
| 24 | 213 | MIC (Staphylococcus epidermidis*, Streptococcus mitis*) = 8 μg mL−1 (20 μM) | 104 |
| MIC (Cutibacterium acnes*) = 16 μg mL−1 (40 μM) | |||
| MIC (Rothia mucilaginosa*, Salmonella typhimurium**) = 31 μg mL−1 (77 μM) | |||
| 25 | 214 | MIC (MRSA*) = 7.4 μM | 151 |
| MIC (Staphylococcus aureus*) = 15 μM | |||
| 26 | 215 | IC50 (Trypanosoma cruzif) = 4.2 μM, SI = 8 | 152 |
| IC50 (Leishmania donovanif) = 6.6 μM, SI = 14 | |||
| 27 | 216 | IC50 (Trypanosoma cruzif) = 3.9 μM, SI = 20 | |
| IC50 (Leishmania donovanif) = 2.3 μM, SI = 15 | |||
| 28 | 217 | IC50 (Leishmania donovanif) = 9 μM, SI = 33 | |
| 29 | 218 | IC50 (Trypanosoma cruzif) = 1.4 μM, SI = 17 | |
| 30 | 219 | IC50 (Trypanosoma cruzif) = 7.1 μM, SI = 31.4 | 153 |
| 31 | 220 | IC50 (Leishmania infantumg) = 2.5 μM, SI = 51.8 | 154 |
| IC50 (Leishmania amazonensisg) = 11.6 μM, SI = 11.2 | |||
| IC50 (Leishmania guyanensisg) = 14.2 μM, SI = 9.1 | |||
| IC50 (Leishmania donovanig) = 14.8 μM, SI = 8.8 | |||
| IC50 (Leishmania infantumh) = 37.2 μM, SI = 3.5 | |||
| IC50 (Leishmania amazonensisi) = 31.4 μM, SI = 4.1 | |||
| 32 | 221 | IC50 (Leishmania amazonensisg) = 3.9 μM, SI = 8.7 | |
| IC50 (Leishmania infantumg) = 5.0 μM, SI = 6.8 | |||
| IC50 (Leishmania guyanensisg) = 5.9 μM, SI = 5.8 | |||
| IC50 (Leishmania donovanig) = 9.21 μM, SI = 3.7 | |||
| 33 | 222 | MIC (Aspergillus fumigatus, Aspergillus terreus) = 25 μg mL−1 (83 μM) | 155 |
| MIC (Aspergillus niger) = 50 μg mL−1 (165 μM) | |||
| 34 | 223 | EC50 (DENV-2i) = 1.4 μM, SI = 57.7 | 156 and 157 |
| EC50 (ZIKVi) = 6.3 μM | |||
| EC50 (Brazilian ZIKVi) = 7.7 μM | |||
| EC50 (Colombian CHIKVi) = 9.8 μM | |||
| EC50 (HHV-2i) = 19.2 μM | |||
| 35 | 226 | MIC (Staphylococcus aureus*, Escherichia coli**) = 30 μM | 158 |
| 36 | 227 | IC50 (Leishmania donovanif) = 0.06 μM | 159 |
| IC50 (Leishmania donovanih) = 0.37 μM, SI = 63 | |||
| IC50 (Trypanosoma cruzif) = 0.6 μM, SI = 58 | |||
| 37 | 228 | IC50 (Leishmania donovanig) = 2.2 μM, SI > 90 | 160 |
| IC50 (Leishmania infantumg) = 3.1 μM, SI > 64 | |||
| IC50 (Leishmania amazonensisg) = 3.7 μM, SI > 54 | |||
| IC50 (Leishmania infantumh) = 4.7 μM, SI > 42 | |||
| IC50 (Leishmania amazonensish) = 5.0 μM, SI > 40 | |||
| IC50 (Leishmania guyanensisg) = 20.4 μM, SI > 10 | |||
| 38 | 229 | IC50 (Leishmania donovanig) = 3.2 μM, SI > 62 | |
| IC50 (Leishmania infantumg) = 3.3 μM, SI > 18 | |||
| IC50 (Leishmania infantumh) = 3.3 μM, SI > 61 | |||
| IC50 (Leishmania amazonensish) = 3.5 μM, SI > 57 | |||
| IC50 (Leishmania amazonensisg) = 20.7 μM, SI > 100 | |||
| 39 | 230 | IC50 (Leishmania amazonensish) = 3.7 μM, SI > 54 | |
| IC50 (Leishmania donovanig) = 5.4 μM, SI > 37 | |||
| IC50 (Leishmania amazonensisg) = 12.2 μM, SI > 16 | |||
| IC50 (Leishmania infantumh) = 17.5 μM, SI > 11 | |||
| IC50 (Leishmania infantumg) = 23.9 μM, SI > 8 | |||
| IC50 (Leishmania guyanensisg) = 38.5 μM, SI > 5 | |||
| 40 | 231 | IC50 (Leishmania infantumh) = 2.5 μM, SI > 80 | |
| IC50 (Leishmania amazonensish) = 3.0 μM, SI > 67 | |||
| IC50 (Leishmania donovanig) = 4.0 μM, SI > 50 | |||
| IC50 (Leishmania amazonensisg) = 4.9 μM, SI > 41 | |||
| IC50 (Leishmania guyanensisg) = 8.6 μM, SI > 23 | |||
| IC50 (Leishmania infantumg) = 8.7 μM, SI > 23 | |||
| 41 | 232 | IC50 (Plasmodium falciparum) = 0.086 μM, SI > 290 | 125 |
| IC50 (Plasmodium falciparumj) = 0.20 μM, SI > 124 | |||
| 42 | 233 | MIC (Enterococcus casseliflavus*) = 0.98 μg mL−1 (1.9 μM) | 121 |
| MIC (Enterococcus faecium*) = 1.95 μg mL−1 (3.7 μM) | |||
| MIC (Enterococcus faecalisb,*) = 1.95–3.91 μg mL−1 (3.7–7.4 μM) | |||
| MIC (MRSA*)b = 3.91–7.81 μg mL−1 (7.4–15 μM) | |||
| MIC (Staphylococcus aureusb,*) = 3.91–15.63 μg mL−1 (7.4–30 μM) | |||
| 43 | 234 | MIC (Enterococcus faecium*) = 0.98 μg mL−1 (1.9 μM) | |
| MIC (Enterococcus faecalisb,*) = 0.98–1.95 μg mL−1 (1.9–3.7 μM) | |||
| MIC (MRSA*,b, Enterococcus casseliflavus*) = 3.91 μg mL−1 (7.5 μM) | |||
| MIC (Staphylococcus aureusb,*) = 3.91–62.5 μg mL−1 (7.5–119 μM) | |||
| 44 | 235 | MIC (Cryptococcus neoformans var. grubii) = 16 μg mL−1 (29 μM) | 161 |
| MIC (Candida albicans) = 32 μg mL−1 (58 μM) | |||
| 45 | 236 | MIC (Cryptococcus neoformans var. grubii) = 16 μg mL−1 (29 μM) | |
| MIC (Candida albicans) = 32 μg mL−1 (58 μM) | |||
| 46 | 237 | IC50 (Leishmania majorg) = 23.32 μg mL−1 (47 μM) | 157 |
| 47 | 238 | IC50 (Leishmania majorg) = 9.8 μg mL−1 (18 μM) | |
| 48 | 239 | IC50 (H1N1) = 3.5 μM, SI = 200 | 162 |
4.1 Naturally occurring abietane-type diterpenoids
The activity of abietic acid (3) (Fig. 2) against a panel of bacterial strains has been well documented and found to be more significant against Gram-positive bacteria (Table 3, entry 1).103–105 One study determined the susceptibility of standard American Type Culture Collection (ATCC) strains as well as that of multi-resistant Staphylococcus aureus and Escherichia coli strains to abietic acid (3), where this compound was consistently less active against Gram-negative and resistant strains.103 However, in Escherichia coli, the combination of sub-inhibitory concentrations of abietic acid (3) with either the aminoglycoside gentamicin or the fluoroquinolone norfloxacin could decrease the MIC of both antibiotics. Combination regimens of ciprofloxacin or the pump inhibitor ethidium bromide with abietic acid (3) were also effective in inhibiting the growth of Staphylococcus aureus strains overexpressing NorA or MepA, two genes coding for efflux pumps in bacterial cells. Abietic acid (3) was reported to significantly inhibit the growth of the cariogenic bacteria Streptococcus mutans and limit biofilm formation by this species by 2 log units at a concentration of 64 μg mL−1.105 The integrity of the bacterial membrane was compromised after treatment with this compound and under SEM, the bacterial surfaces appeared rough and irregular. Abietic acid (3) was mostly toxic to human cells at concentrations higher than 168 μg mL−1, apart from monocytic cells, where toxicity was observed at 64 μg mL−1, suggesting that oral rinse products will be more suitable for inclusion of abietic acid (3) to lessen its toxicity towards epithelial cells and fibroblasts.105The ability of dehydroabietic acid (4) (Fig. 2) to limit biofilm formation in Staphylococcus aureus strains was first reported following the observation that two abietane-type diterpenoids, namely, 4-epi-pimaric acid and salvipisone, were bacterial biofilm inhibitors.107 This compound was found to prevent biofilm formation in the low micromolar range, displaying a good cytocompatibility index, i.e., being well tolerated by human cells (Table 3, entry 2). With an MIC value of 70 μM, dehydroabietic acid (4) could affect the viability and biomass of established biofilms at only 2- to 4-fold higher concentrations, an effect that could not be observed in the presence of antibiotics such as penicillin G and vancomycin, even at the impractical concentrations of 400 μM.107,108,163 Other studies evaluated the antimicrobial and antibiofilm potential of dehydroabietic acid (4) against several Gram-positive and Gram-negative strains.9,109–111,164 Overall, similar to abietic acid (3), dehydroabietic acid (4) was mostly active against Gram-positive bacteria.
