
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the total synthesis of galantamine, a natural medicine for Alzheimer's disease
Bichu
Cheng
 ac,
Qi
Wang
c,
Yi
An
a and
Fener
Chen
*abc
ac,
Qi
Wang
c,
Yi
An
a and
Fener
Chen
*abc
aEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: rfchen@fudan.edu.cn
bCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang, Jiangxi 330022, China
cSchool of Science, Green Pharmaceutical Engineering Research Center, Harbin Institute of Technology, Shenzhen 518055, China
First published on 7th March 2024
Abstract
Covering: 2006 to 2023
(−)-Galantamine is a natural product with distinctive structural features and potent inhibitory activity against acetylcholine esterase (AChE). It is clinically approved for the treatment of Alzheimer's disease. The clinical significance and scarcity of this natural product have prompted extensive and ongoing efforts towards the chemical synthesis of this challenging tetracyclic structure. The objective of this review is to summarize and discuss recent progress in the total synthesis of galantamine from 2006 to 2023. The contents are organized according to the synthetic strategies for the construction of the quaternary center. Key features of each synthesis have been highlighted, followed by a summary and outlook at the end.
1. Introduction
(−)-Galantamine (1) is a natural product of the Amaryllidaceae alkaloid family. It was first isolated from the plant Caucasian snowdrop in the 1950s by scientists from the Soviet Union, and later from the bulbs of several different species of the Amaryllidaceae family.1,2 These plants have been used in traditional herbal medicine for centuries. (−)-Galantamine (1) was found to be the major active component and has a dual mechanism of action on the cholinergic system.3 It is a centrally acting, selective, reversible, and competitive acetylcholinesterase (AChE) inhibitor and an allosteric modulator of the nicotinic receptor for acetylcholine (Fig. 1). It has 50-fold greater selectivity for human erythrocyte AChE than for plasma BuChE (AChE IC50 = 0.35 μM; BuChE IC50 = 18.6 μM).4 In 2001, after half a century of investigation, (−)-galantamine hydrobromide, commercially known as Razadyne, was approved by the FDA as a drug for the treatment of mild to moderate confusion (dementia) related to Alzheimer's disease (AD). A meta-analysis of randomized controlled trials of galantamine showed that it significantly improved cognitive, behavioral, and global performance in patients with AD.5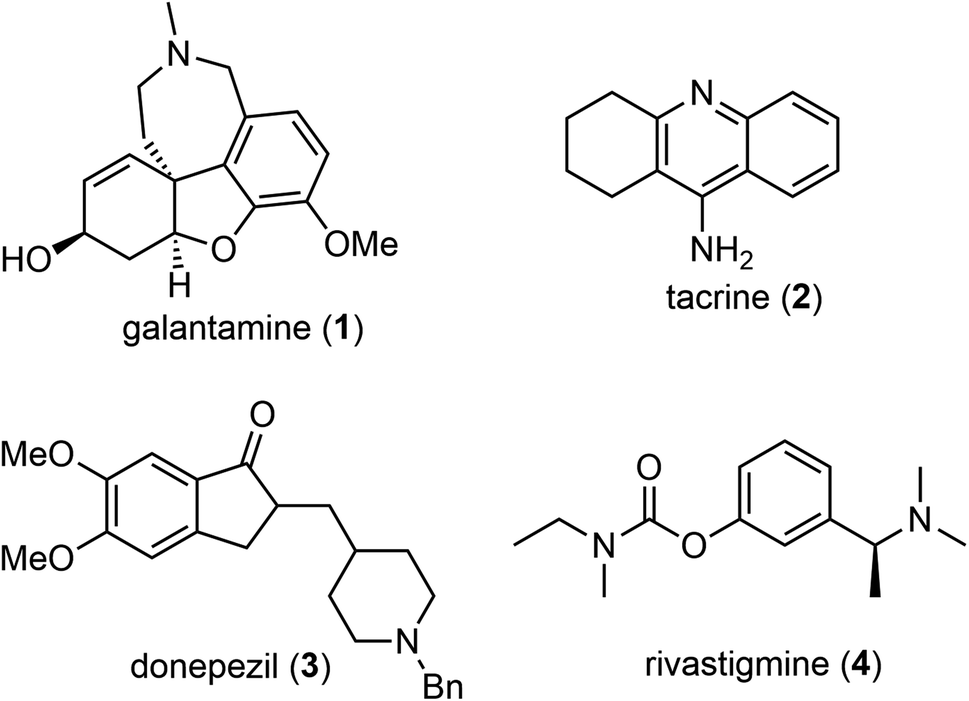 | ||
| Fig. 1 Structure of (−)-galantamine and AChE inhibitors. | ||
(−)-Galantamine (1) has a fascinating and synthetically challenging structure containing a strained tetracyclic framework and three stereocenters. The fused tetracyclic structure consists of an aromatic A ring, a heterocyclic B ring, a cyclohexenol C ring and an azepine D ring. The three stereocenters, including a spiro benzylic quaternary center,6–8 are embedded in the tetracyclic structure (Fig. 2). The structure of (−)-galantamine (1) and its close relatives narwedine (5) and lycoramine (6) is similar to that of morphine (7). These structural challenges, and the demand for an economical and sustainable supply of the natural product to the market have stimulated extensive and ongoing interest in the chemical synthesis of galantamine (1).
 | ||
| Fig. 2 Structure of (−)-galantamine from different perspectives and related natural products. | ||
Numerous synthetic studies of (−)-galantamine (1) and its analogues have been performed and dozens of total syntheses have been reported.9–12 This review aims to summarize the recent synthetic achievements of this fascinating natural product and to discuss the evolution of the synthetic strategies employed during different periods.13–43 Special focus is placed on the construction of the key benzylic quaternary center and the asymmetric synthesis. For the sake of brevity and completeness of this review, only a few selected key early discoveries prior to 2006 will be summarized, followed by more recent developments.
2. Biomimetic oxidative coupling reactions
The biosynthesis of galantamine with oxidative ortho–para phenol coupling as the key step was first proposed by Barton and coworkers and later supported by their own biomimetic synthesis. Since then, oxidative coupling of phenols44 has become the most extensively studied method for the total synthesis of galantamine. The early efforts have been summarized in a previous review.92.1 Biosynthesis of galantamine
Barton and co-workers recognized that Amaryllidaceae alkaloids, including galantamine (1), could be derived from a common precursor, 4′-O-methylnorbelladine (8) (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, or its N-methyl congener) (Scheme 1).45 Oxidative ortho–para phenol coupling of 8 forms the central C–C bond, with simultaneous generation of the quaternary center and the azepine ring. An intramolecular oxa-Michael addition of the phenol to the quinoid system generates product 10 with the tetracyclic scaffold. Further reduction of the keto group and methylation of the amine would afford galantamine (1). The biosynthesis of Amaryllidaceae alkaloids has been investigated biochemically using labelled precursors and intermediates.45–47 More recently, genomic studies have identified several biosynthetic genes that encode enzymes involved in these steps.48,49
H, or its N-methyl congener) (Scheme 1).45 Oxidative ortho–para phenol coupling of 8 forms the central C–C bond, with simultaneous generation of the quaternary center and the azepine ring. An intramolecular oxa-Michael addition of the phenol to the quinoid system generates product 10 with the tetracyclic scaffold. Further reduction of the keto group and methylation of the amine would afford galantamine (1). The biosynthesis of Amaryllidaceae alkaloids has been investigated biochemically using labelled precursors and intermediates.45–47 More recently, genomic studies have identified several biosynthetic genes that encode enzymes involved in these steps.48,49
 | ||
| Scheme 1 Possible biosynthetic pathway for galantamine. | ||
2.2 Previous results of oxidative phenol coupling
Barton and Kirby were the first to report the biomimetic intramolecular oxidative coupling of 11a with potassium ferricyanide (K3Fe(CN)6) as the oxidant, resulting in the desired product 12a, albeit in very low yield (Scheme 2). Subsequent ketone reduction with lithium aluminium hydride (LiAlH4) led to a mixture of (±)-galantamine and (±)-epigalantamine.50 This landmark biomimetic synthesis inspired many groups to make significant contributions to improve the performance of Barton's synthesis in the following years. Subsequent modifications were made to the substrate structure, and alternative oxidants were employed to enhance the efficacy of the intramolecular oxidative phenol coupling reaction.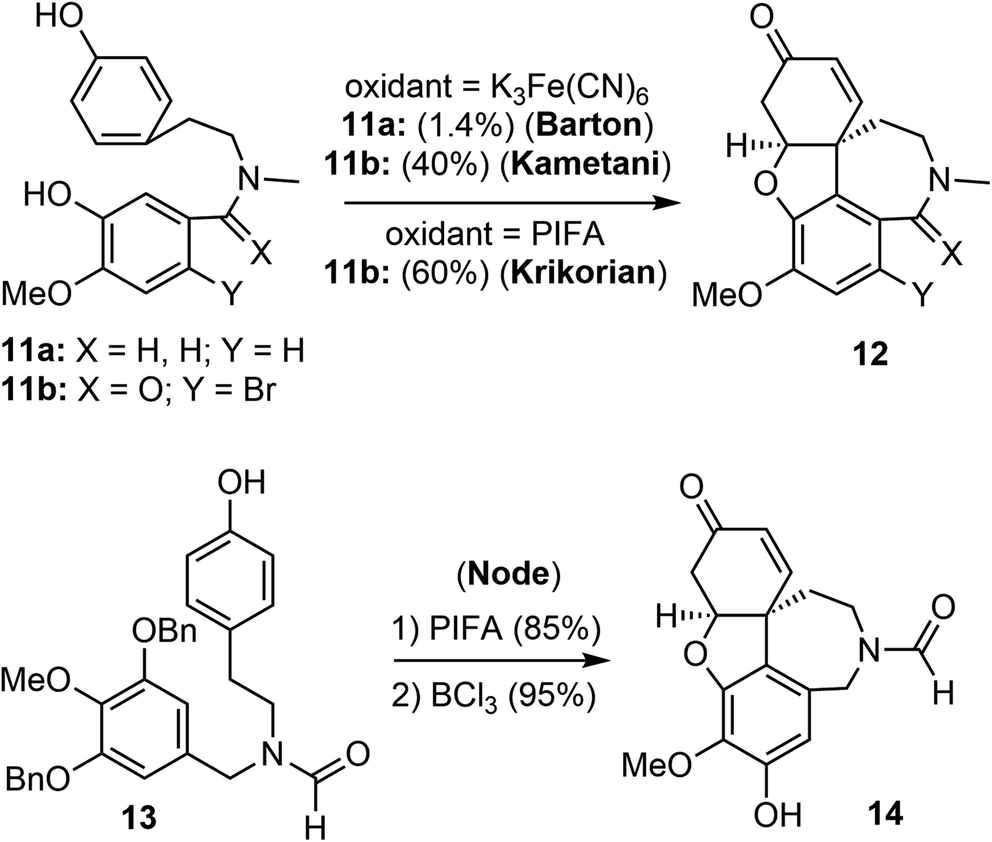 | ||
| Scheme 2 Racemic oxidative coupling reactions. | ||
Kametani and co-workers found that by blocking the para position of the phenol with a bromide and using a lactam instead of an amine significantly increased the yield of the oxidative phenol coupling (11b).51,52 In 1998, Kita and co-workers reported that [bis(trifluoroacetoxy)iodo]benzene (PIFA) was a suitable oxidant for promoting the diphenol coupling.53 Krikorian and co-workers then applied PIFA to the key oxidative coupling reaction of amide 11b and obtained the tetracyclic derivative 12b in 60% yield.54 In 2001, Node and co-workers reported a synthesis of galantamine using the symmetric N-formamide 13 as the substrate.55 The PIFA-promoted oxidative coupling reaction of 13 in trifluoroethanol at room temperature afforded a dienone in 85% yield, a selective O-debenzylation using BCl3 and in situ oxa-Michael addition provided the narwedine-type product 14 in a high yield. The use of the symmetrical substrate 13 as a precursor avoided the use of bromide as a blocking group at the para-phenol position. The extra hydroxy group was later removed by a palladium-catalyzed reduction of the corresponding triflate.
The first asymmetric synthesis of enantiomerically pure (+)- and (−)-galantamine was reported by Koga's laboratory (Scheme 3).56 Compound 15 was derived from L-tyrosine methyl ester. The phenol oxidative ortho–para coupling reaction was carried out with 5 equiv. of manganic tris(acetylacetonate) (Mn(acac)3) in acetonitrile. The resulting tetracyclic compound was protected as the diethyl phosphonate to give 16 in 81% yield. Compound 16 was then converted to (+)-galantamine. Node and co-workers reported in 2004 a creative synthesis of (−)-galantamine based on the concept of remote asymmetric induction.57 Compound 17 was readily obtained from tyramine with (R)-N-BOC-D-phenylalanine. The oxidative phenol coupling reaction of 17 afforded a dienone. O-debenzylation and in situ oxa-Michael addition delivered product 18, which was converted to (−)-galantamine.
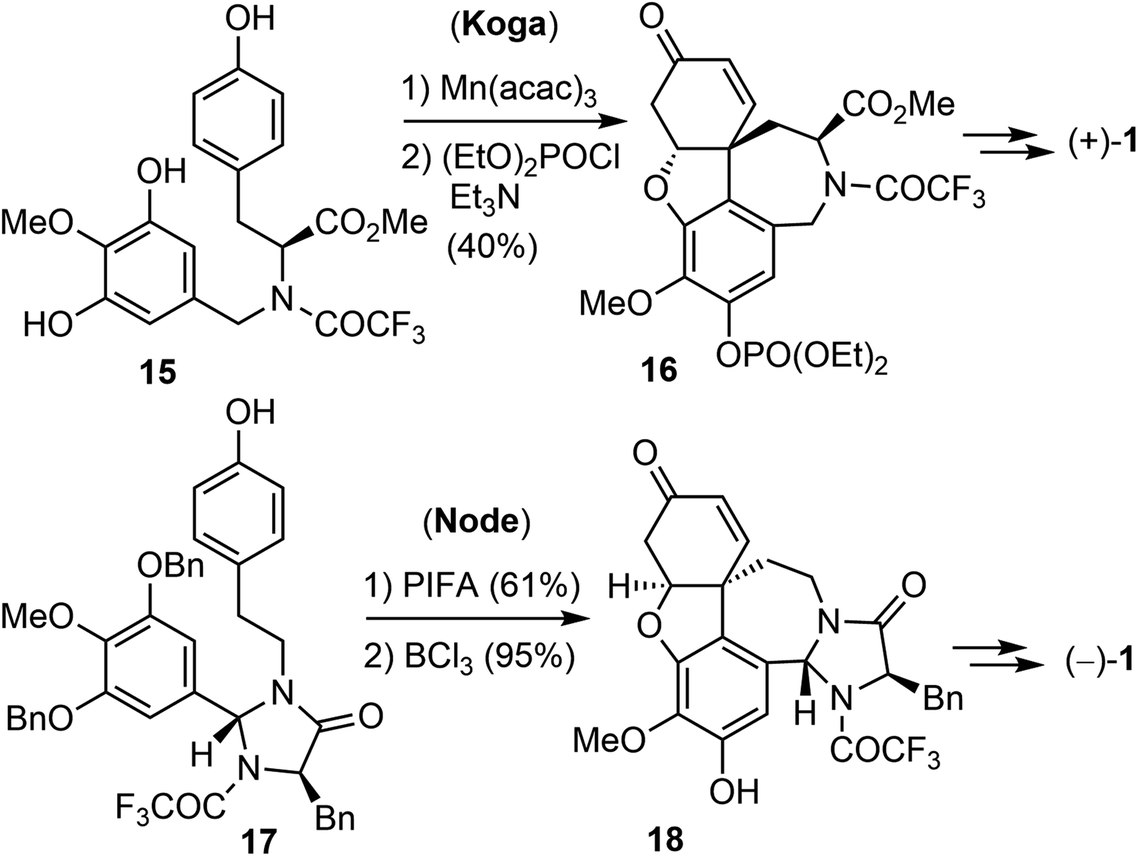 | ||
| Scheme 3 Asymmetric oxidative coupling reactions. | ||
Barton and Kirby also investigated the interconversion of galantamine and narwedine (Scheme 4).50 Oxidation of (−)-galantamine (1) with manganese dioxide, and crystallization from acetone led to (−)-narwedine (5). Serendipitously, they discovered that crystallization from ethanol (EtOH) led to (+)-narwedine. The apparently spontaneous formation of (+)-narwedine during crystallization was most likely due to the presence of small amounts of unoxidized (−)-galantamine. The process must proceed through the symmetrical dienone intermediate 9. Eventually, they obtained (−)-narwedine (5) by crystallization of (±)-narwedine with 0.5 equiv. of (+)-galantamine in a mixture of EtOH/Et3N. Further reduction of the ketone group in 5 with LiAlH4 completed a relay synthesis of (−)-galantamine. Shieh and coworkers reinvestigated this crystallization-induced dynamic resolution of narwedine.58,59 They found that resolution of (±)-narwedine in EtOH/Et3N worked equally well with a catalytic amount of (−)-narwedine (2.5%) or (+)-galantamine (1%). This process is highly efficient, as evidenced by the conversion of 10 g of (±)-narwedine into 9.02 g of (−)-narwedine in just two cycles. Shieh and coworkers also solved the issue of stereoselectivity in the final ketone reduction. The desired product was stereoselectively obtained by employing L-selectride as the reducing agent instead of LiAlH4. These two key improvements are crucial for the pilot scale synthesis of (−)-galantamine (vide infra).
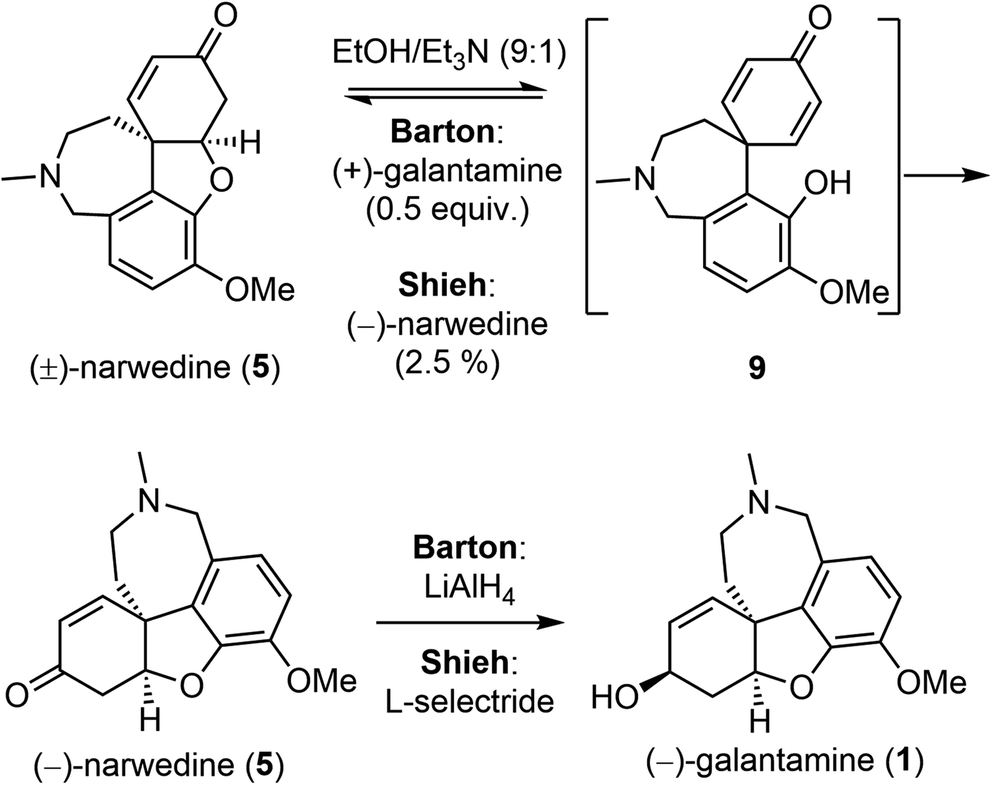 | ||
| Scheme 4 Crystallization-induced dynamic chiral resolution and ketone reduction. | ||
In the late 1990s, a collaboration between the pharmaceutical company Sanochemia and Vienna University of Technology (Johannes Fröhlich and Ulrich Jordis) resulted in a pilot-scale process for the production of (−)-galantamine (Scheme 5).60,61 The phenol 19 was prepared from veratraldehyde in 4 steps on a 100 kg scale. The oxidative phenol coupling reaction was optimized by a multifactorial analysis of reaction parameters, and 40–42% yield of 20 were consistently obtained on scales up to 12 kg. Compound 20 was transformed to racemic narwedine ((±)-5) in 2 steps. Racemic narwedine is then resolved according to Shieh's procedure to give enantiomerically pure (−)-narwedine.58 Using a catalytic amount of seed crystals of (−)-5, a consistently reproducible procedure has been developed for the crystallisation-induce chiral transformation of (±)-5 to (−)-5 on scales up to 7 kg. (−)-Narwedine was stereoselectively reduced with L-selectride to afford (−)-galantamine (1), which was isolated as the salt of hydrobromide in nearly quantitative yield. More than 5 kg of the final product was obtained by this process in 12.4% overall yield.
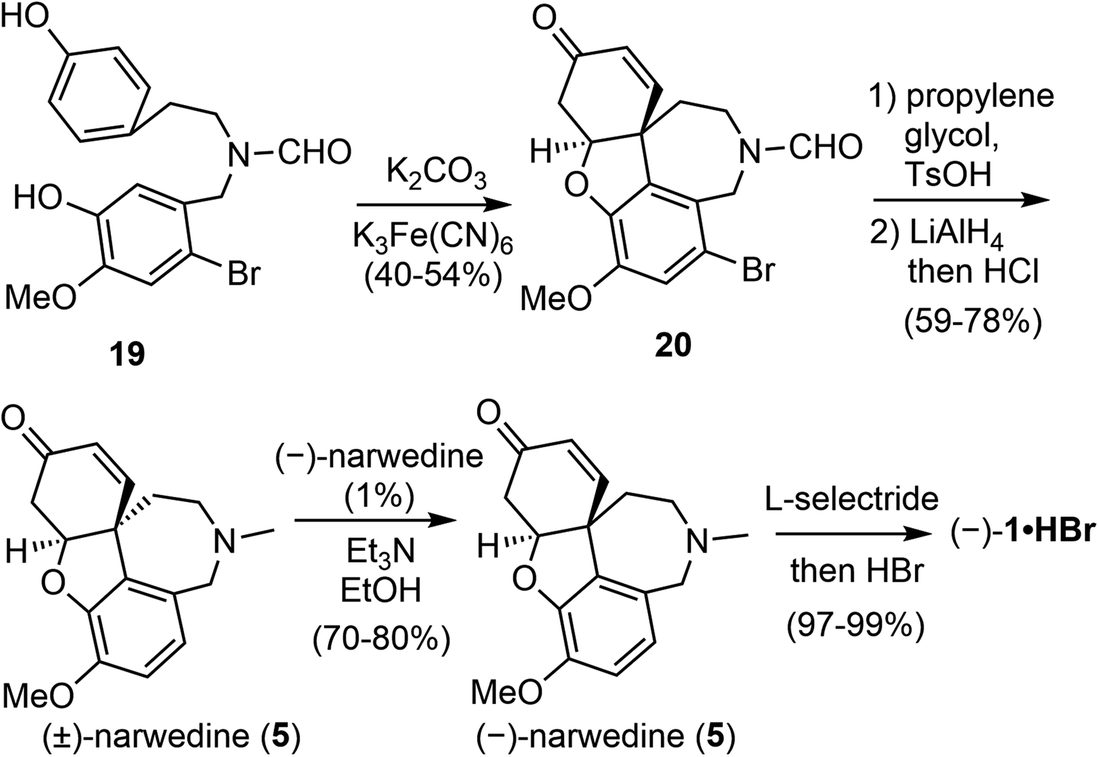 | ||
| Scheme 5 Industrial synthesis of (−)-galantamine by Sanochemia. | ||
From Barton's biomimetic synthesis to Sanochemia's pilot scale synthesis of galantamine, it took generations of chemists and almost four decades of effort to optimize a low-yielding but landmark reaction to an industrial scale reaction with practical values. There are continuous and ongoing interest in this biomimetic intramolecular oxidative coupling reaction, especially with the advent of new technologies. The clinical demand for (−)-galantamine also requires a sustainable and economical supply to complement the isolation of (−)-galantamine from its natural source.62
2.3 Chemoenzymatic approach (Saladino)
Biocatalysis is especially useful for the preparation of novel building blocks or the late-stage modification of complex molecules, due to the high selectivity profiles of enzymes. Chemoenzymatic synthesis allows the organic chemists to combine the versatility of synthetic organic transformations with the unparalleled selectivity of biocatalysis, providing efficient access to important bioactive small molecules.63Laccases (EC 1.10.3.2) are a class of multicopper-containing oxidoreductase that catalyze the one-electron oxidation in nature (E0 = 0.5 to 0.8 V vs. normal hydrogen electrode NHE), using air as the primary oxidant.64,65 In 2020, Saladino and coworkers reported a novel and biomimetic synthesis of galantamine (Scheme 6).36 They developed a laccase catalyzed para–ortho oxidative radical coupling of a norbelladine derivative 19 to a spirocyclohexadienone 21 in the presence of the redox mediator 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO). The phenol 19 was prepared in two steps from commercially available tyramine and 2-bromoisovanillin via reductive amination and N-formylation. Treatment of 19 with laccase from Trametes versicolor (1000 U mmol−1) and 0.6 equivalents of TEMPO as the redox mediator and oxygen as the primary oxidant led to the oxidative radical coupling product 21 in 70% yield. Product 21 is a tricyclic fused system, containing the azepine D ring and two hexadienone rings. Surprisingly, in contrast to the spontaneous oxa-Michael addition to form the B ring in the biomimetic chemical synthesis of galantamine (vide supra),60,61 this enzyme catalyzed reaction stopped at the oxidative coupling stage, reflecting the mildness of the reaction conditions. Tautomerization of 21 with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) induced an intramolecular oxa-Michael addition to give the tetracyclic product 20 in 91% yield. Final sequential reduction of the ketone with L-selectride and reduction of the amide and debromination with LiAlH4 afforded galantamine in 61% yield.
