The Kornblum DeLaMare rearrangement in natural product synthesis: 25 years of innovation
Marc C.
Kimber
*a and
Darren S.
Lee
*b aDepartment of Chemistry, School of Science, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: M.C.Kimber@lboro.ac.uk bCentre for Green Chemistry and Green Engineering at Yale, Yale University, New Haven, CT 06511, USA. E-mail: Darren.lee@yale.edu
Received
30th October 2023
First published on 31st January 2024
Abstract
Covering: 1998 up to the end of 2023
Since its initial disclosure in 1951, the Kornblum DeLaMare rearrangement has proved an important synthetic transformation and has been widely adopted as a biomimetic step in natural product synthesis. Utilising the base catalysed decomposition of alkyl peroxides to yield a ketone and alcohol has found use in many syntheses as well as a key strategic step, including the unmasking of furans, as a biomimetic synthetic tool, and the use of the rearrangement to install oxygen enantioselectively. Since ca. 1998, its impact as a synthetic transformation has grown significantly, especially given the frequency of use in natural product syntheses, therefore this 25 year time period will be the focus of the review.
Marc C. Kimber
Marc C. Kimber's undergraduate and PhD studies (1998) were undertaken at University of Adelaide. After postdoctoral positions at the Universities of Sydney and Adelaide he moved in 2002 to the UK as a postdoc with Prof. J. Stephen Clark at the University of Nottingham. After briefly working in industry, he returned to Nottingham in 2006 to work with Prof. Christopher J. Moody. In 2008 he began a Lectureship in Organic Chemistry at Loughborough University and was promoted to Senior Lecturer in 2015. Current research focuses on synthetic photochemistry, singlet oxygen in natural product and materials synthesis, and continuous flow organophotocatalysis.
Darren S. Lee
Darren S. Lee received an MChem (2009) and PhD (2013) at Loughborough University, UK. His postdoctoral career began in 2014 with Prof. Simon Woodward at the University of Nottingham. In 2015, he moved groups at Nottingham to work with Professors Sir Martyn Poliakoff and Michael George. In 2022, he moved to Yale University in the US as an Associate Research Scientist at the Center for Green Chemistry and Green Engineering with Professors Paul T. Anastas and Julie B. Zimmerman. In 2024, Darren returned to the UK as a senior lecturer in sustainable chemistry at Nottingham Trent University.
1 Introduction
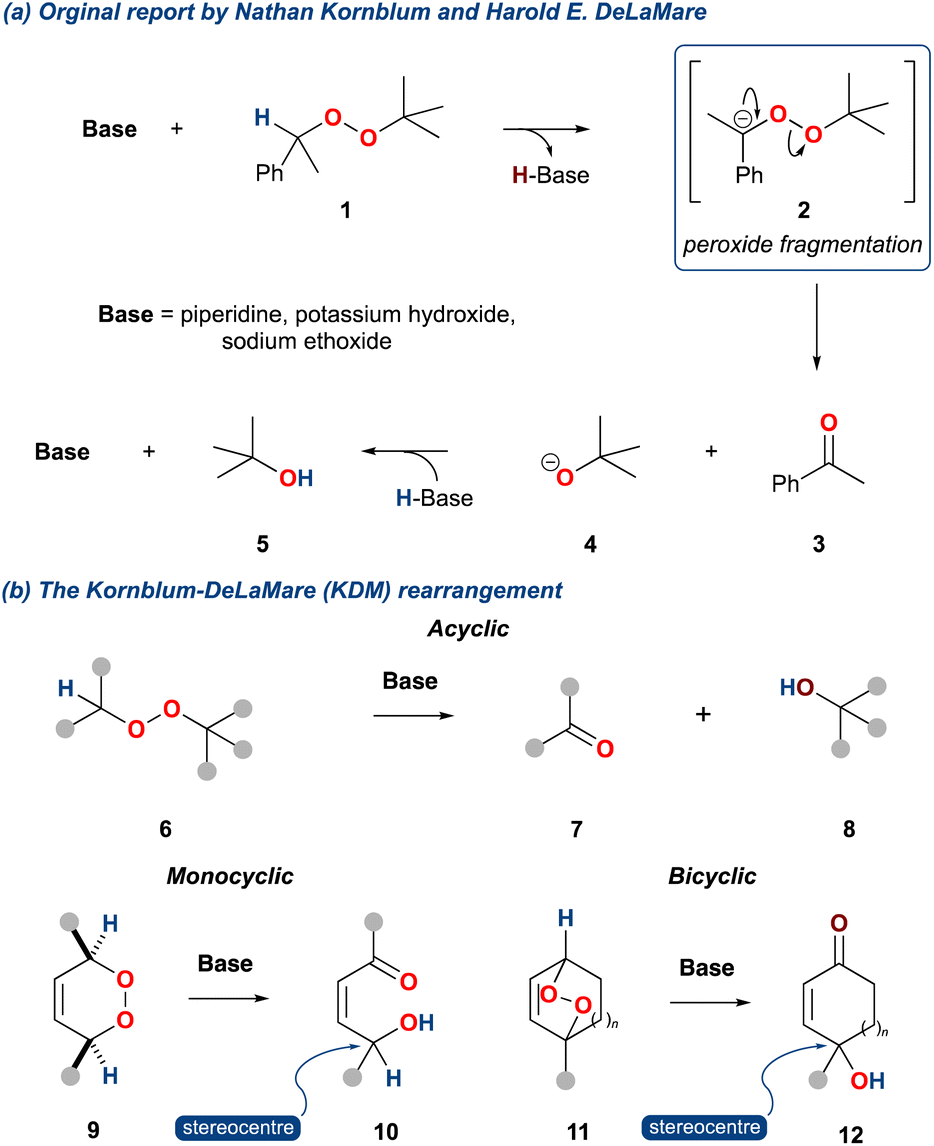
The base catalysed decomposition of an alkyl-peroxide to give a ketone and alcohol was first reported by Nathan Kornblum and Harold DeLaMare in 1952.1 Unlike previous work by Milas and Surgenor,2 who had demonstrated the stability of t-butyl hydroperoxide to base, Kornblum and DeLaMare exposed 1-phenylethyl-t-butyl peroxide 1 to piperidine giving acetophenone (3) and t-butyl alcohol (5) in 79% and 25%, respectively (Scheme 1a). This reaction was also found to be consistent with potassium hydroxide and sodium ethoxide as the base. Significantly, the authors identified the importance of an α-hydrogen relative to the peroxide, and accordingly they proposed an elimination mechanism as outlined in Scheme 1a. This mechanism was found to have similarities with the base mediated decomposition of nitrate esters3 and has subsequently become the bedrock of what is now known as the Kornblum DeLaMare rearrangement of peroxides, which we will now refer to as the KDM rearrangement within this review.
Scheme 1 (a) The original report of Nathan Kornblum and Harold DeLaMare; (b) product outcome of the Kornblum DeLaMare rearrangement for acyclic (6), monocyclic (7), and bicyclic peroxides (10).
In the KDM rearrangement, acyclic alkyl peroxides (6) upon treatment with base give rise to a ketone and an alcohol (Scheme 1b, 7 and 8, respectively), while cyclic peroxides such as 9 give rise to γ-hydroxyenone 10, and bicyclic peroxides (11) give rise γ-hydroxyenone such as 12. The former variant can be used to access carbonyl products, while the later variant provides a very useful functional group possessing a stereocentre.4 Since the disclosure of the KDM rearrangement considerable focus has been on this later variant, as the functional group, a γ-hydroxyenone, is present in several natural product classes. Furthermore, this functionality is a valuable synthon which can be harnessed in subsequent synthetic transformations. Notable examples of the use of the KDM rearrangement over the past 70 years are its application in tropone and cycloheptatriene chemistry,5–11 its appearance in prostaglandin biosynthesis,12,13 and more recently, its employment as a tool in biomimetic natural product synthesis.
The scope of this review is to show-case the increasing use of the KDM rearrangement in natural product synthesis over the past 25 years. A recent general review on peroxide rearrangements,14,15 of which the KDM rearrangement sits, and the use of singlet oxygen in target synthesis16 provides some excellent context, but the aim of this treatise is to equip the reader with the necessary tools to successfully incorporate the KDM rearrangement in their own natural product syntheses.
2 Mechanism
We will begin with a discussion of the underpinning mechanism of the KDM rearrangement. This is essential when considering the selectivity of the initial base catalysed deprotonation event and is informative when designing and implementing enantioselective variants of the KDM rearrangement.
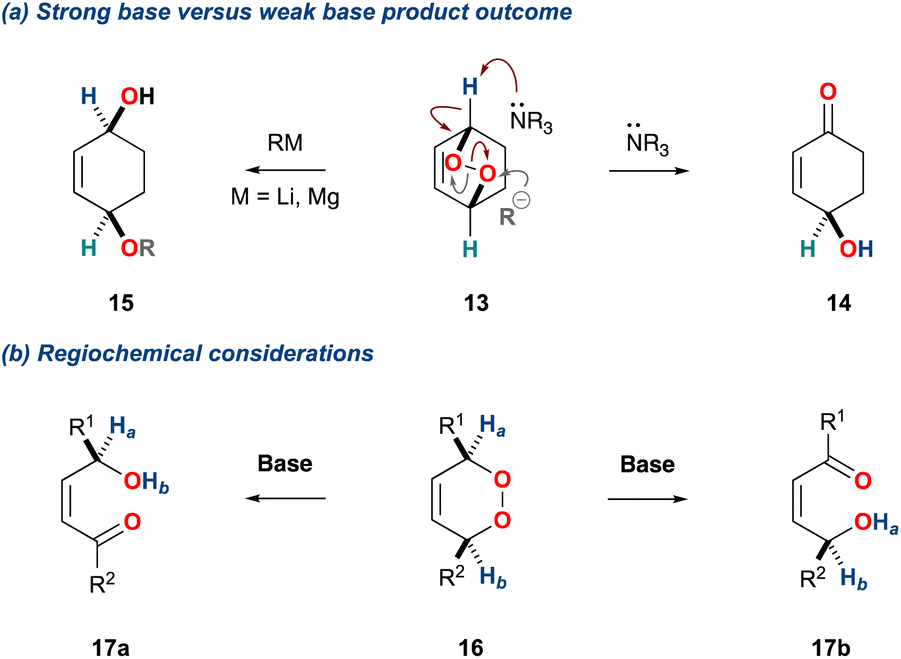
The mechanism is centred on the bond dissociation energy of the oxygen–oxygen σ-bond in the peroxide; the bond energy of organic peroxides is ca. 45 kcal mol−1.17 The KDM rearrangement is initiated by treatment of an endoperoxide with a base. Non-nucleophilic bases promote an initial deprotonation of an α-hydrogen, leading to breakage of the weak oxygen–oxygen σ-bond; an example is shown for the bicyclic endoperoxide 13 (Scheme 2a).18,19 Conversely, when an endoperoxide is treated with a nucleophilic base a competing process can occur. For example, treatment of the bicyclic endoperoxide 13 with an organolithium leads to direct reaction onto one of the oxygen centres and subsequent peroxide cleavage providing the diol 15 (Scheme 2a).
