
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the total synthesis of bis- and tris-indole alkaloids containing N-heterocyclic linker moieties
Kyra R.
Dvorak
a and
Jetze J.
Tepe
 *b
*b
aMichigan State University, USA. E-mail: dvorakky@msu.edu
bUniversity of Virginia, USA
First published on 26th April 2024
Abstract
The past several years have seen an increase in the discovery and isolation of natural products of the indole alkaloid class. Bis- and tris-indole alkaloids are classes of natural products that have been shown to display diverse, potent biological activities. Of particular interest are bis- and tris-indole alkaloids containing N-heterocyclic linker moieties. It has been reported that more than 85% of biologically active compounds contain one or more heterocyclic moieties; of these, N-heterocycles have been identified as the most prevalent. The goal of this review is to provide a detailed overview of the recent advances in isolation and total synthesis of bis- and tris-indole alkaloids that contain N-heterocyclic linker moieties. The known biological activities of these natural products will also be discussed.
 Kyra R. Dvorak | Kyra Dvorak is currently a graduate student in Dr Jetze Tepe's lab at Michigan State University. Her research interests focus on the synthesis of nitrogen-containing heterocycles and their application in the total synthesis of bis- and tris-indole natural products and 20S proteasome modulation. |
 Jetze J. Tepe | Jetze J. Tepe is currently a professor of chemistry and director of drug discovery at the University of Virginia. Prior to his position at the University of Virginia, he was a professor of chemistry at Michigan State University. His research program is focused on the synthesis and biological evaluations of natural products and derivatives thereof, as leads for drug development. |
1. Introduction
As their name suggests, indole alkaloids are natural products that contain one or more indole structural moieties. They are commonly isolated from a variety of marine sources, including sponges, tunicates, red algae, acorn worms, and symbiotic bacteria, and they represent the largest, and among the most complicated, class of the marine alkaloids.1 Indole alkaloids have been shown to display diverse biological activities, including cytotoxic, antitumor, antiviral, antibacterial, and anti-inflammatory activities.2 The structure, activity, and synthesis of indole alkaloids have been discussed in several reviews over the years.3 The goal of this review is to provide a detailed overview on recent advances in isolation and total synthesis of bis- and tris-indole alkaloid natural products that contain N-heterocyclic linker moieties. These heterocyclic moieties are of particular interest as it has been reported that more than 85% of biologically active compounds contain one or more heterocyclic moieties with N-heterocycles being the most prevalent.4 These heterocyclic moieties are of particular interest as it has been reported that more than 85% of biologically active compounds contain one or more heterocyclic moieties.5 The biological activities of these natural products will also be discussed.2. Bis-indole alkaloids
2.1 Imidazole and imidazoline linker moieties
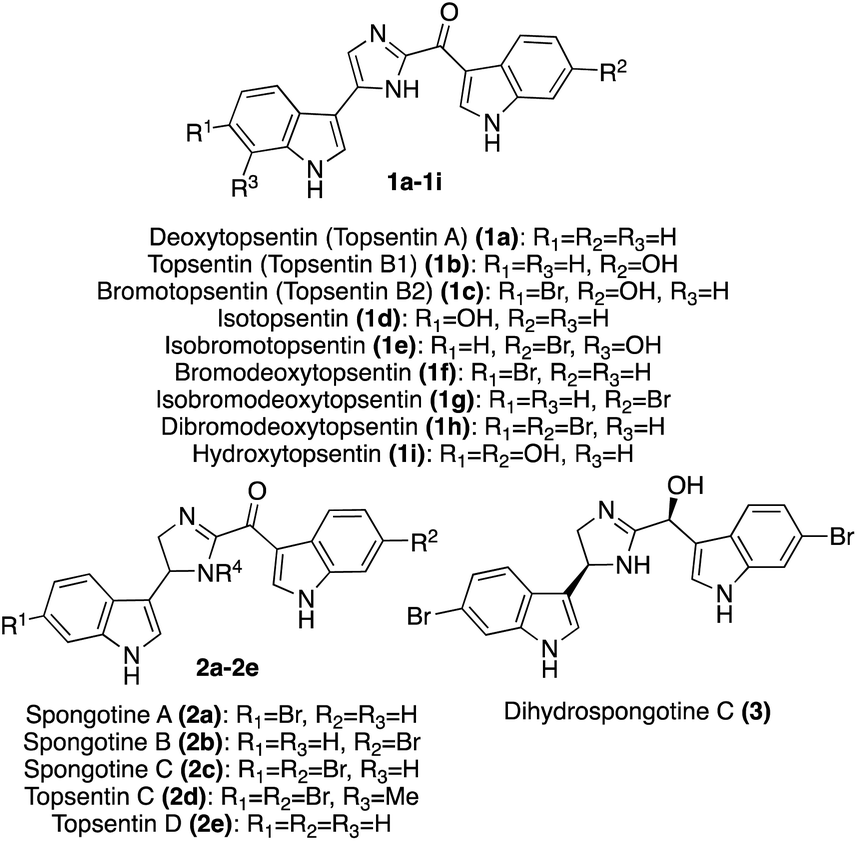 | ||
| Fig. 1 Chemical structures of Topsentin and Spongotine natural products (1a–h, 2a–e, 3) and the synthetic Topsentin analogue Hydroxytopsentin (1i). | ||
In addition to these previously discussed natural products, the most recently isolated natural product in the Spongotine class was Dihydrospongotine C (3), which was isolated as a single enantiomer in 2017 from the Topsentia sp. marine sponge. Its structure was elucidated via spectroscopic methods. Both stereocenters were determined to have the S-stereochemistry configurations consistent with experimental and calculated circular dichroism (CD) data (MPW1PW91/6-31G(d,p)).13 In addition, 3 displayed antibacterial activity toward S. aureus (MIC: 3.7 μg mL−1), anti-HIV activity (IC50 (YU2): 3.5 μM; IC50 (HxB2): 4.5 μM), and displayed no evidence of cytotoxic activity toward mammalian cells.13 Dihydrospongotine C (3) has yet to be accessed via total synthesis.
The first natural product of the Topsentin and Spongotine classes to be accessed via total synthesis was Deoxytopsentin (Topsentin A) (1a) by Braekman, J., et. al. in 1988.15 As shown in Scheme 1, 3-(bromoacetyl)indole (4) was reacted with 1,1-dimethylhydrazine in acidic conditions to afford the amine salt (5). Then, 5 was refluxed in isopropanol (IPA) to prompt migration of the methyl groups and elimination of dimethylamine to afford the imine intermediate 6, which immediately dimerized to afford 1a in 27% yield from 5.15
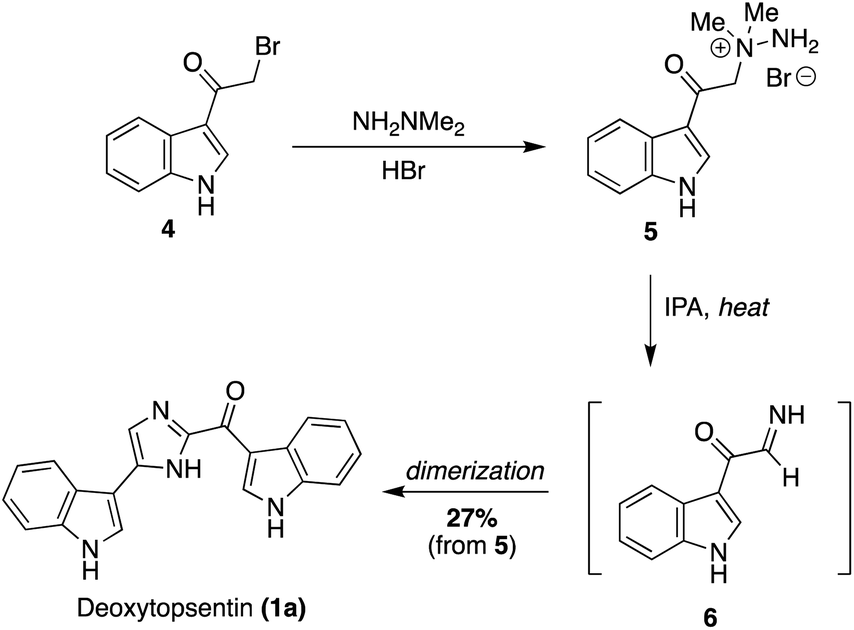 | ||
| Scheme 1 The first total synthesis of Deoxytopsentin (1a). | ||
Additional progress was then made toward the synthesis of the Topsentin natural products 1a–d, and the natural product analogue 1i. The synthetic route began with the synthesis of key indole-3-keto–aldehyde fragments 7a and 7bvia a subsequent oxidative step that was carried out on the indole-3-α-chloro-ketone starting material. The penultimate step of this synthesis was the condensation and cyclization of 7a and 7b to ideally afford Topsentin (1b), as shown in Scheme 2. However, this cyclization proved to be unselective, affording a mixture of 1a (26%), 8b (9%), 8c (10%), and 8d (8%). Each of these intermediates were isolated before undergoing quantitative hydrogenolysis to remove the benzyl group, affording 1b, 1d, and the natural product analogue 1i. The same condensation/cyclization reaction was carried out to dimerize and condense two equivalents of 7a, rather than the mixed keto–aldehyde intermediates, and the desired product 1a was accessed in 63% yield. This emphasized that the selectivity was the major issue with this approach.16 Over the years, additional dimerization-cyclization approaches toward the total synthesis of 1a have also been completed, such as the dimerization of an indolic α-amino-ketone fragment by Miyake et. al.17
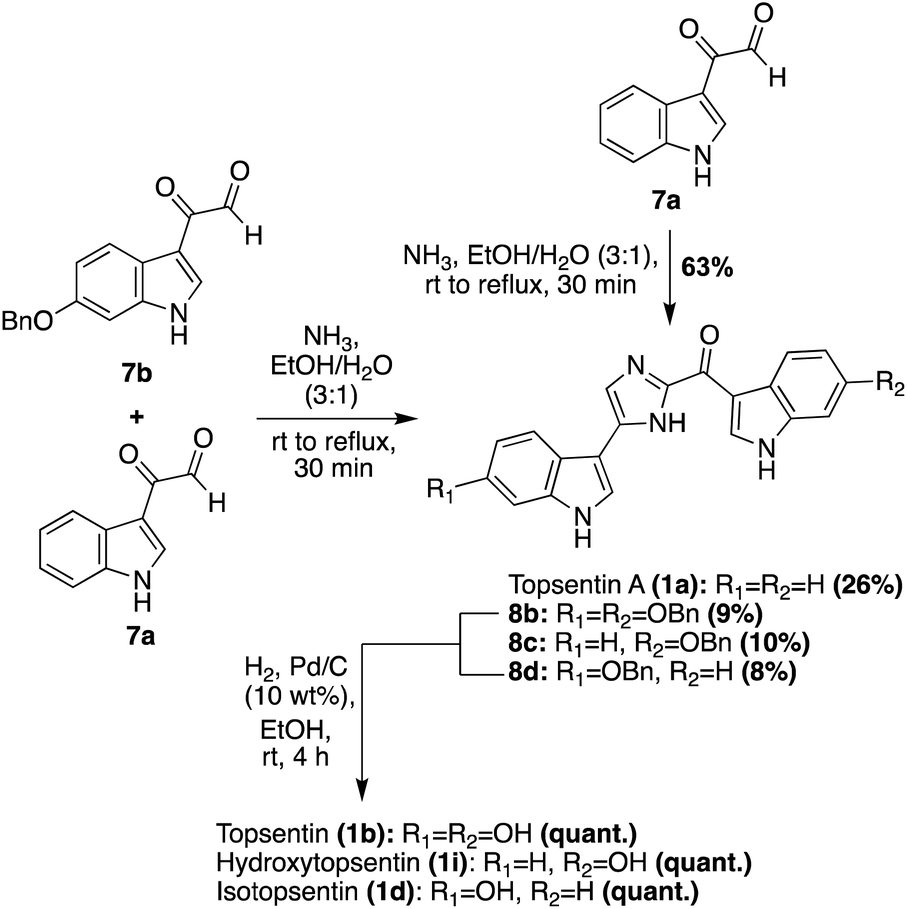 | ||
| Scheme 2 Total syntheses of Topsentins 1a–d, and the natural product analogue 1i. | ||
Considering the selectivity issues of these dimerization-like approaches toward the Topsentin natural products, additional synthetic approaches toward these scaffolds were developed to improve this selectivity, such as the use of lithiation and subsequent cross coupling reactions. For example, in this approach, the imidazole core was the starting point, and the indole-3-aldehyde and indole moieties were subsequently added to the imidazole ring (Scheme 3).18,19 There were two lithiation and subsequent nucleophilic addition approaches to access the key imidazoline intermediate (13). In the first synthetic strategy, di-iodo-imidazole (9) was first lithiated with nBuLi, which subsequently underwent nucleophilic addition to indole-3-aldehydes (10) to access 11 in high yields. In the second approach, the electronics of the reaction were reversed in which the 3-iodo-indoles (11) underwent lithium–halogen exchange and were subsequently added to the imidazole-2-aldehydes (12) to afford the key intermediate 13 in good yield. To access the Topsentin natural products 1a–c, and 1f, the alcohol 13 was oxidized to the ketone 14 using MnO2 and coupled with the tributyltin indole 15via the Stille method in high yield. After removal of protecting groups, the desired products 1a–c, and 1f were isolated in 57–92% yield.18,19 This approach proved to be a much more efficient and highly selective approach toward installing two indole fragments on the imidazole core that bear different substituents. Considering the efficiency of cross-coupling, additional cross-coupling approaches toward these natural products were completed.
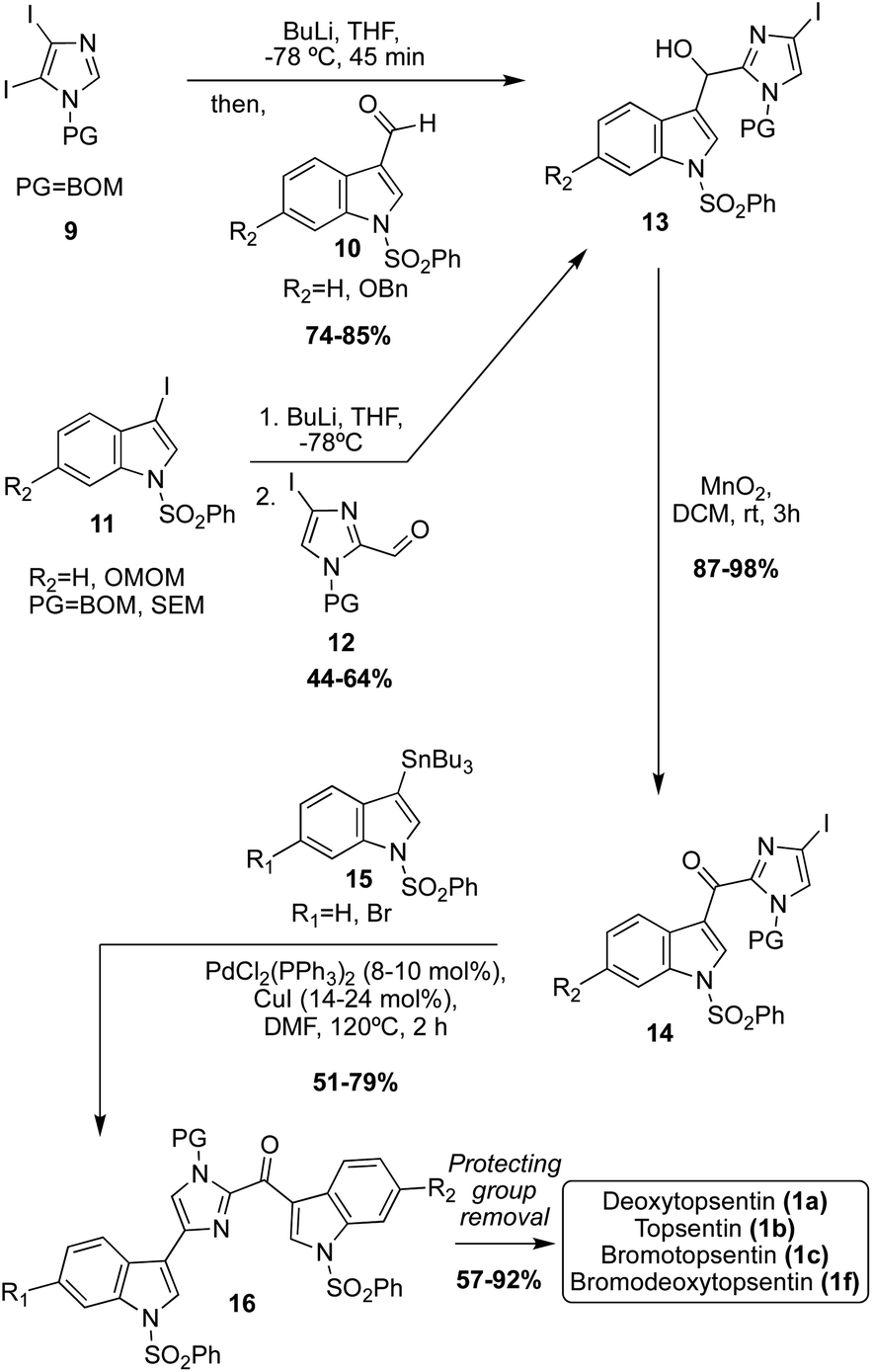 | ||
| Scheme 3 Lithiation and cross-coupling synthetic approach toward 1a–c, and 1f. | ||
For example, Kawasaki et al. coupled a borylated indole 17 with N-SEM-di-iodoimidazole 18 and subsequently de-iodinated to afford the imidazole intermediate 19 in high yields (Scheme 4). A late-stage lithiation of 19 was then carried out and subsequently reacted with the indole-3-amide intermediate 20. Lastly, the protecting groups were removed using BBr3, to render Topsentin (1b) in 69% yield.20
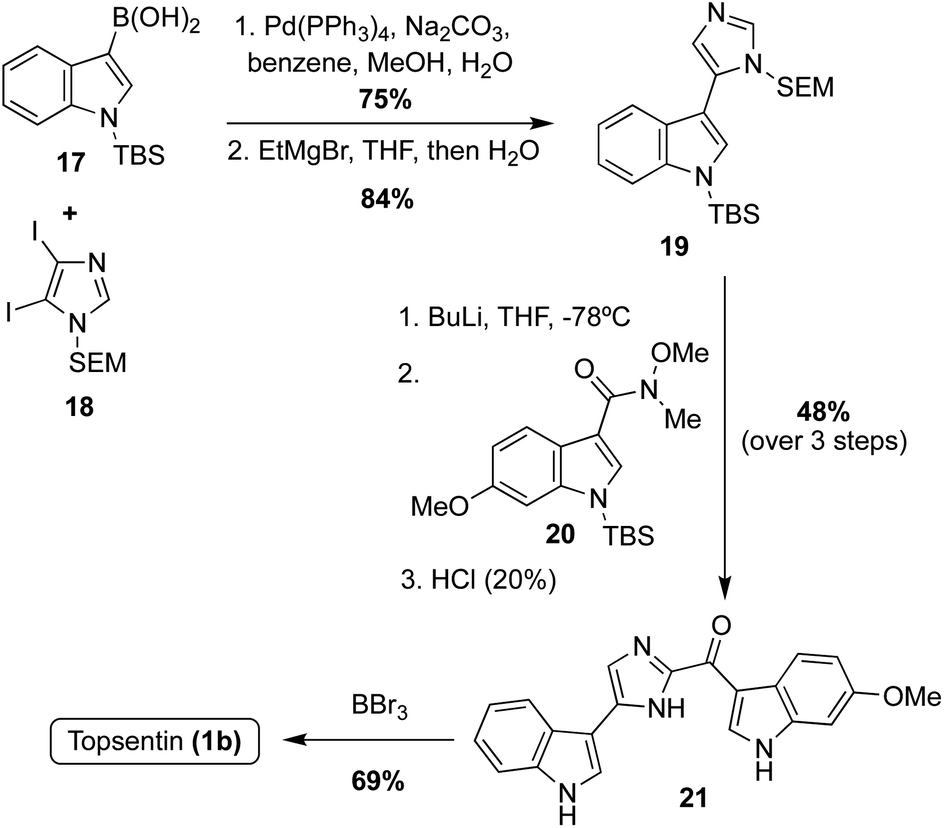 | ||
| Scheme 4 Cross-coupling and late-stage lithiation approach to Topsentin (1b). | ||
The newer isolated Spongotine natural products were accessed via total synthesis more recently. The first total syntheses of several Spongotine and Topsentin natural products were achieved via a common key cyclization approach toward the imidazoline core, in which a keto–thioimidate (25a–c) fragment and diamine (29a–b) fragment were cyclized.21,22 Intermediates 25a–c were synthesized in four steps from commercially available indole starting materials (22a–c) in good yields (Scheme 5). There were two different approaches for accessing the indolic diamine fragments (29a–b). The first approach was used to synthesize the indolic diamine (29a). In this approach, an indolic hydroxylamine intermediate (26a) underwent reductions and hydrogenolysis via hydrogen and Pearlman's catalyst to access the debenzylated intermediate in 90% yield. Then, the Boc protecting group was quantitatively removed under acidic conditions to access 29a.23 The second approach was a little more step-intensive to access the indolic diamine intermediate 29b as it could not be synthesized via the hydrogenative method of 29a due to problematic dehydrohalogenation of indoles under hydrogenative conditions.23 In order to avoid hydrogenation conditions, the 6-bromo-indolic hydroxylamine intermediate 26b was oxidized to 27via MnO2 and subsequently reacted with hydroxylamine hydrochloride to remove the benzyl group and reduced via TiCl3 to achieve 28 in high yields. From there, 28 was subjected to acidic conditions to remove the Boc group and access 29b in 92% yield.22 The desired natural products were then accessed via the key base-catalyzed imidazoline cyclization between 25a–c and 29a–b. This cyclization was carried out with either Et3N or Amberlyst A21 resin to access the desired natural products (2a (74%), 2b (46%),2c (65%), 2e (72%)) and the natural product analogue (2f (63%)) in good yields. Then, the imidazoline cores of 2a–c, 2e, and 2f were oxidized via IBX to the imidazole of Topsentins 1f (78%), 1g (34%), 1h (90%), 1a (91%), and Topsentin analogue 1j (quant). This was the first total synthesis of Spongotine A–C (2a–c), Bromodeoxytopsentin (1f), Isobromodeoxytopsentin (1g), and Dibromodeoxytopsentin (1h).21,22 The natural product analogue 1j was also synthesized and could be de-methylated in the future with BBr3 to access Hydroxytopsentin (Scheme 5).
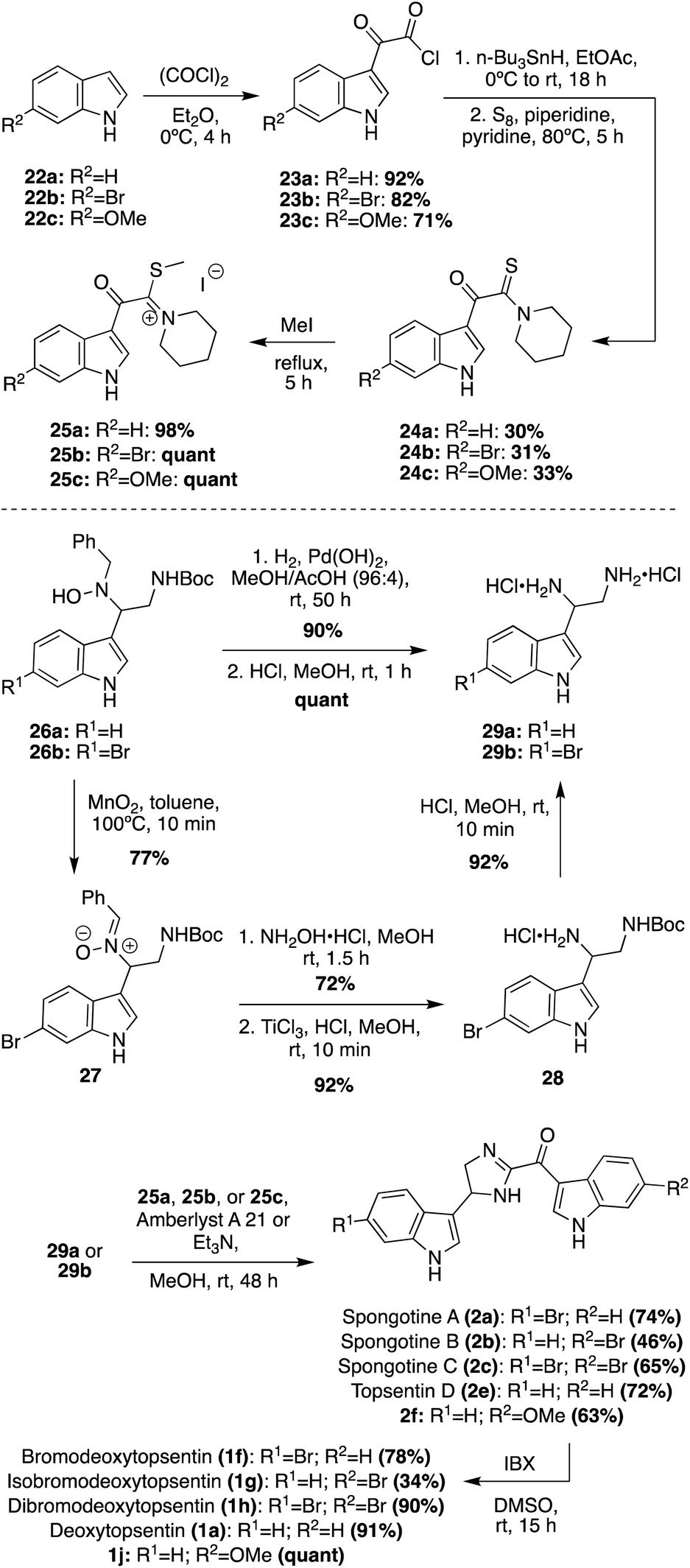 | ||
| Scheme 5 Total syntheses of 1a, 1f–h, 2a–c and 2e. | ||
The first enantioselective total synthesis of Spongotine A (2a) was achieved via a key imidazoline cyclization between a keto–aldehyde fragment (34) and an optically active diamine fragment (33), as shown in Scheme 6.24 First, 34 was synthesized in three steps from 1H-indole, via a keto–acyl chloride intermediate, according to literature procedures.25 Fragment 33 was synthesized via Sharpless dihydroxylation of a 3-vinyl indole intermediate 30 to access (R)-31 in high yield and enantioselectivity. Then, (R)-31 underwent a stereospecific Mitsunobu reaction to access the diazide intermediate (S)-32 in 95% yield, in which the (S)-stereochemistry was set via inversion of the chiral center. The diazide (S)-32 was then reduced to access the optically active indolic diamine intermediate (S)-33 in high yields with 98% ee. To synthesize the imidazoline core and achieve Spongotine A (2a), (S)-33 and 34 underwent condensation, cyclization, and oxidation to the imidazoline via NCS. Subsequent removal of the tosyl protecting group with base achieved Spongotine A (2a) in 51% yield. Through these final steps, the stereochemistry was retained, allowing for the first enantioselective total synthesis of Spongotine A (2a).24 In addition, the specific optical rotation of the synthesized (S)-Spongotine A (2a) matched that of the natural Spongotine A (2a), allowing for the establishment of its previously unknown absolute configuration as (S)-Spongotine A.
