
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBacillus subtilis as a host for natural product discovery and engineering of biosynthetic gene clusters
Hanne
Put†
 ab,
Hans
Gerstmans†
bcd,
Hanne
Vande Capelle†
bc,
Maarten
Fauvart‡
abe,
Jan
Michiels‡
ab and
Joleen
Masschelein‡
bc
ab,
Hans
Gerstmans†
bcd,
Hanne
Vande Capelle†
bc,
Maarten
Fauvart‡
abe,
Jan
Michiels‡
ab and
Joleen
Masschelein‡
bc
aCentre of Microbial and Plant Genetics, KU Leuven, 3001 Leuven, Belgium
bVIB-KU Leuven Center for Microbiology, Flanders Institute for Biotechnology, 3001 Leuven, Belgium. E-mail: joleen.masschelein@kuleuven.be
cLaboratory for Biomolecular Discovery & Engineering, KU Leuven, 3001 Leuven, Belgium
dBiosensors Group, KU Leuven, 3001 Leuven, Belgium
eimec, 3001 Leuven, Belgium
First published on 11th March 2024
Abstract
Covering: up to October 2023
Many bioactive natural products are synthesized by microorganisms that are either difficult or impossible to cultivate under laboratory conditions, or that produce only small amounts of the desired compound. By transferring biosynthetic gene clusters (BGCs) into alternative host organisms that are more easily cultured and engineered, larger quantities can be obtained and new analogues with potentially improved biological activity or other desirable properties can be generated. Moreover, expression of cryptic BGCs in a suitable host can facilitate the identification and characterization of novel natural products. Heterologous expression therefore represents a valuable tool for natural product discovery and engineering as it allows the study and manipulation of their biosynthetic pathways in a controlled setting, enabling innovative applications. Bacillus is a genus of Gram-positive bacteria that is widely used in industrial biotechnology as a host for the production of proteins from diverse origins, including enzymes and vaccines. However, despite numerous successful examples, Bacillus species remain underexploited as heterologous hosts for the expression of natural product BGCs. Here, we review important advantages that Bacillus species offer as expression hosts, such as high secretion capacity, natural competence for DNA uptake, and the increasing availability of a wide range of genetic tools for gene expression and strain engineering. We evaluate different strain optimization strategies and other critical factors that have improved the success and efficiency of heterologous natural product biosynthesis in B. subtilis. Finally, future perspectives for using B. subtilis as a heterologous host are discussed, identifying research gaps and promising areas that require further exploration.
 Hanne Put | Hanne Put graduated with a MSc in Bioscience Engineering (2019) and is currently undertaking a PhD at VIB and KU Leuven under the supervision of Prof. Jan Michiels. In her PhD research, she is exploring novel heterologous expression systems in B. subtilis for natural product discovery. |
 Hans Gerstmans | Hans Gerstmans is a postdoctoral researcher at the KU Leuven – VIB Center for Microbiology in the group of Prof. Joleen Masschelein. He obtained his Master's degree in Bioscience engineering from KU Leuven in 2015. His joint PhD (Ghent University and KU Leuven) focused on the high-throughput engineering of endolysins. Currently, his research focuses on the high-throughput engineering of RiPPs from host-associated bacteria to improve their therapeutic properties. |
 Hanne Vande Capelle | Hanne Vande Capelle earned her MSc in Bioscience Engineering in 2022. She is now pursuing her PhD at the VIB-KU Leuven Center for Microbiology under the guidance of Prof. Joleen Masschelein. Her research focuses on the engineering of sactipeptides, produced by Bacillus species, to obtain compounds with novel-to-nature properties. |
 Maarten Fauvart | Maarten Fauvart obtained his PhD in 2008 at KU Leuven, having studied determinants of host range specificity in Rhizobium. As a postdoc, his research focused mainly on the molecular biology of antibiotic resistance and tolerance in Gram-negative pathogenic bacteria. In 2015 he joined imec, a world-leading R&D and innovation hub in nanoelectronics and digital technologies, to work on lab-on-chip solutions for molecular biology applications. He is now a Principal Scientist in imec's Life Science Technologies department, leading efforts on nucleic acid analysis. One of the main aims of his current research is to exploit these technologies for advancing natural product discovery. |
 Jan Michiels | Jan Michiels is full Professor at the KU Leuven Faculty of Bioscience Engineering and Director of the Centre of Microbial and Plant Genetics. In 2017 he became a group leader at the Flemish Institute for Biotechnology (VIB). He teaches courses on biochemistry, molecular biology, genetics, and bacterial physiology. His research focuses on molecular aspects of microbe–host interactions and stress resistance in bacteria, in particular the antibiotic tolerance of pathogenic bacteria. A second line of research is directed towards the discovery of novel antibiotics and the development of microbial cell factories using metabolic engineering and synthetic biology. |
 Joleen Masschelein | Joleen Masschelein has a PhD in Bioscience Engineering from KU Leuven. She carried out postdoctoral research at the University of Warwick in the group of Prof. Greg Challis and at the KU Leuven Rega Institute for Medical Research in the group of Prof. Piet Herdewijn. She is currently an Assistant Professor at the Biology Department at KU Leuven, Group Leader at VIB and Visiting Professor at the Tianjin Institute for Industrial Biotechnology. Her research focuses on diverse aspects of microbial bioactive natural products, including genomics-driven discovery, biosynthetic pathway elucidation and engineering, and molecular mode of action and resistance studies. |
1 Introduction
Bacillus subtilis is a Gram-positive bacterial species that belongs to the versatile B. subtilis group, along with Bacillus pumilus, Bacillus licheniformis and Bacillus amyloliquefaciens.1,2 It exhibits a remarkable genetic diversity which allows it to adapt to diverse ecological niches, ranging from deep-sea hydrothermal vents to the soil and the human gastrointestinal tract.3,4 Its ability to form spores that are resilient to harsh conditions further contributes to the survival of B. subtilis in these challenging environments.5 To gain a selective advantage in its particular habitat, B. subtilis also produces an extensive repertoire of bioactive metabolites, including polyketides (PKs), nonribosomal peptides (NRPs), ribosomally synthesized and post-translationally modified peptides (RiPPs) and terpenes with potent antimicrobial properties.6,7 The species is also used as a biocontrol agent in agriculture for combatting plant pathogens and promoting plant growth.6,8In industrial settings, B. subtilis has established itself as a reliable and versatile workhorse for the production of a wide array of compounds, ranging from enzymes to high-purity fine chemicals.1,9,10 Its natural competence for DNA uptake,11 high secretion capacity,12 generally recognized as safe (GRAS) status,13 lack of endotoxins,14 and remarkable genetic diversity among closely related strains,15 coupled with its well-described gene expression system12 are all features that underpin its popularity.16 In academia, B. subtilis continues to serve as a model organism for studying diverse physiological processes, such as protein secretion, cell motility and division, biofilm formation, minimal cell development, secondary metabolite biosynthesis, and molecular interactions with plants and fungi.13,17
Despite its promising biotechnological potential, the use of B. subtilis for heterologous secondary metabolite production lags behind other microbial hosts, such as Escherichia coli, Streptomyces spp. and Saccharomyces cerevisiae. However, over the past 15 years, significant advances have been made in the development of synthetic biology and genome engineering tools for Bacillus species.11,14 With the growing availability of such tools, Bacillus is now gaining increasing interest as a heterologous host for natural product biosynthesis.11 Heterologous expression is particularly valuable when tools for the genetic manipulation of the native host are limited or unknown, e.g., for metagenome-derived gene clusters.11 In the case of B. subtilis, successful heterologous production of NRPs,18 PKs,19 terpenoids,14 and other small molecules and peptides20,21 has been reported.
This review presents an overview of the natural products that have been successfully produced heterologously in Bacillus species, organized by natural product class. It also offers insights into the inherent advantages of Bacillus species as heterologous expression hosts and highlights diverse tools that can be employed for strain engineering and biosynthetic pathway manipulation. It explores potential biosynthetic gene clusters (BGCs) that may be suitable for expression in Bacillus, and provides recommendations for the most optimal (engineered) host strain for this purpose. With the ongoing advancement of molecular engineering tools, B. subtilis promises to emerge as a powerful player in the field of heterologous natural product biosynthesis.
2 Bacillus as a host for heterologous expression of natural product BGCs
2.1 Favourable characteristics of B. subtilis for heterologous BGC expression
Various characteristics have been put forward to describe the ideal host strains for heterologous expression of natural product BGCs.22–24 These include (1) easy cultivation and lab handling, (2) broad availability of gene editing tools and expression vectors, (3) thorough genetic characterization, (4) adaptable metabolism with adequate availability of (uncommon) precursors to support natural product biosynthesis, (5) clean genetic background, (6) promiscuous host transcriptional machinery, (7) high levels of self-resistance and efficient compound secretion systems, (8) ease of fermentation and upscaling, (9) Generally Recognized As Safe (GRAS) status and absence of endotoxins. In this section, we discuss these characteristics with respect to B. subtilis and other Bacillus spp., and compare them with other commonly used heterologous hosts, such as Streptomyces spp. and E. coli.(1) Easy cultivation and efficient growth of the host strain are important factors for minimizing experimental workload and fermentation times. B. subtilis and E. coli both benefit from fast growth on standard nutrient agar, doubling in number every 20 to 30 minutes under ideal circumstances in the exponential phase. In contrast, Streptomyces strains take several days to form visible colonies. Handling Streptomycetes in the lab is also complicated due to their complex life cycle, which involves mycelial growth, hyphae formation, and sporulation.25,26 For example, high-level expression of polyketide synthase (PKS) genes in Streptomyces typically occurs at the onset of the stationary phase of mycelial growth.27B. subtilis exhibits endospore formation, a well-studied trait regulated primarily by spo0A and triggered in response to starvation and other stresses.28–30 These highly resistant spores facilitate survival in harsh conditions but may prevent cells from reaching high densities in bioreactors due to nutrient limitations in fed-batch systems.31 Several mutants have been constructed and tested to circumvent this challenge, as discussed in Section 4.1. The Bacillus life cycle also involves biofilm formation and swarming on surfaces. The former has been studied extensively in Bacillus32 and a potential link between native secondary metabolism and biofilm differentiation has been proposed.33 Intriguingly, heterologous production of the NRP iturin A in B. subtilis was found to be significantly enhanced during biofilm fermentation compared to planktonic cultures.34 Notably, no biofilm-related problems have been reported during heterologous expression of BGCs in Bacillus. Swarming in B. subtilis has also been associated with the production of secondary metabolites, particularly biosurfactants like the cyclic lipopeptide surfactin.35 However, swarming is primarily observed in undomesticated Bacillus strains due to frameshift mutations in the sfp and swrA genes in the majority of widely-used B. subtilis lab strains, e.g. B. subtilis 168.36
(2) A versatile set of genetic parts and gene editing tools is fundamental for the precise genomic integration and robust expression and engineering of natural product BGCs, which often span tens of kb in size and are frequently cryptic in nature. Like in E. coli, an elaborate molecular toolbox is available for B. subtilis, comprising standardized genetic elements,37,38 a CRISPR-Cas system,39 and markerless genome modification systems.40–42 For a comprehensive overview of the Bacillus synthetic biology toolbox, the reader is referred to Section 4. The growing interest in Streptomyces as a host for natural product biosynthesis has recently fuelled the design and development of synthetic parts, chassis strains and genetic tools for this genus, bringing its toolbox closer to the level of more stablished model organisms. Recent examples include regulatory sequences for promoter engineering,43 CRISPR-based tools for genome editing and silencing,44,45 and specific vectors for targeted BGC capture, refactoring and expression.46 Overall, B. subtilis offers a broader range of general engineering tools, while the tools available in Streptomycetes are more specifically designed for the expression of natural product BGCs. However, the cloning of BGCs and their introduction into the host remains a significant challenge. A particular advantage of B. subtilis over other hosts lies in its natural competence combined with its efficient homologous recombination machinery, allowing fast, straightforward and stable integration of large BGCs in the genome.47 Contrary to E. coli, autonomous plasmids are unstable and prone to recombination in B. subtilis, especially when carrying very large inserts.48–50 For a description of different cloning strategies for BGCs, the reader is referred to Section 4.3.
(3) To efficiently express natural product BGCs in a heterologous host, a thorough understanding of its metabolism and physiology is needed. Detailed analysis of the host's metabolic potential and genomic characteristics not only provides important information about precursor and cofactor availability, but also serves as a valuable resource for metabolic engineering, e.g. to limit the metabolic burden and enhance fermentation yields. The diversity and accessibility of genome modification tools in B. subtilis have substantially advanced its genetic characterization, making it one of the best-studied species to date. Nevertheless, despite decades of research, approximately one in four B. subtilis proteins remain uncharacterized.51 The MiniBacillus project aims to reduce that number by creating a B. subtilis strain with a minimal genome. So far, approximately 42% of the genome has been successfully deleted as part of this initiative.52,53 The absence of extracellular serine protease activity in this strain has enabled the successful production of subtilin, nisin and flavucin hybrids.54 A comprehensive overview of B. subtilis genes, proteins and their interactions, expression networks, and other related information has been compiled in the SubtiWiki database.17,55
(4) Natural products are constructed from a diverse range of building blocks, which is reflected in their remarkable structural diversity and complexity. PKSs are known to use a variety of coenzyme A (CoA)-activated substrates, such as acetyl-, propionyl-, (methyl)malonyl-, and isovaleryl-CoA.56 In E. coli, the metabolism controlling the fluxes of these substrates is under tight regulation, which can limit precursor supply during heterologous expression.57 A variety of metabolic engineering strategies have been applied to increase cellular levels of acetyl- and malonyl-CoA in E. coli, including deletion of competing pathways and overexpression of the enzyme acetyl-CoA carboxylase, respectively.58 The supply of methyl-malonyl-CoA, on the other hand, has been improved by expressing the Streptomyces coelicolor propionyl-CoA carboxylase and the Propionibacteria shermanii methylmalonyl-CoA mutase/epimerase pathways.59 Nonribosomal peptide synthetases (NRPSs) are able to incorporate an even broader range of substrates, including (non-) proteinogenic amino acids, α-hydroxy acids and glycosylated and methylated residues. In E. coli, the number of native NRP pathways is limited, and they show only minimal diversity. Furthermore, heterologous NRP production in this species has been successful in only a limited number of cases.60,61 Overall, the success rate for PK and NRP production in E. coli remains low, mainly because its metabolism is not well adapted for secondary metabolite production and because of problems with folding and stability of the large PKS and NRPS multienzyme complexes.62–64 To enable post-translational activation of the acyl and peptidyl carrier proteins domains within PKSs and NRPSs, respectively, an engineered E. coli BAP-1 strain has been constructed, which expresses the sfp phosphopantetheinyl transferase gene from B. subtilis. Sfp is a phosphopantetheinyl transferase with an exceptionally broad substrate tolerance, capable of modifying ACP and PCP domains from almost any organism. Unlike E. coli, B. subtilis is a prolific producer of natural products, harbouring a great diversity of PK, NRP, and hybrid NRP-PK pathways.65–68 While there is no doubt that Streptomyces strains contain the highest number of genomic BGCs on average, Bacillus species are not far behind. Nevertheless, genomic integration of additional precursor biosynthesis genes or feeding of uncommon amino acid building blocks has been proven necessary for the heterologous production of some NRPs.18,69,70 For example, the supplementation of D-hydroxyisovalerate in the growth medium was shown to increase heterologous enniatin production,69 while L-2,4-diaminobutyric acid feeding or heterologous expression of the ectoine biosynthetic gene ectB was required for heterologous polymyxin production.18,70 Additionally, it is worth mentioning that several commonly used B. subtilis lab strains are auxotrophic for specific amino acids. For example, B. subtilis 168 requires exogenously supplied tryptophan for growth.71 Taken together, the metabolism of B. subtilis is well adapted for natural product biosynthesis, although feeding of specific building blocks is necessary in certain cases.
(5) While the native ability to produce many structurally diverse secondary metabolites signifies a well-adapted metabolism, it can also impede the production and detection of heterologous compounds. The metabolic burden associated with natural product biosynthesis is significant, and the presence of native compounds with similar biological activities can interfere with biological activity screenings. Heterologous hosts with ‘clean’ genetic backgrounds are therefore preferred. In Streptomycetes, such genome-reduced host strains, like S. coelicolor M1152 and M1154, have been successfully used for heterologous expression.72–74 For B. subtilis, a markerless gene deletion system was used for genomic removal of all known antibiotic-producing BGCs, including those responsible for subtilosin, plipastatin, and bacilysin biosynthesis.40 The resulting strain was evaluated for the production of surfactin. Although the growth rate was improved, overall surfactin biosynthesis was reduced compared to the control strain, and more research was deemed necessary to further optimize this strain.75
(6) Having promiscuous transcriptional machinery is beneficial for a heterologous host since it allows expression of diverse BGCs originating from a wide range of genetic backgrounds. The use of rare codons is one of the influencing factors in this context. B. subtilis is proposed to have a relatively low codon bias,76,77 which in turn facilitates the expression of directly cloned BGCs. Additionally, Bacillus genomes have a low GC content. This makes cloning of BGCs faster and easier than in Streptomyces spp., whose genomes are very GC rich (>70% GC). This creates a specific niche for B. subtilis as a favourable host for the heterologous expression of low-GC BGCs.
(7) Efficient secretion of heterologously produced compounds and proteins allows for higher production titers and facilitates downstream purification processes. B. subtilis has a very efficient secretion system, comprising multiple pathways.78,79 This feature has been exploited for industrial applications and is one of the reasons this species has become a popular workhouse for the industrial production of enzymes and chemicals. Prominent examples include α-amylases and riboflavin, produced through one of the most efficient fermentation processes in the world.80,81 For heterologous production of secondary metabolites, which often have antimicrobial activity, rapid and efficient secretion is necessary to avoid potential toxicity to the host and to ensure self-resistance.
(8) Another quality of a good heterologous host is its compatibility with fermentation and upscaling processes. B. subtilis, as outlined above, has a long and successful track record of use in industrial settings. As a result, a wealth of experience has been gained in the upscaling of these processes.12 Likewise, E. coli easily reaches high-cell density fermentations and the species has been widely used in industrial processes for the production of biofuels, amino acids, biopolymers, and many more.82 While mycelial growth makes this process harder for Streptomycetes, effective industrial fermentation methods have also been established for this genus.83
(9) Lastly, host strains that do not produce endotoxins facilitate the use of fermentation in industrial production. Bacillus and Streptomyces both meet this criterion84–86 in contrast to E. coli, whose lipopolysaccharides in the outer membrane need to be meticulously removed in a challenging and costly process.87 Therefore, the RiPPs microcin J25 and Y were recombinantly produced in Bacillus instead of E. coli.88 The absence of exotoxins and the non-pathogenic nature of B. subtilis has further led to the approval of the GRAS status for this species by the FDA.
In summary, B. subtilis, as non-pathogenic species, inherently possesses many traits that make it a favourable host for the heterologous expression of natural product BGCs (Fig. 1).89–91 The combination of straightforward laboratory handling, decades of molecular characterization research and its natural competence and ability for natural product biosynthesis, suggest a promising future for this species as a host for heterologous BGC expression, a field that currently remains underexplored. The longstanding use of Bacillus species in industrial applications, along with the absence of exo- and endotoxins, and its extensive secretion system collectively highlight the potential in fermentation processes. Research gaps that remain to be studied include the design of more efficient expression systems for large BGCs, and the development of specific host strains with clean backgrounds for high yield secondary metabolite production.
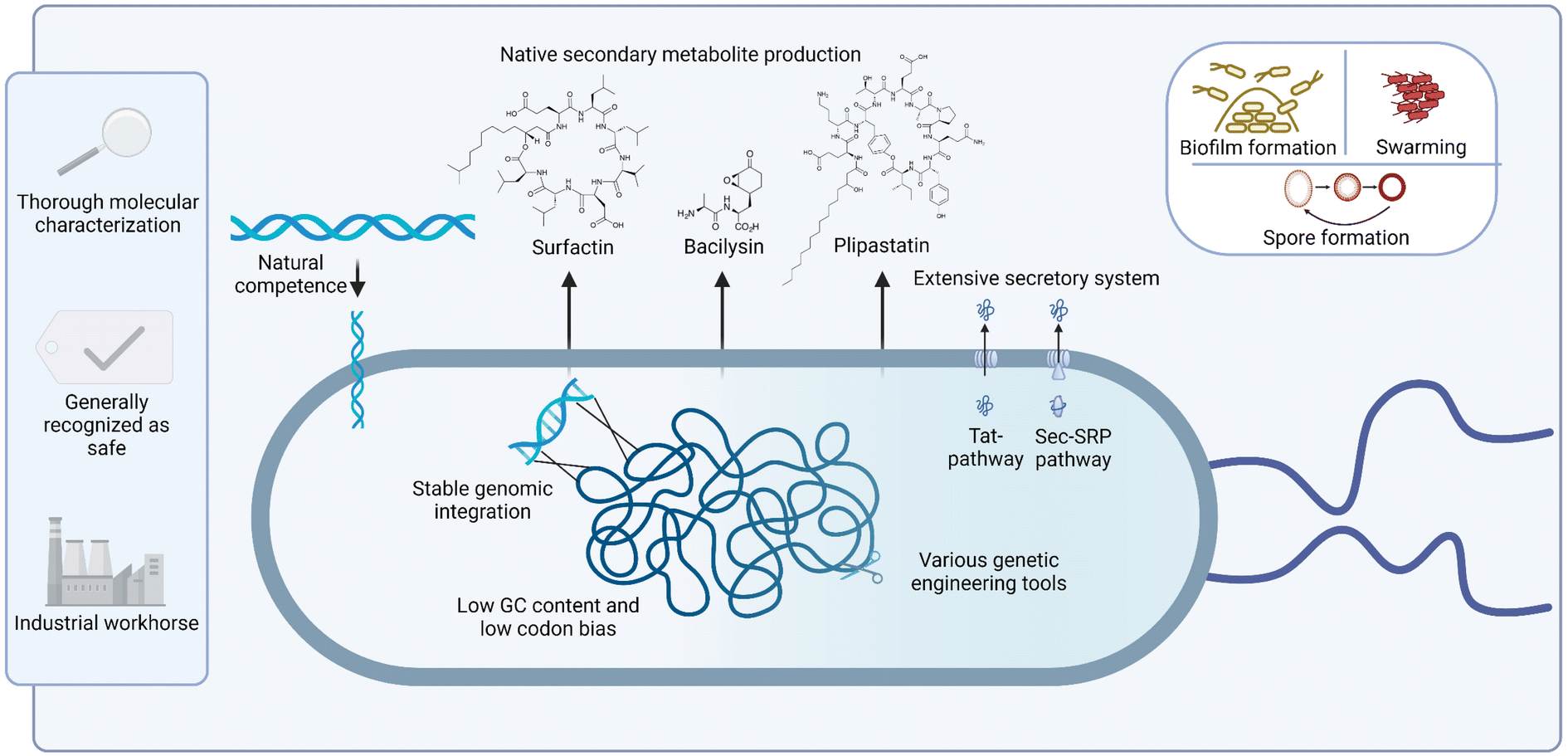 | ||
| Fig. 1 Graphic representation of inherent advantages of using B. subtilis as heterologous production host for NPs. Figure created with BioRender, adapted from ref. 89–91. | ||
2.2 BGC expression in other Bacillus species
Apart from B. subtilis, other Bacillus species have also been used for heterologous secondary metabolite production. A well-known example is Bacillus velezensis FZB42, formerly known as Bacillus amyloliquefaciens FZB42. This strain was originally isolated from the beet rhizosphere and its genome was first sequenced in 2007.92B. velezensis FZB42 is a model biocontrol agent and plant growth-promoting rhizobacterium. It shares many inherent characteristics with B. subtilis. Both are capable of sporulation and biofilm formation, have a natural competence for DNA uptake, possess efficient homologous recombination systems which facilitate genomic integration, and have received the GRAS status for specific industrial applications. The ongoing characterization of the strain has led to the creation of AmyloWiki, the B. velezensis counterpart of SubtiWiki, where information about gene functions, protein interactions and related details are collected.93 Several genetic modification tools for this strain have been developed, including Cre-lox94 and a CRISPR-Cas9 nickase.100 However, they often suffer from low efficiencies. Different constitutive and inducible promoters have also been described,96,97 but the development of other regulatory elements, such as ribosome binding sites (RBSs) and terminators, is lagging behind.98Interest in B. velezensis as a host for heterologous production has mainly arisen from its extensive secondary metabolism, with over 10% of its genome dedicated to the synthesis of antimicrobial compounds.99,100 Consequently, B. velezensis FZB42 has been used industrially as a biocontrol agent in different pesticides.101 Successful examples of heterologous expression of BGCs in this strain remain scarce but have been reported in two studies. The first example is mersacidin, a lanthipeptide originally produced by a related Bacillus isolate. B. velezensis was chosen as a host because it already harboured a partial mersacidin (mrs) BGC, encoding an ATP binding cassette (ABC) transporter that provides self-resistance. The mrs BGC was completed via natural competence transformation and genomic integration of the missing genes, while the mrsA structural gene was expressed from an autonomously-replicating plasmid. Mersacidin production was confirmed through HPLC and MALDI-TOF analysis. Functional evaluation of heterologous mersacidin biosynthesis by monitoring the antibiotic activity of the B. velezensis host was more difficult due to the background production of other antimicrobial compounds.102
Another study has reported the heterologous expression of the BGC that directs the biosynthesis of the locillomycin lipopeptides.99 This loc cluster originates from a closely related B. velezensis strain and was cloned in a fosmid vector. After exchanging the native promoter with an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible Pspac promoter, the entire loc BGC was integrated in the genome of the host via homologous recombination, exploiting sequence homology in flanking genomic regions. RT-PCR analysis revealed that the expression pattern of the loc genes was comparable in both the wild-type strain and the heterologous host, with two-fold higher expression levels in the latter. Also here, biological activity testing of the locillomycin-producing host strain was hindered by native lipopeptide production. This indicates the importance of having host strains with a clean genetic background. Recently, a genome-reduced B. amyloliquefaciens strain that lacks several non-essential genes, including those involved in antibiotic production and prophage regions, was constructed.103 This strain was shown to have a higher growth rate, reached higher biomass levels, and showed increased transformation efficiency. Surprisingly, however, no significant increase in surfactin production levels was observed. Nevertheless, this genome-reduced strain represents an interesting starting point for further engineering and optimization.