Scheme 2 (a) Product outcome of bicyclic peroxides with MgR/LiR versus tertiary amines; (b) regiochemical considerations with monocyclic peroxides.
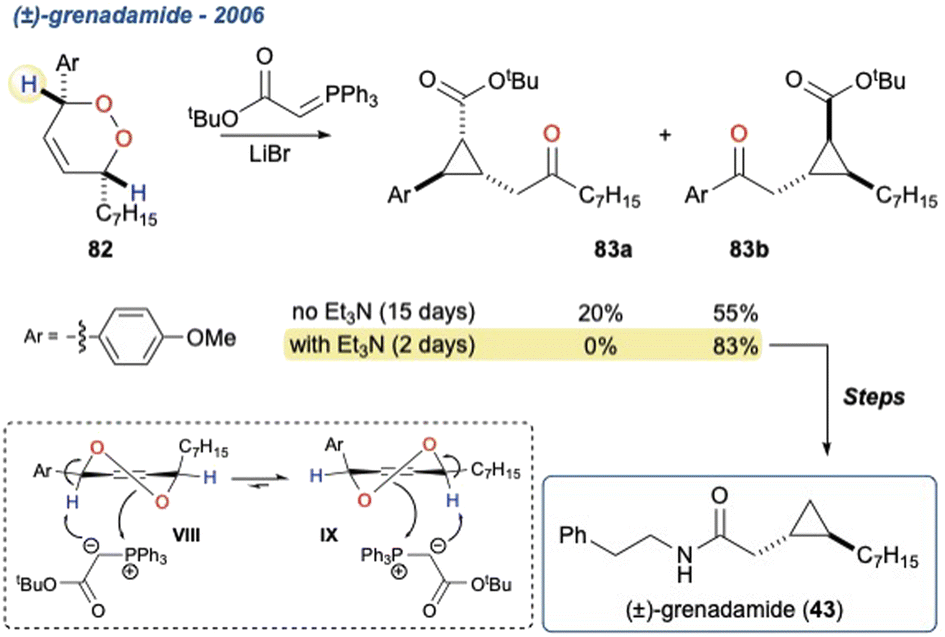
Additionally, the initial α-hydrogen deprotonation becomes crucial when considering the regiochemical outcome. For example, monocyclic peroxides of the type 16, provide two potential α-hydrogens (Scheme 2b, 16 Ha and Hb) which can then deliver to isomeric γ-hydroxyenones 17a and 17b, respectively. This regiochemical complexity in the KDM rearrangement was observed by Taylor and co-workers in their first total synthesis of grenadamide 43, a cyclopropyl amide from the marine cyanobacterium, Lyngbya majuscula;20 this will be discussed later within this review.
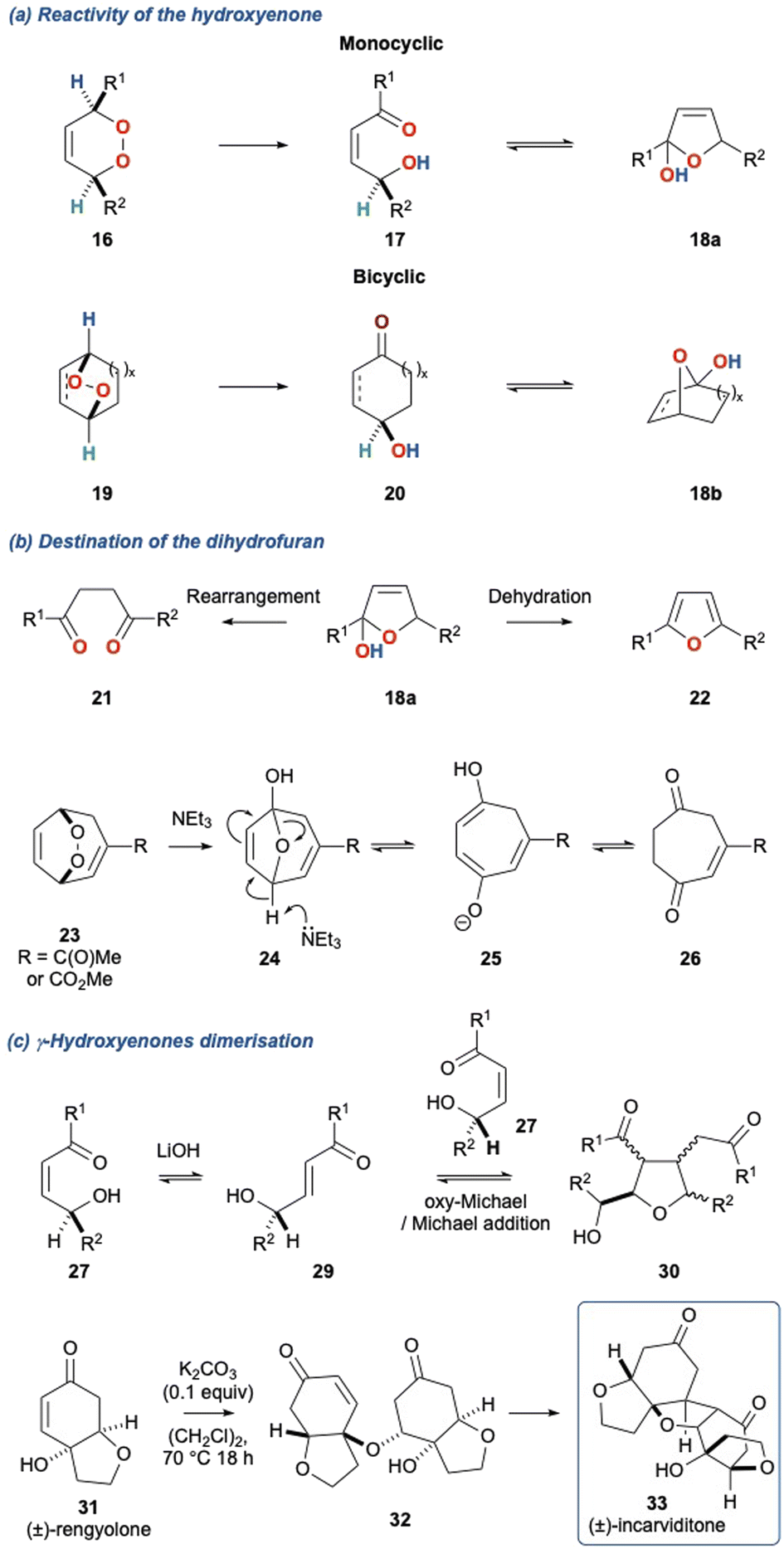
Tertiary amines have been the predominant bases employed in the KDM rearrangement, and include Et3N,19,21,22 DIPEA,23 DABCO,24,25 DBU26–28 and piperidine.29 Other bases to be used in the KDM rearrangement include, LiOH,30,31 phosphorus ylides,30 stabilised phosphates,32 malonates,33 SiO2,34 and more recently bases derived from haloforms.35 Each of these bases gives the key product of the KDM rearrangement for cyclic peroxides, the γ-hydroxyenone, whose fate is often defined by the base used in the initial rearrangement. Tertiary amines commonly provide the γ-hydroxyenone which then cyclises to give the dihydrofuran 20 (Scheme 3a).
Scheme 3 (a) Equilibrium between the γ-hydroxyenone and dihydrofuran for KDM rearranged mono- and bicyclic peroxide; (b) dihydrofuran dehydration to give furans, and the base catalysed rearrangement to 1,4-diketones; (c) the base catalysed dimerisation of γ-hydroxyenones and basis for the biomimetic synthesis of natural products.
This also occurs with monocyclic peroxides (16 to 17 to 18a) and bicyclic peroxides (19 to 20 to 18b); note that in each case the γ-hydroxyenone and dihydrofuran are in equilibrium. In the case of dihydrofuran 18 derived from monocyclic peroxides, this can be readily aromatised via dehydration to provide the furan 22; alternatively, depending on the nature of the substituents, this dihydrofuran can rearrange to give a 1,4-diketone 21. Bicyclic peroxides can also undergo this alternate rearrangement, for example peroxide 23 can undergo a NEt3 mediated KDM rearrangement to provide dihydrofuran 24 which then undergoes a subsequent fragmentation to ultimately provide diketone 26. Under basic conditions monocyclic and bicyclic γ-hydroxyenones can also undergo conjugate addition. This is particularly pertinent for γ-hydroxyenones such as 27 which can readily dimerise in the presence of LiOH to provide tetrasubstituted tetrahydrofurans such as 30.31 This transformation is reminiscent of the proposed biomimetic synthesis of (±)-incarviditone (33) through the oxa-Michael/Michael dimerization of (±)-rengyolone (31) described by Sherburn and co-workers (Scheme 3c).23,36 There are sparce examples of alternate bases such as acetates37,38 as well as weak acids such as thiourea.6,39 Bach and co-workers40 described an acid mediated KDM rearrangement via protonation of the peroxide bridge and this report highlights an alternate mechanistic pathway for the KDM rearrangement.