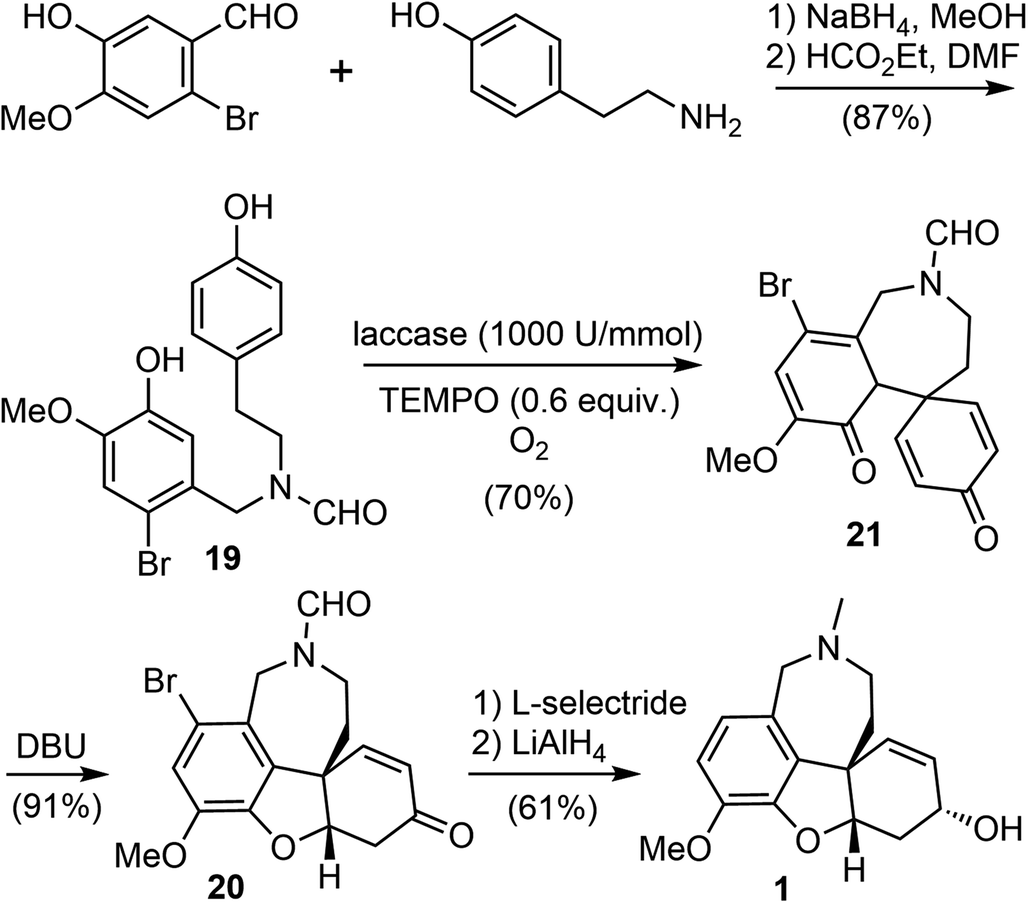 | ||
| Scheme 6 Saladino's chemoenzymatic synthesis of galantamine. | ||
The entire process of this total synthesis required only 6 steps from commercial compounds, with an overall yield of 34%. Further optimization of the reaction conditions, e.g., reducing the amount of TEMPO and using supported laccase for recycling, would make this route a potential candidate for scaling up the synthesis of galantamine.
2.4 Electrosynthesis approach (Wirth, Opatz and Waldvogel)
Organic electrosynthesis is an old and rich discipline that has experienced a significant renaissance in recent years. It provides a customizable, cost effective and environmentally benign alternative for conducting redox reactions using only electrons as traceless reagents, thereby eliminating the need for dangerous and toxic stoichiometric oxidants.66 With the recent renaissance of the field, two independent reports on the electrochemical synthesis of galantamine were published almost simultaneously.39,40In 2022, Wirth and coworkers reported a new biomimetic total synthesis of (−)-galantamine (1), using an anodic aryl–aryl coupling as the key synthetic step, which was further optimized in a flow electrochemical setup (Scheme 7a).39 The substrate 22 was synthesized from methyl D-tyrosine and methyl gallate in 6 steps. Various parameters of the electrolysis were screened and optimized. It was found that RVC anode and platinum cathode is the best electrode material combination, trifluoroethanol with 0.1 M nBu4NClO4 as supporting electrolyte is a very good solvent, the addition of an acidic additive such as trifluoroacetic acid successfully increased the yield of the electrolysis product 23 to 40% in batch mode. This reaction was then examined using a flow electrochemical reactor. After optimizing the flow system, the yield of 23 increased to 55%. Subsequently, product 23 was converted to (−)-galantamine by adapting the strategy from Koga's synthesis.56
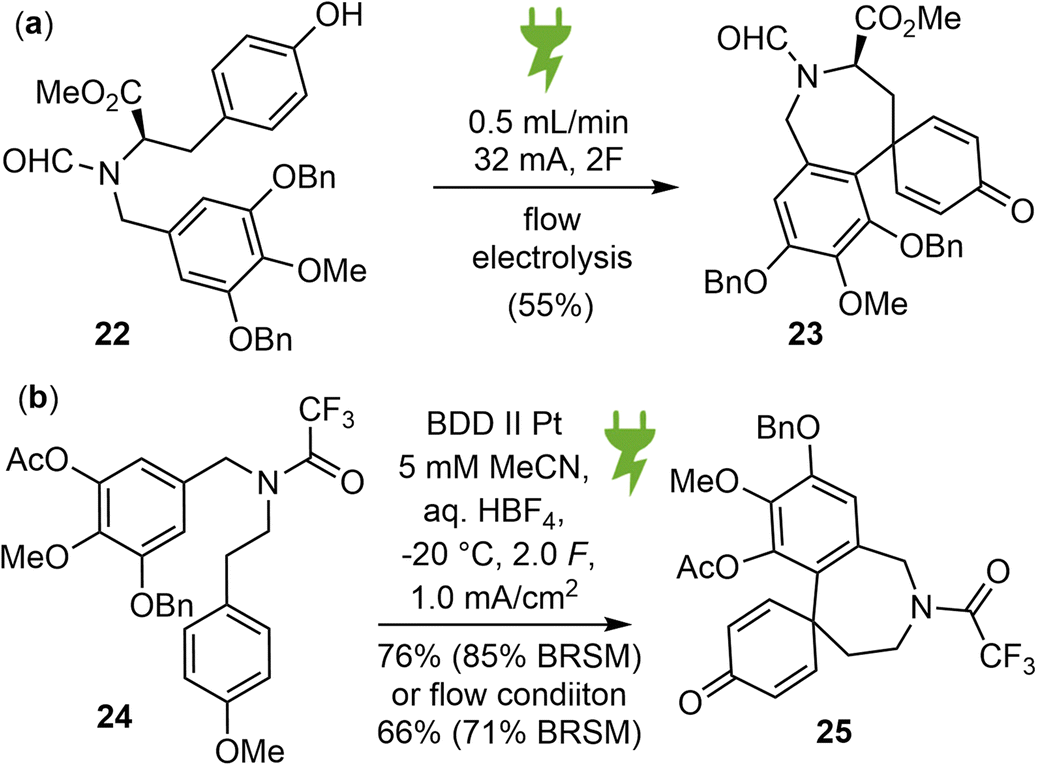 | ||
| Scheme 7 Electro-synthesis of galantamine. (a) Wirth and (b) Opatz and Waldvogel's work. | ||
In the same year, Opatz and Waldvogel reported a biomimetic approach towards the synthesis of Amaryllidaceae alkaloids using a highly versatile anodic key transformation to spirodienones (Scheme 7b).40 The substrate 24 for the electro oxidative coupling was prepared from methyl gallate in 11 steps. The metal-free, high performance boron-doped diamond (BDD) electrode was found to be the anode material of choice. Acetonitrile with an acid additive gave the most promising results. To prevent the rearrangement of the sensitive spirodienone moiety, a continuous flow setup was refined to neutralize the electrolyte immediately after electrolysis. Product 25 was then converted to Node's intermediate,55 which represents a formal total synthesis of galantamine.
Electrosynthesis provided an alternative approach to traditional oxidants for this key biomimetic oxidative coupling in the total synthesis of galantamine. However, both the substrates and the reaction parameters for this key reaction required a lot of effort for careful optimization in the above two cases, making the overall syntheses less efficient.
2.5 Chemical resolution (Bandichhor)
The process for the scalable chemical synthesis of (−)-galantamine by Sanochemia60,61 was adapted or modified on certain steps for further improvement by all subsequent production procedures. In 2008, Bandichhor and coworkers reported an alternative scalable synthesis of (−)-galantamine via Kametani's intermediate 11b (Scheme 8).17 The acid 26 and amine 27 were both obtained in 3 steps from readily available starting materials. Coupling of these two fragments with 10 mol% of ecofriendly phenyl boronic acid afforded the amide 28 in 93% yield.67 The O-benzyl groups was removed under acidic condition to give the known phenol 11b.51,52 The incremental addition of 11b to Kametani's conditions (K3Fe(CN)6, NaHCO3) led to the key intramolecular oxidative coupling followed by a spontaneous oxa-Michael addition to give the tetracyclic product 12b in 62% yield, much higher than those reported by Kametani and the Sanochemia process. The enone was reduced with L-selectride to afford an alcohol in good yield (80%) and diastereoselectivity (de 99.6%). The reduction of the lactam and concomitant debromination with vitride led to the racemic galantamine ((±)-1). The racemic mixture was resolved using (+)-di-p-tolyl tartaric acid ((+)-DPTTA) to produce the enantiomerically pure product (−)-galantamine (1) with good yield (70%, based on available enantiomer in the racemate) and chiral purity (99.91%).68 The HBr salt of (−)-galantamine was formed conventionally using aq. HBr. All reactions in the process were performed on a decagram scale, and an overall yield of 9% was obtained from isovanillin.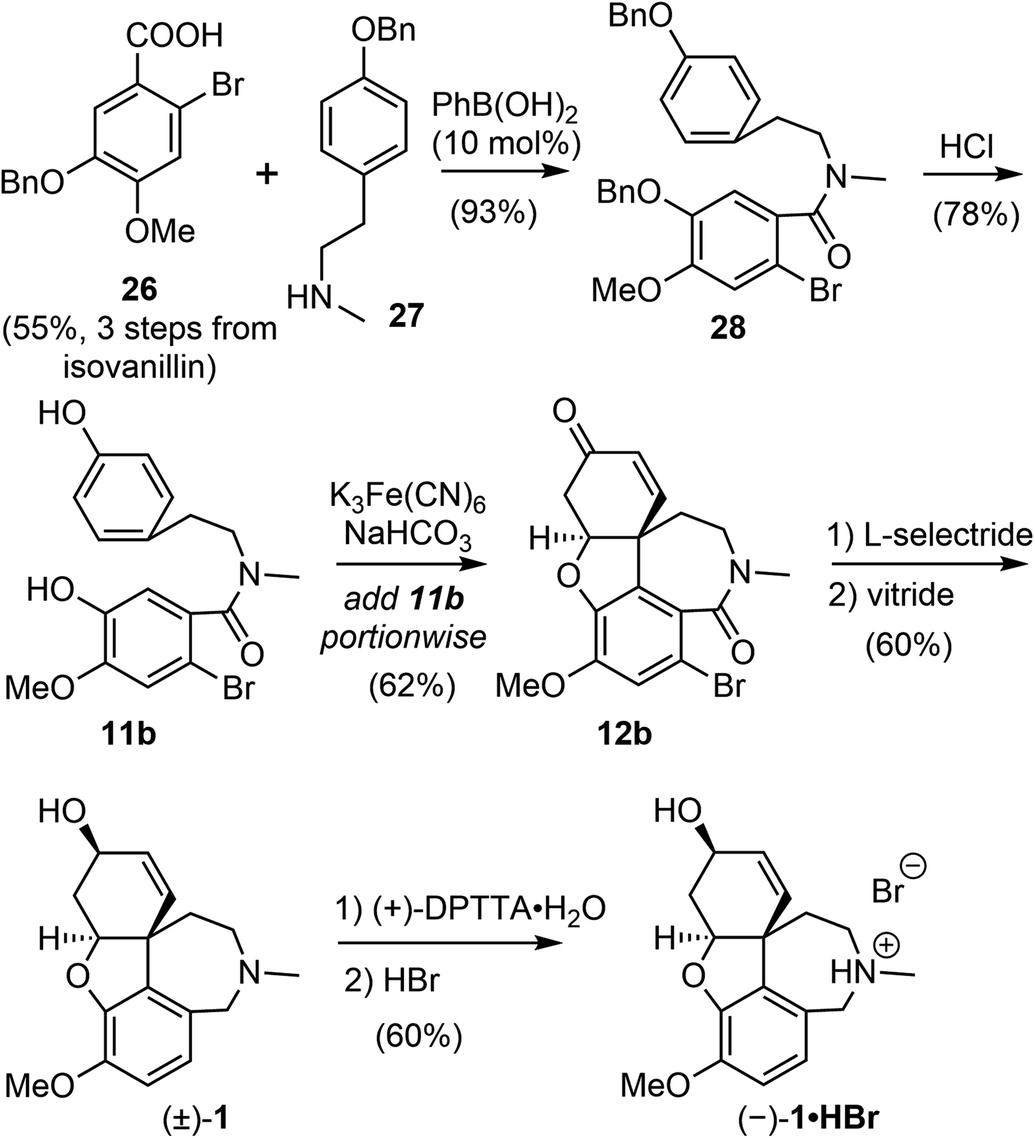 | ||
| Scheme 8 Bandichhor's chemical resolution approach. | ||
By modifying the substrate and reaction conditions, they were able to obtain a good yield for the key intramolecular oxidative coupling reaction. However, half of the valuable late-stage product was lost using the classical resolution method, and they did not demonstrate the recycling of the other enantiomer, which makes the route less cost effective for scalable production of (−)-galantamine.
3. Transition metal catalyzed reactions
Transition metal catalyzed reactions play an indispensable role in modern synthetic chemistry. They have been awarded the Nobel Prize in Chemistry three times in the 21st century. For decades, oxidative phenol coupling was the only way to construct the benzylic quaternary center in the total synthesis of galantamine. Trost and coworkers were the first to break this paradigm and use a palladium catalyzed intramolecular Heck reaction.69–71 Since then, numerous applications of transition metal catalyzed reactions, including all three Nobel Prize-winning reactions (the Heck coupling reaction, asymmetric hydrogenation reaction22 and olefin metathesis reaction14,23,41), have been employed in the total synthesis of galantamine.14,23,24,28,293.1 Heck reaction (Trost and Guillou)
In 2000, Trost and Toste reported the first catalytic asymmetric synthesis of (−)-galantamine (Scheme 9).69 They used sequential palladium-catalyzed asymmetric allylic alkylation (AAA) and intramolecular Heck reaction to build the key vicinal stereocenters with high enantioselectivity. This is the first total synthesis that does not use an oxidative phenol coupling to construct the quaternary center of (−)-galantamine (1).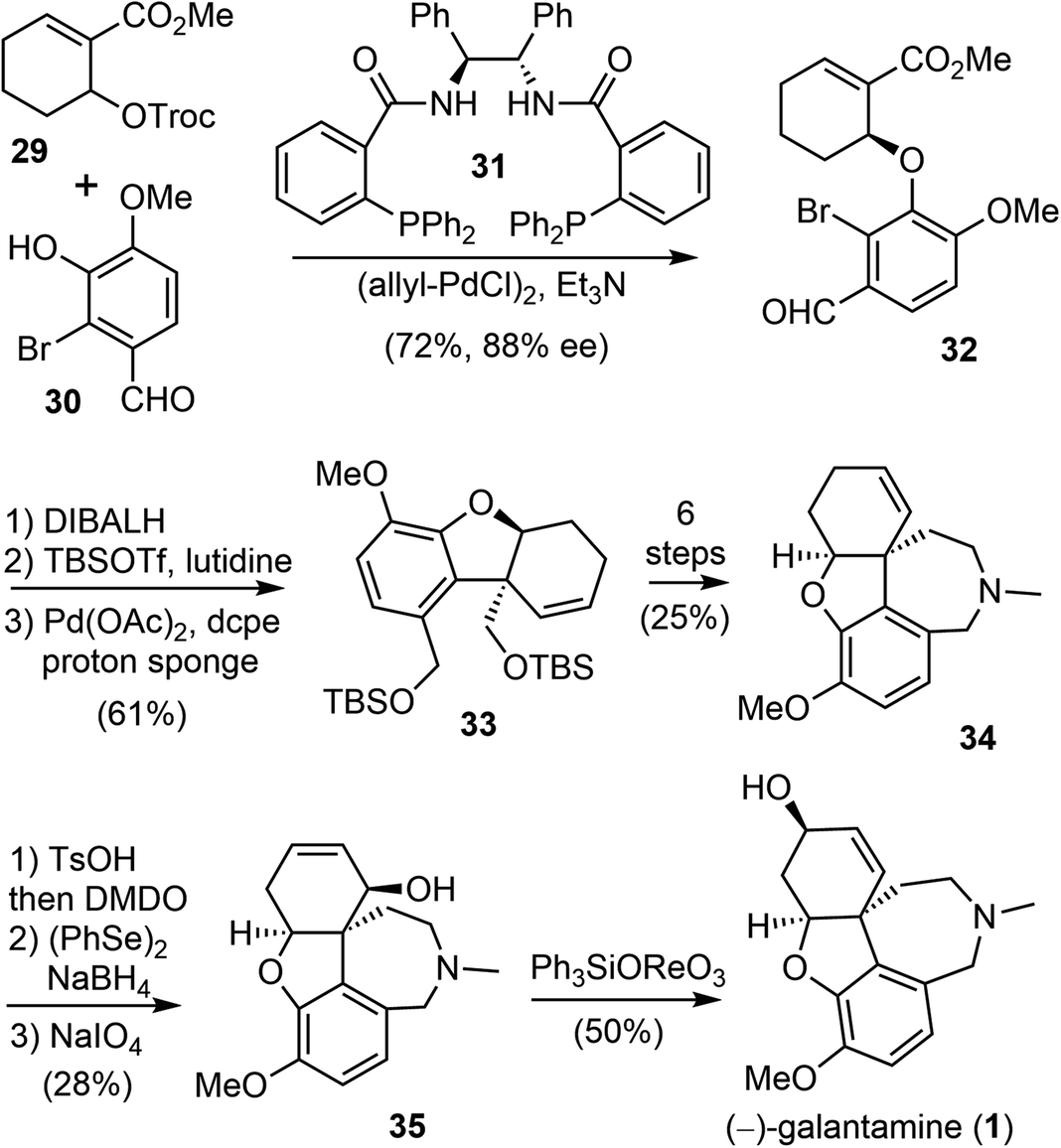 | ||
| Scheme 9 Trost and Toste's total synthesis of (−)-galantamine. | ||
First, a palladium-catalyzed AAA reaction of allylic carbonate 29 with 2-bromovanillin (30) in the presence of the chiral ligand 31 afforded the required chiral aryl ether 32 in 72% yield and with 88% enantiomeric excess (ee). The attempted intramolecular Heck reaction of the electron-poor olefin 32 to construct the crucial quaternary center failed. Therefore, 32 was transformed to an electron-rich bis-TBS ether. This compound underwent the palladium-catalyzed intramolecular Heck reaction smoothly with the electron-rich bidentate ligand 1,2-bis(dicylohexylphosphino)ethane (dcpe). The reaction yielded benzofuran 33 in good yield, together with some monodeprotected product. Both the olefin isomerization reaction and the phenol ionization reaction were suppressed under this condition. Compound 33 was then converted to 34 in 6 steps, including a reductive amination to form the azepine D ring. Compound 34 was converted to allylic alcohol 35 in a further 3 steps, through an epoxide to allylic alcohol isomerization. Finally, a 1,3-transposition of the allylic alcohol provided (−)-galantamine (1).
Two years later, Trost and Tang improved the total synthesis of (−)-galantamine (1) to an 8-step process (Scheme 10).72 The same arylether 32 was used in their second generation total synthesis. The α,β-unsaturated ester was homologated to a β,γ-unsaturated nitrile 36 in four steps, including a Mitsunobu reaction with acetone cyanohydrin. The enantiomeric excess of 36 was improved to 96% ee after recrystallization. The palladium-catalyzed intramolecular Heck cyclization proceeded smoothly in the presence of silver carbonate (Ag2CO3) and provided the product 37 in high yield (91%). An allylic oxidation mediated by SeO2 introduced the allylic hydroxy group and selectively delivered the product 38 (dr 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The condensation of aldehyde 38 with methylamine, followed by subsequent reduction and intramolecular cyclization, formed the hydrobenzazepine D ring and gave (−)-galantamine (1) in a one-pot process.
:1). The condensation of aldehyde 38 with methylamine, followed by subsequent reduction and intramolecular cyclization, formed the hydrobenzazepine D ring and gave (−)-galantamine (1) in a one-pot process.
 | ||
| Scheme 10 Trost and Tang's total synthesis of (−)-galantamine. | ||
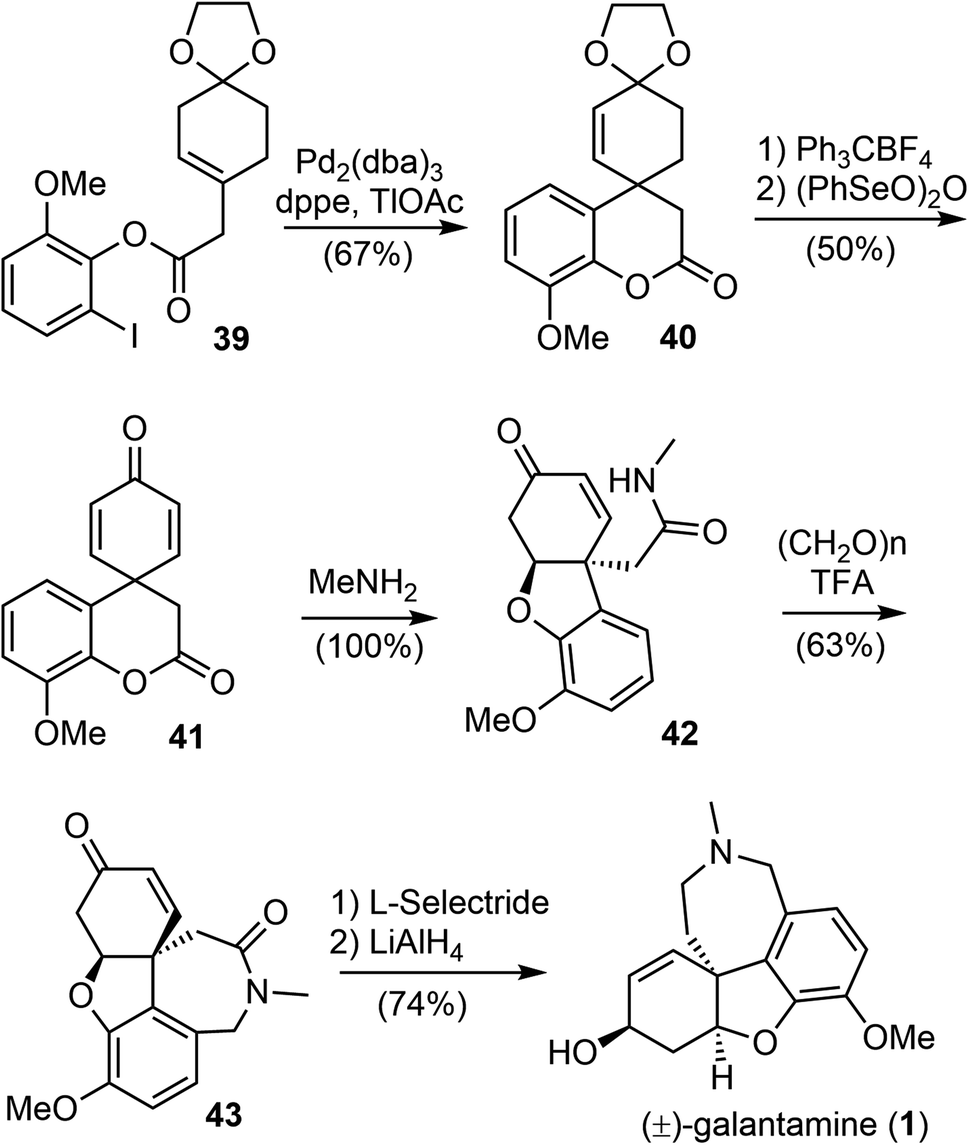
In 2001, Guillou and Thal reported an efficient synthesis of (±)-galantamine from a readily available ester 39 (Scheme 11).73 The palladium-catalyzed intramolecular Heck cyclization of 39 afforded the tricyclic compound 40 in 67% yield. Removal of the dioxolane group, followed by oxidation with (PhSeO)2O afforded the key spirocyclohexadienone intermediate 41 in 50% yield. The ester amidation reaction of 41 with 40% aqueous methylamine was carried out at room temperature and resulted in the spontaneous phenolic oxa-Michael addition to cyclohexadienone to afford amide 42. Compound 42 was then subjected to Pictet–Spengler cyclization with paraformaldehyde to form the azepine D ring and give tetracyclic product 43 in 63% yield. Successive stereoselective reduction of the enone 43 with L-selectride and reduction of the lactam with LiAlH4 afforded (±)-galantamine in good yield.
 | ||
| Scheme 11 Guillou and Thal's total synthesis of (±)-galantamine. | ||
The above three total syntheses of galantamine formed the basis for many subsequent total syntheses. The intramolecular Heck reaction has been used many times to construct the quaternary center. The reductive amination method of Trost and the Pictet–Spengler cyclization method of Guillou were employed to form the azepine D ring. The key intermediates in their total syntheses became the targets of later syntheses.