Bacillus licheniformis is another industrial workhorse that has been used for decades for the production of amylase104 and the synthesis of bacitracin for veterinary applications.105 The strain has similar characteristics as B. subtilis, but is particularly superior under anaerobic conditions. Several B. licheniformis strains have been isolated from oil reservoirs and have therefore adapted to oxygen-limiting conditions and higher temperatures.106–109 An important factor behind this adaptation is the presence of an oxygen-independent ribonucleotide reductase, which is missing in B. subtilis.110 This feature is especially interesting for the production of biosurfactants, like surfactin, fengycin, and lichenysin, which are natively produced by Bacillus species. Due to the need for oxygen supply in aerobic bioreactors, excessive foam formation occurs during surfactant production, a phenomenon that can be avoided under anaerobic conditions. An overview of the different tools and metabolic engineering strategies that have been applied to increase lichenysin surfactant titers in B. licheniformis was recently published.111 Overall, heterologous BGC expression in this strain has garnered little interest up to now, even though it provides interesting opportunities for the production of oxidation-sensitive compounds.
3 Lessons from successful heterologous BGC expression in Bacillus
3.1 Nonribosomal peptides, polyketides and hybrids
NRPSs are large multimodular megaenzymes that operate in a stepwise assembly line fashion to construct structurally diverse peptide natural products. Each module consists of a set of discrete catalytic domains responsible for the incorporation of one amino acid building block and a variable range of modifications to the growing peptide chain. Adenylation (A) domains select a specific amino acid building block, activate it via an adenylation reaction, and load it onto an adjacent thiolation (T) domain. T domains, also referred to as peptidyl carrier protein (PCP) domains, are post-translationally modified with a coenzyme A (CoA)-derived phosphopantetheinyl (Ppant) prosthetic group, which is required for covalent attachment of the aminoacyl and peptidyl intermediates. Following the loading reaction, condensation (C) domains couple the PCP-bound extender units to the growing peptide chain bound to the PCP domain of the preceding module by catalysing peptide bond formation. Throughout the assembly process, all building blocks and biosynthetic intermediates remain covalently bound to the PCP domains as thioesters. In most NRPSs, a terminal thioesterase (TE) domain catalyzes the release of the fully assembled peptide from the multienzyme complex via macrocyclization or hydrolysis. Additional domains, such as epimerization or methylation domains, and trans-acting tailoring enzymes can also be present, further increasing the structural diversity and complexity of NRPs.112–114PKSs are another class of modular multienzyme complexes responsible for polyketide natural product biosynthesis. These biosynthetic assembly lines have evolved from fatty acid synthases and share functional and architectural similarities with NRPSs. In PKSs, acyltransferase (AT) domains specifically select small, CoA-activated acyl thioester starter and extender units, such as acetyl- and malonyl-CoA respectively, and load them onto the terminal thiol group of T domains, or acyl carrier protein (ACP) domains. Ketosynthase (KS) domains receive the growing polyketide chain from ACP domains in the upstream module and subsequently catalyse a decarboxylative Claisen condensation with the extender unit loaded by the AT domain. Additional auxiliary domains, such as ketoreductase, dehydratase, or enoyl reductase domains, can modify the α- and β-carbons in the ACP-bound intermediates, further increasing the structural diversity of polyketide natural products.114,115 After the polyketide chain is fully assembled, the release of the product is frequently mediated by a TE domain which catalyses macrolactonization or hydrolysis. The resulting polyketide products can then undergo a range of post-assembly tailoring reactions, such as glycosylation, oxidation, hydroxylation, and halogenation, to attain their biological activity.
NRPSs, PKSs, and hybrid combinations of these biosynthetic assembly lines are designated as megasynth(et)ases. Common characteristics include the large size of these multifunctional enzyme complexes, the selection of specific building blocks, and the post-translational activation of the carrier protein (CP) domains with a Ppant prosthetic group in a reaction catalyzed by a 4′-phosphopantetheinyl transferase.116 Successful heterologous expression of such BGCs in B. subtilis has been reported to varying extent for the different classes: expression of only two PKS BGCs has been described to date, in contrast to nearly a dozen PKS-NRPS hybrids and NRPSs (Table 1).
| Compound | Class | Native producer | Bacillus host | BGC size | Yield | MiBIG | Ref. |
|---|---|---|---|---|---|---|---|
| a Exact size is not mentioned. | |||||||
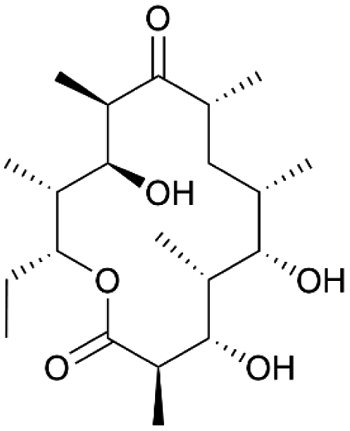
| 6-Deoxyerythronolide B | PKS | Saccharopolyspora erythraea | B. subtilis JK13 (168 derivative) | 30 kb | 2.6 ± 0.3 μg l−1 | BGC0000055 | 19 |
|
|||||||
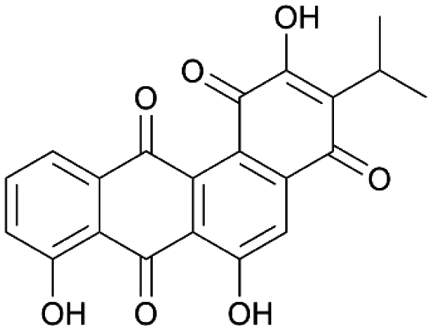
| Wexrubicin | PKS | Blautia wexlerae DSM 19850 | B. subtilis 168-sfp | 7.7 kb | 0.06 mg l−1 | 120 | |
|
|||||||
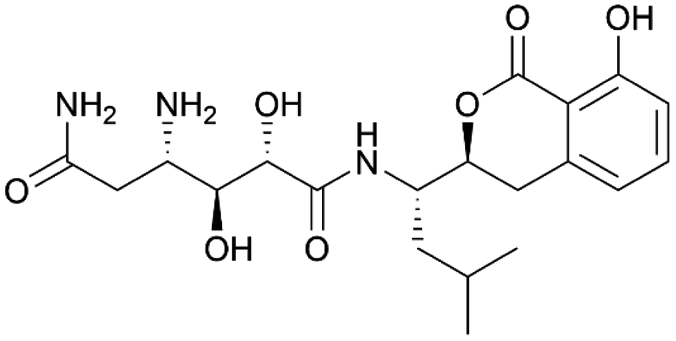
| Amicoumacin | NRPS/PKS | Bacillus subtilis 1779 | B. subtilis JH642+sfp | 47.4 kb | Comparable to native producer | 50 | |
|
|||||||
| Iturin A | NRPS/PKS | Bacillus subtilis RB14 | B. subtilis RM125 (168 derivative) | 38 kb | 51–64 μg ml−1 | BGC0001098 | 34 and 124 |
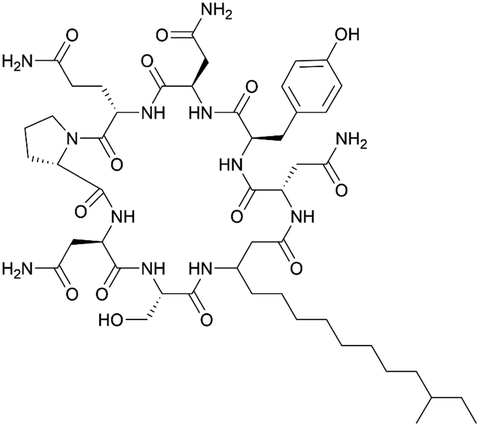
|
|||||||
| Bacillomycin D | NRPS/PKS | Bacillus velezensis FZB42 | B. subtilis 1A751 | 37.2 kb | Not reported | BGC0001090 | 20 |
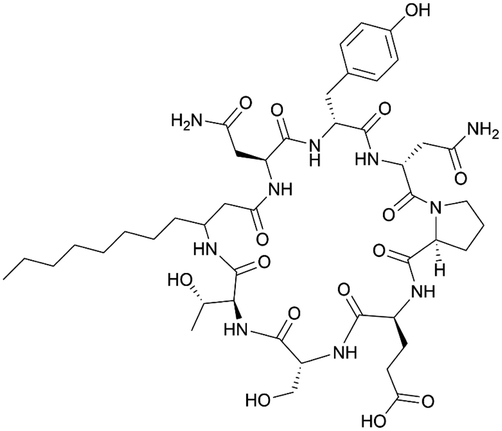
|
|||||||
| Bacitracin | NRPS | Bacillus licheniformis ATCC 10716 | B. subtilis TS30 (ATCC 21332 derivative) | 49 kb | Higher than native producer | BGC0000310 | 121 |
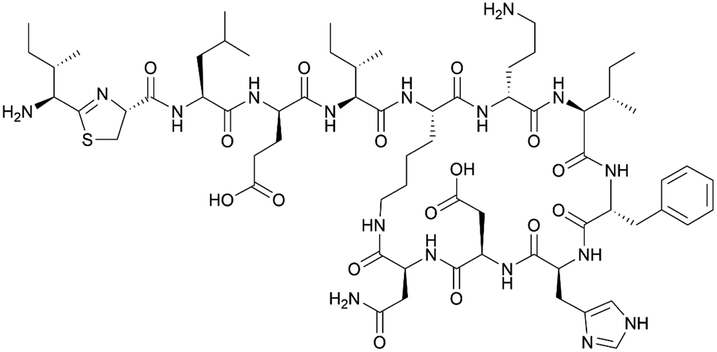
|
|||||||
| Two modules of surfactin BGC | NRPS | Bacillus subtilis ATCC 21332 | B. subtilis KE30 (TS30 derivative) | 6.2 kb | Not reported | 64 | |
| Polymyxin A | NRPS | Paenibacillus polymyxa E681 | B. subtilis 168 | 40.6 kb | Not reported | BGC0000408 | 18, 70 and 128 |
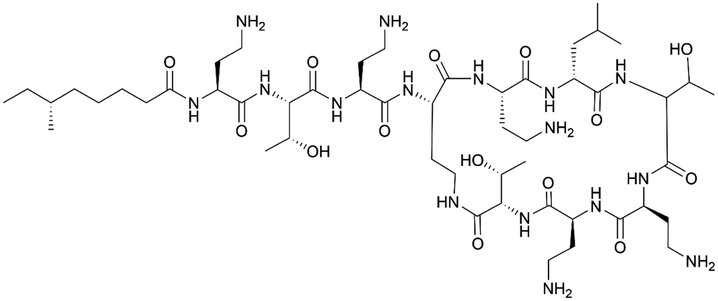
|
|||||||
| Surfactin | NRPS | Bacillus subtilis 1779 | B. subtilis JH642 | 38 kb | Comparable to native producer | 50 | |
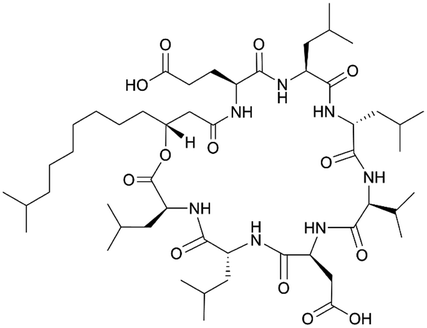
|
|||||||
| Enniatin | NRPS | Fusarium oxysporum | B. subtilis 168 | 10 kb | 1.1 mg l−1 | BGC0000342 | 69 |
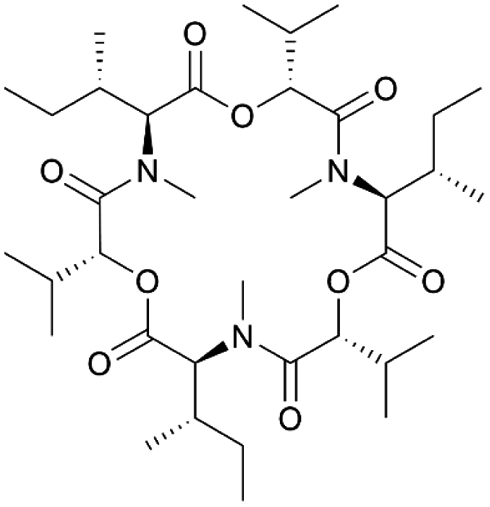
|
|||||||
| Edeine | NRPS | Brevibacillus brevis X23 | B. subtilis 1A751 | 48.9 kb | Compound not detected | 20 | |
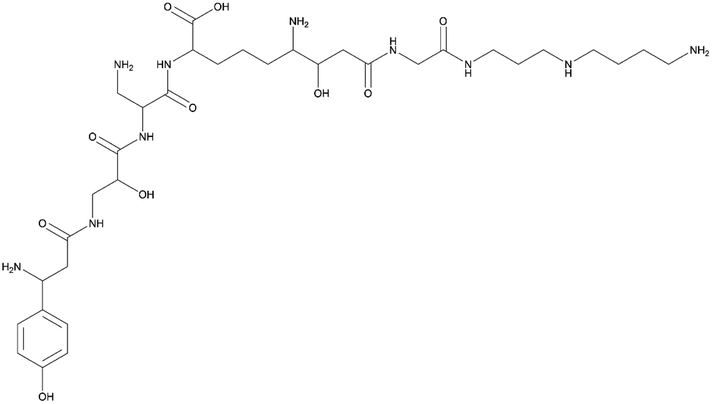
|
|||||||
| PZN12 | NRPS | Blautia producta ATCC 27340 and Clostridium sp. D5 | B. subtilis 168 | 10 kb | Not reported | 122 | |
|
|||||||
| Plipastatin | NRPS | Bacillus amyloliquefaciens HYM12 | B. subtilis 1A751 | 38.4 kb | 1182.5 ± 62.1 mg surfactin equivalents per l | 123 | |
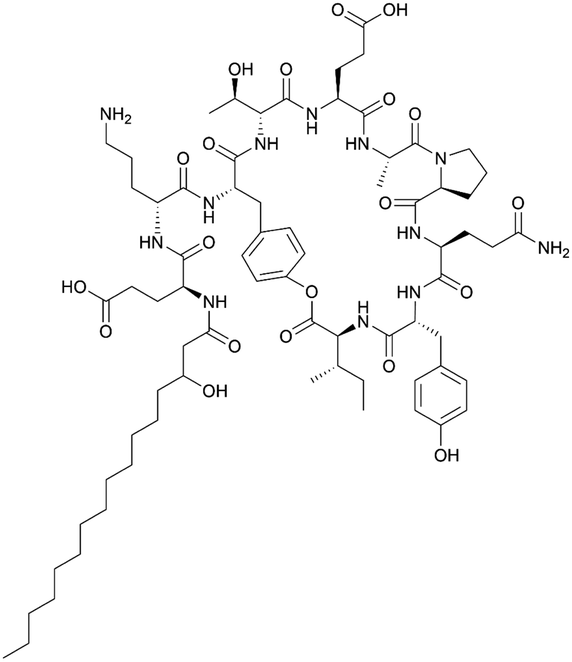
|
|||||||
| Bacillothiazols | NRPS | Bacillus velezensis FZB42 | B. subtilis 1A751-Sfp | 20 kb | Higher than native producer | BGC0002641 | 21 |
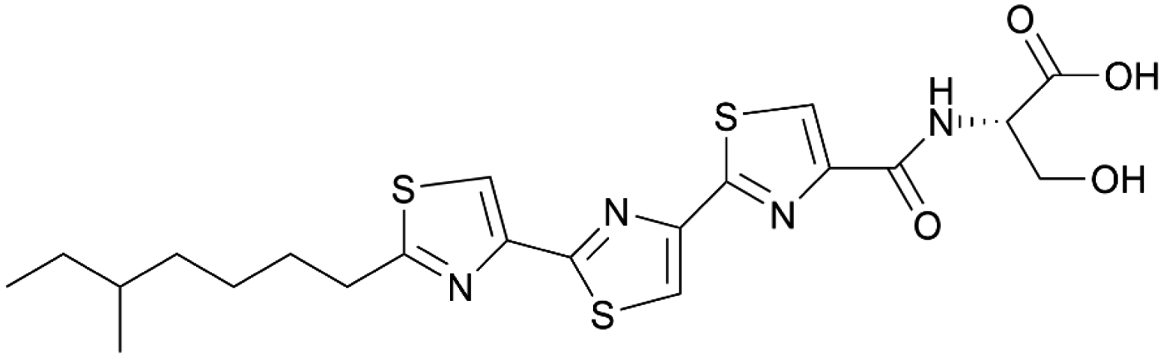
|
|||||||
| Micrococcin P1 | RiPP (thiopeptide) | Macrococcus caseolyticus 115 | B. subtilis BS 168R (168 derivative) | Not reported | Not reported | 129 | |
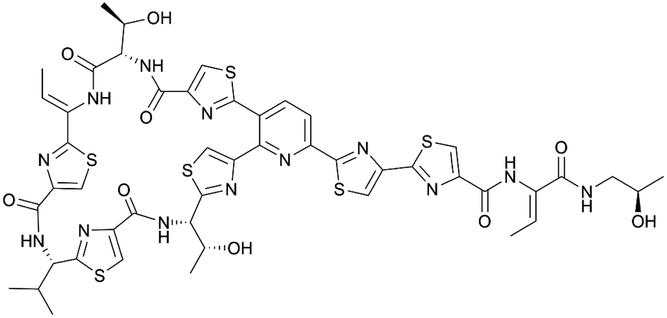
|
|||||||
| Nisin | RiPP (lanthi-peptide) | Lactococcus lactis ATCC 11454 | B. subtilis 168 ermΔsunA (168 derivative) | 16.5 kb | Only transcription | BGC0000535 | 130 |
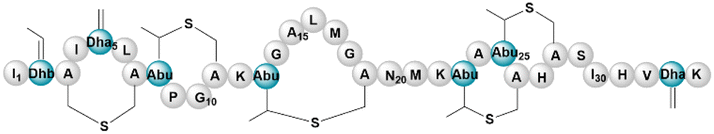
|
RiPP (lanthi-peptide) | Lactococcus lactis | B. subtilis 168 | Not reported | Only increase of resistance | BGC0000535 | 131 |
| RiPP (lanthi-peptide) | Lactococcus lactis ATCC 11454 | B. subtilis BRB779 (ATCC 6633 derivative) | Not reported | Not reported | BGC0000535 | 132 | |
| Subtilin | RiPP (lanthi-peptide) | Bacillus subtilis ATCC 6633 | B. subtilis BR151 (168 derivative) | <45a kb | Not reported | BGC0000559 | 133 |
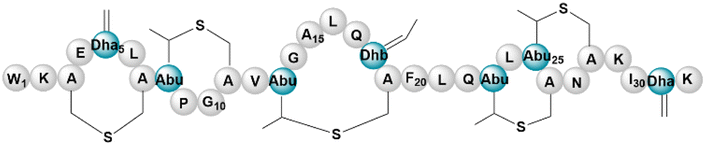
|
RiPP (lanthi-peptide) | Bacillus subtilis ATCC 6633 | B. subtilis 168 | 12 kb | Comparable to native producer | BGC0000559 | 134 |
| RiPP (lanthi-peptide) | Bacillus subtilis ATCC 6633 | B. subtilis ATCC 6633, B. subtilis 168, B. subtilis WB800, B. subtilis 168 strain PG10 | Not reported | Not reported | BGC0000559 | 54 | |
| Bacinapeptin A | RiPP (lanthi-peptide) | Bacillus nakamurai NRRL B-41092 | B. subtilis 168 | <15a kb | Not reported | BGC0002700 | 135 |
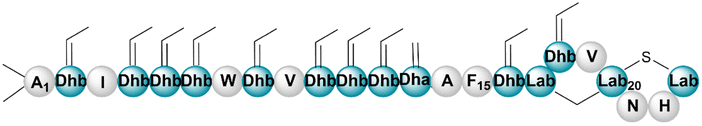
|
|||||||
| Paenithopeptin A | RiPP (lanthi-peptide) | Paenibacillus thiaminolyticus NRRL B-4156 | B. subtilis 168 | <15a kb | Not reported | 135 and 136 | |
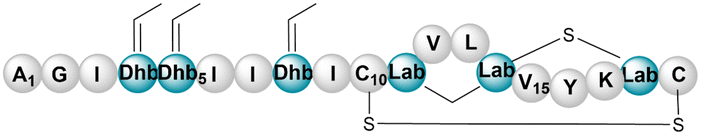
|
|||||||
| Andalusicin A | RiPP (lanthi-peptide) | Bacillus thuringiensis sv. andalousiensis NRRL B23139 | B. subtilis 168 | Not reported | Not reported | BGC0002111 | 137 |
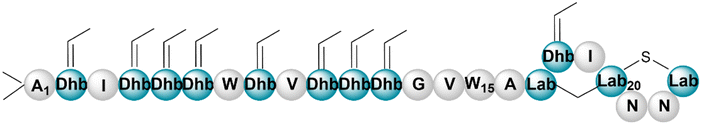
|
|||||||
| Subtilin-nisin hybrid | RiPP (lanthi-peptide) | Not reported | B. subtilis ATCC 6633, B. subtilis 168, B. subtilis WB800, B. subtilis 168 strain PG10 | Not reported | Not reported | 54 | |
| Subtilin-flavucin hybrid | RiPP (lanthi-peptide) | Not reported | B. subtilis ATCC 6633, B. subtilis 168, B. subtilis WB800, B. subtilis 168 strain PG10 | Not reported | Not reported | 54 | |
| Mersacidin | RiPP (lanthi-peptide) | Bacillus sp. HIL Y-8554728 | B. amyloliquefaciens FZB42 | 12.3 kb | Not reported | BGC0000527 | 102 |
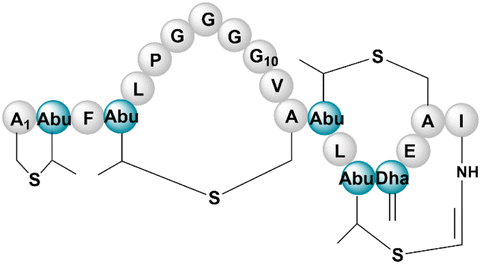
|
|||||||
| Microcin J25 | RiPP (lasso peptide) | Not reported | B. subtilis WB800N, B. subtilis 168 | Not reported | 5.968 mg l−1 | 88 | |
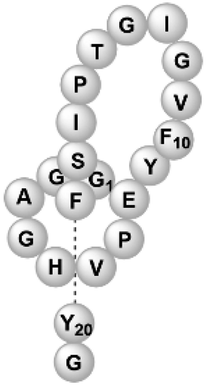
|
|||||||
| Microcin Y | RiPP (lasso peptide) | Not reported | B. subtilis WB800N, B. subtilis 168 | Not reported | 3.303 mg l−1 | 88 | |
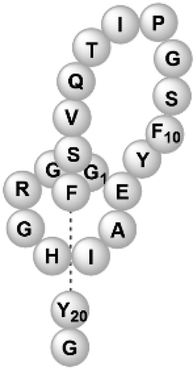
|
|||||||
| Mechercharmycin A | RiPP (cyclic azol(in)e-containing peptide) | Thermoactinomyces sp. YM3-251 | B. subtilis 168 | Not reported | 2–4 mg l−1 | 138 | |
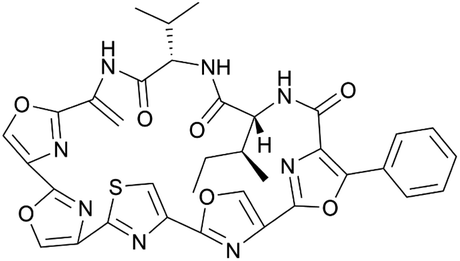
|
|||||||
| 4,4′-Diaponeurosporene and 4,4′-diapolycopene | Terpene (carotenoids) | Staphylococcus aureus NCTC 50581 | B. subtilis JH642 | Not reported | Higher than native producer | 139 | |
|
Terpene (carotenoids) | Staphylococcus aureus NCTC 50581 and Bacillus subtilis | B. subtilis 168 trpC2 | Not reported | 10.65 mg g−1 dry cell weight | 140 | |
| Terpene (carotenoids) | Staphylococcus aureus NCTC 50581 and Bacillus subtilis | B. subtilis 168 trpC2 | Not reported | 21 mg g−1 dry cell weight | 141 | ||
| Isoprene | Terpene (carotenoids) | Bacillus subtilis DSM 10 | B. subtilis DSM 10 (ATCC 6051) | Not reported | Higher than native producer | 142 | |
|
|||||||
| Amorphadiene | Terpene (carotenoids) | Bacillus subtilis 1A1 | B. subtilis 1A1 | Not reported | 20 mg l−1 | 143 | |
|
Terpene (carotenoids) | Not reported | Not reported | Not reported | 416 mg l−1 | 14 | |
| Terpene (carotenoids) | Bacillus subtilis | B. subtilis 168 trpC2 | Not reported | 116 mg l−1 | 144 | ||
| Taxadiene | Terpene (carotenoids) | Taxadiene synthase from Taxus baccata and other genes from Bacillus subtilis | B. subtilis 168 trpC2 | MEP pathway – 16.4 kb | 17.8 mg l−1 | 145 | |
|
|||||||
| Menaquinone-7 (MK-7) | Terpene (carotenoids) | Bacillus subtilis natto | B. subtilis 168 trpC2 | Not reported | 69.5 mg l−1 | 146 | |
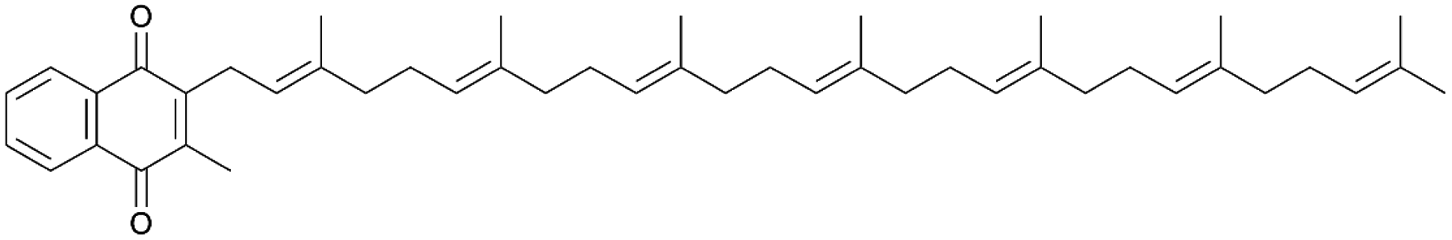
|
Terpene (carotenoids) | Not reported | B. subtilis 168 | Not reported | 360 mg l−1 | 147 | |
| Pediocin | Bacteriocin | Lactobacillus plantarum Zhang-LL | B. subtilis WB800N, B. subtilis 168 | Not reported | Not reported | 148 | |
|
|||||||
| Rhizocticin B | Phosphonate oligopeptide antibiotics | Bacillus subtilis ATCC 6633 | B. subtilis 168 | 13 kb | Not reported | 149 | |
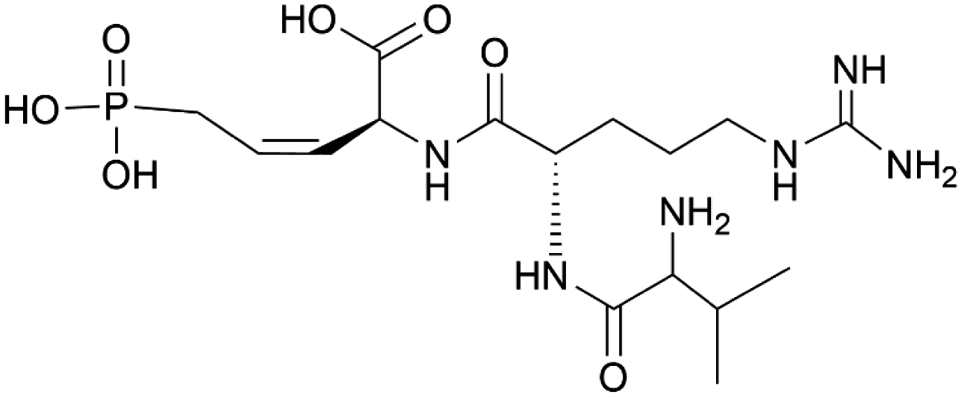
|
|||||||
The polyketide erythromycin is a widely used broad-spectrum antibiotic, originally isolated from the actinomycete Saccharopolyspora erythraea.117 6-Deoxyerythronolide (6dEB) is the PK macrolide core of this compound, and the corresponding BGC has been heterologously expressed in S. coelicolor, B. subtilis and E. coli.27,118,119 In B. subtilis, production levels only reached μg l−1 levels, while in the other hosts, much higher titers in the range of tens of mg l−1 were achieved.19 Promising, however, is that production levels in the Bacillus fed-batch system remained high with stable cell densities, indicating that there is no lack of precursors in the cell and that proteins are expressed at constant levels. Furthermore, 6dEB was only detected in culture supernatants, and not in cell pellet extracts, which confirms that the compound is efficiently secreted out of the cell even in the absence of a dedicated transport gene in the BGC. Remarkably, 6dEB production in B. subtilis was detected only when each of the three core biosynthetic genes, ∼10 kb in size, were expressed individually from separate cassettes under the control of an acetoin-inducible promoter and with an optimized ribosome binding site. mRNA analysis suggested that the production issues were likely caused by mRNA instability due to secondary structures, rather than transcriptional challenges.