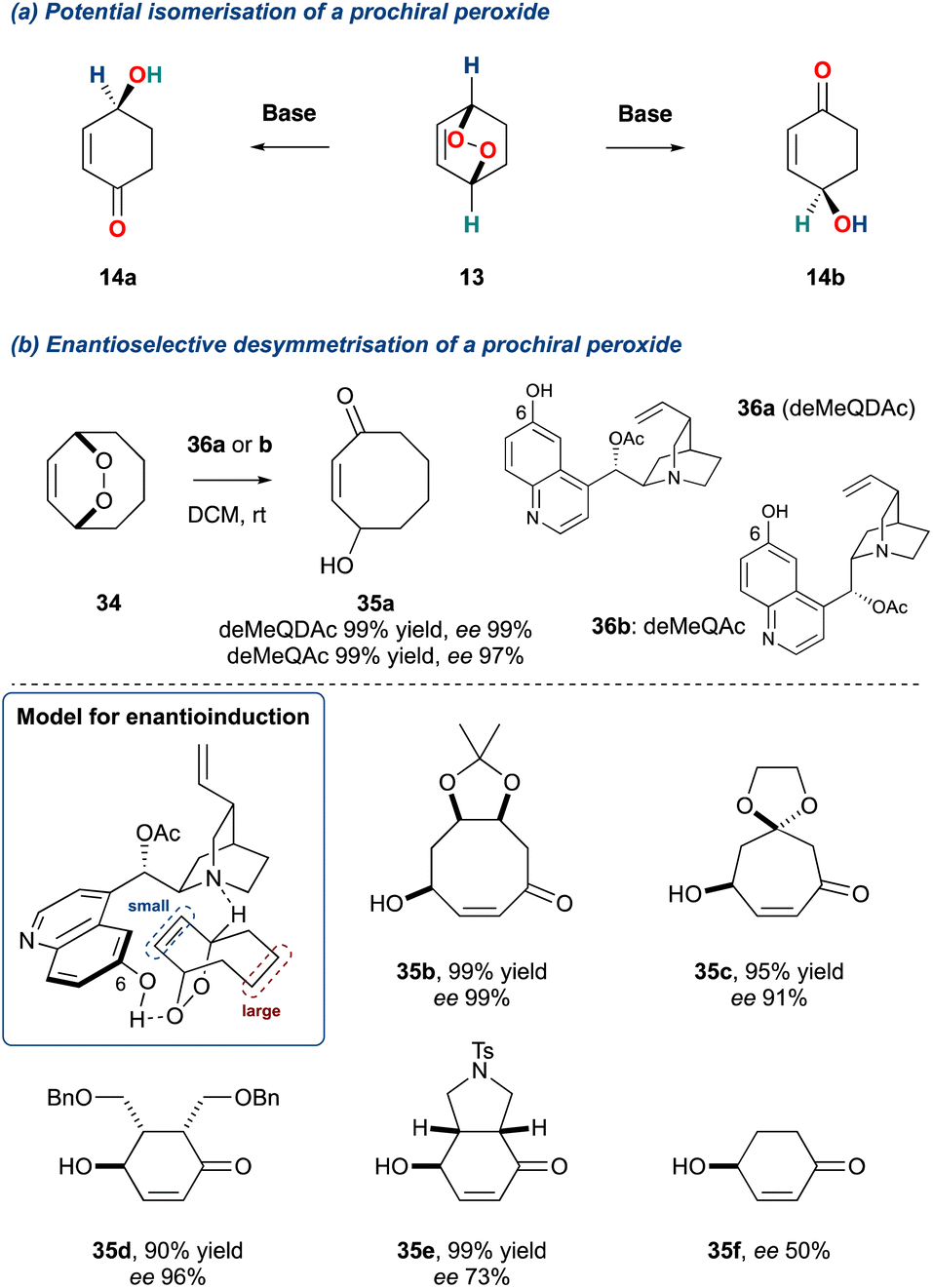
The use of tertiary amine bases in the KDM rearrangement has led to it becoming an increasingly important tool in probing plausible biomimetic pathways in natural product synthesis. This stems from the work of Brame, Morrow and co-workers41,42 who investigated the KDM reaction in the isoprostane pathway, followed by work by Kelly and co-workers19 who identified the potential of an enantioselective isomerisation of a prochiral endoperoxide, and was subsequently realised in 2006 by Staben and Toste (Scheme 4).43 This was achieved by desymmetrisation of meso-endoperoxides using cinchona-alkaloid catalysed enantio-determining deprotonation (Scheme 4b). The model proposed for achieving enantioselective induction is shown in Scheme 4b, where the cinchona alkaloid acts as a bifunctional catalyst providing the γ-hydroxyenone through an E2-elimination. The arrangement of the tertiary amine and the hydroxyl at the 6-position of the quinoline were key for this reaction, facilitating the heterolytic oxygen–oxygen σ-bond cleavage as well as providing a chiral pocket for enantio-discrimination. To achieve high enantio-induction, the prochiral endoperoxide must possess a size differential between the two bridges flanking the oxygen–oxygen bond; this was evidenced by the poor enantio-induction when forming 4-hydroxycyclohex-2-ene-1-one 35f. Additionally, this protocol was shown to give poor enantio-induction for monocyclic endoperoxides.44 It must be noted that the desymmetrisation of prochiral endoperoxides can also be achieved through a homolytic cleavage of the weak oxygen–oxygen σ-bond.45 This also provides a route to enantioenriched γ-hydroxyenones, however, this is not technically a KDM rearrangement and will not be covered within this review.
Scheme 4 (a) Potential desymmetrisation of a prochiral bicyclic peroxides; (b) Toste and co-workers desymmetrisation protocol.
This important disclosure by Stanben and Toste provides a general approach to the synthesis γ-hydroxyenones, crucial chiral building blocks, and significantly, this method has been harnessed by several groups to synthesise natural products, examples of which are discussed further within this review.
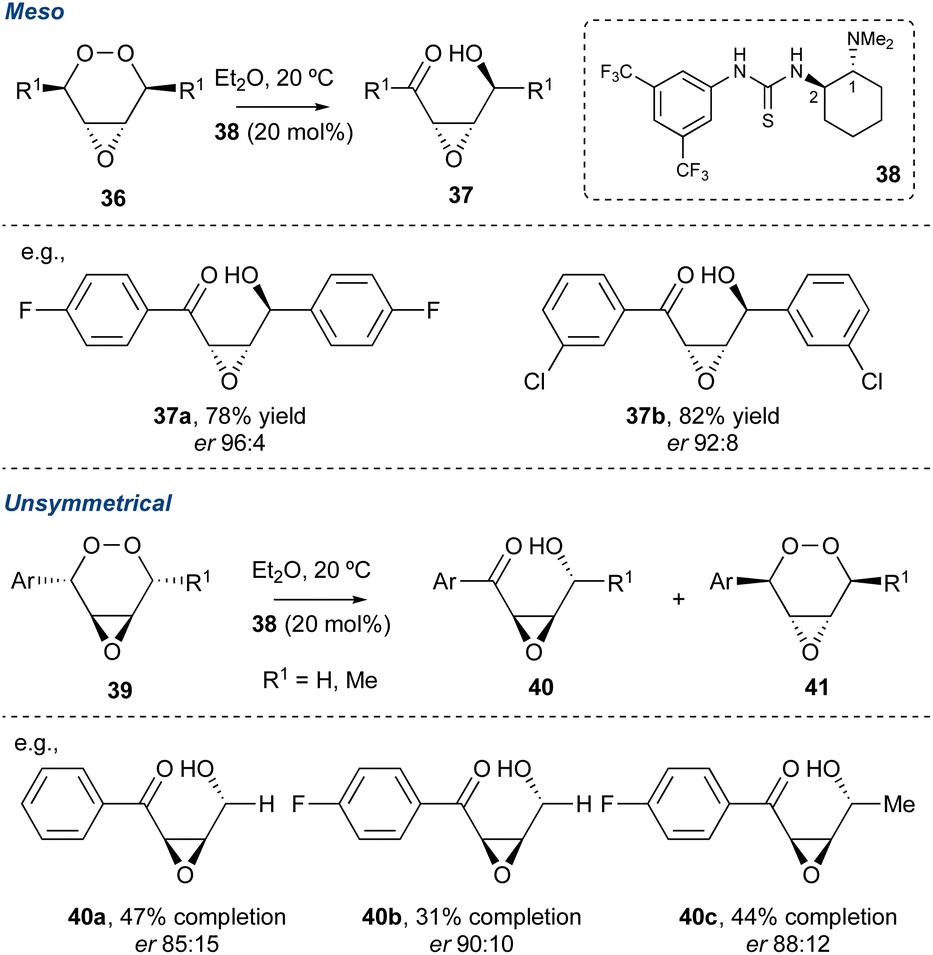
Recently, Greatrex and co-workers divulged an innovative thiourea/amine organocatalytic system to desymmetrise meso-endoperoxide of the type shown in Scheme 5.46 Key to the success of this transformation is the steric requirement of the catalyst, particularly size of the dialkylamine at C1 (cat. 38). The authors observed that if the steric bulk of the amine at C1 increased this was detrimental to the er of the product γ-hydroxyenones. Additionally, for unsymmetrical peroxides such as 39, a kinetic resolution could be realised. The regiochemistry of the KDM rearrangement in this kinetic resolution is confined due to the benzylic proton, which are far more acidic. Therefore, to achieve a satisfactory er in the product γ-hydroxyenones (40a–c) the progress of each reaction required monitoring, and this is highlighted in the percentage completions. However, while this methodology is very recent, it should find applicability within future natural product syntheses.
Scheme 5 Greatrex and co-workers use of an organocatalysis in the desymmetrisation of endoperoxides.
3 Endoperoxides: the KDM rearrangement precursors
The precursors for a KDM rearrangement are endoperoxides, and their synthesis is achieved by treatment of a 1,3-diene with singlet oxygen, with the latter being generated from oxygen in the presence of a photosensitiser and a visible light source (there are examples of generating singlet oxygen through ‘dark’ processes this has yet to be employed in the synthesis of natural products).47–52 This transformation is well established and there are several reviews that describe the underlining mechanism, reaction scope and limitations.15,16 Over recent decades there have been significant improvements in the practicality of this transformation, which has led to improved yields and more importantly improved safety, given the reputation of endoperoxides for their instability, and the challenges of using molecular oxygen at scale.53,54 This includes the use of continuous flow methodology16,53–55 and reactor design.56–64 Often in natural product syntheses, the formation of the endoperoxide is undertaken in batch conditions. However, improvements in yield should be possible given the increasing availability of commercial continuous flow equipment and accessibility of simple homemade flow reactors.