Taxodone (111) (Fig. 12) was reported to be a moderate antistaphylococcal and antifungal agent, with MIC values of 31.25 and 62.5 μg mL−1 against Staphylococcus aureus and Candida albicans, respectively (Table 3, entry 3).113 Taxodione (112) displayed an IC50 value of 0.05 μM against Trypanosoma brucei rhodesiense trypomastigotes, with very high selectivity, and was also active against Trypanosoma cruzi amastigotes and Plasmodium falciparum, but with low selectivity (Table 3, entry 4).114 Taxodone (111) and 7-(2′-oxohexyl)-taxodione (113) were both less potent and selective than taxodione (112) (Table 3, entry 5). Taxodione (112) was also active against Leishmania donovani, Leishmania amazonensis and Leishmania infantum and exhibited antifungal and antimicrobial activities.115–117 Horminones 114 and 115 (Fig. 12) displayed antistaphylococcal activity (Table 3, entries 6 and 7, respectively) and were synergistic (FIC value of 0.2) against MRSA when combined.115,118 Horminone 115 displayed antimycobacterial activity, with MIC90 values ranging from 11.93 to 44.19 μM, and was active against Leishmania donovani promastigotes.115,118 Plectranthroyleanones B (116) and C (117) (Fig. 12) had moderate activity against the Gram-negative Klebsiella pneumoniae, with MIC values of 37.5 μg mL−1 (Table 3, entries 8 and 9, respectively).119 A paper disk test revealed that compounds 118–121 (Fig. 12) from Caryopteris mongolica were potentially active against Gram-positive bacteria including Staphylococcus aureus, Enterococcus faecalis and Micrococcus luteus.165
 | ||
| Fig. 12 Naturally occurring abietane-type diterpenoids. | ||
The presence of inhibition zones also indicated the potential antimicrobial activity of royleanones 122–125 (Fig. 12), isolated from Plectranthus punctatus, against Escherichia coli, Bacillus subtilis, Micrococcus luteus, Pseudomonas agarici and Staphylococcus warneri.166 Compound (126) (Fig. 12), isolated from Kaempferia roscoeana, displayed an MIC value of 25 μg mL−1 against both Staphylococcus epidermidis and Bacillus cereus, whereas 127 (Fig. 12) was only active against Staphylococcus aureus (Table 3, entries 10 and 11, respectively).120 Compounds 128 and 129 (Fig. 12) showed antimycobacterial activity, with MIC90 values of 5.61 to 45.41 μM, against Mycobacterium tuberculosis H37Rv (Table 3, entries 12 and 13, respectively).118 Compound 128 was also active against Enterococcus species.121 Torganol E (130), 6,7-seco-abietane (131) and compound 132 (Fig. 12) displayed antimycobacterial activity against Mycobacterium tuberculosis H37Rv (Table 3, entries 14–16, respectively).122 Compound (132) was also active against Staphylococcus aureus, with an MIC value of 16 μg mL−1.122
Ferruginol (133) (Fig. 12) had antimicrobial activity against Staphylococcus aureus including MRSA strains, but was devoid of antifungal activity.115 Ferruginol (133) was also reported as an inhibitor of Leishmania major promastigotes, with an IC50 value of 12.1 μg mL−1 (Table 3, entry 17),123 and Leishmania donovani promastigotes and Leishmania amazonensis amastigotes, although with low SI values.115,117 Among the abietane diterpenoids isolated from the roots of Salvia sahendica, ferruginol (133) and Δ6,7-ferruginol (134) (Fig. 12) were potent antimalarial agents against Plasmodium falciparum with moderate selectivity (Table 3, entry 18).124,126 Miltiodiol (135) and 7α-ethoxyrosmanol (136) (Fig. 12) were active against Trypanosoma brucei rhodesiense, but inactive against Trypanosoma cruzi (Table 3, entries 19 and 20, respectively).124 6α-Hydroxysugiol (137) and 7,8-seco abietane (138) (Fig. 12), isolated from Taxodium distichum, were active against Leishmania species (Table 3, entries 21 and 22, respectively).117 6α-Hydroxysugiol (137) was also active against Mycobacterium tuberculosis H37v, with an MIC value of 16 μg mL−1 and Staphylococcus aureus, with an MIC value of 4 μg mL−1.122 12-Methoxycarnosic acid (139) (Fig. 12) inhibited the growth of axenic Leishmania donovani amastigotes with an IC50 value of 0.75 μM and SI value of 23.2 (Table 3, entry 23).127 Royleanones 140–142 (Fig. 12) completely (∼100%) inhibited the growth of both Trypanosoma cruzi epimastigotes and amastigotes at a concentration of 5 μg mL−1 with low selectivity.167 Leriifoliol (143) and leriifolione (144) (Fig. 12), two rearranged abietanes isolated from Salvia leriifolia, displayed good antiprotozoal properties (Table 3, entries 24 and 25, respectively).128 Leriifoliol (143) was very effective against Plasmodium falciparum, with an IC50 value of 0.4 μM and SI value of 84.128 Mangiolide (145) and compound (146) (Fig. 12) were also potent antimalarial agents against both chloroquine-sensitive and -resistant Plasmodium falciparum strains (Table 3, entries 26 and 27, respectively).129 In addition, 145 inhibited the growth of Cryptococcus neoformans, MRSA and vancomycin-resistant Enterococcus (VRE); however, its selectivity was poor.129 Among the panel of compounds isolated from Plectranthus barbatus Andr., compound 147 (Fig. 12) was both a potent and selective antiprotozoal agent, especially against macrophages infected with Trypanosoma brucei amastigotes, where it displayed an IC50 of 1.9 μM, with a high SI of 50.5 (Table 3, entry 28).116 Abietanes 148–152 (Fig. 12 and 13) were moderate inhibitors of Giardia lamblia and Entamoeba histolytica (Table 3, entries 29–33, respectively).130
The three ent-abietanes (153–155) (Fig. 13), isolated from the leaves of Croton cascarilloide, were modest antimicrobial agents against only Gram-positive bacteria (Table 3, entries 34–36, respectively).131 Eupholides F (156), G (157), and H (158) (Fig. 13) were the only active compounds against Mycobacterium tuberculosis, among the 15 abietanes isolated from the roots of Euphorbia fischeriana, with MIC values of 50 μM (Table 3, entries 37–39, respectively).132 The ent-abietane derivatives jolkinolide B (159) and 17-hydroxyjolkinolide (160) (Fig. 13), also isolated from Euphorbia fischeriana, were active against Mycobacterium smegmatis with MIC values of 25 and 1.5 μg mL−1 (Table 3, entries 40 and 41),133 whereas ent-abietanes 161 and 162 (Fig. 13), isolated from a different Euphorbia species, were only active against Gram-positive bacteria (Table 3, entries 42 and 43), respectively.134
 | ||
| Fig. 13 Naturally occurring abietane-type diterpenoids (Cont.). | ||
4.2 Naturally occurring pimarane-type diterpenoids
Icacinlactones H (163) and B (164) (Fig. 14) inhibited the growth of both standard and multi-drug resistant strains of Helicobacter pylori, with MIC values ranging from 8 to 16 μg mL−1 (Table 3, entries 44 and 45, respectively).135 Icancinlactone B (164) had an additive effect against this bacterium when used in combination with metronidazole or clarithromycin. Pimaranes 165 and 166 (Fig. 14), isolated from the arctic fungus Eutypella spp. D-1, were active against Escherichia coli, Staphylococcus aureus, Bacillus subtilis and Vibrio vulnificus, but cytotoxic (Table 3, entries 46 and 47, respectively).136,137 Compound 166 also inhibited the growth of Streptococcus agalactiae and Aeromonas hydrophila and displayed antifungal activity against a panel of fungal strains (Table 3, entry 47). Eutypellenoid C (167) and Eutypenoid C (168) (Fig. 14) were overall less potent and devoid of antifungal activity (Table 3, entries 48 and 49, respectively).137 | ||
| Fig. 14 Naturally occurring pimarane-type diterpenoids. | ||
Isopimaranes 169 and 170 (Fig. 14), isolated from the fungus Xylaria spp., inhibited the growth of Pleomorphomonas oryzae with MIC values of 32 and 16 μg mL−1 (Table 3, entries 50 and 51), respectively.138 Both compounds also displayed antifungal activity.