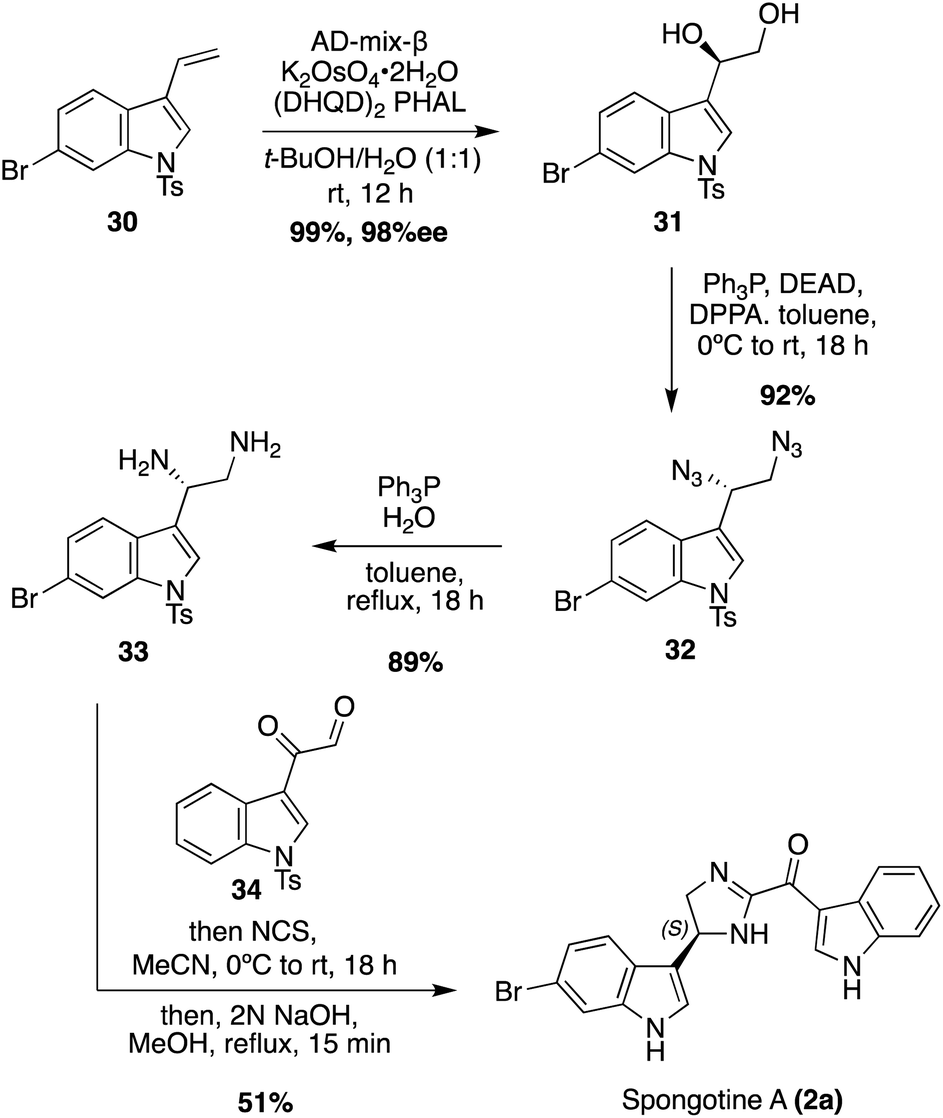 | ||
| Scheme 6 The First enantioselective total synthesis of (S)-Spongotine A (2a). | ||
 | ||
| Fig. 2 Chemical structures of Nortopsentins A–D (35a–c, 36) and Nortopsentin synthetic analogue (35d). | ||
The first total syntheses of Nortopsentins A–C (35a–c), as well as the synthetic analogue 35d, were achieved via a successive Pd-catalyzed cross-couplings of indole fragments to the imidazole core, as shown in Scheme 7.30 The synthetic analogue 35d was the first of these to be accessed via successive cross coupling reactions of a tri-brominated imidazole (38) with an N-TBS-protected indole-3-boronic acid (37a), using Pd(PPh3)4 as a catalyst. The protecting groups and remaining bromide were then removed to access 35d in high yields. During route development, SEM- and MOM-protecting groups were also explored for 37a, yet the TBS-protecting group resulted in the highest yield of the coupling reaction.30 More recently, an adapted method for palladium-catalyzed cross coupling of unprotected imidazoles was developed and utilized in the synthesis of 35d, as shown in Scheme 7.31 This Suzuki–Miyaura cross-coupling method allowed for a significantly expedited approach toward 35d, as compared to previous reports. However, this method is likely only efficient for natural products bearing symmetrical indole moieties. In a similar manner, the first total syntheses of Nortopsentin A (35a) and C (35c) were achieved, as shown in Scheme 7. The efficient synthesis of 35c was very similar to that of 35d. Iodide was utilized as a coupling partner here, rather than bromide. Iodide was likely used here to prevent potential selectivity issues given the bromide substituents on the boronic acid coupling partners. The other major difference was the removal of the additional iodide before the second indole coupling (Scheme 7, synthesis of 35c). It is noteworthy to mention that the selective de-iodination to access intermediate 43 was confirmed via a NOESY correlation between the SEM group on the nitrogen and the C5–H of the imidazole.30 The approach toward the first total synthesis of Nortopsentin A (35a) and Nortopsentin B (35b), which also bear bromide substituents on their indoles, implemented a later stage iodination approach in the synthesis of 46 before the second cross coupling reaction to access 35a and 35b in moderate and good yield, respectively (Scheme 7).30
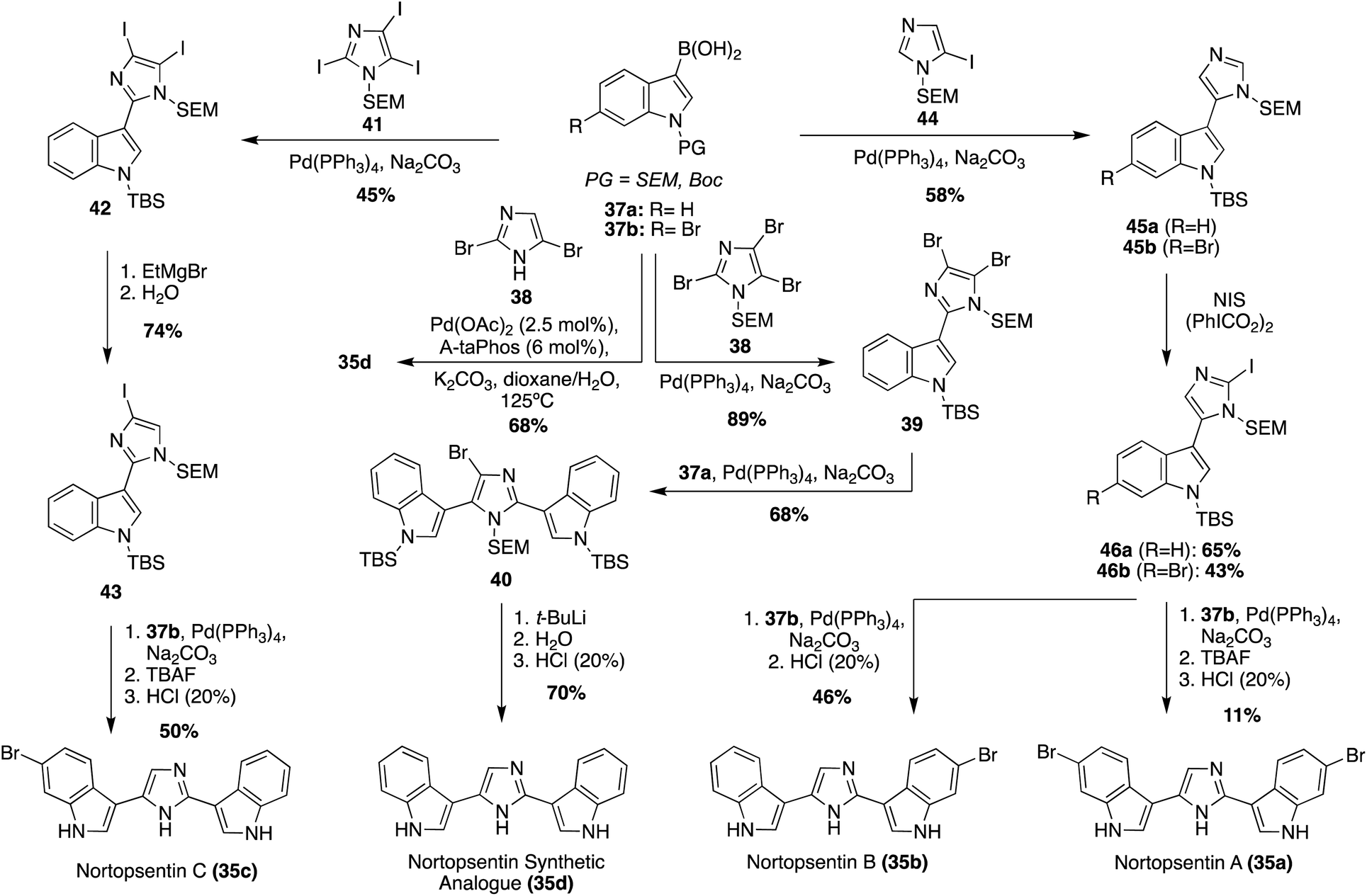 | ||
| Scheme 7 The first total syntheses of Nortopsentin A–C (35a–c), and the Nortopsentin synthetic analogue (35d). | ||
In addition to these cross-coupling approaches, a different non-cross-coupling approach was used to access the Nortopsentin natural products 35b and 35d. This method utilized the condensation of a nitrile with an α-amino-ketone fragment and subsequent cyclization and aromatization of the imidazole at high temperatures to achieve Nortopsentin B (35b) and the Nortopsentin D synthetic analogue 35d in good yields (Scheme 8).17 This method was not viable for the total synthesis of Nortopsentins A (35a) and C (35c). This was likely due to the necessary hydrogenation step for the synthesis of intermediate 48, which would not tolerate a bromide substituent on the indole (48), as has been previously discussed.
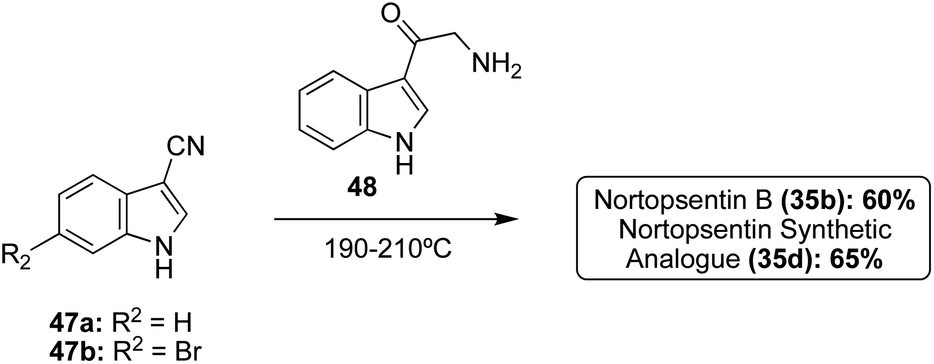 | ||
| Scheme 8 Syntheses of Nortopsentin B (35b) and its synthetic analogue (35d) via thermal condensation–cyclization. | ||
The first total synthesis of the more structurally complex Nortopsentin D (36) was completed by K. Keel, et. al., in 2021, as shown in Scheme 9.32 The complex tri-substituted imidazolinone ((4H)-imidazol-4-one) core was constructed via a late-stage Pinacol-like rearrangement. Two key fragments were utilized in this approach; an alkyne fragment (49) and amidine fragment (51). The alkyne (49) and amidine (51) were accessed in two and five steps, respectively from the commercially available 6-bromo-1H-indole.32 The alkyne fragment (49) was oxidized to the di-ketone intermediate (50) via mercuric nitrate monohydrate. The key condensation–cyclization and subsequent Pinacol-like rearrangement steps were then carried out to form the tri-substituted imidazolinone core of Nortopsentin D (36), accessing 52 in 52% yield. The N-tosyl and N-boc protecting groups were deprotected during this reaction. Precise deprotection was key for optimization, as successful cyclization was dependent upon electronics. It was found that the N-boc protecting group of 51 had to first be deprotected to allow for sufficient nucleophilicity of the amidine (51) to condense and cyclize with the di-ketone (50). The presence of the N-tosyl protecting group of 50 was also found to contribute to increased electrophilicity of the ketone. Therefore, initial acidic conditions were implemented to remove the N-boc group of 51, followed by the strategic use of a mildly nucleophilic base and solvent to allow for cyclization to occur prior to de-tosylation of 50 (Scheme 9). After removal of the remaining protecting group to access the 2-amino-imidazole substituent, Nortopsentin D (36) was accessed in 70% yield.32
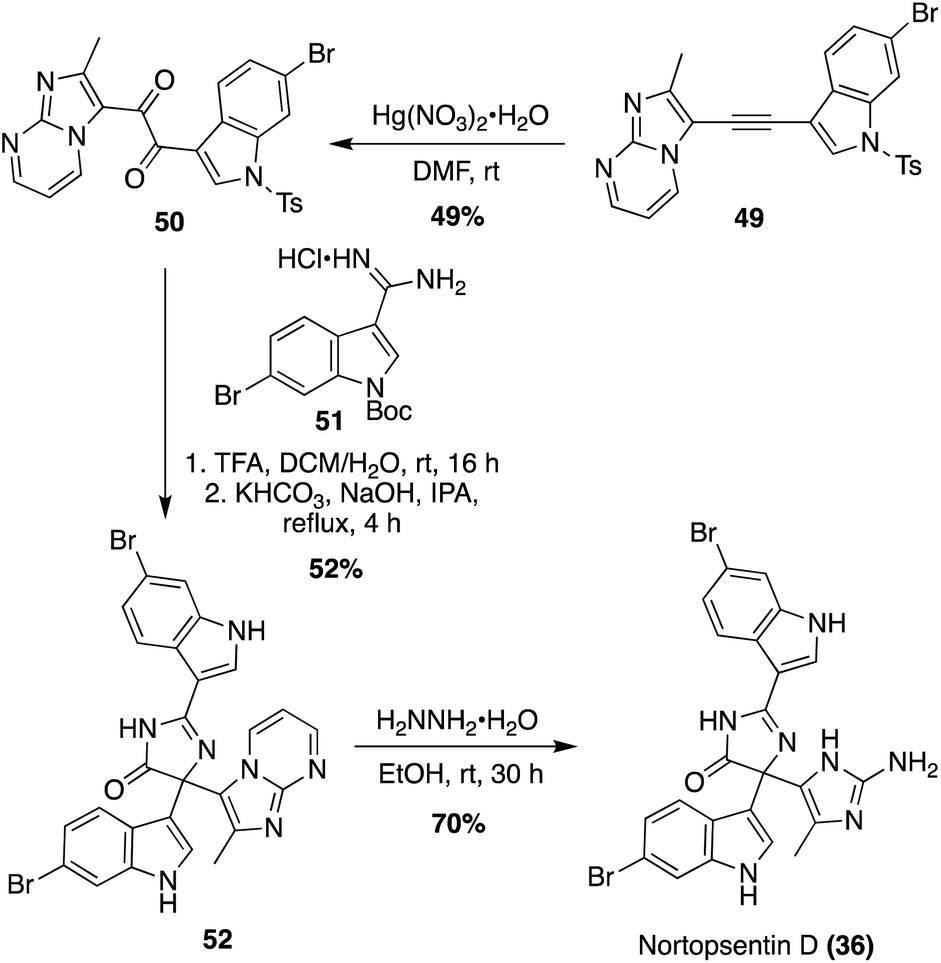 | ||
| Scheme 9 First total synthesis of Nortopsentin D (36). | ||
2.2 Imidazolinone linker moieties
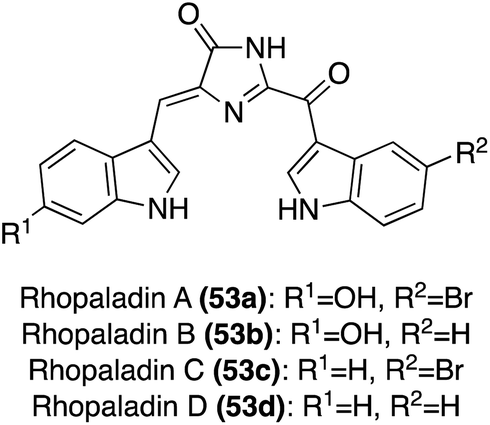 | ||
| Fig. 3 Chemical structure of Rhopaladins A–D (53a–d). | ||
The first total synthesis of Rhopaladin D (53d) was reported in 2000, in which a key intermolecular aza-Wittig reaction was utilized, followed by subsequent condensation and cyclization to form the imidazolinone core of 53d.34 To perform the aza-Wittig reaction, the azide intermediate (56) was first accessed in three steps in high yields from indolyl-3-aldehyde 54 (Scheme 10). Then, the aza-Wittig reaction was performed on azide intermediate 56 to access intermediate 57, which was immediately condensed and cyclized with keto–acyl chloride 23a to access imidazolinone 58 in 56% yield over two steps. Lastly, the protecting group was removed to afford Rhopaladin D (53d) in 60% yield. However, 58 was isolated as a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 mixture of E/Z isomers and after chromatographic separation, the (Z) isomer isomerized to the (E) isomer upon sunlight irradiation. Therefore, after the last step, Rhopaladin D (53d) was also isolated as a mixture of E/Z isomers.34
:4 mixture of E/Z isomers and after chromatographic separation, the (Z) isomer isomerized to the (E) isomer upon sunlight irradiation. Therefore, after the last step, Rhopaladin D (53d) was also isolated as a mixture of E/Z isomers.34
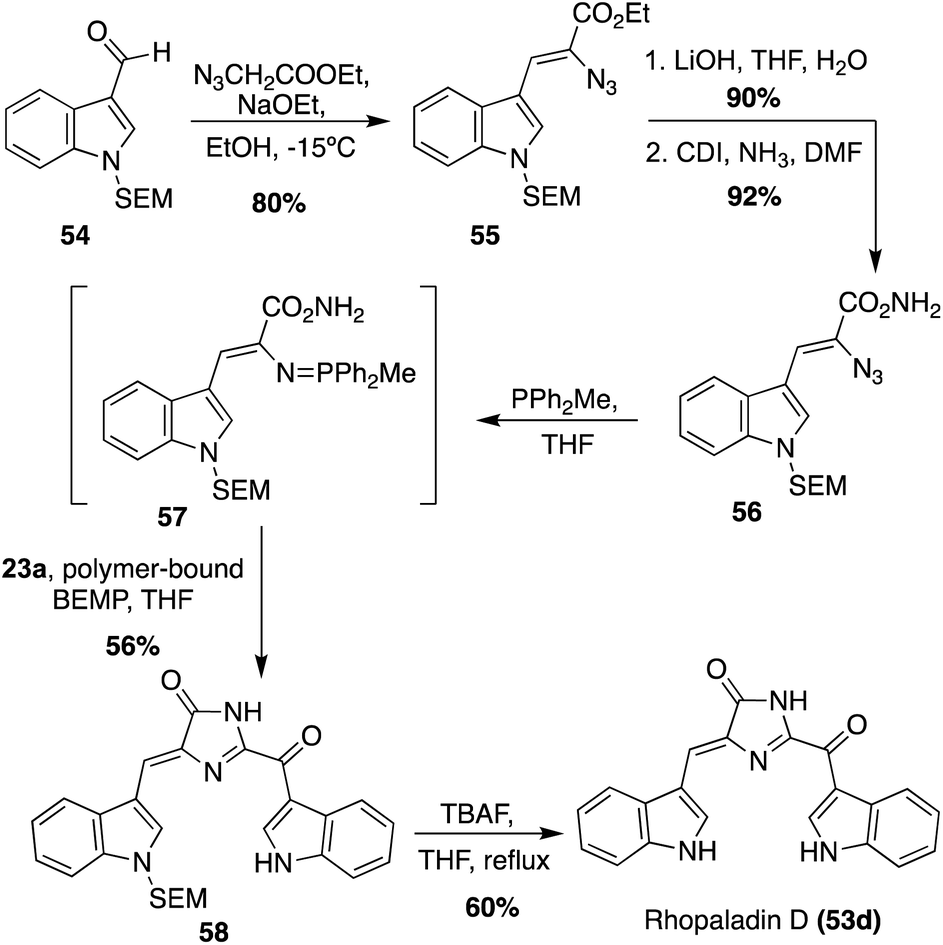 | ||
| Scheme 10 First total synthesis of Rhopaladin D (53d). | ||
A couple years later, the total syntheses of all four Rhopaladins A–D (53a–d) were achieved via a key imidate-based cyclization with tryptophan esters to form the imidazolinone core of these natural products, as shown in Scheme 11.35 First, the carbonyl nitrile intermediate (60a–b) was synthesized via a TMS-cyanohydrin intermediate from aldehyde 59a–b, followed by subsequent oxidation to the carbonyl nitrile 60a–b in high yields. Then, 60a–b underwent a Pinner reaction with gaseous hydrochloric acid and ethanol to form the imidate intermediate (61a–b). Compounds 61a–b were immediately condensed and cyclized with tryptophan methyl ester hydrochloride (62a) in the presence of triethylamine to access Rhopaladin C (53b) and D (53d) in 38% and 35% yield, respectively. Interestingly, the dehydrogenation to form the alkene of the Rhopaladins occurs spontaneously after the cyclization has occurred. This is likely due to the highly conjugated nature of the Rhopaladins, contributing to high stability. Interestingly, this cyclization and spontaneous dehydrogenation specifically produced (Z)-isomers of the Rhopaladins, as was confirmed via NOESY correlations, including a NOESY correlation between the C2–H's of the indole moieties. The presence of the (E)-isomers could not be detected.35 Thus, this method was proven to be very selective to the (Z)-isomer, which is a major advantage compared to the previous synthesis of Rhopaladin D (53d). Rhopaladins A and B (53a and 53b) were synthesized in the same manner as Rhopaladin C and D (53c and 53d), as shown in Scheme 11. The only difference was the identity of the tryptophan methyl ester (62b). In addition, after the key cyclization reaction and spontaneous dehydrogenation to access 63a and 63b, an additional de-methylation step via BBr3 was required to access Rhopaladins A (53a) and B (53b) in 81% and 62% yields, respectively. In addition, similarly to 53c and 53d, the Rhopaladins A (53a) and B (53b) were confirmed with NOESY as the desired (Z)-isomer.35
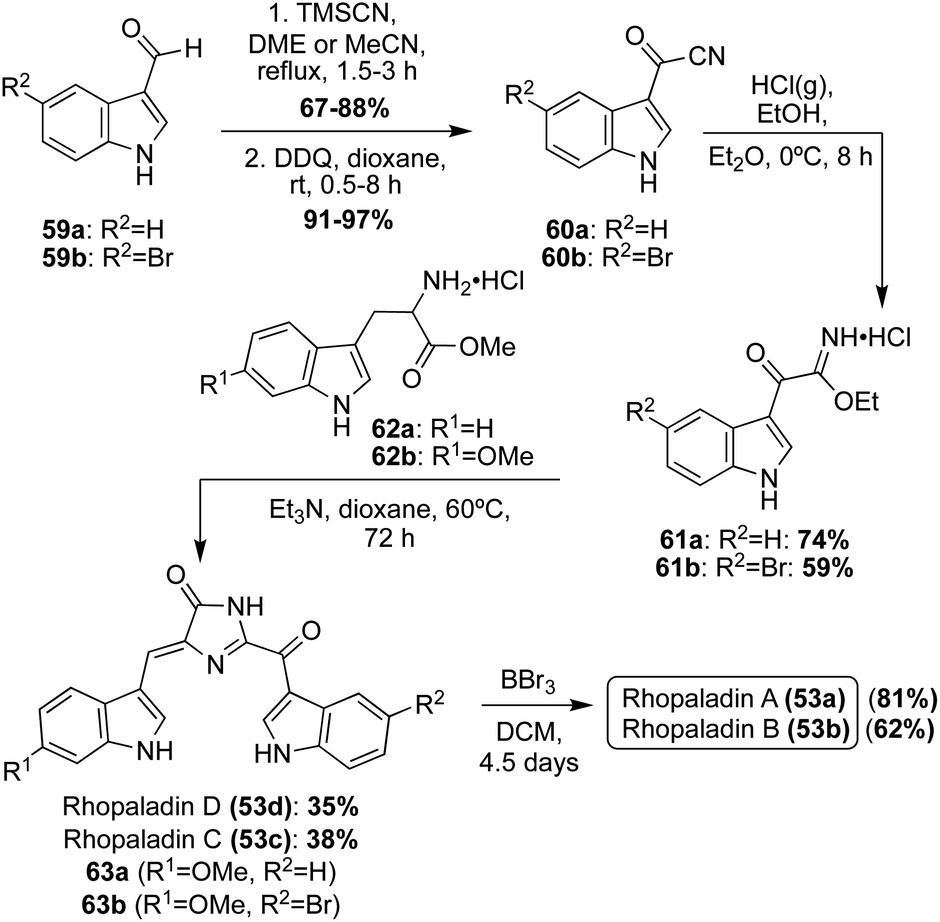 | ||
| Scheme 11 Total syntheses of Rhopaladins A–D (53a–d). | ||
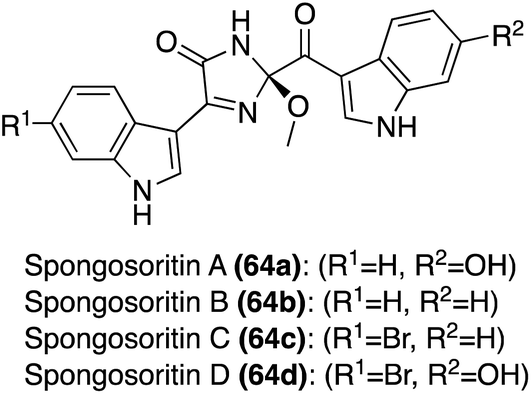 | ||
| Fig. 4 Chemical structures of Spongosoritins A–D (64a–d). | ||
Initial biological activity exploration of the Spongosoritins (64a–d) was conducted and it was found that they exhibited moderate inhibition against transpeptidase sortase A and weak inhibition against human pathogenic bacteria and A549 and K562 cancer cell lines. The biological activity findings are detailed in Table 1, including the known antibiotic, ampicillin, for comparison.36 Though some information is now known, much of the Spongosoritins' biological activities remain elusive. Furthermore, Spongosoritins A–D (64a–d) have yet to be accessed via total synthesis.
| Gram (+) MIC (μg mL−1) | Gram (−) MIC (μg mL−1) | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| # | S. aureus | Enterococcus faecalis | Enterococcus faecium | Klebsiella pneumonia | Salmonella enterica | E. coli | Srt A | A549 | K562 |
| 64a | >128 | >128 | >128 | >128 | >128 | >128 | >329.8 | 77.3 | 24.2 |
| 64b | 64 | >128 | >128 | 128 | 128 | >128 | 62.7 | 55.7 | 28.2 |
| 64c | 32 | 128 | >128 | >128 | 64 | >128 | 43.9 | 61.2 | 37.7 |
| 64d | 16 | 120 | 120 | >128 | 64 | >128 | >274.7 | 70.9 | 54.2 |
| Ampicillin | 0.13 | 0.5 | 1 | — | 0.25 | 8 | |||
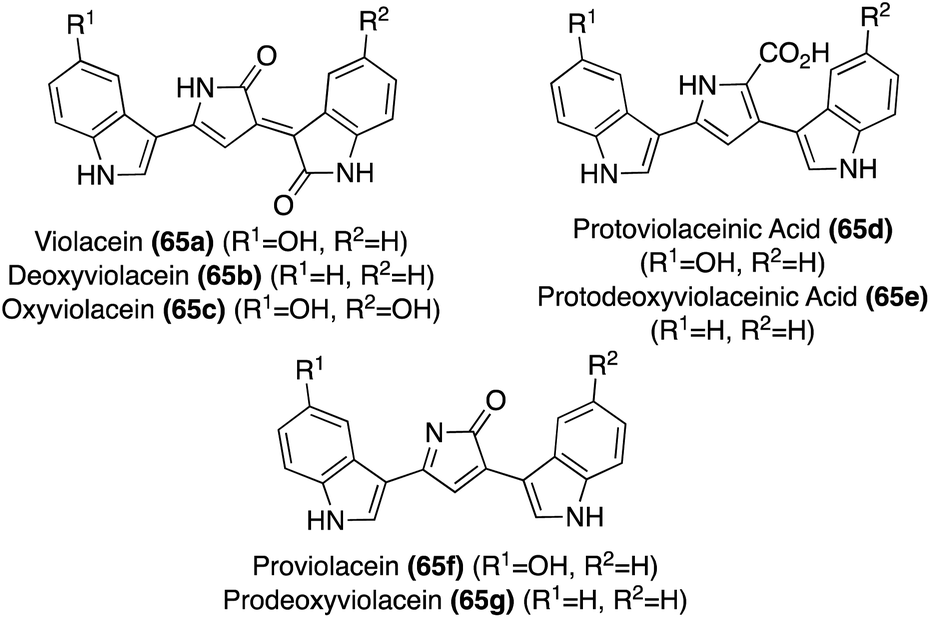 | ||
| Fig. 5 Chemical structures Violacein (65a) and related natural product analogues (65b–g). | ||
Natural products of the Violacein class (65a–g) have been most widely accessed via exploration of biosynthetic pathways.43 However, these biosynthetic pathways fall outside the scope of this review. Progress has also been made toward the total synthesis of these compounds. The first total synthesis of Violacein and Deoxyviolacein was achieved by Ballantine, et. al.45via a key reaction of the lactone intermediates 66a or 66b with ammonia under heating to replace the oxygen atom of the lactone with a nitrogen atom to afford the desired 1,3-dihydro-2H-pyrrol-2-one core of Deoxyviolacein (65b) and imidazolinone intermediate (67) in good yields, as is shown in Scheme 12. Violacein was then accessed after an additional de-methylation step of 67 to complete the first total synthesis of Violacein (65a).45
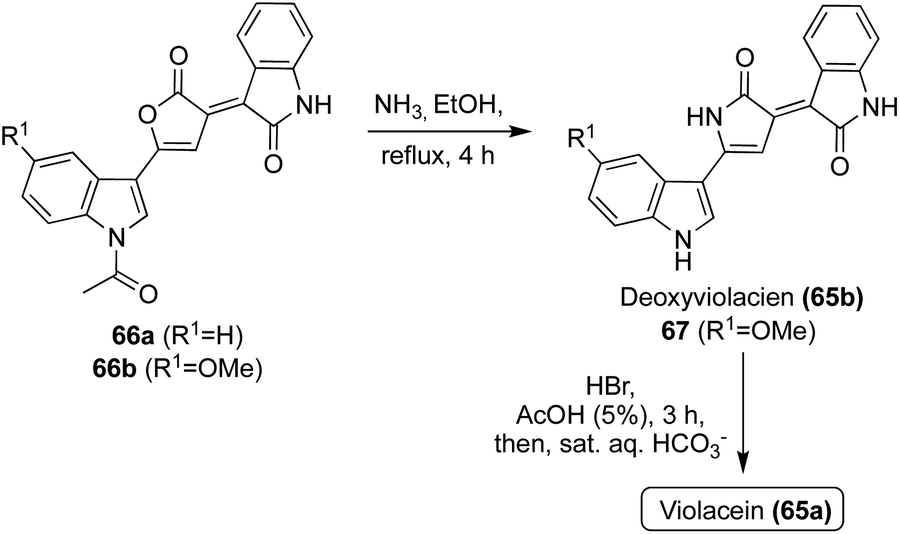 | ||
| Scheme 12 Total syntheses of Violacein (65a) and Deoxyviolacein (65b). | ||
Violacein and Deoxyviolacein were also accessed via total synthesis in 2001, in which Steglich, et. al. implemented a strategy where the two indole groups were attached stepwise to an already formed pyrrolinone core, as shown in Scheme 13.46 First, pyrrolinone 68 underwent an acid-catalyzed reaction with 22a or 22d, and subsequent protection of both the nitrogens via Boc-anhydride, to access intermediates 69a–b in high yields. Then, 69a–b were converted to their corresponding enolates using LiHMDS as the base, followed by subsequent reaction with N-Boc-isatin (70) to afford the aldol condensation intermediates (71a–b). Then, 71a–b were immediately heated to afford Deoxyviolacein (65b) and intermediate 72 in 30% and 24%, respectively. It is noteworthy to mention that upon adsorption to silica gel and heating for 15–20 minutes, dehydration, dehydrogenation, and cleavage of the Boc-protecting groups all occurred simultaneously. Lastly, 72 underwent debenzylation to access Violacein (65a) in 82% yield. The configurations of 65a–b were confirmed as (E)-isomers via NMR.48 This approach was advantageous as it was significantly higher yielding than the first reported total synthesis of these natural products (65a–b).