For the production of the NRP-PK hybrid amicoumacin, opposing results have been described. When the entire ∼47 kb amicoumacin BGC was expressed in B. subtilis, yields close to those of the native producer were achieved. In contrast, production levels in E. coli were 100-fold lower.50 Furthermore, heterologous expression in B. subtilis under a strong, constitutive promoter of a PKS BGC from a Blautia wexlerae gut isolate resulted in the successful discovery of wexrubicin.120 However, it should be noted that original producer and host strain were more closely related, with similar GC levels, in these last two cases. These examples indicate that heterologous PKS expression is feasible in B. subtilis, though it remains highly understudied.
Compared to PKS pathways, the successful heterologous expression of NRPS clusters in B. subtilis has been reported in multiple studies, spanning a diverse range of compounds, including bacitracin,121 polymyxin A,18 surfactin,50 enniatin,69 PZN12 pyrazonine,122 plipastatin,123 and bacillothiazols.21 Additionally, B. subtilis has served as a host for the hybrid NRP-PK BGCs of iturin A124 and bacillomycin D,20 both BGCs composed of mostly NRP modules and only one PK module. The majority of these BGCs originate from species closely related to B. subtilis, and in these studies, the choice was made to clone the BGCs with their native promoters. Overall, the yields of the natural products in the B. subtilis host were comparable to those in the native producer or even exceeded them significantly. Edeine was one exception where, unexpectedly, no production could be detected in the host strain.20 The authors suggested that the issue could lie with the supply of the uncommon building blocks spermidine and 1,3-diaminopropane, or with the potential inactivity of the native promoter from Brevibacillus brevis in the B. subtilis host strain. Generally, heterologous expression of closely related BGCs in B. subtilis has proven to be a successful strategy, giving rise to high production levels.
NRPS BGCs from taxonomically distant species have also been successfully expressed in Bacillus. Examples include the enniatin BGC from the fungus Fusarium oxysporum69 and the pyrazinone BGCs from Blautia producta and Clostridium gut species.122 Although the production level of enniatin in B. subtilis was lower in comparison with that reported in the native producer,125 it is remarkable that the expression of this native BGC was achieved in the heterologous host using an acetoin-inducible promoter. The pyrazinone clusters were codon-optimized and their expression was regulated by the hyper-Pspac promoter. Since the native producer strains for these BGCs were not available, these examples highlight the potential of synthetic biology in combination with heterologous expression of uncharacterized BGCs in B. subtilis, even from distantly related strains. However, it has to be noted that in the same study, five other uncharacterized BGCs were cloned analogously to the pyrazinone BGCs, but no heterologous products were detected. This reinforces the common notion that closely related heterologous hosts have a higher success rate.
3.2 Ribosomally synthesized and post-translationally modified peptides
RiPPs represent a group of post-translationally modified peptide natural products with highly diverse chemical structures and biological activities. RiPPs are first synthesized by ribosomes as inactive, linear precursor peptides consisting of a leader and a core region. RiPP biosynthesis involves recognition of the leader peptide by enzymes that post-translationally modify the core peptide. In the final step, the precursor peptide is typically processed by a protease that cleaves off the leader peptide. The modified and bioactive core peptide is then exported out of the cell.126,127Although E. coli and Streptomyces spp. are currently the most commonly used heterologous expression hosts for RiPP BGCs,126Bacillus species have been put forward as a promising alternative for various reasons. First, a wide range of native RiPP BGCs have been discovered in the genome of Bacillus spp., indicating that this genus is well equipped to produce this class of compounds.150,151 This proficiency has been demonstrated by the successful recombinant production of micrococcin P1, a thiopeptide produced by Macrococcus caseolyticus 115. tRNAGlu played a pivotal role as a glutamate donor during the biosynthesis of this RiPP. Since the sequence of tRNAGlu from B. subtilis BS 168R differed by only a single base pair from that of the original host, this B. subtilis strain was considered to be the ideal heterologous host for the purpose of discovering essential biosynthetic genes and studying promoter activities within the micrococcin P1 BGC.129 Secondly, Bacillus has been used for the heterologous production of RiPPs to study their biosynthetic pathway or examine the effect of regulators on pathway expression when genetic modification in the original, closely-related host was difficult.102,129,134
The majority of RiPP BGCs are introduced into the genome of a Bacillus host through homologous recombination, although some are expressed in trans from a plasmid. One method that has been used to genomically integrate RiPP BGCs involves fragmenting the genomic DNA and adding homologous arms to incorporate the genomic region of interest into the genome of the heterologous host. Examples that have used this technique include the heterologous production of the broad-spectrum lanthipeptides subtilin133 and nisin130 in B. subtilis BR151 and B. subtilis 168, respectively. However, this method has yielded mixed results. In the case of subtilin, recombinant production was successful,133 while for nisin, BGC expression was verified by RT-PCR, but the active RiPP could not be detected.130 A possible explanation for this discrepancy is the absence of a nisin resistance mechanism in the host. In one study, this problem was solved by engineering B. subtilis 168 to increase its resistance in two ways. First, self-resistance genes found in the nisin BGC were integrated into the genome of B. subtilis 168 under the control of a synthetic promoter. Secondly, genes associated with nisin resistance were identified via transcriptomics and proteomics analysis of nisin-treated cells. The genes upregulated in this experiment were overexpressed in B. subtilis 168 to enhance its nisin resistance. Most of these genes were related to a general cell surface or membrane stress response rather than being specific to nisin. However, despite these efforts, the resistance of this engineered Bacillus host was not sufficient to allow commercial production of nisin.131 In another study, switching to a heterologous host that naturally possessed resistance to nisin enabled heterologous production.132 The same technique was also applied for fruitful recombinant production of the lanthipeptide mersacidin, which is naturally synthesized by B. velezensis.102
In addition to increasing host resistance, engineering the regulatory mechanisms that control the expression of RiPP BGCs has had a positive impact on yield. For example, production of the lanthipeptide subtilin is repressed during the exponential growth phase due to the presence of the transition state regulator AbrB. Deletion of the gene encoding this regulator led to a remarkable increase in recombinant subtilin production. Moreover, altered medium composition further increased the production levels to yields comparable to those of the wild type.134
The heterologous expression of RiPP BGCs in Bacillus has also led to the discovery of novel class III lanthipeptides from Firmicutes.135–137 Most of these lanthipeptide BGCs lack a protease gene. Through a combination of correlation network and co-expression analysis, possible proteases for these lanthipeptides were predicted. As a proof of concept, two previously unreported class III lanthipeptide BGCs were heterologously expressed in Bacillus, together with their predicted protease genes. This approach resulted in the discovery of bacinapeptins A and B, and paenithopeptins A-E136. Additionally, heterologous expression in Bacillus revealed an uncommon N-terminal dimethylation in a subset of class III lanthipeptides.136,137 This modification was linked to a methyltransferase that utilizes S-adenosyl methionine as a donor. This dimethylation, found in andalusicin137 and variants of paenithopeptin,136 was found to increase their antibacterial activity136,137 and is dependent on specific amino acids present at the first two positions of the core peptide.136
RiPPs show a remarkable biosynthetic malleability thanks to the tolerance of many RiPP modifying enzymes to amino acid changes in the core peptide sequence. The organization of precursor peptides in two distinctive parts (leader and core peptide) greatly facilitates this promiscuity as it allows leader peptide-dependent tailoring enzymes to remain substrate-specific while processing a wide variety of core sequences. Generating RiPP analogues by implementing these principles has led to the discovery of RiPPs with completely new characteristics and functions. For an overview of the engineering potential of RiPPs, the reader is referred to interesting reviews by Hudson and Mitchell (2018) and Montalbán-López et al. (2021).152,153 Various studies have taken advantage of this relaxed substrate specificity during heterologous expression in Bacillus. In a first study, nisin was recombinantly produced using the subtilin modification machinery present in the host strain. Therefore, the nisA gene encoding the nisin precursor peptide was introduced into the Bacillus genome containing the subtilin modification machinery. To ensure leader peptide recognition by this machinery, a part of the nisin leader peptide was replaced with that of subtilin, encoded by the spaS gene. However, most of the original nisin leader peptide needed to be retained to ensure efficient nisin production.132 In a second study, this principle was further exploited to develop a robust platform for the production of various unnatural lanthipeptide analogues. The subtilin modification and transport machinery, encoded by spaBTC, were combined with hybrid peptides composed of the leader peptide of subtilin, encoded by spaS, and the core peptide of either subtilin, nisin, or flavucin, encoded by spaS, nisA and flaA, respectively. Additionally, the subtilin leader peptide cleavage site was replaced with that of nisin to prevent in vivo leader peptide removal and resulting toxicity of the active RiPP. This approach resulted in a higher yield of inactive lanthipeptides, which were subsequently subjected to in vitro leader peptide removal for activation.54
Bacillus has also served as a heterologous host for RiPP BGCs from taxonomically unrelated species. Examples include the lasso peptides microcin J25 and microcin Y originating from Enterobacteriaceae. Together, they exhibit antibiotic activity against food-born Salmonella. To optimize their production in Bacillus, the difference between expression from a plasmid and the integration of one or more copies of the BGC in the genome was evaluated. In the end, production of both microcins was achieved with comparable yields to E. coli.88 Another example involves mechercharmycin A, a recently discovered polyazole cyclopeptide naturally produced by a Thermoactinomyces sp. This azol(in)e-containing RiPP exhibits strong cytotoxicity against various cancer cell lines. To discover more about the biosynthetic pathway of this RiPP, its BGC was integrated into the Bacillus genome under the control of a strong constitutive promoter, while an extra copy of the mechercharmycin A precursor peptide gene was expressed from a plasmid. A yield of 2–4 mg l−1 was achieved, surpassing the yield of 0.035 mg l−1 obtained from the native producer.138
In summary, heterologous production in Bacillus has been established for certain classes of RiPPs, including thiopeptides, lanthipeptides, lasso peptides, and azol(in)e-containing RiPPs. However, it remains largely unexplored territory for the majority of RiPP families. Successful recombinant production has been achieved for compounds originating from closely related species, as well as from some more distantly related ones. Key factors that need to be taken into account in this process are immunity of the host strain to the RiPP being produced and the regulatory mechanisms governing RiPP BGC expression.
3.3 Terpenoids
Bacillus has proven particularly successful as a heterologous host for terpene and terpenoid biosynthesis, owing to its inherent ability to produce isoprene in high concentrations via the methylerythritol phosphate (MEP) pathway. This pathway leads to the production of isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), which serve as the precursors for most terpenoids. By combining this inherent characteristic with the introduction of relevant terpene synthases and metabolic engineering optimizations, B. subtilis has been engineered to produce a variety of terpenoids, including carotenoids, amorphadiene, taxadiene, and menaquinone-7.144 It is worth noting that while Bacillus spp. natively possess the MEP pathway, other Gram-positive cocci and Lactobacillus spp. exclusively rely on the mevalonate (MVA) pathway, while Listeria spp. and a subset of Actinobacteria, such as Streptomyces, utilize both the MVA and MEP pathway.14,154–160As B. subtilis is recognized for its ability to produce significant quantities of isoprene, it has long been considered a promising microbial platform for terpenoid production. However, the progress in developing B. subtilis as a robust terpenoid cell factory has lagged behind that of E. coli and S. cerevisiae, primarily due to the slower development of molecular tools tailored specifically for B. subtilis. However, recent studies focusing on B. subtilis have yielded promising results, underscoring its potential as an efficient terpenoid production platform (Table 1).14
Carotenoids are widely used in the food, pharmaceutical, and health protection industries and researchers have turned to micro-organisms for their production. Early metabolic engineering efforts in B. subtilis involved the introduction of two genes from Staphylococcus aureus (crtM and crtN) to produce C30 carotenoids, enhancing the organism's resistance to oxidative stress.139 The enzyme CrtM catalyzes the transformation of farnesyl pyrophosphate (FPP) into dehydrosqualene. Subsequently, CrtN, acting as a dehydrosqualene desaturase, facilitates the conversion of dehydrosqualene into a vibrant yellow C30 carotenoid, known as 4,4′-diaponeurosporene.139 Later engineering strategies focused on increasing isoprene production and overexpressing the 1-deoxy-D-xylulose-5-phosphate synthase (dxs) gene, which is responsible for condensing pyruvate and glyceraldehyde-3-phosphate (G3P) to form 1-deoxy-D-xylulose-5-phosphate (DXP). DXP is subsequently processed into IPP and its isomer DMAPP.142 Further improvements were achieved by overexpressing multiple MEP pathway genes, resulting in high levels of C30 carotenoid production in B. subtilis.140 The upregulation of the whole MEP pathway further increased carotenoid production by up to around 20 mg g−1 dry cell weight.141
The crucial precursor for the highly effective antimalarial drug artemisinin is amorphadiene. Currently, microbes are responsible for producing this precursor, which is then converted into artemisinin through chemical methods. Researchers have constructed the amorphadiene biosynthetic pathway in B. subtilis by co-expressing the amorphadiene synthase gene (ads) with dxs and idi, two genes in the DXP pathway.143 ADS is required to convert FPP into amorphadiene, while Idi is essential in balancing the high flux of IPP generated by the MVA pathway.161 Increasing ADS translation and the expression of active terpene synthases can significantly improve production.143B. subtilis has shown promising results in producing the artemisinin precursor (416 mg l−1) through growth medium optimization and additional expression of the FPP synthase gene ispA and the entire MEP pathway.14 In a parallel study, a CRISPR-Cas9 editing system has been used to engineer three additional modules for improving amorphadiene production. This involved enhancing the MEP pathway, mutagenizing the terpene synthase module for overexpression, attenuating the branch pathway module and regulating the TCA cycle metabolism through the use of various weak and strong promoters. These modifications increased extracellular amorphadiene production from 81 to 116 mg l−1.144
Taxadiene is the crucial precursor for paclitaxel, a well-known anticancer drug marketed under the brand name Taxol. In B. subtilis, functional production of taxadiene was achieved by overexpressing the complete MEP pathway, along with the introduction of the taxadiene synthase (Txs) enzyme. Co-expression of the geranylgeranyl pyrophosphate synthase gene crtE from Pantoea ananatis led to 17.8 mg l−1 of taxadiene in B. subtilis, surpassing the yield achieved in yeast.145 Fine-tuning the expression of the MEP pathway genes in E. coli resulted in even higher yields. This success has inspired further improvements in B. subtilis taxadiene production through promoter and RBS optimization.14,162
MK-7, a major vitamin K2 compound recognized for promoting bone growth and cardiovascular health, has traditionally been produced through the fermentation of B. subtilis natto strains.163 However, researchers have engineered B. subtilis 168 for significantly increased MK-7 production by overexpressing endogenous modular pathways (MK-7, shikimate, MEP, and glycerol metabolism). A yield of 12.0 mg l−1 of MK-7 was achieved through the overexpression of the 1,4-dihydroxy-2-naphthoyl-CoA hydrolase gene (menA) under the control of the Plaps promoter, along with four MEP pathway genes. Production levels were further increased by enhancing glycerol metabolism and reducing intermediate metabolite consumption.14,146 The integration sites for overexpression of the MEP pathway genes were also found to affect MK-7 production.146 Finally, dynamic regulation of pathway expression through quorum sensing led to a remarkable increase in production, reaching 360 mg l−1 in B. subtilis, the highest reported yield.147
While substantial progress has been made toward optimizing terpenoid production in B. subtilis, there is still ample room for further improvement. First of all, there is an opportunity to harness the inherent assets that B. subtilis possesses, such as cytochrome P450s and glycosyltransferases, to further diversify the range of terpenoids produced.14 Secondly, current engineering efforts primarily focus on overexpressing genetic components of the pathway due to limited information on the steady-state kinetic parameters of the endogenous enzymes. While this approach is common in metabolic engineering, it can impose a burden on the cells and incur energy costs.164 Protein engineering techniques, such as directed evolution, can enhance enzyme catalytic activity and reduce negative feedback inhibition.161 Promiscuity in terpene synthases can also be minimized to streamline precursor utilization and reduce metabolic burden. In addition, protein fusions and synthetic protein scaffolds can be employed to improve enzyme expression, solubility, stability, and localization to substrates or cofactors.165–168 Therefore, a holistic approach involving multilevel engineering, including gene expression manipulation, protein engineering, and comprehensive omics data analysis, is necessary. Collectively, these strategies will be essential for developing optimal B. subtilis strains for terpenoid production.14,169
3.4 Other bioactive natural products
Most heterologous expression efforts have focused on well-known classes of natural products such as RiPPs, NRPs, PKs, and terpenoids. However, B. subtilis has also proven to be a suitable platform for the production of less-known natural product classes. For example, pediocin, a group IIα bacteriocin with antimicrobial activity against Listeria spp., is naturally produced by Pediococcus acidilactici and Lactobacillus plantarum.170,171 Heterologous production of this bacteriocin has been achieved in various hosts, such as E. coli,172,173Lactobacillus lactis,174,175 and Corynebacterium glutamicum.176 A study by Wang et al. investigated if the pediocin peptide (PapA) could be heterologously produced in B. subtilis WB800N and secreted without the need for accessory protein PedC/PapC and ABC transporter PedD/PapD, as previous research had shown that these proteins impose a metabolic burden to the host148,176 (Table 1). The study used a shuttle vector to express both the original and a codon-optimized version of the papA gene as a fusion protein with an α-amylase signal peptide for secretion.177–179 The resulting supernatants, obtained after induction of expression, both displayed anti-Listeria activity, confirming successful secretion and bioactivity of the fused pediocin peptides. This finding was significant as it represented the first time actively secreted pediocin was obtained without the presence of the cognate secretion machinery. Notably, these results differed from previous findings in E. coli., where the fused protein did not exhibit any antibacterial activity.172,173,180 Surprisingly, the inhibitory activity of the non-codon-optimized fused peptide was stronger than that of codon-optimized one. The authors speculated that the difference in codon usage did not significantly affect expression levels in B. subtilis, at least for the papA sequence. They acknowledged the need for future research to explore the influence of the His-tag on the anti-Listeria activity and to optimize the induction conditions for pure PapA/pediocin. The pediocin produced in B. subtilis showed similar thermotolerant properties as reported in previous studies.148,181,182Rhizocticins are antifungal phosphonate oligopeptide antibiotics produced by B. subtilis ATCC 6633 (Table 1).149,183 These antibiotics consist of di- and tripeptides with the unique non-proteinogenic amino acid (Z)-L-2-amino-5-phosphono-3-pentenoic acid (APPA) at their C-terminus. Rhizocticins enter fungal cells through an oligopeptide transport system. Within the cell, they are cleaved by fungal peptidases to release APPA, which inhibits threonine synthase, leading to inhibition of protein synthesis and growth.184,185 Similarly, plumbemycins, tripeptide antibiotics with a C-terminal APPA residue from Streptomyces plumbeus, also use an oligopeptide transport system to enter bacterial cells and inhibit cell growth by releasing APPA. The selectivity of these antibiotics is determined by the recognition of the specific amino acids attached to APPA by the transport system and peptidases. Since threonine synthase is not present in mammals, the cytotoxicity of these antibiotics is limited.149,186–188 While the rhizocticins where discovered in 1949,183 the rhizocticin biosynthetic cluster was only identified in the genome of B. subtilis ATCC 6633 in 2010 via genome mining. The rhi cluster was heterologously expressed in B. subtilis 168 and experimentally shown to produce rhizocticin B using a combination of phosphorus-31 (31P) NMR spectroscopy and LC-MS.149 To date, only a few phosphonate biosynthetic pathways have been studied in detail.149 Elucidating and understanding the biosynthetic pathways of phosphonates can offer valuable insights into unique enzymatic transformations and novel biochemistry.
These examples demonstrate that B. subtilis can also be used to produce less well-known classes of natural products. However, the suitability of B. subtilis as an expression host for such metabolites needs to be assessed on a case-by-case basis.
4 B. subtilis toolbox for strain and biosynthetic pathway engineering
4.1 Host strain engineering
| Pathways/gene functions | Genes | Effect on NP production | Other effects | Ref. |
|---|---|---|---|---|
| Natural competence | BsuM restriction/modification system | — | KO increases transformation efficiency | 18 |
| comSK | — | Induced expression increases transformation efficiency | 69 and 191 | |
| Sporulation | spo0A | KO decreases heterologous subtilin production, and abolishes heterologous polymyxin production and native surfactin production | KO abolishes sporulation and reduces competence levels | 70, 134, 193 and 194 |
| spoIIGA | KO decreases heterologous enniatin production and increases heterologous 6-deoxyerythronolide B production | KO abolishes sporulation and affects cell density | 19, 69 and 192 | |
| spoIIIE, spoIVB | KO increases native surfactin production | KO abolishes sporulation and cells lack cell integrity | 193 | |
| General regulator | abrB | KO increases heterologous polymyxin and subtilin production. abrB elongation increases native surfactin production | abrB mutants reach lower cell densities | 70, 134 and 194 |
| degQ | DegQ is essential for native plipastatin production. Functional DegQ increases heterologous iturin, heterologous plipastatin and native fengycin production, and decreases native surfactin production | KO or downregulation of degQ reduces protease activity | 123, 124, 195 and 196 | |
| Biofilm | epsA-O tasA-sipW-yqxM | KO increases native surfactin and fengycin production | KO decreases biofilm formation | 197 and 198 |
| Autolysis | lytC | KO increases heterologous enniatin and 6dEB production | KO reduces autolysis and increases cell densities | 19, 69 and 192 |
| Protease activity | nprE, aprA | Successful heterologous production of bacillomycin D, plipastatin and bacillothiazols in KO strain | — | 20, 21, 123 and 199 |
| nprE, aprE, epr, bpr, mpr, nprB, vpr, wprA | Successful heterologous production of microcins in KO strain | — | 88 and 200 | |
| PPTase | sfp | Sfp is essential for the production of native lipopeptides. It is compatible with a wide range of heterologous NRPSs and PKSs | — | 18–20 and 50 |
4.1.1.1 DNA uptake. Due to its natural competence, transformation of B. subtilis requires relatively little effort. However, the presence of the intrinsic BsuM restriction–modification (R–M) system in the widely used lab strain B. subtilis 168 causes plasmid instability and reduced transformation efficiencies for foreign DNA and large sequences.189 The process of cloning and heterologously expressing BGCs, which commonly faces these challenges, therefore remains a challenging task. Encouragingly, deletion of the ydiO-ydiP and ydiR-ydiS-ydjA operons, thereby disrupting the R–M system, has been reported to increase transformation efficiencies for 30–70 kb fragments by at least 100-fold.18 A complementary strategy involves engineering the comSK system.190 Overexpressing these central competence regulator genes under the control of the mannitol-inducible PmltA promoter has been shown to increase transformation efficiencies of plasmid DNA by 6.7 fold, resulting in a so-called super-competent B. subtilis 168 derivative.191 Another strain, B. subtilis JK3, in which comS was placed under the control of an IPTG-inducible Pspac promoter in the sacA locus, was used for heterologous enniatin production, but no quantification of competence was described.69 Both of these competence-enhancing approaches seem valid to use in B. subtilis host strains to facilitate cloning.