3.2 Enyne RCM and Heck reaction (Brown)
In 2007, Brown and coworkers reported their first-generation enantioselective total synthesis of (−)-galantamine in 11 linear steps from commercially available materials.14 An enantioselective reduction of a propargylic ketone 44 with (R)-alpine borane afforded the chiral alcohol 45. Coupling of the alcohol 45 with phenol 47via Mitsunobu reaction afforded enyne 48. The C ring was closed by an efficient enyne metathesis reaction of 48 in the presence of 3 mol% of the first-generation Grubbs' catalyst, generating the diene product 49 in 85% yield at ambient temperature (Scheme 12).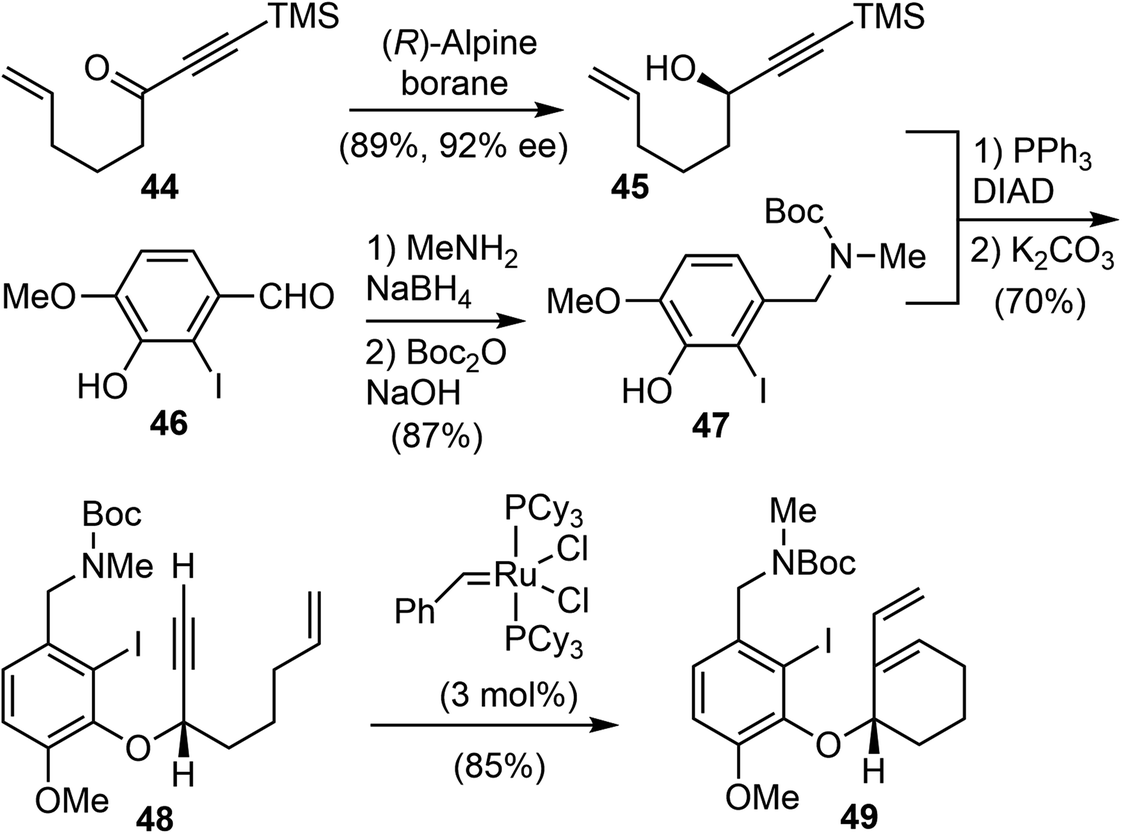 | ||
| Scheme 12 Enyne metathesis reaction. | ||
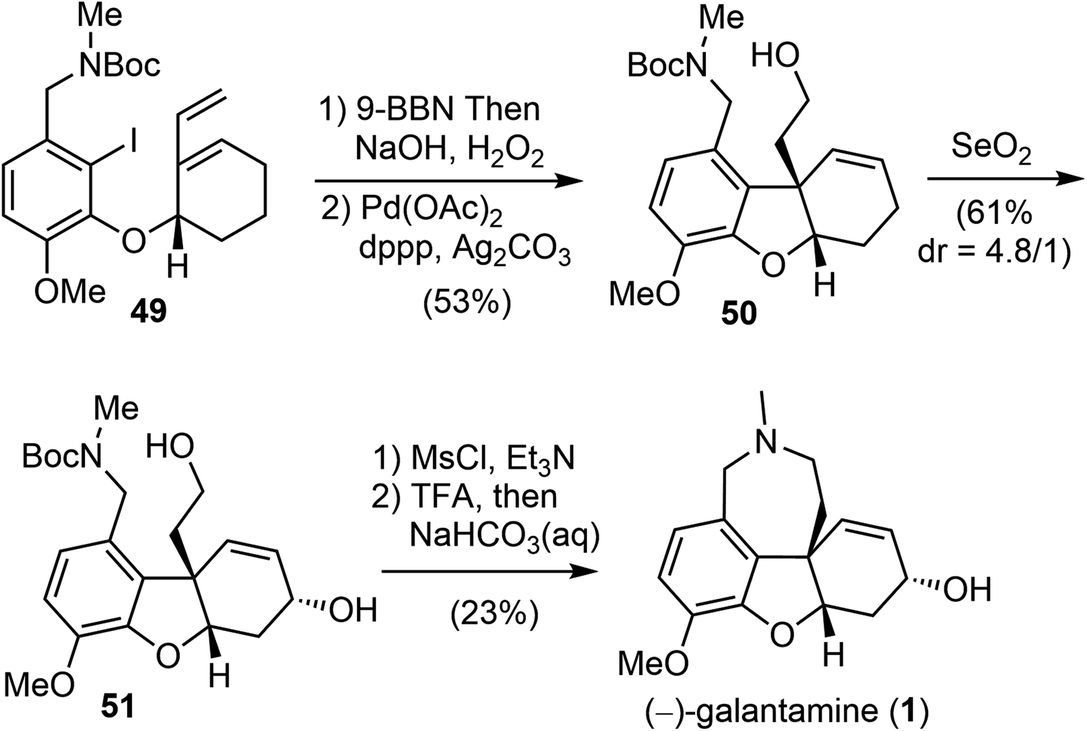
The terminal olefin of 49 was selective hydroborated and oxidized to yield a homoallylic alcohol (Scheme 13). Subsequently, an intramolecular Heck reaction successfully formed the central heterocyclic five-membered B ring to give tricyclic compound 50. Allylic oxidation of 50 using Trost's procedure led to the desired (R)-allylic alcohol 51 with modest selectivity.69 The azepine D ring was generated by selectively activating the primary hydroxyl group through mesylation, followed by a one-pot Boc deprotection/N-alkylation process by sequential treatment with TFA and neutralization with NaHCO3 (aq). (−)-Galantamine and its epimer were obtained after separation by column chromatography.
 | ||
| Scheme 13 Brown's first-generation total synthesis of (−)-galantamine. | ||
Fifteen years later, Brown and coworkers reported their second-generation asymmetric synthetic route to (−)-galantamine with very high levels of stereocontrol. Several transition-metal mediated reactions were pivotal in achieving high levels of chemo and stereo-control, including the use of an enyne ring closing metathesis (RCM), two asymmetric allylation reactions and an intramolecular Heck reaction.41
Asymmetric allylation of aldehyde 52 with Hafner's titanium-TADDOL reagent (S,S)-53 provided the chiral homoallylic alcohol 54 with excellent enantioselectivity (Scheme 14).74 The Mitsunobu coupling of phenol 47 and alcohol 54 afforded an olefin as a single enantiomer in high yield. A two-step dihydroxylation/oxidative cleavage process transformed the terminal olefin to aldehyde 55. A second aldehyde allylation reaction with TADDOL reagent (R,R)-53 gave another homoallylic alcohol in good yield and excellent diastereoselectivity. Functional group manipulation provided enyne 56, which contained the full scaffold of the alkaloid and the necessary stereochemical information, ready for the key reactions.
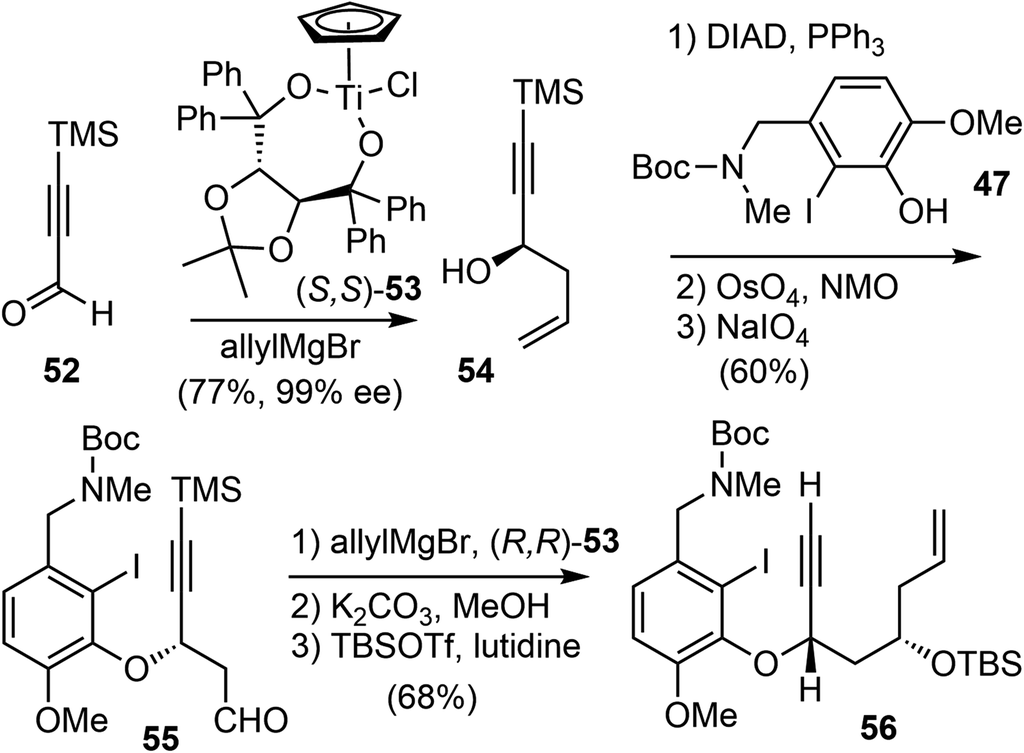 | ||
| Scheme 14 Synthesis of 48 by two asymmetric allylation reactions. | ||
In the presence of Grubbs' first-generation catalyst, enyne 56 underwent an RCM reaction to generate the C ring and afforded the cyclized product 57 (Scheme 15). Selective hydroboration of the terminal olefin with 9-BBN and oxidation, followed by an intramolecular Heck cyclization generated the tricyclic compound 58.70 Finally, formation of the azepine D ring by an N-alkylation reaction and in situ deprotection delivered (−)-galantamine (1). The overall process of their total synthesis of (−)-galantamine is 11 steps with 7.3% overall yield from the known enantiomerically enriched alcohol 54.
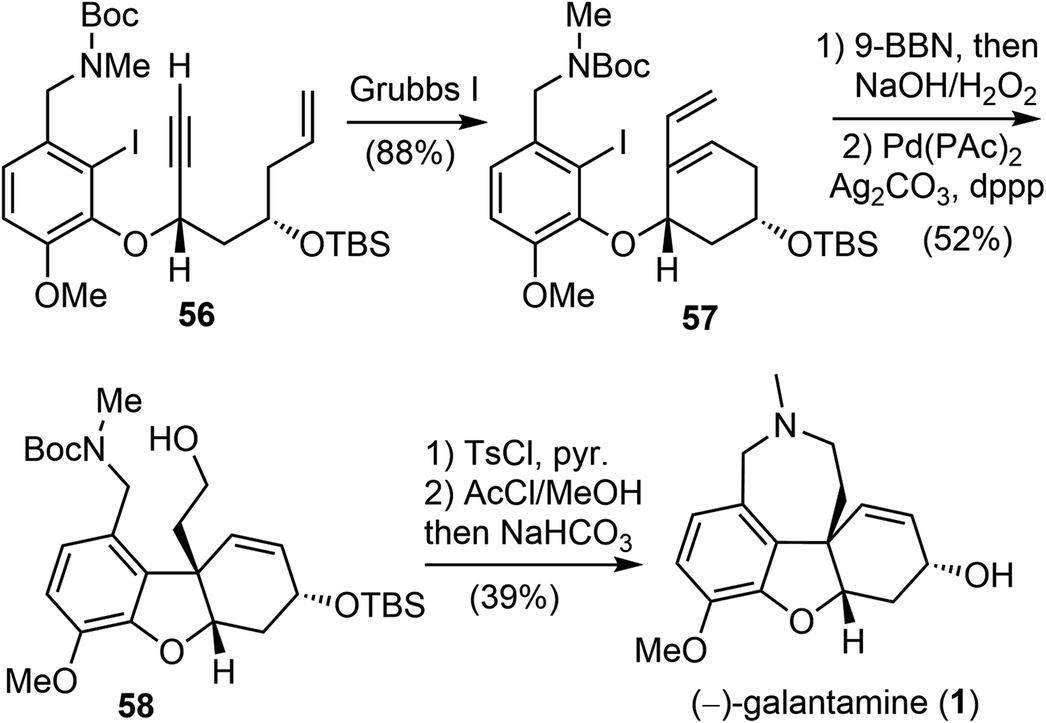 | ||
| Scheme 15 Brown's second-generation total synthesis of (−)-galantamine. | ||
3.3 Dynamic kinetic resolution and reductive Heck (Zhou and Xie)
In 2012, Zhou and Xie reported an asymmetric total synthesis of (−)-galantamine (20.1%, 12 steps).22 Their synthetic strategy features an efficient ruthenium-catalyzed asymmetric hydrogenation of a racemic α-aryloxy cyclic ketone via dynamic kinetic resolution (DKR), and a palladium-catalyzed intramolecular reductive Heck cyclization to construct the benzylic quaternary center.Previously, they developed a highly efficient ruthenium-catalyzed asymmetric hydrogenation of racemic α-aryloxy cyclohexanones via DKR to produce chiral β-aryloxy cycloalkanols with excellent enantioselectivity.75 Using this methodology, the racemic α-aryloxy cyclic ketone 59 was hydrogenated with the chiral ruthenium catalyst RuCl2-(S)-SDP/(R,R)-DPEN (60) to afford the chiral β-aryloxy cyclohexanol 61 in high yield (99%), excellent enantioselectivity (97% ee) and cis-selectivity (cis/trans > 99:1) (Scheme 16). The aryloxy cyclohexanol 61 was then converted by Swern oxidation and Horner–Wadsworth–Emmons reaction (HWE) to give the α, β-unsaturated ester 62 as an inconsequential olefin mixture (Z/E 1:5).
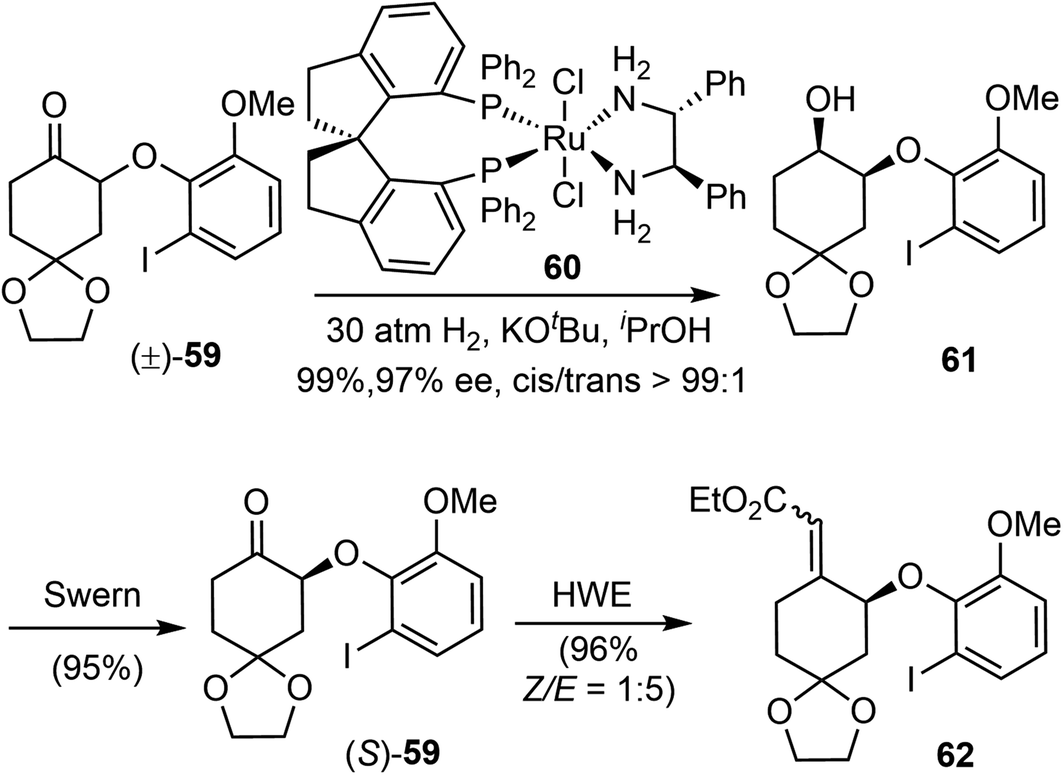 | ||
| Scheme 16 A DKR reaction to generate the oxygenated stereocenter. | ||
The construction of the key quaternary center via the intramolecular Heck reaction of aryl allyl ethers is often impeded by a competitive palladium-catalyzed ionization of the aryloxy group, leading to the formation of a phenol.69 To address this issue, a reductive Heck reaction was investigated. After screening and optimization, it was found that the intramolecular reductive Heck cyclization successfully formed the desired product 63 in 95% yield when the Z, E-mixture of ester (S)-62 was subjected to the catalyst [Pd2(dba)3] in the presence of sodium formate (HCO2Na) (Scheme 17). The ester 63 was then transformed to an amide, followed by Pictet–Spengler cyclization with paraformaldehyde according to Guillou's procedure73 to give the tetracyclic intermediate 64. Stereoselective reduction of the ketone group to the alcohol with K-selectride and a further reduction of the lactam to the amine by Beller's method76 (triethoxysilane ((EtO)3SiH)/Zn(OAc)2) afforded the natural product lycoramine (6). On the other hand, a Sagusa oxidation of the ketone 64 gave the known enone product 43,73 and successive reduction with K-selectride and (EtO)3SiH)/Zn(OAc)2 gave the natural product (−)-galantamine (1). (−)-Galantamine (1) was synthesized in twelve steps with 20.1% overall yield and lycoramine (6) was synthesized in ten steps with 40.2% overall yield from commercially available starting materials.
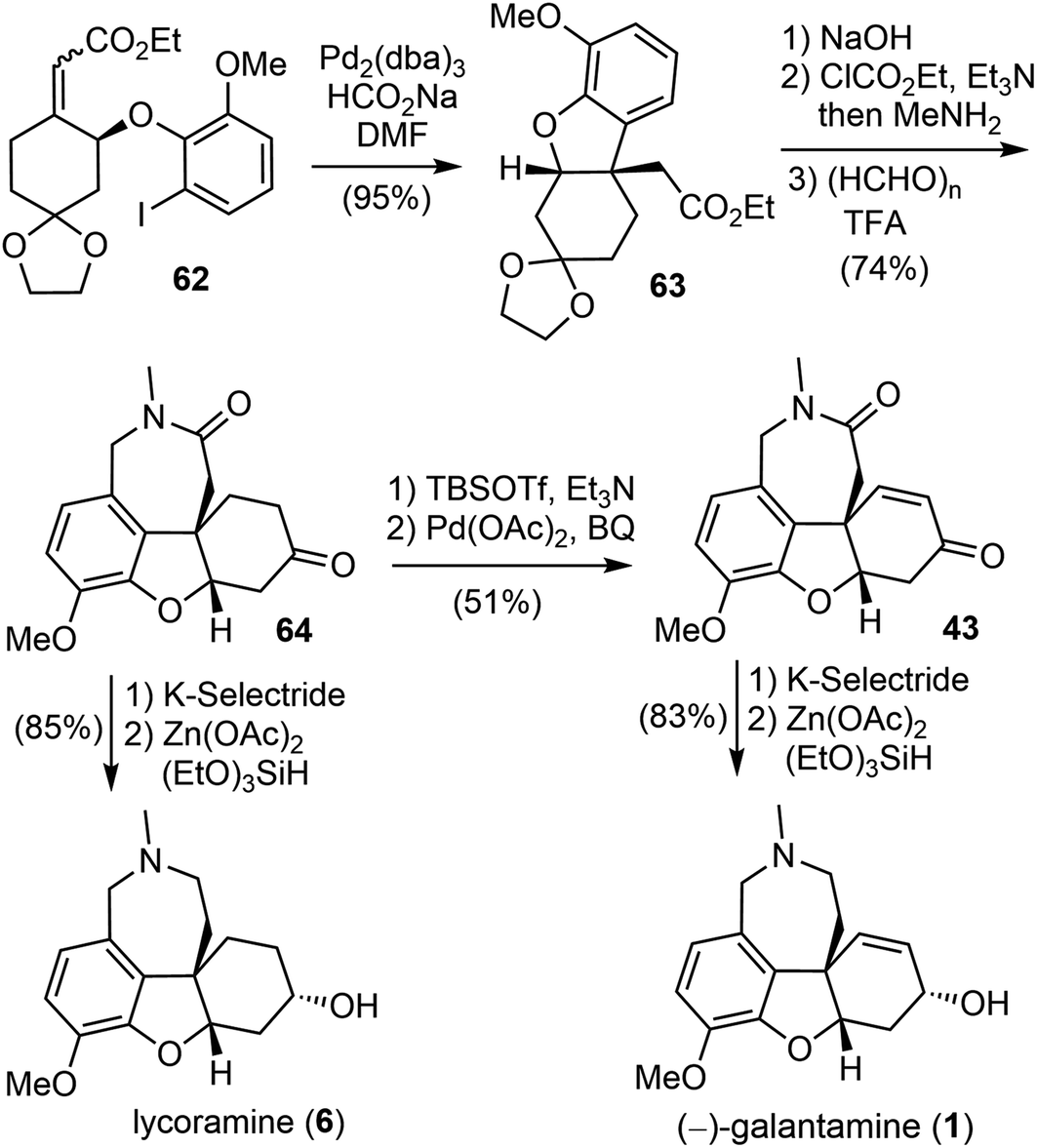 | ||
| Scheme 17 Zhou and Xie's total synthesis of (−)-galantamine. | ||
3.4 Alder-ene, Diels–Alder and Heck reaction (Banwell)
The Banwell group has been working on the total synthesis of galantamine for a long time, and has published several generations of total syntheses with different strategies.19,27,28,42,77 In 2015, they reported a new and distinct total synthesis of galantamine,27 in which the aromatic A ring was made by a Diels–Alder reaction, the heterocyclic B ring and associated quaternary carbon center were constructed by a palladium-catalyzed intramolecular Alder-ene (IMAE) reaction, and the seven-membered D ring was then formed by a modified Bischler–Napieralski reaction.Following a literature method,78 the commercial ketone 65 underwent an α-acyloxylation reaction with N-methyl-O-benzoylhydroxylamine (MeNHOBz) at room temperature to give the product 66 (Scheme 18). This reaction presumably proceeds via a concerted pericyclic rearrangement of the intermediate 65′ to give the α-benzoyloxy imine 66′, with further in situ hydrolysis to give the observed product. The ketone 66 was regioselectively converted to an enol triflate 67, which underwent a B-alkyl Suzuki coupling reaction with the in situ generated boron reagent 68 to afford the coupling product alcohol 69 after ester hydrolysis. The incorporation of the alkyne moiety was completed in 3 steps to give the enyne 70. The palladium-catalyzed IMAE reaction of enyne 70 was best performed with a strong σ-donating ligand N,N′-bis(benzylidene)ethylenediamine (BBEDA), to successfully form the heterocyclic B ring and the quaternary center and provide the desired cyclization product 72.79,80
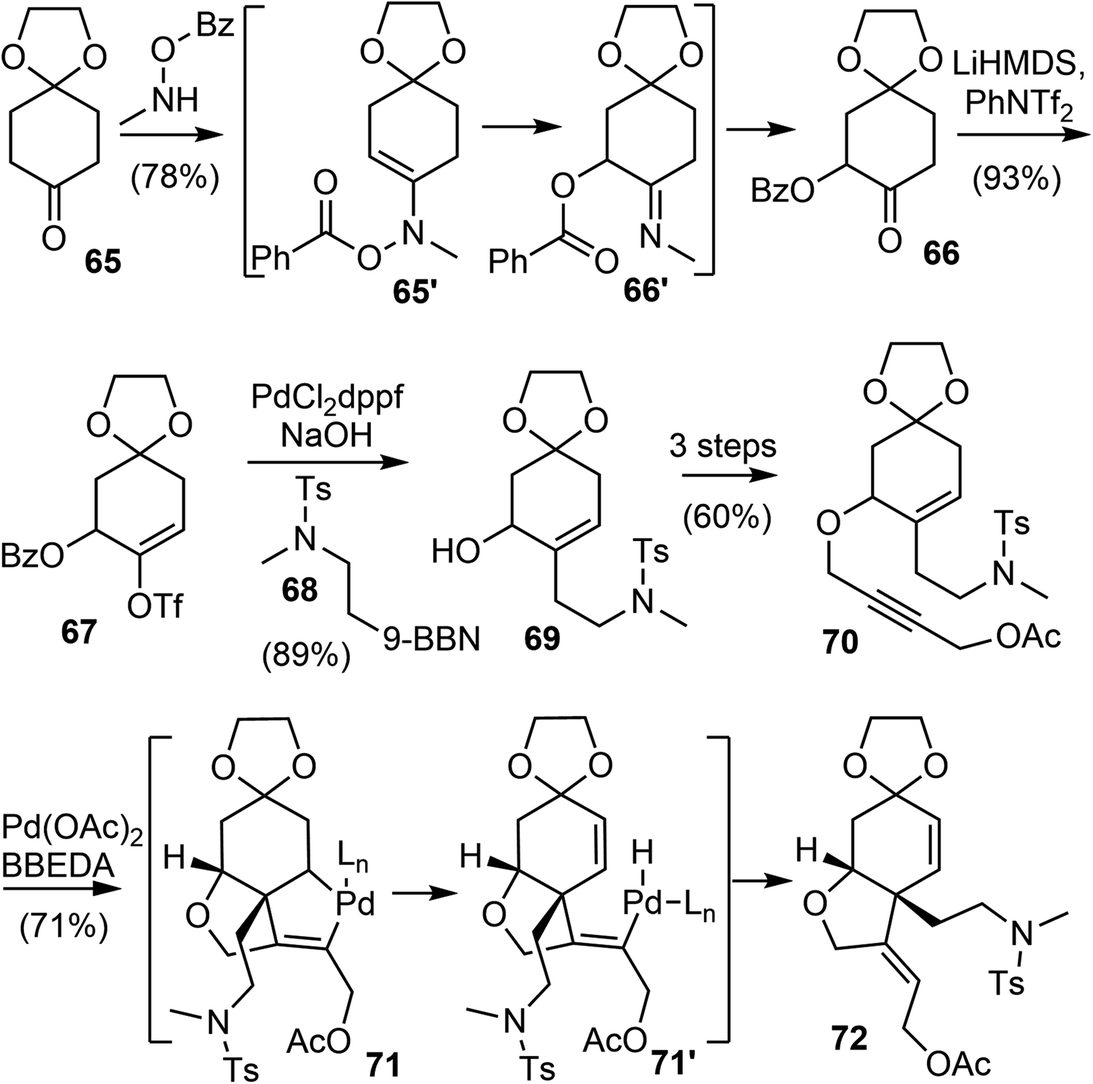 | ||
| Scheme 18 Palladium-catalyzed intramolecular Alder-ene reaction. | ||
The allylic acetate 72 underwent a palladium-catalyzed elimination reaction under basic conditions to give a diene 73 (Scheme 19). Compound 73 then participated in a regioselective Diels–Alder cycloaddition reaction with propynal, followed by immediate oxidation with manganese dioxide (MnO2) to form the aromatic A ring and afford the aldehyde 75. Compound 75 was converted in 5 steps to formamide 76. A modified Bischler–Napieralski cyclodehydration reaction of 76 under Movassaghi's conditions (Tf2O/2-Cl pyridine) formed the D ring,81 and subsequent reduction of the resulting iminium with sodium triacetoxyborohyride (NaBH(OAc)3) afforded narwedine. A diastereoselective reduction of the ketone group with L-selectride afforded (±)-galantamine (1). The longest linear sequence of the total synthesis is 18 steps from commercial compound 66.