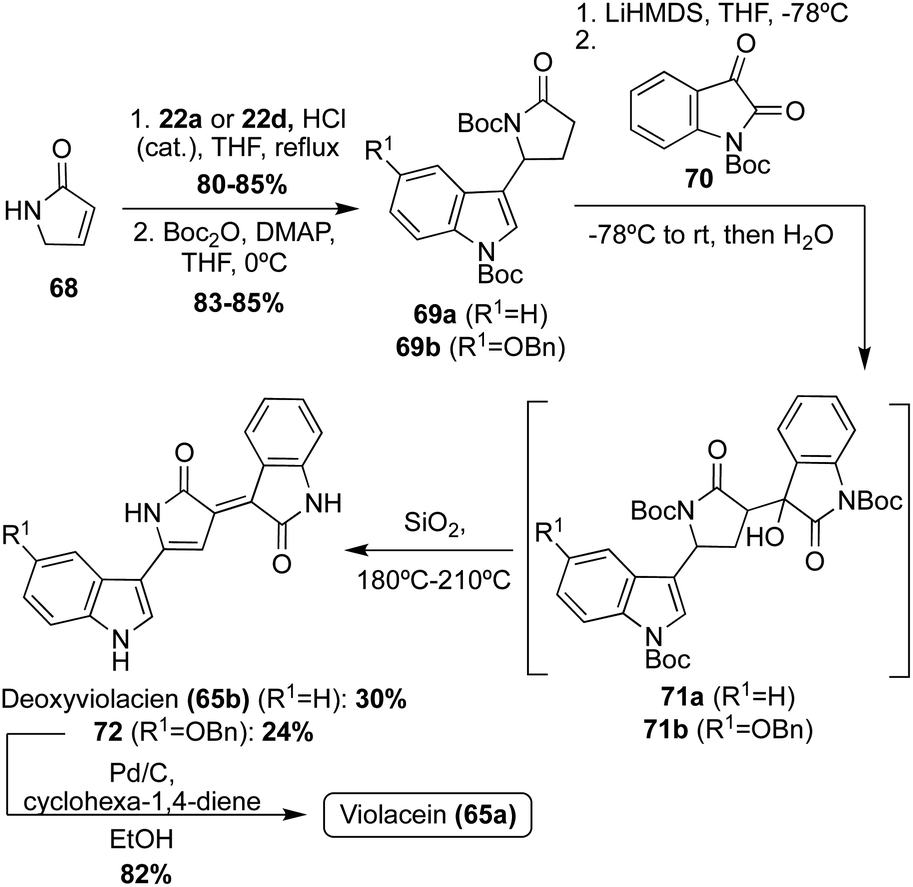 | ||
| Scheme 13 Total syntheses of Violacein (65a) and Deoxyviolacein (65b). | ||
A more recent approach used a tandem ring-closing metathesis (RCM)/isomerization/nucleophilic addition sequence toward the total synthesis of Violacein, as shown in Scheme 14.47 This tandem RCM/isomerization/nucleophilic addition was the first step used in the total synthesis, followed by elimination to intermediate 74 in good yields. The oxindole moiety was then attached via a Ti-catalyzed tautomerization/aldol condensation with isatin to access 75 in high yield. After acid-mediated removal of protecting groups, Violacein (65a) was achieved in 49% yield. The authors mentioned that the final product 65a was very difficult to handle and had very minimal solubility in common solvents. Therefore, it was suspected that the crude yield was significantly higher than the isolated yield.47
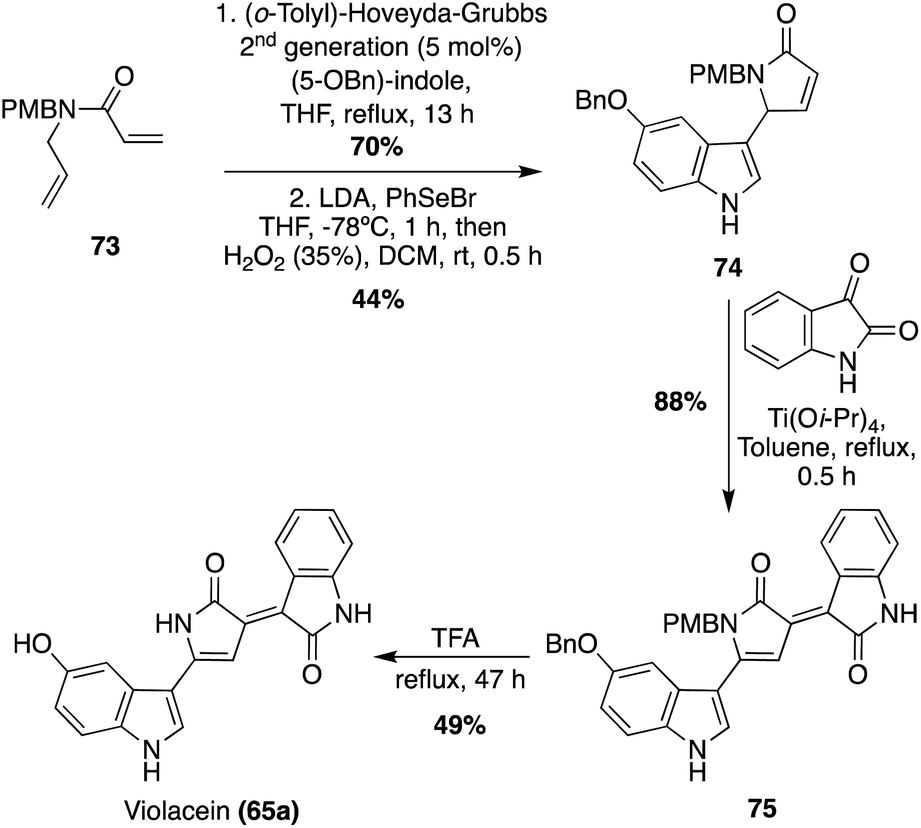 | ||
| Scheme 14 Total synthesis of Violacein (65a) Via a tandem RCM/isomerization/nucleophilic addition sequence. | ||
2.3 Pyrrole and pyrrole-dione linker moieties
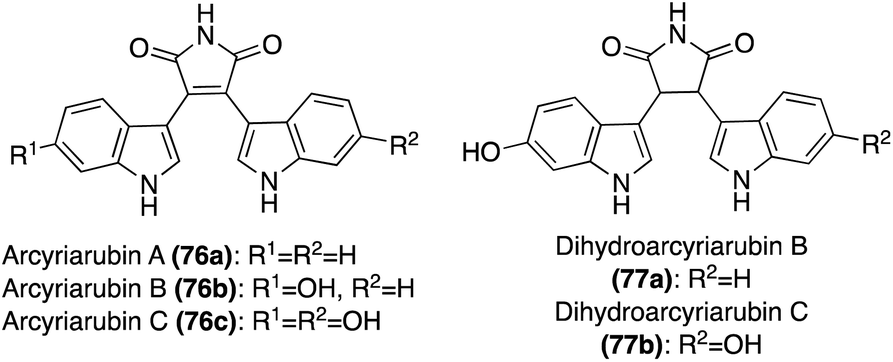 | ||
| Fig. 6 Chemical structures of Arcyriarubins A–C (76a–c) and Dihydroarcyriarubins A–B (77a–b). | ||
In addition, two structurally related natural products that have also been isolated include Dihydroarcyriarubins B (77a) and C (77b). As shown in Fig. 6, these natural products are incredibly similar to the Arcyriarubins (76a–c), however, the reduced pyrrolidine-2,5-dione (succinimide) core of the Dihydroarcyriarubins (77a–b) sets them apart. Dihydroarcyriarubin B (77a) was first isolated by Steglich, et. al. from the slime mold, Arcyria denudate, alongside the Arcyriarubins (76a–b).48 Dihydroarcyriarubin C (77b) was first isolated in 2003 by Ishibashi, et. al. from Arcyria ferruginae, alongside Arcyriarubin C (76b).49 The biological activities of the Dihydroarcyriarubins B (77a) and C (77b) are very similar to that of 76a–c, including antibiotic, antifungal, cytotoxic, and kinase inhibition activities.48,49,52
The first natural products of the Arcyriarubin class to be accessed via total synthesis were Arcyriarubin A (76a) and Arcyriarubin B (76b) in 1988 by Steglich, et. al.53 These total syntheses were accomplished using Grignard reactions to install the indole moieties on the maleimide core, as shown in Scheme 15. In the first total synthesis of Arcyriarubin A (76a), a Grignard reaction was carried out via an indole Grignard reagent and 3,4-dibromo-N-methylmaleimide to access bisindolylmaleimide intermediate (78) in high yield. To remove the methyl group of 78, a maleic anhydride intermediate (79) was accessed and then subsequently reacted with ammonium acetate at high temperatures to access Arcyriarubin A (76a) in high yields.53 Due to the asymmetric nature of the indole moieties in Arcyriarubin B (76b), the synthetic approach to this natural product was slightly different. As shown in Scheme 15, the first total synthesis of 76b was achieved via a reaction of an indole Grignard reagent (77b) with a mono-indolic maleimide intermediate (80) to access intermediate 81 in 89% yield. This approach allowed for the synthesis of a maleimide intermediate with asymmetric indole moieties (81) in a stepwise manner. After de-methylation and removal of protecting groups, Arcyriarubin B (76b) was accessed in high yield.53
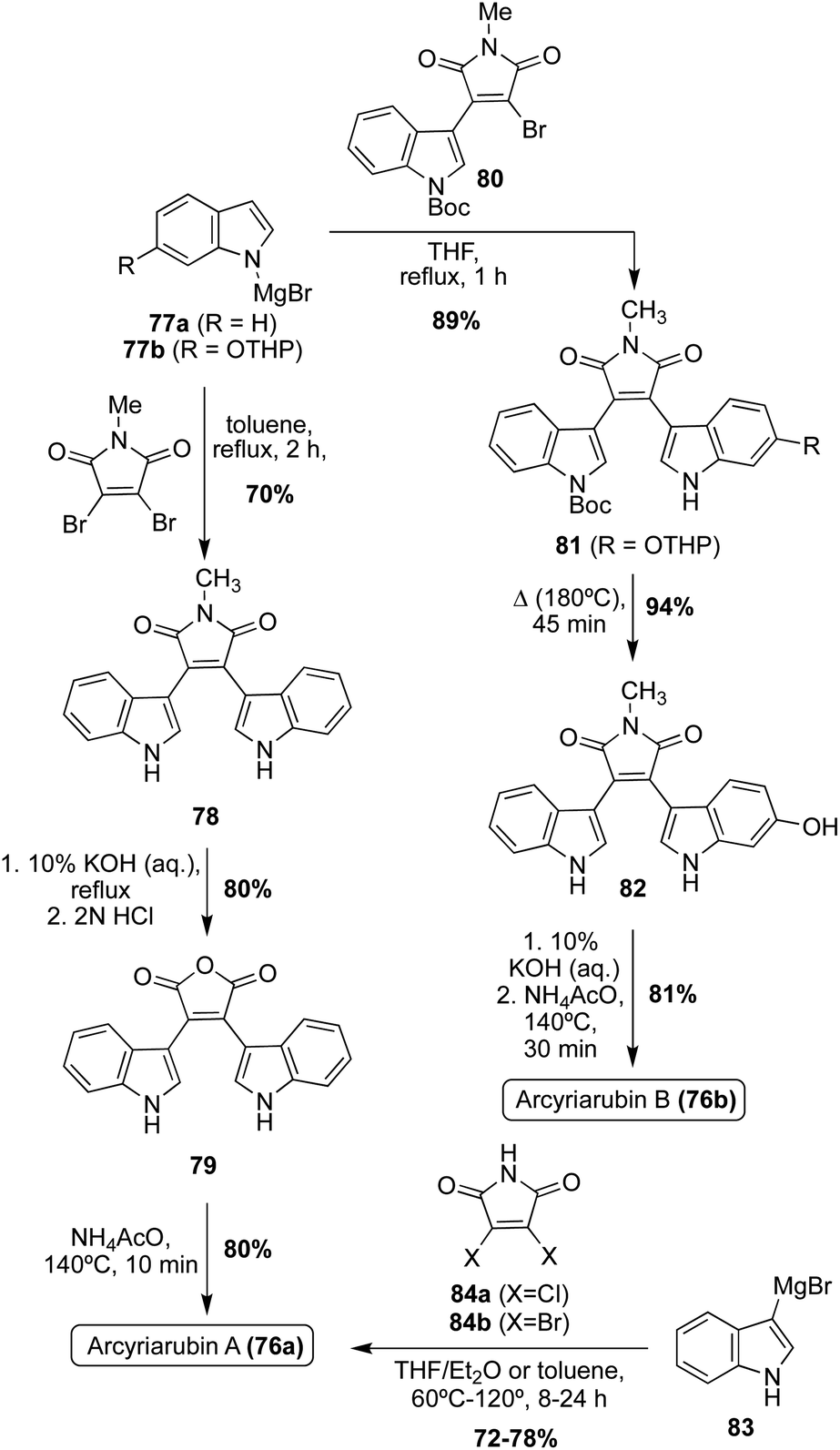 | ||
| Scheme 15 Total synthesis of Arcyriarubins A–B (76a–b). | ||
Several years later, a similar Grignard approach to the total synthesis of Arcyriarubin A (76a) was utilized in 1995 (Scheme 15).54 Here, an indole Grignard (83) was reacted with 3,4-dichloromaleimide (84a) at high temperature to access Arcyriarubin A (76a) in 72% yield. This approach was advantageous in that it allowed for the synthesis of Arcyriarubin A (76a) without the need for protecting groups, which greatly expediting its synthesis.54 However, the intolerance for asymmetric indole moieties in this method makes it unsuitable for application to other Arcyriarubins. This Grignard strategy was again slightly improved upon in 2017 for the total synthesis of Arcyriarubin A (76a), as shown in Scheme 15.55 Utilizing the same indole Grignard (83) and 3,4-dibromomaleimide (84b) at a higher temperature, for a shorter reaction time, Arcyriarubin A (76a) was synthesized in 78% yield.55 It is also noteworthy to mention that Arcyriarubin C (76c) has yet to be accessed via total synthesis.
In addition to this Grignard approach, a more biomimetic approach has also been explored toward the total synthesis of Arcyriarubin A (76a), as shown in Scheme 16. This approach consisted of a base-mediated condensation-cyclization reaction between an indole-3-acetamide intermediate (85) and an indole-3-oxo-acyl chloride intermediate (86a) to access Arcyriarubin A (76a), although in a low 11% yield. Recently, an adapted and improved biomimetic total synthesis of Arcyriarubin A (76a) was developed, as shown in Scheme 16. For this approach, intermediate 85 was condensed and cyclized with an indole-3-oxo-ester intermediate (86b) to access Arcyriarubin A (76a) in an excellent 96% yield.56
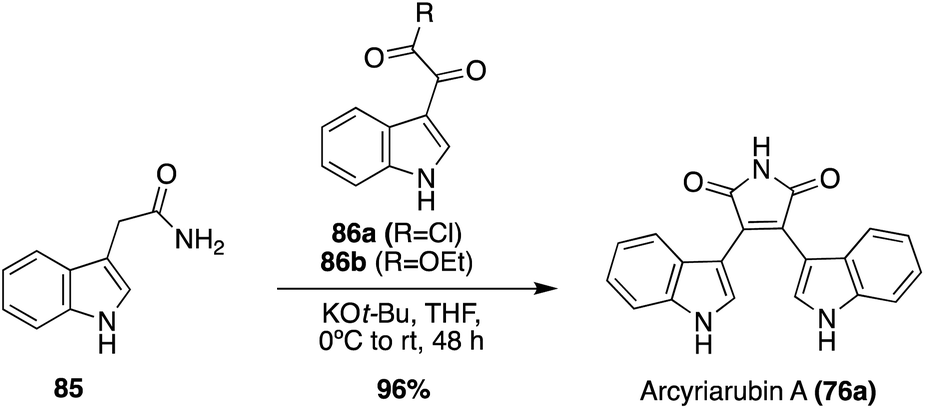 | ||
| Scheme 16 Total synthesis of Arcyriarubin A (76a). | ||
In terms of the Dihydroarcyriarubins (77a–b), Dihydroarcyriarubin B (77a) has yet to be accessed via total synthesis. On the other hand, the first total synthesis of Dihydroarcyriarubin C (77b) was completed in 2007 by Ishibashi, et. al., as shown below in Scheme 17.52 First, the bis-indolyl-N-methylmaleimide intermediate (87) was synthesized in four steps from 3,4-dibromomaleimide and 6-benzyloxyindole, following the same Grignard approach as was previously published by Steglich, et. al.53 To remove the N-methyl group, intermediate 87 was converted to the maleic anhydride intermediate (88), which was subsequently reacted with ammonium acetate at high temperatures to access the bis-indolylmaleimide intermediate (89) in very high yields. Then, 89 underwent hydrogenation to reduce the maleimide core of 89 to the succinimide core of Dihydroarcyriarubin C (77b). In addition to the desired reduction of 89, both benzyl groups were also removed. While this was beneficial for the total synthesis of Dihydroarcyriarubin C (77b); it was also the reason why this approach was not viable for synthesizing Arcyriarubin C (76c). It is noteworthy to mention that at a 12 hours hydrogenation reaction time, cis- Dihydroarcyriarubin C (77b) was accessed in 95% yield, but at a 5 hours reaction time, trans- Dihydroarcyriarubin C (77b) was accessed in 98% yield. In addition, cis- Dihydroarcyriarubin C (77b) could be isomerized to trans-Dihydroarcyriarubin C (77b) either by magnesium in methanol or DBU in tetrahydrofuran in 81% and 93% yield, respectively. To accomplish the first total synthesis of Dihydroarcyriarubin C (77b), both the cis-(77b) and trans-(77b) isomers were synthesized to allow for the stereochemical determination of the isolated Dihydroarcyriarubin C (77b). Comparison of their NMR data allowed for confirmation of the isolated 77b as trans-Dihydroarcyriarubin C (77b).52
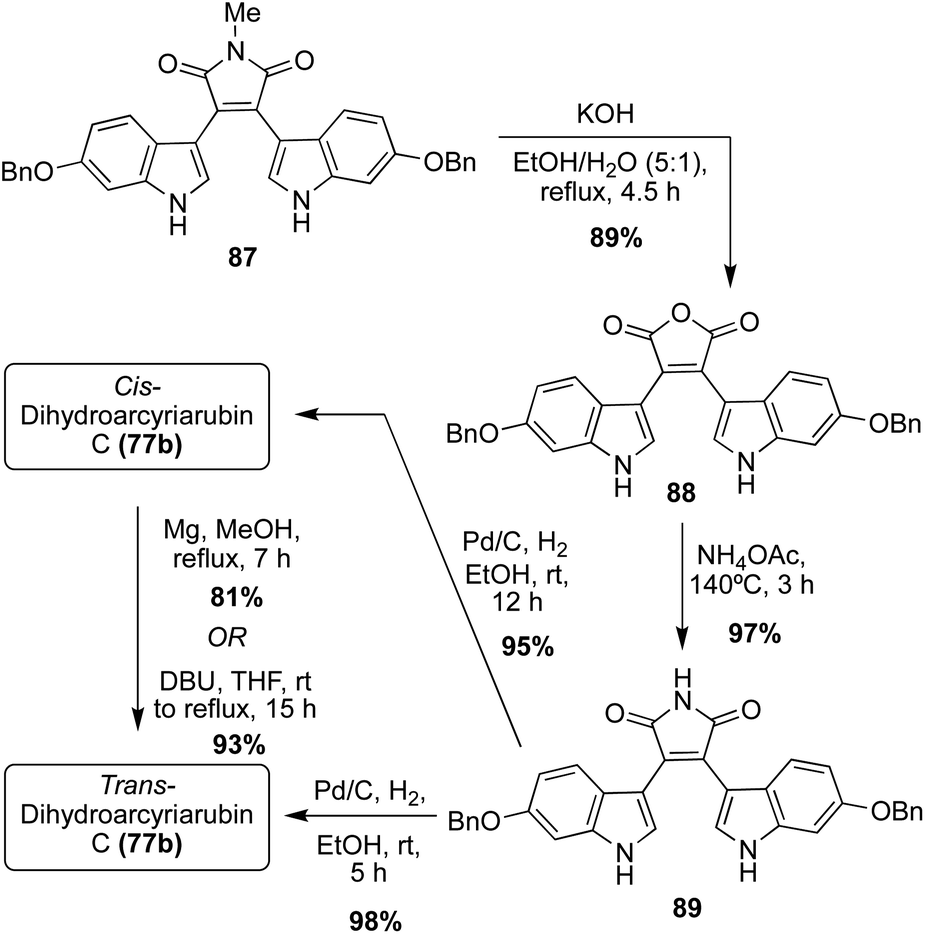 | ||
| Scheme 17 Total synthesis of Dihydroarcyriarubin C (77b). | ||
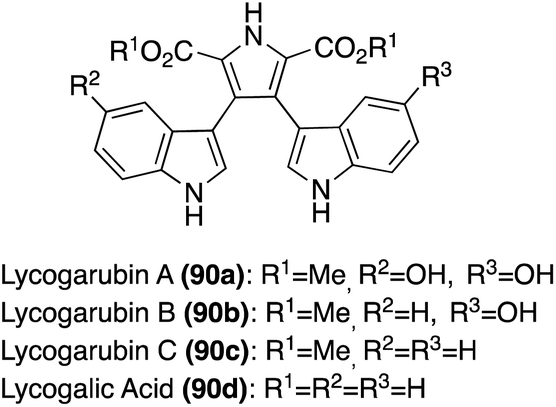 | ||
| Fig. 7 Chemical structures of Lycogarubins A–C (90a–c) and Lycagolic acid (90d). | ||
Many of the early syntheses of the Lycogarubins (90a–c) and Lycagolic acid (90d) were carried out via the exploration of the biosynthetic pathway to these natural products.59 However, the first of these natural products to be accessed via total synthesis was Lycogarubin C (90c) using a palladium-catalyzed Suzuki coupling reaction of dimethyl 3,4-dibromo-1H-pyrrole-2,5-dicarboxylate (91) with an indole-3-boronic acid (92) and subsequent removal of the protecting group via TBAF to access Lycogarubin C (90c) in 81% yield, as shown in Scheme 18.35 This Suzuki method has been implemented in recent literature as well in the synthesis of 90c and similar structures.60 Considering the ongoing study of the biosynthesis of the Lycogarubin natural products, several biomimetic syntheses have been developed as well. One of the earliest of these was completed in 2006 in which a methyl 3-(1H-indol-3-yl)-2- oxopropanoate intermediate (93) underwent a dimerization/cyclization sequence over three steps to access Lycogarubin C (90c) in 42% yield (Scheme 19).59,61
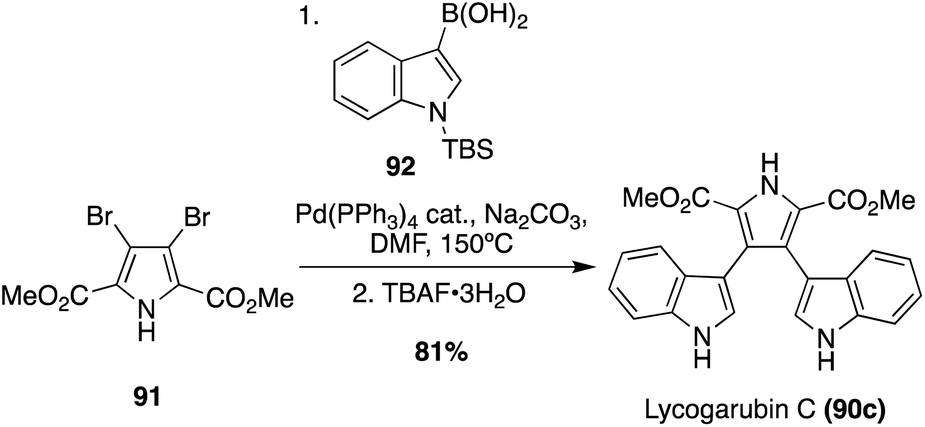 | ||
| Scheme 18 Total synthesis of Lycogarubin C (90c). | ||
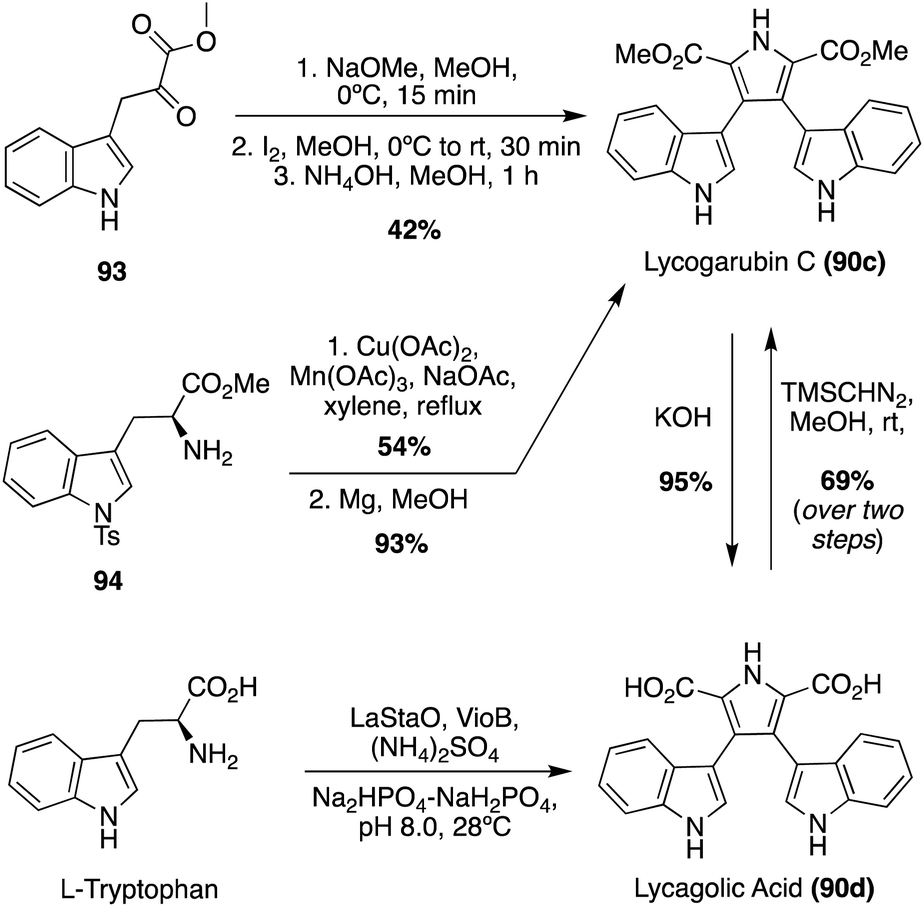 | ||
| Scheme 19 Biomimetic total syntheses of Lycogarubin C (90c) and Lycagolic acid (90d). | ||
Another biomimetic synthesis of Lycogarubin C (90c), as well as Lycagolic acid (90d), was carried out via a Cu/Mn co-oxidized dimerization/cyclization of a Tosyl-tryptophan methyl ester (94) with another molecule of itself (Scheme 19). It is noteworthy to mention that biosynthesis of the Lycogarubins also begins with tryptophan starting materials. After removal of the protecting groups, Lycogarubin C (90c) was accessed in high yields. Lycogarubin C (90c) was then converted to Lycagolic acid (90d) in 95% yield via base-catalyzed ester hydrolysis.62
Recently, a semi-total synthesis of Lycogarubin C (90c) was completed via Lycagolic acid (90d) as an intermediate (Scheme 19). First, enzymatic biological reactions were conducted to accomplish the dimerization/cyclization of the L-Tryptophan (L-Trp) starting material to access Lycagolic acid (90d). Then, 90d was immediately reacted with TMSCHN2 in methanol to access Lycogarubin C (90c) in 69% over two steps from the simple and readily accessible L-Trp starting material.63
Another synthetic method that has been used to synthesize Lycogarubin C (90c) and Lycagolic acid (90d) was the use of key Diels–Alder and Kornfeld–Boger ring contraction reactions. One example utilizing this method, as shown in Scheme 20, began with the synthesis of the di-protected-bis-indole alkene intermediate (97) via a Wittig reaction between the bromide intermediate (95) and the aldehyde intermediate (96). Then, 97 underwent a Diels–Alder reaction with the tetrazine intermediate (98), followed by a subsequent Kornfeld–Boger ring contraction to access the pyrrole intermediate (99) in 54% yield over two steps. After removal of the protecting groups, Lycogarubin C (90c) was accessed in 91% yield. Protection of both indole moieties was necessary to access intermediate 99 in good yields and to avoid side product formation via the Diels–Alder/Kornfeld–Boger reaction sequence.64
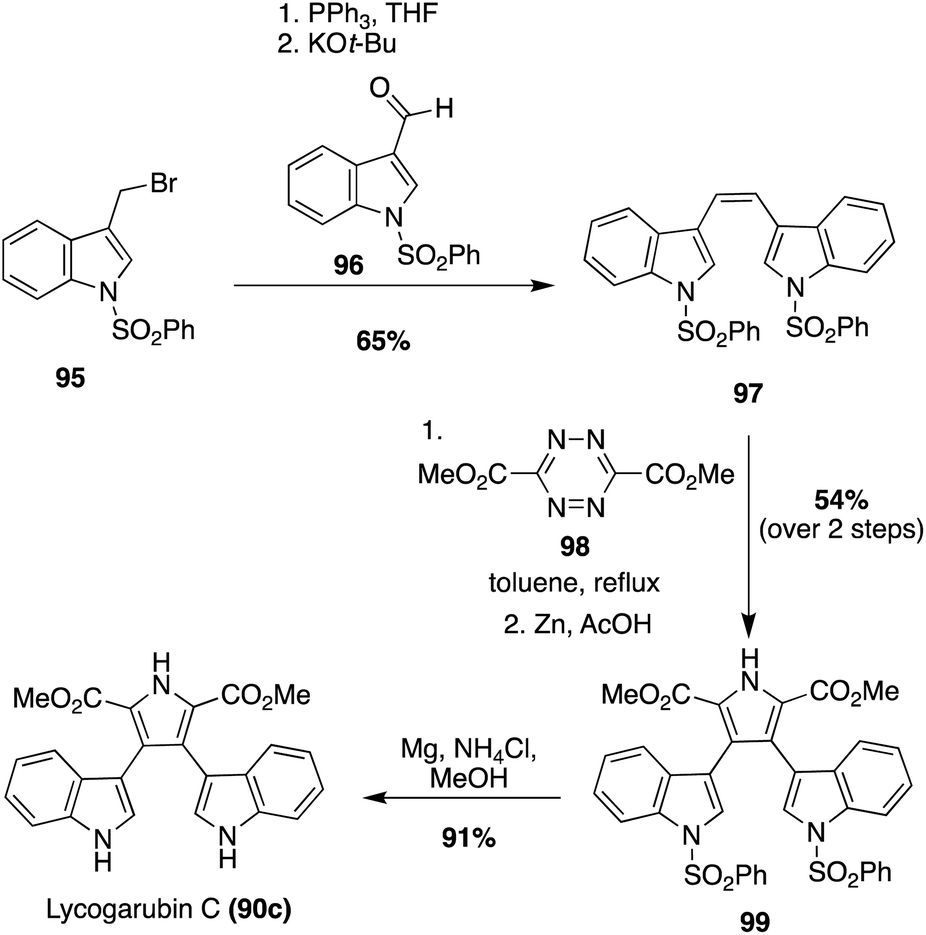 | ||
| Scheme 20 Total synthesis of Lycogarubin C (90c). | ||
Another example in which this method was implemented, as shown in Scheme 21, began with the Diels–Alder reaction between 98 and 1,2-bis-(tributylstannyl)-acetylene to access diazine intermediate (100) in very high yield. Stille coupling of 100 with the 3-iodo-indole intermediate (101) and a subsequent Kornfeld–Boger ring contraction of the diazine core led to the desired pyrrole core of 103 in good yields. Then, 103 could be subjected to LiOH or KOH to access Lycogarubin C (90c) or Lycagolic acid (90d), respectively, in very high yields. Furthermore, Lycogarubin C (90c) could also be converted to Lycogolic acid (90d) via KOH in very high yields.65 This approach (Scheme 21) is favorable in comparison to the previously discussed Diels–Alder/Kornfeld Boger approach (Scheme 20), as it not only allowed for the total synthesis of Lycogarubin C (90c) in good yields, but also allowed for the total synthesis of Lycagolic acid (90d) in good yields.