4 The KDM rearrangement and biomimetic relationship
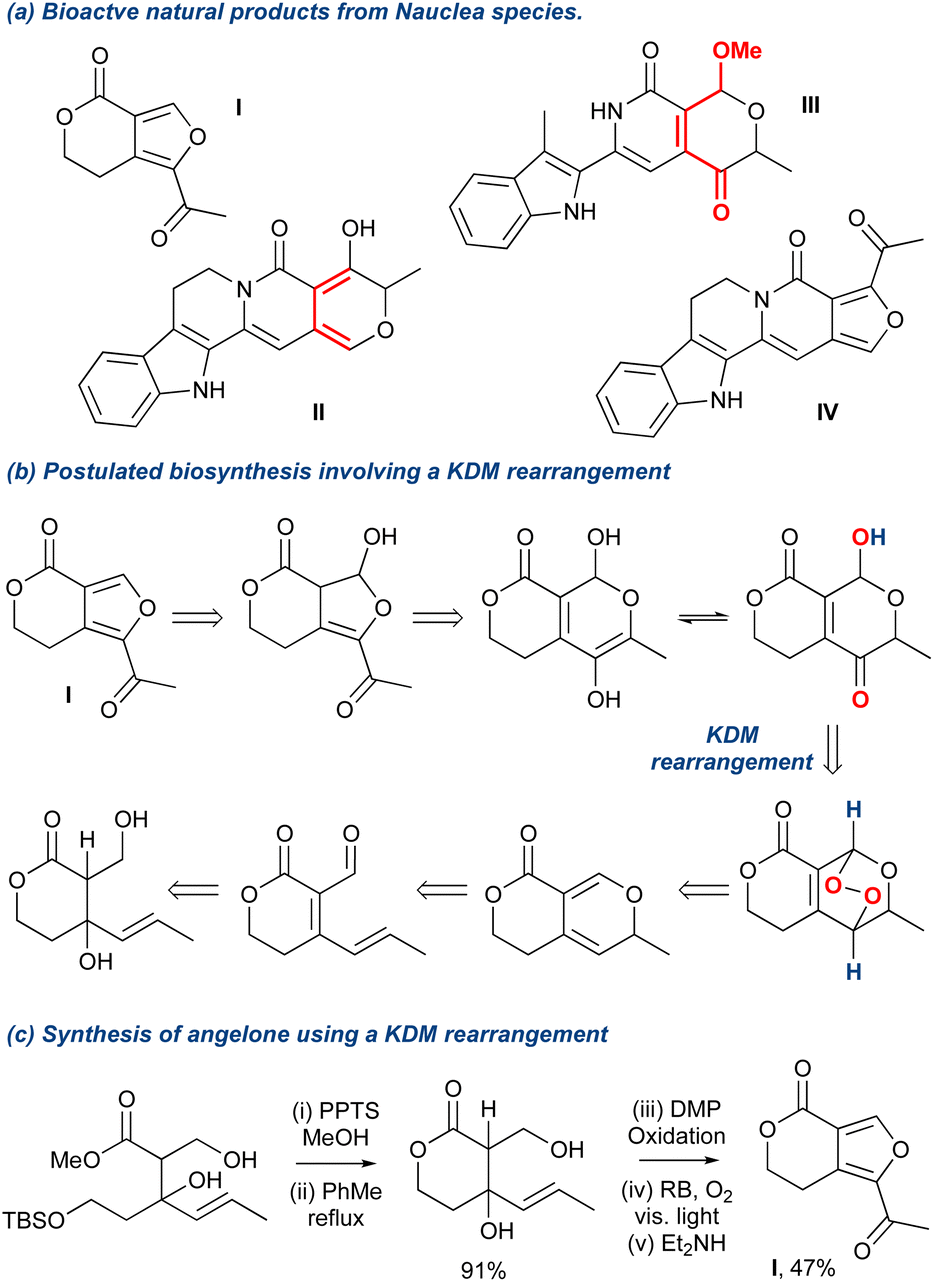
The KDM rearrangement is often described as a biomimetic reaction and as such features in several bioinspired routes to different natural products. The incorporation of the KDM in such a route undoubtedly must proceed through an endo-peroxide intermediate and have an accessible, acidic hydrogen atom that can be removed, see Section 2 for more details on the mechanism. For example, in the synthesis of angelone I, Tan et al. identified that several natural products isolated from Nauclea feature an “oxygenation–deoxygenation relationship” that could be attributed to the reaction of singlet oxygen with diene II and its subsequent KDM rearrangement and O-methylation into III (Scheme 6a).65 The presence of IV was postulated as a ring contraction step going through a 6π-electrocyclic ring opening. With these key steps identified, they targeted I as their synthetic goal and designed their biomimetic retrosynthetic route based around the installation of a peroxide bridge, its subsequent KDM rearrangement and then the ring contraction step (Scheme 6b). They also hypothesised that should the forward route indeed mimic the postulated biomimetic conditions, then the reaction steps could be truncated into a facile process. Indeed, they completed the synthesis of I in a 3-pot procedure with 6 overall steps (Scheme 6c), and a full discussion of this natural product synthesis can be found in Section 5 of this review. Crucially, this example nicely highlights the relevance of the KDM rearrangement as a biomimetic tool and showcases the importance of recognising where it could occur in a biosynthetic pathway, exemplified in the above case between the isolated diene II and the γ-hydroxyenone derivative III.
Scheme 6 An example of a KDM rearrangement used as a key biomimetic step in the total synthesis of angelone.
5 Natural products
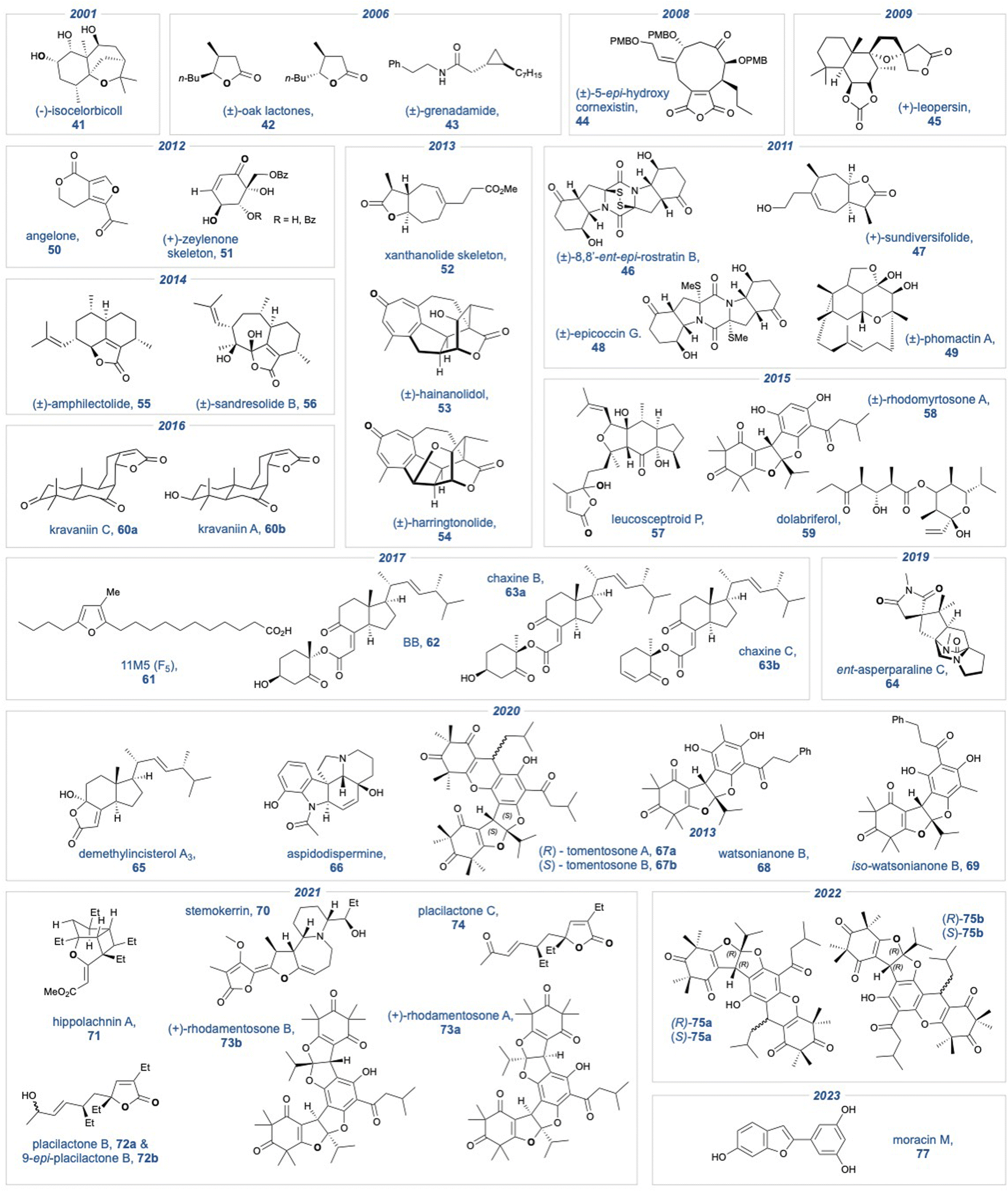
A summary of all natural product targets that have employed a KDM rearrangement over the past 25 years is shown in Fig. 1.
Fig. 1 Natural product targets that have used the KDM rearrangement as a key intermediate step.
We have divided the analysis into two parts. The first section will describe the use of the KDM rearrangement as a key step. We have examined each natural product that uses a KDM rearrangement in chronological order. Where necessary we have provided clarification on the regiochemistry of the products that result from the KDM rearrangement, whether the key KDM rearrangement was enantioselective, and if the KDM rearrangement was employed as a biomimetic tool. The second section of this review describes natural products that specifically contain a butenolide. This structural motif, common to diterpene natural products, can be installed using a late stage KDM rearrangement when performed on a 3-substituted furan.