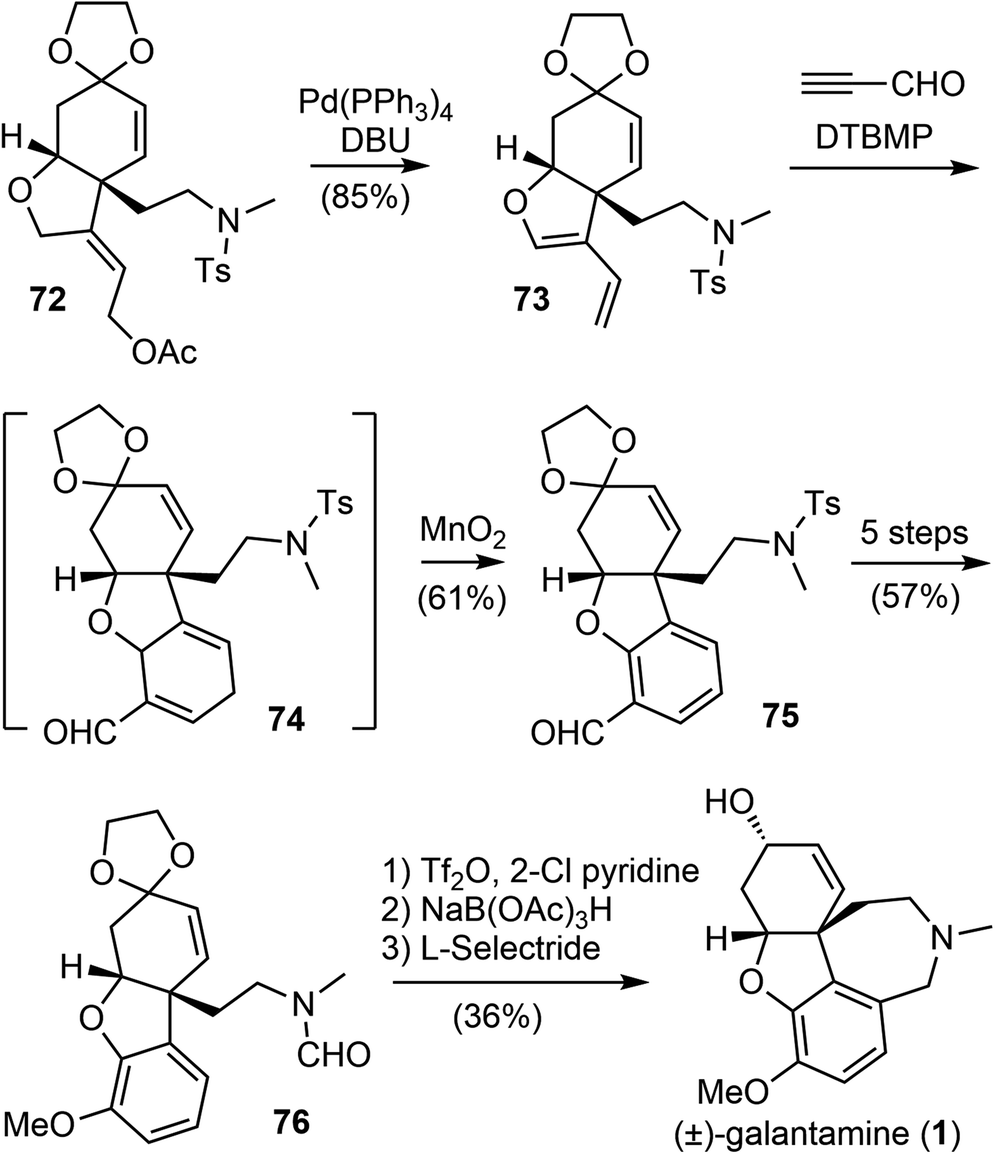 | ||
| Scheme 19 Banwell's total synthesis of (±)-galantamine. | ||
Although several distinctive strategies were used, the installation of the aromatic moiety in the above synthesis was laborious, making the overall process less efficient. After several generations of evolution,19,28 Banwell and coworkers published a concise total synthesis of (−)-galantamine in 2022 (Scheme 20).42 The synthesis commenced from the known chiral allylic alcohol 79, which was readily available from an asymmetric ketone reduction reaction with (L)-TarB-NO2 reagent (78) derived from (L)-tartaric acid.82 Compound 79 was then subjected to a B-alkyl Suzuki cross-coupling reaction to introduce the alkyl side chain and a Mitsunobu reaction with commercial iodoisovanillin 46 to afford the coupling product 80, containing all the carbons of the natural product. An intramolecular Heck reaction of 80 under the standard conditions afforded the cyclohexene derivative 81, with concomitant formation of the quaternary center and the heterocyclic B ring. Allylic oxidation under Trost's conditions with SeO2 diastereoselectively generated the allylic alcohol 82.72 It was proposed that SeO2 reacted with the olefin and the axial proton Hax through an ene mechanism, with Hax perfectly aligned with the π-system. Finally, closure of the azepine D ring via a double reductive amination reaction in the presence of formaldehyde afforded the final target (−)-galantamine (1). In the absence of formaldehyde, a related natural product (−)-norgalantamine was obtained. The whole process for the total synthesis of (−)-galantamine (1) took only 6 steps from the known chiral compound 79.
 | ||
| Scheme 20 Banwell's improved total synthesis of (−)-galantamine. | ||
3.5 Microbial dihydroxylation/Heck reaction (Hudlicky)
In 2016, Hudlicky and coworkers reported a ten-step chemoenzymatic synthesis of ent-galantamine from phenethyl acetate (Scheme 21).29 The synthesis began with the microbial dihydroxylation of phenethyl acetate 83 to afford an intermediate cyclohexadiene diol 84. The less hindered alkene was selective reduced, followed by a selective Mitsunobu reaction of the more reactive allylic alcohol with phenol 47, resulting in the coupling product 85. The tricyclic ABC rings of galantamine was assembled by a subsequent intramolecular Heck reaction of 85. The alcohol 86 was converted to the diene 87 by mesylation, followed by elimination with DBU. A Prevost reaction of the diene 87 gave a diastereomeric mixture of iodoacetates, which were immediately subjected to reduction with sodium borohydride in DMSO to give a 2:1 mixture of diastereomeric acetates 88. Formation of the azepine D ring and adjustment of the stereochemistry of the allylic alcohol was achieved in 5 steps from 88, resulting in the synthesis of (+)-galantamine.
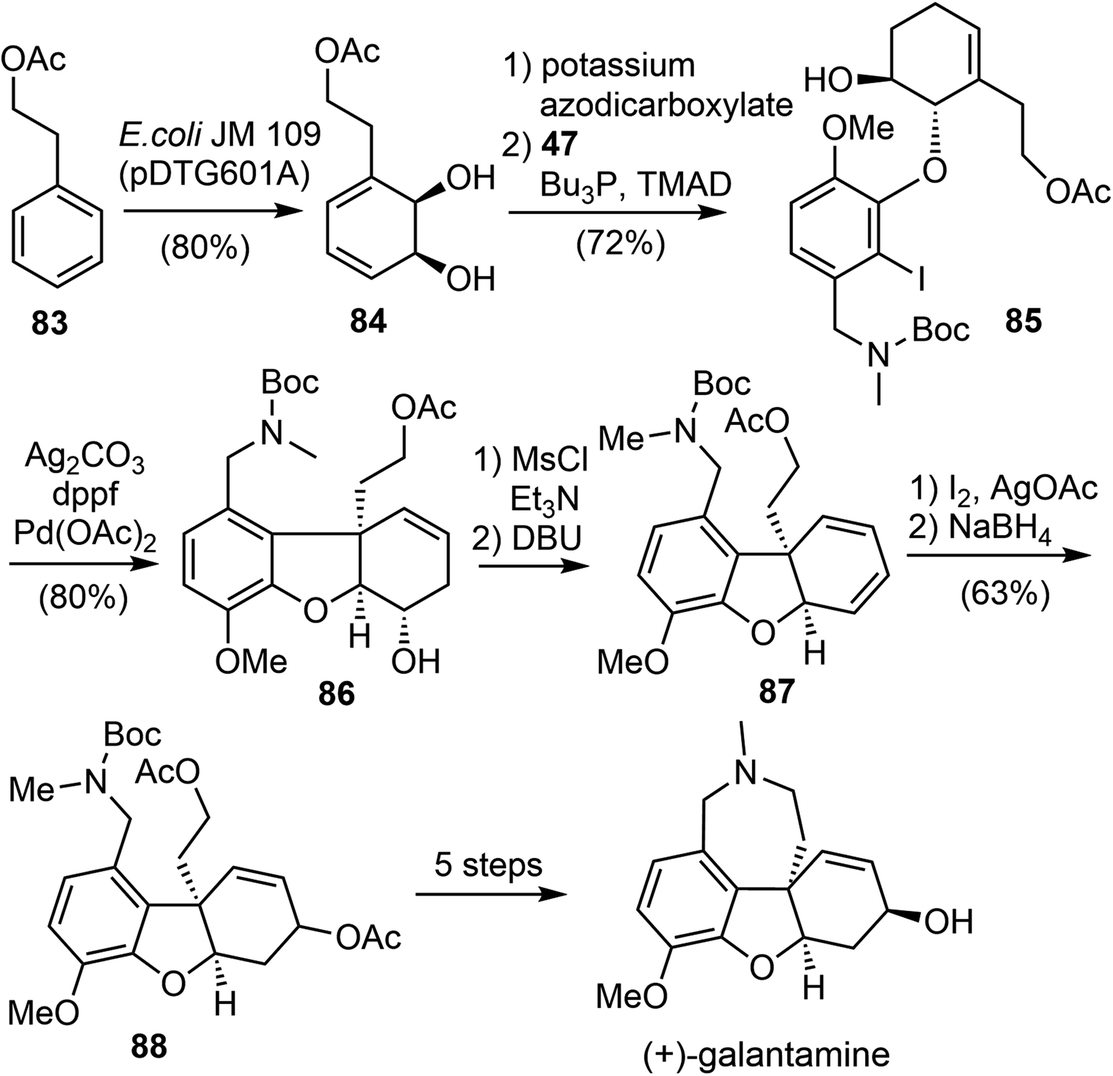 | ||
| Scheme 21 Hudlicky's total synthesis of (+)-galantamine. | ||
3.6 3.6 Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition (Yu)
Vinylcyclopropanes (VCPs) have been widely used in transition-metal-catalyzed cycloaddition reactions.83 The presence of an olefin ligand directs the transition-metal for the selective C–C bond cleavage of the highly strained cyclopropane ring. In 2010, Yu and coworkers developed a Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition reaction,84 which could be regarded as a homologous Pauson–Khand reaction (Scheme 22). Highly substituted bicyclic cyclohexenones and cyclohexanones could be obtained by this method. | ||
| Scheme 22 (a) An intramolecular Pauson–Khand reaction; (b) Rh-catalyzed [(3 + 2) + 1] cycloaddition reaction. | ||
In 2015, they extended the scope of this methodology further and reported a Rh(I)-catalyzed [(3 + 2) + 1] cycloaddition reaction of 1-ene–vinylcyclopropane and CO to build the cis-hydrodibenzofuran skeleton. This strategy was then applied to the formal synthesis of (±)-galantamine and (±)-lycoramine (Scheme 23).26 The substituted vinylcyclopropane substrate 92 was prepared from 89 in 3 steps. The phenol 89 and alcohol 90 underwent a Mitsunobu reaction to form the allylic ether 91 in 71% yield. A Claisen rearrangement in naphthane transformed 91 to a phenol, which underwent a Cu- catalyzed O-vinylation with 2,4,6-trivinylcyclotriboroxane–pyridine complex85 to give the 1-ene-VCP 92. The key Rh-catalyzed [(3 + 2) + 1] cycloaddition reaction under 1 atm of CO gas generated the desired tricyclic cis-hydrodibenzofuran product 93. The yield dropped slightly when the reaction was scaled up. Compound 93 was transformed to aldehyde 94 in 3 steps. Following Tu's precedent,13,86 aldehdye 94 was converted to amide 95 under radical oxidation conditions (vide infra). Install the D ring by a Pictet–Spengler cyclization using Guillou's procedure generated the tetracyclic compound 64,73 which is a key intermediate in Zhou and Xie's total synthesis of galantamine.22
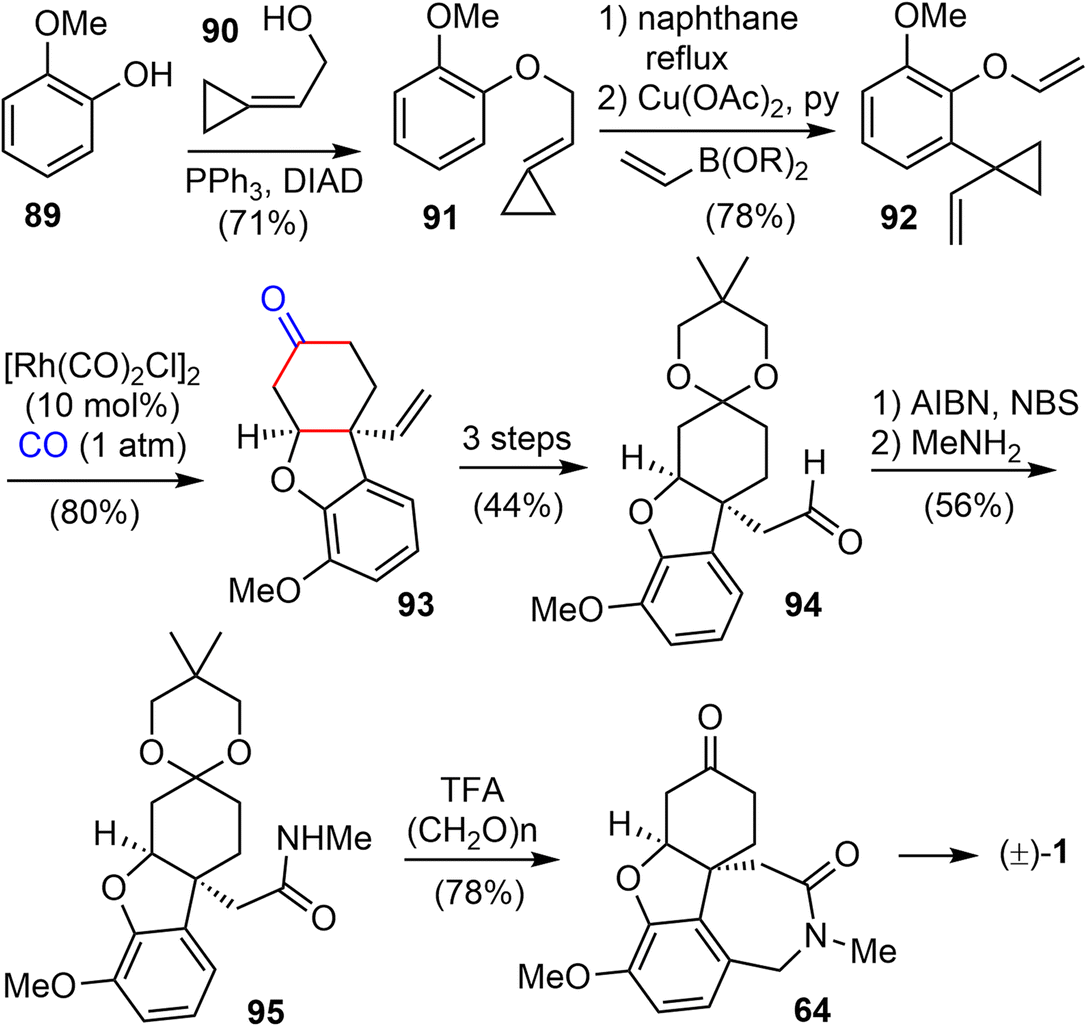 | ||
| Scheme 23 Yu's formal total synthesis of galantamine. | ||
3.7 “Cut and sew” carboacylation reaction (Xu)
Transition metal-catalyzed carbon–carbon bond activation has been increasingly used as a powerful tool for devising unusual bond-disconnecting strategies. The “cut and sew” strategy uses strained rings (benzocyclobutenones and cyclobutanones) with a tethered unsaturated moiety as substrates. It starts with the oxidative addition of a transition metal into the cyclic C–C bond (the “cut” step) to give a reactive metallacycle, followed by intramolecular migratory insertion of the unsaturated unit and reductive elimination to furnish the ring (the “sew” step) (Scheme 25).87,88In 2020, Xu and coworkers reported a novel strategy for the formal total synthesis of galantamine and lycoramine.35 The concise synthesis was enabled by a Rh-catalyzed intramolecular “cut and sew” carboacylation reaction for the formation of the tetracyclic framework and a regioselective Pd-catalyzed C–H activation for double-bond introduction.
Their synthesis commenced with the preparation of substituted benzocyclobutenone 102 (Scheme 24). Lithium enolate 98 was first prepared in situ from THF and n-BuLi. Slow addition of a freshly prepared solution of lithium tetramethylpiperidide (LiTMP) to a mixture of aryl bromide 96 and lithium enolate 98 generated the highly reactive substituted aryne 97, which underwent the [2 + 2] cycloaddition reaction with 98 to afford the benzocyclobutanol 99 regioselectively in 64% yield on a decagram scale.89 The adjacent methoxymethyl ether (MOM) group on benzyne allowed the production of 99 as a single regioisomer through the negative inductive effect.90 Dess–Martin oxidation of the alcohol followed by removal of the MOM group under acidic conditions afforded benzocycloubutenone 100 in 79% overall yield on a decagram scale. Alkylation of 100 with the known bromide 101 followed by a selective Wittig olefination provided the key C–C activation precursor 102 in 45% yield over 2 steps.
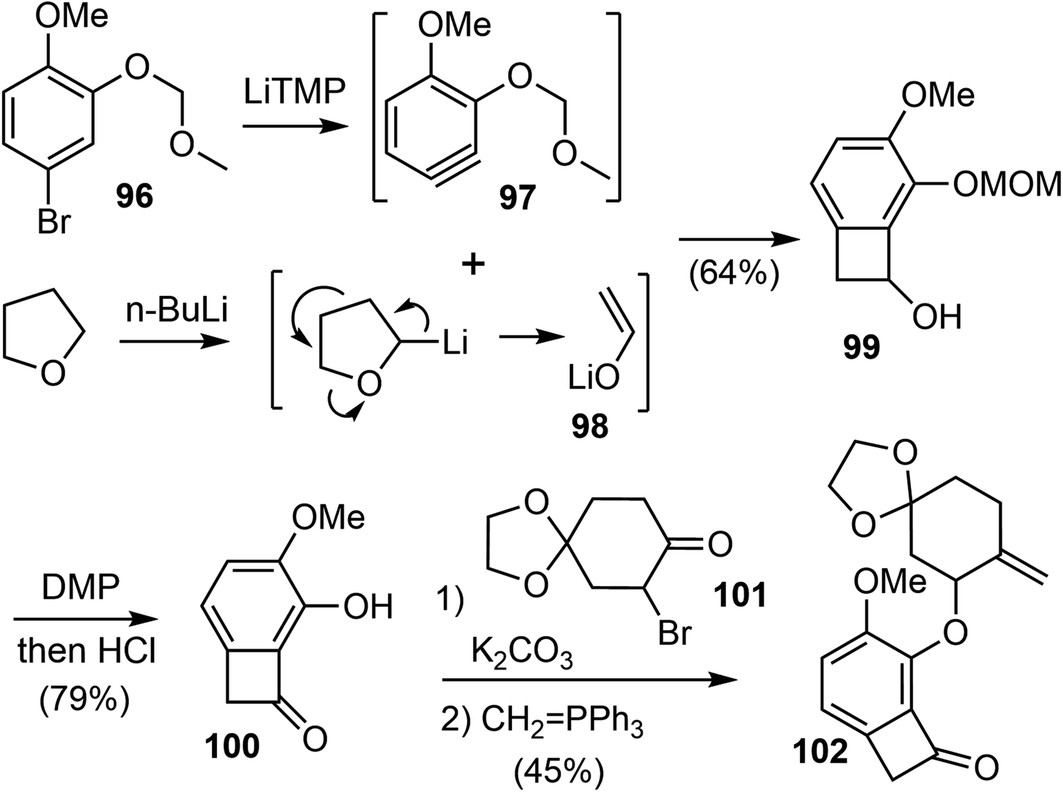 | ||
| Scheme 24 Preparation of benzylcyclobutenone 102. | ||
After screening and optimization of the reaction parameters, it was found that the key “cut and sew” carboacylation reaction87 of 102 was best performed with the catalysis of [Rh(CO)2Cl]2 (5 mol%) in combination with an electronically poor monodentate phosphine ligand P(C6F5)3 (22 mol%). Ring opening of the strained cyclobutenone via C–C bond cleavage, followed by cyclization with the tethered olefin generated the desired product 105 in 77% yield on a gram scale (Scheme 25).
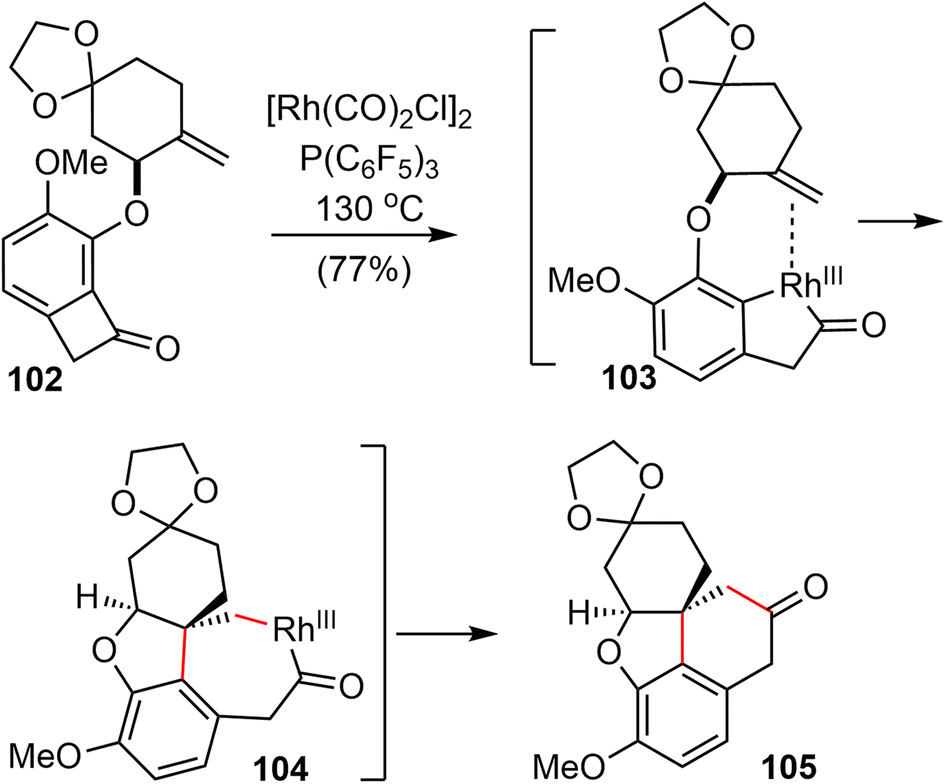 | ||
| Scheme 25 The “cut and sew” carboacylation reaction of 102. | ||
Having successfully established the core structure of the target, the next goal was to form the azepine D ring by an N-insertion reaction of the tetracyclic ketone 105 (Scheme 26). They first tried the Schmidt rearrangement, but it was unsuccessful. Instead, condensation of 105 with hydroxylamine gave the corresponding oxime quantitatively as a separable mixture of E/Z isomers (E/Z = 1:1.2). The E-isomer 106 was isolated and tosylated, and the Beckmann rearrangement occurred spontaneously at 30 °C in THF/water without any additives to give the desired amide. Subsequent N-methylation generated Zhou and Xie's intermediate 64,22 which underwent a Pd-catalyzed regioselective dehydrogenation reaction according to Stahl's procedure91 to give Guillou's enone 43.73 Sequential reduction of the ketone and lactam groups with L-selectride and LiAlH4 successfully elaborated intermediates 64 and 43 to (±)-lycoramine (6) and (±)-galantamine (1), respectively. The longest linear sequences of the total synthesis of galantamine and lycoramine are 11 and 10 steps, respectively.
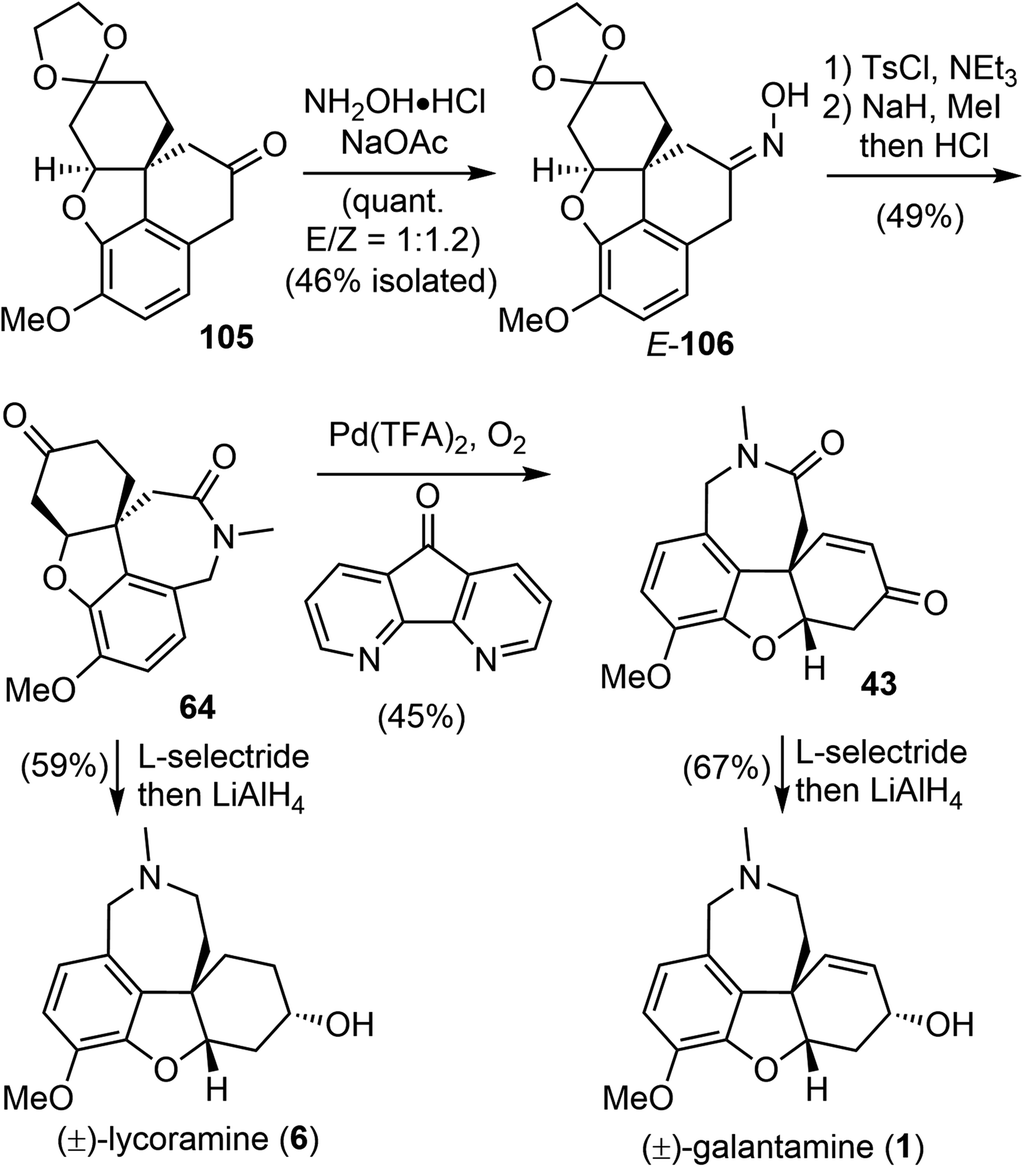 | ||
| Scheme 26 Xu's formal total synthesis of (±)-galantamine and (±)-lycoramine. | ||
3.8 Olefin carbonylative annulation (Zhao)
In 2021, Zhao and coworkers reported a highly efficient formal total synthesis of galantamine and lycoramine.37 A two-phase approach was used in their total synthesis, with a palladium-catalyzed carbonylative cascade annulation and a DDQ-mediated regioselective intramolecular oxidative lactamization to generate the tetracyclic skeleton in the early phase, followed by a BF3·OEt2-promoted selective reorganization of the bridged tetracyclic skeleton in the late phase.Their synthesis started with the deprotonation of racemic allylic alcohol 107, followed by intermolecular O-alkylation with freshly prepared benzylic bromide 108 in the presence of a catalytic amount of NaI to provide the desired 109 in 82% yield (Scheme 27). The crucial palladium catalyzed carbonylative lactonization reaction was then investigated under a variety of conditions.92,93 It was found that the reaction could be catalyzed by Pd(OAc)2 without additional ligand, in the presence of AgOTf and a sterically hindered base 2,6-di-tert-butylpyridine (DTBP) under 1 atm of carbon monoxide. Loss of the MOM group in situ under the reaction conditions led directly to the desired lactonization product 110 in 61% yield. In contrast to the cis-5,6-bicyclic products obtained in the previous total syntheses, only the trans-6,6-bicyclic product was obtained in this case. Treatment of 110 with MeNH2·HCl in the presence of K2CO3, followed by one-pot deprotection of the ketal, afforded the amide 111 in 76% yield. The X-ray crystal structure of 111 confirmed the trans-6,6-bicyclic structure of 110. Although there are many precedents for similar transformations, the structural rearrangement of compound 111 to the galantamine skeleton was unsuccessful under a variety of conditions. Instead, oxidation of 111 with DDQ proceeded smoothly to afford the bridged tetracyclic aza-acetal 112 in excellent yield.