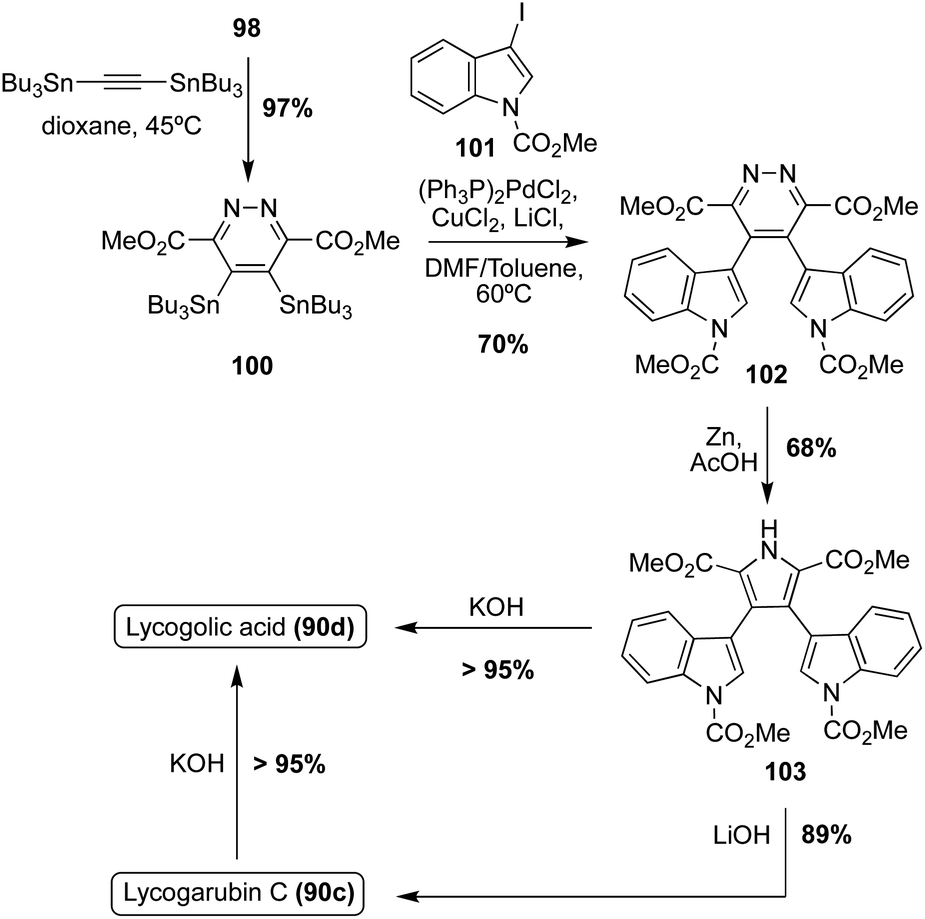 | ||
| Scheme 21 Total syntheses of Lycogarubin C (90c) and Lycagolic acid (90d). | ||
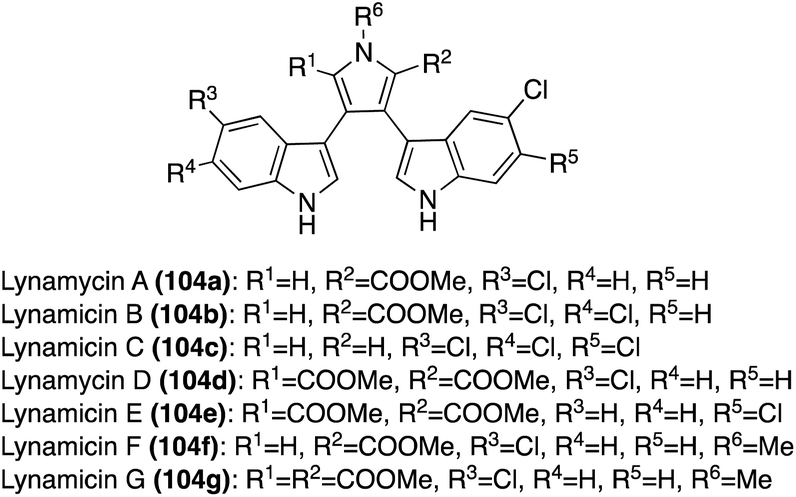 | ||
| Fig. 8 Chemical structures of Lynamicin A–G (104a–g). | ||
Two additional natural products of the Lynamicin class, Lynamicin F (104f) and Lynamicin G (104g), were isolated in 2014 by Zhang, et. al.68 from the deep-sea-derived actinomycete, Streptomyces sp. The chemical structures of Lynamicin F (104f) and Lynamicin G (104g), as shown in Fig. 8, were elucidated via spectroscopic methods. The characteristic N-methyl group in 104f and 104g was confirmed via key HMBC correlations between the hydrogens on the methyl group and the C-2 and C-5 carbons of the pyrrole ring. Interestingly, the presence of this methyl group seems to have a significant effect on biological activity because, unlike Lyncamicins A–E (104a–e), Lynamicin F (104f) and Lynamicin G (104g) showed no significant antibiotic activity against Staphylococcus aureus, Escherichia coli, etc. and no significant cytotoxic activity toward cancer cell lines.68
The first of the Lycogarubin class to be accessed via total synthesis was Lynamicin D (104d) by Nikolakaki, et. al. in 2017, which utilized a Suzuki coupling approach to install the two indole moieties to the pyrrole core.68 As shown in Scheme 22, first, the 3,4-dibromopyrrole intermediate 106 was initially synthesized in two steps from the corresponding pyrrole-2-methylester in good yields. An indolic boronic ester intermediate (105) then underwent a palladium-catalyzed Suzuki reaction with 106 to access intermediate 107 in 76% yield. After removal of the Boc protecting groups via acidic conditions, Lynamicin D (104d) was achieved in 97% yield.68 Recently, a semi-total synthesis of Lynamicin D (104d), resembling that of Lycogarubin C (90c), was also completed (Scheme 22).63 First, enzymatic biological reactions were conducted to accomplish the dimerization/cyclization of the 5-chloro-L-Tryptophan (5-Cl-L-Trp) starting material (108), following the biosynthetic path, to access the carboxylic acid analogue of Lynamicin D (109). Then, 109 was immediately reacted with TMSCHN2 in MeOH to access Lynamicin D (104d) in 37% over two steps from the simple 5-Cl-L-Trp (108) starting material.63
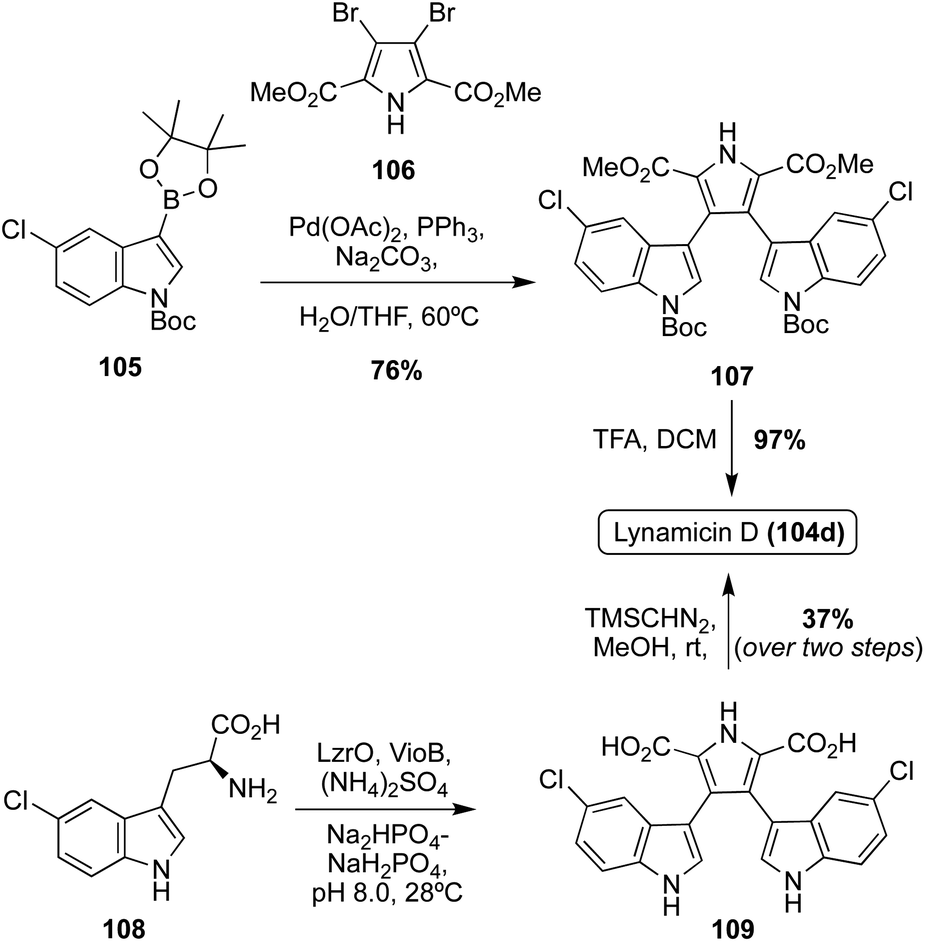 | ||
| Scheme 22 Total synthesis of Lynamicin D (104d). | ||
The first total synthesis of Lynamicin A (104a) was completed in 2021 by Smith, et. al., as well as a slightly adapted total synthesis of Lynamicin D (104d).69 As shown in Scheme 23, these total syntheses follow a very similar Suzuki cross-coupling to that of Nikolakaki's previously discussed method to install the indole moieties onto the pyrrole core of these natural products.67 However, the Suzuki cross coupling step between the boronic ester intermediate (110) and the 3,4-dibromo pyrrole intermediate (106) utilized different palladium catalysts, as shown in Scheme 23, and featured a subsequent deprotection of the protecting groups to access Lynamicin D (104d) in 67% yield. Then, Lynamicin D (104d) was subjected to a basic environment, at high temperatures, which led to the removal of one of the ester groups on the pyrrole ring, accessing Lynamicin A (104a) in 67% yield.69 Lynamicins B, C, and E–G (104b, c, e–g) have yet to be accessed via total synthesis.
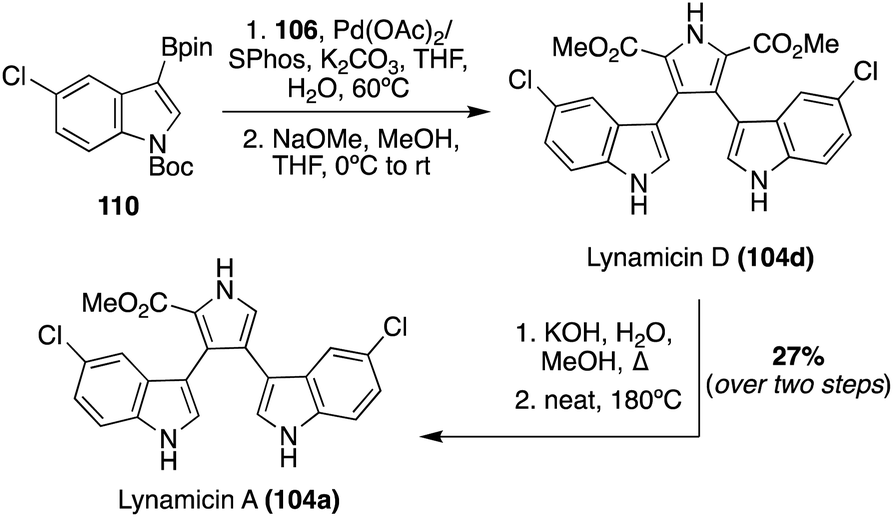 | ||
| Scheme 23 Total synthesis of Lynamicin A (104a). | ||
2.4 Piperazine, piperazinone, and pyridone linker moieties
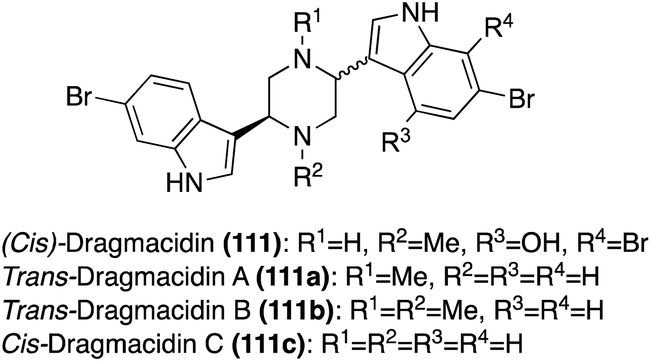 | ||
| Fig. 9 Chemical structures of Dragmacidin (111) and Dragmacidins A–C (111a–c). | ||
The second sub-class of the Dragmacidins is comprised of more structurally complex natural products, including Dragmacidins D (111d), E (111e), and F (111f). These bis-indole natural products (111d–f) contain a 2-pyridone core that links their two indole, or modified indole, moieties, as shown in Fig. 10. Another structural distinction of this sub-class is the presence of a guanidine moiety. Dragmacidin D (111d) and Dragmacidin E (111e) were isolated in 1988 by Carroll, et. al. from Spongosorites and Hexadella marine sponges and their structures were elucidated via spectroscopic means.73 In addition, Dragmacidin D (111d) and Dragmacidin E (111e) were found to be potent inhibitors of serine–threonine protein phosphatases.73 Dragmacidin F (111f) was isolated a couple years later, in 2000, by Riccio, et. al. from a Halicortex marine sponge. The complex ring structure of Dragmacidin F (111f) was elucidated via extensive spectroscopic analysis and its unprecedented carbon skeleton was proposed to result from the cyclization of a partially oxidized form of Dragmacidin D (111d). Dragmacidin F (111f) was found to display antiviral activity against HSV-1 and HIV-1.74 Dragmacidin E (111e) and Dragmacidin F (111f) have both been accessed via total synthesis; however, these syntheses will not be discussed in detail as these chemical structures fall outside the scope of this review.75,76
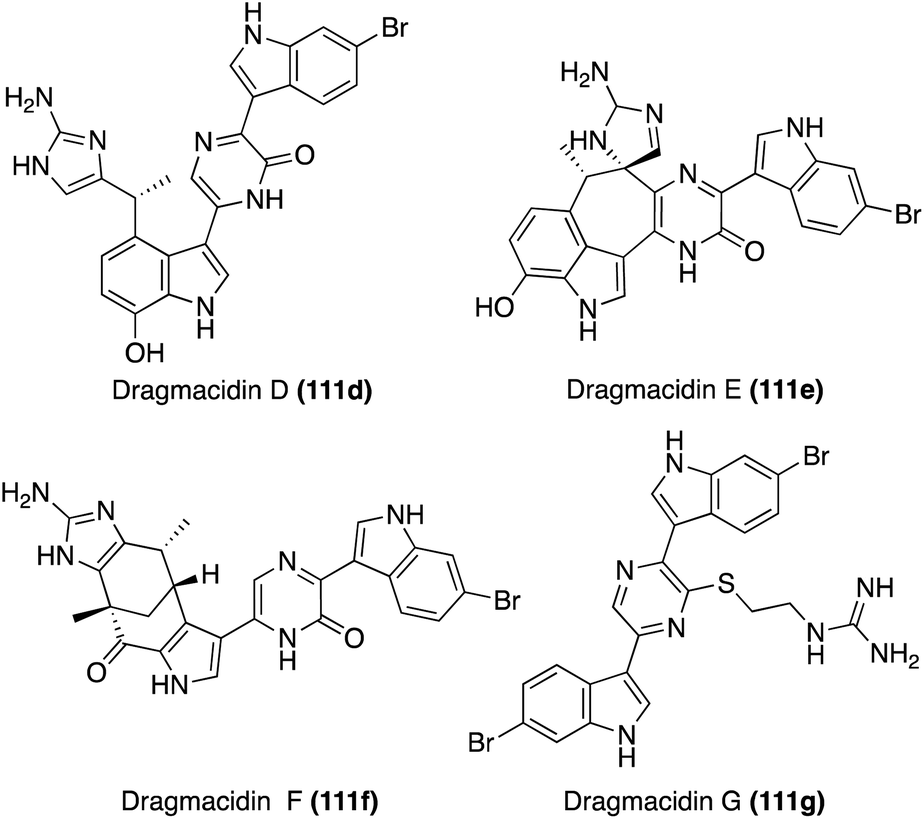 | ||
| Fig. 10 Chemical structures of Dragmacidins D–G (111d–g). | ||
Recently, Dragmacidin G was isolated by Wright, et. al. from Spongosorites marine sponge. As is shown in Fig. 10, the chemical structure of Dragmacidin G (111g) does not exactly fit into the second sub-class of the Dragmacidins, as it is the only natural product in its class to contain a piperazine (1,4-diazine) core linking the two indole moieties. However, Dragmacidin G (111g) does contain a characteristic guanidine moiety, thus enabling It to still be considered part of the second structural sub-class of the Dragmacidins. In addition, Dragmacidin G (111g) was found to exhibit antibacterial activity against Stapphylococcus aureus, Mycobacterium tuberculosis, Plasmodium falciparum, etc., and cytotoxic activity against a panel of pancreatic cell lines.77 Dragmacidin G has yet to be accessed via total synthesis.
Dragmacidin (111) was the first natural product of this class to be accessed via total synthesis. In 1994, Wuonola et. al. completed the first racemic total synthesis of Dragmacidin (111) via an initial condensation of an oxo-acyl chloride (112) and an amino nitrile (113) (synthesized via a Strecker method), followed by subsequent oxidation and cyclization, as shown in Scheme 24. These transformations proceeded in high yields. The cyclized intermediate was then subjected to reductive conditions to access 115a and 115b. This reduction, specifically the loss of the hydroxyl group, was not stereoselective at room temperature, resulting in a ratio of (115a:115b = 1:1). However, significant selectivity in favor of the desired trans isomer (115b) was achieved by lowering the temperature to 0 °C for this reduction reaction, as shown in Scheme 24, resulting in 37% of the trans isomer (115b) compared to 9% of the undesired cis isomer (115a). Then, 115b was de-methylated to afford Dragmacidin (111).78
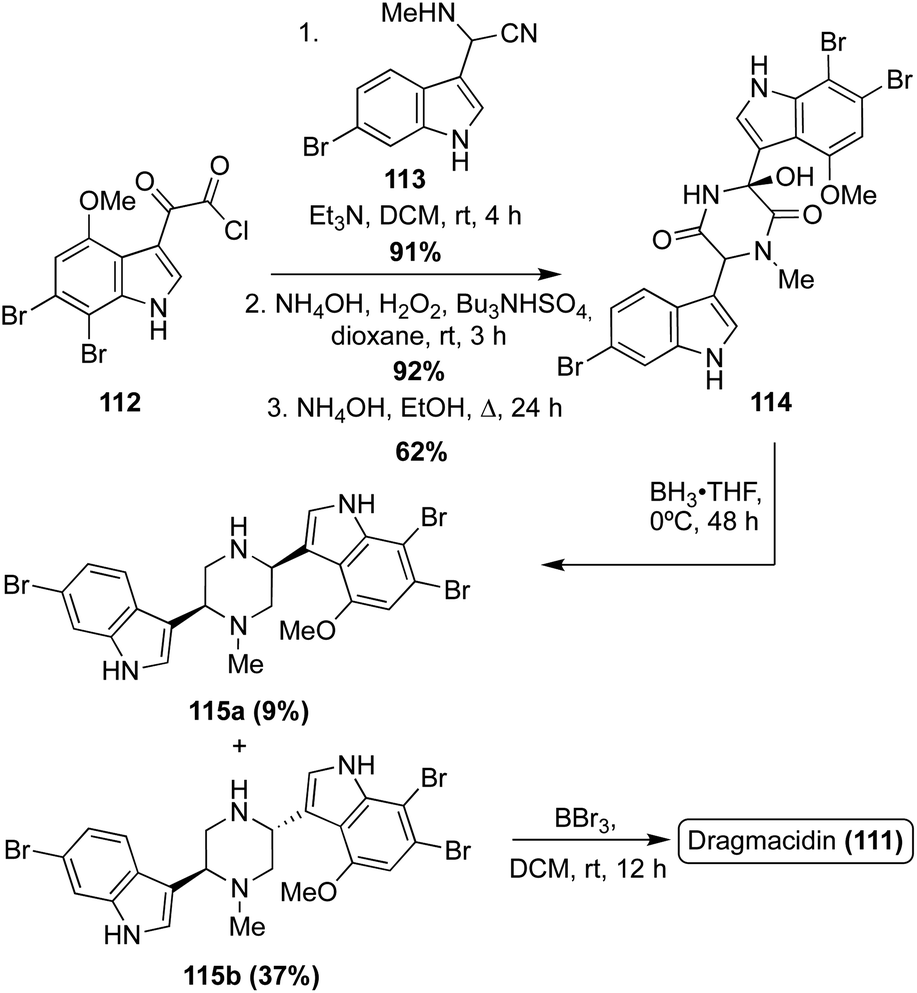 | ||
| Scheme 24 First total synthesis of Dragmacidin (111). | ||
The first total synthesis of Dragmacidin B (111b) was achieved in the same year as Dragmacidin (111). However, considering its less substituted indole moieties, the approach toward Dragmacidin B (111b) was less step intensive, as shown in Scheme 25. First, an N-dimethylated piperazinedione (116) was brominated before undergoing a double nucleophilic attack with 6-bromo-1H-indole to access intermediate 117 in 57% yield. The bromination and nucleophilic attacks were conducted in a one-pot manner and no acid or base was necessary for the nucleophilic step. Then, 117 was reduced to access Dragmacidin B (111b) in moderate yield. This made for a very expeditious two-step approach to complete the total synthesis of Dragmacidin B (111b) in 14% yield overall (Scheme 25).79
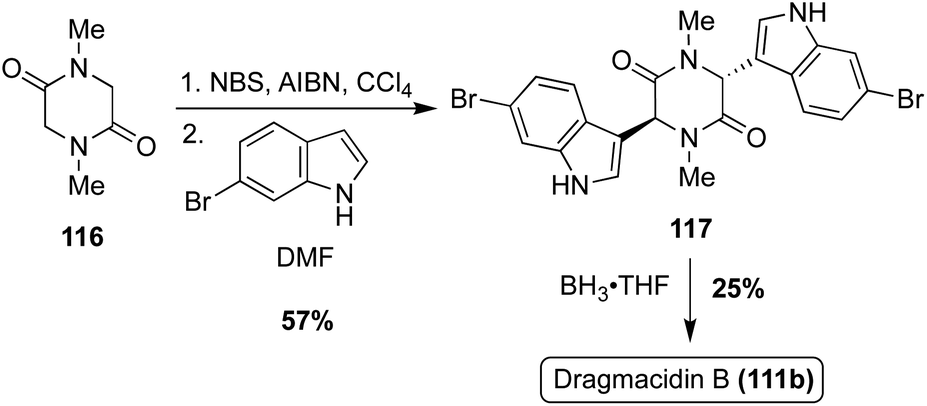 | ||
| Scheme 25 First total synthesis of Dragmacidin B (111b). | ||
The first total synthesis of Dragmacidin A (111a) was completed in 2000 via a condensation/cyclization sequence of methylated (118) and free (119) indolic α-amino carboxylic acids to access the piperazinedione intermediate (121) in good yields, as shown in Scheme 26. The condensation step was diastereoselective and the desired isomer (120b) predominated. After the cyclization, the carbonyl groups of intermediate (121a) were removed via reductive conditions to access Dragmacidin A (111a) in 45% yield. The total yield of Dragmacidin A (111a) after this efficient three-step approach was 21% overall.80
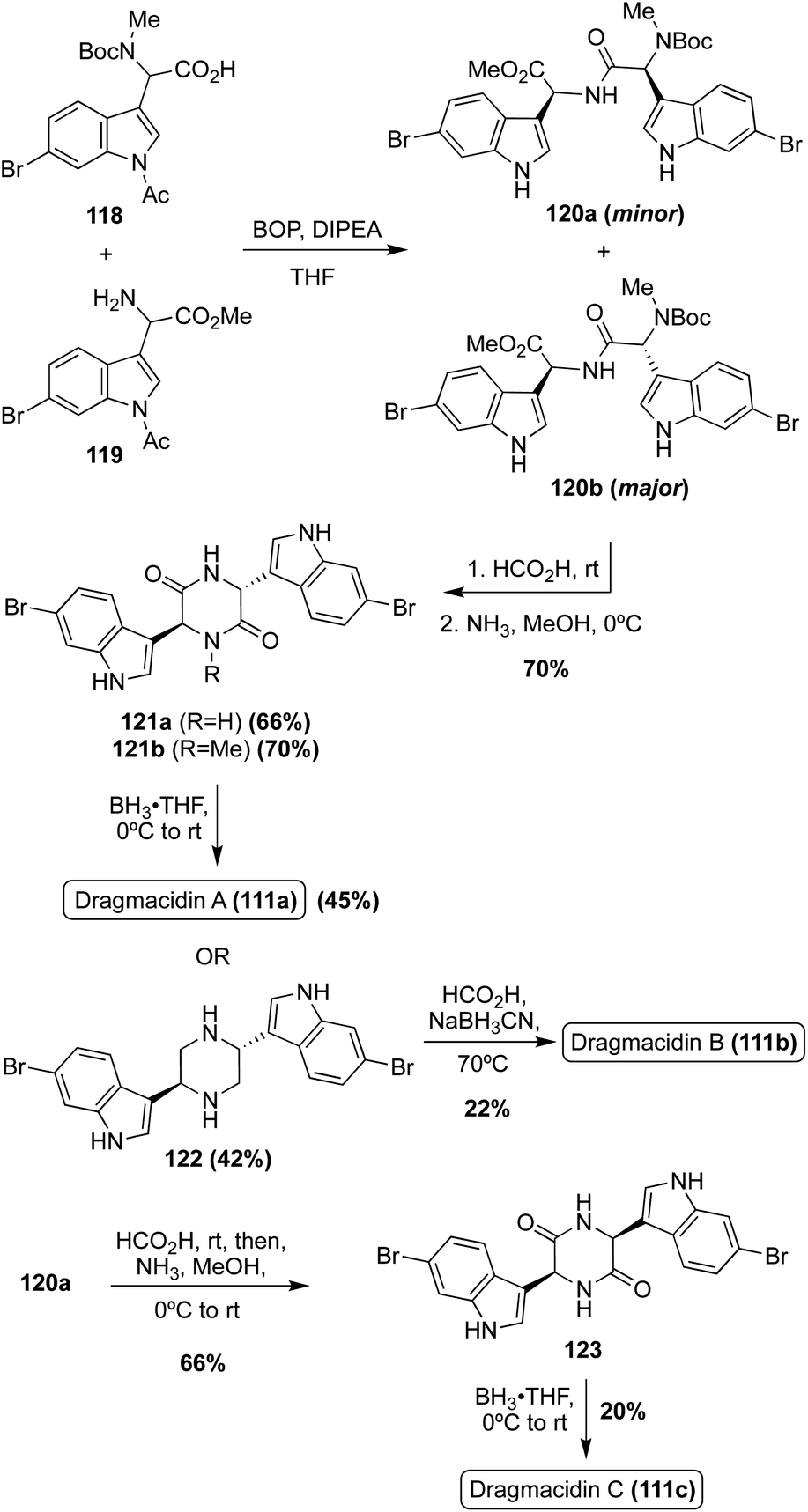 | ||
| Scheme 26 Total syntheses of Dragmacidins A–C (111a–c). | ||
The first total synthesis of Dragmacidin C (111c) was completed in 2002 via the same three step method (condensation/cyclization/oxidation) that was previously discussed for the first total synthesis of Dragmacidin A (111a), and the relative cis configuration of Dragmacidin C (111c) was confirmed (Scheme 26). As shown in Scheme 26, Dragmacidin B (111b) was also synthesized using this method, though incomplete reduction of 121b resulted in a prominent side product in which one carbonyl remained on the ring. This accounts for the lower yield of 122. In the synthesis of Dragmacidin B (111b) a late-stage methylation step was carried out on the piperazine intermediate (122) to install the two methyl groups on the piperazine ring of Dragmacidin B (111b).72