Ent-pimarane 171 (Fig. 14) showed the best activity against Plasmodium falciparum compared to other protozoal parasites, with an IC50 value of 3.8 μM and SI of >10 (Table 3, entry 52).139 Compound 172 (Fig. 14), obtained by fungal biotransformation, was active against a panel of bacterial strains, with MIC values ranging from 8 to 25 μg mL−1 (Table 3, entry 53).140 Talascortenes C-G (173–177) (Fig. 14) from the endozoic fungus Talaromyces scorteus displayed antibacterial activity against several strains (Table 3, entries 54–58, respectively). Talascortene G (177) was active against the Gram-negative Pseudomonas aeruginosa, with an MIC value of 32 μg mL−1.70 Pimaranes 178–180 (Fig. 14) were modest antiprotozoal agents, with poor selectivity (Table 3, entries 59–61, respectively).124,139
4.3 Naturally occurring cassane-type diterpenoids
Compounds 181 and 182 (Fig. 15), isolated from the root bark of Swartzia simplex, could inhibit the growth of Candida albicans, with MIC values of 32 μg mL−1, and limit its ability to form mature biofilms with MIC values of 50 and 25 μg mL−1 (Table 3, entries 62 and 63, respectively).141 Treatment of Candida albicans with 182 resulted in alterations and breakage zones of the plasma membrane, which were evident disorganisation of the cytoplasm and the nuclear membrane. This compound also hampered the budding ability of the fungus. The cassane-type diterpenoid bokkosin (183) (Fig. 15) was reported as a potent antiprotozoal agent targeting Trypanosoma brucei, Trypanosoma congolense and Leishmania mexicana, with low EC50 values, and no cross-resistance to pentamidine or diminazene in the case of Trypanosoma species (Table 3, entry 64).142 The best selectivity index (>200) was obtained for wild-type strains of Trypanosoma brucei. The effect of 183 was dose-dependent, leading to growth arrest and cell death, after exposure to 2- or 4-times the EC50 value, after 2 h. Compounds 184–188 (Fig. 15), isolated from Caesalpinia sappan, were active against the chloroquine-resistant K1 strain of Plasmodium falciparum, with IC50 values ranging from 0.52 and 15.7 μM, and SI values above 10, with the exception of compound 185, which was poorly selective (Table 3, entries 65–69, respectively).143 6β-Cinnamoyl cassane (189) (Fig. 15), isolated from Caesalpinia pulcherrima, was moderately active against the promastigotes of Leishmania major (Table 3, entry 70).144 | ||
| Fig. 15 Naturally occurring cassane-type diterpenoids. | ||
4.4 Semi-synthetic tricyclic diterpenoids
Several abietane-type derivatives have been prepared starting from the parent abietic (3) and dehydroabietic (4) acids (Fig. 2), dehydroabietylamine and ferruginol (133) (Fig. 12), given that they are available in greater amounts and high purity for chemical synthesis. The most accessible positions of the abietane scaffold for modification are the carboxylic acids attached to ring A, C7 on ring B, C12 on ring C, and C15 on the isopropyl side chain. | ||
| Fig. 16 Abietic acid derivatives. | ||
 | ||
| Fig. 17 Dehydroabietic acid derivatives. | ||
The presence of an oxime on the isopropyl side chain alongside a hydroxyl group at C12 was also noted as important for the activity of derivatives 200–203 (Fig. 17) against staphylococci (Table 4, entries 11–14, respectively).168 The activity of compound 201 was particularly noteworthy, given that its MIC values on all strains, both drug-resistant and drug-sensitive, ranged from 7.8 to 15.6 μg mL−1, with some values being lower than that of penicillin and gentamicin. Compound 201 was not cytotoxic at concentrations of up to 250 μg mL−1 and no haemolysis was observed after treatment of peripheral blood mononuclear cells (PBMC) with 4–16 times its MIC value.168
Among a library of 86 abietanes, compounds 204 and 205 (Fig. 17), bearing triazole rings substituted with pyridyl or pyrimidyl groups, displayed MIC values as low as 1.6 and 12.5 μg mL−1 against both Gram-positive and Gram-negative bacteria (Table 4, entries 15 and 16, respectively), with low cytotoxicity against normal foreskin fibroblasts and liver cells.106
1,3,4-Oxadiazin-5(6H)-one derivatives 206 and 207 (Fig. 17) were active against both Gram-positive and Gram-negative bacteria, and fungi (Table 4, entries 17 and 18, respectively).148 However, compound 206, bearing a chlorine substituent, was cytotoxic, as opposed to 207, bearing a cyano group. A C–H activation protocol allowed the functionalization of the otherwise difficult to modify C19 of abietanes.149 Among a series of C-19 arylated derivatives of dehydroabietic acid (4), compound 208 (Fig. 17) displayed desirable antimicrobial activity against MRSA, with an MIC value of 32 μg mL−1 (Table 4, entry 19).149 The presence of a free carboxylic acid on the aryl moiety was relevant for the observed activity, and also for the solubility of the compound to allow the screening of its bioactivity.149
In a series of N-substituted 1H-dibenzo[a,c]carbazole derivatives of dehydroabietic acid (4), the compounds bearing piperazine or azole heterocyclic moieties bridged by flexible ethyl chains were active against Bacillus subtilis, Staphylococcus aureus, Escherichia coli and Pseudomonas fluorescens, with MIC values ranging from 0.9 to 15.6 μg mL−1.150
Derivative 209 (Fig. 17), bearing an N-methyl piperazine and derivative 210, with a 2-methyl-5-nitro-imizazole moiety, were the most active, with 210 being as potent against Bacillus subtilis as the reference drug amikacin (Table 4, entries 20 and 21, respectively). Compound 209 was also active against the fungi Aspergillus niger, Candida albicans and Candida tropicalis, with a low MIC value of 7.8 μg mL−1.150
Among a panel of hybrid compounds bearing the dehydroabietic acid (4) scaffold and an amino acid side chain, compounds 211 and 212 (Fig. 17) displayed improved antimicrobial and antibiofilm activity against staphylococci compared to the parent compound (Table 4, entries 22 and 23, respectively).108,163 Both compounds could affect pre-formed biofilms at concentrations only 3-fold higher than that required to limit biofilm formation by Staphylococcus aureus. Compound 212 was particularly active, displaying an MIC value of 15 μM against planktonic Staphylococcus aureus and reducing the viability and biomass of the cells in pre-formed biofilms by 50% at a concentration of 27.9 μM, an effect that could not be achieved with either penicillin G or vancomycin at a concentration of 400 μM. Moreover, it was deemed relatively safe given that no significant reduction in the viability of HL cells was observed after treatment with 212 at concentrations of up to 100 μM. Compound 212 has an unusual cyclohexyl -L-alanine side chain attached to ring A and a free carboxylic acid, which was found to be essential for the antibiofilm activity of the prepared hybrid compounds.108 However, in the case of derivative 213, it was observed to exhibit activity against several strains of bacteria including Rothia mucilaginosa (Table 4, entry 24), even if the methyl ester was not converted into a free carboxylic acid.104 Compound 214 (Fig. 17), also devoid of a free carboxylic acid, was highly potent against the virulent Staphylococcus aureus strain UAMS-1 and the MRSA strain Mu50, with MIC values of 7.4 μM (Table 4, entry 25).151 Compound 212 and a few other hybrids namely, 215–218 (Fig. 17) were also potent antiprotozoal agents against Trypanosoma cruzi or Leishmania donovani, without significant general toxicity (Table 4, entries 26–29, respectively).152 In the case of compounds 212 and 217, inhibition of the growth of Leishmania donovani residing inside human macrophages was observed with IC50 values of 5 and 9 μM and high SI values of 24 and 33, respectively. In the case of compound 218, a 3-pyridyl-D-alanine methyl ester derivative, a 1.5-fold increase in potency was observed compared to the reference compound benznidazole for inhibiting the growth of Trypanosoma cruzi residing inside rat skeletal muscle myoblasts (L6 cells), with an SI of 17.152 Derivative 219 (Fig. 17), synthesised from abietic acid (3), was 3 times more potent than benznidazole against the amastigote forms of T cruzi, with an IC50 value of 7.1 μM and very low cytotoxicity (Table 4, entry 30).153 When tested in vivo on BALB/c albino mice infected with the parasite for 5 consecutive days at a dose of 5 mg per kg per body mass per day, it could reduce the rate of infection by 80% after 5 days, being more efficient than benznidazole, and to reduce parasitemia and prevent reactivation of the infection. Among the plausible modes of action investigated, the activity of 219 on the parasite glucose metabolism and inhibition of the activity of superoxide dismutase (SOD) were notable. Incubation of 219 with the parasite epimastigotes revealed morphological disturbances such as swollen mitochondria, the presence of small vacuoles in the cytoplasm and a lack of ribosomes.153 The oxidised derivatives of methyl dehydroabietate 220 and 221 (Fig. 17) were potent antiprotozoal agents against the promastigote forms of several Leishmania species (Table 4, entry 31 and 32, respectively), with IC50 values in the low micromolar range, and being more potent than the reference drug miltefosine in some of the tested strains.154 The hydroxyl group at position C12 in compound 221 had a remarkable effect on the SI of the compounds in the case of Leishmania infantum, where compound 220 was deemed exceptionally safe with an SI of 58.1 compared to that of 221, which was 6.8. However, compound 220 was both less potent and more toxic when tested against the amastigote forms of both Leishmania amazonensis and Leishmania infantum (Table 4, entry 31).154
Modest antifungal activity was reported for 12-hydroxyabietane 222 (Fig. 17), with MIC values of 25 and 50 μg mL−1 against Aspergillus fumigatus and Aspergillus terreus, respectively (Table 4, entry 33).155
 | ||
| Fig. 18 Ferruginol derivatives. | ||
 | ||
| Fig. 19 Dehydroabietylamine derivatives. | ||
Compound 227 (Fig. 19), an amide of dehydroabietylamine and acrylic acid, was identified among a library of dehydroabietylamine derivatives as the most promising antiprotozoal agent against Leishmania donovani and Trypanosoma cruzi (Table 4, entry 36).159 This compound displayed an IC50 value of 0.37 μM against Leishmania donovani axenic amastigotes, with an outstanding SI of 63. It could inhibit the growth of intracellular amastigotes in Leishmania donovani-infected human macrophages, with a low IC50 value of 0.06 μM. Compound 227 was 3-times more potent than the reference compound benznidazole in inhibiting the growth of Trypanosoma cruzi residing inside L6 cells, and no general toxicity was observed (SI of 58). A set of benzamide derivatives 228–231 (Fig. 19) had potent antiprotozoal activity against several species of Leishmania (Table 4, entries 37–40, respectively).160 These compounds were more potent than the reference compound miltefosine when tested on Leishmania infantum and Leishmania amazonensis amastigotes and did not display significant cytotoxicity (Table 4, entries 37–40).160
18-Phthalimide derivative 232 (Fig. 19) was reported as a potent antimalarial agent, active against both chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum, with EC50 values of 86 and 201 nM, respectively, and very high selectivity (Table 4, entry 41).125 This study found that the presence of the phthalimide group was better than a free amino group at the same position, and that the presence of chlorine substituents on the phthalimide group was not advantageous for the activity of the compound.