5.1 KDM rearrangement as a key step
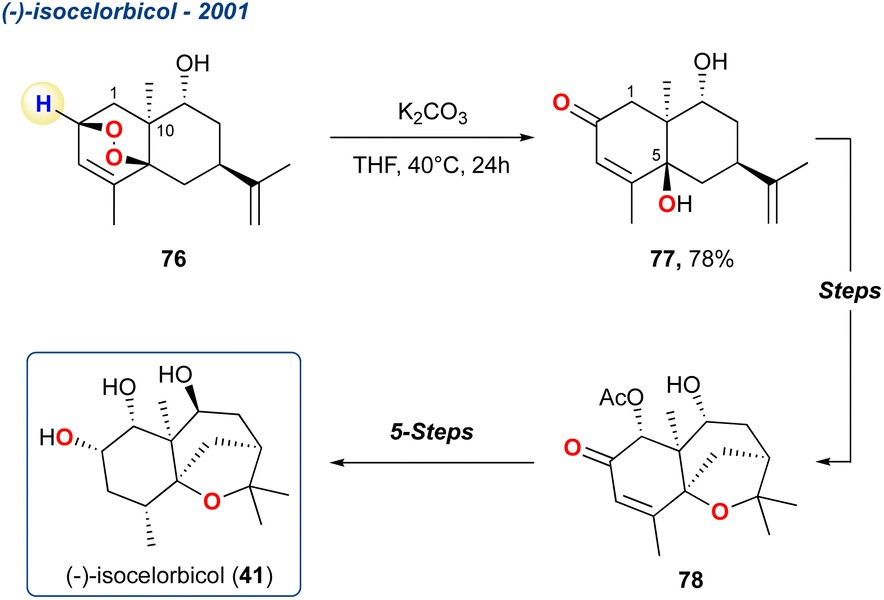
5.1.1 (−)-Isocelorbicol, 2001. The KDM rearrangement was applied to a short, enantioselective synthesis of (−)-isocelorbicol (41), a sesquiterpene polyol of the agarofuran family of natural products (Scheme 7).
Scheme 7 Zhou et al. use of the KDM rearrangement to initially give precursor 77 which was subsequently used to give (−)-isocelorbicol 41.
In their initial report, Zhou et al. prepared peroxide 76 in 8-steps from (−)-carvone and upon treatment with K2CO3 in THF produced diol 77 in high yield, which was subsequently transformed to enone 78 which they suggested was an enantioenriched precursor for polyhydroxylated agarofurans.66 The regiochemistry of the KDM rearrangement is uncomplicated in this example given there is only one available hydrogen. Additionally, the stereochemistry of the OH at C-5 is defined by the initial endoperoxide formation which occurs anti to the methyl group at C-10. In a follow-up report, the group took enone 78 forward and completed the synthesis of 41, which was realised in 5 linear steps.67
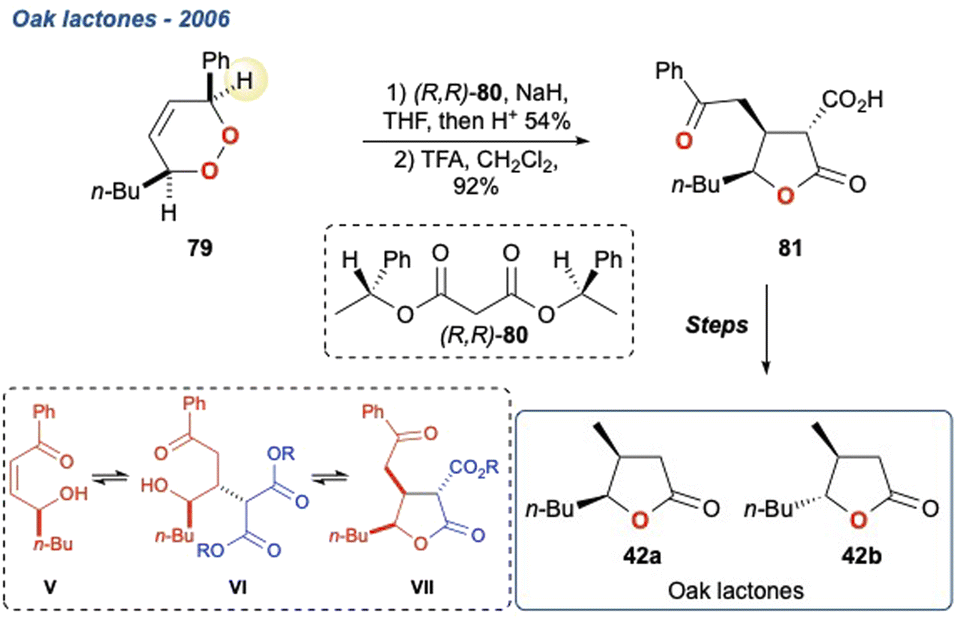
5.1.2 Oak lactones, 2006. The isomers of oak lactone (Scheme 8; 42a,b), an important flavour and fragrance compound often found in alcoholic beverages that have been fermented or matured in wooden vessels, were synthesised by Brown et al. using a KDM rearrangement to construct the enantioenriched lactone.68 Endoperoxide 79 was treated with NaH in the presence of a chiral malonate diester (80) which, after scission of the peroxide, underwent a Michael addition and subsequent lactonisation yielding 81 as a mixture of diastereoisomers This approach to γ-lactones had been developed by the same group.33 In this example the regiochemistry of the KDM is defined by the acidity of the hydrogen adjacent to the phenyl group. The relative stereochemistry observed in the γ-lactone results from an initial conjugate addition to cis-enone V to give VI, with subsequent lactonisation onto the malonate to give VII. Since a chiral malonate is used, this provides a mixture of diastereoisomers which are separated by chromatography, with the 3R,4S,5S-isomer being subsequently treated with TFA to yield the enantiopure acid 81. This was taken forward to give the natural isomers of oak lactone 42a and 42b, respectively.
Scheme 8 The enantioselective total synthesis of oak lactones 42a and 42b, using a KDM rearrangement mediated by a chiral malonate with γ-lactonisation as a key step.
5.1.3 (±)-Grenadamide, 2006. Avery et al. reported the first synthesis of (±)-grenadamide (Scheme 9; 43),20 a cyclopropyl amide, which was isolated from a marine cyanobacterium Lyngbya majuscula.69 The KDM rearrangement was employed as a key step in the formation of the cyclopropane ring as part of a diastereoselective cascade reaction.30,70,71 Using the stabilised tert-butyl ester ylide as the base to facilitate the KDM cleavage yielded the desired cyclopropane 83b in 55% yield after 15 day reaction time. The group also observed an unexpected isomer (83a) which arose from deprotonation of the less acidic proton adjacent to the O–O linkage of 82. Upon further investigation they discovered that endoperoxide 82 remained present in solution and was reacting slowly, suggesting that deprotonation was the limiting step. They attributed the slow deprotonation and unexpected isomer to the conformation of 82 being fixed such that the alkyl chain is in a pseudo equatorial position (VIII to IX), limiting the ylides access to the more acidic proton. When they performed the reaction in the presence of Et3N, to access the more acidic proton, then the desired isomer 83b was obtained in 83% yield with a much shorter reaction time. The racemic cyclopropane was then carried through a further 8-steps providing (±)-grenadamide (43).
Scheme 9 The first synthesis of (±)-grenadamide using a phosphorus ylide mediated KDM cascade, with initial conditions providing two isomeric cyclopropanes 83a and 83b.
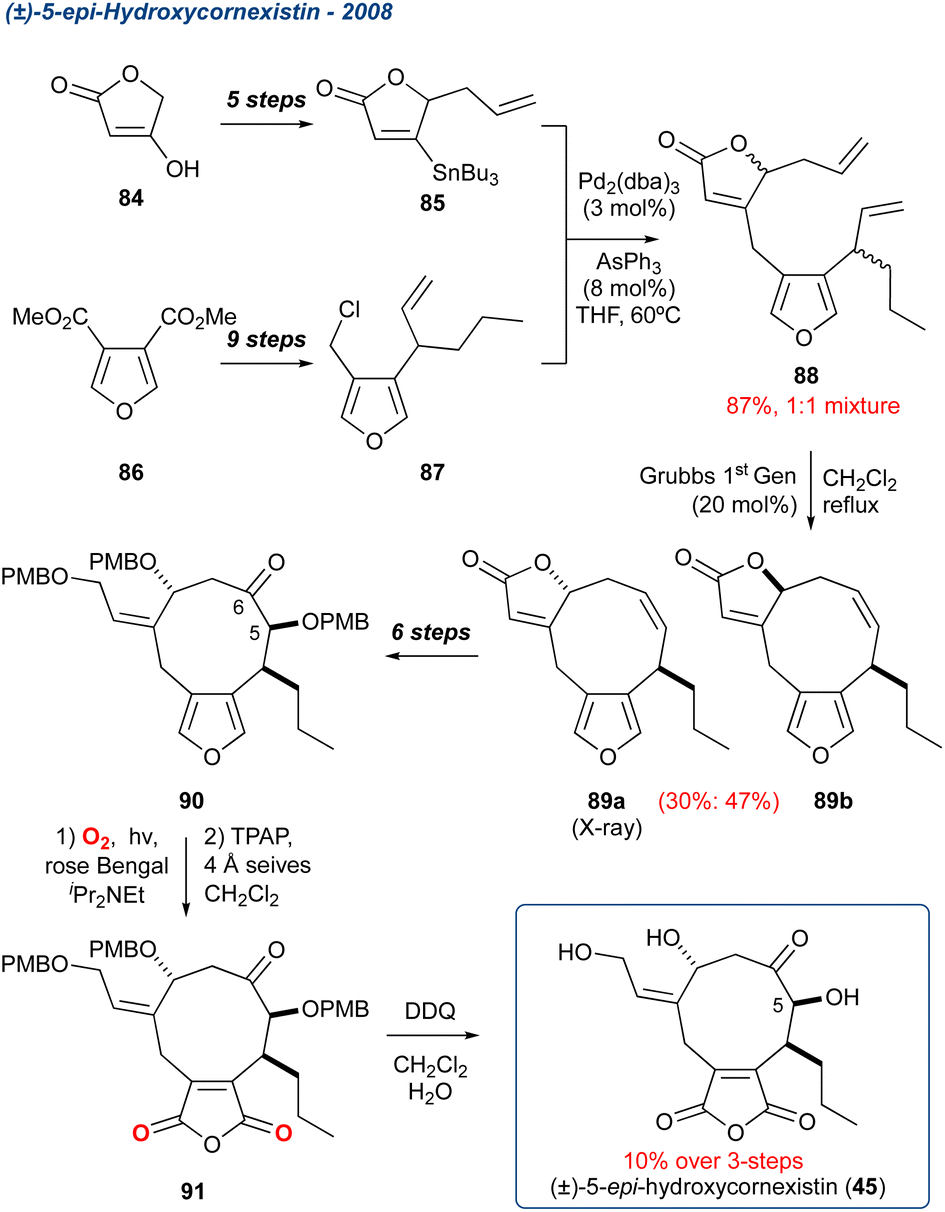
5.1.4 (±)-5-Epi-hydroxycornexistin, 2008. In 2008, Clark et al.72 divulged the synthesis of the 5-epi isomer of hydroxycornexistin, a nonadride natural product isolated from the fungus Paecilomyces variotii,73 that displays (along with cornexistin) potent herbicidal activity (Scheme 10).74 The unusual anhydride unit with hydroxycornexistin presents a significant synthetic challenge due to its reactivity. To overcome this challenge, Clark and co-workers sought to mask the anhydride as a furan. The furan would stay intact until the penultimate steps within the total synthesis, where it would then undergo a KDM rearrangement and subsequent oxidation. The core cyclononane was assembled through a Stille coupling of 85 with racemic chloride 87, providing 88 as a 1:1 mixture of isomers, which subsequently underwent an RCM to provide diastereoisomers 89a and 89b in 30% and 47% yield, respectively. Diastereoisomer 89a was then taken forward in 6-steps, introducing oxidation at C5 and C6, as well as opening of the butenolide. The PMB protected precursor 90 was then exposed to KDM rearrangement conditions followed by TPAP oxidation, and PMB global deprotection with DDQ, to give 5-epi-hydroxycornexistin 45 in 10% over these final three steps. While the sequence was low yielding, this synthesis proves the validity of masking the anhydride as its furan.