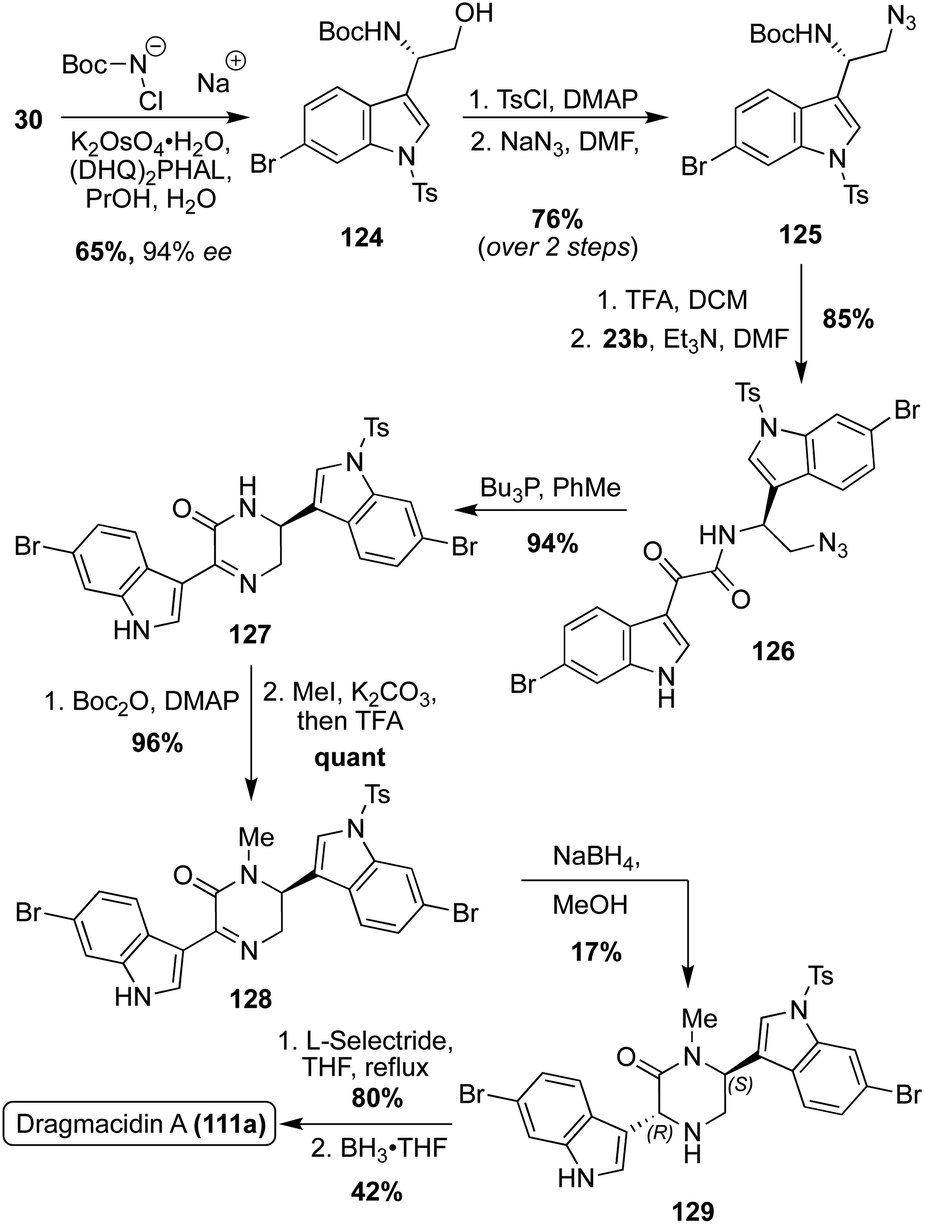
Optically active Dragmacidin A (111a) was synthesized via a Sharpless asymmetric aminohydroxylation of 6-bromo-3-vinylindole (30) to access 124 in 65% yield and 94% enantiomeric excess, as shown in Scheme 27. The hydroxyl group of 124 was then tosylated and subsequently substituted by an azido group in high yield over two steps. The resulting amino azide intermediate (125) was then deprotected and acylated with 23b, followed by reduction of the azide and subsequent cyclization to afford 127. After Boc-protection, 127 was selectively and quantitatively methylated and subsequently deprotected to afford 128. The reduction of intermediate 128 proved to be problematic, as it was nonselective. In fact, the desired isomer (129) was the minor product in just 17% isolated yield (82% of cis isomer isolated). Despite this low yield, 129 was de-tosylated viaL-selectride and the piperazinone was reduced to the desired piperazine core to access (2S, 5R)-Dragmacidin A (111a) in 42% yield over two steps.81
 | ||
| Scheme 27 First total synthesis of optically active Dragmacidin (111). | ||
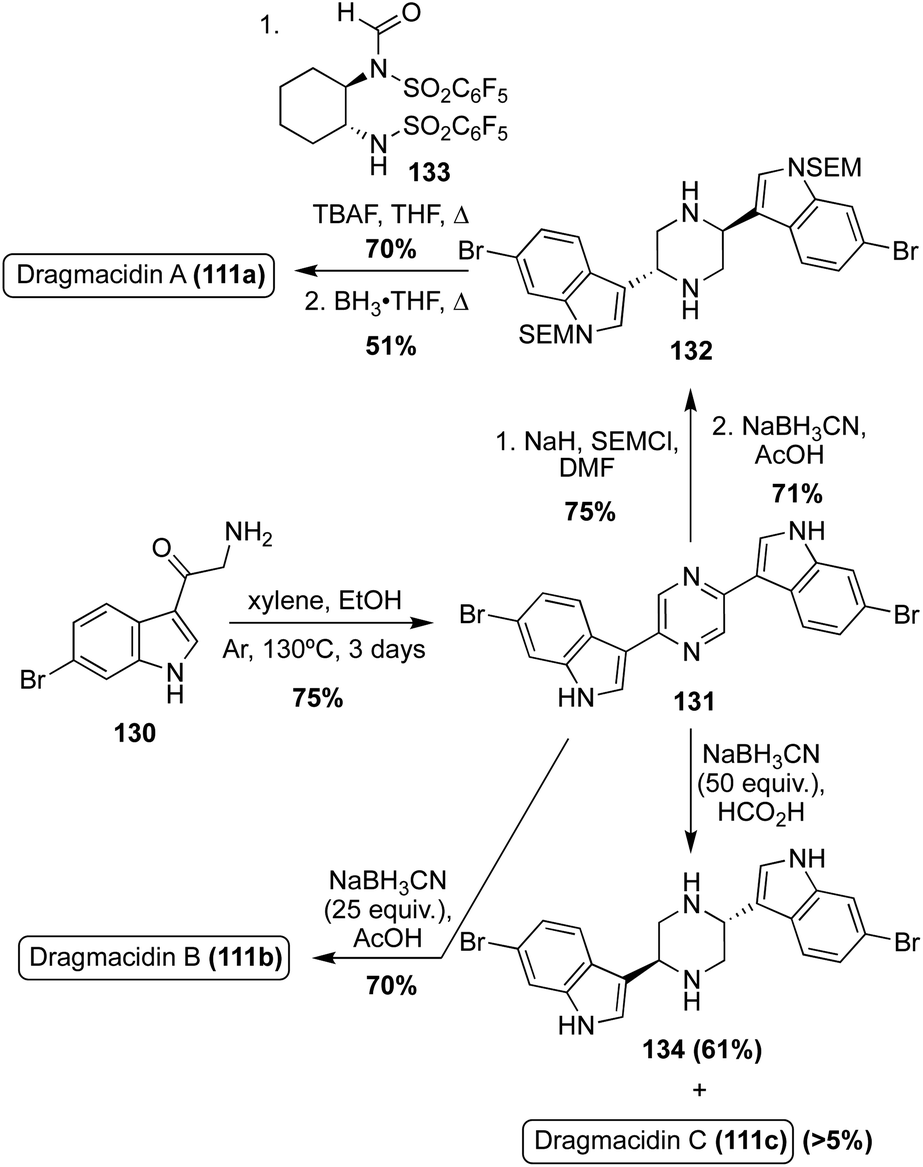
An expedited method of synthesizing optically active Dragmacidin A (111a) was developed in which a bis-indolylpyrazine intermediate (131) was protected and subsequently reduced to the corresponding piperazine intermediate (132) in high yields, as shown in Scheme 28. Then, de-symmetrization of the piperazine (132) was completed via enantioselective formylation using a chiral formylating reagent. This method ultimately proved to be only moderately stereoselective, affording the dextrorotatory isomer of Dragmacidin A (111a) in 44% enantiomeric excess after transformation of the aldehyde to a methyl group and subsequent deprotection.82 Using a similar reductive approach to that used on the bis-indolylpyrazine intermediate (131), an efficient synthesis of Dragmacidin B (111b) was achieved in 53% overall yield over two steps, as shown in Scheme 28. It should be noted that this was not an efficient method for synthesizing Dragmacidin C (111c), as the undesired isomer predominated and less than 5% yield of Dragmacidin C (111c) was isolated.83,84
 | ||
| Scheme 28 Total syntheses of Dragmacidins A–C (111a–c). | ||
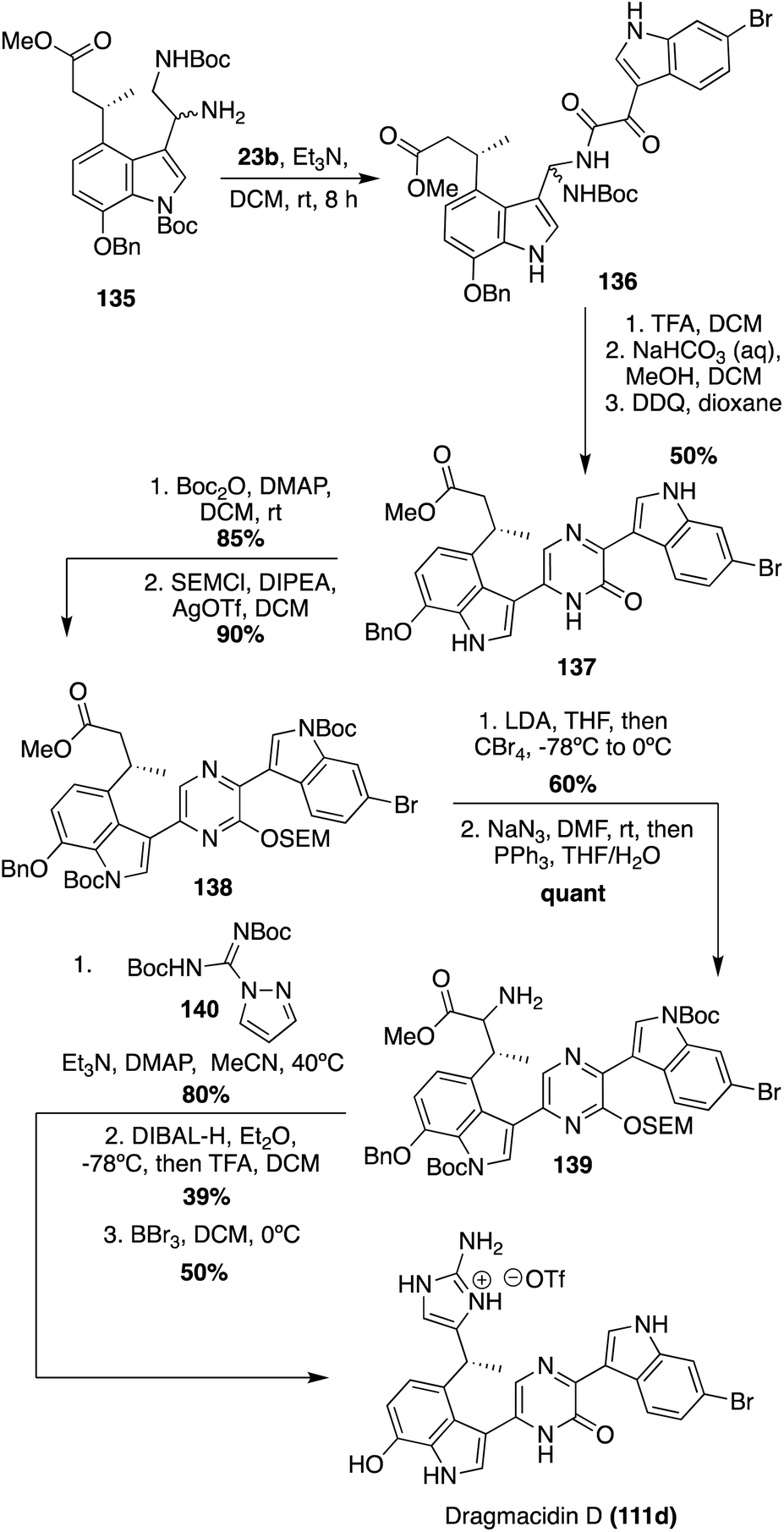
Recently, the first total synthesis of the more structurally complex Dragmacidin D (111d) was completed.85 First, the iodinated indolic diamine intermediate (135) was synthesized in 11 linear steps from commercially available starting materials. Here, the desired stereoisomer of 135 was accessed via the use of Evan's oxazolidinone chiral auxiliary ((S)-3-acryloyl-4-phenyloxazolidin-2-one) and column chromatography.85 After the key diamine intermediate (135) was synthesized, it was condensed with 6-bromo-1H-indole-3-oxo-acyl chloride and subsequently cyclized and oxidized to access 2-piperazinone intermediate (137) in 50% yield over multiple steps, as shown in Scheme 29. Then, 137 was protected to allow for selective α-bromination, followed by a Staudinger reaction to access the α-amino ester (139) in high yields. Treatment of this α-amino ester (139) with pyrazole-1-carboxamidine afforded a guanidine intermediate, which underwent subsequent reduction and cyclization to construct the 2-aminoimidazole moiety of Dragmacidin D in good yields. Deprotection then afforded Dragmacidin D (111d) in 50% yield. This synthesis revised the earlier stereochemical assignment that was based on biosynthetic considerations, assigning the absolute configuration as (R)-(+)-Dregmacidin D (111d). Furthermore, chiral HPLC-DAD methodology was developed and utilized to confirm, for the first time, that naturally occurring Dragmacidin D (111d) was isolated as either a racemate or a scalemic mixture (39% ee), which has prompted questions regarding the absolute configurations of Dragmacidins D-F (111d–f).85
 | ||
| Scheme 29 First total synthesis of Dragmacidin D (111d). | ||
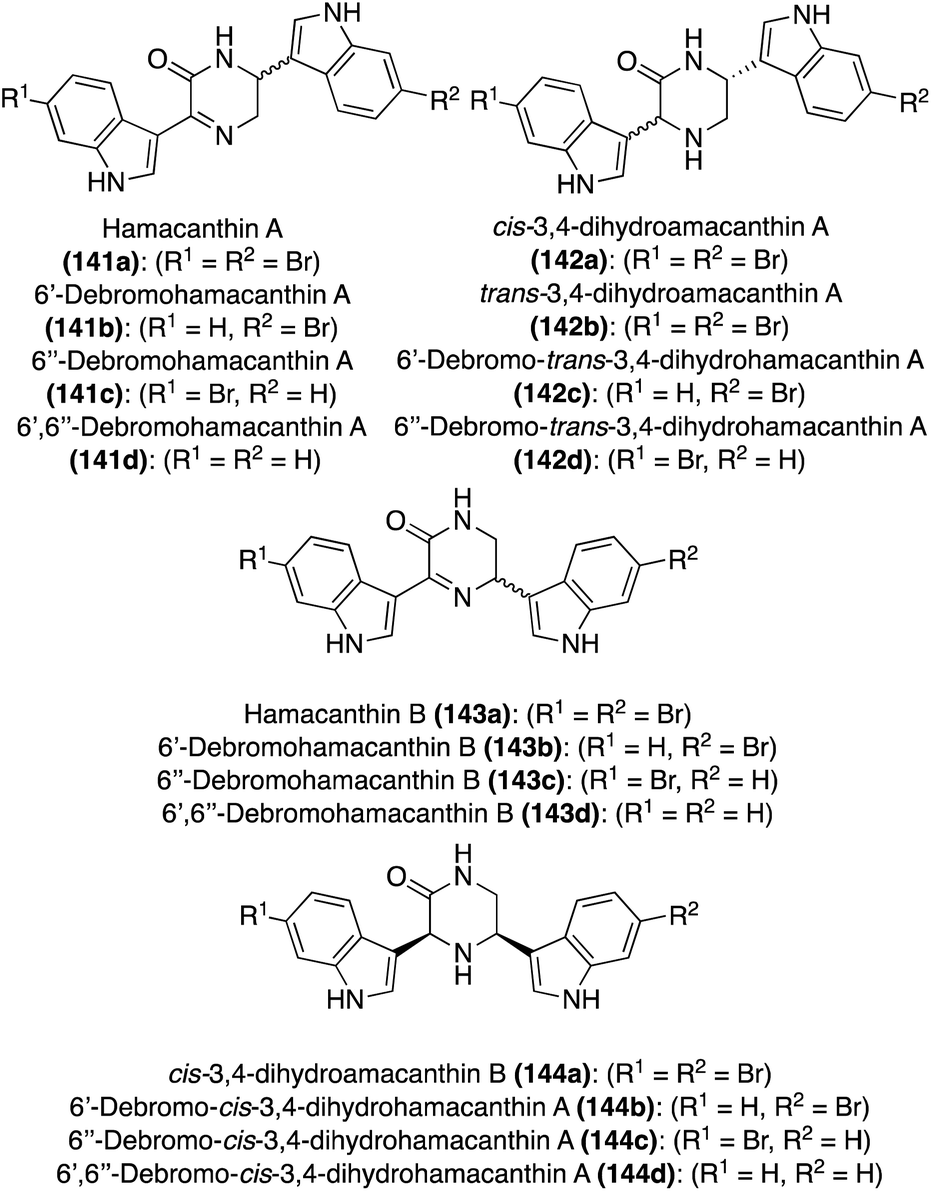 | ||
| Fig. 11 Chemical structures of Hamacanthins A–B (141a and 143a), Dihydrohamacanthins A–B (142a and 144a), and related debrominated natural products. | ||
| Compound | Configuration | Natural source |
|---|---|---|
| 141a | S | Hamacantha, Spongosorites, Disodermia calyx |
| 141b | R | Spongosorites |
| 141c | R | Spongosorites, Discodermia calyx |
| 141d | S | Spongosorites |
| 143a | S | Hamacantha, Spongosorites, Disodermia calyx |
| 143b | R | Spongosorites, Discodermia calyx |
| 143c | R | Spongosorites |
| 143d | R | Spongosorites |
| 142a | 3R, 6R | Rhaphizia lacazei |
| 142b | 3S, 6R | Rhaphizia lacazei, Spongosorites |
| 142c | 3S, 6R | Rhaphizia lacazei, Spongosorites |
| 142d | 3S, 6R | Rhaphizia lacazei, Spongosorites |
| 144a | 3S, 5R | Rhaphizia lacazei, Spongosorites, Discodermia calyx |
| 144b | 3S, 5R | Rhaphizia lacazei, Spongosorites |
| 144c | 3S, 5R | Rhaphizia lacazei, Spongosorites |
| 144d | 3S, 5R | Spongosorites |
The first enantioselective total synthesis of the unnatural (R)-Hamacanthin A (141a) was completed in 2001 by Jiang, et. al. via the coupling of a 3-indolyl-α-oxo-acetyl chloride intermediate (23b) and 3-indolyl azidoethylamine intermediate (R)-(146), prior to an intramolecular aza-Wittig type cyclization to access (R)-Hamacanthin A (141a) in high yields, as shown in Scheme 30.87 The 3-indolyl azidoethylamine intermediate was synthesized in four steps from N-tosylated-6-bromo-3-vinyl indole in high yields. The stereocenter of (141a) was established via a Sharpless asymmetric dihydroxylation reaction, followed by a stereospecific azidation. In spectroscopic comparison of the naturally isolated Hamacanthin A (141a) and the synthesized (R)-Hamacanthin A (141a), it was confirmed that the natural Hamacanthin A (141a) existed as the (S)-isomer.87,88
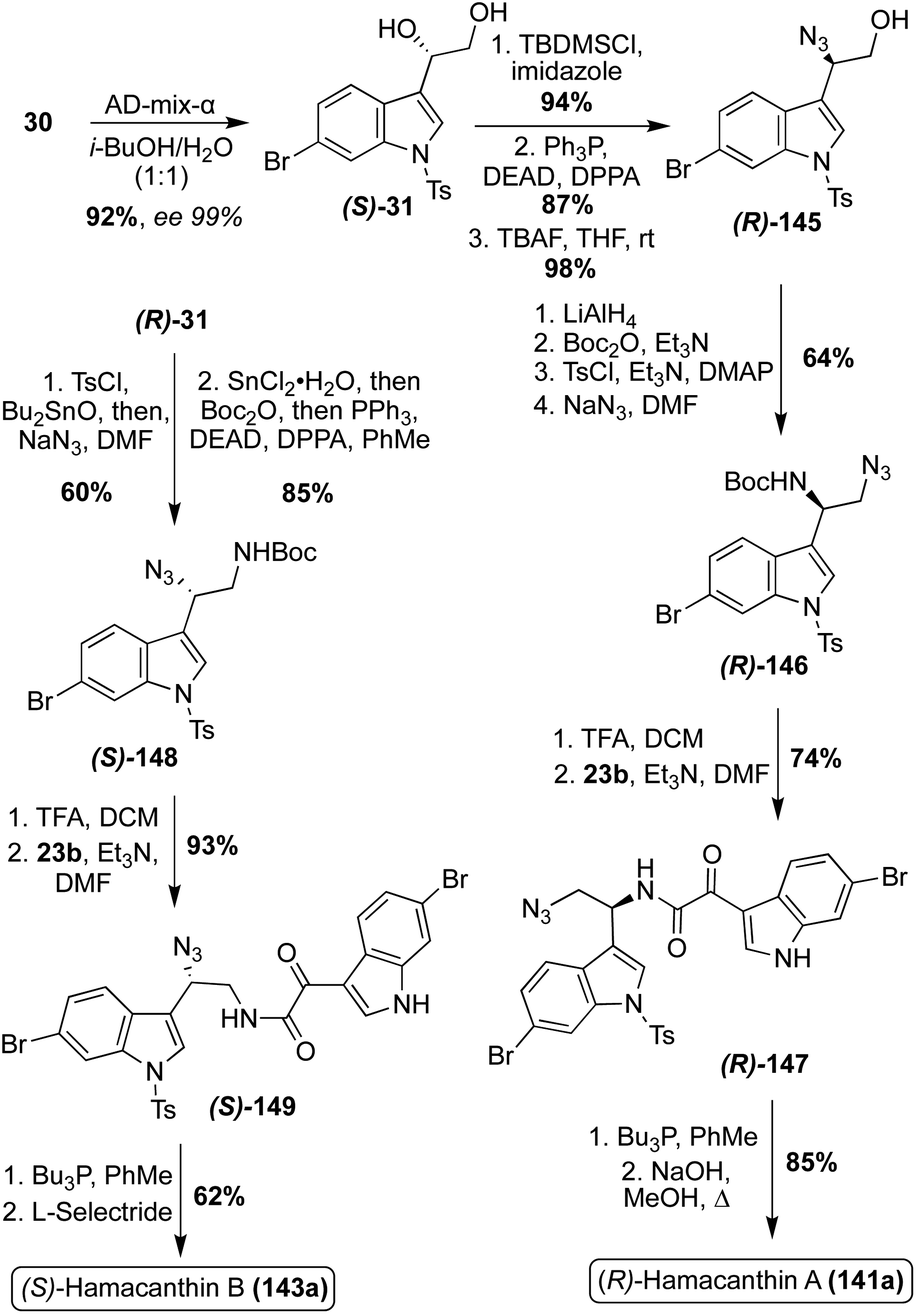 | ||
| Scheme 30 Enantioselective total syntheses of Hamacanthins A and B (141a and 143a). | ||
A year later, the same research team completed the first enantioselective total synthesis of Hamacanthin B (143a). As shown in Scheme 30, this synthesis was completed via a similar approach as was previously discussed for (R)-Hamacanthin A (141a). Considering the established (S)-configuration of (141a), (S)-Hamacanthin B (143a) was synthesized. When compared to the naturally isolated Hamacanthin B (143a), the synthesized (S)-143a confirmed the configuration of the natural 143a as the (S)-isomer.88
With the elucidation of the absolute configuration of (S)-Hamacanthin A (141a), the first enantioselective total synthesis of the desired (S)-Hamacanthin A (141a) was completed via deprotection and simultaneous inversion of the stereocenter of (R)-150 to access (S)-141a in 78% yield, as shown in Scheme 31. This allowed for additional confirmation that this was the correct configuration of the naturally occurring (S)-Hamacanthin A (141a). The synthetic approach toward (R)-150 was previously discussed in the Dragmacidin section (Scheme 27).81 Also, using intermediate (R)-150, the first total syntheses of the unnatural (3S, 6S)-cis-3,4-dihydrohamacanthin A (142a) and (3R, 6S)-trans-3,4-dihydrohamacanthin A (142b) were both completed. The reduction of (R)-150 afforded 62% yield of (3S, 6S)-cis-151 and 36% yield of (3R, 6S)-trans-151. These 2-piperidone intermediates (151) were then deprotected viaL-selectride to access (3S, 6S)-cis-3,4-dihydrohamacanthin A (142a) and (3R, 6S)-trans-3,4-dihydrohamacanthin A (142b) in 87% and 88% yields, respectively (Scheme 31). Via comparison of the naturally isolated cis-3,4-dihydrohamacanthin A (142a) and trans-3,4-dihydrohamacanthin A (142b) with the synthesized (3S, 6S)-cis-142a and (3S, 6S)-trans-142b, it was found that the configurations of these synthesized natural products did not match that of the isolated optical rotation data. Therefore, it was concluded that the absolute configurations of cis-3,4-dihydrohamacanthin A (142a) and cis-3,4-dihydrohamacanthin A (142a) are (3R, 6R) and (3S, 6R), respectively.81 Additionally, racemic cis- and trans-3,4-dihydrohamacanthins can be accessed, albeit in low yields, via partial reduction of cyclized dipeptides (121 and 123), as was also discussed in the Dragmacidin section (Scheme 26).72
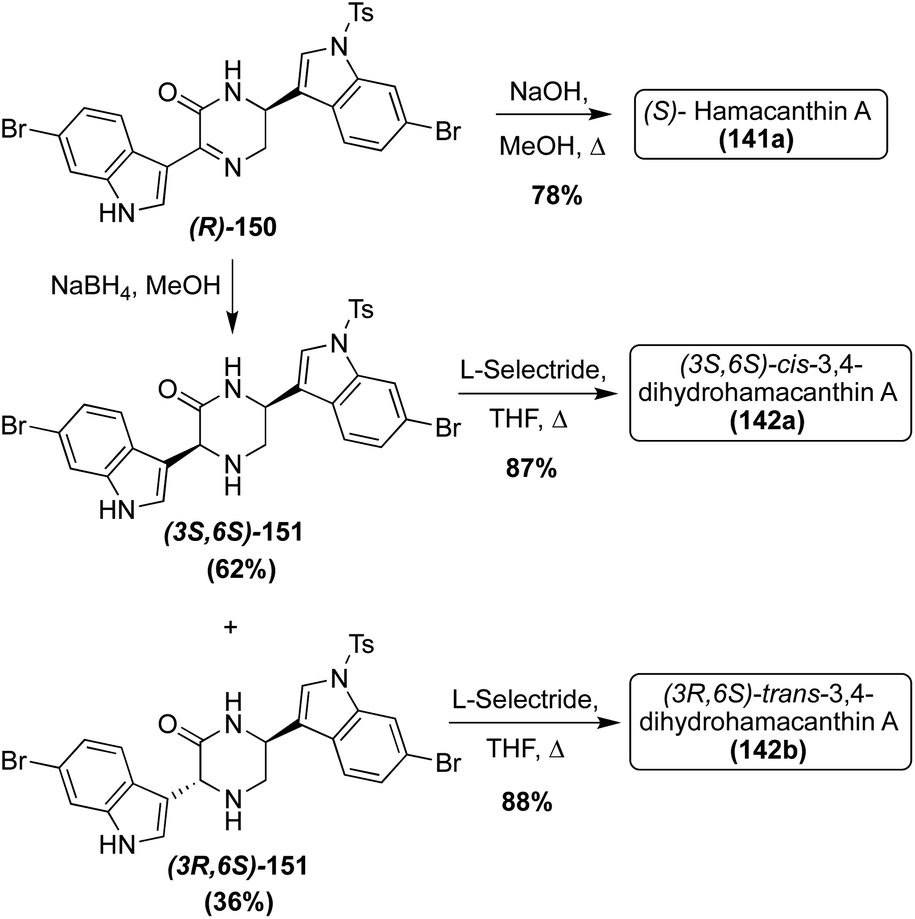 | ||
| Scheme 31 Enantioselective total syntheses of Hamacanthin A (141a) and Dihydrohamacanthin A–B (142a–b). | ||
The first total syntheses of racemic 6′-debromo-trans-3,4-dihydrohamacanthin A (142c) and the unnatural 6′′-debromo-cis-3,4-dihydrohamacanthin A (142e) were carried out via the condensation and subsequent cyclization of an indolic α-amino ketone intermediate (48) with an indolic keto–amide intermediate (153). After formation of this bis-indolylpyrazinone intermediate, it underwent reduction to access 6′-debromo-trans-3,4-dihydrohamacanthin A (142c) and the unnatural 6′′-debromo-cis-3,4-dihydrohamacanthin A (142e) in 70% yield as a 1:1 mixture (Scheme 32). While it was not enantioselective, this method proved to be a very expeditious synthesis of these natural products.90 This method also allowed for the first racemic total synthesis of 6′′-debromo-cis-3,4-dihydrohamacanthin B (144c) and 6′,6′′-debromo-cis-3,4-dihydrohamacanthin B (144d). As shown in Scheme 32, this method was adapted to achieve the 3,5-disubstituted cyclized product by condensing and cyclizing an indolic oxo-acyl chloride intermediate (23a–b) with an indolic α-amino ketone intermediate (48). In addition to being an efficient and high yielding route, this method was selective for the desired cis-isomers of 6′′-debromo-cis-3,4-dihydrohamacanthin B (144c) and 6′,6′′-debromo-cis-3,4-dihydrohamacanthin B (144d), respectively.90