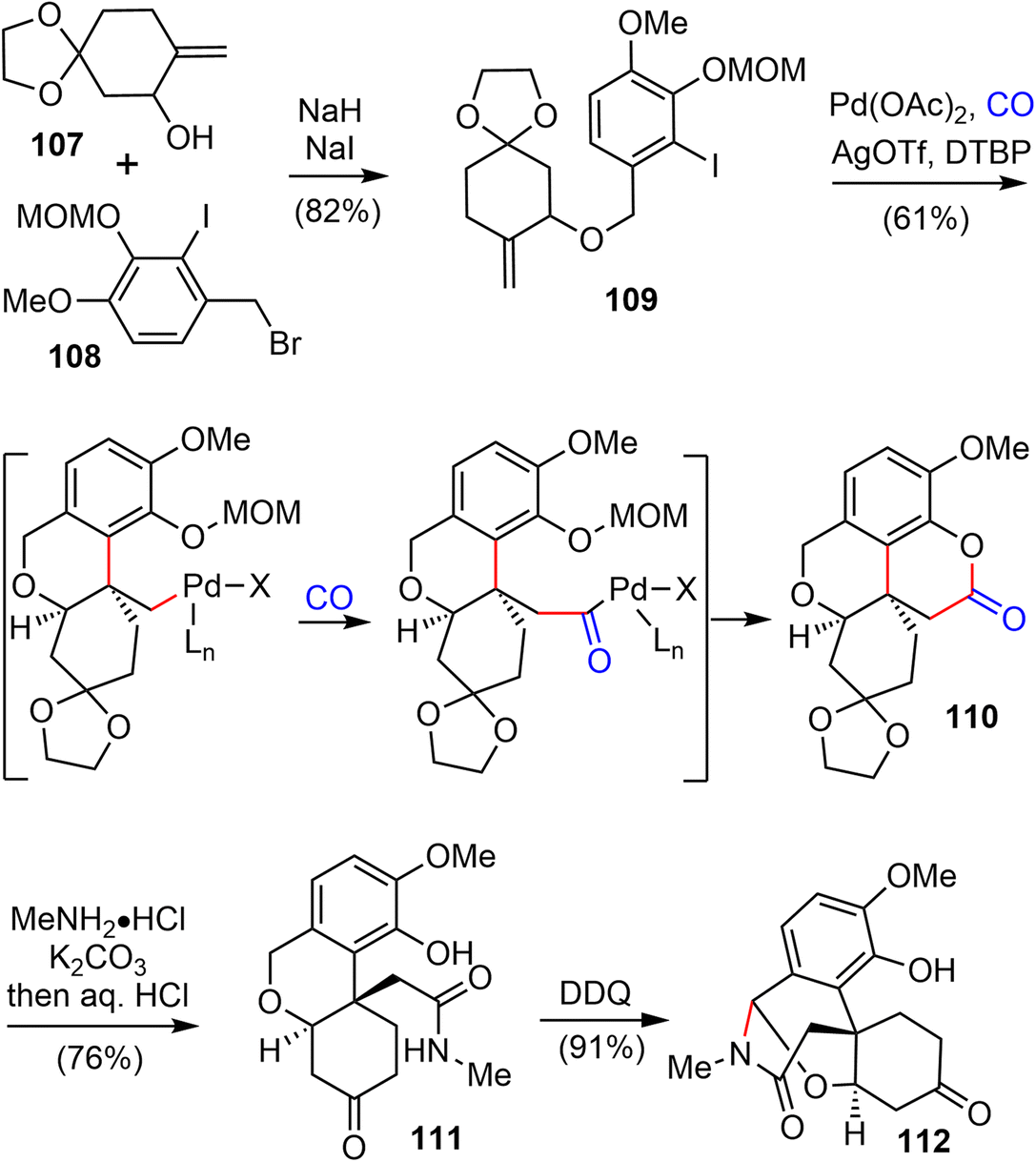 | ||
| Scheme 27 Palladium catalyzed carbonylative esterification. | ||
Compound 112 was found to be a suitable substrate for the desired skeletal rearrangement reaction (Scheme 28). They were pleased to find that treatment of 112 with BF3·Et2O, followed by the addition of (EtO)3SiH as the reductant, successfully converted 112 to the desired tetracyclic lactam 64 in 56% yield. Presumably, the Lewis acid BF3·Et2O initiated the aza-acetal cleavage, a retro-oxa-Michael addition and a phenolic oxa-Michael addition cascade to afford the tetracyclic iminium, followed by reduction with (EtO)3SiH to give the known lactam 64.22 Remarkably, this sequence allowed the preparation of hundreds of milligrams of late intermediate 64 in 19% overall yield in five steps from compounds 107 and 108 in a single batch. As in the previous total synthesis, further oxidation state adjustment of 64 completed the seven- and six-step syntheses of (±)-galantamine (1) and (±)-lycoramine (6), respectively.
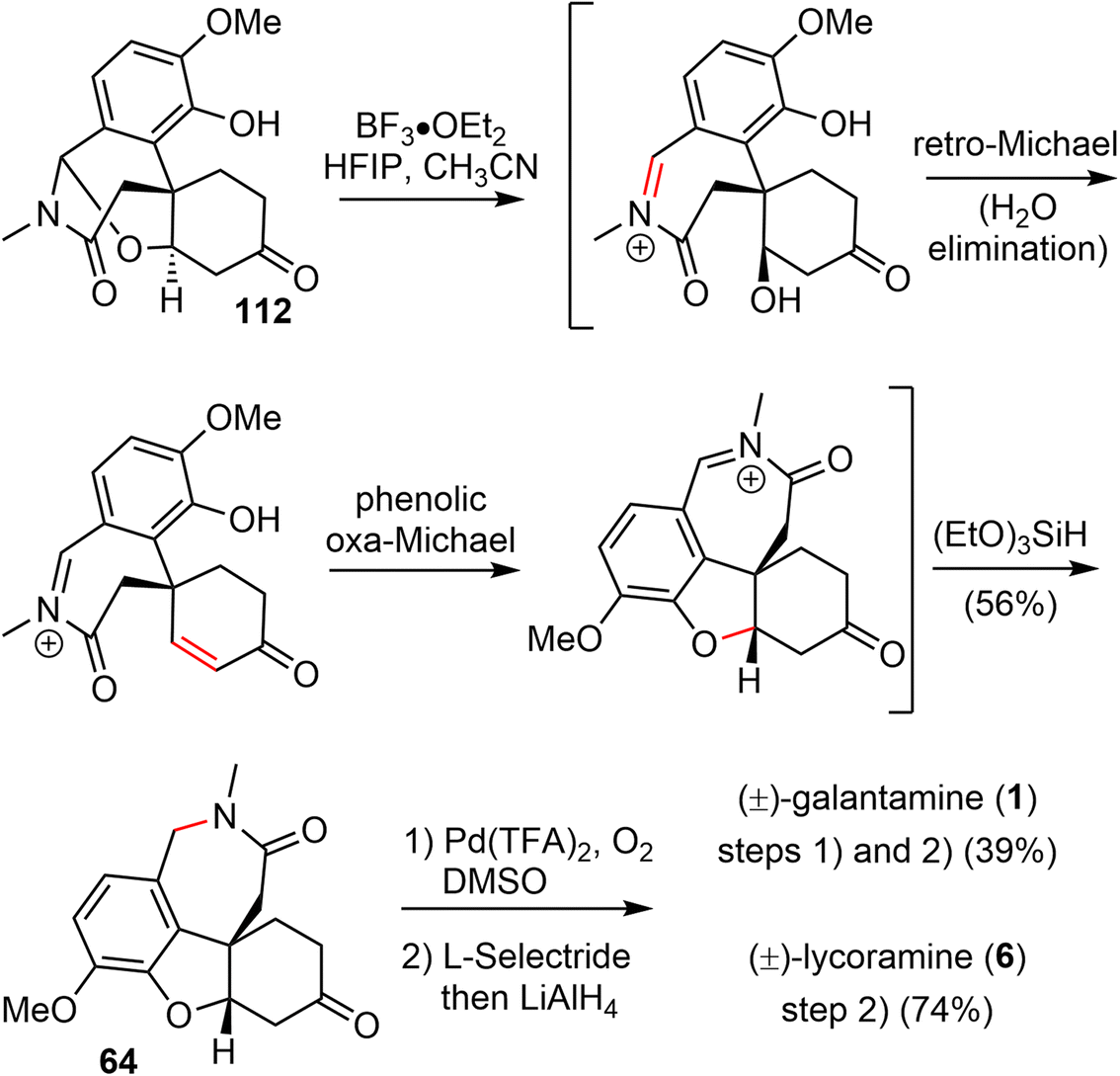 | ||
| Scheme 28 Zhao's formal total synthesis of (±)-galantamine and (±)-lycoramine. | ||
4. Rearrangement reactions
Intramolecular rearrangement reactions are efficient ways to build sterically hindered quaternary centers. Stereospecific rearrangement reactions, such as the sigmatropic rearrangement, can transfer the chirality from a less hindered stereocenter to a sterically congested one with high fidelity. These reactions have been extensively employed in the total syntheses of natural products with quaternary centers. Several rearrangement reactions have been successfully applied in the synthesis of galantamine.13,15,19,38,434.1 Semipinacol rearrangement (Tu)
In 2006, Tu and coworkers reported their total synthesis of (±)-galantamine in 13 steps from the commercially available compounds.13 The overall yield of their synthesis is 12%. Similar to their previous work on the total synthesis of lycoramine,94 a semi-pinacol rearrangement reaction of an allylic alcohol was used to build the benzylic quaternary center.A modified Shapiro reaction of hydrazone 113 with n-BuLi generated a vinyl lithium,95 which reacted a o-vanillin derivative to give an allylic alcohol 114 in 85% yield (Scheme 29). Upon treatment of 114 with NBS in DCM at 0 °C, the aldehyde 116 with a quaternary center was obtained in 95% yield as a single isomer. Presumably, a bromonium ion 115 was formed first, followed by a stereospecific semipinacol rearrangement of the aromatic group to generate the quaternary center. Reaction of 116 with DBU in DMSO initiated a one-pot desilylation and intramolecular SN2 type cyclization reaction to form the B ring and gave the tricyclic product 117 in 90% yield.94 Compound 117 was then transformed into aldehyde 118 in 6 steps, including a one-carbon homologation of the aldehyde and oxidation state adjustment of the C ring. A radical oxidation of the aldehyde generated the corresponding acid bromide, which reacted with MeNH2 to give an amide 119.86 A further Pictet–Spengler reaction of 119 with paraformaldehyde gave the known lactam,73 and reduction of the lactam afforded (±)-galantamine.
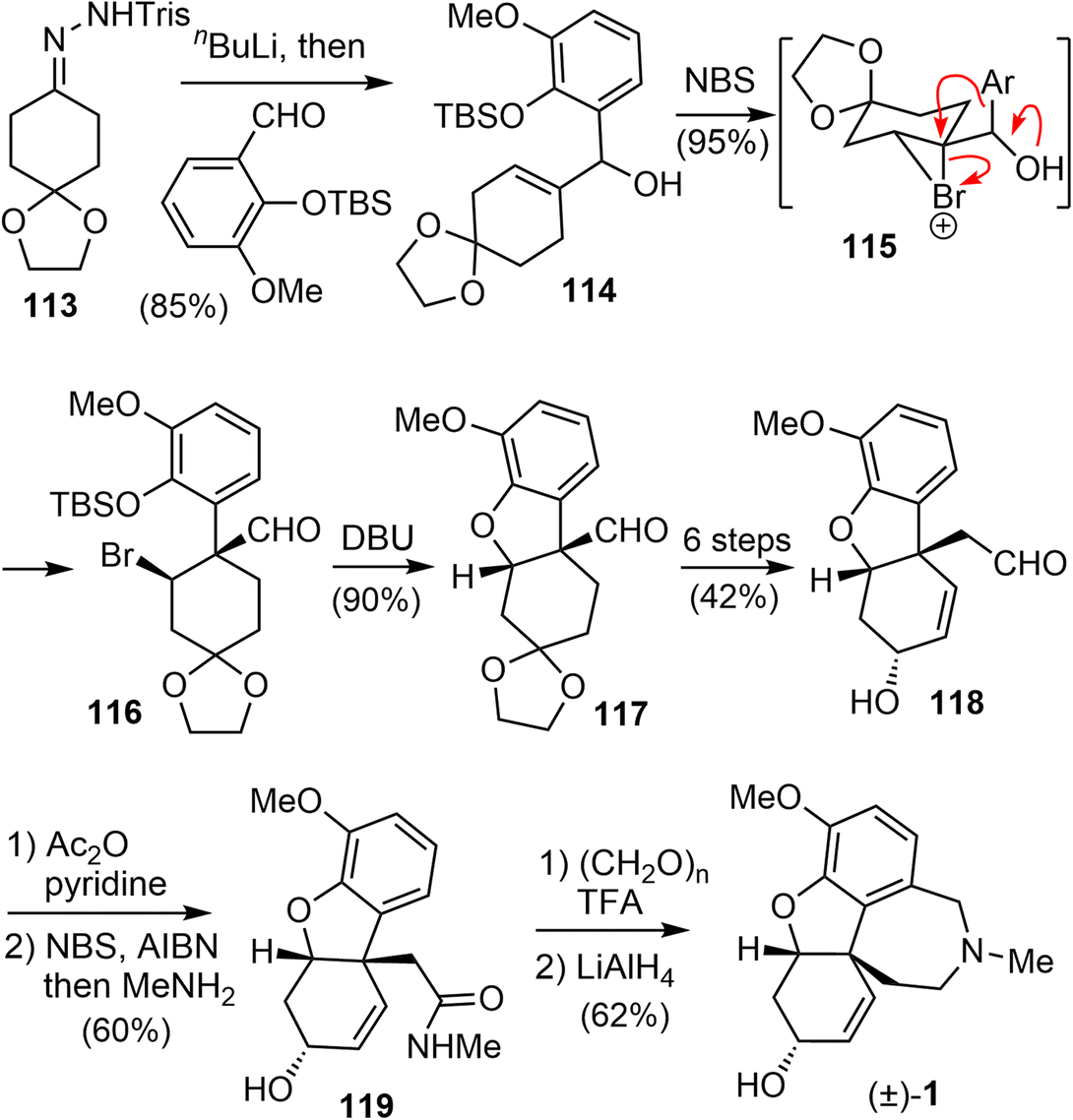 | ||
| Scheme 29 Tu's total synthesis of (±)-galantamine. | ||
4.2 Johnson–Claisen rearrangement (Bisai)
In 2022, Bisai and coworkers reported asymmetric total synthesis of naturally occurring Amaryllidaceae alkaloids sharing dihydrobenzofuran scaffolds, including (−)-galantamine (1), (−)-lycoramine (6), and (−)-narwedine (5).38 The synthesis used a key catalytic enantioselective Corey–Bakshi–Shibata (CBS) reduction of α-bromoenone (99% ee), the chirality of the resulting secondary alcohol was transferred to an all-carbon quaternary center via an orthoester Johnson–Claisen rearrangement.The α-bromo-3-arylcyclohexenone 120 was synthesized by a Stork–Danheiser reaction over a 10 g scale.96 CBS reduction of 120 was carried out using the 20 mol% (S)-Me-CBS catalyst at 25 °C for 3 h to afford allyl alcohol 121 in 99% ee on gram scale (Scheme 30).97 The bromo atom at the α-position was essential for the high enantioselectivity. Two sets of conditions were identified for the crucial Johnson–Claisen rearrangement.98,99 In the first approach, debromination with n-BuLi afforded the allylic alcohol 122, Conventional weak acidic conditions for the Johnson–Claisen rearrangement of 122 led to the product with decreased ee. Further optimization revealed that reaction in a basic medium suppressed the loss of optical purity. Under the optimized conditions, heating a solution of 122 (98% ee) and triethyl orthoacetate in DIPEA promoted the orthoester Johnson–Claisen rearrangement to produce the desired ester 123 in 68% yield in 48 h without the loss of optical purity (97.4% ee). Microwave irradiation shortened the reaction time to 20 min at 210 °C. In the second approach, the orthoester Johnson–Claisen rearrangement of 121 was carried out in the presence of o-nitrophenol using 20 equivalents of triethyl orthoacetate at 220 °C for 36 h. This reaction afforded the desired product without the loss of optical purity. A radical-initiated debromination with tributyltin hydride (Bu3SnH) in the presence of catalytic AIBN provided product 123.
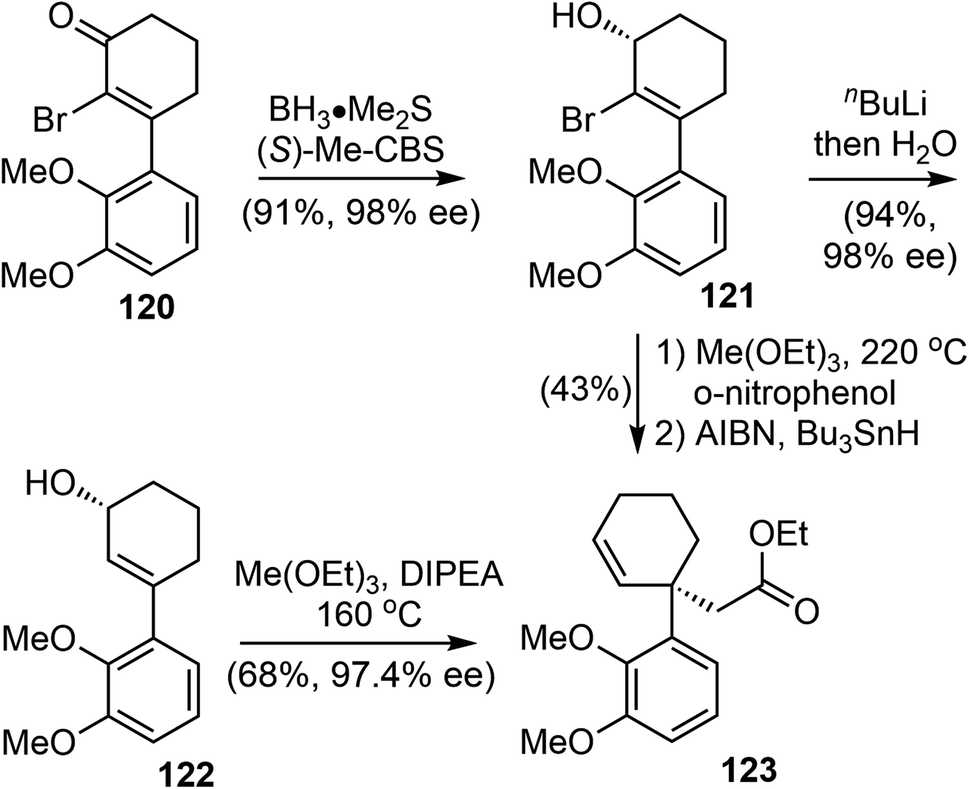 | ||
| Scheme 30 The orthoester Johnson–Claisen rearrangement. | ||
An allylic oxidation of 123 using CrO3 in the presence of 3,5-dimethylpyrazole (DMP) afforded the enone 124 in 71% yield (Scheme 31).100 A chemoselective demethylation of 124 with 1.05 equivalent of BBr3 resulted in a concomitant oxa-Michael cyclization to form the tricyclic structure and delivered 125 in 91% yields in a one-pot fashion. The ester was transformed to an amide, which underwent a Pictet–Spengler reaction with paraformaldehyde to generate the azepine D ring and gave compound 64, an intermediate in Zhou and Xie's total synthesis.22 The tetracyclic compound 64 was a versatile intermediate and was converted to four Amaryllidaceae alkaloids in 2–5 steps, including (−)-galantamine.
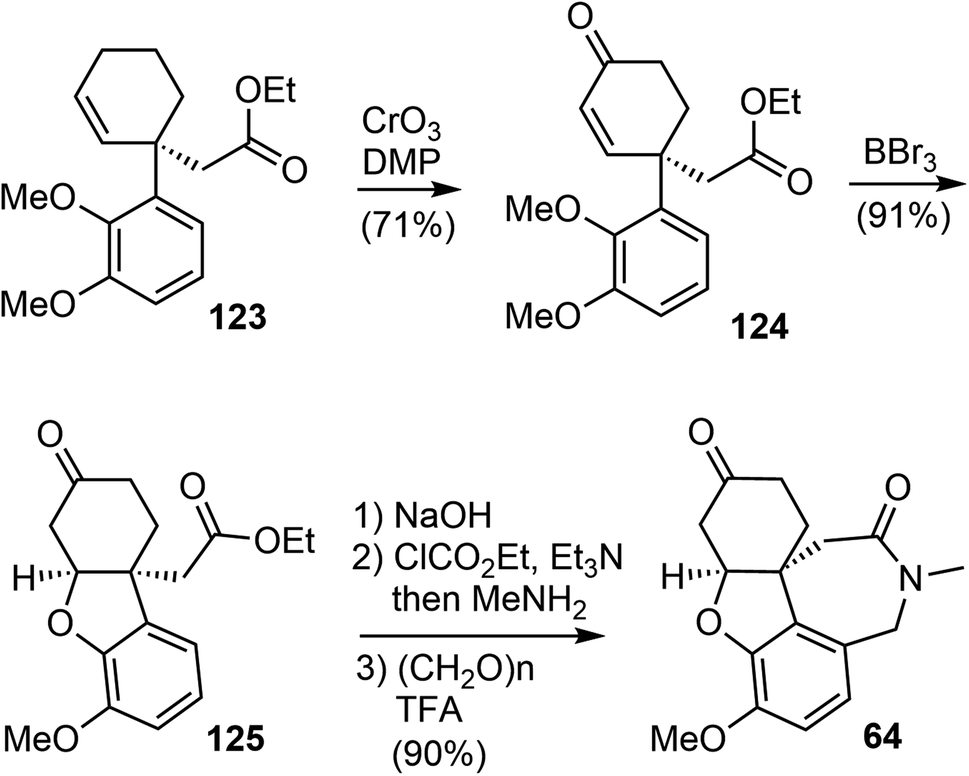 | ||
| Scheme 31 Bisai's formal total synthesis of (−)-galantamine via64. | ||
4.3 Eschenmoser–Claisen rearrangement (Li)
In 2023, Li and coworkers reported a chemo- and regioselective 1,2-reduction of arenes via η6-coordination to chromium (126), providing rapid access to 1,3-cyclohexadienes 128 (Scheme 32).43 The process begins with the in situ generation of the hydride ion from Ph3SiH and KOt-Bu or Me4NF. This ion then attacks the arene ring to form the η5-cyclohexadienyl anion intermediate. Protonation of this intermediate results in the formation of the Cr–H species 127. Reductive elimination completes the 1,2-reduction process, and the resulting η4-cyclohexadiene complex quickly undergoes solvolysis to release the final 1,3-cyclohexadiene product. The reaction conditions are mild and can tolerate various reduction-sensitive functional groups. More than 50 examples have been developed to demonstrate the versatility of the reaction. Additionally, it allows for regiodivergent deuteration, with precise control over the position of deuteration and the degree of deuterium incorporation by using different sequences of (non)deuterated hydride and acid reagents. | ||
| Scheme 32 Selective 1,2-reduction of chromium-bound arenes. | ||
The reaction was then applied in a formal total synthesis of galantamine (Scheme 33). The η6-coordinated chromium complex 130 was prepared by refluxing 4-methoxydibenzo[b,d]furan 129 with chromium hexacarbonyl. Following the standard conditions developed above, the chromium-bound complex 130 was reduced to a diene on a gram-scale. The crude 1,2-reduction product was subjected to a hydroboration/oxidation process to give the alcohol product 131. A novel dearomative Eschenmoser–Claisen rearrangement was used to convert 131 to amide 132 with a quaternary center in 78% yield. Compound 132 was further transformed to compound 94, an intermediate in Yu's synthesis of (±)-galantamine.26
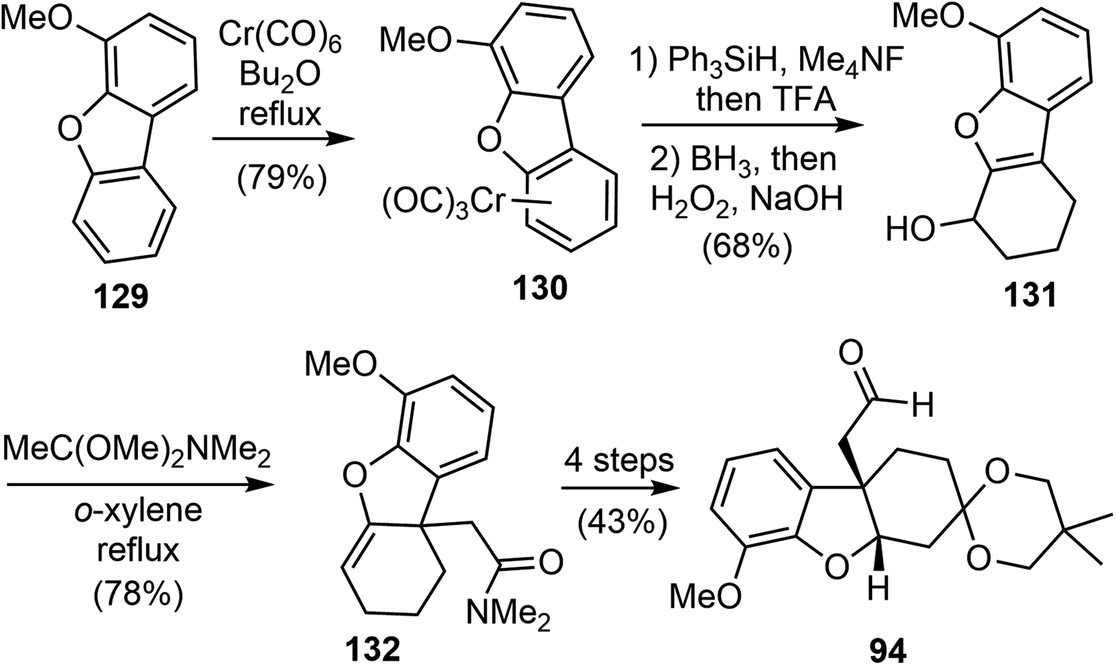 | ||
| Scheme 33 Li's formal total synthesis of (±)-galantamine via94. | ||
5. Alkylation reactions
The enolate alkylation reaction is a fundamental and well-established process for C–C bond formation, including the creation of bonds involving highly congested quaternary centers. The control of the stereoselectivity of enolate alkylation using chiral auxiliaries, chiral bases, and chiral catalysts has received considerable attention, resulting in many innovative methodologies. The enolate alkylation reaction has been successfully implemented in the construction of the congested quaternary center and C ring of galantamine. Highly efficient asymmetric enolate alkylation reactions with organocatalysts and Lewis acid catalyst were identified for the asymmetric total synthesis of (−)-galantamine.5.1 Cascade double Michael addition and Dieckmann reaction (Ishikawa)
In 2008, Ishikawa and Saito developed a practical method for synthesizing 4,4-disubstituted cyclohexane-1,3-diones from nonactivated simple ketones (Scheme 34).16 The process involved a double Michael addition of substituted acetone derivatives 133 with excess acrylic acid ester under basic conditions, followed by a Dieckmann cyclization, resulting in the formation of 4,4-disubstituted cyclohexane-1,3-diones 134 in a single pot. Interestingly, only highly substituted enolates participate as Michael donors in the cascade process. Enol ethers 135 were selectively formed from diketones 134 under acidic conditions. Alkyl and aryl groups were successfully introduced at the quaternary stereogenic center, which is typically challenging to achieve.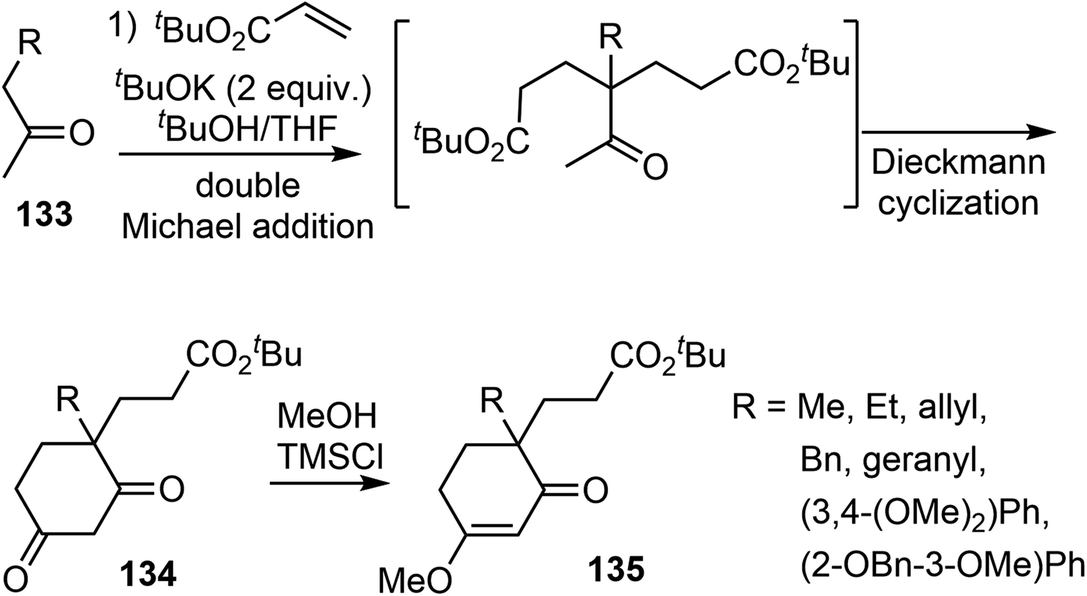 | ||
| Scheme 34 The cascade double Michael addition and Dieckmann reaction. | ||
Enol ethers 135 were found to be useful intermediates for the synthesis of sterically congested natural products (Scheme 35). A hydroxy enone was obtained by reduction of the ester and ketone groups of 136. When treated with TsOH, it underwent an intramolecular oxy-Michael addition reaction, and the olefin was temporarily protected. Subsequent O-debenzylation under hydrogenolysis conditions gave a phenol derivative 137. A MgCl2-mediated retro-Michael and a phenolic hydroxy-Michael addition reaction of 137 afforded the thermodynamically more stable dihydrobenzofuran 138. The primary hydroxy group of 138 was transformed to a carbamate 139 in 3 steps. A Pictet–Spengler cyclization of 139 with paraformaldehyde generated the D ring and gave the key tetracyclic product 140.73 As in the previous total syntheses, compound 140 served as a common intermediate for the total syntheses of (±)-galantamine and (±)-lycoramine.