Scheme 10 Unveiling of the anhydride of the cornexistin's using a KDM/oxidation protocol.
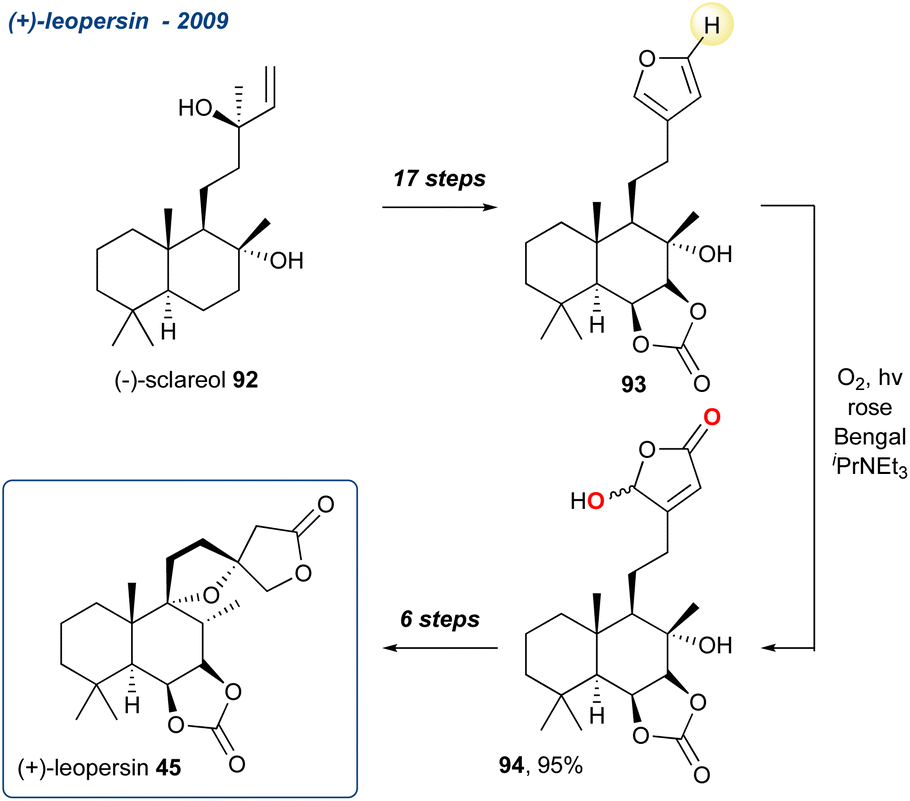
5.1.5 (+)-Leopersin, 2009. Marcos et al. reported the synthesis of (+)-leopersin D (Scheme 11; 45), a naturally occurring spirolabdanolide first isolated in 1996 from the aerial parts of Leonurus persicus.75
Scheme 11 Semi-synthesis of (+)-leopersin (45) from (−)-sclareol (92).
Starting their synthesis from (−)-sclareol (92) they accessed furan intermediate 93 in 17-steps and treated it with singlet oxygen in the presence of DIPEA yielding the γ-hydroxybutenolide 94 in 95% yield. The unmasking of a butenolide using a KDM rearrangement is discussed in more detail in Section 5.2. The synthesis was completed in a further 6-steps with 45 isolated with its C-13 epimer in a 1:1 ratio.
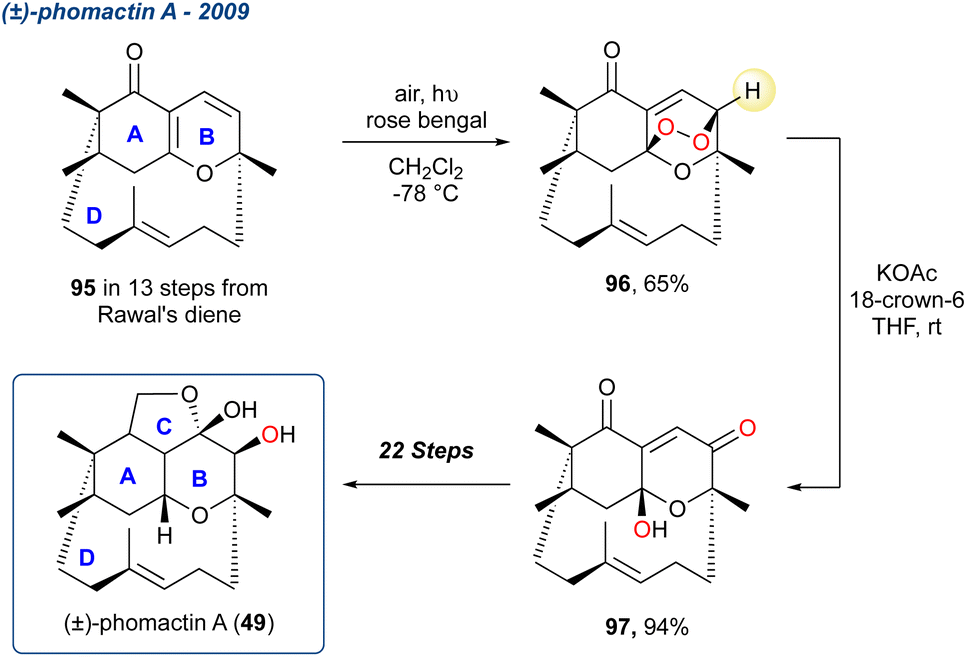
5.1.6 (±)-Phomactin A, 2009–2011. Tang et al. completed the total synthesis of (±)-phomactin A in 2009,38,76 a complex structure consisting of 4-rings (Scheme 12; 49). It was originally isolated by Sugano et al. from a culture of parasitic marine fungus Phoma sp. (SANK 11486) that was collected from the shell of a crab Chionoecetes opilio off the Japanese coast in the Fukui prefecture.77 Following their earlier work into the synthesis of the ABD-tricycle,78 the group utilised the KDM rearrangement after construction of the ABD-tricycle.79 During several attempts to cleave the remarkably stable endoperoxide bridge of 96 they attempted to install an acetate using a Michael addition with KOAc/18-crown-6; however, instead they found peroxide 96 was converted cleanly to the KDM product 97 with no hydrolysis to the corresponding ene-trione. As with the synthesis of (−)-isocelorbicol (41) (Scheme 6), the configuration of the resultant hydroxy group within 97 is controlled by the initial endoperoxide formation, which occurs on the top face of 96 due to ring-D. However, this stereocentre is inconsequential, and 97 was carried forward to give (±)-phomactin A 49 in a further 22-steps.
Scheme 12 Tang and co-workers' racemic total synthesis of (±)-phomactin A (49) using the unexpected KDM rearrangement using KOAc and 18-crown-6.
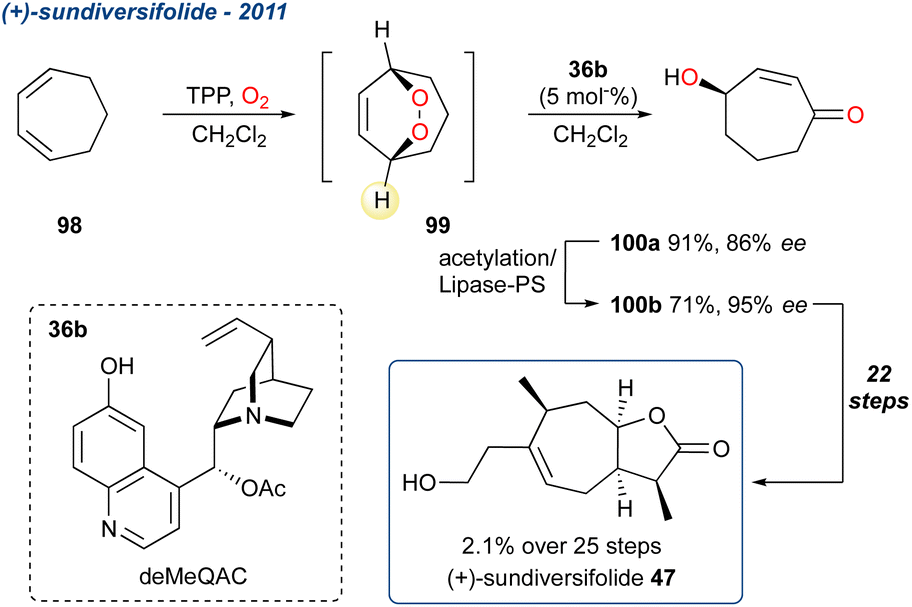
5.1.7 (+)-Sundiversifolide, 2011, enantioselective. Kawasumi et al. reported the formal synthesis of (+)-sundiversifolide 47, a 7,5-bicyclic lactone containing 4-stereocenters (Scheme 13).80 It was originally isolated from exudates of germinating sunflower seeds (Helianthus annuus L.) and demonstrated allelopathic activity.81 Following on the work of Shishido and co-workers, who used ring-closing metathesis to construct the 7-membered ring,82 the group opted for an oxidative approach, using cycloheptadiene (98) as the starting point. Oxidation with singlet oxygen yielded the symmetrical endoperoxide 99 which was then treated with catalytic deMeQAc to at ambient temperature, yielding the (R)-γ-hydroxyenone 100a in 91% yield and 86% ee. This represents the first use of Toste and co-workers organocatalytic desymmetrisation described in Scheme 4. The ee of 100a could be improved to 95% through a 2-step acetylation/lipase-PS protocol. Enantioenriched γ-hydroxyenone 100b was then taken forward to (+)-sundiversifolide 47 in an additional 22-steps and an overall yield of 2.1%.