4.1.1.2 Sporulation. To improve lab handling, boost production levels and increase cell densities in bioreactors, efforts have been made to disable the sporulation pathway. However, the outcomes have been mixed. Knocking out the spo0A gene, which encodes a general regulator for initiating the sporulation process,28 resulted in reduced heterologous production of subtilin134 and even complete abolition of heterologous polymyxin production.70 Modification of the signal-transducing protease gene spoIIGA, on the other hand, increased levels of heterologous 6dEB production,19 but unexpectedly led to significantly reduced enniatin titers.69 Notably, significantly reduced cell density levels were reported in both publications, while the spoIIGA deletion in the undomesticated B. subtilis ATCC 6051 strain resulted in increased cell densities.192 In a recent study, the effect of single sporulation gene deletions on enzyme and secondary metabolite production levels in B. subtilis was investigated.193 It was found that single deletions of either spo0A, spoIIIE, or spoIVB were sufficient to abolish sporulation, but each had different effects on physiology and production. Specifically, native surfactin production was abolished in the spo0A mutant but increased in the other two mutants. Since the spoIIIE deletion compromised cell integrity, the spoIVB mutant was identified as the best secondary metabolite producer. The positive effect was suggested to be linked to metabolism, with increased synthesis of branched-chain fatty acids, glutamate, and aspartate, even though valine and leucine levels were decreased. Furthermore, spo0A deficient strains show greatly reduced competence levels.194 Partial reversal of this effect can be achieved through the modification of AbrB, a small regulatory protein negatively regulated by Spo0A.201 AbrB has been shown to downregulate or abolish the expression of heterologous BGCs,134 even by directly binding upstream of the heterologous polymyxin BGC, native to Paenibacillus spp.70 Both studies suggest, through the analysis of double mutants, that modifying spo0A leads to derepression of abrB, which, in turn, results in reduced production levels. The combination of a spo0A nonsense mutation and a stop codon mutation in abrB, leading to an 11 bp extension of the open reading frame, is naturally present in B. subtilis 3NA.202 The effect of these mutations on surfactin production was recently studied in a B. subtilis 168-derived strain. The findings indicated that spo0A mutants can reach higher cell densities with low surfactin production, while abrB-elongated mutants attained lower cell densities but high surfactin production. The combination of both modifications had overall beneficial effects.194
4.1.1.3 Biofilm formation. Biofilm formation is another trait of Bacillus that can complicate lab handling and requires significant metabolic capacity. The deletion of the epsA-O and tasA-sipW-yqxM operons, responsible for biofilm formation,32 leads to increased levels of native surfactin production in B. subtilis by allowing the reallocation of energy and substrates.197 For native fengycin titers, only the tasA deletion has been shown to improve production, while the other mutants did not show significant differences.198
4.1.1.4 Autolysis. Additional yield improvements for secondary metabolites have been achieved by deleting lytC, which encodes an autolysin responsible for flagellar function. Inactivation of lytC increased heterologous enniatin and 6dEB production.19,69 Furthermore, this modification is associated with reduced autolysis and the ability to reach higher cell densities compared to the wildtype strain.192
4.1.1.5 Growth phase regulation and proteolysis. The DegS–DegU two-component system in B. subtilis is involved in regulating the transition to the stationary growth phase, affecting various processes, including competence initiation, motility, and poly-γ-glutamate synthesis.203 DegQ promotes the activation of this system via phosphorylation reactions, and this regulatory protein is therefore essential for plipastatin production in B. subtilis 168.204,205 Functional expression of DegQ also led to increased production levels in a B. subtilis 3NA derivative, but interestingly, promoter exchange in the plipastatin BGC to the strong constitutive Pveg promoter abolished this increase.195 The native degQ gene in the B. subtilis 168 lab strain has a one base pair substitution in the promoter region, causing greatly reduced expression of the gene.196,206 The impact of degQ on secondary metabolite production appears to be variable. Heterologous production of the NRP iturin increased to half of the wildtype producer levels upon degQYB8 expression124 and degQ expression was also successfully used for heterologous plipastatin production.123 However, for the production of native lipopeptides, detrimental effects were observed for surfactin levels, while fengycin production was high.196 Additionally, the direct effect of DegQ on the production of extracellular proteases was analyzed. This is especially important for RiPP-like natural products to avoid degradation. No effect on NRP-derived lipopeptides was detected, but degQ downregulation or deletion significantly reduced protease activity.196 An alternative and more straightforward approach to avoid proteolysis is the use of protease-deficient heterologous hosts. A well-known example is the two-protease deficient B. subtilis 1A751 strain,199 which has been used for heterologous expression of several different classes of BGCs.20,21,123 Another option is the WB800 strain, in which eight proteases have been disrupted200 and successful heterologous microcin production has been achieved.88
4.1.1.6 PKS and NRPS biosynthesis. Phosphopantetheinyl transferases are essential for the posttranslational modification and functionality of PKSs, NRPSs, and fatty acid synthases by converting the inactive apo-enzymes to their active holo forms. In B. subtilis, the native sfp gene encodes such an enzyme, which has served as a model for the entire enzyme class due to its widespread use and remarkable substrate promiscuity. Sfp is essential for the production of the native lipopeptides surfactin and plipastatin in B. subtilis.204,207 However, the most widely used lab strains, like B. subtilis 168, contain a frameshift mutation in the sfp coding sequence, resulting in premature translation termination.20 To overcome this problem, functional sfp genes from other Bacillus species have been integrated into the genome,18,50 or the frameshift mutation in the native gene has been reversed.19,20 Sfp is able to interact productively with an exceptionally broad range of NRPSs and PKSs from both Gram-positive and -negative bacteria, making it a crucial component of a heterologous expression platform.
In order to increase the production levels of secondary metabolites, metabolic engineering is a valid strategy. Even though the thorough characterization of Bacillus provides a great starting point, little effort has been put into metabolic engineering strategies to increase heterologous production of natural products in B. subtilis. One successful example is the heterologous biosynthesis of the polyketide 6dEB. In this study, the prpBD operon responsible for the conversion of propionyl-CoA to succinate was deleted, resulting in a 75% increase in 6dEB production, but only in medium supplemented with propionate.19
Currently, the majority of metabolic engineering studies have focused on enhancing native surfactin production in B. subtilis. Several recent studies have successfully engineered the fatty acid biosynthetic pathway, resulting in increased production levels of this lipopeptide natural product. Strategies, such as increasing the pool of CoA-linked precursors by inhibiting their utilization for branched-chain fatty acid biosynthesis, overexpression of the rate-limiting enzyme in fatty acid biosynthesis to increase malonyl-ACP formation, and relieving the feedback inhibition on fatty acid synthesis by inhibiting degradation steps have all been shown to be effective.197,208,209 However, it is important to consider the potential impact on the production profile of different surfactin variants from the same BGC, as metabolic adaptations can shift the relative production levels.209 Similar results have been reported for fengycin biosynthesis.198 In another elegant study, CRISPRi was used to investigate the influence of amino acid metabolism on surfactin biosynthesis, allowing the identification of several genes that could be targeted for improved yields.210
4.1.1.7 Terpenoid biosynthesis. Regarding terpenoids, different genes can be overexpressed or downregulated, depending on the desired final product, as they are all produced from the same precursor and pathway. General engineering strategies include enhancing the MEP pathway through enzyme overexpression, attenuating the branch pathways, (down)regulating the TCA cycle, and engineering specific terpene synthases.144
For example, researchers have used metabolic engineering to enhance carotenoid production in B. subtilis by manipulating various biosynthetic genes, such as crtM, crtN, dxs, ispD, ispF, ispH, ispC, ispE, ispG and ispA. Overexpression of certain genes, such as dxs or dxr, has been found to increase carotenoid production. Additionally, by modifying the growth medium conditions and supplementing it with increased levels of salt, hydrogen peroxide and heat, the production of isoprene, the precursor to carotenoids, can also be improved.14
Amorphadiene, the precursor to the antimalarial drug artemisinin, can be produced in B. subtilis by introducing the amorphadiene biosynthetic pathway. This involves co-expression of the amorphadiene synthase gene (ads) with other genes involved in the MEP pathway, such as dxs and idi. Increasing the expression of ads, ispA and providing additional precursors through the MEP pathway has been shown to further enhance amorphadiene production.14
Similarly, taxadiene, the precursor to the anticancer drug paclitaxel, can be produced in B. subtilis by combining the expression of the taxadiene synthase gene (txs) with the regulated overexpression of the MEP pathway, including the FDP synthase gene ispA. Finetuning the expression of MEP pathway genes has been demonstrated to improve taxadiene production.14
The vitamin K2 menaquinone-7 (MK-7) has been synthesized in B. subtilis through modular pathway engineering. To boost MK-7 production, several approaches have been employed, including overexpression of genes involved in the MK-7 pathway (menA) and the MEP pathway (dxs, dxr/ispC, yacM/ispD and yacN/ispF together with menA), along with enhancement of glycerol metabolism by overexpressing glpD and downregulating dhhB. The choice of integration sites for overexpressing MEP pathway genes, with menA, dxs and dxr inserted into yxlA, yjoB and ydeO, respectively, has also been shown to impact MK-7 production. Additionally, dynamically balancing cell growth via the Phr60-Rap60-Spo0A quorum-sensing molecular switch has further contributed to enhanced MK-7 production.14
Genome reduction is a valuable strategy to facilitate bioactivity-guided screenings. Through the use of the markerless manP counter-selection system, a reduction of the B. subtilis genome to 3.6 Mbps has been achieved.40 This reduction involved the deletion of various genetic elements, including antibiotic-producing BGCs (the sublancin 168, subtilosin, plipastatin, surfactin, bacilysin, bacilysocin, 3,3′-neotrehalosadiamine pathways and a cryptic PKS cluster), phages and cryptic prophages, ICEBs1, sporulation delaying protein toxin genes sdpABC and sdpRI, sigma factors σE and σF, and genes like spoGA and bpr. The resulting strain, named IIG-Bs20-4, did not show any growth defects compared to B. subtilis 168. In follow-up research, surfactin production in this strain was assessed.75 Interestingly, in terms of growth rate and glucose to biomass conversion efficiency, the genome-reduced strain outperformed the reference JABs24 strain, while the overall surfactin yield and surfactin production per unit of biomass, unfortunately, lagged behind. Nevertheless, such clean-background strains remain valuable for characterizing cryptic BGCs and simplifying downstream processing.
An additional genome reduction of 36% has been achieved in the miniBacillus PG10 strain.212 This strain has demonstrated the ability to successfully produce lanthipeptides heterologously. Notably, the absence of extracellular proteases in this strain leaves these RiPPs in an inactive state by inhibiting leader peptide processing. This provides an advantage by preventing toxicity to the host without the need for resistance or immunity genes. On the other hand, for the production of unknown RiPPs, additional maturation proteases will be necessary to obtain and identify the final bioactive peptide.54 These examples highlight the significant potential of genome-reduced strains in simplifying the screening and production of heterologous natural products. However, there are currently limited studies that have used such strains compared to Streptomyces hosts with clean backgrounds.72,213,214
4.2 BGC refactoring elements
Standardized and well-characterized genetic building blocks play a crucial role in simplifying and finetuning heterologous BGC expression. Examples of such refactoring elements include promoters, terminators, RBSs, and protein tags (Table 3). It is important to note that these elements have organism-specific requirements, such as GC content, regulatory signals, and codon preference.37,38 In 2013, Radeck et al. laid the initial groundwork for establishing a repository of readily useable genetic elements specifically tailored for Bacillus species by developing the Bacillus BioBrick Box,38 which later underwent further updates and refinements.37 In addition to the refactoring elements in this BioBrick Box, this section will also explore other genetic components that have been used to optimize heterologous expression in Bacillus spp.| Constitutive promoters | Strength | Ref. |
|---|---|---|
| PJ23101 | Weak | 38 |
| PliaG | Intermediate | 38 |
| PlepA | Strong | 38 |
| Pveg | Very strong | 38, 88 and 219 |
| Pbsp | Not reported | 88 |
| P43 | Not reported | 88, 227 and 228 |
| P43p | Not reported | 88 |
| PrepU | Strong | 216–218 |
| PhpaII | Strong | 95 |
| Pamy | Intermediate | 95 |
| PsrfA | Weak | 95 and 229 |
| Psig | Not reported | 95 |
| Pvgb | Not reported | 95 |
| PA2cup | Strong | 96 |
| PC2up | Strong | 96 |
| PpenP | Not reported | 230 |
| PBH4 | Three times stronger than PsrfA | 229 |
| PylbP | Not reported | 229 |
| Inducible promoters | Inducer | Ref. |
|---|---|---|
| PliaI | Bacitracin | 38 |
| PxylA | Xylose | 38, 54, 95, 96, 129, 228, 229 and 231 |
| Pgrac | IPTG | 95 and 96 |
| PsacB | Sucrose | 96 |
| Pspank | IPTG | 54 |
| Pspank-hy | IPTG | 54 and 230 |
| Pspac | IPTG | 222 and 232 |
| Pspac-hy | IPTG | 122 |
| PacoA | Acetoin | 69 and 220 |
| Pglv | Maltose | 223 and 224 |
| opuAA | Salt | 225 |
| PspaS | Subtilin | 222 |
| PspaS-mut | Subtilin | 222 |
| PN25 | IPTG | 64 |
| PqdoI | Flavonoid | 233 |
| PT7lac | IPTG | 230 |
| P2 | 45 °C | 226 |
| P7 | 45 °C | 226 |
| Phase-dependent auto-inducible promoters | Phase of induction | Ref. |
|---|---|---|
| PabrB | Exponential | 234 |
| PvalS | Exponential | 234 |
| Phag234 | Exponential | 234 |
| Pasd | Middle-log and early stationary | 234 |
| PspoVG | Middle-log and early stationary | 234 |
| PlytR | Lag–log and stationary | 88 and 234 |
| PgsiB | Lag–log and stationary | 95 and 234 |
| PmmgA | Stationary | 234 |
| PsigW | Stationary | 234 |
| PyafD | Stationary | 234 |
| Reporter gene | Reporter protein | Ref. |
|---|---|---|
| lacZ | β-Galactosidase | 38 |
| luxABCDE | Luciferase | 38 |
| mCherry | Red fluorescent protein | 37 |
| mTagBFP | Blue fluorescent protein | 37 |
| eCFP | Cyan fluorescent protein | 37 |
| sfGFP | Green fluorescent protein | 37 |
| GFPmut1 | Green fluorescent protein | 37 |
| mEYFP | Yellow fluorescent protein | 37 |
| SYFP2 | Yellow fluorescent protein | 37 |
Inducible promoters can be used to generate a two-phase system, involving bacterial growth followed by the addition of an inducer to activate the production system.220 The original Bacillus BioBrick Box includes two inducible promoters: PliaI and PxylA. PliaI is induced by the antibiotic bacitracin, while PxylA can be induced by adding xylose to the growth medium. For the optimal functioning of PxylA, fructose has been suggested as the best carbon source.38 The PxylA promoter has been used in combination with the XylR repressor for heterologous production of the thiopeptide micrococcin P1.129 Another study, aimed at enhancing poly-γ-glutamic acid, evaluated two inducible promoters for overexpression of the inulin hydrolase gene cscA: PxylA and the IPTG-inducible promoter Pgrac.95 Both of these promoters were also tested alongside the sucrose-inducible promoter PsacB to boost levansucrase expression for enhanced levan production, with Pgrac yielding the best results.96,221 Furthermore, van Tilburg et al. established a heterologous expression system to generate various hybrid lanthipeptides by controlling the expression of the modification and export genes with PxylA and regulating the expression of the precursor peptide genes with two IPTG-inducible promoters, Pspank and Pspank-hy.54 The latter is the hyper version of Pspank and is six times stronger.54 Another inducible promoter, Pspac-hy, was used for heterologous expression of various gut microbiota-derived NRPS clusters in B. subtilis.122 This promoter is a derivative of the Pspac promoter, which combines elements from a B. licheniformis penicillinase promoter and the E. coli lac promoter.222 PacoA, which is repressed by glucose and induced by acetoin, has been used for the recombinant production of the fungal NRP enniatin. However, excessive levels of the acetoin inducer resulted in cell lysis.69 In 2006, Ming-Ming et al. developed the maltose-inducible promoter Pglv, which is repressed in the presence of glucose.223 This promoter has been successfully used in B. subtilis WB800N for recombinant production of T9W, a variant of the pig myeloid antimicrobial peptide-36 effective against Pseudomonas aeruginosa. Optimal production was achieved with the addition of 5% maltose as an inducer.224 For a more cost-effective and straightforward induction, the salt-inducible promoter opuAA has been employed. This promoter is part of the natural stress-response system in B. subtilis. When tested by expressing a protease gene in B. subtilis WB800, a nine-fold increase in expression was observed upon induction with 4% NaCl compared to non-inducing conditions.225 Some promoters can also be induced by a change in temperature. Examples include P2 and P7, which show higher expression levels at 45 °C compared to 37 °C in both E. coli and B. subtilis.226
To enable a more dynamic production of subtilin in B. subtilis, a system for subtilin-regulated gene expression (SURE) was established, analogous to the nisin-controlled gene expression (NICE) system in Lactococcus lactis. The promoter of the subtilin BGC (PspaS) is regulated by the two-component system SpaKR, which is autoactivated by subtilin. Based on this principle, vectors were designed for subtilin-inducible heterologous expression: one vector for integration of the spaKR regulatory system into amyE, along with expression vectors containing the PspaS promoter and a mutant derivative PspaS-mut. The PspaS promoter was shown to be more strictly controlled, while the PspaS-mut resulted in higher expression levels. The SURE system has the potential to be used for the heterologous expression of any gene of interest.222
Certain promoters can be induced based on the growth phase of the bacteria. Such phase-dependent auto-inducible promoters are classified in four categories depending on the phase during which they are activated: class I (exponential phase), class II (middle-log and early stationary phase), class III (lag–log and stationary phase), and class IV (stationary phase). In one study, 114 endogenous B. subtilis promoters were selected from the DBTBS database and experimentally characterized to classify them into these four classes. Class I promoters include PabrB, PvalS, and Phag. Class II is represented by Pasd, with PspoVG showing the highest activity among this class. Class III promoters exhibit linearly increasing activity as cells grow, with PlytR being the promoter with the highest activity in this class.234 This promoter has been used for the successful heterologous production of microcin J25 and Y.88 PgsiB234 is another representative of class III and has been used to overproduce inulin hydrolase for enhanced poly-γ-glutamic acid yield.95 Examples of class IV promoters include PmmgA, PsigW, and PyqfD. Some of these promoters also exhibit pH and temperature dependencies.234
The promoters listed here have been successfully used for the heterologous expression of natural product BGCs. For a more general overview of effective promoters in Bacillus, the reader is referred to an excellent review by de Souza et al.215
For the purification of the P. aeruginosa antimicrobial peptide T9W, a His6-tag was appended to the N-terminus of the peptide. Additionally, a SUMO-tag was inserted between the His6-tag and T9W to enable efficient removal of the His6-tag. This SUMO-tag also helps to improve the stability and expression level of recombinant proteins.224
To increase the production of poly-γ-glutamic acid, inulinases involved in the production process have to be exported via a peptide-dependent signalling pathway. Therefore, five signal peptides were assessed: SPyncM, SPamyQ, SPsacB, SPsacC, and SPnpr, with SPnpr showing the best results.95 A similar strategy has been employed to enhance levan production by adding signal peptides to the N-terminus of levansucrase. Various signal peptides, including SPamyE, SPbglS, SPbpr, SPnprB, SPnprE, and SPwapA were used, in addition to SPsacB and SPyncM.96 For the purification of the thiopeptide micrococcin P1, a His6- or glutathione S-transferase (GST)-tag was added to the N-terminus of the compound. Additionally, Strep-tags were attached to either the N- or C-termini of the modifying enzymes.129
| Vector | Type | Resistance | Additional information | Ref. |
|---|---|---|---|---|
| pDG1662 | Integrates in amyE | Chloramphenicol | 235 | |
| pDG1730 | Integrates in amyE | Spectinomycin | 235 | |
| pDG1664 | Integrates in thrC | Macrolide-lincosamide-streptogramin B | 235 | |
| pDG1731 | Integrates in thrC | Spectinomycin | 235 | |
| pBS1C | Integrates in amyE | Ampicillin and chloramphenicol | 38 | |
| pBS1E | Integrates in amyE | Ampicillin and macrolide-lincosamide-streptogramin B | 37 | |
| pBS1K | Integrates in amyE | Ampicillin and kanamycin | 37 | |
| pBS2E | Integrates in lacA | Ampicillin and macrolide-lincosamide-streptogramin B | 38 | |
| pBS4S | Integrates in thrC | Ampicillin and spectinomycin | 38 | |
| pSD193 | Integrates in srfA | Ampicillin and kanamycin | 64 | |
| pSD270 | Integrates in srfA | Ampicillin and kanamycin | C-Terminal His6-tag | 64 |
| pKE151 | Integrates in srfA | Ampicillin and kanamycin | 64 | |
| pKE170 | Integrates in srfA | Ampicillin and kanamycin | C-Terminal His6-tag | 64 |
| pBS0E | Replicative vector with ori1030 | Ampicillin and macrolide-lincosamide-streptogramin B | 37 | |
| pBS1ClacZ | Integrates in amyE | Ampicillin and chloramphenicol | β-Galactosidase reporter | 38 |
| pBS3Clux | Integrates in sacA | Ampicillin and chloramphenicol | Luciferase reporter | 38 |
| pBS3Elux | Integrates in sacA | Ampicillin and macrolide-lincosamide-streptogramin B | Luciferase reporter | 37 |
| pBS3Klux | Integrates in sacA | Ampicillin and kanamycin | Luciferase reporter | 37 |
| pBS3Ecatlux | Integrates in sacA | Ampicillin and macrolide-lincosamide-streptogramin B | Luciferase reporter combined with chloramphenicol resistance | 37 |
| pBS3Kcatlux | Integrates in sacA | Ampicillin and kanamycin | Luciferase reporter combined with chloramphenicol resistance | 37 |
| pBS1CαlacZ | Integrates in amyE | Ampicillin and chloramphenicol | LacZ-α part of β-galactosidase reporter, for screening of RBS libraries | 37 |
| pBS3Cαlux | Integrates in sacA | Ampicillin and chloramphenicol | LacZ-α part of β-galactosidase reporter, for screening of RBS libraries | 37 |
| pBS2EPxylA | Integrates in lacA | Ampicillin and macrolide-lincosamide-streptogramin B | Xylose-inducible | 37 |
| pBS2EPliaI | Integrates in lacA | Ampicillin and macrolide-lincosamide-streptogramin B | Bacitracin-inducible | 37 |
| pBS2EXylRPxylA | Integrates in lacA | Ampicillin and macrolide-lincosamide-streptogramin B | Xylose-inducible, XylR repressor present | 37 |
| pBS0EPliaI | Replicative vector with ori1030 | Ampicillin and macrolide-lincosamide-streptogramin B | Bacitracin-inducible | 37 |
| pBS0EXylRPxylA | Replicative vector with ori1030 | Ampicillin and macrolide-lincosamide-streptogramin B | Xylose-inducible, XylR repressor present | 37 and 136 |
| pMSE3 | Replicative vector with pAMβ1 ori | Kanamycin | 69 and 220 | |
| pDR111 | Integrates in amyE | Ampicillin and spectinomycin | IPTG-inducible | 136 |
It is common practice to integrate heterologous BGCs into the genome of B. subtilis. Examples of vectors suitable for integration in the amyE locus are pDG1662 and pDG1730, which confer chloramphenicol and spectinomycin resistance, respectively. For integration into the thrC locus, pDG1664 and pDG1731, which provide MLS (macrolide-lincosamide-streptogramin B) and spectinomycin resistance, respectively, can be used.235 The original Bacillus BioBrick Box consists of five BioBrick-compatible integrative B. subtilis vectors, three of which are equipped with compatible resistance markers and integration sites. These vectors are derived from the B. subtilis vectors pDG1662, pAX01 and pDG1731, which were modified to be BioBrick-compatible, resulting in pBS1C, pBS2E and pBS4S, respectively. They can be used to introduce any construct of interest into the amyE, lacA, and thrC loci, respectively. The integrative part of these vectors consists of flanking homology regions, a chloramphenicol, MLS or spectinomycin resistance cassette for selection in Bacillus and a MCS containing type II restriction enzyme recognition sites. Situated between these recognition sites in the MCS is an rfp-cassette. During the cloning process, this cassette is replaced with the desired insert, resulting in a white colour instead of a red when screening for positive hits in E. coli.38 In the updated version of the BioBrick Box, new pBS1C derivatives were included, in which the chloramphenicol resistance gene was replaced with either an MLS or kanamycin resistance cassette.37
The original Bacillus BioBrick Box also includes two reporter vectors derived from pAC6 and pAH328. One contains the lacZ reporter gene, known as pBS1ClacZ, while the other carries the luxABCDE operon, known as pBS3Clux. They are designed for integration into the amyE and sacA loci, respectively, and they both harbour a selection marker conferring chloramphenicol resistance.38 In the updated version of the BioBrick Box, the chloramphenicol resistance gene of pBS3Clux was replaced with either an MLS or kanamycin resistance cassette, resulting in the derivative vectors pBS3Elux and pBS3Klux, respectively. These vectors were further equipped with a chloramphenicol resistance gene as co-selection marker in front of the luxABCDE operon. This allows for a more robust screening of promoter libraries and facilitates the analysis of promoter strengths. Moreover, reporter vectors designed to screen RBS libraries were generated as derivatives of pBS1ClacZ and pBS3Clux. For this purpose, a lacZα fragment was inserted downstream of the MCS. This arrangement allows for red/blue/white screening in E. coli upon introduction of a promoter in the MCS and an RBS at the position of lacZα. Five different RBS variants, differing by a few point mutations from the consensus sequence AA AGG were screened using this system.37
As an alternative to integrative vectors, replicative vectors that do not integrate themselves in the genome can be used for heterologous BGC expression in Bacillus. The updated Bacillus BioBrick Box was expanded to include such a replicative vector, known as pBS0E, which is derived from the medium-copy vector pGP380. Also here, an rfp-cassette was introduced into the MCS to facilitate red-white screening during cloning in E. coli.37 Another example is pMSE3, a replicative shuttle vector compatible with both E. coli and B. subtilis.220 The use of this vector has resulted in higher yields during recombinant enniatin production compared to integration in the genome. This replicative plasmid led to the presence of about 200 copies of the enniatin biosynthetic genes in the Bacillus strain.69
To ensure reproducibility, it is important to have vectors that are already equipped with promoters, so that genes of interest can be introduced directly in the MCS for expression. Therefore, the BioBrick Box was updated to contain redesigned versions of pBS2E and pBS0E that carry an inducible promoter, such as PliaI, PxylA, or a combination of PxylA with its repressor xylR, upstream of the MCS.37 This latter vector was used in combination with the integrative vector pDR111 to overexpress the paenithopeptin precursor peptide gene from the replicative vector, while integrating one copy of the post-translational modification genes for this lanthipeptide into the genome.136 An exhaustive list of all vectors that are available for heterologous BGC expression in Bacillus can be found on SubtiWiki.17,55
In another study, a system was devised in which a cryptic promoter was activated by a spontaneous mutation to enable inducer-free activation of gene expression. The bacteria harbouring this mutation were selectively enriched due to a growth advantage linked to glutamate homeostasis. To simplify the detection of active mutations, a gfp gene was integrated into the construct.237