 | ||
| Fig. 20 Other semi-synthetic tricyclic diterpenoids. | ||
5. Structure–activity considerations
The outstanding chemical diversity of the labdanes that stems from their biosynthetic origin is well portrayed across the naturally occurring anti-infective labdane-type diterpenoids. Semi-synthetic derivatives tested for anti-infective properties remain scarce, with the exception of a few derivatives of salvic (13), ent-polyalthic (57) and ent-copalic (14) acids, where a modest improvement concerning bioactivity/selectivity has been achieved compared to their parent compounds. Among the anti-infective labdanes, stachyonic acid A (42) (Fig. 4), belonging to the “normal” series of labdanes, with the particular E-configuration on the diene side chain, has promising broad-spectrum antiviral properties worth further investigation, where chemical derivatization should establish important SAR in this field. Among the tricyclic diterpenoids, phthalimide derivative 223 (Fig. 18) showed the best potential to affect several viruses in the post-infection stage, and its hydroxyl group at position 12 was very important for this particular activity (Fig. 21). | ||
| Fig. 21 Relevant SAR for labdane-type (A) and tricyclic (B) diterpenoids. | ||
Concerning antifungal compounds, the labdane-type diterpenoids produced by fungal biotransformation stand out as compounds among the diterpenoid classes portrayed herein with the most promising activity. The ent-labdane di-acids with acyclic side chains (15) and (19) (Fig. 4), 77 (Fig. 10) and derivative 78 (Fig. 10) were all active, but the presence of the additional hydroxymethyl groups introduced by biotransformation in 77 and 78 was particularly relevant for the antifungal activity. Biotransformation of labdanes with a furan ring on the side chain at C9 was not as beneficial, which is consistent with the more modest antifungal activity of the furan labdane ent-polyalthic acid (57).
Overall, both the naturally occurring labdanes and the tricyclic diterpenoids of all classes are mostly active against Gram-positive bacteria with the significant exception of the highly oxygenated pimaranes talascortenes (173–177) (Fig. 14), isolated not from plants but from fungal sources, which can target important Gram-negative pathogens including Escherichia coli and Pseudomonas aeruginosa. Their highly oxygenated structures suggest that a decrease in lipophilicity is advantageous when targeting Gram-negative bacteria because this facilitates permeation through the outer membrane via hydrophilic β-barrel protein pores,171 which is consistent with the common knowledge in the antibiotics field. Despite the focus on Gram-positive bacteria, the activity of dehydroabietic acid (4) and of the semi-synthetic derivatives bearing amino acid side chains 211 and 212 (Fig. 17) to target bacterial biofilms has been well documented.108,163 The ability to limit biofilm formation and/or to affect bacterial biofilms once they are fully established can be advantageous, for instance, when looking for synergistic effects to revive the action of traditional antibiotics. Finally, the presence of a lactone in both labdanes 65 and 66 (Fig. 7) and abietane 60 (Fig. 13) seems relevant for their activity against mycobacteria.
The tricyclic diterpenoids should be regarded as an indisputable source of potential leads for new antiprotozoal agents, especially among the abietanes. Several naturally occurring abietanes display potent activity against parasites causing malaria, Chagas disease and leishmaniasis, with very high selectivity indexes. The presence of a quinone moiety or a highly oxygenated ring as in 112, 133–135, 139, 143 and 147 (Fig. 12) seems very important for this activity. In this regard, chemical derivatization work has already allowed the confirmation of the potential of abietanes to produce new leads for the treatment of diseases caused by protozoan parasites. Several amides prepared from dehydroabietic acid (4) and dehydroabietylamine, namely compounds 215–218 (Fig. 17) and 227–231 (Fig. 19), are very potent antiprotozoal agents, with outstanding selectivity, which can target parasites inside infected cells, i.e., during the most relevant stages of the disease. In addition, phthalimide derivative 232 (Fig. 19) is an outstanding antimalarial agent.
6. Conclusions and future perspectives
This review clearly shows that new drug leads for infection can be found among the bi- and tricyclic diterpenoids of the classes covered herein. The road to the clinic is a long one, with the final goal still out of reach. In this regard, the economic burden of anti-infective drug development on major pharmaceutical companies, which fail to see return for their investment, is still a significant roadblock. As humanity becomes increasingly aware of the global impact of AMR on human health, we hope that this issue will be overcome. In fact, finding new molecules with original modes of action that can either act as self-standing agents or revive the action of clinically used anti-infectives through synergy will be key for future breakthroughs to overcome resistance.Therefore, additional work is needed for deconvolution of the primary targets of the most promising bi- and tricyclic diterpenoids, which will prompt medicinal chemistry optimisation campaigns to drive the compounds forward along the pipeline. It is also relevant to go beyond in vitro assessment of pathogen growth inhibition/death alone to report the actions of the compounds as broadly as possible, i.e., including effects on biofilms, mixed species communities, synergy studies, and effects inside infected cells. New developments in pathogen biology, druggable targets, and infection models are currently emerging, especially in the case of bacteria, which are worth following closely, and will surely aid in improving the quality of future bioactivity data produced.
The overwhelming majority of the compounds reported herein originate from plant sources; however, the few originating from fungi, and especially that produced by biotransformation with fungi display very interesting anti-infective activity. This suggests that not only should the kingdom of fungi be investigated in more detail in search for novel bioactive compounds, but also exploited as an outstanding biotransformation factory to allow the unusual functionalization of diterpenoids of several classes and sources. Biotransformation is an important source of chemical diversity, which allows access to scaffold positions otherwise very difficult to modify via common synthetic chemistry, and the introduction of polar groups that make the compounds overall more hydrophilic. This hydrophilicity can be advantageous, for instance, in making the diterpenoids more “drug-like” in the long run, which is desirable for any anti-infective activity, with the goal of developing oral drugs. Designing novel bi- or tricyclic diterpenoids that are more hydrophilic should also not be overlooked for trying to shift their predominant antibacterial activity against Gram-positive bacteria towards a more broad-spectrum effect, given that the harder infections to treat at present are mostly caused by resistant Gram-negative bacteria.
We envision that interesting new diterpenoids will continue to be investigated during the next decade, with important disclosures on the more precise modes of antiprotozoal and antibacterial actions of the tricyclic diterpenoids, which should continue to steer research in this field. Among the natural sources portrayed in this review, half of the compounds reported were tested as antibacterials and one quarter as antiprotozoal agents, whereas their antifungal and antiviral activities remained far less exploited. We encourage all researchers in this field to bridge this gap and focus on all anti-infective activities when possible, harmonising the presentation of the results for the sake of producing data that is fully comparable, i.e., including the reported activities in molar units, with reference to a positive control, and including the selectivity index.
7. Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.8. Author contributions
Conceptualization: VMM, OA. Data curation & Formal Analysis: OA, AM, VMM. Writing – original draft: All authors. Writing – review & editing: All authors.9. Conflicts of interest
There are no conflicts to declare.10. Acknowledgements
OA acknowledges the Portuguese Science and Technology Foundation (FCT) for a doctoral scholarship (2023.02956.BD). AM acknowledges junior researcher contract under PTDC/BIA-MIC/0122/2021 (https://doi.org/10.54499/PTDC/BIA-MIC/0122/2021) and 2022.06809.PTDC (https://doi.org/10.54499/2022.06809.PTDC). VMM, JS and NE acknowledge UIDB/04539/2020 (https://doi.org/10.54499/UIDB/04539/2020), UIDP/04539/2020 (https://doi.org/10.54499/UIDP/04539/2020), LA/P/0058/2020 (https://doi.org/10.54499/LA/P/0058/2020).11. Notes and references
- R. Li, S. L. Morris-Natschke and K.-H. Lee, Nat. Prod. Rep., 2016, 33, 1166–1226 RSC.
- A. M. Roncero, I. E. Tobal, R. F. Moro, D. Díez and I. S. Marcos, Nat. Prod. Rep., 2018, 35, 955–991 RSC.
- I. Chinou, Curr. Med. Chem., 2005, 12, 1295–1317 CrossRef CAS.
- M. Singh, M. Pal and R. P. Sharma, Planta Med., 1999, 65, 002–008 CrossRef CAS PubMed.
- C. Demetzos and K. S. Dimas, Stud. Nat. Prod. Chem., 2001, 25, 235–292 CAS.