Scheme 13 Kawasumi et al. approach to (−)-sundiversifolide (47) using an enantio-enriched y-hydroxyenone synthesised using Toste's organocatalytic symmetrisation of a centro-symmetric endoperoxide.
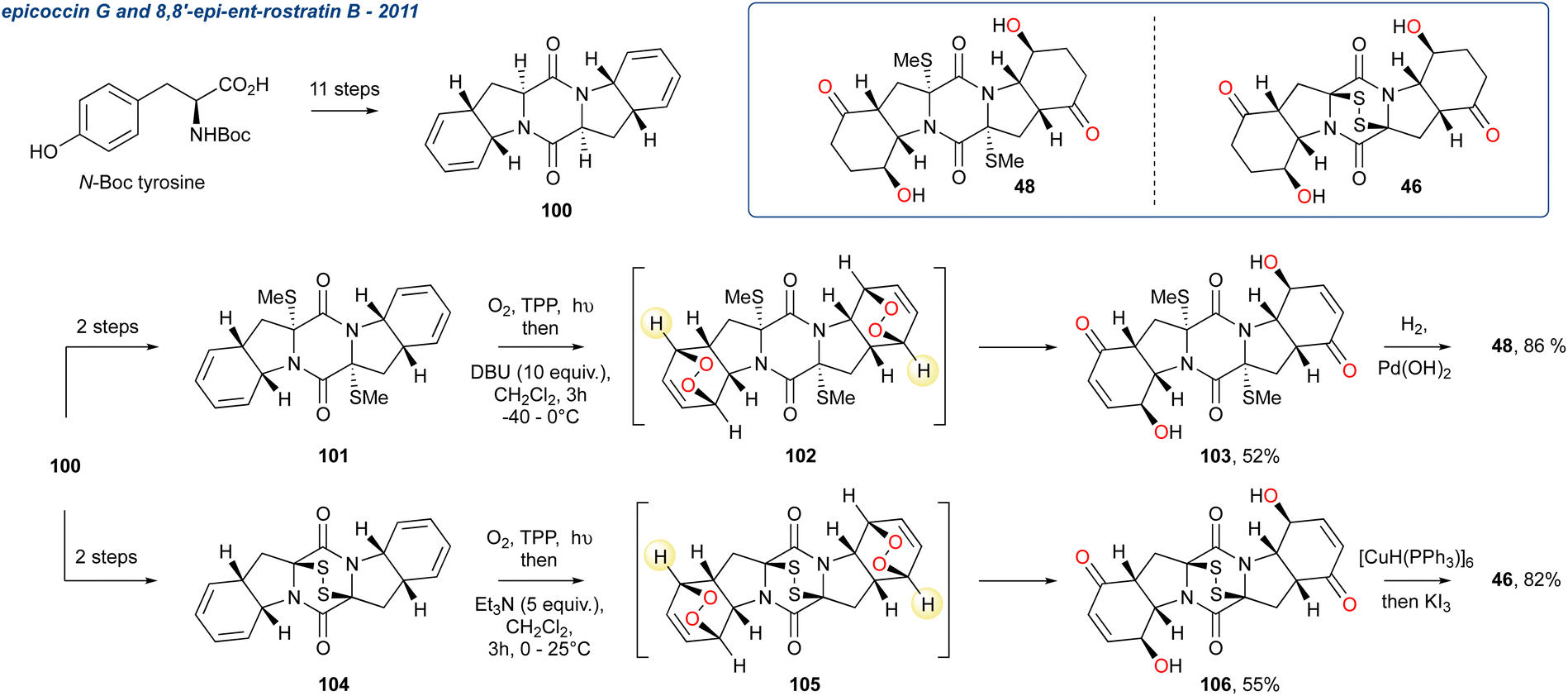
5.1.8 Epicoccin G and 8,8′-epi-ent-rostratin B, 2011. Two structurally similar diketopiperazines, epicoccin G (48) and 8,8′-epi-ent-rostratin B (46), were synthesised by Nicolaou et al. from a common intermediate 100 that was accessed from N-Boc tyrosine in 11-steps (Scheme 14).83,84 Using different sulfenylation strategies 100 was transformed into either epidithiodiketopiperazine 104 or the bis-methylthio compound 101 that were reacted with singlet oxygen, generating the endoperoxide intermediates, which were used in the subsequent KDM step without purification. The KDM rearrangement was regioselective, initiated using DBU or Et3N, and this gave 106 and 103 in 52 and 55% yields, respectively. The completion of each synthesis was via reduction of 103 to give epicoccin G (48), while a reduction/oxidation strategy was used to access 8,8′-epi-ent-rostratin B (48). Uniquely, the authors recognised that the γ-hydroxyenone present in 48 and 46 could be accessed efficiently by the rarely used, at the time of disclosure, KDM rearrangement.
Scheme 14 Use of a singlet oxygen formation/KDM rearrangement protocol to install the γ-hydroxyenones within epicoccin G and 8′8′-epi-entrostratin B.
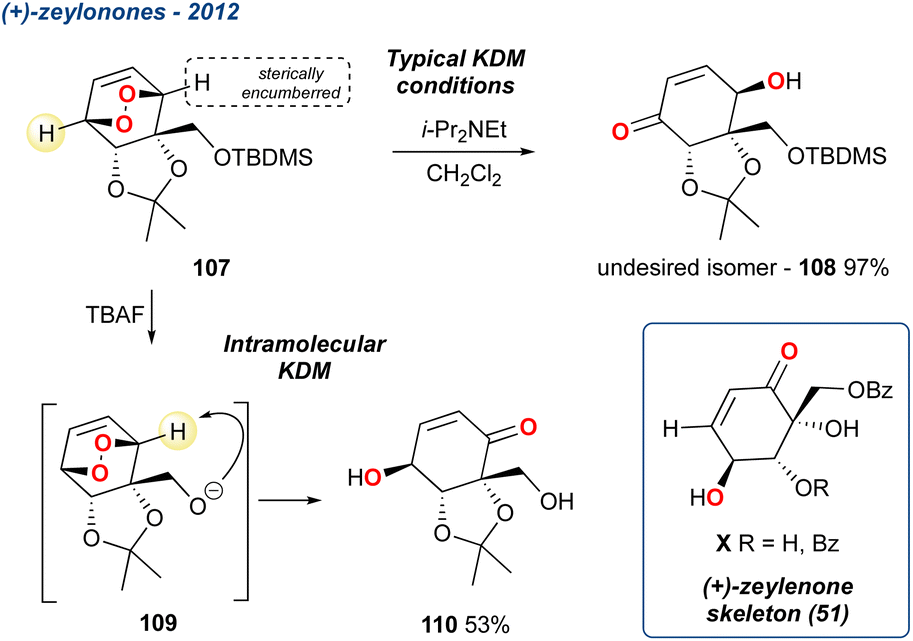
5.1.9 Toward (+)-zeylenones, 2012. In 2012, Palframan et al. reported the synthesis of a range of zeylenols85,86 and zelenones87,88 (Scheme 15, skeleton 51) using a common endoperoxide intermediate 107, by either reduction or KDM rearrangement, respectively.39 The KDM rearrangement, initiated by Hünigs's base, was used to access the zeylenone family, however the intrinsic regioselectivity, attributed to the steric approach of the base, prevented formation of the desired isomer providing, instead, the enone 108. To address the undesired regioselectivity, the group developed a neighbouring-group participation/directed approach to trigger an intramolecular KDM rearrangement using the neighbouring silyl protected alcohol.
Scheme 15 An intramolecular KDM rearrangement, through TBAF deprotection, reported by Palframan et al. in their synthesis of the zeylenone skeleton (51).
When treated with tetrabutylammonium fluoride (TBAF), the resultant alkoxide triggered an intramolecular KDM rearrangement giving the desired isomer in 53% yield, with oxy-anion 109 serving as the base; notably none of the undesired regioisomer 108 was observed. However, the KDM rearrangement product 110 was not taken forward in the synthesis due to its instability and instead the group used additional steps from the diol obtained from the reduced endoperoxide. Nevertheless, this methodology is a useful addition, demonstrating that the regioselectivity of the reaction can be tuned in some circumstances.
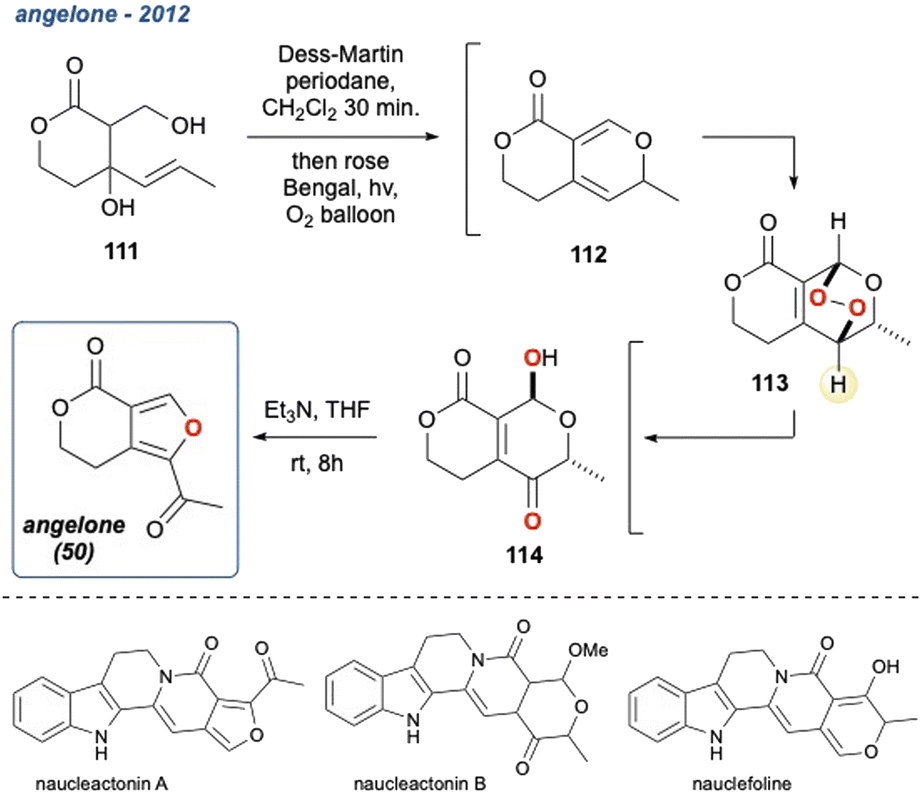
5.1.10 Angelone, 2012. A small bicyclic natural product angelone (Scheme 16; 50)89 was synthesised by Tan et al. using a biomimetic approach, where the team drew similarities between the biosynthesis of other indole alkaloids and devised a “three-pot” strategy that consisted of 6-steps.65 Lactone 114, was formed in 3-steps and the final one-pot reaction started with the treatment of 111 with Dess–Martin periodinane forming cyclic diene 112, followed by endoperoxide formation from singlet oxygen. The KDM rearrangement occurred spontaneously with excellent regioselectivity to the desired γ-hydroxyenone (114) and it was noted that no deprotonation at the α-position of cyclic oxygen was observed. It was speculated that the source of base for the KDM rearrangement arose from residual acetate as the Dess–Martin periodinane reaction and the one-pot nature of the reaction. To complete the synthesis, 114 was treated with Et2NH to initiate a 6π-disrotatory electrocyclic ring-opening cascade that yielded angelone 50 in 47% over the 3-steps and 35% overall. This synthesis was inspired by the biomimetic sequence postulated in the synthesis of structurally similar indole analogues such as naucleactonin A and B90 as well as nauclefoline,91 although the total synthesis of any these three intriguing natural products has yet to be divulged.