Han et al. developed a tool in B. subtilis named ‘Stepwise Evolution Targeting the Spacer region of Core Promoter’ (SETarSCoP).229 This method offers an effective way for evolving the strength of bacterial promoters. In a first step, the spacer sequences between the −35 and −10 box were compared in strong and weak native promoters of B. subtilis 168. This analysis revealed a conserved region of seven bp upstream of the −10 box. In a second step, this insight was applied to PsrfA, a constitutive promoter with higher strength than P43. Via a two-step, random mutagenesis process, the mutant promoter PBH4 was created, which exhibits threefold higher strength compared to PsrfA. To further demonstrate its applicability, the same approach was used to improve the strength of the constitutive promoter PylbP and the xylose-inducible promoter PxylA.229
In the first group, the gene of interest is expressed under the control of a T7 promoter on a replicative plasmid, while the T7 RNA polymerase is integrated into the chromosome.227,228,231,233 Conrad et al. pioneered the development of a T7 RNA polymerase-dependent system for heterologous expression in Bacillus.231 In this system, the T7 RNA polymerase gene rpoT7 was integrated into the amyE gene of the B. subtilis chromosome, under the regulation of the XylR-PxylA xylose-inducible system. In addition, a replicative plasmid was constructed for inserting the gene of interest between a T7 promoter and terminator. However, rifampicin had to be added to the medium to repress the host RNA polymerase. Moreover, the presence of the T7 RNA polymerase appeared to affect the processing and/or the secretion of the recombinant proteins.231 More recently, an analogous system was implemented in B. subtilis 164S, which is derived from the industrial strain B. subtilis ATCC 6051a. The same xylose-inducible regulatory cassette was used but integrated in the aprE gene instead of amyE.228 Consequently, the protease encoded by aprE was no longer functional.227 When comparing GFP production in this system to GFP production driven by the constitutive P43 promoter, a ten-fold increase in yield was observed. This system was also employed to recombinantly produce an α-L-arabinofuranosidase enzyme, which degrades arabinoxylan polymers to release D-xylose, making the system self-inducible.228 However, a limitation of this system is that the inducer, xylose, is continuously degraded by the host.227,228 To circumvent this issue, Ye et al. developed an IPTG-inducible system in B. subtilis.227 This strain was equipped with a xylose-inducible comK gene to facilitate biotransformation. It also featured deletions in protease genes like aprE and nprE. Additionally, genes related to sporulation (spoIIAC) and surfactin synthesis (srfAC), which can cause foam formation during fermentation, were inactivated to ensure optimal recombinant production and high cell densities during fermentation. The T7 RNA polymerase gene was integrated into the amyE locus on the B. subtilis SCK6 chromosome, under the control of the constitutive P43 promoter. To express genes of interest, a plasmid, named pHT7, was constructed, featuring a hybrid T7-lac promoter, a T7 terminator, and a B. subtilis RBS. Genes can be efficiently introduced in this plasmid using prolonged overlap extension polymerase chain reaction (POE-PCR), followed directly by transformation. The effectiveness of this engineered system was demonstrated through the expression of various fluorescent proteins and enzymes.227
Recently, a flavonoid-inducible system was developed. In this system, the T7 RNA polymerase gene was integrated into the amyE gene of the B. subtilis 168 chromosome under the control of PqdoI. This promoter contains two boxes to which the repressors LmrA and QdoR bind in the absence of flavonoids, such as quercetin and fisetin. The gene of interest was expressed on a multicopy plasmid under the control of the T7 promoter. The functionality of the system was tested by heterologous production of EGFP. No leaky expression was observed and induction was dependent on the specific flavonoid used. Additionally, an engineered version of PqdoI was implemented, which exhibited increased, but also more leaky expression.233
In the second group of T7 RNA polymerase-dependent systems, the gene of interest is integrated into the chromosome to enhance genetic stability.230,232 In one such system, the T7 RNA polymerase gene, T7 gene 1, was placed under the control of Pspac, and integrated into the wprA gene of B. subtilis DB428, which encodes a cell wall-associated protease. The T7 promoter and the gene of interest were introduced in the mpr gene, which encodes an extracellular protease. By strategically targeting these genes for integration, six proteases were eliminated from the producer strain, which had a positive effect on recombinant production.232 In an IPTG-inducible system established in B. subtilis PY79, the gene of interest was placed under the control of PT7lac, a chimeric promoter controlled by both the T7 RNA polymerase and the presence of IPTG. This design significantly reduced the leakiness of the system. The T7 RNA polymerase gene was driven by a Phy-spank promoter, while the LacI repressor was constitutively expressed from a PpenP promoter.230 This system showed a dynamic range of over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, surpassing the capabilities of the previously described systems,231,232 which had dynamic ranges up to 50.230
000, surpassing the capabilities of the previously described systems,231,232 which had dynamic ranges up to 50.230
4.3 BGC cloning strategies and engineering tools
Despite the increasing number of BGCs identified through genome sequencing, only a small fraction has been experimentally characterized and linked to a metabolic product.238 Silent BGCs, which are not expressed under standard laboratory conditions, hold significant potential for the discovery of novel drugs.239–242 However, the large-scale activation of silent BGCs poses challenges due to the lack of universal strategies and enabling technologies.242 Two main strategies are employed to tackle this problem: native host-based and heterologous host-based approaches. Native host-based methods maintain BGC integrity but rely on strain-specific genetic tools, making them unsuitable for genetically intractable or uncultivated strains. Heterologous host-based methods offer more flexibility, allowing for the cloning, heterologous expression, and functional characterization of diverse types of natural product BGCs, including those identified through metagenomics. Cloning large BGCs remains a limiting step, but various methods, including library construction, PCR-based assembly, and direct cloning, have been developed (Fig. 2).243,244 Direct cloning offers advantages by eliminating the need for library construction and minimizing random mutations. It holds promise for large-scale discovery of bioactive natural products. In this section, we will focus on methods that are specifically developed for heterologous expression of natural product BGCs in Bacillus. A method that has not yet been widely used for BGC cloning, but is making fast progress, is de novo DNA synthesis. While fragments of 5–7 kb can now be ordered routinely for less than $0.10/bp, some manufacturers offer even longer fragments of 50 kb and more. However, at a current price of $0.50–1/bp and lead times of several months, direct BGC synthesis remains out of reach for most researchers in the natural product discovery field.245 Conversely, so-called biofoundries, highly automated facilities for synthetic biology that largely rely on synthetic DNA, are becoming increasingly popular and accessible. Recent efforts to harness their capabilities for natural product discovery have yielded promising results.246 It can therefore be anticipated that in the future, when prices drop, natural product discovery will be able to benefit from complete, de novo BGC synthesis combined with bottom-up refactoring techniques.242 | ||
| Fig. 2 Overview of Bacillus toolboxes for strain and BGC engineering discussed in this review. Different targeted mutagenesis approaches for strain engineering are depicted on the left panel, together with the exchange units (XUs) approach for specific NRPS engineering. Genetic elements for BGC refactoring described in this review are grouped on the right upper panel. Below, two techniques for the direct capture of BGCs are depicted; transformation-associated recombination (TAR) and Cas9-assisted targeting of chromosome segments (CATCH). Figure was created with Biorender, and adapted from ref. 50, 243, 244, 268 and 292. | ||
A major advantage of clone libraries is that, once constructed, they can be screened numerous times in search of diverse BGCs of interest. However, despite improvements in the library construction process, the identification and recovery of clones carrying target BGCs from millions of unrelated clones remains a major bottleneck. This typically involves iterative rounds of sequential serial dilution and PCR, a highly laborious task.253 In this context, it is interesting to highlight a recently developed platform for plasmid library enrichment that combines microfluidics and droplet PCR.254 Using this platform, each clone in a library can be individually interrogated for the presence of a target insert at a throughput of thousands per second, yielding a pool of purified and sequence-enriched plasmid DNA. While only demonstrated as a means to achieve targeted sequencing of rare clones, it may also be possible to recover and amplify the enriched plasmids by transformation, offering an attractive and cost-effective route to rapidly deliver BGCs into the hands of researchers.254
A unique and undervalued feature of PCR-based screening of metagenomic libraries is the capability to isolate completely novel BGCs, even if the PCR primers are designed based on known genetic elements. This has most convincingly and extensively been demonstrated by Sean Brady and colleagues, who have used degenerate primers and amplicon sequencing to identify previously unknown BGCs that assemble structurally distinct natural products with novel modes of action.253,255 A well-known example is the discovery of the malacidins, relatively common calcium-dependent antibiotics that nevertheless were not previously discovered by culture-dependent methods. This distinctive class of antibiotics was identified using primers that target highly conserved NRPS domains.251 The potential of rationally designed PCR assays, utilizing degenerate primers to target class-defining enzymes for novel BGC identification from metagenomic libraries, combined with a plasmid enrichment platform as described above for physical BGC isolation, and subsequent transfer into appropriate heterologous expression systems, offers exciting prospects for future drug discovery efforts.
So far, the use of library-based techniques for heterologous expression of BGCs in Bacillus has been relatively limited. Luo et al. constructed a fosmid library from B. velezensis 916 to isolate the BGC for the NRP locillomycin.99 The library was introduced into E. coli using the lambda phage and PCR analysis identified the fosmid that contained the locillomycin BGC. Next, the fosmid was modified to enable the heterologous expression of the BGC in B. velezensis FZB42. This was done by introducing a spectinomycin resistance cassette and the IPTG-inducible Pspac promoter using λ Red recombination. The authors showed that locillomycin production was increased by more than 15-fold compared to the native B. velezensis 916 strain.99 The same approach was also applied to produce the enniatin synthetase gene of Fusarium oxysporum in B. subtilius.69
In 2014, a new vector known as pCAP01 was developed for TAR cloning, which can exist in multiple copies in E. coli. It was used to clone a genomic region containing the taromycin BGC from Saccharomonospora sp. CNQ490. Although the initially cloned tar cluster was not efficiently expressed in S. coelicolor M1146, subsequent gene remodelling led to successful activation and production of taromycin A, a compound highly similar to but structurally distinct from daptomycin. The pCAP01 vector can be used to clone various types of natural product BGCs.46 To improve the efficiency of TAR cloning, a counter-selectable marker gene, URA3, was introduced, resulting in pCAP03. This vector showed higher capture rates and reduced self-circularization. Additionally, a ready-to-use version of pCAP03 (RTU-pCAP03) was designed, eliminating the need for PCR in capture plasmid construction and thereby increasing capture efficiency.73,259 In 2019, a new vector called pCL01 was developed to facilitate the capture of larger BGCs in a single-copy form. It also allows higher copy numbers of cloned BGCs within E. coli cells. This vector was used to capture the 5-oxomilbemycin BGC and subsequently enhance its production using genome engineering.260 In the meantime, various derivative TAR cloning vectors have been developed to expand the range of heterologous hosts for natural product biosynthesis, including the pCAPB02 vector for B. subtilis50,259 and the pCAP05 vector for broad-host-range expression in Gram-negative hosts.261
The pCAPB02 vector combines yeast elements from pCAP01, E. coli elements from pBR322, and a homologous recombination-based integration system for B. subtilis from pDR111. It can be selected in E. coli and B. subtilis using ampicillin and spectinomycin. The insertion site for the BGC is located between the 5′ and 3′ gene fragments of the conserved B. subtilis amyE gene, allowing for double-crossover recombination into the genomes of various B. subtilis strains.259 So far, the vector has been used to successfully clone and express the amicoumacin BGC (PKS-NRPS hybrid, 47.4 kb) from B. subtilis 1779,50 the plipastatin BGC (NRPS, 39 kb) from B. amyloliquefaciens HYM12 (ref. 123) and the iturin BGC (NRPS, 38 kb) from B. amyloliquefaciens HYM12.262
A further advancement was made by combining CRISPR/Cas9-mediated in vitro digestion of DNA with TAR cloning.263 This method, reported by Lee, Larionov, and Kouprina, has led to a significant increase in the success rate of obtaining positive clones. By using CRISPR/Cas9, DNA can be precisely cut at specific locations, thereby enhancing the efficiency of TAR cloning.263 While this approach has not yet been applied to TAR cloning of microbial BGCs for heterologous expression, it has the potential to be widely applicable to various direct cloning methods. It has already been used in combination with other methods, such as RecE-catalyzed linear–linear homologous recombination (LLHR) and Gibson assembly. It offers a promising avenue for improving the efficiency of TAR cloning and expanding its applications in the future.247
Another direct capture technique that has been optimized for heterologous expression of BGCs in B. subtilis is LLHR. In 2012, Fu et al. introduced the LLHR strategy, which relies on homologous recombination between linear DNA molecules facilitated by RecE and RecT Rac prophage proteins.264 The procedure involves designing PCR primers, generating a linear capture vector, digesting genomic DNA, and co-electrotransformation of an engineered E. coli strain GB05-dir, where RecET-Redγ-RecA are integrated in the genome under the control of a PBAD promoter. This method has been successfully used to clone PKS-NRPS BGCs from Photorhabdus luminescens. However, the LLHR strategy showed limitations when cloning larger gene clusters due to self-circularization of the capture vector. To address this challenge, the authors combined LLHR with Redαβ-mediated linear plus circular homologous recombination (LCHR) in a two-step cloning approach, allowing them to clone the 52 kb plu2670 gene cluster.242,264 Another example of the successful use of LLHR is the cloning of the cryptic nrs BGC.21 Heterologous expression in B. subtilis of this BGC led to the discovery of the bacillothiazoles, illustrating again the potential of B. subtilis as host for the mining of new compound classes. In the meantime, several groups have updated the method to accommodate specific needs or to generally improve the method. These updated versions include the ExoCET platform (Exonuclease Combined with RecET recombination)265,266 and RedEx method.242,267
Liu et al. further adapted the LLHR method in 2016 to enable cloned gene clusters to be directly integrated into the genome of B. subtilis without the need for further modification.20 They developed a simplified method in which a pair of primers with 70 bp homologous arms is used to generate a linear cloning vector through PCR. This vector, containing the desired gene cluster, can be directly integrated into the B. subtilis chromosome. The entire process can be completed within a week if the gene cluster is intact and lacks homologous regions with the host strain. Overall, there are three main advantages of this adapted LLHR method, compared to TAR cloning. Firstly, it relies on Red/ET recombineering in E. coli, which simplifies the process by using shorter homologous arms and eliminating several steps associated with the TAR method. Secondly, the use of a shorter linear cloning vector, coupled with a ccdB toxin gene, reduces the rate of negative clones and minimizes vector self-circularization. Finally, the recombinase system in Red/ET recombineering is strictly regulated by an arabinose-inducible promoter, which reduces unintended recombination events within the cloned gene cluster. To demonstrate the feasibility of their adapted approach, the researchers successfully cloned two gene clusters: the edeine biosynthetic pathway (49 kb) from Brevibacillus brevis X2340 and the bacillomycin biosynthetic pathway (37 kb) from B. amyloliquefaciens FZB42.20 Both gene clusters were directly cloned and integrated into the chromosome of B. subtilis within a week. Remarkably, the researchers successfully achieved heterologous production of bacillomycin in the host strain. However, they encountered difficulties in heterologous expression of the edeine BGC in B. subtilis, presumably due to a mutation in the cloned gene cluster, the lack of certain essential precursors, or potential incompatibility between the promoter of the edeine BGC and B. subtilis.20
In 2015, Jiang et al. introduced a method termed CATCH (Cas9-assisted targeting of chromosome segments) for targeted cloning of bacterial genomic regions.268 It involves cleaving specific DNA regions from intact bacterial chromosomes embedded in agarose plugs and ligating them with capture plasmids using Gibson assembly. CATCH demonstrates good efficiency in cloning genomic regions up to 100 kb in E. coli. The advantages of CATCH include reduced background DNA fragments due to in-gel Cas9 cleavage, which protects chromosomal DNA from mechanical shearing, and time-efficient in vitro circularization through Gibson assembly. Even though CATCH is less efficient for DNA segments longer than 100 kb, it is still suitable for cloning the majority of natural product BGCs since most are smaller than 100 kb.242,269 This method has been used to successfully clone the 78 kb bacillaene BGC from B. subtilis. However, it is not yet compatible with heterologous expression. Over the past few years, several derivatives of the technique have been developed, some of which also directly enable heterologous expression of the captured BGCs, such as iCATCH270 and CAT-FISHING.271
CAPTURE, or Cas12a-assisted precise targeted cloning using in vivo Cre-lox recombination, is another direct cloning technique that utilizes Cas12a instead of Cas9.272 The process involves digesting genomic DNA with Cas12a-sgRNA, amplifying two capture plasmid fragments, each carrying a loxP site, assembling the digested genomic DNA and plasmid fragments using T4 polymerase exo + fill-in DNA assembly in vitro, and then introducing the pre-assembled linear products into an engineered E. coli strain for in vivo DNA circularization via Cre-lox recombination. Enghiad et al. successfully used this technique to clone 43 uncharacterized BGCs from various natural product classes, with sizes ranging from 10–113 kb.272 These BGCs were subsequently heterologously expressed in either S. avermitilis, S. lividans, or B. subtilis, depending on the original host of the BGC, which was either a Streptomyces or Bacillus species. However, it is noteworthy that the heterologous expression of the five BGCs originating from Bacillus species did not yield the expected new compounds. The authors suggested that simply cloning the whole BGC was not sufficient to activate the silent gene clusters and that additional measures, such as introducing strong promoters or optimizing precursor availability would be needed for successful heterologous production. For the BGCs captured from Streptomyces, seven were detected by HPLC, and five of them were produced in sufficient quantities for structural characterization.272
A recently developed recombineering system for B. subtilis, based on the recombinase pair YqaJ/YqaK from a B. subtilis 168 prophage, can be used to insert large DNA sequences as well as introduce gene deletions in BGCs.273 To achieve this, the transformation efficiency of B. subtilis was enhanced through overexpression of the ComK regulator. The system relies on the co-expression of yqaJK and comK, and utilizes a double-stranded DNA substrate with 100 base pair homology arms and a phosphorothioate modification at the 5′-end of the lagging targeting strand to improve recombination efficiency. This method offers a simpler and faster alternative to the labour-intensive preparation of single-stranded DNA substrates that are used in other recombineering methods.274 Taken together, the YqaJK system proved to be superior to other recombinase systems previously used in B. subtilis, making it an efficient genome manipulation tool for this species.273
CRISPR-Cas technology has revolutionized the field of genome engineering by offering high editing efficiency and precision, cost-effectiveness, and ease of manipulation. While many CRISPR tools were initially developed for B. subtilis, their applications are expanding to other Bacillus species as well. Here, we will only briefly highlight the different systems that have been developed so far and provide some examples of their application in natural product BGC engineering. For more detailed reviews about CRISPR in Bacillus, we recommend the following reviews.278,279
Since its initial adaptation in 2016 as a genome-editing tool, CRISPR-Cas has undergone specific developments for metabolic engineering in B. subtilis.39,280 This includes tools for generating both double-strand and single-strand breaks, which has enabled the introduction of point mutations, gene insertions, and gene deletions up to 38 kb. It has been successfully used in CRISPR interference (CRISPRi)39 and CRISPR activation (CRISPRa)281 systems for regulation of gene expression. Notable innovations include the Cas9 nickase (Cas9n)-variant for genome editing with reduced lethality282,283 and the use of CRISPR effector proteins Cpf1 and MAD7 for genome and metabolic engineering purposes.281,284 Additionally, the dCas9-AID tool enables single-base editing without the need for homologous recombination.285 Multiplex CRISPR editing has been achieved with success, targeting up to six loci simultaneously for point mutations.286 However, the current toolbox still lacks efficient multiplex CRISPR tools for other functionalities, such as knock-ins.281
Lui et al. recently showcased the application of CRISPR-Cas9 in cloning and enhancing the expression of a natural product BGC.287 Although B. subtilis does not naturally produce lycopene, it possesses a native MEP pathway. This pathway assembles IPP and its isomer DMAPP, which are essential precursors for the biosynthesis of various compounds, including carotenoids like lycopene. Lui et al. first developed a versatile CRISPR/Cas9-based cloning toolkit for B. subtilis, comprising an optimized artificial exogenous gene insertion box, promoters, terminators, and guide RNA targets. The toolkit features six promoters of varying strengths, ranging from 0.9 to 23 times the potency of the commonly used promoter P43. Additionally, seven highly efficient terminators were identified. A total of 13 key genes involved in the lycopene biosynthetic pathway were integrated into the B. subtilis genome at six specific sites and heterologously expressed to successfully produce lycopene. The researchers were even able to improve lycopene production further, achieving a 278.2-fold enhancement, amounting to 1.12 mg l−1.287
In another example, the production of plipastatin in B. subtilis 1A751 was enhanced by disrupting the surfactin operon using CRISPR/Cas9. Plipastatin is synthesized by a NRPS and is considered to be a promising candidate for a range of applications, including biopesticides, fruit and vegetable preservation, cosmetics, and pharmaceuticals. Plipastatin and surfactin, which are both produced by Bacillus strains, share common biosynthetic precursors, such as glutamate, valine, and fatty acids.288 By targeting and disrupting the srfAB and srfAC surfactin biosynthetic genes, more of these precursors were diverted to the plipastatin biosynthetic pathway, enhancing its yield.289 While the gene editing efficiency was higher in the model strain B. subtilis 168, it was comparatively lower in the engineered strain 1A751-pps-srf, possibly due to its instability as a result of its extensive genomic modifications. Two novel plipastatins, along with nine known variants or derivatives, were identified and characterized. Moreover, a yield of 1600 mg l−1 plipastatin was reached, which is the highest reported yield to date.289
Engineering of NRPSs has been a longstanding goal in the field of natural products. Historically, strategies to engineer NRPSs, such as A domain modification and module exchanges, have often resulted in impaired or non-functional pathways. This recurring issue, often manifesting as undesirable assemblies, has been partly attributed to the extensive sequence repeats that are often present in NRPSs, leading to unwanted homologous recombination events. To overcome these challenges, structure-based approaches were developed, such as the Seamed Express Assembly Method (SEAM)” coupled with Ordered Gene Assembly in B. subtilis (OGAB) method.290 The SEAM-OGAB method was applied to the plipastatin NRPS gene cluster from B. subtilis, which contains some of the most extensive direct-repeat sequences observed in any NRPS gene cluster, with 97% identity between repeating enzyme units. These repeated sections appear as module sequences throughout the A-T-C catalytic domains. In order to assemble the plipastatin BGC using SEAM-OGAB, each module was defined as an A-T-C/A-T-E-C unit. Extremely small (3 bp) seam overhangs were introduced into the C-A linkers based on homology. The seams were generated by introducing SfiI restriction enzyme recognition sites, which then enabled gene assembly via the first-generation OGAB method. Since the plipastatin BGC consists of 10 C-A linker sequences, a total of 10 seams were required to reassemble the complete nucleotide sequence. Upon construction, the SEAM-OGAB-assembled plipastatin cluster, featuring multiple seams, was compared to the plipastatin BGC assembled without seams. Importantly, the introduction of seams was found to not impair the function of the NRPS. In a next step, modules were swapped between the plipastatin and surfactin NRPS, generating chimeric assembly lines capable of synthesizing hybrid lipopeptides. Overall, this approach enabled efficient and precise assembly of the gene cluster, paving the way for further studies on the functional characterization and modification of the plipastatin NRPS, as well as other NRPS gene clusters with repetitive sequences.290
Bozhüyük et al. developed the concept of eXchange units (XUs)291 and eXchange unit condensation domain (XUC)292 to achieve efficient NRPS engineering. XUs leverage the modular structure of NRPSs but redefine modules as A-T-C units with a fusion site in the C-A linker region. The fusion site, typically an α-helix, can be identified via sequence alignment. Successful NRPS engineering through XU shuffling follows three key rules. First, XUs should ideally be mixed and matched within the same genera, as attempts to recombine NRPSs from distinct genera often lead to non-functional or impaired assembly lines. Second, the type of C domain should be considered, as changing the stereochemistry of a substrate significantly affects the productivity of engineered NRPSs. Finally, the acceptor-site specificity of the upstream C domain must be respected. While recent studies have questioned its role, adhering to this rule can guide engineering attempts. These rules improve the efficiency of engineering, although they become less relevant as the number of XUs decreases. However, it is important to note that while these rules enhance success rates, they are not absolute requirements for generating functional, engineered NRPSs.293
Another study has demonstrated the power of CRISPR-Cas9 gene editing for engineering complex NRPS assembly lines. Thong et al. successfully engineered NRPSs that were highly selective for specific amino acid substrates, producing new lipopeptides at levels comparable to those of the wild-type strain.294 They found that different FSDs (Fused Salinomycin Domains) with identical selectivity but from different sources had varying effects on production titers, suggesting that subtle sequence and structural differences may influence compatibility. CRISPR-Cas9-mediated replacements proved to be faster, more efficient, and more accurate compared to conventional methods, making it feasible to screen and optimize multiple FSD exchanges.294 So far, these methods have not been used in Bacillus species or used on NRPSs from Bacillus.