- R. J. Peters, Nat. Prod. Rep., 2010, 27, 1521 RSC.
- P. Saha, F. I. Rahman, F. Hussain, S. M. A. Rahman and M. M. Rahman, Front. Pharmacol., 2021, 12, 820312 CrossRef CAS PubMed.
- K. O. Ndjoubi, R. Sharma and A. A. Hussein, Curr. Pharm. Des., 2020, 26, 2909–2932 CrossRef CAS PubMed.
- M. Hao, J. Xu, H. Wen, J. Du, S. Zhang, M. Lv and H. Xu, Toxins, 2022, 14, 632 CrossRef CAS PubMed.
- M. A. González, Nat. Prod. Rep., 2015, 32, 684–704 RSC.
- Institute for Health Metrics and Evaluation (IHME). Global Burden of Disease, GBD Results, IHME, University of Washington, Seattle, WA, 2019. https://vizhub.healthdata.org/gbd-results, (accessed April 2024) Search PubMed.
- G. B. D. A. R. Collaborators, Lancet, 2022, 400, 2221–2248 CrossRef.
- World Health Organization (WHO), The top 10 causes of death, Published online, 2020, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death, accessed April 2024.
- Institute for Health Metrics and Evaluation (IHME), Measuring Infectious Causes and Resistance Outcomes for Burden Estimation (MICROBE), IHME, University of Washington, Seattle, WA, 2022, https://vizhub.healhtdata.org/microbe, (accessed April 2024) Search PubMed.
- G. B. D. A. R. Collaborators, Lancet, 2022, 399, 629–655 CrossRef.
- M. Lyman, K. Forsberg, D. J. Sexton, N. A. Chow, S. R. Lockhart, B. R. Jackson and T. Chiller, Ann. Intern. Med., 2023, 176, 489–495 CrossRef.
- Organisation for Economic Co-operation and Development (OECD), 17 Embracing a One Health Framework to Fight Antimicrobial Resistance, 2023. https://www.oecd.org/els/embracing-a-one-health-framework-to-fight-antimicrobial-resistance-ce44c755-en.htm.
- World Health Organization (WHO), Prioritization of Pathogens To Guide Discovery, Research And Development of New Antibiotics For Drug-Resistant Bacterial Infections, Including Tuberculosis, World Health Organization, https://www.who.int/publications/i/item/WHO-EMP-IAU-2017.12.
- M. C. Fisher and D. W. Denning, Nat. Rev. Microbiol., 2023, 21, 211–212 CrossRef CAS.
- World Health Organization (WHO), Fungal Priority Pathogens List to Guide Research, Development and Public Health Action, https://www.who.int/publications/i/item/9789240060241.
- K. I. Kasozi, E. T. MacLeod and S. C. Welburn, Pathogens, 2022, 11, 1100 CrossRef CAS PubMed.
- A. Ponte-Sucre, F. Gamarro, J.-C. Dujardin, M. P. Barrett, R. López-Vélez, R. García-Hernández, A. W. Pountain, R. Mwenechanya and B. Papadopoulou, PLoS Neglected Trop. Dis., 2017, 11, e0006052 CrossRef PubMed.
- J. Hoefle-Bénard and S. Salloch, BMJ Glob. Health, 2024, 9, e013439 CrossRef PubMed.
- P. J. Rosenthal, V. Asua, J. A. Bailey, M. D. Conrad, D. S. Ishengoma, M. R. Kamya, C. Rasmussen, F. G. Tadesse, A. Uwimana and D. A. Fidock, Lancet Infect. Dis., 2024, 9, e591–e600 CrossRef.
- R. J. Melander, A. K. Basak and C. Melander, Nat. Prod. Rep., 2020, 37, 1454–1477 RSC.
- A. Uberoi, A. McCready-Vangi and E. A. Grice, Nat. Rer. Microbiol., 2024, 8, 507–521 CrossRef.
- C. S. Thornton, M. Mellett, J. Jarand, L. Barss, S. K. Field and D. A. Fisher, Eur. Respir. rev., 2021, 30, 200299 CrossRef.
- P. Chakraborty, S. Bajeli, D. Kaushal, B. D. Radotra and A. Kumar, Nat. Commun., 2021, 12, 1606 CrossRef CAS.
- M. Usui, Y. Yoshii, S. Thiriet-Rupert, J.-M. Ghigo and C. Beloin, Commun. Biol., 2023, 6, 275 CrossRef CAS PubMed.
- K. Abel, E. Agnew, J. Amos, N. Armstrong, D. Armstrong-James, T. Ashfield, S. Aston, J. K. Baillie, S. Baldwin, G. Barlow, V. Bartle, J. Bielicki, C. Brown, E. Carrol, M. Clements, G. Cooke and A. Dane, Lancet Microbe, 2024, 5, e500–e507 CrossRef.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlén, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef.
- U. Theuretzbacher, K. Bush, S. Harbarth, M. Paul, J. H. Rex, E. Tacconelli and G. E. Thwaites, Nat. Rev. Microbiol., 2020, 18, 286–298 CrossRef CAS.
- U. Theuretzbacher, E. Baraldi, F. Ciabuschi and S. Callegari, Clin. Microbiol. Infect., 2023, 29, 610–615 CrossRef.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- J. Sucher, A. Thai, C. Tran, N. Mantena, A. Noronha and E. B. Chahine, Am. J. Health-Syst. Pharm., 2022, 79, 2208–2221 CrossRef PubMed.
- T. P. McCarty, C. M. White and P. G. Pappas, Infect. Dis. Clin. North Am., 2021, 35, 389–413 CrossRef.
- S. Krishna, L. Bustamante, R. K. Haynes and H. M. Staines, Trends Pharmacol. Sci., 2008, 29, 520–527 CrossRef CAS PubMed.
- J. Zi, S. Mafu and R. J. Peters, Annu. Rev. Plant Biol., 2014, 65, 259–286 CrossRef CAS.
- J. S. Dickschat, Angew. Chem., Int. Ed., 2019, 58, 15964–15976 CrossRef CAS.
- L. Zhao, W. Chang, Y. Xiao, H. Liu and P. Liu, Annu. Rev. Biochem., 2013, 82, 497–530 CrossRef CAS PubMed.
- M. Sapir-Mir, A. Mett, E. Belausov, S. Tal-Meshulam, A. Frydman, D. Gidoni and Y. Eyal, Plant Physiol., 2008, 148, 1219–1228 CrossRef CAS.
- R. C. Misra, A. Garg, S. Roy, C. S. Chanotiya, P. G. Vasudev and S. Ghosh, Plant Sci., 2015, 240, 50–64 CrossRef CAS.
- M. J. Smanski, R. M. Peterson and B. Shen, in Methods in Enzymology, ed. D. A. Hopwood, Academic Press, 2012, vol. 515, pp. 163–186 Search PubMed.
- S. Kugler, P. Ossowicz, K. Malarczyk-Matusiak and E. Wierzbicka, Molecules, 2019, 24, 1651 CrossRef PubMed.
- A. San Feliciano, M. Gordaliza, M. Salinero and J. del Corral, Planta Med., 1993, 59, 485–490 CrossRef CAS PubMed.
- Mordor Intelligence, Tall Oil Rosin Market Size & Share Analysis – Growth Trends & Forecasts (2024–2029), Source: https://www.mordorintelligence.com/industry-reports/tall-oil-rosin-market, (Acessed on July 22nd, 2024).
- N. B. Cech and N. H. Oberlies, Nat. Prod. Rep., 2023, 40, 1153–1157 RSC.
- A. M. Erlendsson, U. H. Olesen, M. Haedersdal and A. M. Rossi, Adv. Drug Delivery Rev., 2020, 153, 185–194 CrossRef CAS PubMed.
- S. Paukner and R. Riedl, Cold Spring Harbor Perspect. Med., 2017, 7, a027110 CrossRef PubMed.
- A. Kameyama, A. Saito, A. Haruyama, T. Komada, S. Sugiyama, T. Takahashi and T. Muramatsu, Materials, 2020, 13, 1700 CrossRef CAS PubMed.
- A. Sipponen, O. Kuokkanen, R. Tiihonen, H. Kauppinen and J. J. Jokinen, Int. J. Dermatol., 2012, 51, 726–732 CrossRef PubMed.
- Y. R. Lee and K. L. Jacobs, Drugs, 2019, 79, 1867–1876 CrossRef CAS.
- A. San Feliciano, M. Gordaliza, M. Salinero and J. del Corral, Planta Med., 1993, 59, 485–490 CrossRef CAS PubMed.
- A. Fonseca, F. Estrela, T. Moraes, L. Carneiro, J. Bastos, R. Santos, S. Ambrósio, C. Martins and R. Veneziani, Molecules, 2013, 18, 7865–7872 CrossRef CAS PubMed.
- A. Bisio, A. M. Schito, F. Pedrelli, O. Danton, J. K. Reinhardt, G. Poli, T. Tuccinardi, T. Bürgi, F. De Riccardis, M. Giacomini, D. Calzia, I. Panfoli, G. C. Schito, M. Hamburger and N. De Tommasi, J. Nat. Prod., 2020, 83, 1027–1042 CrossRef CAS.
- V. Iobbi, P. Brun, G. Bernabé, R. A. Dougué Kentsop, G. Donadio, B. Ruffoni, P. Fossa, A. Bisio and N. De Tommasi, Molecules, 2021, 26, 6681 CrossRef CAS.