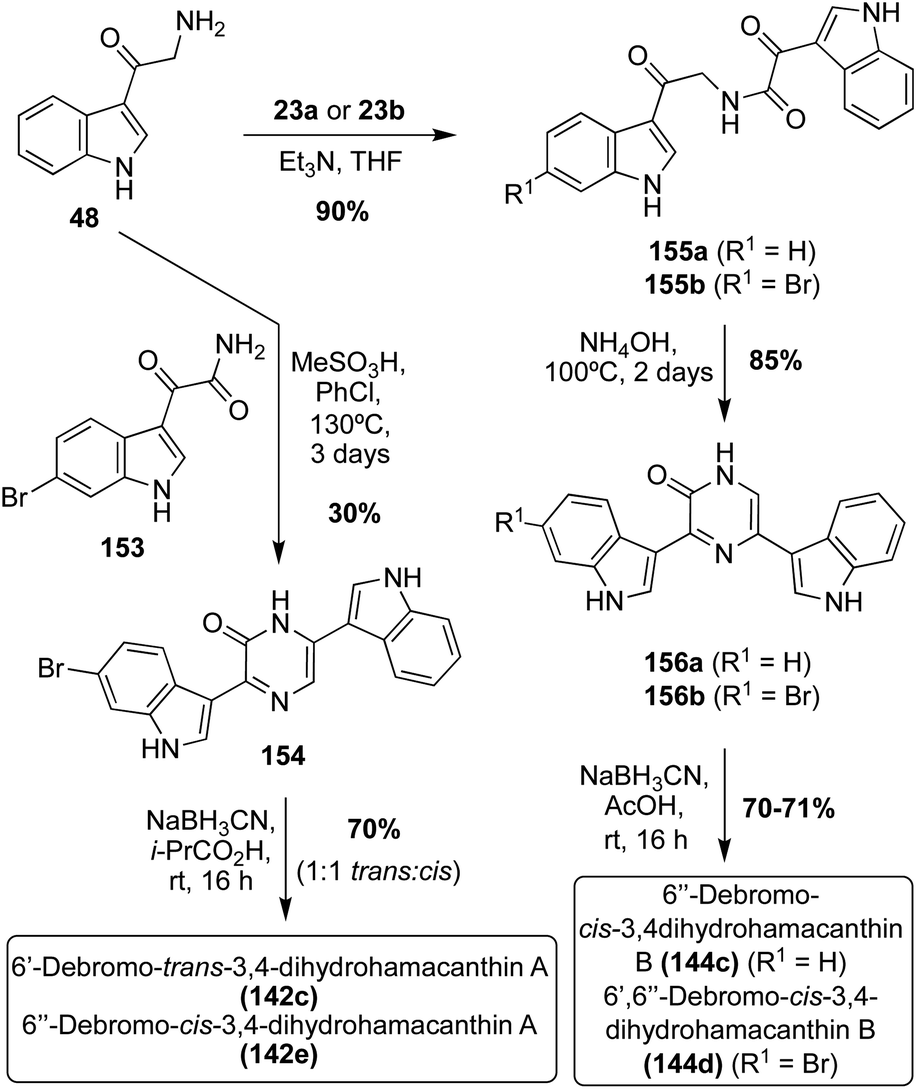 | ||
| Scheme 32 Enantioselective total syntheses of Debromo-Dihydrohamacanthins (142c) and (144c–d). | ||
A similar bio-mimetic cyclization of a ketoamide (157) was proven efficient for the synthesis of both racemic Hamacanthin A (141a) and Hamacanthin B (143a). As shown in Scheme 33, to access Hamacanthin B (143a), nucleophilic attack of the primary amine on the indolic (C3) ketone and subsequent dehydration led to intermediate (II), which was deprotected to access 143a. On the other hand, nucleophilic attack of the primary amine on the amide carbonyl to form a five-membered ring intermediate (III), followed by a ring opening that resulted in a rearrangement of the original ketoamide to intermediate (IV). Cyclization of intermediate (IV) then led to Hamacanthin A (141a) after removal of protecting groups.90 Through mechanistic study, it was determined that the ratio of 141a:143a was heavily dependent on the identity of the solvent, and the protecting groups (R′ and R′′), as indicated by the results shown in Table 3. In this case, more polar, protic solvents, such as ethanol, led to predominant formation of intermediate (II) while more non-polar solvents, such as dichloroethane (DCE), led to predominant formation of intermediate (V). In addition, the presence of stronger electron-withdrawing groups, such as a tosyl group, on the indole nitrogen led to predominant formation of intermediate (II) over intermediate (V) due to the resulting increased electrophilicity of the carbonyl group adjacent to the indole, making nucleophilic attack on this carbonyl preferable.90
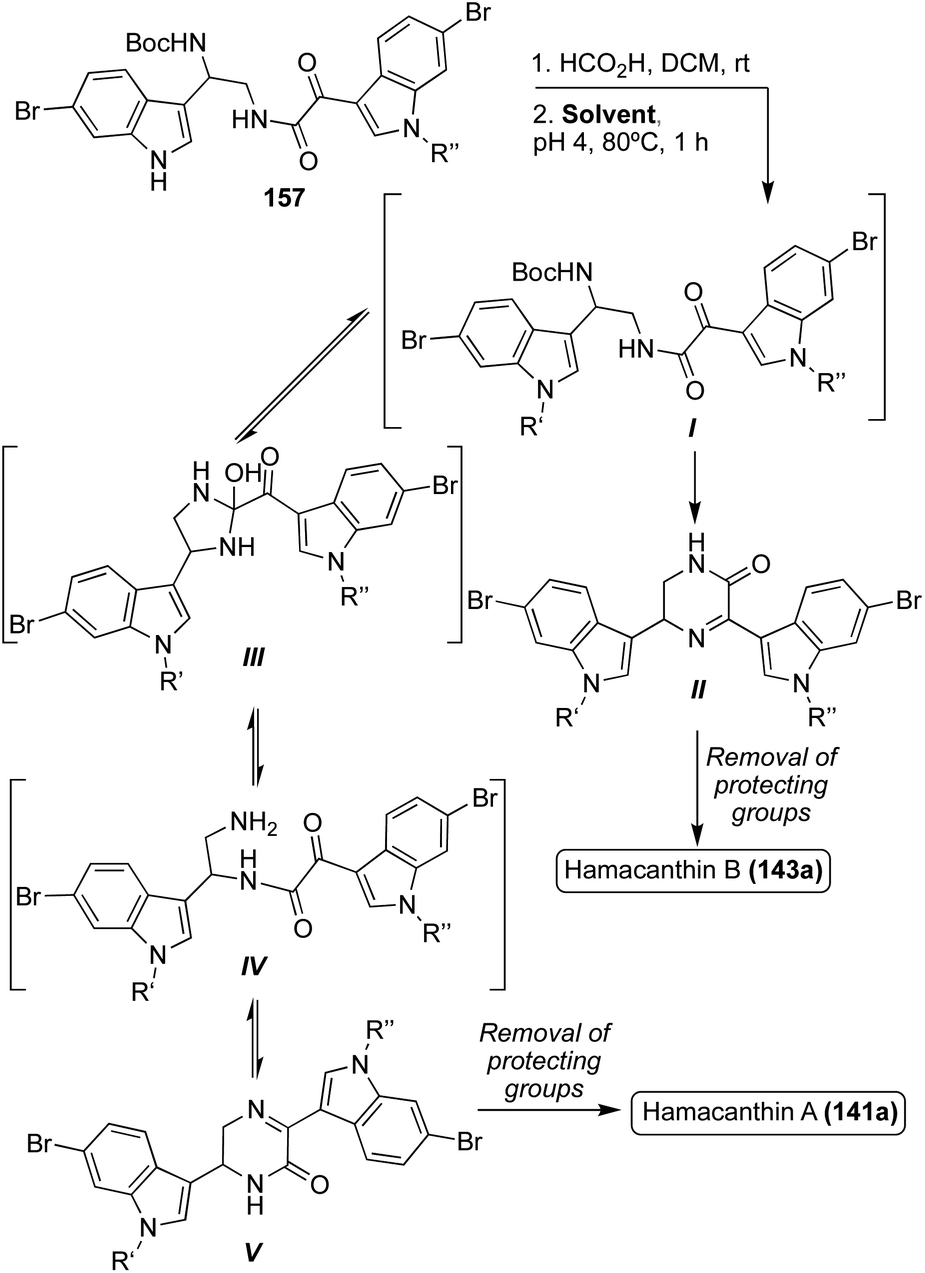 | ||
| Scheme 33 Total syntheses of Hamacanthins A and B (141a) (143a). | ||
| R′ | R′′ | Solvent | Yield (V) | Yield (II) |
|---|---|---|---|---|
| Ts | H | DCE | 42% | 35% |
| Ts | H | 1,4-Dioxane | 30% | 38% |
| Ts | H | EtOH | 15% | 75% |
| Ts | H | DMF | 7% | 46% |
| Ts | Ac | DCE | 42% | 55% |
| Ts | Ts | DCE | 18% | 68% |
In 2007, along with enantioselective total synthesis of (S)-Hamacanthins A and B (141a and 143a), the first enantioselective total synthesis of cis-3,4-dihydrohamacanthin B (144a) was also completed.91 As shown in Scheme 34, this synthesis proceeded via the condensation of an indolic amino–alcohol intermediate (162) and an indolic oxo-acyl chloride intermediate (23b) to access a similar keto–amide intermediate (163), as was previously discussed. Subsequent cyclization of the keto–amide intermediate (163) and stereospecific reduction afforded (3S-,5R)-cis-3,4-dihydrohamacanthin B (144a) in high yields. Here, the stereochemistry was set via the installation of an (S)-phenyloxazolidone chiral auxiliary (S)-(159).91
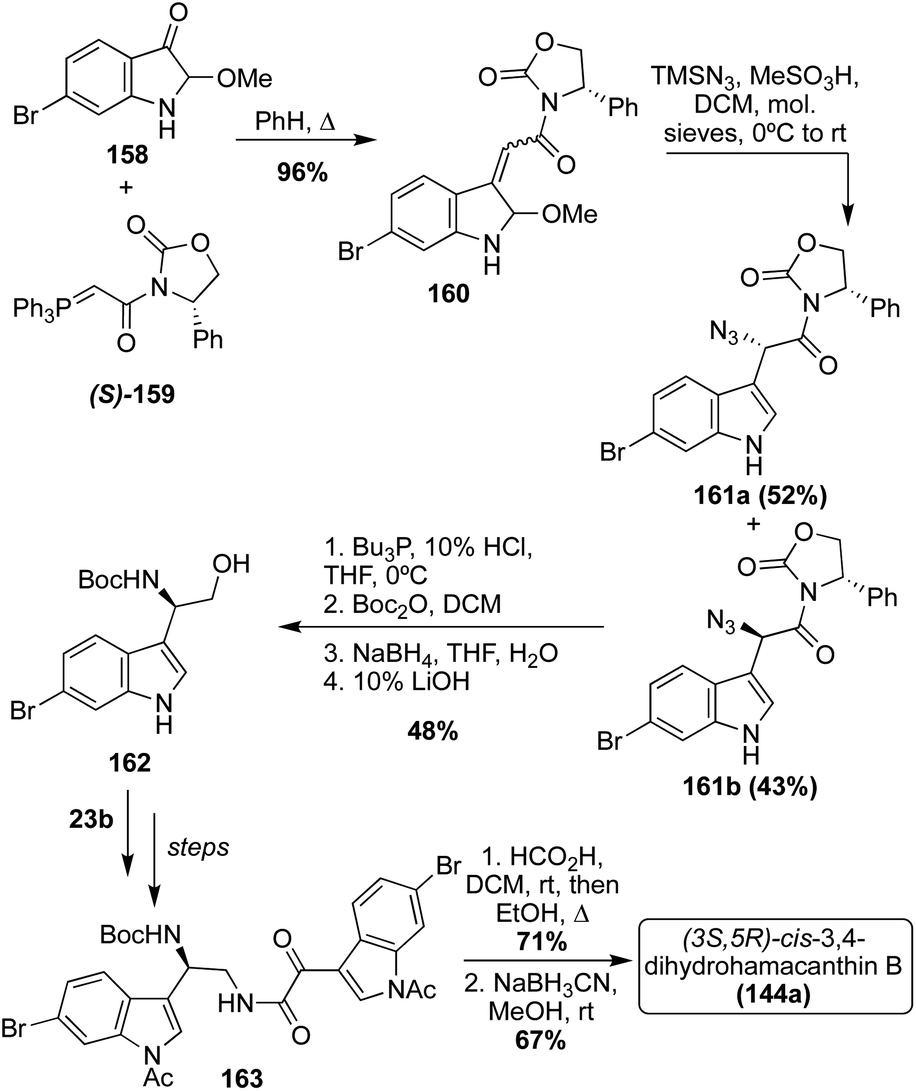 | ||
| Scheme 34 First enantioselective total synthesis of Dihydrohamacanthin B (144a). | ||
In a slightly adapted, but very similar condensation of an indolic diamine intermediate (164) and an indolic oxo-acyl chloride intermediate (23a–b) and following cyclization, racemic Hamacanthin A (141a) and its related Debromohamacanthins (141b–d) were accessed in moderate to high yields, as shown in Scheme 35.92 The indolic diamine intermediates (164a–b) were synthesized using the same method discussed previously in the Spongotine section (Scheme 5). At present, several debromo analogues of Hamacanthin B and Dihydrohamacanthin B have yet to be accessed via total synthesis.
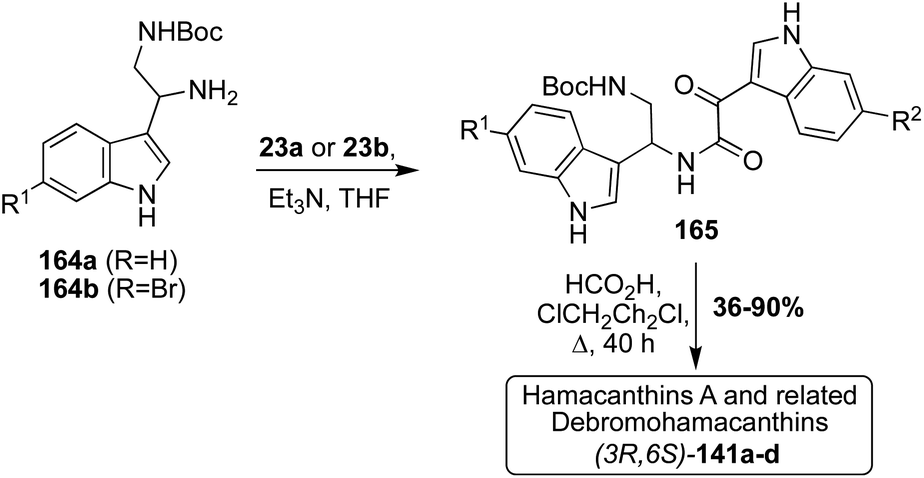 | ||
| Scheme 35 Total syntheses of Hamacanthin A (141a) and related Debromohamacanthins (141b–d). | ||
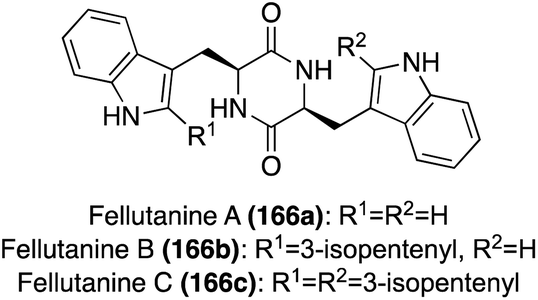 | ||
| Fig. 12 Chemical structures of Fellutanines A–C (166a–c). | ||
Interestingly, the first total synthesis of Fellutanine C (166c) was completed before its first isolation, as it was an intermediate that was accessed in the 1995 total synthesis of another related fused bicyclic natural product, Gypsetin.95 As is shown in Scheme 36, this total synthesis began with indolic C-2 reverse prenylation of N-phthaloyl-L-tryptophan methyl ester (167) via prenyl-9-BBN in high yield, followed by hydrazinolysis and subsequent Boc-protection and saponification to access intermediate (170) in high yields. Then, 170 and 168 were coupled to access the desired cis-Fellutanine C (166c) in a very high 87% yield over three steps.95
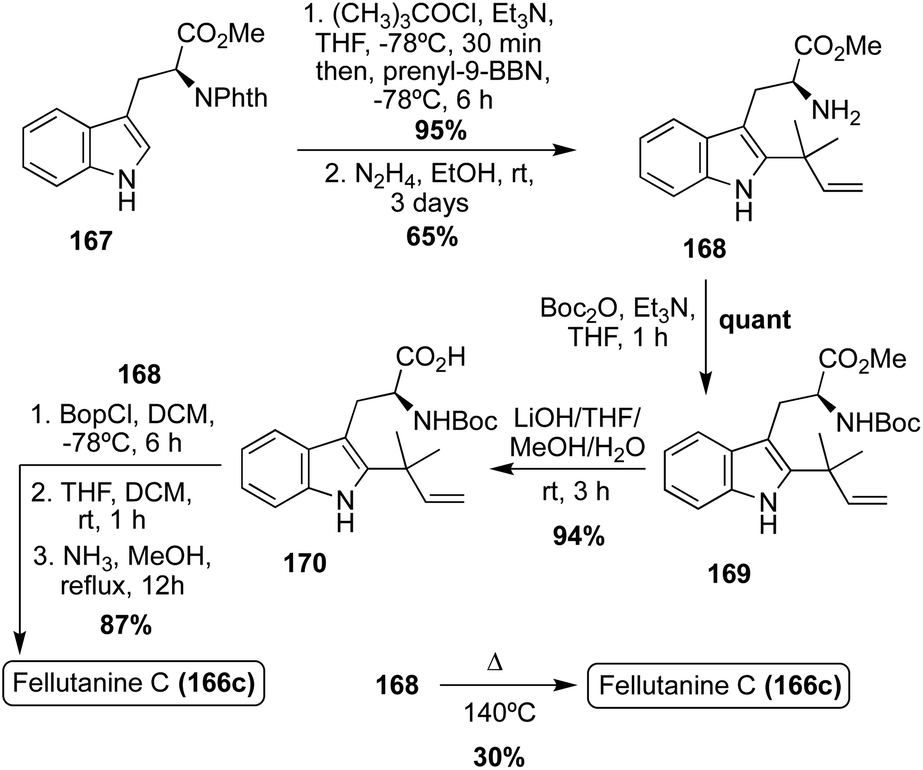 | ||
| Scheme 36 First total synthesis of Fellutanine C (166c). | ||
In a similar approach using L-tryptophan analogues as starting materials, Fellutanine C (166c) was synthesized via the same intermediate (168) used previously, though here undergoing an expeditious dimerization and cyclization via pyrolysis conditions to access cis-Fellutanine C (166c) in 30% yield.96 This proved to be an advantageous process in terms of step count by saving four steps compared to the previous synthesis, yet the yield decreased significantly in spite of this.
The first total synthesis of Fellutanine A (166a) was completed in 2008 in a similar method to that of Fellutanine C (166c). Here, a tryptophan analogue (169) and TrpOMe·HCl were condensed and subsequently cyclized to access the desired cis-Fellutanine A (166a) in 76% yield (Scheme 37).97
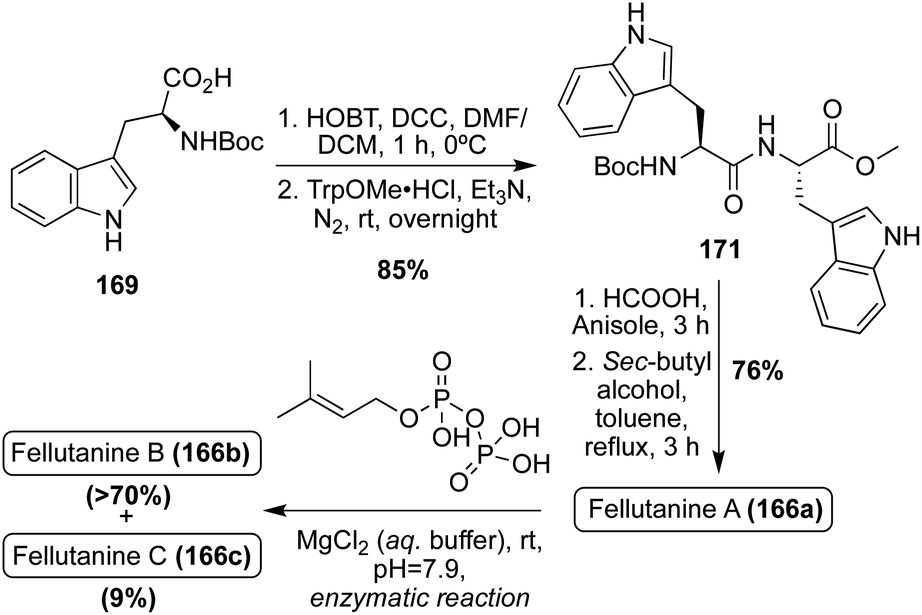 | ||
| Scheme 37 First total syntheses of Fellutanine A–B (166a–b). | ||
In addition to the previously discussed synthetic methods of accessing these Fellutanine natural products, many enzymatic biosynthetic methods have also been utilized over the years, using L-tryptophan as the starting material to access these molecules98,99 Recently, a semi-total synthesis was developed and conducted to achieve all three natural products, Fellutanines A–C (166a–c), via the same route. As shown in Scheme 37, the same condensation and subsequent cyclization of tryptophan derivatives (169) and TrpOMe·HCl was conducted to access cis-Fellutanine A (166a) in 76% yield. Then, the enzymatic C2-reverse-prenylation of indole was performed via DMAPP prenyltransferase and aqueous MgCl2 under slightly basic conditions, to access the mono-reverse prenylated cis-Fellutanine B (166b) as the major product in >70% yield. It is noteworthy to mention that, while not the major product, cis-Fellutanine C (166c) was also accessed via this method in 9% yield. The most significant achievement of this work was the first total synthesis of Fellutanine B (166b) and the development of a late stage selective reverse prenylation methodology of indole. It was also a significant that all three Fellutanines A–C (166a–c) could be accessed via this same route (Scheme 37).100
2.5 Pyridine and diazine linker moieties
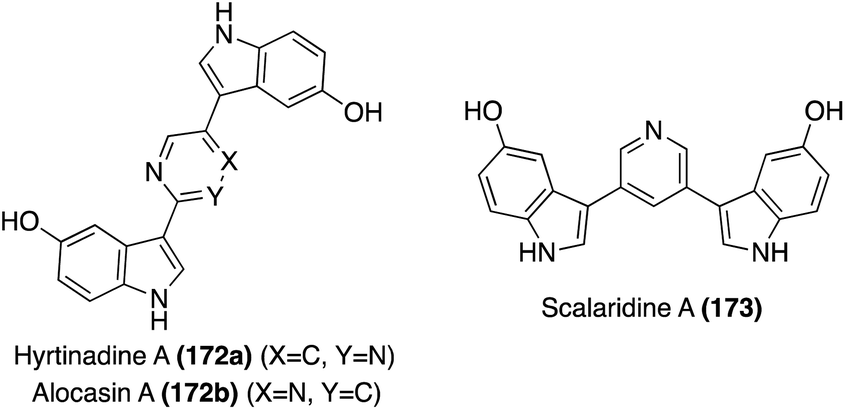 | ||
| Fig. 13 Chemical structures of Hyrtinadine A (172a), Alocasin A (172b), and Scalaridine A (173). | ||
The first of these natural products to be accessed via total synthesis was Hyrtinadine A (172b) in 2011, by Müller, et. al. As shown in Scheme 38, the first total synthesis of Hyrtinadine A (173) was achieved via sequential palladium-catalyzed Masuda borylation and double Suzuki cross-coupling reactions to install the two indole moieties on the 2- and 5-positions of the pyrimidine core. Subsequent de-methylation via BBr3 achieved Hyrtinadine A (172a) in 78% yield.104
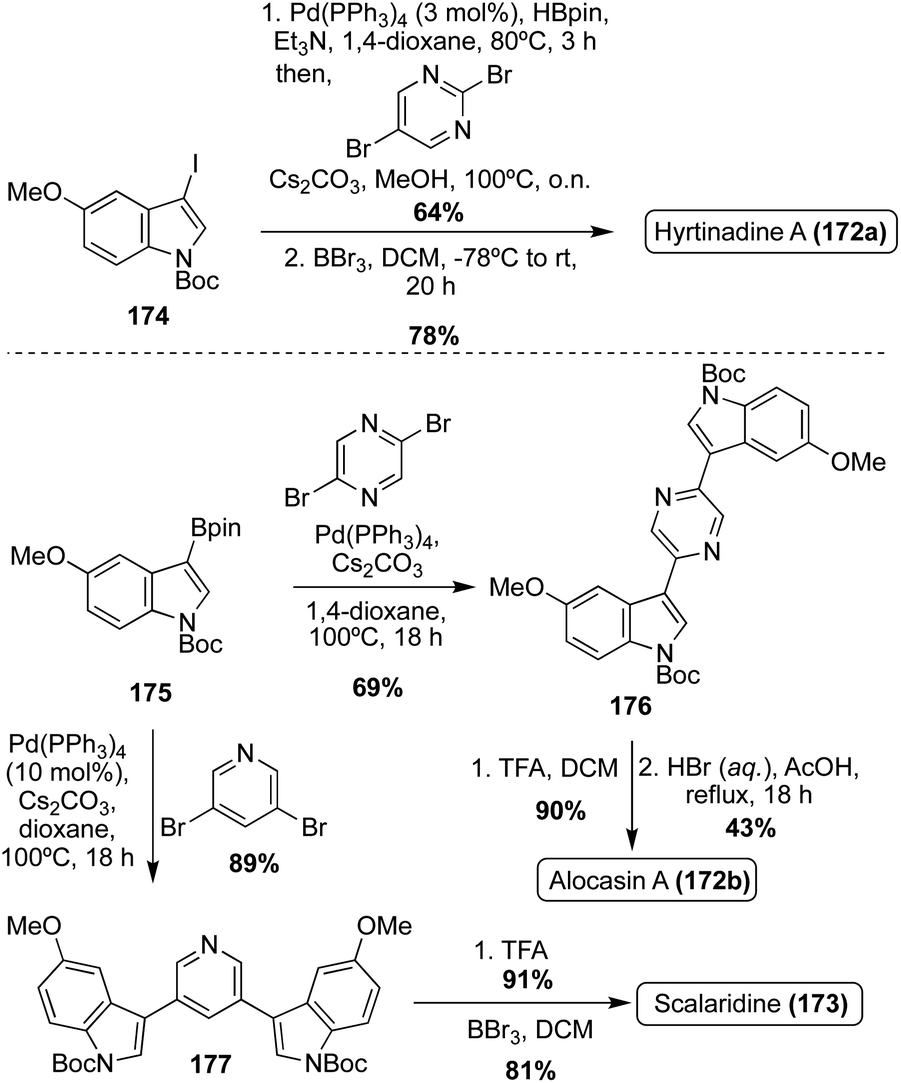 | ||
| Scheme 38 First total syntheses of Hyrtinadine A (172a), Alocasin A (172b), and Scalaridine A (173). | ||
In 2015, the first total syntheses of Alocasin A (172b) and Scalaridine A (173) were completed via a similar borylation and subsequent double Suzuki coupling route as was used in the first total synthesis of Hyrtinadine A (172a), followed by de-methylation of the indolic hydroxyl groups to access Alocasin A (172b) and Scalaridine A (173) in high yields.105,106
One year later, a different approach to the total synthesis of all three natural products (172a, 172b, and 173), was conducted via successive palladium-catalyzed reactions; a Kosugi–Migita–Stille cross coupling reaction to install the aromatic substituents on the heterocyclic linker, a reductive N-heterocyclization to form the indole moieties, and a final hydrogenolysis to access 172a, 172b, and 173 in high yields, as shown in Scheme 39.107
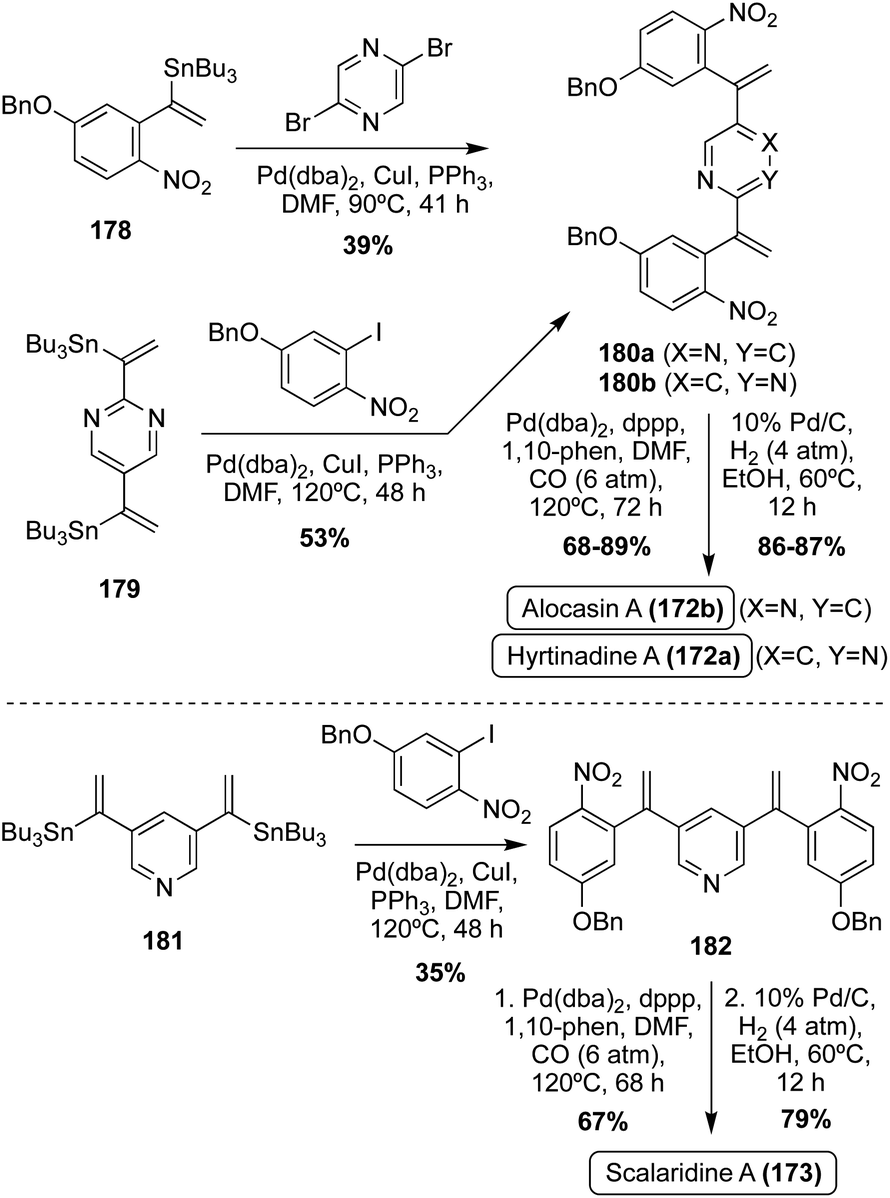 | ||
| Scheme 39 Total syntheses of Hyrtinadine A (172a), Alocasin A (172b), and Scalaridine A (173). | ||
As can be concluded from the previously discussed syntheses of Hyrtinidine A (172a), Alocasin A (172b), and Scalaridine A (173), the common method of synthesis for these natural products is the use of palladium-catalyzed cross-coupling reactions to install the indole moieties on the aromatic N-heterocyclic cores.