5 Conclusions and future perspectives
Over the past few decades, the identification of uncharacterized BGCs from various sources has become more easy due to the advances in sequencing technology and genome mining tools, such as antiSMASH.295,296 However, accessing these BGCs presents many challenges, and heterologous expression has emerged as a successful strategy to circumvent some of these challenges. Until now, efforts have primarily focused on a subset of heterologous hosts, such as Streptomyces spp. and E. coli. However, there is significant potential in diversifying the pool of available host species and strains. This diversity can be valuable, especially when dealing with unknown mechanisms that impact heterologous expression. B. subtilis, with its inherent characteristics, is a promising host for heterologous expression of BGCs. In this final part, we aim to provide some guidelines for heterologous expression in Bacillus. First, we discuss the selection of an appropriate heterologous host. Secondly, we assess the suitability of different classes of BGCs for heterologous expression in B. subtilis. Lastly, we summarize the currently available and optimized B. subtilis host strains.5.1 Where to find suitable BGCs for heterologous expression in B. subtilis
The selection of an appropriate host can significantly influence the success of heterologous expression. However, this choice is not always a straightforward decision. A key factor in maximizing the likelihood of successful heterologous expression is the close phylogenetic relationship between the host and the original producer. Bacillus species are prolific producers of bioactive natural products and show a high level of biosynthetic diversity, even among closely related strains.297 A comprehensive analysis of Bacillus genomes, spanning 139 species, revealed an average presence of 11.6 putative BGCs per genome.298 Marine-derived Bacillus and Paenibacillus strains in particular represent a rich source of novel chemistry.299–301 Although interesting new secondary metabolites are discovered frequently,302–305 research suggests that many Bacillales natural products remain undiscovered.306,307 Furthermore, it has been shown that Firmicutes species, including many Bacillus strains, are often underrepresented in metagenomics libraries due to their ability to form endospores, which hinders the efficacy of standard DNA extraction techniques.308 As a result, there is a vast untapped potential for discovering new chemistry within these taxonomic classes.On the other hand, the taxonomic distance hypothesis, which suggests that the phylogenetic distance between the host and the producer predicts heterologous expression success, has been challenged.261 While further research is necessary to understand the underlying mechanisms, it opens up a wider range of BGCs that can potentially be heterologously expressed. Low-GC BGCs from unrelated species may thus be feasible candidates for heterologous expression in Bacillus. In summary, the potential for heterologous expression in Bacillus remains largely untapped, although many research questions remain to be answered. Therefore, in the next two sections, we propose a framework of guidelines and conclusions addressing two important questions: which cryptic BGCs would be suitable for heterologous expression in Bacillus? Which Bacillus strain is the best host for a given BGC?
5.2 Assessing suitability of cryptic BGCs for heterologous expression in Bacillus
Currently, there is no universal one-size-fits-all host species for heterologous expression of all types of BGCs. In order to assess the suitability of a BGC for expression in B. subtilis, we summarize the current research findings for each class of natural products and identify areas where further pioneering studies are needed.Firstly, we see many clear advantages for heterologous expression of terpenoid BGCs in B. subtilis due to its inherent ability to produce large amounts of isoprene via the MEP pathway. This metabolic pathway is responsible for producing isopentenyl diphosphate and dimethylallyl diphosphate, essential precursors for the biosynthesis of terpenoids (Fig. 3).144
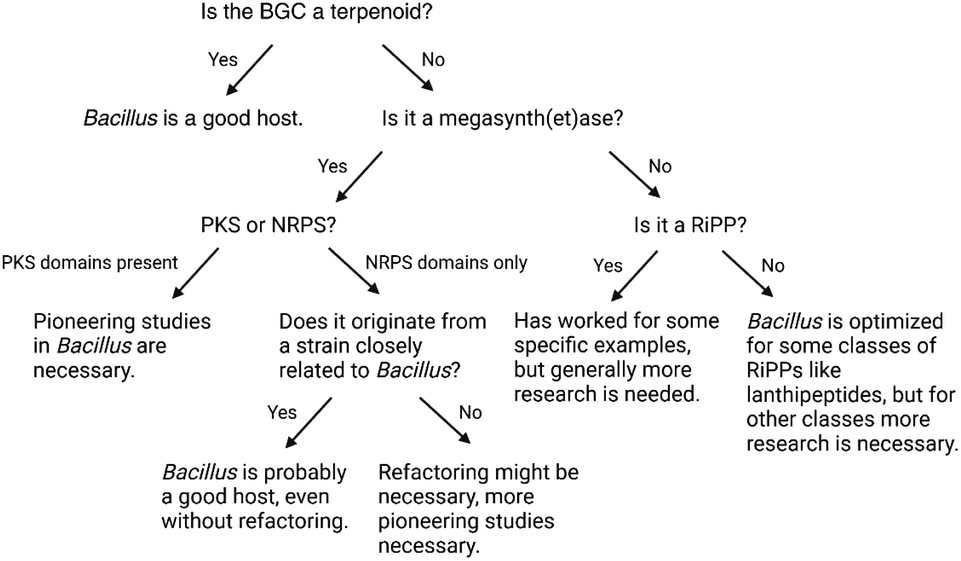 | ||
| Fig. 3 Decision tree to verify whether Bacillus could be suitable for the expression of a given BGC. | ||
For other natural product classes, selecting the right host strain is less clear-cut, although the inherent traits of Bacillus species provide a promising foundation for heterologous expression, regardless of the type of BGC. These include the ease of lab handling, natural competence, high transformation efficiency, thorough characterization of the metabolism, including native natural product biosynthesis, efficient secretion systems, and a strong track record in industrial production applications. Despite these advantages, B. subtilis has been underexplored as a host for heterologous BGC expression. Especially for PKS pathways, further research is required to uncover both the opportunities and challenges. On the other hand, heterologous expression of NRPS BGCs has been attempted and successfully achieved in several reported cases. Specifically, BGCs originating from taxonomically closely related species have shown high success rates and yields, often without the need for promoter replacement or other refactoring strategies. Additionally, BGCs from more distantly related gut species and even a fungus have been successfully expressed in Bacillus, albeit with some optimization. Overall, there is significant potential in using B. subtilis as a host for heterologous natural product BGC expression, though further research is needed to fully understand and address its limitations. One particular aspect is the size of the BGCs, as currently there has been no successful expression in Bacillus for BGCs larger than 50 kb.
With respect to RiPPs, B. subtilis has proven to be a very efficient host, particularly for lanthipeptide BGCs. Since the vast majority of these BGCs originate from Gram-positive species, using a B. subtilis host is a logical choice. However, one element that needs to be taken into account to ensure high yields is that the heterologous host must be resistant to the lanthipeptide being produced. Also other RiPP classes, naturally produced by both Gram-positive and Gram-negative bacteria, as well as compounds from marine prokaryotes have been successfully expressed in Bacillus. For BGCs that do not belong to any of these major classes, the feasibility of using Bacillus as a host must be evaluated on a case-by-case basis. Examples of some lesser-known natural product BGCs that have been successfully expressed in Bacillus species, include the rhizocticins and bacteriocins (pediocin).
5.3 Selecting the optimal Bacillus host for BGC expression
Various B. subtilis strains have been used for heterologous expression, with optimization efforts yielding significant improvements in lab handling. These improvements include increased transformation efficiencies18,191 and disabled sporulation,193,194 biofilm formation,197,198 and autolysis19,69,192 pathways to reach higher cell densities. In addition, several genes involved in the regulation of secondary metabolite production, such as abrB, degQ and sfp have been identified and studied.20,50,134,196,204 Metabolic engineering strategies for yield optimization have been explored, albeit limitedly for some specific cases. This is particularly true for terpenoid biosynthesis, where efforts have been focused on overproduction of the starter enzymes of the MEP pathway, along with specific enzymes that are needed for the synthesis of specific terpenoids.14,144 In addition, steps towards developing Bacillus strains with clean genetic backgrounds and reduced genomes have been taken.54,75 However, demonstrated applications of these strains are still limited and their full potential is yet to be realized.Different Bacillus strains have been optimized for different goals. Thus, we would like to offer guidance on selecting the optimal Bacillus strains for diverse purposes. For RiPP BGCs, the use of protease-deficient host strains is highly recommended for avoiding proteolytic degradation of the peptide product. Host strain options here include B. subtilis 1A751,199 which is deficient in two proteases, B. subtilis WB800,309 deficient in 8 proteases, and miniBacillus PG10,212 a strain with the added benefit of having a clean genetic background due to substantial genome reduction efforts.
Currently, there are no publicly available Bacillus strains that are specifically designed for terpenoid production. Most researchers start with a well-characterized Bacillus strain, such as B. subtilis 168, and then engineer it to suit their specific needs. This includes overexpression of certain genes or whole pathways, inactivating certain genes, generating mutant enzymes to increase fluxes, or fine-tuning the expression of genes or pathways by using specific promoters.
Up to now, the majority of megasynth(et)ase BGCs have been heterologously expressed in B. subtilis 168 or a derivative thereof, such as B. subtilis 1A751. The high transformation efficiencies of these strains facilitate the integration of these very large clusters. In addition, it is essential to repair the frameshift mutation in the sfp gene or integrate an sfp homologue to enable the production of NRPs, PKs, and hybrid metabolites. Although additional optimization strategies have been explored, this continues to be a topic for future research. The B. subtilis strain IIG-Bs20-4,40 in which all native secondary metabolite BGCs have been removed, could also be a suitable host for heterologous expression. This strain is particularly interesting for uncharacterized BGCs, as it greatly facilitates compound detection and activity testing. However, the strain has so far only been used to assess surfactin production.75
While Bacillus has long been used for industrial enzyme production, there is room for optimization in the large-scale heterologous production of secondary metabolites. Known high-yield strains have not yet been widely applied for heterologous BGC expression, and the currently-used strains may require additional optimization for this purpose. The body of research on how to increase surfactin yields in Bacillus can serve as a good starting point here. An example of an interesting strain in this context is the undomesticated B. subtilis ATCC 6051. This strain offers several advantages, such as high genomic stability during production due to its highly compromised natural competence levels, non-auxotrophy, the ability to reach high cell densities, and improved growth characteristics in complex media compared to B. subtilis 168.192
Furthermore, Bacillus strains that can be used for screening purposes, such as those designed for bacterial two-hybrid systems to study protein–protein interactions, have not yet been developed. Generating strains for these specific purposes in the future will further enhance the utility of Bacillus as a heterologous host for various applications.
In summary, B. subtilis has been shown to be a versatile host for natural product discovery through the heterologous expression of BGCs from various (meta)genomic sources. While specific classes of natural products, such as terpenoids, are particularly suited for expression in Bacillus, other classes require closer attention when selecting an appropriate expression host. In general though, it is clear that advances in host strain engineering and BGC cloning and refactoring have vastly improved the potential of using B. subtilis as a host for heterologous BGC expression.
6 Author contributions
H. P., H. G., H. V. C., M. F., Ja. M. and Jo. M. conceptualised the manuscript. H. P., H. G., and H. V. C. wrote the original draft. M. F., Ja. M. and Jo. M. reviewed and edited the manuscript.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
This work was supported by the Research Foundation–Flanders (FWO) under the scope of strategic funding of SB scholarships (1S45722N, H. P.; 1S28323N, H. V. C.), junior postdoctoral fellowship (1229222N, H. G.), FWO projects (G0B0420N and G0C4322N), FWO-SBO project (S001421N), EOS project (40007496); by KU Leuven, project C16/17/006 and C14/20/088; by VIB. We thank Dries De Vadder for designing the figures.9 References
- S. Iqbal, F. Begum, A. A. Rabaan, M. Aljeldah, B. R. Al Shammari, A. Alawfi, A. Alshengeti, T. Sulaiman and A. Khan, Molecules, 2023, 28, 927 CrossRef CAS PubMed.
- B. Fan, J. Blom, H.-P. Klenk and R. Borriss, Front. Microbiol., 2017, 8, 1–15 Search PubMed.
- H. A. Hong, R. Khaneja, N. M. K. Tam, A. Cazzato, S. Tan, M. Urdaci, A. Brisson, A. Gasbarrini, I. Barnes and S. M. Cutting, Res. Microbiol., 2009, 160, 134–143 CrossRef CAS PubMed.
- R. M. Martinez, in Brenner's Encyclopedia of Genetics, ed. S. Maloy and K. Hughes, Academic Press, San Diego, 2nd edn, 2013, pp. 246–248 Search PubMed.
- A. M. Earl, R. Losick and R. Kolter, Trends Microbiol., 2008, 16, 269–275 CrossRef CAS PubMed.
- F. Kaspar, P. Neubauer and M. Gimpel, J. Nat. Prod., 2019, 82, 2038–2053 CrossRef CAS PubMed.
- S. Caulier, C. Nannan, A. Gillis, F. Licciardi, C. Bragard and J. Mahillon, Front. Microbiol., 2019, 10, 302 CrossRef PubMed.
- T. Stein, Mol. Microbiol., 2005, 56, 845–857 CrossRef CAS PubMed.
- K. Y. Hara, M. Araki, N. Okai, S. Wakai, T. Hasunuma and A. Kondo, Microb. Cell Factories, 2014, 13, 173 CrossRef PubMed.
- E. B. Drejer, S. Hakvåg, M. Irla and T. Brautaset, Microorganisms, 2018, 6, 42 CrossRef PubMed.
- J. Liu, X. Wang, G. Dai, Y. Zhang and X. Bian, Biotechnol. Adv., 2022, 59, 107966 CrossRef CAS PubMed.
- Y. Su, C. Liu, H. Fang and D. Zhang, Microb. Cell Factories, 2020, 19, 173 CrossRef PubMed.
- J. Stülke, A. Grüppen, M. Bramkamp and S. Pelzer, J. Bacteriol., 2023, 205, e00102 CrossRef PubMed.
- H. Pramastya, Y. Song, E. Y. Elfahmi, S. Sukrasno and W. J. Quax, J. Appl. Microbiol., 2021, 130, 1839–1856 CrossRef CAS PubMed.
- P. H. Brito, B. Chevreux, C. R. Serra, G. Schyns, A. O. Henriques and J. B. Pereira-Leal, Genome Biol. Evol., 2018, 10, 108–124 CrossRef CAS PubMed.
- T. Miyazawa, C. Abe, M. Bhaswant, R. Ikeda, O. Higuchi and T. Miyazawa, Food Bioeng., 2022, 1, 241–251 CrossRef.
- T. Pedreira, C. Elfmann and J. Stülke, Nucleic Acids Res., 2022, 50, D875–D882 CrossRef CAS PubMed.
- S. K. Choi, S. Y. Park, R. Kim, S. B. Kim, C. H. Lee, J. F. Kim and S. H. Park, J. Bacteriol., 2009, 191, 3350–3358 CrossRef CAS PubMed.
- J. Kumpfmüller, K. Methling, L. Fang, B. A. Pfeifer, M. Lalk and T. Schweder, Appl. Microbiol. Biotechnol., 2016, 100, 1209–1220 CrossRef PubMed.
- Q. Liu, Q. Shen, X. Bian, H. Chen, J. Fu, H. Wang, P. Lei, Z. Guo, W. Chen, D. Li and Y. Zhang, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- Q. Shen, H. Zhou, G. Dai, G. Zhong, L. Huo, A. Li, Y. Liu, M. Yang, V. Ravichandran, Z. Zheng, Y.-J. Tang, N. Jiao, Y. Zhang and X. Bian, ACS Catal., 2022, 12, 3371–3381 CrossRef CAS.
- J. J. Hug, D. Krug and R. Müller, Nat. Rev. Chem, 2020, 4, 172–193 CrossRef CAS PubMed.
- M. Myronovskyi and A. Luzhetskyy, Nat. Prod. Rep., 2019, 36, 1281–1294 RSC.
- B. Baral, A. Akhgari and M. Metsä-Ketelä, Synth. Syst. Biotechnol., 2018, 3, 163–178 CrossRef PubMed.
- E. A. Barka, P. Vatsa, L. Sanchez, N. Gaveau-Vaillant, C. Jacquard, H.-P. Klenk, C. Clément, Y. Ouhdouch and G. P. van Wezel, Microbiol. Mol. Biol. Rev., 2016, 80, 1–43 CrossRef PubMed.
- P. Yagüe, J. Willemse, R. I. Koning, B. Rioseras, M. T. López-García, N. Gonzalez-Quiñonez, C. Lopez-Iglesias, P. V. Shliaha, A. Rogowska-Wrzesinska, A. J. Koster, O. N. Jensen, G. P. van Wezel and Á. Manteca, Nat. Commun., 2016, 7, 12467 CrossRef PubMed.
- C. M. Kao, L. Katz and C. Khosla, Science, 1994, 265, 509–512 CrossRef CAS PubMed.
- V. Molle, M. Fujita, S. T. Jensen, P. Eichenberger, J. E. González-Pastor, J. S. Liu and R. Losick, Mol. Microbiol., 2003, 50, 1683–1701 CrossRef CAS PubMed.
- P. Setlow, J. Appl. Microbiol., 2006, 101, 514–525 CrossRef CAS PubMed.
- J. Errington, Nat. Rev. Microbiol., 2003, 1, 117–126 CrossRef CAS PubMed.
- P. Klausmann, K. Hennemann, M. Hoffmann, C. Treinen, M. Aschern, L. Lilge, K. Morabbi Heravi, M. Henkel and R. Hausmann, Appl. Microbiol. Biotechnol., 2021, 105, 4141–4151 CrossRef CAS PubMed.
- S. Arnaouteli, N. C. Bamford, N. R. Stanley-Wall and Á. T. Kovács, Nat. Rev. Microbiol., 2021, 19, 600–614 CrossRef CAS PubMed.
- A. A. Schoenborn, S. M. Yannarell, E. D. Wallace, H. Clapper, I. C. Weinstein and E. A. Shank, J. Bacteriol., 2021, 203, 003377 CrossRef PubMed.
- M. S. Rahman, T. Ano and M. Shoda, J. Biotechnol., 2007, 127, 503–507 CrossRef CAS PubMed.
- D. B. Kearns and R. Losick, Mol. Microbiol., 2004, 49, 581–590 CrossRef PubMed.
- D. B. Kearns, F. Chu, R. Rudner and R. Losick, Mol. Microbiol., 2004, 52, 357–369 CrossRef CAS PubMed.
- P. F. Popp, M. Dotzler, J. Radeck, J. Bartels and T. Mascher, Sci. Rep., 2017, 7, 15058 CrossRef PubMed.
- J. Radeck, K. Kraft, J. Bartels, T. Cikovic, F. Dürr, J. Emenegger, S. Kelterborn, C. Sauer, G. Fritz, S. Gebhard and T. Mascher, J. Biol. Eng., 2013, 7, 29 CrossRef PubMed.
- A. W. Westbrook, M. Moo-young and C. P. Chou, Appl. Environ. Microbiol., 2016, 82, 4876–4895 CrossRef CAS PubMed.
- M. Wenzel and J. Altenbuchner, Microbiology, 2015, 161, 1942–1949 CrossRef CAS PubMed.
- C. Fabret, S. Dusko Ehrlich and P. Noirot, Mol. Microbiol., 2002, 46, 25–36 CrossRef CAS PubMed.
- J. E. Patrick and D. B. Kearns, Mol. Microbiol., 2008, 70, 1166–1179 CrossRef CAS PubMed.
- C.-H. Ji, J.-P. Kim and H.-S. Kang, ACS Synth. Biol., 2018, 7, 1946–1955 CrossRef CAS PubMed.
- R. E. Cobb, Y. Wang and H. Zhao, ACS Synth. Biol., 2015, 4, 723–728 CrossRef CAS PubMed.
- M. M. Zhang, F. T. Wong, Y. Wang, S. Luo, Y. H. Lim, E. Heng, W. L. Yeo, R. E. Cobb, B. Enghiad, E. L. Ang and H. Zhao, Nat. Chem. Biol., 2017, 13, 607–609 CrossRef CAS PubMed.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS PubMed.
- J. Spizizen, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 1072–1078 CrossRef CAS PubMed.
- H. Zhang, B. A. Boghigian, J. Armando and B. A. Pfeifer, Nat. Prod. Rep., 2011, 28, 125–151 RSC.
- S. Bron, S. Holsappel, G. Venema and B. P. H. Peeters, J. Biol. Eng., 1991, 226–226, 88–96 Search PubMed.
- Y. Li, Z. Li, K. Yamanaka, Y. Xu, W. Zhang, H. Vlamakis, R. Kolter, B. S. Moore and P. Y. Qian, Sci. Rep., 2015, 5, 9383 CrossRef CAS PubMed.
- D. Wicke, J. Meißner, R. Warneke, C. Elfmann and J. Stülke, Mol. Microbiol., 2023, 120, 8–19 CrossRef CAS PubMed.
- D. R. Reuß, F. M. Commichau, J. Gundlach, B. Zhu and J. Stülke, Microbiol. Mol. Biol. Rev., 2016, 80, 955–987 CrossRef PubMed.
- S. Michalik, A. Reder, B. Richts, P. Faßhauer, U. Mäder, T. Pedreira, A. Poehlein, A. J. van Heel, A. Y. van Tilburg, J. Altenbuchner, A. Klewing, D. R. Reuß, R. Daniel, F. M. Commichau, O. P. Kuipers, L. W. Hamoen, U. Völker and J. Stülke, ACS Synth. Biol., 2021, 10, 2767–2771 CrossRef CAS PubMed.
- A. Y. van Tilburg, A. J. van Heel, J. Stülke, N. A. W. de Kok, A.-S. Rueff and O. P. Kuipers, ACS Synth. Biol., 2020, 9, 1833–1842 CrossRef CAS PubMed.
- C. P. Zschiedrich, R. H. Michna, F. M. Commichau and D. To, Nucleic Acids Res., 2014, 42, 692–698 CrossRef PubMed.
- B. A. Pfeifer and C. Khosla, Microbiol. Mol. Biol. Rev., 2001, 65, 106–118 CrossRef CAS PubMed.
- J. T. Kealey, L. Liu, D. V. Santi, M. C. Betlach and P. J. Barr, Proc. Natl. Acad. Sci. U.S.A., 1998, 95, 505–509 CrossRef CAS PubMed.
- W. Zha, S. B. Rubin-Pitel, Z. Shao and H. Zhao, Metab. Eng., 2009, 11, 192–198 CrossRef CAS PubMed.
- S. Murli, J. Kennedy, L. C. Dayem, J. R. Carney and J. T. Kealey, J. Ind. Microbiol. Biotechnol., 2003, 30, 500–509 CrossRef CAS PubMed.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–429 CrossRef CAS PubMed.
- Y. Tang, S. Frewert, K. Harmrolfs, J. Herrmann, L. Karmann, U. Kazmaier, L. Xia, Y. Zhang and R. Müller, J. Biotechnol., 2015, 194, 112–114 CrossRef CAS PubMed.
- K. Watanabe, M. A. Rude, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 9774–9778 CrossRef CAS PubMed.
- S. C. Mutka, J. R. Carney, Y. Liu and J. Kennedy, Biochemistry, 2006, 45, 1321–1330 CrossRef CAS PubMed.
- S. Doekel, K. Eppelmann and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 216, 185–191 CrossRef CAS PubMed.
- K. Gustafson, M. Roman and W. Fenical, J. Am. Chem. Soc., 1989, 111, 7519–7524 CrossRef CAS.
- W. Li, X. X. Tang, X. Yan, Z. Wu, Z. W. Yi, M. J. Fang, X. Su and Y. K. Qiu, Nat. Prod. Res., 2016, 30, 2777–2782 CrossRef CAS PubMed.
- P. S. Patel, S. Fisher, D. Pirnik, C. Aklonis and E. Meyers, J. Antibiot., 1995, 48, 997–1003 CrossRef CAS PubMed.
- H. Chen, L. Wang, C. X. Su, G. H. Gong, P. Wang and Z. L. Yu, Lett. Appl. Microbiol., 2008, 47, 180–186 CrossRef CAS PubMed.
- S. Zobel, J. Kumpfmüller, R. D. Süssmuth and T. Schweder, Appl. Microbiol. Biotechnol., 2015, 99, 681–691 CrossRef CAS PubMed.
- S.-Y. Park, S.-K. Choi, J. Kim, T.-K. Oh and S.-H. Park, Appl. Environ. Microbiol., 2012, 78, 4194–4199 CrossRef CAS PubMed.