- C. Kim, J.-G. Kim and K.-Y. Kim, J. Fungi, 2023, 9, 98 CrossRef CAS PubMed.
- J. Echeverría, A. Urzúa, L. Sanhueza and M. Wilkens, Molecules, 2017, 22, 1039 CrossRef.
- M. G. M. de Souza, L. F. Leandro, T. da S. Moraes, F. Abrão, R. C. S. Veneziani, S. R. Ambrosio and C. H. G. Martins, Anaerobe, 2018, 52, 43–49 CrossRef.
- M. Nakamura, E. Endo, J. P. de Sousa, D. Callejon, T. Ueda-Nakamura, B. Dias Filho, O. de Freitas, C. Nakamura and N. Lopes, J. Braz. Chem. Soc., 2017, 28, 08 Search PubMed.
- A. L. Pfeifer Barbosa, A. Wenzel-Storjohann, J. D. Barbosa, C. Zidorn, C. Peifer, D. Tasdemir and S. S. Çiçek, J. Ethnopharmacol., 2019, 233, 94–100 CrossRef CAS PubMed.
- Y. Qiao, Y. Liu, X. Duan, C. Chen, J. Liu, H. Zhu, Y. Xue and Y. Zhang, Tetrahedron, 2018, 74, 3852–3857 CrossRef CAS.
- I. P. Sousa, M. V. De Sousa Teixeira, J. A. Freitas, A. G. Ferreira, L. M. Pires, R. A. Santos, V. C. G. Heleno and N. A. J. C. Furtado, Chem. Biodiversity, 2022, 19, e202100757 CrossRef CAS PubMed.
- P. Chawengrum, J. Boonsombat, P. Kittakoop, C. Mahidol, S. Ruchirawat and S. Thongnest, Phytochem. Lett., 2018, 24, 140–144 CrossRef CAS.
- M. K. Langat, A. Helfenstein, C. Horner, P. Tammela, H. Hokkanen, D. Izotov and D. A. Mulholland, Chem. Biodiversity, 2018, 15, e1800056 CrossRef.
- J. Yu, Z. Yu, D. Wu, X. Yan, Y. Wang and H. Zhang, Chem. Biodiversity, 2019, 16, e1900317 CrossRef.
- Y. Sivasothy, H. Ibrahim, A. S. Paliany, S. A. Alias and K. Awang, Bioorg. Med. Chem. Lett., 2013, 23, 6280–6285 CrossRef CAS PubMed.
- I. M. Hulley, S. F. van Vuuren, N. J. Sadgrove and B.-E. van Wyk, J. Ethnopharmacol., 2019, 228, 92–98 CrossRef CAS.
- N. Corlay, M. Lecsö-Bornet, E. Leborgne, F. Blanchard, X. Cachet, J. Bignon, F. Roussi, M.-J. Butel, K. Awang and M. Litaudon, J. Nat. Prod., 2015, 78, 1348–1356 CrossRef CAS PubMed.
- L.-H. Meng, X.-M. Li, F.-Z. Zhang, Y.-N. Wang and B.-G. Wang, J. Nat. Prod., 2020, 83, 2528–2536 CrossRef CAS.
- R. R. Kulkarni, K. Shurpali, V. G. Puranik, D. Sarkar and S. P. Joshi, J. Nat. Prod., 2013, 76, 1836–1841 CrossRef CAS.
- S. Ghosh, K. Indukuri, S. Bondalapati, A. K. Saikia and L. Rangan, Eur. J. Med. Chem., 2013, 66, 101–105 CrossRef CAS.
- D. Daniel-Jambun, J. Dwiyanto, Y. Y. Lim, J. B. L. Tan, A. Muhamad, S. W. Yap and S. M. Lee, J. Appl. Microbiol., 2017, 123, 810–818 CrossRef CAS PubMed.
- M. Farimani, A. Taleghani, A. Aliabadi, A. Aliahmadi, M. Esmaeili, N. Namazi Sarvestani, H. Khavasi, M. Smieško, M. Hamburger and S. Nejad Ebrahimi, Planta Med., 2016, 82, 1279–1285 CrossRef CAS PubMed.
- M. Afolayan, R. Srivedavyasasri, O. T. Asekun, O. B. Familoni, A. Orishadipe, F. Zulfiqar, M. A. Ibrahim and S. A. Ross, Med. Chem. Res., 2018, 27, 2325–2330 CrossRef CAS PubMed.
- K. Mahadeo, G. Herbette, I. Grondin, O. Jansen, H. Kodja, J. Soulange, S. Jhaumeer-Laulloo, P. Clerc, A. Gauvin-Bialecki and M. Frederich, J. Nat. Prod., 2019, 82, 1361–1366 CrossRef CAS PubMed.
- Y. P. Tan, S. D. Houston, N. Modhiran, A. I. Savchenko, G. M. Boyle, P. R. Young, D. Watterson and C. M. Williams, Chem.–Eur. J., 2019, 25, 5664–5667 CrossRef CAS PubMed.
- Y. P. Tan, Y. Xue, A. I. Savchenko, S. D. Houston, N. Modhiran, C. L. D. McMillan, G. M. Boyle, P. V. Bernhardt, P. R. Young, D. Watterson and C. M. Williams, J. Nat. Prod., 2019, 82, 2828–2834 CrossRef CAS.
- K.-L. Xiang, R.-X. Liu, L. Zhao, Z.-P. Xie, S.-M. Zhang and S.-J. Dai, Phytochemistry, 2020, 173, 112298 CrossRef CAS PubMed.
- L. Zhao, K.-L. Xiang, R.-X. Liu, Z.-P. Xie, S.-M. Zhang and S.-J. Dai, Bioorg. Chem., 2020, 96, 103651 CrossRef CAS PubMed.
- F. Abrão, J. A. Alves, G. Andrade, P. F. de Oliveira, S. R. Ambrósio, R. C. S. Veneziani, D. C. Tavares, J. K. Bastos and C. H. G. Martins, Front. Microbiol., 2018, 9, 201 CrossRef.
- M. B. Santiago, V. C. O. dos Santos, S. C. Teixeira, N. B. S. Silva, P. F. de Oliveira, S. D. Ozelin, R. A. Furtado, D. C. Tavares, S. R. Ambrósio, R. C. S. Veneziani, E. A. V. Ferro, J. K. Bastos and C. H. G. Martins, Pharmaceuticals, 2023, 16, 1357 CrossRef CAS.
- S. C. Teixeira, A. M. Rosini, G. de Souza, A. F. Martínez, R. J. Silva, S. R. Ambrósio, R. C. Veneziani, J. K. Bastos, C. H. Martins, B. F. Barbosa and E. A. Ferro, Exp. Parasitol., 2023, 250, 108534 CrossRef CAS PubMed.
- I. Pontes de Sousa, A. G. Ferreira, A. E. Miller Crotti, R. Alves dos Santos, J. Kiermaier, B. Kraus, J. Heilmann and N. A. Jacometti Cardoso Furtado, Bioorg. Chem., 2020, 95, 103560 CrossRef CAS.
- C. S. Mizuno, A. B. Souza, B. L. Tekwani, S. R. Ambrósio and R. C. S. Veneziani, Bioorg. Med. Chem. Lett., 2015, 25, 5529–5531 CrossRef CAS PubMed.
- S. Songsri and N. Nuntawong, Molecules, 2016, 21, 749 CrossRef.
- N. Intakhan, W. Chanmol, P. Somboon, M. D. Bates, V. Yardley, P. A. Bates and N. Jariyapan, Pathogens, 2020, 9, 49 CrossRef CAS.
- M. Banerjee, D. Parai, P. Dhar, M. Roy, R. Barik, S. Chattopadhyay and S. K. Mukherjee, Acta Trop., 2017, 176, 58–67 CrossRef CAS PubMed.
- P. Wintachai, P. Kaur, R. C. H. Lee, S. Ramphan, A. Kuadkitkan, N. Wikan, S. Ubol, S. Roytrakul, J. J. H. Chu and D. R. Smith, Sci. Rep., 2015, 5, 14179 CrossRef CAS PubMed.
- J. Lee, C. Tseng, K. Young, H. Sun, S. Wang, W. Chen, C. Lin and Y. Wu, Br. J. Pharmacol., 2014, 171, 237–252 CrossRef CAS.
- P. Panraksa, S. Ramphan, S. Khongwichit and D. R. Smith, Antiviral Res., 2017, 139, 69–78 CrossRef CAS.
- E. F. Martínez-Pérez, E. Burgueño-Tapia, S. Roa-Flores, A. E. Bendaña-Piñeiro, E. Sánchez-Arreola, H. Bach and L. R. Hernández, Nat. Prod. Res., 2022, 36, 71–78 CrossRef.
- A. Paemanee, A. Hitakarun, P. Wintachai, S. Roytrakul and D. R. Smith, Biomed. Pharmacother., 2019, 109, 322–332 CrossRef CAS.
- K. Du, M. De Mieri, M. Neuburger, P. C. Zietsman, A. Marston, S. F. van Vuuren, D. Ferreira, M. Hamburger and J. H. van der Westhuizen, J. Nat. Prod., 2015, 78, 2494–2504 CrossRef CAS.
- N. Tiwari, J. Thakur, D. Saikia and M. M. Gupta, Phytomedicine, 2013, 20, 605–610 CrossRef CAS PubMed.