In recent years, additional total syntheses of 172a–b and 173 have been reported using similar palladium-catalyzed cross coupling approaches.108–110 A couple examples of these are shown in Scheme 40, such as the 2022 total synthesis of Scalaridine A (173) and the significantly expedited one-pot approach using a similar palladium-catalyzed methodology in the total synthesis of Alocasin A (172b). The latter achieved Alocasin A (172b) in 81% from the iodoindole intermediate (183), using a three phase, one-pot method.111
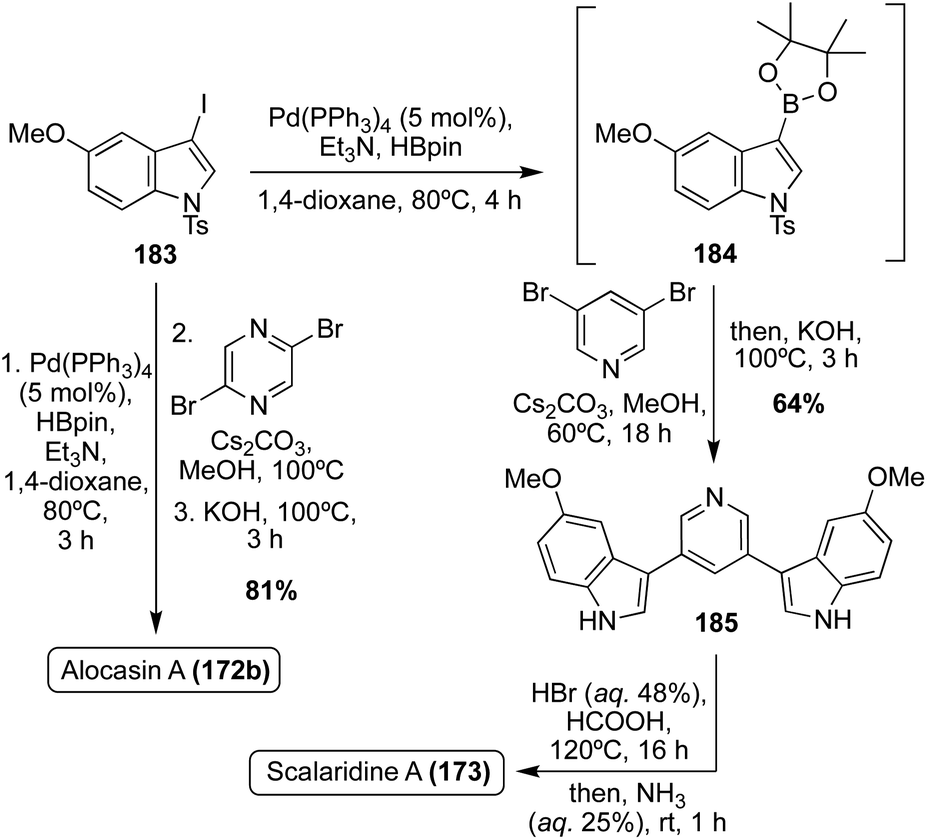 | ||
| Scheme 40 Total syntheses of Alocasin A (172b) and Scalaridine A (173). | ||
3. Tris-indole alkaloids
Compared to their previously discussed bis-indole alkaloid counterparts, tris-indole alkaloids are rarer and more elusive in nature. However, in recent years, discovery and synthesis of these natural products has become more prevalent.3.1 Aromatic N-heterocyclic linker moieties
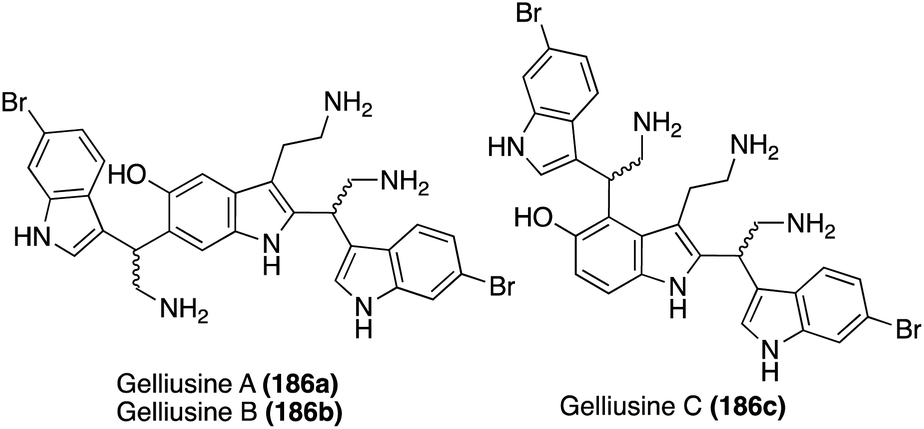 | ||
| Fig. 14 Chemical structures of Gelliusines A–C (186a–c). | ||
One year later, an additional structurally similar natural product of this class, Gelliusine C (186c) was isolated by the same group from the deep-water marine sponge Gellius sp. (also known as Orina sp). As shown in Fig. 14, the chemical structure of Gelliusine C (186c) was determined to be a similar tris-indole alkaloid containing in indole core connecting two additional indole moieties, as well as containing three amine substituents. The main difference was, instead of the 2,6-substituted indole core of 186a–b, the indole core of Gelliusine C (186c) had the two additional indole moieties connected at the C2 and C4 positions. Similar to 186a–b, the absolute configuration of Gelliusine C (186c) was inconclusive via obtained data and, because Gelliusines C (186c) have yet to be accessed via total synthesis, its absolute configuration also remains unknown. In terms of their biological activity, Gelliusine C (186c) exhibited cytotoxic activity, as well as anti-serotonin activity.113
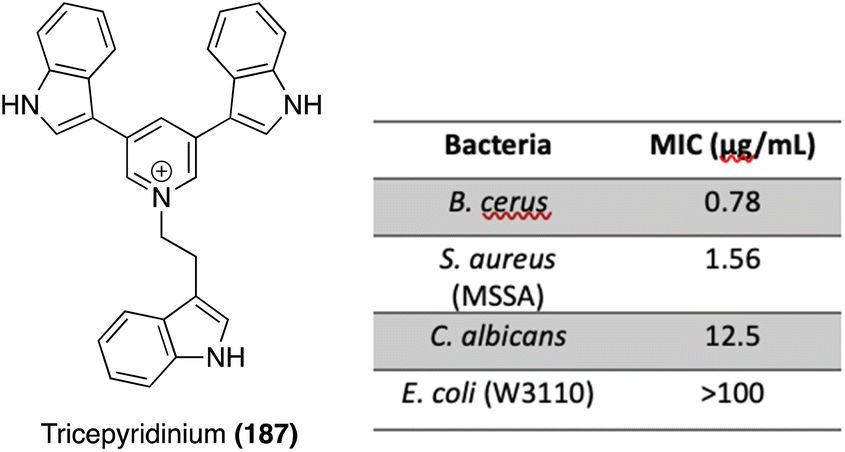 | ||
| Fig. 15 Chemical structure of Tricepyridinium (187) and its related biological activity data. | ||
The first total synthesis of Tricepyridinium (187) was completed in 2017 by Abe, et. al. at the time of its isolation and structural elucidation to confirm its structure. As shown in Scheme 41, a bis-borylation of 3,5-dibromopyridine was completed to access intermediate 188, followed by a subsequent palladium-catalyzed double Suzuki coupling to install two of the indole moieties on the pyridine ring. Boc deprotection and late-stage alkylation of the pyridine nitrogen via191 to install the third indole moiety afforded Tricepyridinium (187) in high yields.114
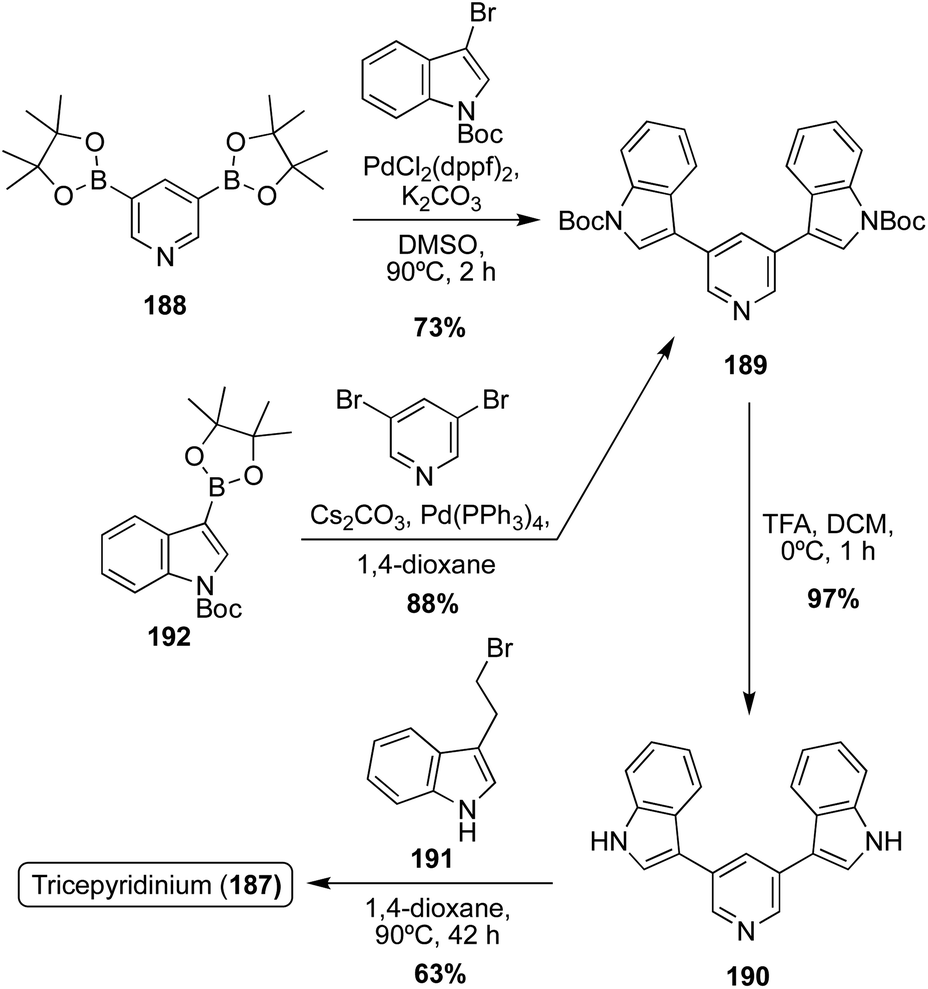 | ||
| Scheme 41 Total synthesis Tricepyridinium (187). | ||
In 2021, the total synthesis of Tricepyridinium (187) was completed via a similar approach as previously described. However, the major difference in this approach was the palladium catalyzed Suzuki coupling reaction, in which the electronics of the coupling partners were swapped. In this case, an indolic boronic ester (192) was coupled with 3,5-dibromopyridine, similar to the previously discussed approaches toward Scalaridine A (173). This approach led to a slightly higher yield of intermediate (189) of 88% compared to the previous 73% yield. Then, 189 was deprotected and acylated via alkyl bromide 191 to access Tricepyridinium (187) in 59% yield over two steps (Scheme 41).115
3.2 Non-aromatic N-heterocyclic linker moieties
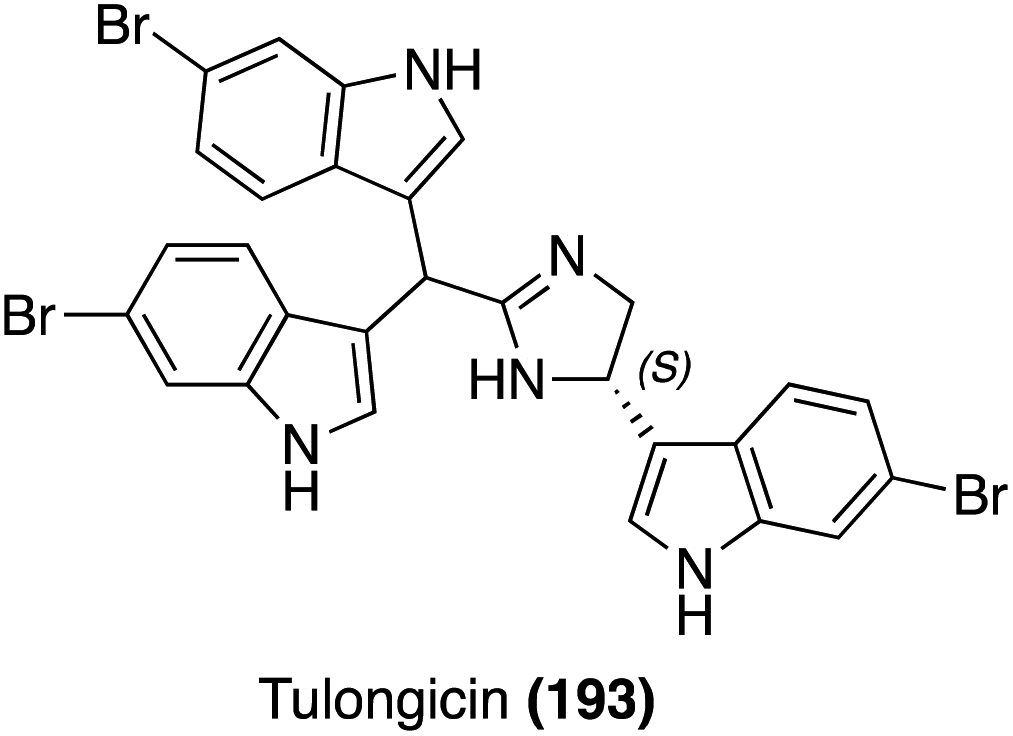 | ||
| Fig. 16 Chemical structure of Tulongicin (193). | ||
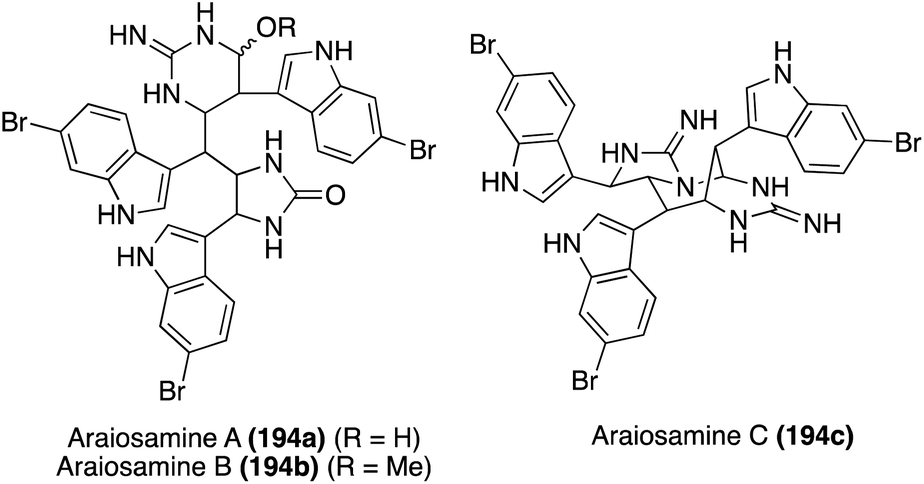 | ||
| Fig. 17 Chemical structures of Araiosamines A–C (194a–c). | ||
Of these Araiosamine natural products, Araiosamine C (194c) is the only one to have been accessed via total synthesis. In 2016, Baran, et. al. completed the first total synthesis via key guanidine installation and selective C–H functionalization steps driving toward a biomimetic final step to construct the complex core of 194c. As is shown in Scheme 42, the first total synthesis of Araiosamine C (194c) began with formation of 195, followed by deprotection and subsequent guanidinylation. Many guanidinylation reagents that were screened led to no reaction and it was hypothesized that the TFA group would impart adequate reactivity for the guanidinylation reagent to react with hindered or electron deficient amines. Reduction of the ester and subsequent cyclization led to 198 in 36% yield over three steps. The oxime intermediate (199) was then synthesized and subsequently reduced to the primary amine intermediate via SmI2. The reduction of the oxime (199) was very challenging because the oxime moiety is sterically encumbered by two adjacent indole groups and other functionalities such as the N,O-acetal or three aryl bromides are likely more prone toward reduction. After reaction of the resulting amine with N,N-di-Boc-S-methylisothiourea, the guanidine intermediate (200) was accessed in 53% over two steps.118 As shown in Scheme 42, after deprotection and elimination of the methoxy group via acidic conditions and high temperatures, 201 was setup for the pivotal cyclization of the guanidine nitrogen and the fused cyclic imine/enamine to form the characteristic bridgehead of 194c. After this sequence, Araiosamine C (194c) was accessed in a very high 81% yield.118
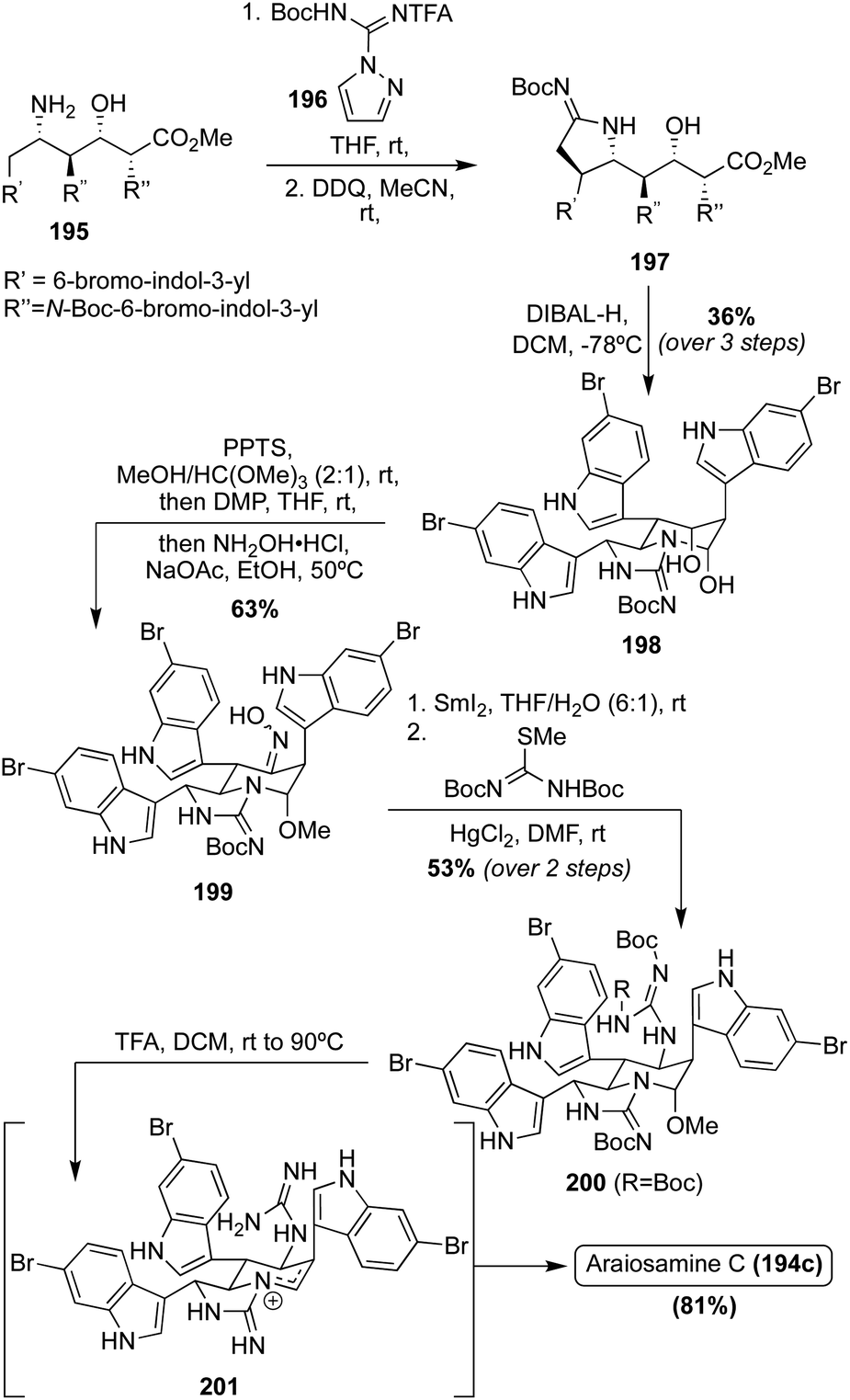 | ||
| Scheme 42 First total synthesis of Araiosamine C (194c). | ||
Since the absolute configuration of Araiosamine C (194c) was not known, the researchers adapted the previously discussed racemic path to 194c to gain access to a stereoselective synthesis. As shown in Scheme 43, this was done by utilizing Ellman's Auxiliary to synthesize the optically active 203 in 76% yield as the favored diastereomer (d.r. = 7:1:2:0.5). This stereochemistry was then retained through to 205, and using the same methodology as was previously discussed, the optically active (+)-Araiosamine C (194c) was synthesized, which was found to match the naturally isolated natural product based on optical rotation data.118 It is noteworthy to mention that Araiosamines A and B (194a–b) have yet to be accessed via total synthesis.
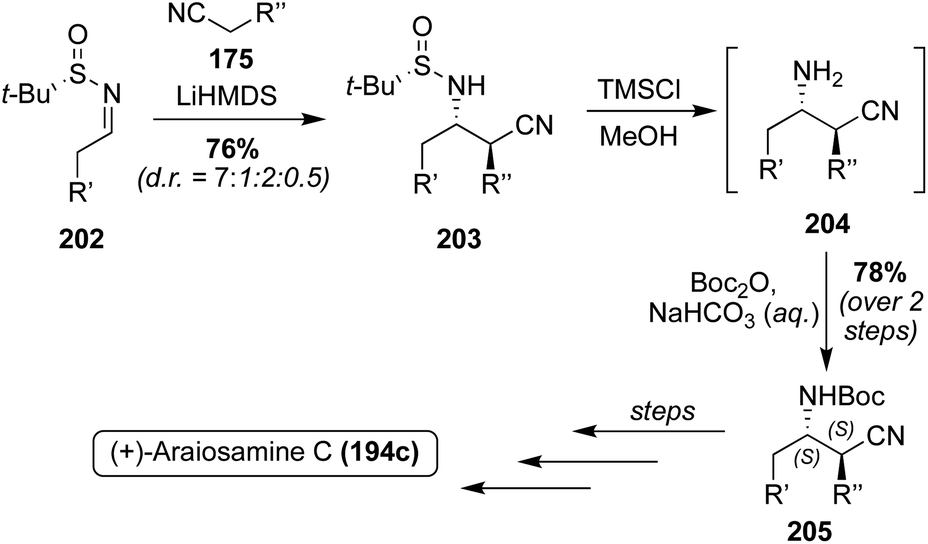 | ||
| Scheme 43 Enantioselective total synthesis of Araiosamine C (194c). | ||
4. Conclusions
To conclude, the examples discussed in this review summarize the wide range of bis- and tris-indole alkaloids containing N-heterocyclic linker moieties that have been identified and isolated in recent years. These molecules exhibit potent biological activities, such as antibacterial, antiviral, cytotoxic, and anti-inflammatory activities. Natural products of this kind seem to share significant antibacterial activity overall. This could prove to be very advantageous in the future, as antibiotic resistance continues to be a major global issue. However, many of these previously discussed natural products have yet to be accessed via total synthesis, preventing their detailed biological evaluations. Development of synthetic methodologies to access these natural products and explore their promising biological activities will likely be of significant interest to the field in the future. It is also likely that the class of bis- and tris-indole natural products containing N-heterocyclic linker moieties will continue to expand as additional novel alkaloids are isolated.5. Author contributions
Kyra R. Dvorak: conceptualization, writing original draft, writing, reviewing, and editing; Jetze, J. Tepe: supervision, reviewing, and editing.6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
The authors gratefully acknowledge the National Institutes of Health, National Institutes of Aging R01 AG076994 for financial support.8. Notes and references
- J. Kobayashi, T. Murayama, M. Ishibashi, S. Kosuge, M. Takamatsu, Y. Ohizumi, H. Kobayashi, T. Ohta, S. Nozoe and S. Takuma, Hyrtiosins A and B, New Indole Alkaloids from the Okinawan Marine Sponge Hyrtios Erecta, Tetrahedron, 1990, 46(23), 7699–7702, DOI:10.1016/S0040-4020(01)90065-1.
- B. Bao, Q. Sun, X. Yao, J. Hong, C.-O. Lee, H. Y. Cho and J. H. Jung, Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from a Marine Sponge Spongosorites Sp, J. Nat. Prod., 2007, 70(1), 2–8, DOI:10.1021/np060206z.
- N. E. Golantsov, A. A. Festa, A. V. Karchava and M. A. Yurovskaya, Marine Indole Alkaloids Containing an 1-(Indol-3-Yl)Ethane-1,2-Diamine Fragment (Review), Chem. Heterocycl. Compd., 2013, 49(2), 203–225, DOI:10.1007/s10593-013-1238-9.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications, Molecules, 2020, 25(8), 1909, DOI:10.3390/molecules25081909.
- K. Bartik, J.-C. Braekman, D. Daloze, C. Stoller, J. Huysecom, G. Vandevyver and R. Ottinger, Topsentins, New Toxic Bis-Indole Alkaloids from the Marine Sponge Topsentia Genitrix, Can. J. Chem., 1987, 65(9), 2118–2121, DOI:10.1139/v87-352.
- J. Shin, Y. Seo, K. W. Cho, J.-R. Rho and C. J. Sim, New Bis(Indole) Alkaloids of the Topsentin Class from the Sponge Spongosorites Genitrix, J. Nat. Prod., 1999, 62(4), 647–649, DOI:10.1021/np980507b.
- B. Bao, Q. Sun, X. Yao, J. Hong, C.-O. Lee, C. J. Sim, K. S. Im and J. H. Jung, Cytotoxic Bisindole Alkaloids from a Marine Sponge Spongosorites Sp, J. Nat. Prod., 2005, 68(5), 711–715, DOI:10.1021/np049577a.
- K.-B. Oh, W. Mar, S. Kim, J.-Y. Kim, M.-N. Oh, J.-G. Kim, D. Shin, C. J. Sim and J. Shin, Bis(Indole) Alkaloids as Sortase A Inhibitors from the Sponge Spongosorites Sp, Bioorg. Med. Chem. Lett., 2005, 15(22), 4927–4931, DOI:10.1016/j.bmcl.2005.08.021.
- S. A. Morris and R. J. Andersen, Nitrogenous Metabolites from the Deep Water Sponge Hexadella Sp, Can. J. Chem., 1989, 67(4), 677–681, DOI:10.1139/v89-102.
- S. A. Morris and R. J. Andersen, Brominated Bis(Indole) Alkaloids from the Marine Sponge Hexadella Sp, Tetrahedron, 1990, 46(3), 715–720, DOI:10.1016/S0040-4020(01)81355-7.
- J. H. Hung, P. B. Shinde, J. Hong, Y. Liu and C. J. Sim, Secondary metabolites from a marine sponge Discodermia calyx, Biochem. Syst. Ecol., 2007, 35, 38 Search PubMed.
- A. Casapullo, G. Bifulco, I. Bruno and R. Riccio, New Bisindole Alkaloids of the Topsentin and Hamacanthin Classes from the Mediterranean Marine Sponge Rhaphisia Lacazei, J. Nat. Prod., 2000, 63(4), 447–451, DOI:10.1021/np9903292.
- H.-B. Liu, G. Lauro, R. D. O'Connor, K. Lohith, M. Kelly, P. Colin, G. Bifulco and C. A. Bewley, Tulongicin, an Antibacterial Tri-Indole Alkaloid from a Deep-Water Topsentia Sp, J. Nat. Prod., 2017, 80(9), 2556–2560, DOI:10.1021/acs.jnatprod.7b00452.
- H. Sun, K. Sun and J. Sun, Recent Advances of Marine Natural Indole Products in Chemical and Biological Aspects, Molecules, 2023, 28(5), 2204, DOI:10.3390/molecules28052204.
- J. C. Braekman, D. Daloze, B. Moussiaux, C. Stoller and F. Deneubourg, Sponge Secondary Metabolites: New Results, Pure Appl. Chem., 1989, 61(3), 509–512, DOI:10.1351/pac198961030509.
- S. Tsujii, K. Rinehart and B. Topsentin, Dihydrodeoxytopsentin: Antiviral and Antitumor Bis(Indolyl)Imidazoles from Caribbean Deep-Sea Sponges of the Family Halichondriidae, Structural and Synthetic Studies, J. Org. Chem., 1988, 53(23), 5446–5453 CrossRef CAS.
- F. Y. Miyake, K. Yakushijin and D. A. Horne, A Concise Synthesis of Topsentin A and Nortopsentins B and D, Org. Lett., 2000, 2(14), 2121–2123, DOI:10.1021/ol000124g.
- S. Achab, A Three-Component Coupling Approach to the Marine Bis-Indole Alkaloids: Topsentin, Deoxytopsentin and Bromotopsentin, Tetrahedron Lett., 1996, 37(31), 5503–5506, DOI:10.1016/0040-4039(96)01148-3.
- S. K. Mal, L. Bohé and S. Achab, Convenient Access to Bis-Indole Alkaloids. Application to the Synthesis of Topsentins, Tetrahedron, 2008, 64(25), 5904–5914, DOI:10.1016/j.tet.2008.04.045.
- I. Kawasaki, H. Katsuma, Y. Nakayama and M. Yamashita, Total Synthesis of Topsentin, Antiviral and Antitumor Bis(Indolyl)Imidazole, Heterocycles, 1998, 48(9), 1887–1901 CrossRef CAS.