- D. R. Zeigler, Z. Prágai, S. Rodriguez, B. Chevreux, A. Muffler, T. Albert, R. Bai, M. Wyss and J. B. Perkins, J. Bacteriol., 2008, 190, 6983–6995 CrossRef CAS PubMed.
- J. P. Gomez-Escribano and M. J. Bibb, Microb. Biotechnol., 2011, 4, 207–215 CrossRef CAS PubMed.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS PubMed.
- Z. Qian, T. Bruhn, P. M. D'Agostino, A. Herrmann, M. Haslbeck, N. Antal, H.-P. Fiedler, R. Brack-Werner and T. A. M. Gulder, J. Org. Chem., 2020, 85, 664–673 CrossRef CAS PubMed.
- M. Geissler, I. Kühle, K. M. Heravi, J. Altenbuchner, M. Henkel and R. Hausmann, AMB Express, 2019, 9, 84 CrossRef PubMed.
- O. Naotake, Gene, 1985, 40, 145–150 CrossRef PubMed.
- D. C. Shields and P. M. Sharp, Nucleic Acids Res., 1987, 15, 8040 CrossRef PubMed.
- H. Tjalsma, H. Antelmann, J. D. H. Jongbloed, P. G. Braun, E. Darmon, R. Dorenbos, J. F. Dubois, H. Westers, G. Zanen, W. J. Quax, O. P. Kuipers, S. Bron, M. Hecker and J. M. V. Dijl, Microbiol. Mol. Biol. Rev., 2004, 68, 207–233 CrossRef CAS PubMed.
- J. M. van Dijl and M. Hecker, Microb. Cell Factories, 2013, 12, 1–6 CrossRef PubMed.
- S. Yan and G. Wu, Microb. Cell Fact., 2017, 16, 124 CrossRef PubMed.
- J. L. Revuelta, R. Ledesma-Amaro, P. Lozano-Martinez, D. Díaz-Fernández, R. M. Buey and A. Jiménez, J. Ind. Microbiol. Biotechnol., 2017, 44, 659–665 CrossRef CAS PubMed.
- X. Chen, L. Zhou, K. Tian, A. Kumar, S. Singh, B. A. Prior and Z. Wang, Biotechnol. Adv., 2013, 31, 1200–1223 CrossRef CAS PubMed.
- S. Yin, Z. Li, X. Wang, H. Wang, X. Jia, G. Ai, Z. Bai, M. Shi, F. Yuan, T. Liu, W. Wang and K. Yang, Appl. Microbiol. Biotechnol., 2016, 100, 10563–10572 CrossRef CAS PubMed.
- L. Liu, Y. Liu, H. Shin, R. R. Chen, N. S. Wang, J. Li, G. Du and J. Chen, Appl. Microbiol. Biotechnol., 2013, 97, 6113–6127 CrossRef CAS PubMed.
- M. Schallmey, A. Singh and O. P. Ward, Can. J. Microbiol., 2004, 50, 1–17 CrossRef CAS PubMed.
- F. Berini, F. Marinelli and E. Binda, Front. Microbiol., 2020, 11, 1958 CrossRef PubMed.
- U. Mamat, K. Wilke, D. Bramhill, A. B. Schromm, B. Lindner, T. A. Kohl, J. L. Corchero, A. Villaverde, L. Schaffer, S. R. Head, C. Souvignier, T. C. Meredith and R. W. Woodard, Microb. Cell Fact., 2015, 14, 57 CrossRef PubMed.
- G. Zhang, M. Lin, M. Qin, Q. Xie, M. Liang, J. Jiang, H. Dai, S. Xu, S. Feng and M. Liao, J. Agric. Food Chem., 2023, 71, 5600–5613 CrossRef CAS PubMed.
- Á. T. Kovács, Trends Microbiol., 2019, 27, 724–725 CrossRef PubMed.
- J. Errington and L. T. van der Aart, Microbiology, 2020, 166, 425–427 CrossRef CAS PubMed.
- E. P. Riley, C. Schwarz, A. I. Derman and J. Lopez-Garrido, Microb. Cell, 2021, 8, 1–16 CrossRef CAS PubMed.
- X. H. Chen, A. Koumoutsi, R. Scholz, A. Eisenreich, K. Schneider, I. Heinemeyer, B. Morgenstern, B. Voss, W. R. Hess, O. Reva, H. Junge, B. Voigt, P. R. Jungblut, J. Vater, R. Süssmuth, H. Liesegang, A. Strittmatter, G. Gottschalk and R. Borriss, Nat. Biotechnol., 2007, 25, 1007–1014 CrossRef CAS PubMed.
- B. Fan, C. Wang, X. Ding, B. Zhu, X. Song and R. Borriss, Database, 2019, 2019, baz071 Search PubMed.
- L. Wu, H. Wu, L. Chen, L. Lin, R. Borriss and X. Gao, Appl. Microbiol. Biotechnol., 2015, 99, 4255–4263 CrossRef CAS PubMed.
- Y. Qiu, Y. Zhu, Y. Sha, P. Lei, Z. Luo, X. Feng, S. Li and H. Xu, ACS Sustainable Chem. Eng., 2020, 8, 9763–9774 CrossRef CAS.
- Y. Gu, J. Zheng, J. Feng, M. Cao, W. Gao, Y. Quan, Y. Dang, Y. Wang, S. Wang and C. Song, Appl. Microbiol. Biotechnol., 2017, 101, 4163–4174 CrossRef CAS PubMed.
- Y. Liao, L. Huang, B. Wang, F. Zhou and L. Pan, Gene, 2015, 571, 252–262 CrossRef CAS PubMed.
- Z. Luo, Y. Yan, S. Du, Y. Zhu, F. Pan, R. Wang, Z. Xu, X. Xu, S. Li and H. Xu, Crit. Rev. Biotechnol., 2022, 43, 1073–1091 CrossRef PubMed.
- C. Luo, Y. Chen, X. Liu, X. Wang, X. Wang, X. Li, Y. Zhao and L. Wei, Appl. Microbiol. Biotechnol., 2019, 103, 4467–4481 CrossRef CAS PubMed.
- B. Fan, C. Wang, X. Song, X. Ding, L. Wu, H. Wu, X. Gao and R. Borriss, Front. Microbiol., 2018, 9, 2491 CrossRef PubMed.
- M. S. Ngalimat, R. S. R. Yahaya, M. M. A. Baharudin, S. M. Yaminudin, M. Karim, S. A. Ahmad and S. Sabri, Microorganisms, 2021, 9, 614 CrossRef CAS PubMed.
- A. M. Herzner, J. Dischinger, C. Szekat, M. Josten, S. Schmitz, A. Yakéléba, R. Reinartz, A. Janse, H.-G. Sahl, J. Piel and G. Bierbaum, PLoS Genet., 2011, 6, e22389 CrossRef CAS PubMed.
- F. Zhang, K. Huo, X. Song, Y. Quan, S. Wang, Z. Zhang, W. Gao and C. Yang, Microb. Cell Fact., 2020, 19, 223 CrossRef CAS PubMed.
- Ikram-ul-Haq, H. Ashraf, J. Iqbal and M. A. Qadeer, Bioresour. Technol., 2003, 87, 57–61 CrossRef CAS PubMed.
- J. Zhu, S. Wang, C. Wang, Z. Wang, G. Luo, J. Li, Y. Zhan, D. Cai and S. Chen, Synth. Syst. Biotechnol., 2023, 8, 314–322 CrossRef CAS PubMed.
- M. M. Yakimov, K. N. Timmis, V. Wray and H. L. Fredrickson, Appl. Environ. Microbiol., 1995, 61, 1706–1713 CrossRef CAS PubMed.
- M. M. Yakimov, M. M. Amro, M. Bock, K. Boseker, H. L. Fredrickson, D. G. Kessel and K. N. Timmis, J. Pet. Sci. Eng., 1997, 18, 147–160 CrossRef CAS.
- S. Joshi, S. Yadav and A. J. Desai, Biochem. Eng. J., 2008, 41, 122–127 CrossRef CAS.
- I. A. Purwasena, D. I. Astuti, M. Syukron, M. Amaniyah and Y. Sugai, J. Pet. Sci. Eng., 2019, 183, 106383 CrossRef CAS.
- B. Veith, C. Herzberg, S. Steckel, J. Feesche, K. H. Maurer, P. Ehrenreich, S. Bäumer, A. Henne, H. Liesegang, R. Merkl, A. Ehrenreich and G. Gottschalk, Microb. Physiol., 2004, 7, 204–211 CrossRef CAS PubMed.
- E. J. Gudiña and J. A. Teixeira, Biotechnol. Adv., 2022, 60, 108013 CrossRef PubMed.
- G. L. Challis and J. H. Naismith, Curr. Opin. Struct. Biol., 2004, 14, 748–756 CrossRef CAS PubMed.
- M. Strieker and A. Tanovic, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468–3496 CrossRef CAS PubMed.
- C. T. Walsh, Science, 2004, 303, 1805–1810 CrossRef CAS PubMed.
- K. J. Weissman and R. Müller, ChemBioChem, 2008, 9, 826–848 CrossRef CAS PubMed.
- V. A. Mironov, O. V. Sergienko, I. N. Nastasyak and V. N. Danilenko, Appl. Biochem. Microbiol., 2004, 40, 531–541 CrossRef CAS.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1793 CrossRef CAS PubMed.
- B. Pfeifer, Z. Hu, P. Licari and C. Khosla, Appl. Environ. Microbiol., 2002, 68, 3287–3292 CrossRef CAS PubMed.
- Y. Sugimoto, F. R. Camacho, S. Wang, P. Chankhamjon, A. Odabas, A. Biswas, P. D. Jeffrey and M. S. Donia, Science, 2019, 366, eaax9176 CrossRef CAS PubMed.
- K. Eppelmann, S. Doekel and M. A. Marahiel, J. Biol. Chem., 2001, 276, 34824–34831 CrossRef CAS PubMed.
- C.-J. Guo, F.-Y. Chang, T. P. Wyche, K. M. Backus, T. M. Acker, M. Funabashi, M. Taketani, M. S. Donia, S. Nayfach, K. S. Pollard, C. S. Craik, B. F. Cravatt, J. Clardy, C. A. Voigt and M. A. Fischbach, Cell, 2017, 168, 517–526 CrossRef CAS PubMed.
- Y. Hu, F. Nan, S. W. Maina, J. Guo, S. Wu and Z. Xin, J. Biotechnol., 2018, 288, 1–8 CrossRef CAS PubMed.
- K. Tsuge, S. Inoue, T. Ano, M. Itaya and M. Shoda, Antimicrob. Agents Chemother., 2005, 49, 4641–4648 CrossRef CAS PubMed.
- N. Madry, R. Zocher and H. Kleinkauf, Eur. J. Appl. Microbiol. Biotechnol., 1983, 17, 75–79 CrossRef CAS.
- Y. Zhang, M. Chen, S. D. Bruner and Y. Ding, Front. Microbiol., 2018, 9, 1801 CrossRef PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- S.-Y. Kim, S.-Y. Park, S.-K. Choi and S.-H. Park, J. Microbiol. Biotechnol., 2015, 25, 1015–1025 CrossRef CAS PubMed.
- P. R. Bennallack, K. D. Bewley, M. A. Burlingame, R. A. Robison, S. M. Miller and J. S. Griffitts, J. Bacteriol., 2016, 198, 2431–2438 CrossRef CAS PubMed.
- S. Yuksel and J. N. Hansen, Appl. Microbiol. Biotechnol., 2007, 74, 640–649 CrossRef CAS.
- M. E. Hansen, R. Wangari, E. B. Hansen, I. Mijakovic and P. R. Jensen, Appl. Environ. Microbiol., 2009, 75, 6688–6695 CrossRef CAS.
- H. Rintala, T. Graeffe, L. Paulin, N. Kalkkinen and P. E. J. Saris, Biotechnol. Lett., 1993, 15, 991–996 CrossRef CAS.
- W. Liu and J. N. Hansen, J. Bacteriol., 1991, 173, 7387–7390 CrossRef CAS PubMed.
- Q. Zhang, C. M. Kobras, S. Gebhard, T. Mascher and D. Wolf, Microb. Cell Factories, 2022, 21, 57 CrossRef CAS PubMed.
- D. Xue, E. A. Older, Z. Zhong, Z. Shang, N. Chen, N. Dittenhauser, L. Hou, P. Cai, M. D. Walla, S.-H. Dong, X. Tang, H. Chen, P. Nagarkatti, M. Nagarkatti, Y.-X. Li and J. Li, Nat. Commun., 2022, 13, 1647 CrossRef CAS PubMed.
- D. Xue, Z. Shang, E. A. Older, Z. Zhong, C. Pulliam, K. Peter, M. Nagarkatti, P. Nagarkatti, Y.-X. Li and J. Li, ACS Chem. Biol., 2023, 18, 508–517 CrossRef CAS PubMed.
- A. Grigoreva, J. Andreeva, D. Bikmetov, A. Rusanova, M. Serebryakova, A. H. Garcia, D. Slonova, S. K. Nair, G. Lippens, K. Severinov and S. Dubiley, iScience, 2021, 24, 102480 CrossRef CAS PubMed.
- Z.-F. Pei, M.-J. Yang, K. Zhang, X.-H. Jian and G.-L. Tang, Cell Chem. Biol., 2022, 29, 650–659 CrossRef CAS PubMed.
- K. Yoshida, S. Ueda and I. Maeda, Biotechnol. Lett., 2009, 31, 1789–1793 CrossRef CAS PubMed.
- D. Xue, I. I. Abdallah, I. E. M. de Haan, M. J. J. B. Sibbald and W. J. Quax, Appl. Microbiol. Biotechnol., 2015, 99, 5907–5915 CrossRef CAS PubMed.
- I. I. Abdallah, D. Xue, H. Pramastya, R. van Merkerk, R. Setroikromo and W. J. Quax, J. Ind. Microbiol. Biotechnol., 2020, 47, 243–249 CrossRef CAS PubMed.
- J. Xue and B. K. Ahring, Appl. Environ. Microbiol., 2011, 77, 2399–2405 CrossRef CAS PubMed.
- K. Zhou, R. Zou, C. Zhang, G. Stephanopoulos and H.-P. Too, Biotechnol. Bioeng., 2013, 110, 2556–2561 CrossRef CAS.
- Y. Song, S. He, I. I. Abdallah, A. Jopkiewicz, R. Setroikromo, R. van Merkerk, P. G. Tepper and W. J. Quax, J. Agric. Food Chem., 2021, 69, 4785–4794 CrossRef CAS PubMed.
- I. I. Abdallah, H. Pramastya, R. van Merkerk, Sukrasno and W. J. Quax, Front. Microbiol., 2019, 10, 218 CrossRef PubMed.
- S. Yang, Y. Cao, L. Sun, C. Li, X. Lin, Z. Cai, G. Zhang and H. Song, ACS Synth. Biol., 2019, 8, 70–81 CrossRef CAS PubMed.
- S. Cui, X. Lv, Y. Wu, J. Li, G. Du, R. Ledesma-Amaro and L. Liu, ACS Synth. Biol., 2019, 8, 1826–1837 CrossRef CAS PubMed.
- G. Wang, Z. Guo, X. Zhang, H. Wu, X. Bai, H. Zhang, R. Hu, S. Han, Y. Pang, Z. Gao, L. Yan, C. Huang, L. Zhang, C. Pan and X. Liu, Microb. Cell Factories, 2022, 21, 104 CrossRef CAS PubMed.
- S. A. Borisova, B. T. Circello, J. K. Zhang, W. A. V. D. Donk and W. W. Metcalf, Chem. Biol., 2010, 17, 28–37 CrossRef CAS.
- A.-C. Letzel, S. J. Pidot and C. Hertweck, BMC Genomics, 2014, 15, 983 CrossRef PubMed.
- N. Poorinmohammad, R. Bagheban-Shemirani and J. Hamedi, Antonie van Leeuwenhoek, 2019, 112, 1477–1499 CrossRef CAS PubMed.
- G. A. Hudson and D. A. Mitchell, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS PubMed.
- M. Montalbán-López, T. A. Scott, S. Ramesh, I. R. Rahman, A. J. van Heel, J. H. Viel, V. Bandarian, E. Dittmann, O. Genilloud, Y. Goto, M. J. G. Burgos, C. Hill, S. Kim, J. Koehnke, J. A. Latham, A. James Link, B. Martínez, S. K. Nair, Y. Nicolet, S. Rebuffat, H.-G. Sahl, D. Sareen, E. W. Schmidt, L. Schmitt, K. Severinov, R. D. Süssmuth, A. W. Truman, H. Wang, J.-K. Weng, G. P. van Wezel, Q. Zhang, J. Zhong, J. Piel, D. A. Mitchell, O. P. Kuipers and W. A. van der Donk, Nat. Prod. Rep., 2021, 38, 130–239 RSC.
- B. M. Lange, T. Rujan, W. Martin and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13172–13177 CrossRef CAS PubMed.
- E. I. Wilding, J. R. Brown, A. P. Bryant, A. F. Chalker, D. J. Holmes, K. A. Ingraham, S. Iordanescu, C. Y. So, M. Rosenberg and M. N. Gwynn, J. Bacteriol., 2000, 182, 4319–4327 CrossRef CAS PubMed.
- T. Kuzuyama and H. Seto, Nat. Prod. Rep., 2003, 20, 171–183 RSC.
- M. Hedl, A. Sutherlin, E. I. Wilding, M. Mazzulla, D. McDevitt, P. Lane, J. W. Burgner, K. R. Lehnbeuter, C. V. Stauffacher, M. N. Gwynn and V. W. Rodwell, J. Bacteriol., 2002, 184, 2116–2122 CrossRef CAS PubMed.
- N. Campobasso, M. Patel, I. E. Wilding, H. Kallender, M. Rosenberg and M. N. Gwynn, J. Biol. Chem., 2004, 279, 44883–44888 CrossRef CAS PubMed.
- J. Lombard and D. Moreira, Mol. Biol. Evol., 2011, 28, 87–99 CrossRef CAS PubMed.
- S. Heuston, M. Begley, M. S. Davey, M. Eberl, P. G. Casey, C. Hill and C. G. M. Gahan, Microbiology, 2012, 158, 1684–1693 CrossRef CAS PubMed.
- A. Banerjee, Y. Wu, R. Banerjee, Y. Li, H. Yan and T. D. Sharkey, J. Biol. Chem., 2013, 288, 16926–16936 CrossRef CAS PubMed.
- P. K. Ajikumar, W.-H. Xiao, K. E. J. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS PubMed.
- E. Mahdinia, A. Demirci and A. Berenjian, Curr. Pharm. Biotechnol., 2018, 19, 917–924 CAS.
- M. Lynch and G. K. Marinov, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 15690–15695 CrossRef CAS PubMed.
- J. E. Dueber, G. C. Wu, G. R. Malmirchegini, T. S. Moon, C. J. Petzold, A. V. Ullal, K. L. J. Prather and J. D. Keasling, Nat. Biotechnol., 2009, 27, 753–759 CrossRef CAS PubMed.
- P. J. Bakkes, S. Biemann, A. Bokel, M. Eickholt, M. Girhard and V. B. Urlacher, Sci. Rep., 2015, 5, 12158 CrossRef CAS PubMed.
- B. M. Hess, J. Xue, L. M. Markillie, R. C. Taylor, H. S. Wiley, B. K. Ahring and B. Linggi, PLoS One, 2013, 8, e66104 CrossRef CAS PubMed.
- Z. Guan, D. Xue, I. I. Abdallah, L. Dijkshoorn, R. Setroikromo, L. Guiyuan and W. J. Quax, Appl. Microbiol. Biotechnol., 2015, 99, 395–9406 Search PubMed.
- J. Zhao, Q. Li, T. Sun, X. Zhu, H. Xu, J. Tang, X. Zhang and Y. Ma, Metab. Eng., 2013, 17, 42–50 CrossRef CAS PubMed.
- D. Drider, G. Fimland, Y. Héchard, L. M. McMullen and H. Prévost, Microbiol. Mol. Biol. Rev., 2006, 70, 564–582 CrossRef CAS PubMed.
- J. J. Quereda, A. Morón-García, C. Palacios-Gorba, C. Dessaux, F. García-Del Portillo, M. G. Pucciarelli and A. D. Ortega, Virulence, 2021, 12, 2509–2545 CrossRef CAS PubMed.
- K. W. Miller, R. Schamber, Y. Chen and B. Ray, Appl. Environ. Microbiol., 1998, 64, 14–20 CrossRef CAS PubMed.
- B. Mesa-Pereira, P. M. O'Connor, M. C. Rea, P. D. Cotter, C. Hill and R. P. Ross, Sci. Rep., 2017, 7, 3069 CrossRef PubMed.
- N. Horn, M. I. Martínez, J. M. Martínez, P. E. Hernández, M. J. Gasson, J. M. Rodríguez and H. M. Dodd, Appl. Environ. Microbiol., 1999, 65, 4443–4450 CrossRef CAS PubMed.
- J. L. Arqués, J. M. Rodríguez, M. J. Gasson and N. Horn, J. Dairy Sci., 2008, 91, 2591–2594 CrossRef PubMed.
- O. Goldbeck, D. N. Desef, K. V. Ovchinnikov, F. Perez-Garcia, J. Christmann, P. Sinner, P. Crauwels, D. Weixler, P. Cao, J. Becker, M. Kohlstedt, J. Kager, B. J. Eikmanns, G. M. Seibold, C. Herwig, C. Wittmann, N. S. Bar, D. B. Diep and C. U. Riedel, Metab. Eng., 2021, 68, 34–45 CrossRef CAS PubMed.
- A. Lakowitz, R. Krull and R. Biedendieck, Microb. Cell Factories, 2017, 16, 14 CrossRef PubMed.
- T. Hioki, D. Yamashita, M. Tohata, K. Endo, A. Kawahara and M. Okuda, Microb. Cell Factories, 2021, 20, 231 CrossRef CAS PubMed.
- J. Heinrich, C. Drewniok, E. Neugebauer, H. Kellner and T. Wiegert, Microb. Cell Factories, 2019, 18, 31 CrossRef PubMed.
- A. Allende, B. Martínez, V. Selma, M. I. Gil, J. E. Suárez and A. Rodríguez, Food Microbiol., 2007, 24, 759–766 CrossRef CAS PubMed.
- J. M. Rodríguez, M. I. Martínez and J. Kok, Crit. Rev. Food Sci. Nutr., 2002, 42, 91–121 CrossRef PubMed.
- C. A. van Reenen, L. M. T. Dicks and M. L. Chikindas, J. Appl. Microbiol., 1998, 84, 1131–1137 CrossRef CAS PubMed.
- H. D. Michener and N. Snell, Arch. Biochem., 1949, 22, 208–214 CAS.
- M. Kugler, W. Loeffler, C. Rapp, A. Kern and G. Jung, Arch. Microbiol., 1990, 153, 276–281 CrossRef CAS PubMed.
- B. Laber, S. D. Lindell and H. D. Pohlenz, Arch. Microbiol., 1994, 161, 400–403 CAS.
- H. Diddens, M. Dorgerloh and H. Zähner, J. Antibiot., 1979, 32, 87–90 CrossRef CAS PubMed.
- B. K. Park, A. Hirota and H. Sakai, Agric. Biol. Chem., 1977, 41, 161–167 CAS.
- B. K. Park, A. Hirota and H. Sakai, Agric. Biol. Chem., 1977, 41, 573–579 CAS.
- H. Ohshima, S. Matsuoka, K. Asai and Y. Sadaie, J. Bacteriol., 2002, 184, 381–389 CrossRef CAS PubMed.
- K. Turgay, J. Hahn, J. Burghoorn and D. Dubnau, EMBO J., 1998, 17, 6730–6738 CrossRef CAS PubMed.
- R. Rahmer, K. M. Heravi and J. Altenbuchner, Front. Microbiol., 2015, 6, 1–11 Search PubMed.
- J. Kabisch, A. Thürmer, T. Hübel, L. Popper, R. Daniel and T. Schweder, J. Biotechnol., 2013, 163, 97–104 CrossRef CAS PubMed.
- M. Wang, H. Yu, X. Li and Z. Shen, Metab. Eng., 2020, 62, 235–248 CrossRef CAS PubMed.
- P. Klausmann, L. Lilge, M. Aschern, K. Hennemann, M. Henkel, R. Hausmann and K. Morabbi Heravi, Microb. Cell Fact., 2021, 20, 188 CrossRef CAS PubMed.
- M. Vahidinasab, L. Lilge, A. Reinfurt, J. Pfannstiel, M. Henkel, K. Morabbi Heravi and R. Hausmann, Microb. Cell Factories, 2020, 19, 1–12 CrossRef PubMed.
- L. Lilge, M. Vahidinasab, I. Adiek, P. Becker, C. Kuppusamy Nesamani, C. Treinen, M. Hoffmann, K. Morabbi Heravi, M. Henkel and R. Hausmann, Microbiologyopen, 2021, 10, e1241 CrossRef CAS PubMed.
- Q. Wu, Y. Zhi and Y. Xu, Metab. Eng., 2019, 52, 87–97 CrossRef CAS PubMed.
- G.-R. Gao, Z.-J. Hou, M.-Z. Ding, S. Bai, S.-Y. Wei, B. Qiao, Q.-M. Xu, J.-S. Cheng and Y.-J. Yuan, ACS Synth. Biol., 2022, 11, 4065–4076 CrossRef CAS PubMed.
- R. H. Doi and F. Kawamura, J. Bacteriol., 1984, 160, 442–444 CrossRef PubMed.
- H. Nguyen, T. Phan and W. Schumann, AMB Express, 2011, 1, 22 CrossRef PubMed.
- M. Perego, G. B. Spiegelman and J. A. Hoch, Mol. Microbiol., 1988, 2, 689–699 CrossRef CAS PubMed.
- D. R. Reuß, J. Schuldes, R. Daniel and J. Altenbuchner, Genome Announc., 2015, 3, 000844 Search PubMed.