- W. Li, L. Zhao, L.-T. Sun, Z.-P. Xie, S.-M. Zhang, X.-D. Yue and S.-J. Dai, RSC Adv., 2021, 11, 29684–29689 RSC.
- H. Ikeda, K. Shin-ya, T. Nagamitsu and H. Tomoda, J. Ind. Microbiol. Biotechnol., 2016, 43, 325–342 CrossRef CAS PubMed.
- A. Endale, D. Bisrat, A. Animut, F. Bucar and K. Asres, Phytother. Res., 2013, 27, 1805–1809 CrossRef CAS PubMed.
- M. Argentin, F. Cruz, A. Souza, E. D'Aurea, J. Bastos, S. Ambrósio, R. Veneziani, I. Camargo and C. Mizuno, Antibiotics, 2023, 12, 1202 CrossRef CAS.
- P. Matos, B. Mahoney, Y. Chan, D. Day, M. Cabral, C. Martins, R. Santos, J. Bastos, P. Page and V. Heleno, Molecules, 2015, 20, 18264–18278 CrossRef CAS.
- A. K. Maurya, N. Baliyan, R. Kumar and V. K. Agnihotri, J. Nat. Prod., 2022, 85, 1691–1696 CrossRef CAS PubMed.
- Q. T. N. Tran, R. C. H. Lee, H. J. Liu, D. Ran, V. Z. L. Low, D. Q. To, J. J. H. Chu and C. L. L. Chai, Eur. J. Med. Chem., 2022, 230, 114110 CrossRef CAS PubMed.
- M. G. de Lima Silva, L. Y. S. da Silva, T. S. de Freitas, J. E. Rocha, R. L. S. Pereira, S. R. Tintino, M. R. C. de Oliveira, A. O. B. P. Bezerra Martins, M. C. P. Lima, G. C. Alverni da Hora, C. L. G. Ramalho, H. D. M. Coutinho and I. R. A. de Menezes, Process Biochem., 2022, 122, 363–372 CrossRef CAS.
- A. Helfenstein, M. Vahermo, D. A. Nawrot, F. Demirci, G. İşcan, S. Krogerus, J. Yli-Kauhaluoma, V. M. Moreira and P. Tammela, Bioorg. Med. Chem., 2017, 25, 132–137 CrossRef CAS.
- Y. Ito, T. Ito, K. Yamashiro, F. Mineshiba, K. Hirai, K. Omori, T. Yamamoto and S. Takashiba, Odontology, 2020, 108, 57–65 CrossRef CAS.
- W. Hou, G. Zhang, Z. Luo, D. Li, H. Ruan, B. H. Ruan, L. Su and H. Xu, Bioorg. Med. Chem. Lett., 2017, 27, 5382–5386 CrossRef CAS PubMed.
- A. Fallarero, M. Skogman, J. Kujala, M. Rajaratnam, V. Moreira, J. Yli-Kauhaluoma and P. Vuorela, Int. J. Mol. Sci., 2013, 14, 12054–12072 CrossRef.
- S. Manner, M. Vahermo, M. E. Skogman, S. Krogerus, P. M. Vuorela, J. Yli-Kauhaluoma, A. Fallarero and V. M. Moreira, Eur. J. Med. Chem., 2015, 102, 68–79 CrossRef CAS.
- I. Neto, E. M. Domínguez-Martín, E. Ntungwe, C. P. Reis, M. Pesic, C. Faustino and P. Rijo, Pharmaceutics, 2021, 13, 825 CrossRef CAS PubMed.
- W.-M. Zhang, T. Yang, X.-Y. Pan, X.-L. Liu, H.-X. Lin, Z.-B. Gao, C.-G. Yang and Y.-M. Cui, Eur. J. Med. Chem., 2017, 127, 917–927 CrossRef CAS.
- M.-L. Liu, X.-Y. Pan, T. Yang, W.-M. Zhang, T.-Q. Wang, H.-Y. Wang, H.-X. Lin, C.-G. Yang and Y.-M. Cui, Bioorg. Med. Chem. Lett., 2016, 26, 5492–5496 CrossRef CAS.
- E. Buommino, A. Vollaro, F. P. Nocera, F. Lembo, M. DellaGreca, L. De Martino and M. R. Catania, Antibiotics, 2021, 10, 80 CrossRef CAS PubMed.
- B. Sadowska, Ł. Kuźma, B. Micota, A. Budzyńska, H. Wysokińska, A. Kłys, M. Więckowska-Szakiel and B. Różalska, Microb. Pathog., 2016, 98, 132–139 CrossRef CAS PubMed.
- Ł. Kuźma, M. Kaiser and H. Wysokińska, Prep. Biochem. Biotechnol., 2017, 47, 58–66 CrossRef.
- J. Búfalo, C. Cantrell, M. Jacob, K. Schrader, B. Tekwani, T. Kustova, A. Ali and C. Boaro, Planta Med., 2015, 82, 131–137 CrossRef.
- R. Mothana, M. Al-Said, N. Al-Musayeib, A. Gamal, S. Al-Massarani, A. Al-Rehaily, M. Abdulkader and L. Maes, Int. J. Mol. Sci., 2014, 15, 8360–8371 CrossRef CAS.
- C. B. Naman, A. D. Gromovsky, C. M. Vela, J. N. Fletcher, G. Gupta, S. Varikuti, X. Zhu, E. M. Zywot, H. Chai, K. A. Werbovetz, A. R. Satoskar and A. D. Kinghorn, J. Nat. Prod., 2016, 79, 598–606 CrossRef CAS.
- K. O. Ndjoubi, R. Sharma, J. A. Badmus, A. Jacobs, A. Jordaan, J. Marnewick, D. F. Warner and A. A. Hussein, Plants, 2021, 10, 175 CrossRef CAS PubMed.
- R. Nzogong, B. Nganou, A. Tedonkeu, M. Awouafack, M. Tene, T. Ito, P. Tane and H. Morita, Planta Med., 2018, 84, 59–64 CrossRef CAS.
- J. Boonsombat, C. Mahidol, P. Chawengrum, N. Reuk-Ngam, N. Chimnoi, S. Techasakul, S. Ruchirawat and S. Thongnest, Phytochemistry, 2017, 143, 36–44 CrossRef CAS PubMed.
- P. Rijo, A. Duarte, A. P. Francisco, T. Semedo-Lemsaddek and M. F. Simões, Phytother. Res., 2014, 28, 76–81 CrossRef CAS.
- J.-J. Cui, W.-J. Li, C.-L. Wang, Y.-Q. Huang, W. Lin, B. Zhou and J.-M. Yue, Phytochemistry, 2022, 201, 113278 CrossRef CAS PubMed.
- S. Zare, G. Hatam, O. Firuzi, A. Bagheri, J. N. Chandran, B. Schneider, C. Paetz, S. Pirhadi and A. R. Jassbi, J. Mol. Struct., 2021, 1228, 129447 CrossRef CAS.
- M. Tabefam, M. Farimani, O. Danton, J. Ramseyer, M. Kaiser, S. Ebrahimi, P. Salehi, H. Batooli, O. Potterat and M. Hamburger, Planta Med., 2018, 84, 913–919 CrossRef CAS PubMed.
- M. A. González, J. Clark, M. Connelly and F. Rivas, Bioorg. Med. Chem. Lett., 2014, 24, 5234–5237 CrossRef PubMed.
- S. Ebrahimi, S. Zimmermann, J. Zaugg, M. Smiesko, R. Brun and M. Hamburger, Planta Med., 2013, 79, 150–156 CAS.
- T. A. Mokoka, X. K. Peter, G. Fouche, N. Moodley, M. Adams, M. Hamburger, M. Kaiser, R. Brun, V. Maharaj and N. Koorbanally, S. Afr. J. Bot., 2014, 90, 93–95 CrossRef CAS.
- M. M. Farimani, B. Khodaei, H. Moradi, A. Aliabadi, S. N. Ebrahimi, M. De Mieri, M. Kaiser and M. Hamburger, J. Nat. Prod., 2018, 81, 1384–1390 CrossRef CAS.
- J. Obegi Matundura, J. O. Midiwo, A. Yenesew, L. K. Omosa, M. Kumarihamy, J. Zhao, M. Wang, S. Tripathi, S. Khan, V. M. Masila, V.-A. Nchiozem-Ngnitedem and I. Muhammad, Nat. Prod. Res., 2023, 37, 4008–4012 CrossRef CAS.
- C. Bustos-Brito, P. Joseph-Nathan, E. Burgueño-Tapia, D. Martínez-Otero, A. Nieto-Camacho, F. Calzada, L. Yépez-Mulia, B. Esquivel and L. Quijano, J. Nat. Prod., 2019, 82, 1207–1216 CrossRef CAS.
- W. Wang and L.-B. Dong, J. Asian Nat. Prod. Res., 2023, 25, 68–74 CrossRef CAS PubMed.
- D.-W. Li, X.-P. Deng, X. He, X.-Y. Han, Y.-F. Ma, H.-L. Huang, Z.-L. Yu, L. Feng, C. Wang and X.-C. Ma, Phytochemistry, 2021, 183, 112593 CrossRef CAS PubMed.
- C.-J. Wang, Q.-L. Yan, Y.-F. Ma, C.-P. Sun, C.-M. Chen, X.-G. Tian, X.-Y. Han, C. Wang, S. Deng and X.-C. Ma, J. Nat. Prod., 2017, 80, 1248–1254 CrossRef CAS PubMed.