- X. Guinchard, Y. Vallée and J.-N. Denis, Total Synthesis of Marine Sponge Bis(Indole) Alkaloids of the Topsentin Class, J. Org. Chem., 2007, 72(10), 3972–3975, DOI:10.1021/jo070286r.
- X. Guinchard, Y. Vallée and J. N. Denis, Total Syntheses of Brominated Marine Sponge Alkaloids, Org. Lett., 2007, 9(19), 3761–3764, DOI:10.1021/ol701626m.
- O. Ottoni, A. D. V. F. Neder, A. K. B. Dias, R. P. A. Cruz and L. B. Aquino, Acylation of Indole under Friedel−Crafts Conditions An Improved Method To Obtain 3-Acylindoles Regioselectively, Org. Lett., 2001, 3(7), 1005–1007, DOI:10.1021/ol007056i.
- K. Murai, M. Morishita, R. Nakatani, O. Kubo, H. Fujioka and Y. Kita, Concise Total Synthesis of (−)-Spongotine A, J. Org. Chem., 2007, 72(23), 8947–8949, DOI:10.1021/jo701668s.
- N. K. Garg, R. Sarpong and B. M. Stoltz, The First Total Synthesis of Dragmacidin D, J. Am. Chem. Soc., 2002, 124(44), 13179–13184, DOI:10.1021/ja027822b.
- S. Sakemi and H. H. Sun, Nortopsentins A, B, and C Cytotoxic and Antifungal Imidazolediylbis[Indoles] from the Sponge Spongosorites Ruetzleri, J. Org. Chem., 1991, 56(13), 4304–4307, DOI:10.1021/jo00013a044.
- H. Sun, S. Sakemi, S. Gunasekera, Y. Kashman, M. Lui, N. Burres and P. McCarthy, Bis-Indole Compounds Which Are Useful Antitumor and Antimicrobial Agents, U.S. patent application, US4970226A, 1990 Search PubMed.
- I. Mancini, G. Guella, F. Pietra, C. Debitus and J. Waikedre, From Inactive Nortopsentin D, a Novel Bis(Indole) Alkaloid Isolated from the Axinellid Sponge Dragmacidon Sp. from Deep Waters South of New Caledonia, to a Strongly Cytotoxic Derivative, Helv. Chim. Acta, 1996, 79(8), 2075–2082, DOI:10.1002/hlca.19960790804.
- S. Tilvi, C. Moriou, M.-T. Martin, J.-F. Gallard, J. Sorres, K. Patel, S. Petek, C. Debitus, L. Ermolenko and A. Al-Mourabit, Agelastatin E, Agelastatin F, and Benzosceptrin C from the Marine Sponge Agelas Dendromorpha, J. Nat. Prod., 2010, 73(4), 720–723, DOI:10.1021/np900539j.
- M. Shi, K. Kazuta, Y. Satoh and Y. Masaki, Synthesis and Investigation of C2-Symmetric Optically Active Pyrrolidinium Salts as Chiral Phase-Transfer Catalysts, Chem. Pharm. Bull., 1994, 42(12), 2625–2628, DOI:10.1248/cpb.42.2625.
- J. Tan, Y. Chen, H. Li and N. Yasuda, Suzuki-Miyaura Cross-Coupling Reactions of Unprotected Haloimidazoles, J. Org. Chem., 2014, 79(18), 8871–8876, DOI:10.1021/jo501326r.
- K. L. Keel and J. J. Tepe, Total Synthesis of Nortopsentin D via a Late-Stage Pinacol-like Rearrangement, Org. Lett., 2021, 23(14), 5368–5372, DOI:10.1021/acs.orglett.1c01681.
- H. Sato, M. Tsuda, K. Watanabe and J. Kobayashi, Rhopaladins A–D, New Indole Alkaloids from Marine Tunicate Rhopalaea Sp, Tetrahedron, 1998, 54(30), 8687–8690, DOI:10.1016/S0040-4020(98)00470-0.
- P. Fresneda, P. Molina and M. Sanz, The First Synthesis of the Bis(Indole) Marine Alkaloid Rhopaladin D, Synlett, 2000, 2000(08), 1190–1192, DOI:10.1055/s-2000-6746.
- A. Fürstner, H. Krause and O. R. Thiel, Efficient Relay Syntheses and Assessment of the DNA-Cleaving Properties of the Pyrrole Alkaloid Derivatives Permethyl Storniamide A, Lycogalic Acid A Dimethyl Ester, and the Halitulin Core, Tetrahedron, 2002, 58(32), 6373–6380, DOI:10.1016/S0040-4020(02)00637-3.
- J. S. Park, E. Cho, J.-Y. Hwang, S. C. Park, B. Chung, O.-S. Kwon, C. J. Sim, D.-C. Oh, K.-B. Oh and J. Shin, Bioactive Bis(Indole) Alkaloids from a Spongosorites Sp. Sponge, Mar. Drugs, 2020, 19(1), 3, DOI:10.3390/md19010003.
- P. E. Lecoq de Boisbaudran, Compt. Rendus, 1882,(94), 55–59 Search PubMed.
- N. Durán and C. F. M. Menck, Chromobacterium Violaceum : A Review of Pharmacological and Industiral Perspectives, Crit. Rev. Microbiol., 2001, 27(3), 201–222, DOI:10.1080/20014091096747.
- L. H. Yang, H. Xiong, O. O. Lee, S.-H. Qi and P.-Y. Qian, Effect of Agitation on Violacein Production in Pseudoalteromonas Luteoviolacea Isolated from a Marine Sponge, Lett. Appl. Microbiol., 2007, 44(6), 625–630, DOI:10.1111/j.1472-765X.2007.02125.x.
- S. Yada, Y. Wang, Y. Zou, K. Nagasaki, K. Hosokawa, I. Osaka, R. Arakawa and K. Enomoto, Isolation and Characterization of Two Groups of Novel Marine Bacteria Producing Violacein, Mar. Biotechnol., 2008, 10(2), 128–132, DOI:10.1007/s10126-007-9046-9.
- S. Hakvåg, E. Fjærvik, G. Klinkenberg, S. E. Borgos, K. Josefsen, T. Ellingsen and S. Zotchev, Violacein-Producing Collimonas Sp. from the Sea Surface Microlayer of Costal Waters in Trøndelag, Norway, Mar. Drugs, 2009, 7(4), 576–588, DOI:10.3390/md7040576.
- P. Jiang, H. Wang, C. Zhang, K. Lou and X.-H. Xing, Reconstruction of the Violacein Biosynthetic Pathway from Duganella Sp. B2 in Different Heterologous Hosts, Appl. Microbiol. Biotechnol., 2010, 86(4), 1077–1088, DOI:10.1007/s00253-009-2375-z.
- T. Hoshino, Violacein and Related Tryptophan Metabolites Produced by Chromobacterium Violaceum: Biosynthetic Mechanism and Pathway for Construction of Violacein Core, Appl. Microbiol. Biotechnol., 2011, 91(6), 1463–1475, DOI:10.1007/s00253-011-3468-z.
- M. Durán, A. N. Ponezi, A. Faljoni-Alario, M. F. S. Teixeira, G. Z. Justo and N. Durán, Potential Applications of Violacein: A Microbial Pigment, Med. Chem. Res., 2012, 21(7), 1524–1532, DOI:10.1007/s00044-011-9654-9.
- J. A. Ballantine, C. B. Barrett, R. J. S. Beer, S. Eardley, A. Robertson, B. L. Shaw and T. H. Simpson, The Chemistry of Bacteria Part VII the Structure of Violacein, J. Chem. Soc., 1958, 5, 755–760 RSC.
- G. Wille and W. Steglich, A Short Synthesis of the Bacterial Pigments Violacein and Deoxyviolacein, Synthesis, 2001, 2001(05), 0759–0762, DOI:10.1055/s-2001-12776.
- M. T. Petersen and T. E. Nielsen, Tandem Ring-Closing Metathesis/Isomerization Reactions for the Total Synthesis of Violacein, Org. Lett., 2013, 15(8), 1986–1989, DOI:10.1021/ol400654r.
- W. Steglich, B. Steffan, L. Kopanski and G. Eckhardt, Indolfarbstoffe aus Fruchtkörpern des SchleimpilzesArcyria denudata, Angew. Chem., 1980, 92(6), 463–464, DOI:10.1002/ange.19800920607.
- S. Nakatani, A. Naoe, Y. Yamamoto, T. Yamauchi, N. Yamaguchi and M. Ishibashi, Isolation of Bisindole Alkaloids That Inhibit the Cell Cycle from Myxomycetes Arcyria Ferruginea and Tubifera Casparyi, Bioorg. Med. Chem. Lett., 2003, 13(17), 2879–2881, DOI:10.1016/S0960-894X(03)00592-4.
- A. P. Monte, D. Marona-Lewicka, N. V. Cozzi and D. E. Nichols, Synthesis and Pharmacological Examination of Benzofuran, Indan, and Tetralin Analogs of 3,4-(Methylenedioxy)Amphetamine, J. Med. Chem., 1993, 36(23), 3700–3706, DOI:10.1021/jm00075a027.
- D. Mochly-Rosen and L. M. Kauvar, Adv. Pharmacol., 1998, 44, 91–145 CAS.
- K. Kaniwa, M. A. Arai, X. Li and M. Ishibashi, Synthesis, Determination of Stereochemistry, and Evaluation of New Bisindole Alkaloids from the Myxomycete Arcyria Ferruginea: An Approach for Wnt Signal Inhibitor, Bioorg. Med. Chem. Lett., 2007, 17(15), 4254–4257, DOI:10.1016/j.bmcl.2007.05.033.
- M. Brenner, H. Rexhausen, B. Steffan and W. Steglich, Synthesis of Arcyriarubin b and Related Bisindolylmaleimides, Tetrahedron, 1988, 44(10), 2887–2892, DOI:10.1016/S0040-4020(88)90025-7.
- M. M. Faul, K. A. Sullivan and L. L. Winneroski, A General Approach to the Synthesis of Bisindolylmaleimides: Synthesis of Staurosporine Aglycone, Synthesis, 1995,(12), 1511–1516, DOI:10.1055/s-1995-4146.
- X. Mei, J. Wang, Z. Zhou, S. Wu, L. Huang, Z. Lin and Q. Ling, Diarylmaleic Anhydrides: Unusual Organic Luminescence, Multi-Stimuli Response and Photochromism, J. Mater. Chem., 2017, 5(8), 2135–2141, 10.1039/C6TC05519B.
- Q. Guo, W.-F. Deng, J.-L. Xiao, P.-C. Shi, J.-L. Li, Z.-X. Zhou and C. Ji, Synthesis, Single Crystal X-Ray Analysis and Vibrational Spectral Studies of 3,4-Di(1H-Indol-3-Yl)-1H-Pyrrole-2,5-dione, J. Mol. Struct., 2023, 1281, 135103, DOI:10.1016/j.molstruc.2023.135103.
- R. Fröde, C. Hinze, I. Josten, B. Schmidt, B. Steffan and W. Steglich, Isolation and Synthesis of 3,4-Bis(Indol-3-Yl)Pyrrole-2,5-Dicarboxylic Acid Derivatives from the Slime Mould Lycogala Epidendrum, Tetrahedron Lett., 1994, 35(11), 1689–1690, DOI:10.1016/0040-4039(94)88320-3.
- T. Hashimoto, Y. Akiyo, K. Akazawa, S. Takaoka, M. Tori and Y. Asakawa, Three Novel Dimethyl Pyrroledicarboxylate, Lycogarubins A-C, from the Myxomycetes Lycogala Epidendrum, Tetrahedron Lett., 1994, 35(16), 2559–2560, DOI:10.1016/S0040-4039(00)77170-X.
- T. Nishizawa, S. Grüschow, D.-H. E. Jayamaha, C. Nishizawa-Harada and D. H. Sherman, Enzymatic Assembly of the Bis-Indole Core of Rebeccamycin, J. Am. Chem. Soc., 2006, 128(3), 724–725, DOI:10.1021/ja056749x.
- W. Chen, Z. Feng, X. He, Q. Zhao and Q. Liang, Design and Synthesis of 3,4-Diarylpyrrole Analogues as Potent Topoisomerase Inhibitors, Med. Chem., 2018, 14(5), 485–494, DOI:10.2174/1573406414666180226164049.
- C. Hinze, A. Kreipl, A. Terpin and W. Steglich, Synthesis of Simple 3,4-Diarylpyrrole-2,5-Dicarboxylic Acids and Lukianol A by Oxidative Condensation of 3-Arylpyruvic Acids with Ammonia, Synthesis, 2007, 2007(04), 608–612, DOI:10.1055/s-2007-965876.
- N. Zhou, T. Xie, L. Liu and Z. Xie, Cu/Mn Co-Oxidized Cyclization for the Synthesis of Highly Substituted Pyrrole Derivatives from Amino Acid Esters: A Strategy for the Biomimetic Syntheses of Lycogarubin C and Chromopyrrolic Acid, J. Org. Chem., 2014, 79(13), 6061–6068, DOI:10.1021/jo500740w.
- X. Zheng, Y. Li, M. Guan, L. Wang, S. Wei, Y. Li, C. Chang and Z. Xu, Biomimetic Total Synthesis of the Spiroindimicin Family of Natural Products, Angew. Chem., Int. Ed., 2022, 61(38), e202208802, DOI:10.1002/anie.202208802.
- L. Fu and G. W. Gribble, Total Synthesis of Lycogarubin C Utilizing the Kornfeld–Boger Ring Contraction, Tetrahedron Lett., 2010, 51(3), 537–539, DOI:10.1016/j.tetlet.2009.11.085.
- J. S. Oakdale and D. L. Boger, Total Synthesis of Lycogarubin C and Lycogalic Acid, Org. Lett., 2010, 12(5), 1132–1134, DOI:10.1021/ol100146b.
- K. A. McArthur, S. S. Mitchell, G. Tsueng, A. Rheingold, D. J. White, J. Grodberg, K. S. Lam and B. C. M. Potts, Lynamicins A−E, Chlorinated Bisindole Pyrrole Antibiotics from a Novel Marine Actinomycete, J. Nat. Prod., 2008, 71(10), 1732–1737, DOI:10.1021/np800286d.
- W. Zhang, L. Ma, S. Li, Z. Liu, Y. Chen, H. Zhang, G. Zhang, Q. Zhang, X. Tian, C. Yuan, S. Zhang, W. Zhang, C. Zhang and A.-E. Indimicins, Bisindole Alkaloids from the Deep-Sea-Derived Streptomyces Sp. SCSIO 03032, J. Nat. Prod., 2014, 77(8), 1887–1892, DOI:10.1021/np500362p.
- I. Sigala, G. Ganidis, S. Thysiadis, A. L. Zografos, T. Giannakouros, V. Sarli and E. Nikolakaki, Lynamicin D an Antimicrobial Natural Product Affects Splicing by Inducing the Expression of SR Protein Kinase 1, Bioorg. Med. Chem., 2017, 25(5), 1622–1629, DOI:10.1016/j.bmc.2017.01.025.
- Z. Zhang, S. Ray, L. Imlay, L. T. Callaghan, H. Niederstrasser, P. L. Mallipeddi, B. A. Posner, D. M. Wetzel, M. A. Phillips and M. W. Smith, Total Synthesis of (+)-Spiroindimicin A and Congeners Unveils Their Antiparasitic Activity, Chem. Sci., 2021, 12(30), 10388–10394, 10.1039/D1SC02838C.
- S. Kohmoto, Y. Kashman, O. J. McConnell, K. L. Rinehart, A. Wright and F. Koehn, Dragmacidin, a New Cytotoxic Bis(Indole) Alkaloid from a Deep Water Marine Sponge, Dragmacidon Sp, J. Org. Chem., 1988, 53(13), 3116–3118, DOI:10.1021/jo00248a040.
- E. Fahy, B. C. M. Potts, D. J. Faulkner and K. Smith, 6-Bromotryptamine Derivatives from the Gulf of California Tunicate Didemnum Candidum, J. Nat. Prod., 1991, 54(2), 564–569, DOI:10.1021/np50074a032.
- T. Kawasaki, K. Ohno, H. Enoki, Y. Umemoto and M. Sakamoto, Syntheses of Bis(Indolyl)-Piperazine Alkaloids, Dragmacidin B and C, and Dihydrohamacanthin A, Tetrahedron Lett., 2002, 43(23), 4245–4248, DOI:10.1016/S0040-4039(02)00771-2.
- R. J. Capon, F. Rooney, L. M. Murray, E. Collins, A. T. R. Sim, J. A. P. Rostas, M. S. Butler and A. R. Carroll, Dragmacidins: New Protein Phosphatase Inhibitors from a Southern Australian Deep-Water Marine Sponge, Spongosorites Sp, J. Nat. Prod., 1998, 61(5), 660–662, DOI:10.1021/np970483t.
- A. Cutignano, G. Bifulco, I. Bruno, A. Casapullo, L. Gomez-Paloma, R. Riccio and F. Dragmacidin, A New Antiviral Bromoindole Alkaloid from the Mediterranean Sponge Halicortex Sp, Tetrahedron, 2000, 56(23), 3743–3748, DOI:10.1016/S0040-4020(00)00281-7.
- N. K. Garg, D. D. Caspi and B. M. Stoltz, The Total Synthesis of (+)-Dragmacidin F, J. Am. Chem. Soc., 2004, 126(31), 9552–9553, DOI:10.1021/ja046695b.
- K. S. Feldman and P. Ngernmeesri, Total Synthesis of (±)-Dragmacidin E, Org. Lett., 2011, 13(20), 5704–5707, DOI:10.1021/ol202535f.
- A. Wright, K. Killday, D. Chakrabarti, E. Guzmán, D. Harmody, P. McCarthy, T. Pitts, S. Pomponi, J. Reed, B. Roberts, C. Rodrigues Felix, K. Rohde and G. Dragmacidin, a Bioactive Bis-Indole Alkaloid from a Deep-Water Sponge of the Genus Spongosorites, Mar. Drugs, 2017, 15(1), 16, DOI:10.3390/md15010016.
- B. Jiang, J. M. Smallheer, C. Amaral-Ly and M. A. Wuonola, Total Synthesis of (.+-.)-Dragmacidin: A Cytotoxic Bis(Indole)Alkaloid of Marine Origin, J. Org. Chem., 1994, 59(22), 6823–6827, DOI:10.1021/jo00101a051.
- C. R. Whitlock and M. P. Cava, A Total Synthesis of Dragmacidin B, Tetrahedron Lett., 1994, 35(3), 371–374, DOI:10.1016/0040-4039(94)85056-9.
- T. Kawasaki, H. Enoki, K. Matsumura, M. Ohyama, M. Inagawa and M. Sakamoto, Total Synthesis of Dragmacidin A via Inolylglycines, Org. Lett., 2000, 2(19), 3027–3029, DOI:10.1021/ol006394g.
- C. G. Yang, J. Wang, X. X. Tang and B. Jiang, Asymmetric Aminohydroxylation of Vinyl Indoles: A Short Enantioselective Synthesis of the Bisindole Alkaloids Dihydrohamacanthin A and Dragmacidin A, Tetrahedron: Asymmetry, 2002, 13(4), 383–394, DOI:10.1016/S0957-4166(02)00111-8.
- C. G. Yang, J. Wang, X. X. Tang and B. Jiang, Asymmetric Aminohydroxylation of Vinyl Indoles: a Short Enantioselective Synthesis of the Bisindole Alkaloids Dihydrohamacanthin and Dragmacidin A, Tetrahedron: Asymmetry, 2002, 13, 383, DOI:10.1016/S0957-4166(02)00111-8.
- F. Y. Miyake, K. Yakushijin and D. A. Horne, A Facile Synthesis of Dragmacidin B and 2,5-Bis(6′-Bromo-3′-Indolyl)Piperazine, Org. Lett., 2000, 2(20), 3185–3187, DOI:10.1021/ol0001970.
- F. Tonsiengsom, F. Y. Miyake, K. Yakushijin and D. A. Horne, Synthesis, 2006, 49 CAS.
- F. Zhang, B. Wang, P. Prasad, R. J. Capon and Y. Jia, Asymmetric Total Synthesis of (+)-Dragmacidin D Reveals Unexpected Stereocomplexity, Org. Lett., 2015, 17(6), 1529–1532, DOI:10.1021/acs.orglett.5b00327.
- S. P. Gunasekera, P. J. McCarthy and M. Kelly-Borges, Hamacanthins A and B, New Antifungal Bis Indole Alkaloids from the Deep-Water Marine Sponge, Hamacantha Sp, J. Nat. Prod., 1994, 57(10), 1437–1441, DOI:10.1021/np50112a014.
- B. Jiang, C.-G. Yang and J. Wang, Enantioselective Synthesis for the (−)-Antipode of the Pyrazinone Marine Alkaloid, Hamacanthin A, J. Org. Chem., 2001, 66(14), 4865–4869, DOI:10.1021/jo010265b.
- B. Jiang, C.-G. Yang and J. Wang, Enantioselective Synthesis of Marine Indole Alkaloid Hamacanthin B, J. Org. Chem., 2002, 67(4), 1396–1398, DOI:10.1021/jo0108109.
- J. H. Jung, P. B. Shinde, J. Hong, Y. Liu and C. J. Sim, Secondary Metabolites from a Marine Sponge Discodermia Calyx, Biochem. Syst. Ecol., 2007, 35(1), 48–51, DOI:10.1016/j.bse.2006.08.008.
- T. Kawasaki, T. Kouko, H. Totsuka and K. Hiramatsu, Synthesis of Marine Bisindole Alkaloids, Hamacanthins A and B through Intramolecular Transamidation–Cyclization, Tetrahedron Lett., 2003, 44(49), 8849–8852, DOI:10.1016/j.tetlet.2003.09.184.
- K. Higuchi, R. Takei, T. Kouko and T. Kawasaki, Total Synthesis of Marine Bisindole Alkaloid (+)- Cis -Dihydrohamacanthin B, Synthesis, 2007, 2007(5), 669–674, DOI:10.1055/s-2007-965907.
- X. Guinchard, Total Syntheses of Brominated Marine Sponge Alkaloids, Org. Lett., 2007, 9(19), 3761–3764, DOI:10.1021/ol701626m.
- A. G. Kozlovsky, N. G. Vinokurova, V. M. Adanin, G. Burkhardt, H. M. Dahse and U. Gräfe, New Diketopiperazine Alkaloids from Penicillium Fellutanum, J. Nat. Prod., 2000, 63(5), 698–700, DOI:10.1021/np9903853.
- H. Hayashi, Bioactive Alkaloids of Fungal Origin, Studies in Natural Products Chemistry, Elsevier, 2005, vol. 32, pp. 549–609, DOI:10.1016/S1572-5995(05)80064-X.
- J. M. Schkeryantz, J. C. G. Woo and S. J. Danishefsky, Total Synthesis of Gypsetin, J. Am. Chem. Soc., 1995, 117(26), 7025–7026, DOI:10.1021/ja00131a035.
- J. M. Schkeryantz, J. C. G. Woo, P. Siliphaivanh, K. M. Depew and S. J. Danishefsky, Total Synthesis of Gypsetin, Deoxybrevianamide E, Brevianamide E, and Tryprostatin B: Novel Constructions of 2,3-Disubstituted Indoles, J. Am. Chem. Soc., 1999, 121(51), 11964–11975, DOI:10.1021/ja9925249.
- K. B. Joshi and S. Verma, Participation of Aromatic Side Chains in Diketopiperazine Ensembles, Tetrahedron Lett., 2008, 49(27), 4231–4234, DOI:10.1016/j.tetlet.2008.04.156.
- X. Cheng and L. Ma, Enzymatic Synthesis of Fluorinated Compounds, Appl. Microbiol. Biotechnol., 2021, 105(21), 8033–8058 CrossRef CAS PubMed.
- H. Usuki, Y. Yamamoto, J. Arima, M. Iwabuchi, S. Miyoshi, T. Nitoda and T. Hatanaka, Peptide Bond Formation by Aminolysin-A Catalysis: A Simple Approach to Enzymatic Synthesis of Diverse Short Oligopeptides and Biologically Active Puromycins, Org. Biomol. Chem., 2011, 9(7), 2327, 10.1039/c0ob00403k.
- S. P. Kelly, V. V. Shende, A. R. Flynn, Q. Dan, Y. Ye, J. L. Smith, S. Tsukamoto, M. S. Sigman and D. H. Sherman, Data Science-Driven Analysis of Substrate-Permissive Diketopiperazine Reverse Prenyltransferase NotF: Applications in Protein Engineering and Cascade Biocatalytic Synthesis of (−)-Eurotiumin A, J. Am. Chem. Soc., 2022, 144(42), 19326–19336, DOI:10.1021/jacs.2c06631.
- T. Endo, M. Tsuda, J. Fromont and J. Kobayashi, Hyrtinadine A, a Bis-Indole Alkaloid from a Marine Sponge, J. Nat. Prod., 2007, 70(3), 423–424, DOI:10.1021/np060420n.
- L. Zhu, C. Chen, H. Wang, W. Ye and G. Zhou, Indole Alkaloids from Alocasia Macrorrhiza, Chem. Pharm. Bull., 2012, 60(5), 670–673, DOI:10.1248/cpb.60.670.
- Y.-J. Lee, D.-G. Lee, H. S. Rho, V. B. Krasokhin, H. J. Shin, J. S. Lee and H.-S. Lee, Cytotoxic 5-Hydroxyindole Alkaloids from the Marine Sponge Scalarispongia Sp.: Cytotoxic 5-Hydroxyindole Alkaloids from the Marine Sponge Scalarispongia Sp, J. Heterocycl. Chem., 2013, 50(6), 1400–1404, DOI:10.1002/jhet.1599.
- B. O. A. Tasch, E. Merkul and T. J. J. Müller, One-Pot Synthesis of Diazine-Bridged Bisindoles and Concise Synthesis of the Marine Alkaloid Hyrtinadine A, Eur. J. Org Chem., 2011, 2011(24), 4532–4535, DOI:10.1002/ejoc.201100680.
- S. H. Kim and J. Sperry, Synthesis of Alocasin A, J. Nat. Prod., 2015, 78(12), 3080–3082, DOI:10.1021/acs.jnatprod.5b00853.
- S. H. Kim and J. Sperry, Synthesis of Scalaridine A, Tetrahedron Lett., 2015, 56(43), 5914–5915, DOI:10.1016/j.tetlet.2015.09.033.
- N. H. Ansari and B. C. G. Söderberg, Short Syntheses of the Indole Alkaloids Alocasin A, Scalaridine A, and Hyrtinadine A-B, Tetrahedron, 2016, 72(29), 4214–4221, DOI:10.1016/j.tet.2016.05.057.
- G. Ranjani and R. Nagarajan, Insight into Copper Catalysis: In Situ Formed Nano Cu2O in Suzuki–Miyaura Cross-Coupling of Aryl/Indolyl Boronates, Org. Lett., 2017, 19(15), 3974–3977, DOI:10.1021/acs.orglett.7b01669.
- M. Kruppa, G. A. Sommer and T. J. J. Müller, Concise Syntheses of Marine (Bis)Indole Alkaloids Meridianin C, D, F, and G and Scalaridine A via One-Pot Masuda Borylation-Suzuki Coupling Sequence, Molecules, 2022, 27(7), 2233, DOI:10.3390/molecules27072233.
- J. Dong, H. Ma, B. Wang, S. Yang, Z. Wang, Y. Li, Y. Liu and Q. Wang, Discovery of Hyrtinadine A and Its Derivatives as Novel Antiviral and Anti-Phytopathogenic-Fungus Agents, Molecules, 2022, 27(23), 8439, DOI:10.3390/molecules27238439.
- N. Rehberg, G. A. Sommer, D. Drießen, M. Kruppa, E. T. Adeniyi, S. Chen, L. Wang, K. Wolf, B. O. A. Tasch, T. R. Ioerger, K. Zhu, T. J. J. Müller and R. Kalscheuer, Nature-Inspired (Di)Azine-Bridged Bisindole Alkaloids with Potent Antibacterial In Vitro and In Vivo Efficacy against Methicillin-Resistant Staphylococcus Aureus, J. Med. Chem., 2020, 63(21), 12623–12641, DOI:10.1021/acs.jmedchem.0c00826.
- G. Bifulco, I. Bruno, L. Minale, R. Riccio, A. Calignano and C. Debitus, (±)-Gelliusines A and B, Two Diastereomeric Brominated Tris-Indole Alkaloids from a Deep Water New Caledonian Marine Sponge (Gellius or Orina Sp.), J. Nat. Prod., 1994, 57(9), 1294–1299, DOI:10.1021/np50111a020.
- G. Bifulco, I. Bruno, R. Riccio, J. Lavayre and G. Bourdy, Further Brominated Bis- and Tris-Indole Alkaloids from the Deep-Water New Caledonian Marine Sponge Orina Sp, J. Nat. Prod., 1995, 58(8), 1254–1260, DOI:10.1021/np50122a017.
- M. Okada, T. Sugita, C. P. Wong, T. Wakimoto and I. Abe, Identification of Pyridinium with Three Indole Moieties as an Antimicrobial Agent, J. Nat. Prod., 2017, 80(4), 1205–1209, DOI:10.1021/acs.jnatprod.6b01152.
- M. A. Garrison, A. R. Mahoney and W. M. Wuest, Tricepyridinium-inspired QACs Yield Potent Antimicrobials and Provide Insight into QAC Resistance, Chem. Med. Chem., 2021, 16(3), 463–466, DOI:10.1002/cmdc.202000604.
- H.-B. Liu, G. Lauro, R. D. O'Connor, K. Lohith, M. Kelly, P. Colin, G. Bifulco and C. A. Bewley, Tulongicin, an Antibacterial Tri-Indole Alkaloid from a Deep-Water Topsentia Sp, J. Nat. Prod., 2017, 80(9), 2556–2560, DOI:10.1021/acs.jnatprod.7b00452.
- X. Wei, N. M. Henriksen, J. J. Skalicky, M. K. Harper, T. E. Cheatham, C. M. Ireland and R. M. Van Wagoner, Araiosamines A−D: Tris-Bromoindole Cyclic Guanidine Alkaloids from the Marine Sponge Clathria (Thalysias) Araiosa, J. Org. Chem., 2011, 76(14), 5515–5523, DOI:10.1021/jo200327d.
- M. Tian, M. Yan and P. S. Baran, 11-Step Total Synthesis of Araiosamines, J. Am. Chem. Soc., 2016, 138(43), 14234–14237, DOI:10.1021/jacs.6b09701.
| This journal is © The Royal Society of Chemistry 2024 |