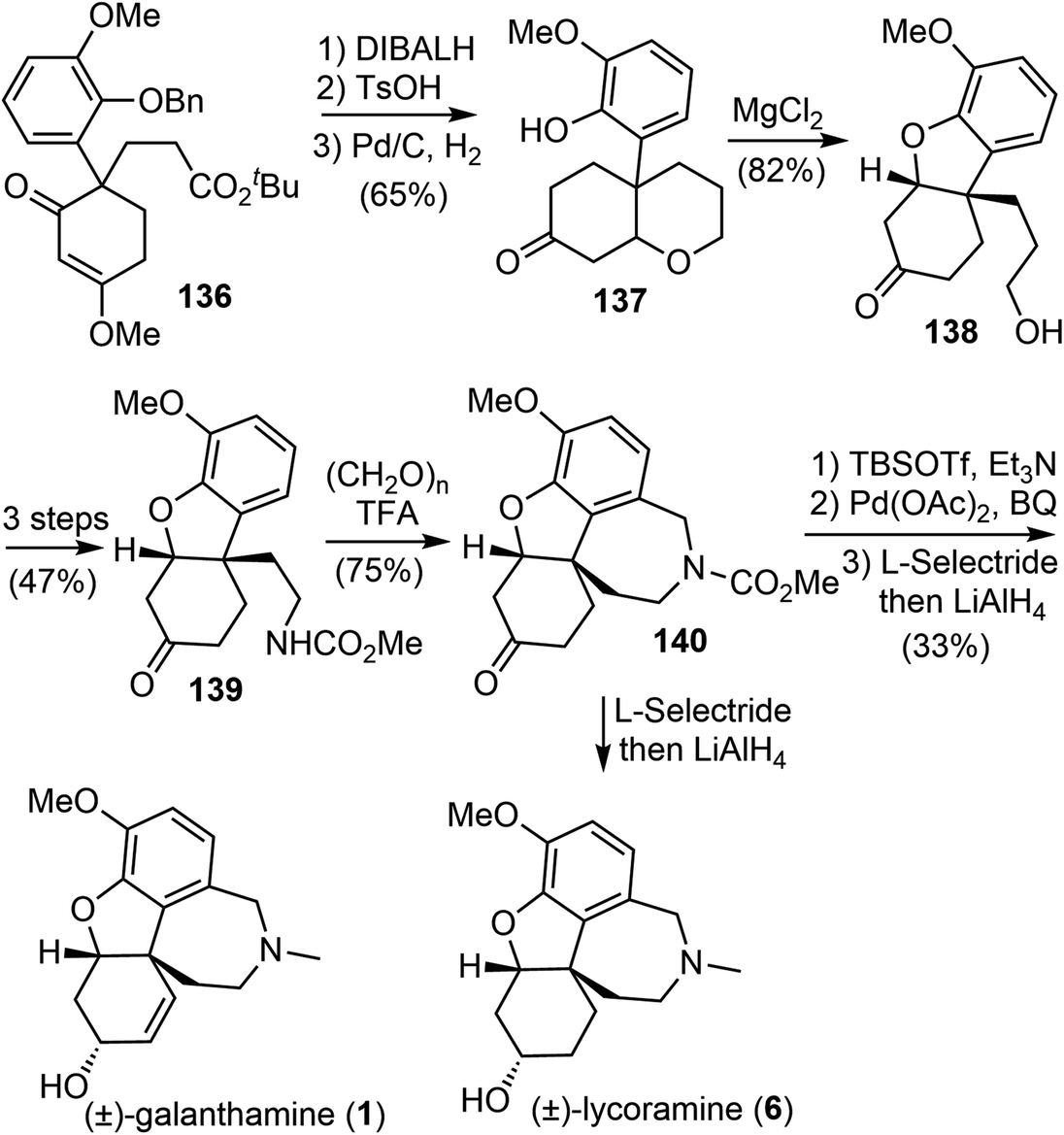 | ||
| Scheme 35 Ishikawa and Saito's synthesis of (±)-galantamine and (±)-lycoramine. | ||
Ishikawa's work opened up a new avenue for the total synthesis of galantamine. In the majority of previous total syntheses, the C ring of galantamine was obtained from commercial supplies. Ishikawa's work was one of the earliest examples to generate the C ring during the total synthesis, and it was also the first example to create the quaternary center by the classic enolate alkylation reaction. The new synthetic strategy provides many opportunities for the development of asymmetric total synthesis of galantamine. Following their racemic synthesis, several groups successfully identified highly efficient asymmetric catalytic systems for similar reactions and completed the enantioselective total synthesis of (−)-galantamine.
5.2 Organocatalyzed asymmetric intermolecular Michael addition (Fan)
In 2011, Fan and coworkers reported total syntheses of several hydrodibenzofuran alkaloids.21 They first developed a method to set up the key sterically congested aryl-substituted quaternary carbon center through an organocatalytic asymmetric intermolecular Michael addition of α-aryl-α-cyanoketones 141 with acrylates 142 (Scheme 36). Compound 141 was readily prepared in 5 steps from o-vanillin in a good overall yield. The Takemoto catalyst 144 (ref. 101) and the cinchonidine-derived bifunctional catalyst 145 (ref. 102) gave the best results and allowed for tuning of the enantioselectivity. Among the solvents tested, p-xylene was found to be the optimal choice with respect to both enantioselectivity as well as the reactivity of the catalyst.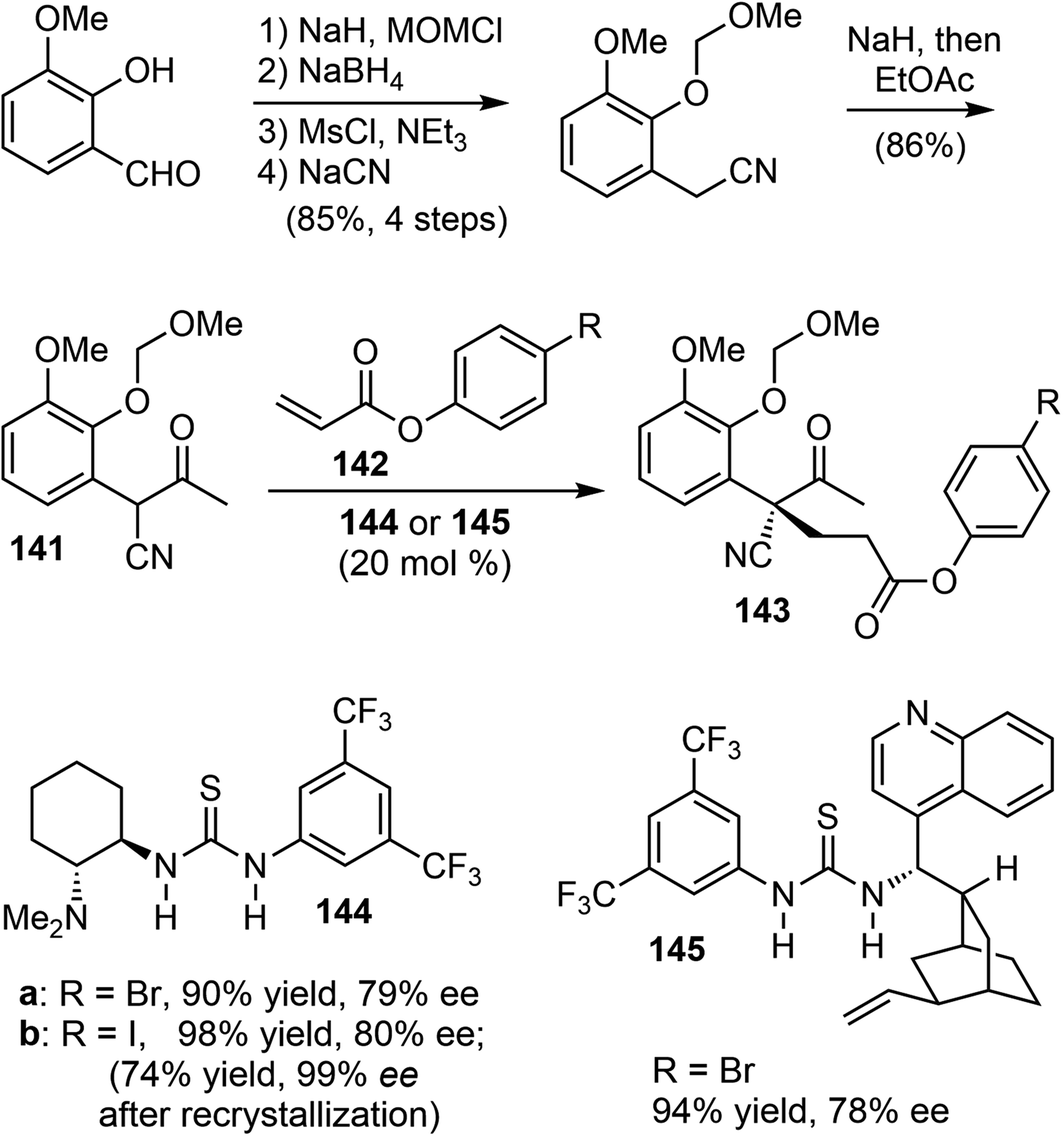 | ||
| Scheme 36 Intermolecular asymmetric Michael addition to form the quaternary center. | ||
Using 144 as the catalyst, the Michael addition of 141 and 142b yielded the functionalized δ-keto ester 143b with an aryl-substituted all-carbon quaternary center. Iodide 143b was selected for the final synthesis because of its ease of crystallization. An optical purity of 99% ee was obtained with a yield of 74% after a single recrystallization. The absolute configuration of the chiral all-carbon quaternary stereocenter was confirmed by X-ray crystallography.
Following Ishikawa's racemic total synthesis, compound 143b underwent a Dieckmann condensation reaction under basic conditions. The resulting 1,3-diketone was regioselectively transformed to enol ether 146 under acidic conditions (Scheme 37). The subsequent Luche reduction of 146, followed by acidic workup led to the formation of an enone that underwent a further intramolecular oxa-Michael addition reaction. The tricyclic intermediate 147 with the key cis-hydrodibenzofuran skeleton was obtained in 40% yield over 4 steps. Subsequently, a one-carbon homologation of the nitrile group was performed in 7 further steps, converting 147 into the carbamate 148. Compound 148 underwent a Pictet–Spengler reaction with paraformaldehyde to form the tetracyclic compound 140, which is a key intermediate in Ishikawa's racemic total synthesis.16 Following similar reaction sequences, 140 was converted to (−)-galantamine and (−)-lycoramine after the oxidation state adjustment.
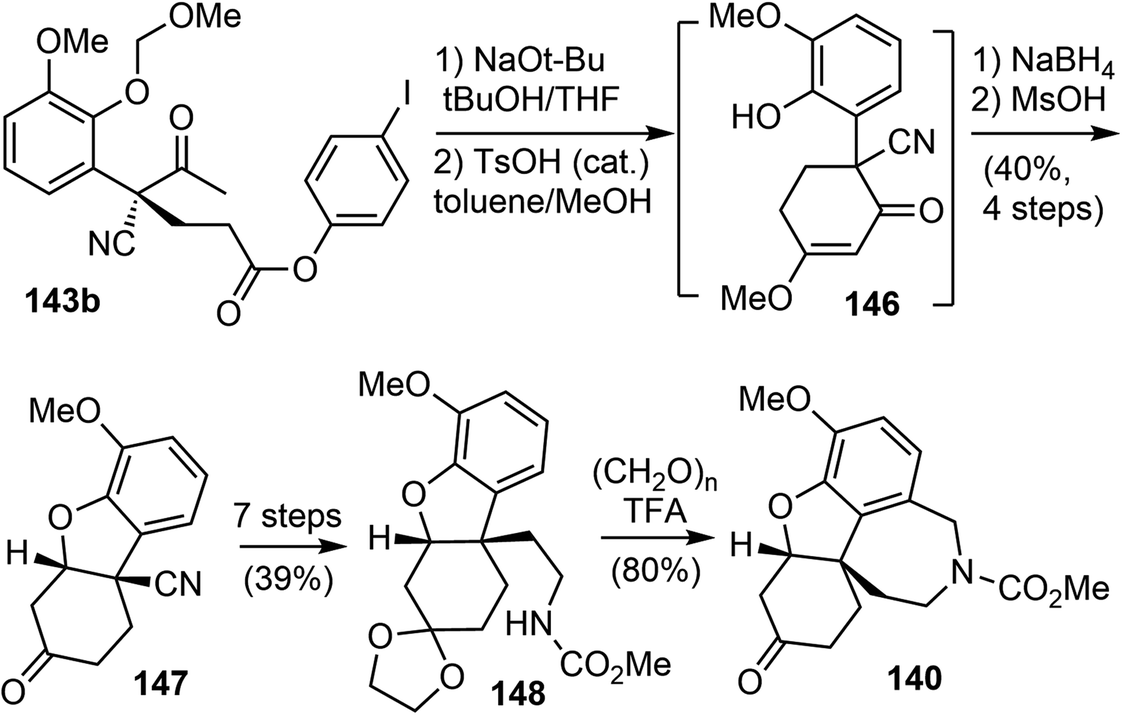 | ||
| Scheme 37 Fan's formal synthesis of (−)-galantamine and (−)-lycoramine via140. | ||
5.3 Lewis acid catalyzed asymmetric intermolecular Michael addition (Jia)
In 2015, Jia and coworkers disclosed a catalytic asymmetric total synthesis of (−)-galantamine (1) and (−)-lycoramine (6).25 Two metal catalyzed reactions were developed to build the core structure of the natural products. Specifically, a palladium-catalyzed intramolecular Larock annulation reaction was employed to construct the 3,4-fused benzofuran, while a Sc(III)/N,N′-dioxide complex catalyzed enantioselective conjugate addition of a 3-alkyl-substituted benzofuranone to methyl vinyl ketone was used to set up the quaternary carbon center.They first investigated the synthesis of 3,4-fused benzofuran by an intramolecular Larock annulation reaction of an ortho-iodophenol tethered with an internal alkyne.103 The reaction of 149 under the catalysis of [Pd2(dba)3] (5 mol%) and P(t-Bu)3·HBF4 (20 mol%) afforded the desired product 150 in 95% yield (Scheme 38). This transformation was found to be quite general, with more than 15 examples of 3,4-fused benzofurans containing either carbon, oxygen, or nitrogen tethers were obtained in reasonable yields.
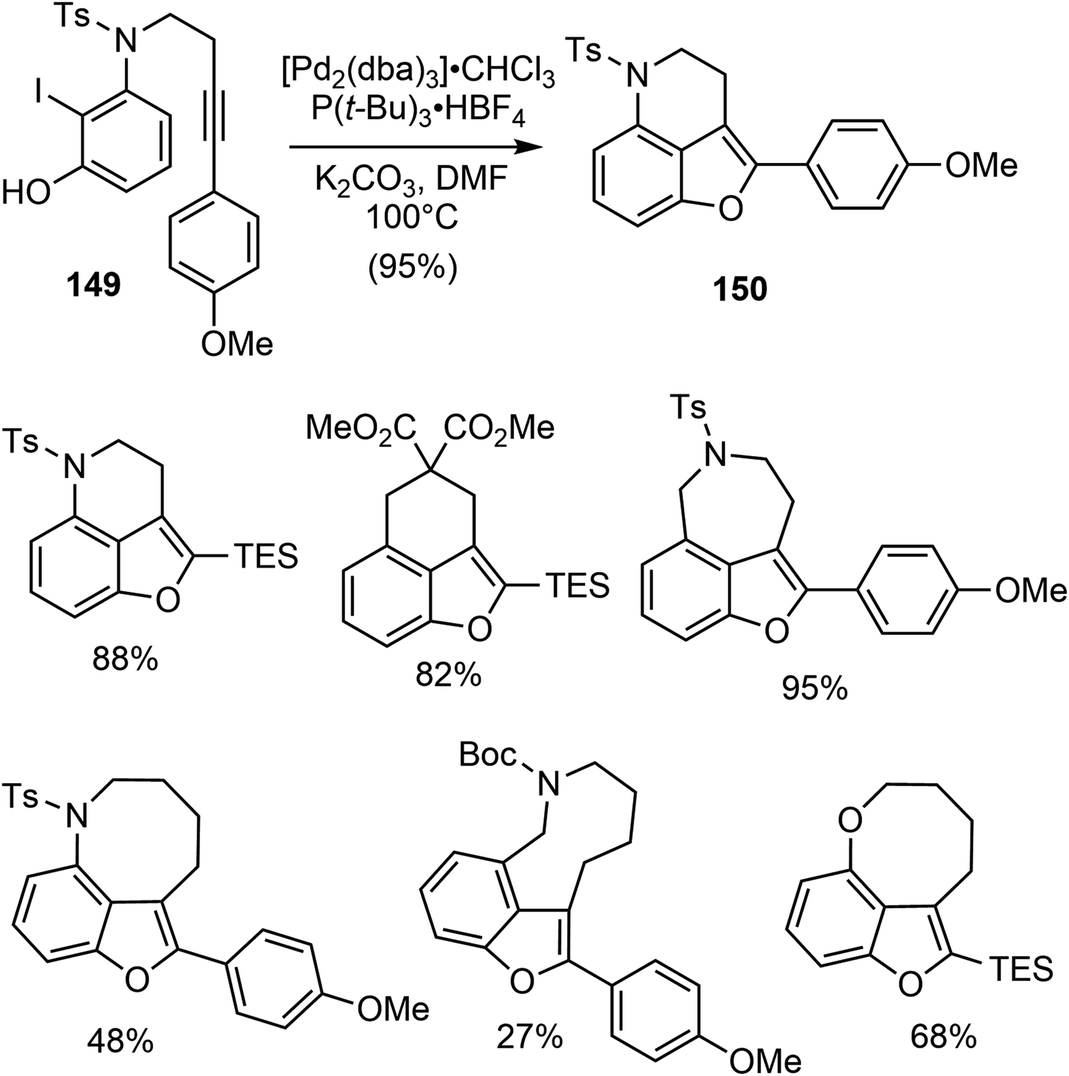 | ||
| Scheme 38 Larock-type annulation for the synthesis of 3,4-fused benzofurans. | ||
Having developed this method, they applied it to the total synthesis of galantamine. The substrate 151 was obtained in 2 steps from readily available materials. Under the reaction conditions developed above, the 3,4-fused benzofuran 152 was obtained from 151 in 89% yield. Removal of the TES group with TBAF followed by oxidation with m-CPBA afforded the lactone 153. They then explored the construction of the chiral all-carbon quaternary stereocenter. They first examined a number of organocatalysts for the reaction of 153 with methyl vinyl ketone (MVK). However, the highest enantioselectivity obtained was 55% ee. Alternatively, other catalytic reaction systems were explored, and it was discovered that a combination of the Lewis acid Sc(OTf)3 with the chiral N,N′-dioxide ligand 154 catalyzed the asymmetric Michael addition reaction of 153 to MVK.104 The reaction proceeded smoothly at room temperature to afford the desired product 155 in 85% yield with 93% ee. An intramolecular cyclization reaction of 155 yielded a hemiketal and generated the tetracyclic core structure 156. Ionic reduction of the hemiketal with Et3SiH resulted in simultaneous reduction of the ketone and removal of the Boc group. Protection of the amine with methyl chloroformate and oxidation of the alcohol delivered 140, the key intermediate in Ishikawa's racemic total synthesis16 and Fan's asymmetric synthesis (Scheme 39).21
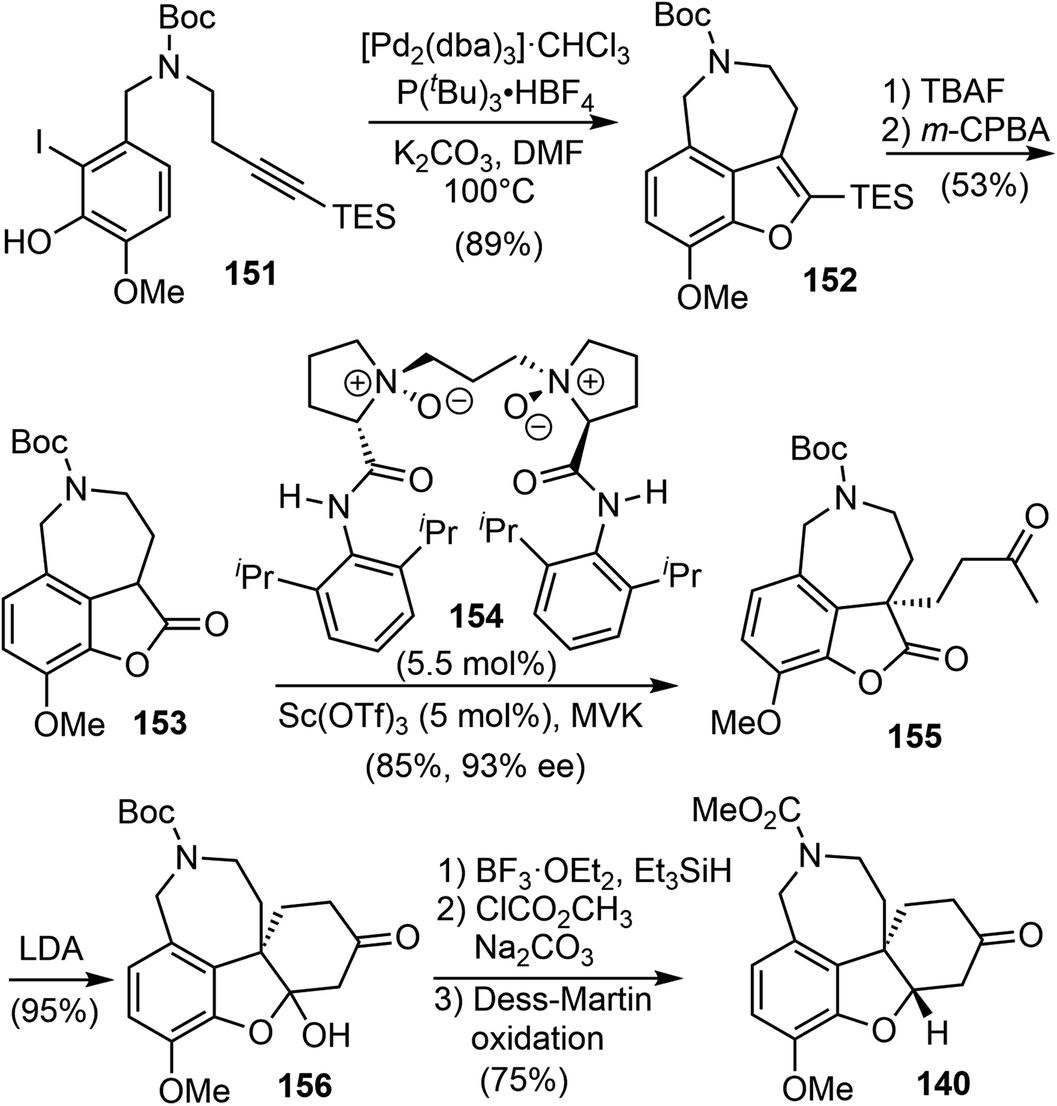 | ||
| Scheme 39 Jia's formal total synthesis of (−)-galantamine via140. | ||
5.4 Organocatalyzed asymmetric Robinson annulation (Tu)
In 2019, Tu and coworkers reported catalytic asymmetric total syntheses of (−)-galantamine (1) and (−)-lycoramine (6).33 They developed a spirocyclic pyrrolidine (SPD)-catalyzed enantioselective Robinson annulation reaction to construct the key cis-hydrodibenzofuran core with the all-carbon quaternary stereocenter.105Their synthesis commenced from 3-butyn-1-ol 157 (Scheme 40). A one-pot double transformation of 157 led to an internal alkyne 158. A copper catalyzed regioselective and stereoselective hydroboration reaction of alkyne 158, followed by a Pd-catalyzed Suzuki coupling reaction with aryl bromide 159, selectively afforded the trisubstituted olefin 160. Compound 160 was then converted to the α, β-unsaturated ketone 161.
 | ||
| Scheme 40 Synthesis of α, β-unsaturated ketone 161. | ||
An initial effort was made to produce the tricyclic compound 164 through a direct intramolecular Robinson annulation of 161. However, the transformation stopped at the Michael addition stage, despite numerous experimental conditions screening. The product was isolated as 163 after a Wittig olefination. Among the reaction conditions screened, it was discovered that the Michael addition/Wittig olefination product 163 was obtained in good yield and selectivity in the presence of the SPD catalyst C and 2,4,6-triisopropylbenzoic acid as an additive. The outcome of the reaction was further improved by decreasing the reaction temperature to −30 °C, resulting in the product 163 being obtained in 87% yield with 96% ee (Scheme 41).
 | ||
| Scheme 41 Intramolecular Michael addition/Wittig olefination of α, β-unsaturated ketone 161. | ||
As a direct intramolecular Robinson annulation of 161 to generate the tricyclic compound 164 was not successful, it was discovered that an acid catalyzed aldol cyclization reaction of crude Michael product 163 afforded the tricyclic compound 164 (>99% ee) (Scheme 42). Compound 164 was used as a key intermediate in their total synthesis of morphine.105 A Rubottom oxidation reaction installed the hydroxy group of 165 in 3 steps from compound 164. Followed by a further oxidation state adjustment, the aldehyde 166 was generated in 5 steps. The tetracyclic product 167 was produced by oxidative amidation of the aldehyde and a Pictet–Spengler reaction with paraformaldehyde, following the protocol from their previous racemic total synthesis,13 Stereo inversion of the alcohol was achieved by Dess–Martin oxidation of the alcohol and L-selectride reduction of the ketone. A final lactam reduction with LiAlH4 completed the total synthesis of (−)-galantamine (1).