- U. Mäder, H. Antelmann, T. Buder, M. Dahl, M. Hecker and G. Homuth, Mol. Genet. Genomics, 2002, 268, 455–467 CrossRef PubMed.
- K. Tsuge, T. Ano, M. Hirai, Y. Nakamura and M. Shoda, Antimicrob. Agents Chemother., 1999, 43, 2183–2192 CrossRef CAS PubMed.
- K. Tsuge, K. Matsui and M. Itaya, J. Biotechnol., 2007, 129, 592–603 CrossRef CAS PubMed.
- N. R. Stanley and B. A. Lazazzera, Mol. Microbiol., 2005, 57, 1143–1158 CrossRef CAS PubMed.
- M. M. Nakano, M. A. Marahiel and P. Zuber, J. Bacteriol., 1988, 170, 5662–5668 CrossRef CAS PubMed.
- D. Dhali, F. Coutte, A. A. Arias, S. Auger, V. Bidnenko, G. Chataigné, M. Lalk, J. Niehren, J. de Sousa, C. Versari and P. Jacques, Biotechnol. J., 2017, 12, 1600574 CrossRef PubMed.
- Y. Jing, H. Wei, F. Liu, Y. Liu, L. Zhou, J. Liu, S. Yang, H. Zhang and B. Mu, Biotechnol. Appl. Biochem., 2023, 70, 238–248 CrossRef CAS PubMed.
- C. Wang, Y. Cao, Y. Wang, L. Sun and H. Song, Microb. Cell Fact., 2019, 18, 90 CrossRef PubMed.
- F. Kunst, N. Ogasawara, I. Moszer, A. M. Albertini, G. Alloni, V. Azevedo, M. G. Bertero, P. Bessières, A. Bolotin, S. Borchert, R. Borriss, L. Boursier, A. Brans, M. Braun, S. C. Brignell, S. Bron, S. Brouillet, C. V. Bruschi, B. Caldwell, V. Capuano, N. M. Carter, S.-K. Choi, J.-J. Codani, I. F. Connerton, N. J. Cummings, R. A. Daniel, F. Denizot, K. M. Devine, A. Düsterhöft, S. D. Ehrlich, P. T. Emmerson, K. D. Entian, J. Errington, C. Fabret, E. Ferrari, D. Foulger, C. Fritz, M. Fujita, Y. Fujita, S. Fuma, A. Galizzi, N. Galleron, S.-Y. Ghim, P. Glaser, A. Goffeau, E. J. Golightly, G. Grandi, G. Guiseppi, B. J. Guy, K. Haga, J. Haiech, C. R. Harwood, A. Hénaut, H. Hilbert, S. Holsappel, S. Hosono, M.-F. Hullo, M. Itaya, L. Jones, B. Joris, D. Karamata, Y. Kasahara, M. Klaerr-Blanchard, C. Klein, Y. Kobayashi, P. Koetter, G. Koningstein, S. Krogh, M. Kumano, K. Kurita, A. Lapidus, S. Lardinois, J. Lauber, V. Lazarevic, S.-M. Lee, A. Levine, H. Liu, S. Masuda, C. Mauël, C. Médigue, N. Medina, R. P. Mellado, M. Mizuno, D. Moestl, S. Nakai, M. Noback, D. Noone, M. O'Reilly, K. Ogawa, A. Ogiwara, B. Oudega, S.-H. Park, V. Parro, T. M. Pohl, D. Portetelle, S. Porwollik, A. M. Prescott, E. Presecan, P. Pujic, B. Purnelle, G. Rapoport, M. Rey, S. Reynolds, M. Rieger, C. Rivolta, E. Rocha, B. Roche, M. Rose, Y. Sadaie, T. Sato, E. Scanlan, S. Schleich, R. Schroeter, F. Scoffone, J. Sekiguchi, A. Sekowska, S. J. Seror, P. Serror, B.-S. Shin, B. Soldo, A. Sorokin, E. Tacconi, T. Takagi, H. Takahashi, K. Takemaru, M. Takeuchi, A. Tamakoshi, T. Tanaka, P. Terpstra, A. Tognoni, V. Tosato, S. Uchiyama, M. Vandenbol, F. Vannier, A. Vassarotti, A. Viari, R. Wambutt, E. Wedler, H. Wedler, T. Weitzenegger, P. Winters, A. Wipat, H. Yamamoto, K. Yamane, K. Yasumoto, K. Yata, K. Yoshida, H.-F. Yoshikawa, E. Zumstein, H. Yoshikawa and A. Danchin, Nature, 1997, 390, 249–256 CrossRef CAS PubMed.
- A. Müller, M. Wenzel, H. Strahl, F. Grein, T. N. V. Saaki, B. Kohl, T. Siersma, J. E. Bandow, H. G. Sahl, T. Schneider and L. W. Hamoen, Genome Res., 2016, 27, 289–299 Search PubMed.
- Y. Ahmed, Y. Rebets, M. R. Estévez, J. Zapp, M. Myronovskyi and A. Luzhetskyy, Microb. Cell Fact., 2020, 19, 5 CrossRef CAS PubMed.
- A. Thanapipatsiri, J. Claesen, J.-P. Gomez-Escribano, M. Bibb and A. Thamchaipenet, Microb. Cell Fact., 2015, 14, 145 CrossRef PubMed.
- C. C. de Souza, J. M. Guimarães, S. dos S. Pereira and L. A. M. Mariúba, Exp. Biol. Med., 2021, 246, 2443–2453 CrossRef PubMed.
- K. Tsuge, T. Akiyama and M. Shoda, J. Bacteriol., 2001, 183, 6265–6273 CrossRef CAS PubMed.
- V. Leclère, M. Béchet, A. Adam, J.-S. Guez, B. Wathelet, M. Ongena, P. Thonart, F. Gancel, M. Chollet-Imbert and P. Jacques, Appl. Environ. Microbiol., 2005, 71, 4577–4584 CrossRef PubMed.
- F. Coutte, V. Leclère, M. Béchet, J. -S. Guez, D. Lecouturier, M. Chollet-Imbert, P. Dhulster and P. Jacques, J. Appl. Microbiol., 2010, 109, 480–491 CrossRef CAS PubMed.
- J. Willenbacher, T. Mohr, M. Henkel, S. Gebhard, T. Mascher, C. Syldatk and R. Hausmann, J. Biotechnol., 2016, 224, 14–17 CrossRef CAS PubMed.
- J. Silbersack, B. Jürgen, M. Hecker, B. Schneidinger, R. Schmuck and T. Schweder, Appl. Microbiol. Biotechnol., 2006, 73, 895–903 CrossRef CAS PubMed.
- S.-L. Liu and K. Du, Protein Expression Purif., 2012, 83, 164–168 CrossRef CAS PubMed.
- R. S. Bongers, J.-W. Veening, M. Van Wieringen, O. P. Kuipers and M. Kleerebezem, Appl. Environ. Microbiol., 2005, 71, 8818–8824 CrossRef CAS PubMed.
- Y. Ming-Ming, Z. Wei-Wei, Z. Xi-Feng and C. Pei-Lin, Biotechnol. Lett., 2006, 28, 1713–1718 CrossRef PubMed.
- L. Zhang, G. Li, N. Zhan, T. Sun, B. Cheng, Y. Li and A. Shan, Process Biochem., 2019, 81, 22–27 CrossRef CAS.
- R. Promchai, B. Promdonkoy, S. Tanapongpipat, W. Visessanguan, L. Eurwilaichitr and P. Luxananil, J. Biotechnol., 2016, 222, 86–93 CrossRef CAS PubMed.
- W. Li, H.-X. Li, S.-Y. Ji, S. Li, Y.-S. Gong, M.-M. Yang and Y.-L. Chen, Biochem. Biophys. Res. Commun., 2007, 358, 1148–1153 CrossRef PubMed.
- J. Ye, Y. Li, Y. Bai, T. Zhang, W. Jiang, T. Shi, Z. Wu and Y.-H. P. J. Zhang, Bioresour. Bioprocess., 2022, 9, 56 CrossRef.
- M. Ji, S. Li, A. Chen, Y. Liu, Y. Xie, H. Duan, J. Shi and J. Sun, Enzyme Microb. Technol., 2021, 144, 109726 CrossRef CAS PubMed.
- L. Han, W. Cui, F. Suo, S. Miao, W. Hao, Q. Chen, J. Guo, Z. Liu, L. Zhou and Z. Zhou, Microb. Cell Fact., 2019, 18, 96 CrossRef PubMed.
- S. M. Castillo-Hair, M. Fujita, O. A. Igoshin and J. J. Tabor, ACS Synth. Biol., 2019, 8, 1673–1678 CrossRef CAS PubMed.
- B. Conrad, R. S. Savchenko, R. Breves and J. Hofemeister, Mol. Gen. Genet., 1996, 250, 230–236 CAS.
- P. T. Chen, J.-F. Shaw, Y.-P. Chao, T.-H. David Ho and S.-M. Yu, J. Agric. Food Chem., 2010, 58, 5392–5399 CrossRef CAS PubMed.
- A. Koreeda, R. Taguchi, K. Miyamoto, Y. Kuwahara and K. Hirooka, Biosci., Biotechnol., Biochem., 2023, 87, 1017–1028 CrossRef PubMed.
- S. Yang, G. Du, J. Chen and Z. Kang, Appl. Microbiol. Biotechnol., 2017, 101, 4151–4161 CrossRef CAS PubMed.
- A.-M. Guérout-Fleury, N. Frandsen and P. Stragier, Gene, 1996, 180, 57–61 CrossRef PubMed.
- S. Guiziou, V. Sauveplane, H.-J. Chang, C. Clerté, N. Declerck, M. Jules and J. Bonnet, Nucleic Acids Res., 2016, 44, 7495–7508 CAS.
- M. Dormeyer, R. Egelkamp, M. J. Thiele, E. Hammer, K. Gunka, L. Stannek, U. Völker and F. M. Commichau, Microbiology, 2015, 161, 354–361 CrossRef CAS PubMed.
- S. A. Kautsar, K. Blin, S. Shaw, J. C. Navarro-Muñoz, B. R. Terlouw, J. J. J. van der Hooft, J. A. van Santen, V. Tracanna, H. G. Suarez Duran, V. Pascal Andreu, N. Selem-Mojica, M. Alanjary, S. L. Robinson, G. Lund, S. C. Epstein, A. C. Sisto, L. K. Charkoudian, J. Collemare, R. G. Linington, T. Weber and M. H. Medema, Nucleic Acids Res., 2020, 48, D454–D458 Search PubMed.
- D. A. Hopwood, Nat. Biotechnol., 2003, 21, 505–506 CrossRef CAS PubMed.
- S. D. Bentley, K. F. Chater, A.-M. Cerdeño-Tárraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C.-H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M.-A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 CrossRef PubMed.
- S. Ōmura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki and M. Hattori, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12215–12220 CrossRef PubMed.
- J. Wan, N. Ma and H. Yuan, Eng. Microbiol., 2023, 3, 100085 CrossRef CAS.
- B. I. Adaikpoh, H. N. Fernandez and A. S. Eustáquio, Curr. Opin. Biotechnol., 2022, 77, 102782 CrossRef CAS PubMed.
- J. Ke and Y. Yoshikuni, Curr. Opin. Biotechnol., 2020, 62, 88–97 CrossRef CAS PubMed.
- GenBrick Gene Synthesis – Long DNA Sequences, GenScript, https://www.genscript.com/synthetic-biology-gene-synthesis-service.html, accessed November 17, 2023 Search PubMed.
- R. S. Ayikpoe, C. Shi, A. J. Battiste, S. M. Eslami, S. Ramesh, M. A. Simon, I. R. Bothwell, H. Lee, A. J. Rice, H. Ren, Q. Tian, L. A. Harris, R. Sarksian, L. Zhu, A. M. Frerk, T. W. Precord, W. A. van der Donk, D. A. Mitchell and H. Zhao, Nat. Commun., 2022, 13, 6135 CrossRef CAS PubMed.
- J. J. Zhang, X. Tang and B. S. Moore, Nat. Prod. Rep., 2019, 36, 1313–1332 RSC.
- I. G. U. Pait, S. Kitani, F. W. Roslan, D. Ulanova, M. Arai, H. Ikeda and T. Nihira, J. Ind. Microbiol. Biotechnol., 2018, 45, 77–87 CrossRef CAS PubMed.
- F. Wolf, F. Leipoldt, A. Kulik, D. Wibberg, J. Kalinowski and L. Kaysser, ChemBioChem, 2018, 19, 1189–1195 CrossRef CAS PubMed.
- F. Leipoldt, J. Santos-Aberturas, D. P. Stegmann, F. Wolf, A. Kulik, R. Lacret, D. Popadić, D. Keinhörster, N. Kirchner, P. Bekiesch, H. Gross, A. W. Truman and L. Kaysser, Nat. Commun., 2017, 8, 1965 CrossRef PubMed.
- B. M. Hover, S.-H. Kim, M. Katz, Z. Charlop-Powers, J. G. Owen, M. A. Ternei, J. Maniko, A. B. Estrela, H. Molina, S. Park, D. S. Perlin and S. F. Brady, Nat. Microbiol., 2018, 3, 415–422 CrossRef CAS PubMed.
- J. W. Bok, R. Ye, K. D. Clevenger, D. Mead, M. Wagner, A. Krerowicz, J. C. Albright, A. W. Goering, P. M. Thomas, N. L. Kelleher, N. P. Keller and C. C. Wu, BMC Genomics, 2015, 16, 343 CrossRef PubMed.
- A. F. Rosenzweig, J. Burian and S. F. Brady, Curr. Opin. Microbiol., 2023, 75, 102335 CrossRef CAS PubMed.
- P. Xu, C. Modavi, B. Demaree, F. Twigg, B. Liang, C. Sun, W. Zhang and A. R. Abate, Nucleic Acids Res., 2020, 48, e48 CrossRef CAS PubMed.
- J. G. Owen, Z. Charlop-Powers, A. G. Smith, M. A. Ternei, P. Y. Calle, B. V. B. Reddy, D. Montiel and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4221–4226 CrossRef CAS PubMed.
- V. Larionov, N. Kouprina, J. Graves, X. N. Chen, J. R. Korenberg and M. A. Resnick, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 491–496 CrossRef CAS PubMed.
- V. Larionov, N. Kouprina, J. Graves and M. A. Resnick, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13925–13930 CrossRef CAS PubMed.
- J. H. Kim, Z. Feng, J. D. Bauer, D. Kallifidas, P. Y. Calle and S. F. Brady, Biopolymers, 2010, 93, 833–844 CrossRef CAS PubMed.
- J. J. Zhang, K. Yamanaka, X. Tang and B. S. Moore, Methods Enzymol., 2019, 621, 87–110 CAS.
- L. Li, K. Wei, X. Liu, Y. Wu, G. Zheng, S. Chen, W. Jiang and Y. Lu, Metab. Eng., 2019, 52, 153–167 CrossRef CAS PubMed.
- J. J. Zhang, X. Tang, M. Zhang, D. Nguyen and B. S. Moore, mBio, 2017, 8, 012911 Search PubMed.
- G. Wu, J. Zhou, J. Zheng, D. Abdalmegeed, J. Tian, M. Wang, S. Sun, R.-C. A.-A. Sedjoah, Y. Shao, S. Sun and Z. Xin, Food Biosci., 2023, 53, 102813 CrossRef CAS.
- N. C. O. Lee, V. Larionov and N. Kouprina, Nucleic Acids Res., 2015, 43, e55 CrossRef PubMed.
- J. Fu, X. Bian, S. Hu, H. Wang, F. Huang, P. M. Seibert, A. Plaza, L. Xia, R. Müller, A. F. Stewart and Y. Zhang, Nat. Biotechnol., 2012, 30, 440–446 CrossRef CAS PubMed.
- H. Wang, Z. Li, R. Jia, J. Yin, A. Li, L. Xia, Y. Yin, R. Müller, J. Fu, A. F. Stewart and Y. Zhang, Nucleic Acids Res., 2018, 46, e28 CrossRef PubMed.
- C. Song, J. Luan, Q. Cui, Q. Duan, Z. Li, Y. Gao, R. Li, A. Li, Y. Shen, Y. Li, A. F. Stewart, Y. Zhang, J. Fu and H. Wang, ACS Synth. Biol., 2019, 8, 137–147 CrossRef CAS PubMed.
- C. Song, J. Luan, R. Li, C. Jiang, Y. Hou, Q. Cui, T. Cui, L. Tan, Z. Ma, Y.-J. Tang, A. F. Stewart, J. Fu, Y. Zhang and H. Wang, Nucleic Acids Res., 2020, 48, e130 CrossRef CAS PubMed.
- W. Jiang, X. Zhao, T. Gabrieli, C. Lou, Y. Ebenstein and T. F. Zhu, Nat. Commun., 2015, 6, 8101 CrossRef PubMed.
- W. Jiang and T. F. Zhu, Nat. Protoc., 2016, 11, 960–975 CrossRef CAS PubMed.
- J. Wang, A. Lu, J. Liu, W. Huang, J. Wang, Z. Cai and G. Zhao, Acta Biochim. Biophys. Sin., 2019, 51, 97–103 CrossRef CAS PubMed.
- M. Liang, L. Liu, F. Xu, X. Zeng, R. Wang, J. Yang, W. Wang, L. Karthik, J. Liu, Z. Yang, G. Zhu, S. Wang, L. Bai, Y. Tong, X. Liu, M. Wu, L.-X. Zhang and G.-Y. Tan, Nucleic Acids Res., 2022, 50, 3581–3592 CrossRef CAS PubMed.
- B. Enghiad, C. Huang, F. Guo, G. Jiang, B. Wang, S. K. Tabatabaei, T. A. Martin and H. Zhao, Nat. Commun., 2021, 12, 1171 CrossRef CAS PubMed.
- Q. Liu, R. Li, H. Shi, R. Yang, Q. Shen, Q. Cui, X. Wang, A. Li, Y. Zhang and J. Fu, Eng. Microbiol., 2023, 3, 100099 CrossRef CAS.
- Z. Sun, A. Deng, T. Hu, J. Wu, Q. Sun, H. Bai, G. Zhang and T. Wen, Appl. Microbiol. Biotechnol., 2015, 99, 5151–5162 CrossRef CAS PubMed.
- K. J. Wozniak and L. A. Simmons, Methods Mol. Biol., 2022, 2479, 159–174 CrossRef CAS PubMed.
- B.-M. Koo, G. Kritikos, J. D. Farelli, H. Todor, K. Tong, H. Kimsey, I. Wapinski, M. Galardini, A. Cabal, J. M. Peters, A.-B. Hachmann, D. Z. Rudner, K. N. Allen, A. Typas and C. A. Gross, Cell Syst., 2017, 4, 291–305 CrossRef CAS PubMed.
- L. M. Cozy and D. B. Kearns, Mol. Microbiol., 2010, 76, 273–285 CrossRef CAS PubMed.
- V. F. B. Zocca, G. G. Corrêa, M. R. d. C. R. Lins, V. N. de Jesus, L. F. Tavares, L. A. d. S. Amorim, G. E. Kundlatsch and D. B. Pedrolli, Crit. Rev. Biotechnol., 2022, 42, 813–826 CrossRef CAS PubMed.
- Y. Song, S. He, A. Jopkiewicz, R. Setroikromo, R. van Merkerk and W. J. Quax, J. Appl. Microbiol., 2022, 133, 2280–2298 CrossRef CAS PubMed.
- J. Altenbuchner, Appl. Environ. Microbiol., 2016, 82, 5421–5427 CrossRef CAS PubMed.
- Y. Wu, Y. Liu, X. Lv, J. Li, G. Du and L. Liu, Biotechnol. Bioeng., 2020, 117, 1817–1825 CrossRef CAS PubMed.
- D. Liu, C. Huang, J. Guo, P. Zhang, T. Chen, Z. Wang and X. Zhao, Biotechnol. Biofuels, 2019, 12, 197 CrossRef PubMed.
- A. A. Toymentseva and J. Altenbuchner, FEMS Microbiol. Lett., 2019, 366, fny284 CrossRef CAS PubMed.
- M. A. Price, R. Cruz, J. Bryson, F. Escalettes and S. J. Rosser, Biotechnol. Bioeng., 2020, 117, 1805–1816 CrossRef CAS PubMed.
- S. Yu, M. A. Price, Y. Wang, Y. Liu, Y. Guo, X. Ni, S. J. Rosser, C. Bi and M. Wang, ACS Synth. Biol., 2020, 9, 1781–1789 CrossRef CAS PubMed.
- W. Hao, F. Suo, Q. Lin, Q. Chen, L. Zhou, Z. Liu, W. Cui and Z. Zhou, Front. Bioeng. Biotechnol., 2020, 8, 524676 CrossRef PubMed.
- Y. Liu, H. Cheng, H. Li, Y. Zhang and M. Wang, Appl. Environ. Microbiol., 2023, 89, 002300 Search PubMed.
- F. Hu, Y. Liu and S. Li, Microb. Cell Factories, 2019, 18, 42 CrossRef PubMed.
- D. Zou, S. W. Maina, F. Zhang, Z. Yan, L. Ding, Y. Shao and Z. Xin, J. Agric. Food Chem., 2020, 68, 11358–11367 CrossRef CAS PubMed.
- V. Jagadeesh, T. Yoshida, M. Uraji, N. Okahashi, F. Matsuda, C. J. Vavricka, K. Tsuge and A. Kondo, ACS Synth. Biol., 2023, 12, 305–318 CrossRef CAS PubMed.
- K. A. J. Bozhüyük, F. Fleischhacker, A. Linck, F. Wesche, A. Tietze, C.-P. Niesert and H. B. Bode, Nat. Chem., 2018, 10, 275–281 CrossRef PubMed.
- K. A. J. Bozhüyük, A. Linck, A. Tietze, J. Kranz, F. Wesche, S. Nowak, F. Fleischhacker, Y.-N. Shi, P. Grün and H. B. Bode, Nat. Chem., 2019, 11, 653–661 CrossRef PubMed.
- N. Abbood, L. Präve, K. A. J. Bozhueyuek and H. B. Bode, in Non-Ribosomal Peptide Biosynthesis and Engineering: Methods and Protocols, ed. M. Burkart and F. Ishikawa, Springer US, New York, NY, 2023, pp. 219–234 Search PubMed.
- W. L. Thong, Y. Zhang, Y. Zhuo, K. J. Robins, J. K. Fyans, A. J. Herbert, B. J. C. Law and J. Micklefield, Nat. Commun., 2021, 12, 6872 CrossRef CAS PubMed.
- K. Blin, S. Shaw, H. E. Augustijn, Z. L. Reitz, F. Biermann, M. Alanjary, A. Fetter, B. R. Terlouw, W. W. Metcalf, E. J. N. Helfrich, G. P. van Wezel, M. H. Medema and T. Weber, Nucleic Acids Res., 2023, 51, W46–W50 CrossRef CAS PubMed.
- M. H. Medema, K. Blin, P. Cimermancic, V. De Jager, P. Zakrzewski, M. A. Fischbach, T. Weber, E. Takano and R. Breitling, Nucleic Acids Res., 2011, 39, 339–346 CrossRef PubMed.
- H. Maughan and G. Van der Auwera, Infect., Genet. Evol., 2011, 11, 789–797 CrossRef PubMed.
- L. Xia, Y. Miao, A. Cao, Y. Liu, Z. Liu, X. Sun, Y. Xue, Z. Xu, W. Xun, Q. Shen, N. Zhang and R. Zhang, Nat. Commun., 2022, 13, 1023 CrossRef CAS PubMed.
- S. Xiao, N. Chen, Z. Chai, M. Zhou, C. Xiao, S. Zhao and X. Yang, Mar. Drugs, 2022, 20, 567 CrossRef CAS PubMed.
- S. A. Cochrane and J. C. Vederas, Med. Res. Rev., 2016, 36, 4–31 CrossRef CAS PubMed.
- P. Baindara, N. Nallabelli and S. Korpole, J. Appl. Microbiol., 2020, 128, 473–490 CrossRef CAS PubMed.
- Z. Wang, B. Koirala, Y. Hernandez, M. Zimmerman, S. Park, D. S. Perlin and S. F. Brady, Nature, 2022, 601, 606–611 CrossRef CAS PubMed.
- K. Chakraborty, V. K. Kizhakkekalam, M. Joy and R. D. Chakraborty, World J. Microbiol. Biotechnol., 2021, 37, 1–12 CrossRef PubMed.
- K. Chakraborty, B. Thilakan, R. D. Chakraborty, V. K. Raola and M. Joy, Appl. Microbiol. Biotechnol., 2017, 101, 569–583 CrossRef CAS PubMed.
- R. Ebrahimi, R. Pournejati and H. R. Karbalaei-Heidari, Iran. J. Sci. Technol. Trans. A-Science, 2021, 45, 1165–1175 CrossRef.
- G. Aleti, A. Sessitsch and G. Brader, Comput. Struct. Biotechnol. J., 2015, 13, 192–203 CrossRef CAS PubMed.
- K. J. Grubbs, R. M. Bleich, K. C. Santa Maria, S. E. Allen, S. Farag, AgBiome Team, E. A. Shank and A. A. Bowers, mSystems, 2017, 2, 000400 CrossRef PubMed.
- S. Filippidou, T. Junier, T. Wunderlin, C.-C. Lo, P.-E. Li, P. S. Chain and P. Junier, Comput. Struct. Biotechnol. J., 2015, 13, 299–306 CrossRef CAS PubMed.
- S.-C. Wu, J. C. Yeung, Y. Duan, R. Ye, S. J. Szarka, H. R. Habibi and S.-L. Wong, Appl. Environ. Microbiol., 2002, 68, 3261–3269 CrossRef CAS PubMed.
Footnotes |
| † These authors contributed equally as first authors. |
| ‡ These authors contributed equally as senior authors. |
| This journal is © The Royal Society of Chemistry 2024 |