- H. Li, P. Yang, E.-H. Zhang, L.-M. Kong and C.-Y. Meng, J. Asian Nat. Prod. Res., 2021, 23, 652–659 CrossRef CAS.
- M.-M. Xu, J. Zhou, L. Zeng, J. Xu, M. M. Onakpa, J.-A. Duan, C.-T. Che, H. Bi and M. Zhao, Org. Chem. Front., 2021, 8, 3014–3022 RSC.
- X. Wang, K. Sun and B. Wang, Chem. Biodiversity, 2018, 15, 7 Search PubMed.
- H.-B. Yu, X.-L. Wang, W.-H. Xu, Y.-X. Zhang, Y.-S. Qian, J.-P. Zhang, X.-L. Lu and X.-Y. Liu, Mar. Drugs, 2018, 16, 284 CrossRef PubMed.
- S.-H. Wu, J. He, X.-N. Li, R. Huang, F. Song, Y.-W. Chen and C.-P. Miao, Phytochemistry, 2014, 105, 197–204 CrossRef CAS.
- M. Nogueira, F. Da Costa, R. Brun, M. Kaiser and T. Schmidt, Molecules, 2016, 21, 1237 CrossRef.
- T. S. Porto, M. R. Simão, L. Z. Carlos, C. H. G. Martins, N. A. J. C. Furtado, S. Said, N. S. Arakawa, R. A. dos Santos, R. C. S. Veneziani and S. R. Ambrósio, Phytother. Res., 2013, 27, 1502–1507 CrossRef CAS.
- Q. Favre-Godal, S. Dorsaz, E. F. Queiroz, L. Marcourt, S. N. Ebrahimi, P.-M. Allard, F. Voinesco, M. Hamburger, M. P. Gupta, K. Gindro, D. Sanglard and J.-L. Wolfender, J. Nat. Prod., 2015, 78, 2994–3004 CrossRef CAS.
- J. B. Nvau, S. Alenezi, M. A. Ungogo, I. A. M. Alfayez, M. J. Natto, A. I. Gray, V. A. Ferro, D. G. Watson, H. P. de Koning and J. O. Igoli, Front. Chem., 2020, 8, 574103 CrossRef CAS.
- G. Ma, H. Wu, D. Chen, N. Zhu, Y. Zhu, Z. Sun, P. Li, J. Yang, J. Yuan and X. Xu, J. Nat. Prod., 2015, 78, 2364–2371 CrossRef CAS.
- O. Erharuyi, A. Adhikari, A. Falodun, R. Imad and M. I. Choudhary, Tetrahedron Lett., 2016, 57, 2201–2206 CrossRef CAS.
- E. V. Tret'yakova, G. F. Zakirova, E. V. Salimova, O. S. Kukovinets, V. N. Odinokov and L. V. Parfenova, Med. Chem. Res., 2018, 27, 2199–2213 CrossRef.
- A. B. Schons, J. S. Correa, P. Appelt, D. Meneguzzi, M. A. A. Cunha, C. Bittencourt, H. E. Toma and F. J. Anaissi, Molecules, 2022, 27, 6679 CrossRef CAS PubMed.
- W.-M. Zhang, Y. Yao, T. Yang, X.-Y. Wang, Z.-Y. Zhu, W.-T. Xu, H.-X. Lin, Z.-B. Gao, H. Zhou, C.-G. Yang and Y.-M. Cui, Bioorg. Med. Chem. Lett., 2018, 28, 1943–1948 CrossRef CAS PubMed.
- X. Jin, K. Zhang, H. Chen, T. Miao, S. Wang and W. Gu, J. Chin. Chem. Soc., 2018, 65, 538–547 CrossRef CAS.
- M. Berger, A. Roller and N. Maulide, Eur. J. Med. Chem., 2017, 126, 937–943 CrossRef CAS.
- W. Gu, C. Qiao, S.-F. Wang, Y. Hao and T.-T. Miao, Bioorg. Med. Chem. Lett., 2014, 24, 328–331 CrossRef CAS.
- G. Hassan, N. Forsman, X. Wan, L. Keurulainen, L. M. Bimbo, S. Stehl, F. van Charante, M. Chrubasik, A. S. Prakash, L.-S. Johansson, D. C. Mullen, B. F. Johnston, R. Zimmermann, C. Werner, J. Yli-Kauhaluoma, T. Coenye, P. E. J. Saris, M. Österberg and V. M. Moreira, ACS Appl. Bio Mater., 2020, 3, 4095–4108 CrossRef CAS PubMed.
- M. Vahermo, S. Krogerus, A. Nasereddin, M. Kaiser, R. Brun, C. L. Jaffe, J. Yli-Kauhaluoma and V. M. Moreira, Medchemcomm, 2016, 7, 457–463 RSC.
- F. Olmo, J. J. Guardia, C. Marin, I. Messouri, M. J. Rosales, K. Urbanová, I. Chayboun, R. Chahboun, E. J. Alvarez-Manzaneda and M. Sánchez-Moreno, Eur. J. Med. Chem., 2015, 89, 683–690 CrossRef CAS.
- D. Hamulić, M. Stadler, S. Hering, J. M. Padrón, R. Bassett, F. Rivas, M. A. Loza-Mejía, M. A. Dea-Ayuela and M. A. González-Cardenete, J. Nat. Prod., 2019, 82, 823–831 CrossRef.
- B. Zapata, M. Rojas, L. Betancur-Galvis, A. C. Mesa-Arango, D. Pérez-Guaita and M. A. González, Medchemcomm, 2013, 4, 1239 RSC.
- M. A. González-Cardenete, D. Hamulić, F. J. Miquel-Leal, N. González-Zapata, O. J. Jimenez-Jarava, Y. M. Brand, L. C. Restrepo-Mendez, M. Martinez-Gutierrez, L. A. Betancur-Galvis and M. L. Marín, J. Nat. Prod., 2022, 85, 2044–2051 CrossRef.
- V. C. Roa-Linares, Y. M. Brand, L. S. Agudelo-Gomez, V. Tangarife-Castaño, L. A. Betancur-Galvis, J. C. Gallego-Gomez and M. A. González, Eur. J. Med. Chem., 2016, 108, 79–88 CrossRef CAS PubMed.
- G. Hassan, N. Forsman, X. Wan, L. Keurulainen, L. M. Bimbo, L.-S. Johansson, N. Sipari, J. Yli-Kauhaluoma, R. Zimmermann, S. Stehl, C. Werner, P. E. J. Saris, M. Österberg and V. M. Moreira, ACS Sustain. Chem. Eng., 2019, 7, 5002–5009 CrossRef CAS.
- M. Pirttimaa, A. Nasereddin, D. Kopelyanskiy, M. Kaiser, J. Yli-Kauhaluoma, K.-M. Oksman-Caldentey, R. Brun, C. L. Jaffe, V. M. Moreira and S. Alakurtti, J. Nat. Prod., 2016, 79, 362–368 CrossRef CAS PubMed.
- M. A. Dea-Ayuela, P. Bilbao-Ramos, F. Bolás-Fernández and M. A. González-Cardenete, Eur. J. Med. Chem., 2016, 121, 445–450 CrossRef CAS.
- E. V. Tret'yakova, E. V. Salimova and L. V. Parfenova, Nat. Prod. Res., 2022, 36, 79–86 CrossRef PubMed.
- E. V. Tretyakova, X. Ma, O. B. Kazakova, A. A. Shtro, G. D. Petukhova, A. A. Smirnova, H. Xu and S. Xiao, Nat. Prod. Res., 2023, 37, 1954–1960 CrossRef CAS PubMed.
- World Intellectual Property Organization, WO pat. 2016/051013A1, 2016.
- R. C. S. Veneziani, S. R. Ambrosio and C. H. G. Martins, J. Med. Microbiol., 2014, 63, 1649–1653 CrossRef.
- E. Saruul, T. Murata, E. Selenge, K. Sasaki, F. Yoshizaki and J. Batkhuu, Bioorg. Med. Chem. Lett., 2015, 25, 2555–2558 CrossRef CAS PubMed.
- N. Abdissa, M. Frese and N. Sewald, Molecules, 2017, 22, 1919 CrossRef PubMed.
- S. Alegre-Gómez, P. Sainz, M. Simões, P. Rijo, C. Moiteiro, A. González-Coloma and R. Martínez-Díaz, Planta Med., 2016, 83, 306–311 CrossRef PubMed.
- M. F. Chabán, A. I Antoniou, C. Karagianni, D. Toumpa, M. B. Joray, J. L. Bocco, C. Sola, C. M Athanassopoulos and M. C. Carpinella, Future Med. Chem., 2019, 11, 3109–3124 CrossRef.
- F. T. G. Sousa, C. Nunes, C. M. Romano, E. C. Sabino and M. A. González-Cardenete, Rev. Inst. Med. Trop. Sao Paulo, 2020, 7(62), e97 CrossRef PubMed.
- M. Varbanov, S. Philippot and M. A. González-Cardenete, Viruses, 2023, 15, 1342 CrossRef CAS PubMed.
- D. Saxena, R. Maitra, R. Bormon, M. Czekanska, J. Meiers, A. Titz, S. Verma and S. Chopra, npj Antimicrob. Resist., 2023, 1, 17 CrossRef.
Footnote |
| † All the reviews on diterpenoids by James R. Hanson (1996–2019) are available from the Nat. Prod. Rep. online database. |
| This journal is © The Royal Society of Chemistry 2024 |