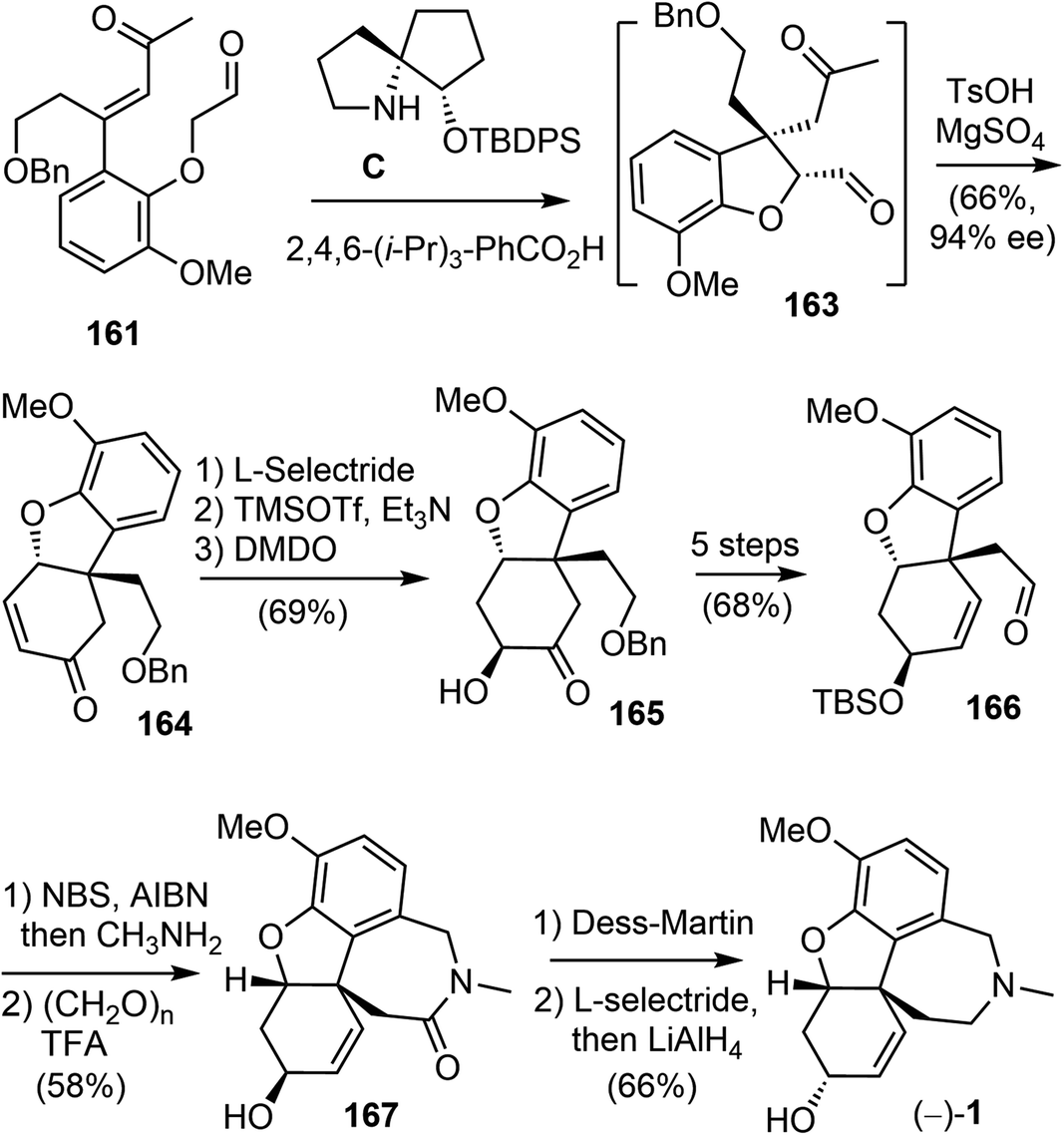 | ||
| Scheme 42 Tu's total synthesis of (−)-galantamine. | ||
5.5 Aryne insertion reaction (Chandrasekhar)
In 2019, Chandrasekhar and coworkers reported a racemic total synthesis of (±)-galantamine (1).34 The process involved only two sub-critical temperature reactions and less than five chromatographic purifications. The synthesis used a key regioselective aryne insertion reaction into a GABA (γ-amino butyric acid) derivative to form the substituted aromatic A ring of galantamine.Aryne is a highly reactive intermediate, it allows rapid functionalization of an aromatic ring by forming multiple carbon–carbon or carbon–heteroatom bonds in a single operation. The acyl–alkylation reaction of aryne with β-ketoester represents a mild and direct aryne insertion into a carbon–carbon bond (Scheme 43). The use of o-silyl aryl triflates, e.g., 168, as aryne precursors has allowed generation of the reactive intermediate under almost neutral conditions. The acyl-alkylation product 170 is the net result of aryne insertion into the α,β C–C single bond of the β-ketoester 169, presumably by a formal [2 + 2] cycloaddition/fragmentation cascade.106
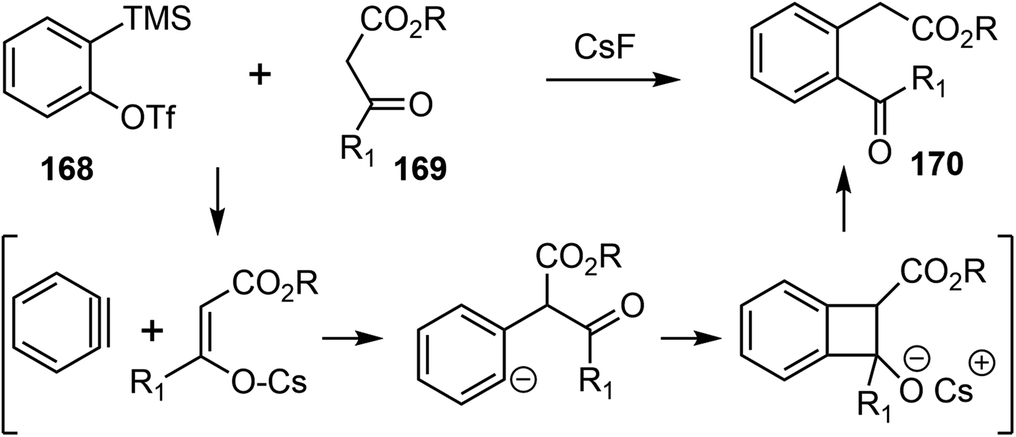 | ||
| Scheme 43 Acyl-alkylation of aryne with β-ketoester. | ||
The synthesis began with the β-formyl ester 171, which was synthesized from GABA in four steps. Upon treatment with the methoxy benzyne precursor 172 and cesium fluoride (CsF), the trisubstituted aryl compound 173 was regioselectively obtained in 62% yield (Scheme 44). The methoxy group on benzyne, through the negative inductive effect,90 provided 173 as a single regioisomer. The product was transformed to the lactone 174 in 3 steps. The lactone was α-alkylated with methyl vinyl ketone, and the azepine D ring was installed with a Pictet–Spengler reaction to generate compound 175. A further intramolecular condensation reaction gave the tetracyclic product 176, which is a close analogue of Jia's intermediate 156.25 By carrying out similar transformations to those in Jia's synthesis, compound 176 was converted to galantamine in a further seven steps.
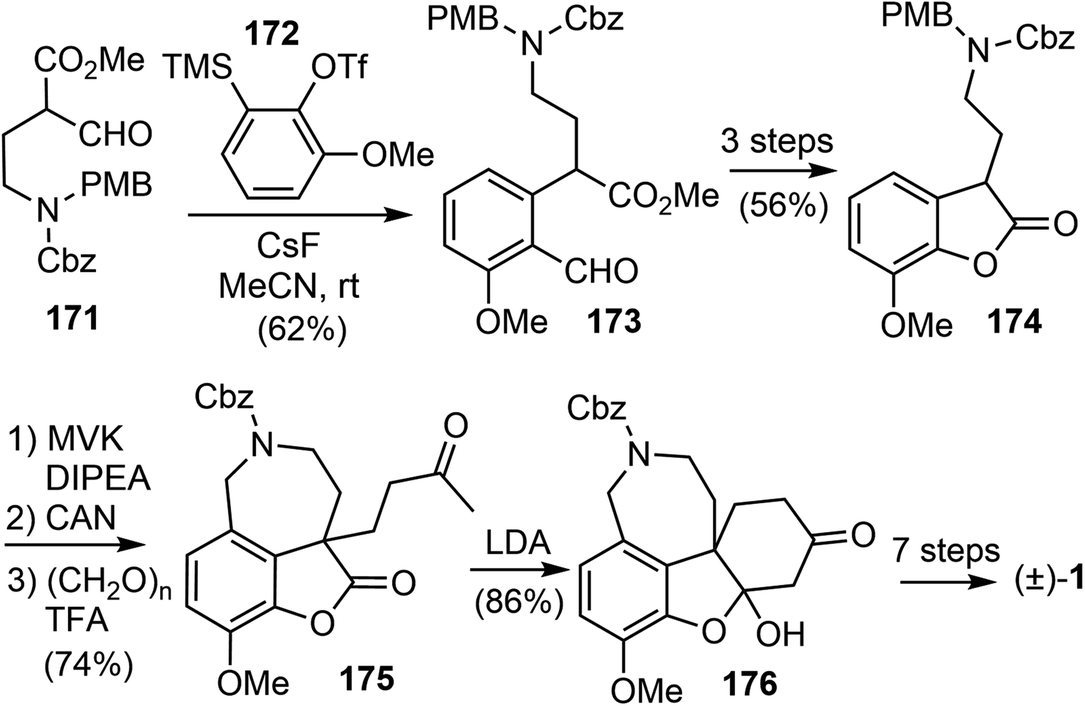 | ||
| Scheme 44 Chandrasekhar's total synthesis of (±)-galantamine. | ||
5.6 Intramolecular phenol alkylation (Magnus)
In 2009, Magnus and coworkers reported a distinct total synthesis of (±)-narwedine (5).18 The synthesis used the para-alkylation of a substituted phenol as the key reaction.31 This reaction generated the cross-conjugated 2,5-cyclohexadienone 179 with a quaternary center and avoided the phenolic oxidation reaction.Their synthesis commenced from a Suzuki cross coupling reaction of commercially available 2-bromovanillin (30) with the boronic acid anhydride 177 (Scheme 45). The resulting biaryl phenol was treated with ethyl vinyl ether and Br2 in the presence of N,N-diisopropylethylamine at 0 °C. The bromoetherification product 178 was obtained in excellent yield (98%). Exposure of 178 to CsF (3 equiv.) in N,N-dimethylformamide (DMF) at 130 °C resulted in a clean desilylation and intramolecular alkylation reaction. The 2,5-cyclohexadienone 179 was obtained in high yield, and thus confirming the viability of this strategy. Acid catalyzed hydrolysis of the acetal in 179 resulted in two oxa-Michael addition reactions to give product 180. Reductive amination of 180 with MeNH2 generated the azepine D ring, which was converted into narwedine (5) by treatment with MeSO3H. The overall process was completed with an impressive yield of 63%, which is approximately five times the yield of the current commercial process.60,61 Since (±)-narwedine (5) has been converted into (−)-galantamine (1) by a crystallization-induced dynamic chiral resolution, and a diastereoselective reduction with L-selectride,58 this completes an eight-step formal total synthesis of (−)-galantamine (1).
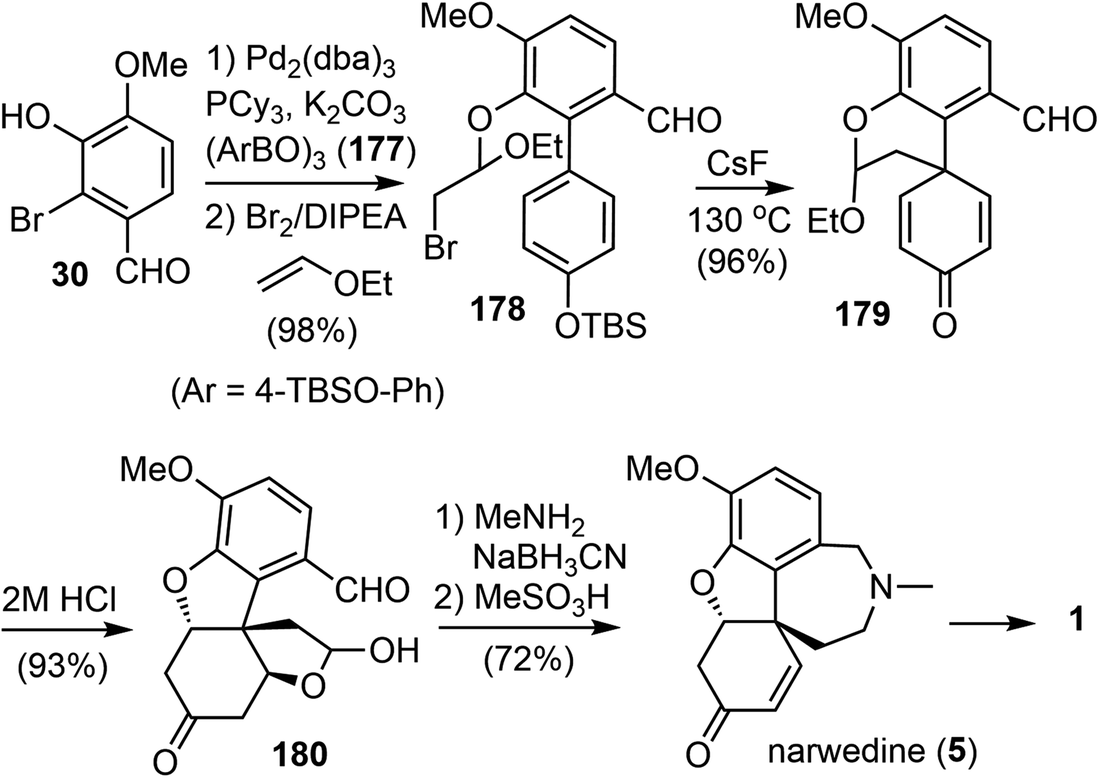 | ||
| Scheme 45 Magnus' total synthesis of (±)-narwedine. | ||
6. Miscellaneous approaches
Other than the above four general approaches to the total synthesis of galantamine, there are several other miscellaneous approaches reported. Yu and coworkers reported a formal synthesis with a radical cyclization to form the quaternary center,30 Cho and coworkers reported a synthesis of (±)-galantamine by a tandem C3-selective Stille coupling-intramolecular Diels–Alder (IMDA) cascade reaction.20 Nagase and coworkers finished a semi synthesis from naltrexone,32 which will not be discussed in this review.6.1 Tandem Stille/IMDA (Cho)
In 2010, Cho and coworkers reported their synthesis of (±)-galantamine by using a tandem C3-selective Stille coupling-intramolecular Diels–Alder (IMDA) cascade of 3,5-dibromo-2-pyrone as a key strategy, which was a novel strategy and a valuable addition to the galantamine synthesis predominated by the approaches involving either oxidative phenol coupling or an intramolecular Heck reaction.20A Cu(II)-promoted O-vinylation of the iodophenol 46 provided the aryl vinyl ether 181 in good yield.107 The Pd-catalyzed stannylation of iodide in 181 proceeded smoothly to afford the aryltin fragment 182. Alternatively, compound 182 could be made in 2 steps from the bromo analogue 183 in a more economical way (Scheme 46).
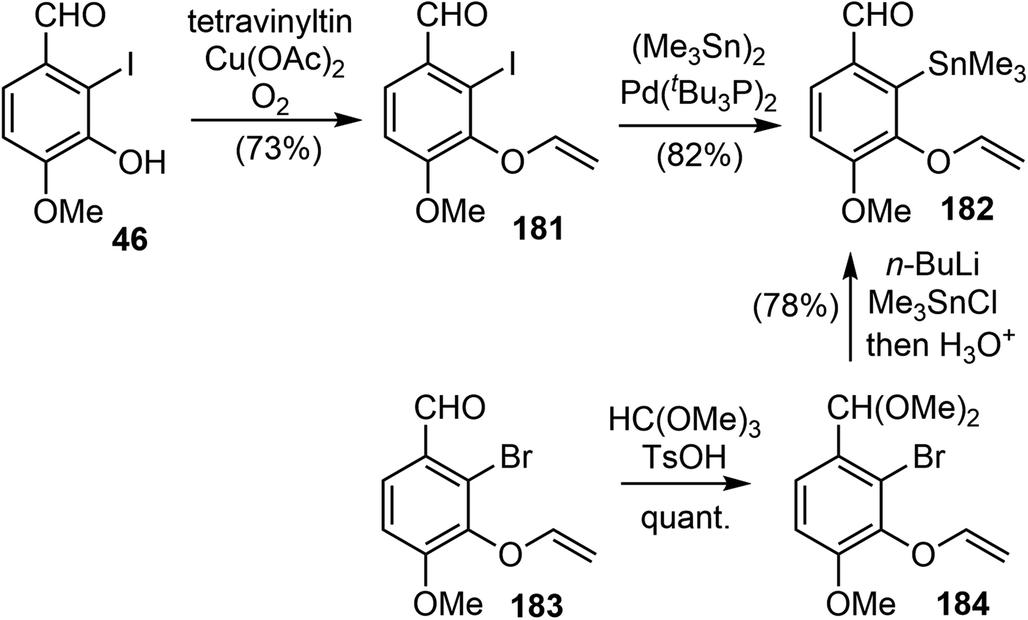 | ||
| Scheme 46 Two approaches to aryltin fragment 182. | ||
A C3-selective Stille coupling reaction of 182 with 3,5-dibromo-2-pyrone was best performed with 5 mol% of Pd(PPh3)4 and 10 mol% of CuI in DMF (Scheme 47). The reaction did not afford the Stille coupling product 185, but instead gave the tandem Stille/IMDA cascade product 186 directly in modest yield and selectivity. The lactone ring opening of 6-endo product 186 with NaOMe and protection of the resultant secondary hydroxyl group as a MOM ether furnished ester 187 in 84% yield over two steps. From this point, they converted compound 187 to the tetracyclic intermediate 188 in 7 steps, adapting the procedures developed by Trost and co-workers for the synthesis of galantamine on the basis of the structural similarity with their tricyclic intermediate.69 Final epimerization of the alcohol and reductive removal of the bromide afforded galantamine (1).
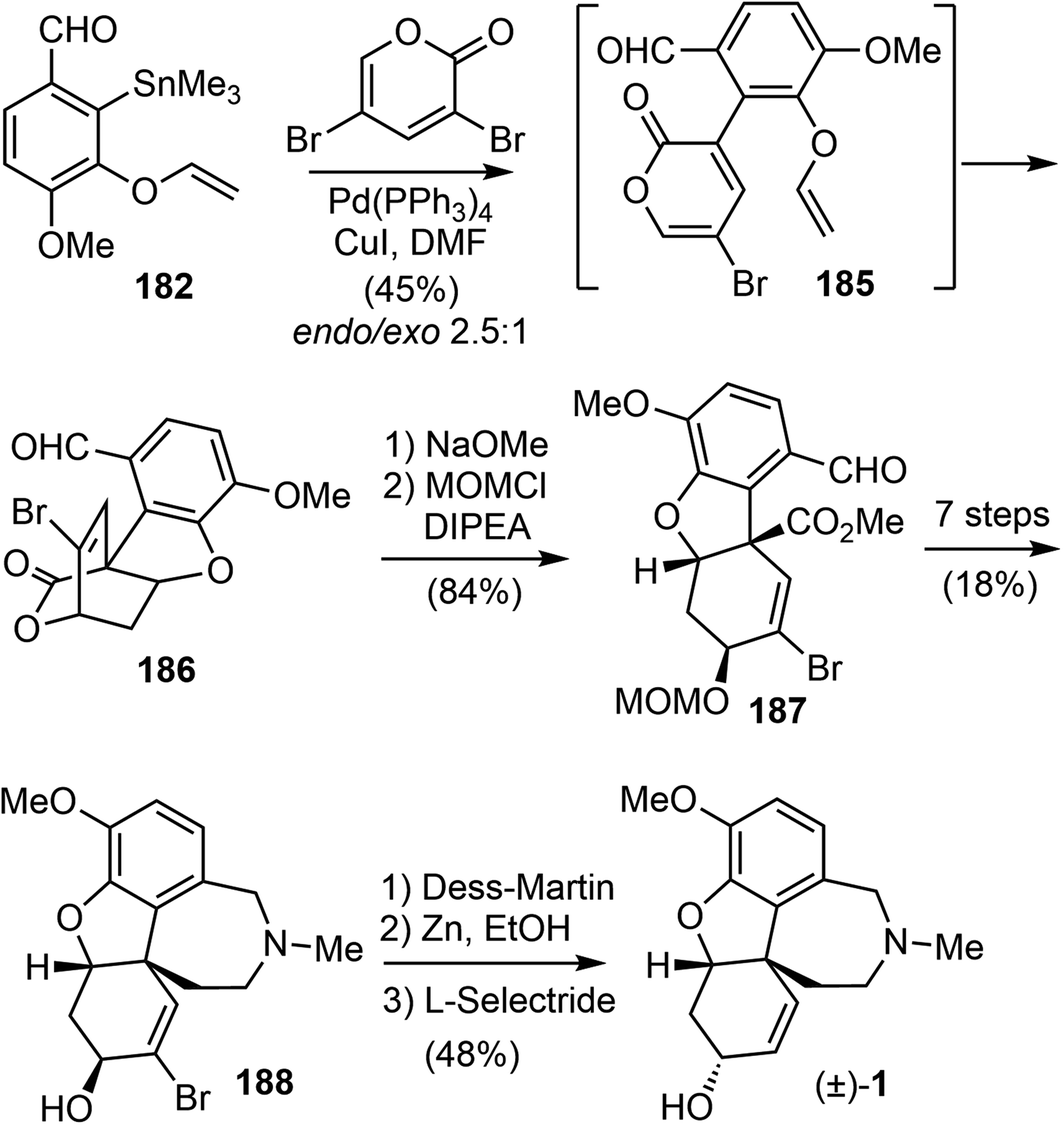 | ||
| Scheme 47 Cho's total synthesis of galantamine. | ||
6.2 Radical cyclization (Yu)
In 2016, Yu and coworkers reported another formal total synthesis of (−)-galantamine,30 shortly after the first generation.26 They first investigated a Rh-catalyzed [5 + 1] cycloaddition of di- and tri-substituted allenylcyclopropanes with CO (Scheme 48). The allene derivative 191 was prepared in 5 steps from the propargylic alcohol 189. Under the optimized conditions, the Rh-catalyzed cycloaddition reaction of 191 with CO afforded the functionalized 2-methylidene-3,4-cyclohexenone 192.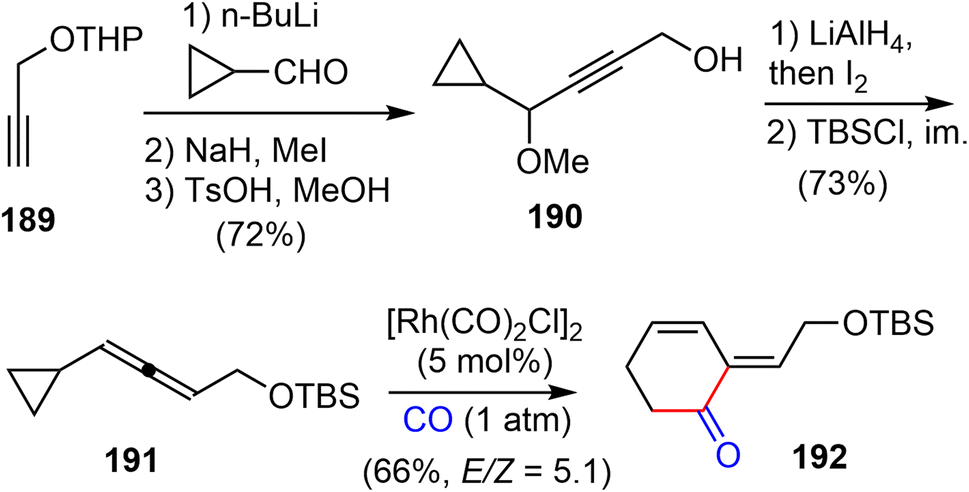 | ||
| Scheme 48 Rh-catalysed [5 + 1] cycloaddition of allenylcyclopropanes and CO. | ||
CBS reduction of enone 192 led to the chiral alcohol product 193 in 79% yield and 97% ee (Scheme 49). Then alcohol 193 and the known phenol underwent the Mitsunobu reaction to form ether 194 in 86% yield. The TBS group was deprotected using TBAF and then the alcohol intermediate was oxidized to aldehyde 195 by activated MnO2 in 86% yield over two steps. The initial attempts to form the five-membered ring in the natural product (−)-galantamine with Heck reaction of 194 or its deprotected alcohol was not successful. It was found that the starting materials decomposed slowly under the reaction conditions. Instead, ring closing cyclization of 195 under the radical conditions led to the desired product in 60% yield. Finally, reduction of the aldehyde led to Brown's intermediate 50 in total synthesis of galantamine,14 which constituted a formal total synthesis of (−)-galantamine.
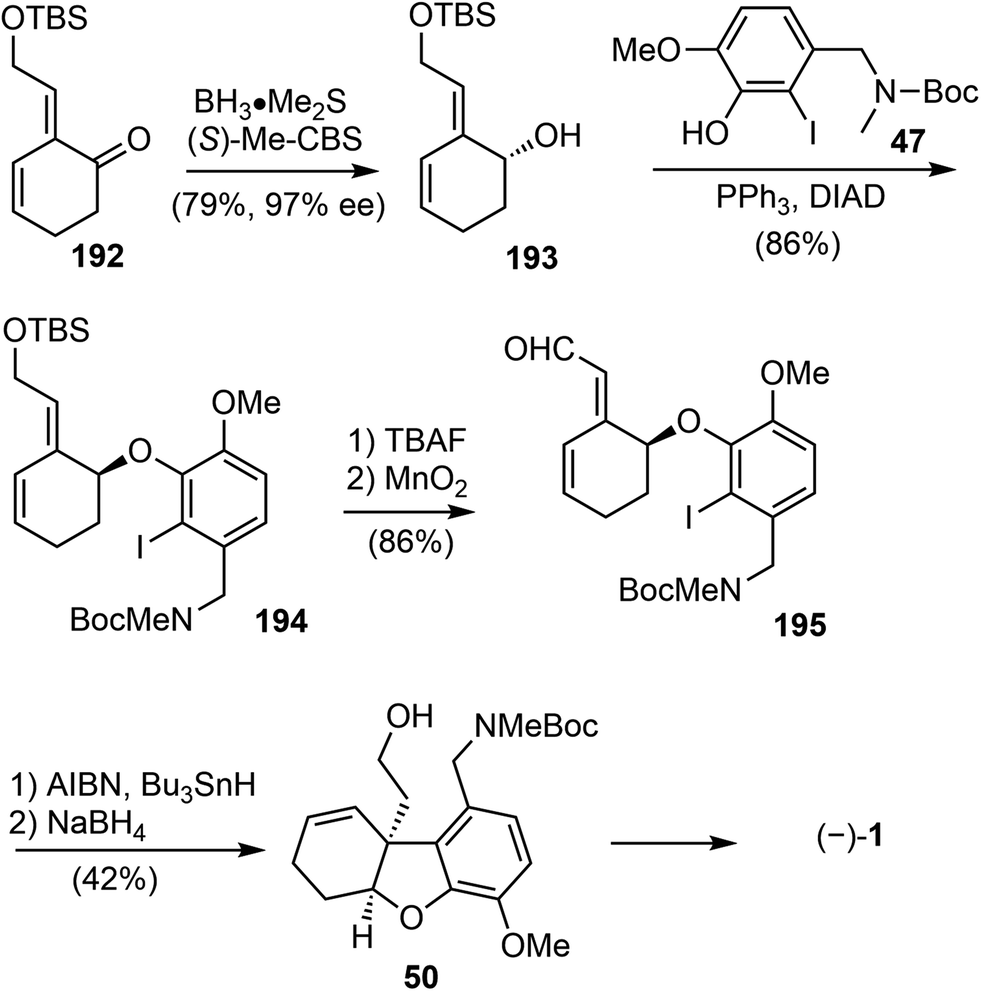 | ||
| Scheme 49 Yu's formal total synthesis of (−)-galantamine via50. | ||
7. Summary of the total syntheses
Prior to 2006, there were only two synthetic strategies available for constructing the quaternary center in the total synthesis of galantamine: oxidative phenol coupling and the intramolecular Heck reaction.9 The asymmetric total synthesis of galantamine relied on chiral auxiliaries or chemical resolution, with only one example of catalytic asymmetric total synthesis coming from the Trost group. However, substantial progress has been made in the synthetic chemistry of galantamine since then, with over 30 new syntheses reported (Table 1).| Strategy | Main author/Year | Key reactions | Ring forming sequence | Source of chirality | Ref. |
|---|---|---|---|---|---|
| Oxidative phenol coupling | Bandichhor/2008 | Phenol oxidative coupling | A + C → DB | Chemical resolution | 17 |
| Wirth/2022 | Electrosynthesis | A + C → D → B | Chiral pool | 39 | |
| Opatz, Waldvogel/2022 | Electrosynthesis | A + C → D → B | Racemic | 40 | |
| Saladino/2022 | Chemoenzymatic phenol oxidative coupling | A + C → D → B | Racemic | 36 | |
| Transition metal catalyzed reaction | Zhou, Xie/2012 | Dynamic kinetic resolution and reductive Heck | A + C → B → D | Dynamic kinetic resolution | 22 |
| Brown/2007 | Enyne RCM and Heck reaction | A → C → B → D | Asymmetric ketone reduction | 14 | |
| Brown/2022 | Enyne RCM and Heck reaction | A → C → B → D | Asymmetric aldehyde allylation | 41 | |
| Banwell/2015 | Alder-ene, Diels–Alder B-alkyl Suzuki | C → B → A → D | Racemic | 27 | |
| Banwell/2022 | B-alkyl Suzuki, Heck | A + C → B → D | Asymmetric ketone reduction | 42 | |
| Hudlicky/2016 | Microbial dihydroxylation/Heck | A + C → B → D | Microbial dihydroxylation | 29 | |
| Yu/2015 | Rh-catalyzed [(3 + 2) + 1] cycloaddition | A → BC → D | Racemic | 26 | |
| Xu/2020 | Rh-catalyzed “cut and sew” reaction | A + C → B → D | Racemic | 35 | |
| Zhao/2021 | Pd-catalyzed olefin carbonylative cyclization | A + C → BD | Racemic | 37 | |
| Enolate alkylation | Ishikawa/2008 | Cascade double Michael addition and Dieckmann reaction | A → C → B → D | Racemic | 16 |
| Fan/2011 | Organocatalyzed asymmetric Michael addition | A → C → B → D | Organocatalysis | 21 | |
| Jia/2015 | Lewis acid catalyzed asymmetric Michael addition | A → BD → C | Lewis acid catalysis | 25 | |
| Tu/2019 | Organocatalyzed asymmetric Michael addition | A → B → C → D | Organocatalysis | 33 | |
| Chandrasekhar/2019 | Aryne insertion | A → B → D → C | Racemic | 34 | |
| Magnus/2009 | Intramolecular phenol alkylation | A + C → B → D | Racemic | 18 | |
| Rearrangement reaction | Tu/2006 | Semipinacol rearrangement | A + C → B → D | Racemic | 13 |
| Bisai/2022 | Johnson–Claisen rearrangement | A + C → B → D | Catalytic asymmetric ketone reduction | 38 | |
| Li/2023 | Eschenmoser–Claisen rearrangement | ABC → D | Racemic | 43 | |
| Miscellaneous approaches | Cho/2010 | Tandem Stille coupling/IMDA | A → B + C → D | Racemic | 20 |
| Yu/2016 | Rh-catalyzed [5 + 1] cycloaddition, radical cyclization | A + C → B → D | Catalytic asymmetric ketone reduction | 30 |
7.1 Evolution of synthetic strategy for the quaternary center
For almost half a century, the field has been dominated by the classical oxidative phenol coupling method, during which time some landmark achievements were discovered. The biomimetic oxidative phenol coupling reaction evolved from a low-yield, proof-of-concept experiment50 to a high-yield, reliable synthetic strategy.53,55 Asymmetric versions of total synthesis were achieved with chiral auxiliary control,56,57 and more significantly, with the discovery of crystallization-induced dynamic resolution of (±)-narwedine.58 Finally, based on these early discoveries, an impressive pilot scale chemical synthesis of (−)-galantamine was developed.60,61 Recent progress on this strategy involves the implementation of new synthetic technologies, including chemoenzymatic synthesis36 and electrosynthesis.39,40The situation changed at the beginning of this century with the advent of transition metal catalyzed reactions.69–72 These reactions not only introduced new synthetic strategies, but also provided new ways for the asymmetric synthesis of (−)-galantamine, which coincides with the rapid development in the field of transition metal catalyzed asymmetric reactions. Since these pioneering works, in addition to the use of the Heck coupling strategy in new circumstances,14,23,24,28,29,41,42 novel transition metal catalyzed reactions, such as the reductive Heck reaction,22 rhodium-catalyzed C–C bond activation,26,30,35 and palladium-catalyzed cyclization reactions,27,37 have been successfully applied in the total synthesis of galantamine.
In addition to the advances in the two classical approaches mentioned above, several more diverse synthetic strategies have emerged in the recent total synthesis of galantamine, including enolate and phenolate alkylation reactions, several types of rearrangement reactions and other miscellaneous approaches. Highly efficient asymmetric total syntheses have been developed based on either traditional stoichiometric chiral reagents38 or asymmetric Lewis acid catalysis25 and organocatalysis.21,33
7.2 Assembly of the tetracyclic structure
The aromatic A ring was obtained from commercial sources and was used as the starting material in most total syntheses of galantamine. The only exception was the total synthesis reported by Banwell in 2015.27 They used a regioselective Diels–Alder reaction of a diene with propynal, followed by oxidation to form the A ring.The heterocyclic B ring was formed either by C–O bond linkage via an intramolecular phenolic oxa-Michael addition in the oxidative coupling strategy and was the last ring formed for the tetracyclic structure, or alternatively, by C–C bond linkage via intramolecular Heck cyclization. Recent developments include the formation of the C–O bond by intramolecular phenolic substitution reaction,13 or the formation of the C–C bond via intramolecular reductive Heck reaction,22 Alder-ene reaction27 or radical cyclization reaction.30 The B ring has also been formed in intramolecular annulation reactions with concomitant formation of D or C ring.25,26,33
The cyclohexene C ring has been obtained from commercial sources in many total syntheses. However, disconnection at the C ring provides new options for diverse and innovative strategies in the total synthesis of galantamine. Recent progress in the synthesis of C ring include Robinson type annulation reactions,16,21,25,33,34 metal catalyzed14,26,30,41 or thermal cycloaddition reactions,20 and arene dearomatization.43
The azepine D ring is typically the last ring to be formed in most total syntheses. Methods for the synthesis of this ring were mostly developed before 2006. The A and C rings were united in the intramolecular oxidative phenol coupling reaction, with simultaneous formation of the D ring. Other methods were the intramolecular C–N bond formation via reductive amination or N-alkylation reaction developed by Trost,69,72 and C–C bond formation via the Pictet–Spengler reaction first used in Guillou's total synthesis.73 A recent development came from Jia's synthesis,25 in which the D ring was formed by a Pd-catalyzed intramolecular Larock annulation reaction with simultaneous formation of the B ring to give 3,4-fused benzofuran. In Xu' total synthesis, a ring expansion reaction via Beckman rearrangement was used to form the D ring.
7.3 Source of chirality
The industrial production of (−)-galantamine currently relies on a highly efficient crystallization-induced dynamic chiral resolution of a racemic intermediate (±)-narwedine (5) with a catalytic amount of chiral mediator. This process may reduce the importance of asymmetric synthesis for (−)-galantamine, but it remains a valuable platform for its application. Various methodologies have been used in the asymmetric total synthesis of (−)-galantamine and its enantiomer. These include chemical resolution,17 biocatalysis,19,29 asymmetric ketone reduction with catalytic30,38 or stoichiometric14,42 chiral reagents, asymmetric aldehyde allylation reaction,41 and asymmetric catalysis with chiral transition metal complexes,22 chiral Lewis acid25 and organocatalysis.21,338. Conclusions
Galantamine is a bioactive natural product that was isolated from a herbal medicine and developed into a clinically approved drug for Alzheimer's disease. It is another gift of nature to mankind.108–110 Because of its important bioactivities, scarcity of the natural resources and intriguing structural features, galantamine has attracted a lot of interest from the synthetic organic chemistry community, and even more so after it became a generic drug. The development of synthetic strategies for the total synthesis of galantamine parallels the advances in modern synthetic organic chemistry. It provides a good opportunity to demonstrate the interplay between target-oriented synthesis and synthetic methodology by summarizing and analyzing the total synthesis of galantamine. It is anticipated that galantamine will remain as a source of inspiration and a test for new synthetic methods and strategies.The demand for galantamine has outstripped its natural supply. Currently, the natural product is synthesized industrially using the classic work of oxidative phenol coupling and crystallization-induced dynamic resolution. While this method is elegant and practical, there is still room for improvement. Several routes in the previously mentioned total syntheses have already surpassed the current industrial pilot-scale synthesis in terms of both step count and overall yield. The development of new chemistry and technology, as well as the pursuit of sales and profit, may lead to newer and more creative total syntheses of the natural product, potentially including a more efficient and scalable route.111,112
9. Author contributions
B. Cheng wrote the manuscript with the assistance from Q. Wang and Y. An. F. Chen supervised the project.10. Conflicts of interest
There are no conflicts to declare.11. Acknowledgements
Financially support from Shenzhen Fundamental Research Program (GXWD20201230155427003-20200822151253001) was acknowledged.12. Notes and references
- D. A. Cozanitis, Wien. Med. Wochenschr., 2021, 171, 205–213 CrossRef PubMed.
- B. Janssen and B. Schäfer, ChemTexts, 2017, 3, 7 CrossRef.
- S. Lilienfeld, CNS Drug Rev., 2002, 8, 159–176 CrossRef CAS PubMed.
- A. L. Harvey, Pharmacol. Ther., 1995, 68, 113–128 CrossRef CAS PubMed.
- D. Jiang, X. Yang, M. Li, Y. Wang and Y. Wang, J. Neural Transm., 2015, 122, 1157–1166 CrossRef CAS PubMed.
- K. W. Quasdorf and L. E. Overman, Nature, 2014, 516, 181–191 CrossRef CAS PubMed.
- C. Li, S. S. Ragab, G. Liu and W. Tang, Nat. Prod. Rep., 2020, 37, 276–292 RSC.
- P.-W. Xu, J.-S. Yu, C. Chen, Z.-Y. Cao, F. Zhou and J. Zhou, ACS Catal., 2019, 9, 1820–1882 CrossRef CAS.
- J. Marco-Contelles, M. do Carmo Carreiras, C. Rodríguez, M. Villarroya and A. G. García, Chem. Rev., 2006, 106, 116–133 CrossRef CAS PubMed.
- U. Rinner, C. Dank and T. Hudlicky, Targets Heterocycl. Syst., 2016, 20, 283 CAS.
- W. Haiming, C. Peng and T. Meng, Chin. J. Org. Chem., 2014, 34, 852 CrossRef.
- F. Lei, G. O. U. Shao-Hua and Z. Yi-Hua, Chin. J. Org. Chem., 2011, 31, 286 Search PubMed.
- X.-D. Hu, Y. Q. Tu, E. Zhang, S. Gao, S. Wang, A. Wang, C.-A. Fan and M. Wang, Org. Lett., 2006, 8, 1823–1825 CrossRef CAS PubMed.
- V. Satcharoen, N. J. McLean, S. C. Kemp, N. P. Camp and R. C. D. Brown, Org. Lett., 2007, 9, 1867–1869 CrossRef CAS PubMed.
- H. Tanimoto, T. Kato and N. Chida, Tetrahedron Lett., 2007, 48, 6267–6270 CrossRef CAS.
- T. Ishikawa, K. Kudo, K. Kuroyabu, S. Uchida, T. Kudoh and S. Saito, J. Org. Chem., 2008, 73, 7498–7508 CrossRef CAS PubMed.
- J. M. Reddy, K. V. Kumar, V. Raju, B. V. Bhaskar, V. Himabindu, A. Bhattacharya, V. Sundaram, R. Banerjee, G. M. Reddy and R. Bandichhor, Synth. Commun., 2008, 38, 2138–2149 CrossRef CAS.
- P. Magnus, N. Sane, B. P. Fauber and V. Lynch, J. Am. Chem. Soc., 2009, 131, 16045–16047 CrossRef CAS PubMed.
- M. G. Banwell, X. Ma, O. P. Karunaratne, A. C. Willis, M. G. Banwell, X. Ma, O. P. Karunaratne and A. C. Willis, Aust. J. Chem., 2010, 63, 1437–1447 CrossRef CAS.
- J. H. Chang, H.-U. Kang, I.-H. Jung and C.-G. Cho, Org. Lett., 2010, 12, 2016–2018 CrossRef CAS PubMed.
- P. Chen, X. Bao, L.-F. Zhang, M. Ding, X.-J. Han, J. Li, G.-B. Zhang, Y.-Q. Tu and C.-A. Fan, Angew. Chem., Int. Ed., 2011, 50, 8161–8166 CrossRef CAS PubMed.
- J.-Q. Chen, J.-H. Xie, D.-H. Bao, S. Liu and Q.-L. Zhou, Org. Lett., 2012, 14, 2714–2717 CrossRef CAS PubMed.
- J. Choi, H. Kim, S. Park and J. Tae, Synlett, 2013, 24, 379–382 CrossRef CAS.
- Y. Zang and I. Ojima, J. Org. Chem., 2013, 78, 4013–4018 CrossRef CAS PubMed.
- L. Li, Q. Yang, Y. Wang and Y. Jia, Angew. Chem., Int. Ed., 2015, 54, 6255–6259 CrossRef CAS PubMed.
- Y. Feng and Z.-X. Yu, J. Org. Chem., 2015, 80, 1952–1956 CrossRef CAS PubMed.
- J. Nugent, E. Matoušová and M. G. Banwell, Eur. J. Org Chem., 2015, 3771–3778 CrossRef CAS.
- J. Nugent and M. G. Banwell, Eur. J. Org Chem., 2016, 5862–5867 CrossRef CAS.
- M. A. A. Endoma-Arias and T. Hudlicky, Chem.–Eur. J., 2016, 22, 14540–14543 CrossRef PubMed.
- C.-H. Liu and Z.-X. Yu, Org. Biomol. Chem., 2016, 14, 5945–5950 RSC.
- B. Dong, B. Zhou, J. Ren, L. Lu, G. Lu, P. Hu and B.-B. Zeng, Tetrahedron, 2017, 73, 4719–4722 CrossRef CAS.
- N. Yamamoto, T. Okada, Y. Harada, N. Kutsumura, S. Imaide, T. Saitoh, H. Fujii and H. Nagase, Tetrahedron, 2017, 73, 5751–5758 CrossRef CAS.
- Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed.
- T. Venkatesh, P. S. Mainkar and S. Chandrasekhar, Org. Biomol. Chem., 2019, 17, 2192–2198 RSC.
- Y. Zhang, S. Shen, H. Fang and T. Xu, Org. Lett., 2020, 22, 1244–1248 CrossRef CAS PubMed.
- C. Zippilli, L. Botta, B. M. Bizzarri, M. C. Baratto, R. Pogni and R. Saladino, RSC Adv., 2020, 10, 10897–10903 RSC.
- Y.-P. Chang, X. Ma, H. Shao and Y.-M. Zhao, Org. Lett., 2021, 23, 9659–9663 CrossRef CAS PubMed.
- S. Majumder, A. Yadav, S. Pal, A. Khatua and A. Bisai, J. Org. Chem., 2022, 87, 7786–7797 CrossRef CAS PubMed.
- Z. Xiong, F. Weidlich, C. Sanchez and T. Wirth, Org. Biomol. Chem., 2022, 20, 4123–4127 RSC.
- D. Pollok, L. M. Großmann, T. Behrendt, T. Opatz and S. R. Waldvogel, Chem.–Eur. J., 2022, 28, e202201523 CrossRef CAS PubMed.
- I. R. Miller, N. J. McLean, G. A. I. Moustafa, V. Ajavakom, S. C. Kemp, R. K. Bellingham, N. P. Camp and R. C. D. Brown, J. Org. Chem., 2022, 87, 1325–1334 CrossRef CAS PubMed.
- N. Hu, Y.-T. He, P. Lan, M. G. Banwell, L. V. White, N. Hu, Y.-T. He, P. Lan, M. G. Banwell and L. V. White, Aust. J. Chem., 2022, 75, 974–982 CrossRef CAS.
- J.-Y. Qiu, W.-L. Zeng, H. Xie, M.-Y. Wang and W. Li, Angew. Chem., Int. Ed., 2023, 62, e202218961 CrossRef CAS PubMed.
- M. C. Carson and M. C. Kozlowski, Nat. Prod. Rep., 2024, 41, 208–227 RSC.
- D. H. R. Barton, G. W. Kirby, J. B. Taylor and G. M. Thomas, J. Chem. Soc., 1963, 4545–4558 RSC.
- A. M. Takos and F. Rook, Int. J. Mol. Sci., 2013, 14, 11713–11741 CrossRef PubMed.
- J. Eichhorn, T. Takada, Y. Kita and M. H. Zenk, Phytochemistry, 1998, 49, 1037–1047 CrossRef CAS.
- B. B. Majhi, S.-E. Gélinas, N. Mérindol, S. Ricard and I. Desgagné-Penix, Front. Plant Sci., 2023, 14, 1231809 CrossRef PubMed.
- T. U. Jayawardena, N. Merindol, N. S. Liyanage and I. Desgagné-Penix, Nat. Prod. Rep., 2024 10.1039/D3NP00044C.
- D. H. R. Barton and G. W. Kirby, J. Chem. Soc., 1962, 806–817 RSC.
- T. Kametani, K. Yamaki, H. Yagi and K. Fukomuto, J. Chem. Soc. Chem. Commun., 1969, 425–426 RSC.
- T. Kametani, K. Yamaki, H. Yagi and K. Fukumoto, J. Chem. Soc. C, 1969, 2602–2605 RSC.
- Y. Kita, M. Arisawa, M. Gyoten, M. Nakajima, R. Hamada, H. Tohma and T. Takada, J. Org. Chem., 1998, 63, 6625–6633 CrossRef CAS.
- D. Krikorian, V. Tarpanov, S. Parushev and P. Mechkarova, Synth. Commun., 2000, 30, 2833–2846 CrossRef CAS.
- M. Node, S. Kodama, Y. Hamashima, T. Baba, N. Hamamichi and K. Nishide, Angew. Chem., Int. Ed., 2001, 40, 3060–3062 CrossRef CAS PubMed.
- K. Koga, K. Shimizu, K. Tomioka and S. Yamada, Heterocycles, 1977, 8, 277 CrossRef.
- S. Kodama, Y. Hamashima, K. Nishide and M. Node, Angew. Chem., Int. Ed., 2004, 43, 2659–2661 CrossRef CAS PubMed.
- W.-C. Shieh and J. A. Carlson, J. Org. Chem., 1994, 59, 5463–5465 CrossRef CAS.
- N. G. Anderson, Org. Process Res. Dev., 2005, 9, 800–813 CrossRef CAS.
- L. Czollner, W. Frantsits, B. Küenburg, U. Hedenig, J. Fröhlich and U. Jordis, Tetrahedron Lett., 1998, 39, 2087–2088 CrossRef CAS.
- B. Küenburg, L. Czollner, J. Fröhlich and U. Jordis, Org. Process Res. Dev., 1999, 3, 425–431 CrossRef.
- 1.64–1.75 kg of galantamine was isolated per hectare of N. pseudonarcissus: M. D. Fraser, H. E. Vallin, J. R. T. Davies, G. E. Rowlands and X. Chang, Sci. Rep., 2021, 11, 1389 CrossRef CAS PubMed.
- J. Li, A. Amatuni and H. Renata, Curr. Opin. Chem. Biol., 2020, 55, 111–118 CrossRef CAS PubMed.
- M. Mogharabi and M. A. Faramarzi, Adv. Synth. Catal., 2014, 356, 897–927 CrossRef CAS.
- N. Cardullo, V. Muccilli and C. Tringali, RSC Chem. Biol., 2022, 3, 614–647 RSC.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- R. K. Mylavarapu, K. GCM, N. Kolla, R. Veeramalla, P. Koilkonda, A. Bhattacharya and R. Bandichhor, Org. Process Res. Dev., 2007, 11, 1065–1068 CrossRef CAS.
- D. A. Chaplin, N. B. Johnson, J. M. Paul and G. A. Potter, Tetrahedron Lett., 1998, 39, 6777–6780 CrossRef CAS.
- B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 11262–11263 CrossRef CAS.
- P. J. Parsons, M. D. Charles, D. M. Harvey, L. R. Sumoreeah, A. Shell, G. Spoors, A. L. Gill and S. Smith, Tetrahedron Lett., 2001, 42, 2209–2211 CrossRef CAS.
- C. Pilger, B. Westermann, U. Flörke and G. Fels, Synlett, 2000, 1163–1165 CAS.
- B. M. Trost and W. Tang, Angew. Chem., Int. Ed., 2002, 41, 2795–2797 CrossRef CAS PubMed.
- C. Guillou, J.-L. Beunard, E. Gras and C. Thal, Angew. Chem., Int. Ed., 2001, 40, 4745–4746 CrossRef CAS.
- A. Hafner, R. O. Duthaler, R. Marti, G. Rihs, P. Rothe-Streit and F. Schwarzenbach, J. Am. Chem. Soc., 1992, 114, 2321–2336 CrossRef CAS.
- W.-J. Bai, J.-H. Xie, Y.-L. Li, S. Liu and Q.-L. Zhou, Adv. Synth. Catal., 2010, 352, 81–84 CrossRef CAS.
- S. Das, D. Addis, S. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770–1771 CrossRef CAS PubMed.
- M. G. Banwell, J. N. Buckler, C. J. Jackson, P. Lan, X. Ma, E. Matoušová and J. Nugent, in Strategies and Tactics in Organic Synthesis, ed. M. Harmata, Academic Press, 2015, vol. 11, pp. 29–50 Search PubMed.
- C. S. Beshara, A. Hall, R. L. Jenkins, K. L. Jones, T. C. Jones, N. M. Killeen, P. H. Taylor, S. P. Thomas and N. C. O. Tomkinson, Org. Lett., 2005, 7, 5729–5732 CrossRef CAS PubMed.
- L. Petit, M. G. Banwell and A. C. Willis, Org. Lett., 2011, 13, 5800–5803 CrossRef CAS PubMed.
- B. M. Trost and C. Pedregal, J. Am. Chem. Soc., 1992, 114, 7292–7294 CrossRef CAS.
- M. Movassaghi and M. D. Hill, Org. Lett., 2008, 10, 3485–3488 CrossRef CAS PubMed.
- S. Eagon, C. DeLieto, W. J. McDonald, D. Haddenham, J. Saavedra, J. Kim and B. Singaram, J. Org. Chem., 2010, 75, 7717–7725 CrossRef CAS PubMed.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- L. Jiao, M. Lin, L.-G. Zhuo and Z.-X. Yu, Org. Lett., 2010, 12, 2528–2531 CrossRef CAS PubMed.
- N. F. McKinley and D. F. O'Shea, J. Org. Chem., 2004, 69, 5087–5092 CrossRef CAS PubMed.
- I. E. Markó and A. Mekhalfia, Tetrahedron Lett., 1990, 31, 7237–7240 CrossRef.
- Y. Xue and G. Dong, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef CAS PubMed.
- P. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed.
- P.-H. Chen, N. A. Savage and G. Dong, Tetrahedron, 2014, 70, 4135–4146 CrossRef CAS PubMed.
- P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed.
- T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed.
- K. Ma, B. S. Martin, X. Yin and M. Dai, Nat. Prod. Rep., 2019, 36, 174–219 RSC.
- P. H. Gehrtz, V. Hirschbeck, B. Ciszek and I. Fleischer, Synthesis, 2016, 48, 1573–1596 CrossRef CAS.
- C.-A. Fan, Y.-Q. Tu, Z.-L. Song, E. Zhang, L. Shi, M. Wang, B. Wang and S.-Y. Zhang, Org. Lett., 2004, 6, 4691–4694 CrossRef CAS PubMed.
- A. R. Chamberlin, J. E. Stemke and F. T. Bond, J. Org. Chem., 1978, 43, 147–154 CrossRef CAS.
- G. Stork and R. L. Danheiser, J. Org. Chem., 1973, 38, 1775–1776 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- W. S. Johnson, L. Werthemann, W. R. Bartlett, T. J. Brocksom, T.-T. Li, D. J. Faulkner and M. R. Petersen, J. Am. Chem. Soc., 1970, 92, 741–743 CrossRef CAS.
- R. A. Fernandes, A. K. Chowdhury and P. Kattanguru, Eur. J. Org Chem., 2014, 2014, 2833–2871 CrossRef CAS.
- W. G. Salmond, M. A. Barta and J. L. Havens, J. Org. Chem., 1978, 43, 2057–2059 CrossRef CAS.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS PubMed.
- J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC.
- R. C. Larock, J. Organomet. Chem., 1999, 576, 111–124 CrossRef CAS.
- X. Liu, L. Lin and X. Feng, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS PubMed.
- Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Nat. Commun., 2019, 10, 2507 CrossRef PubMed.
- U. K. Tambar and B. M. Stoltz, J. Am. Chem. Soc., 2005, 127, 5340–5341 CrossRef CAS PubMed.
- M. Blouin and R. Frenette, J. Org. Chem., 2001, 66, 9043–9045 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- Y. Tu, Angew. Chem., Int. Ed., 2016, 55, 10210–10226 CrossRef CAS PubMed.
- C. A. Kuttruff, M. D. Eastgate and P. S. Baran, Nat. Prod. Rep., 2014, 31, 419–432 RSC.
- X.-Y. Liu and Y. Qin, Nat. Prod. Rep., 2023, 40, 1694–1700 RSC.
| This journal is © The Royal Society of Chemistry 2024 |