Scheme 16 Tan and co-workers' biomimetic synthesis of angelone (50), inspired by the proposed biosynthesis of indole alkaloids, naucleactonin A, B, and nauclefoline.
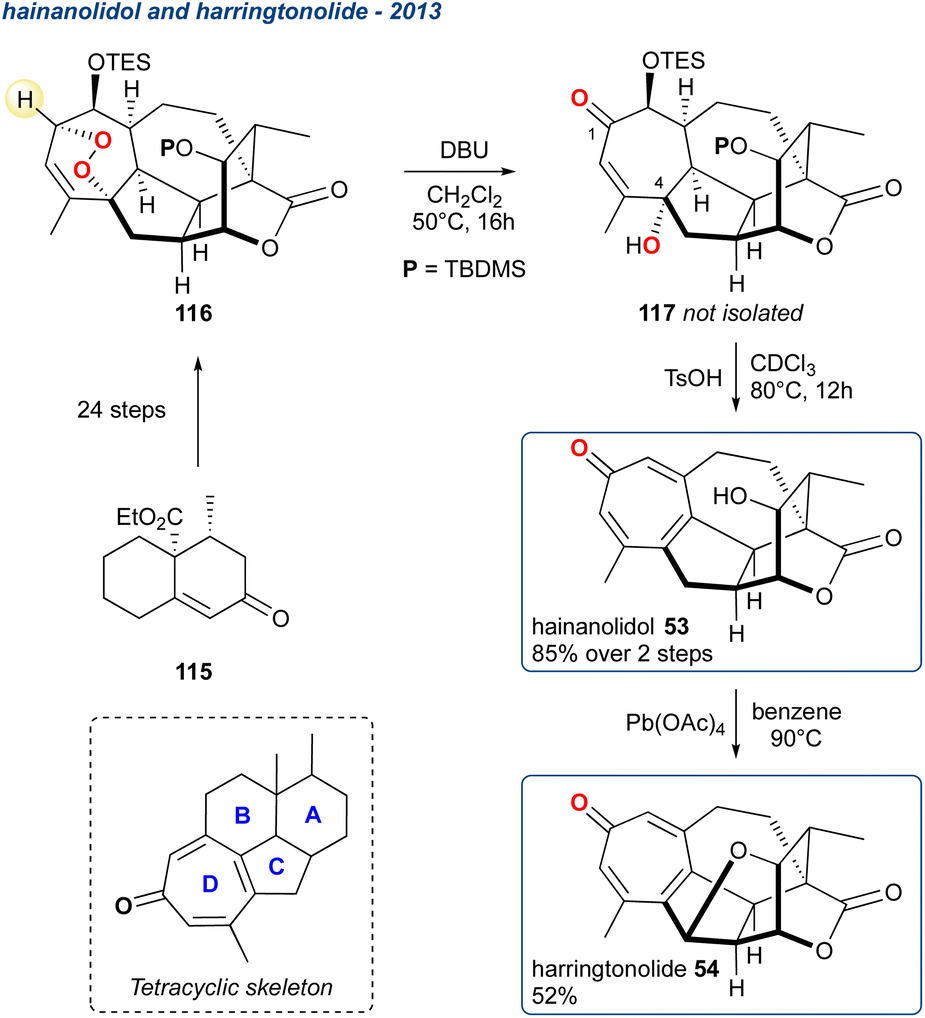
5.1.11 Hainanolidol and harringtonolide, 2013. The complex natural product harringtonolide (Scheme 17; 54) was isolated from C. harringtonia in 1978 where its structure was assigned by X-ray crystallography.92
Scheme 17 Zhang et al. synthesis of hainanolidol and harringtonolide.
A year later, 54 was isolated again but from C. hainanensis and this alongside the related compound 53 (hainanolidol, formerly named hainanolide) which was later proposed as the precursor to harringtonolide. Whilst 54 shows antineoplastic and antiviral activity the proposed precursor 53 is inactive, suggesting the tetrahydrofuran ring is important for their bioactivity. These two related natural products were synthesised in the laboratory by Zhang et al. starting from enone 115 producing peroxide 116 in 24-steps, which was then subjected to KDM conditions using DBU as the base furnishing the γ-hydroxyenone 117.93 The stereochemistry of the hydroxy group at C4 is a consequence of the [4 + 2]-cycloaddition in the preceding endoperoxide forming reaction, although this is inconsequential given it is subsequently eliminated. Additionally, 117 is used without further purification, being treated immediately with TsOH that gave deprotection and elimination forming the key tropone D-ring (see tetracyclic skeleton) and completing the synthesis of hainanolidol 53, which was isolated in 85% over the 2-steps. Treatment of 53 with Pb(OAc)4 produced the tetrahydrofuran ring, thereby forming harringtonolide 54 in 52%.
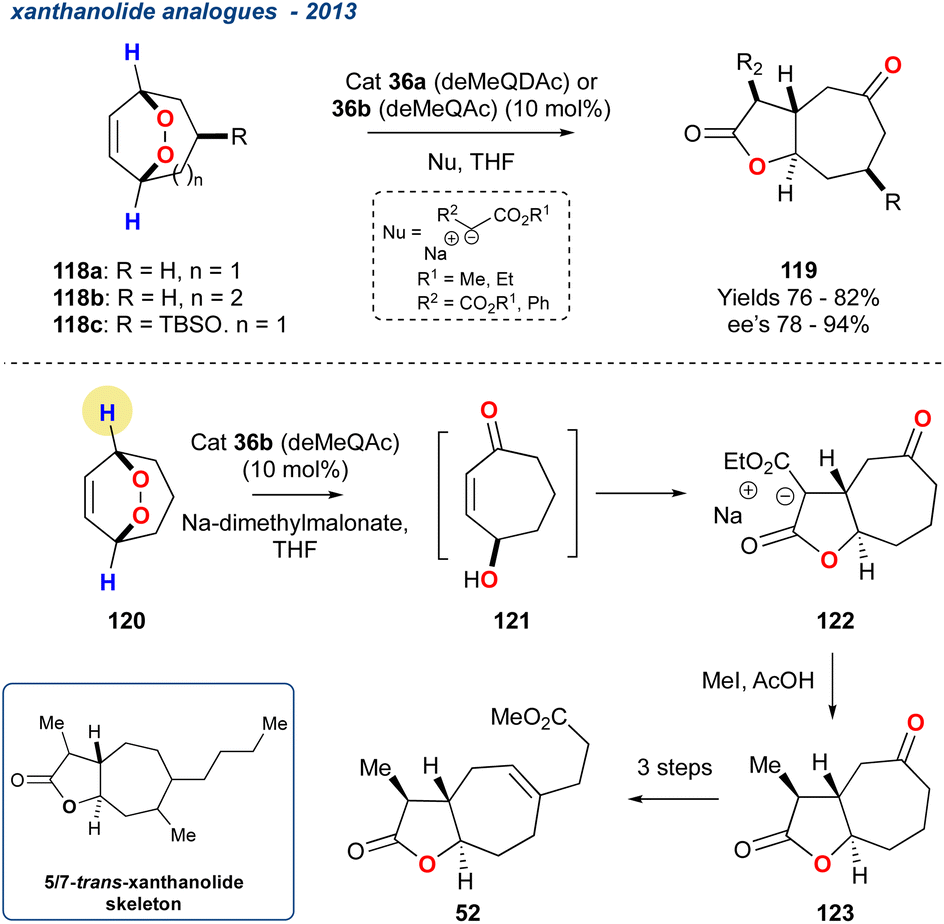
5.1.12
trans-Xanthanolide analogues, 2013. Priest et al. made use of the Toste organocatalysed asymmetric KDM43 rearrangement as an intermediatory step in the formation of trans-fused butyrolactones (Scheme 18; 119) from readily accessible symmetrical endoperoxides (118).44
Scheme 18 Priest and co-workers use of the desymmetrisation of a prochiral endoperoxides to provide a rapid synthesis of the trans-xanthanolide skeleton.
Catalysts 36a or 36b (see Scheme 4) were used to promote the asymmetric reaction leading to the desired γ-hydroxyenones, that were then treated in situ with various nucleophiles followed by spontaneous lactonization to yield the desired butyrolactones (119) in high yields and ee's of up to 94%. The chemistry was then applied to the synthesis of a trans-xanthanolide analogue 52 with the installation of the methyl group with MeI followed by decarboxylation in AcOH to yield the desired butyrolactones 123 in 79% yield and good diastereoselectivity. An alternative strategy using α-methyldiethylsodiomalonate yielded a similar overall yield (69%). The trans-xanthanolide analogue 52 was realised in 51% yield over an additional 3-steps.
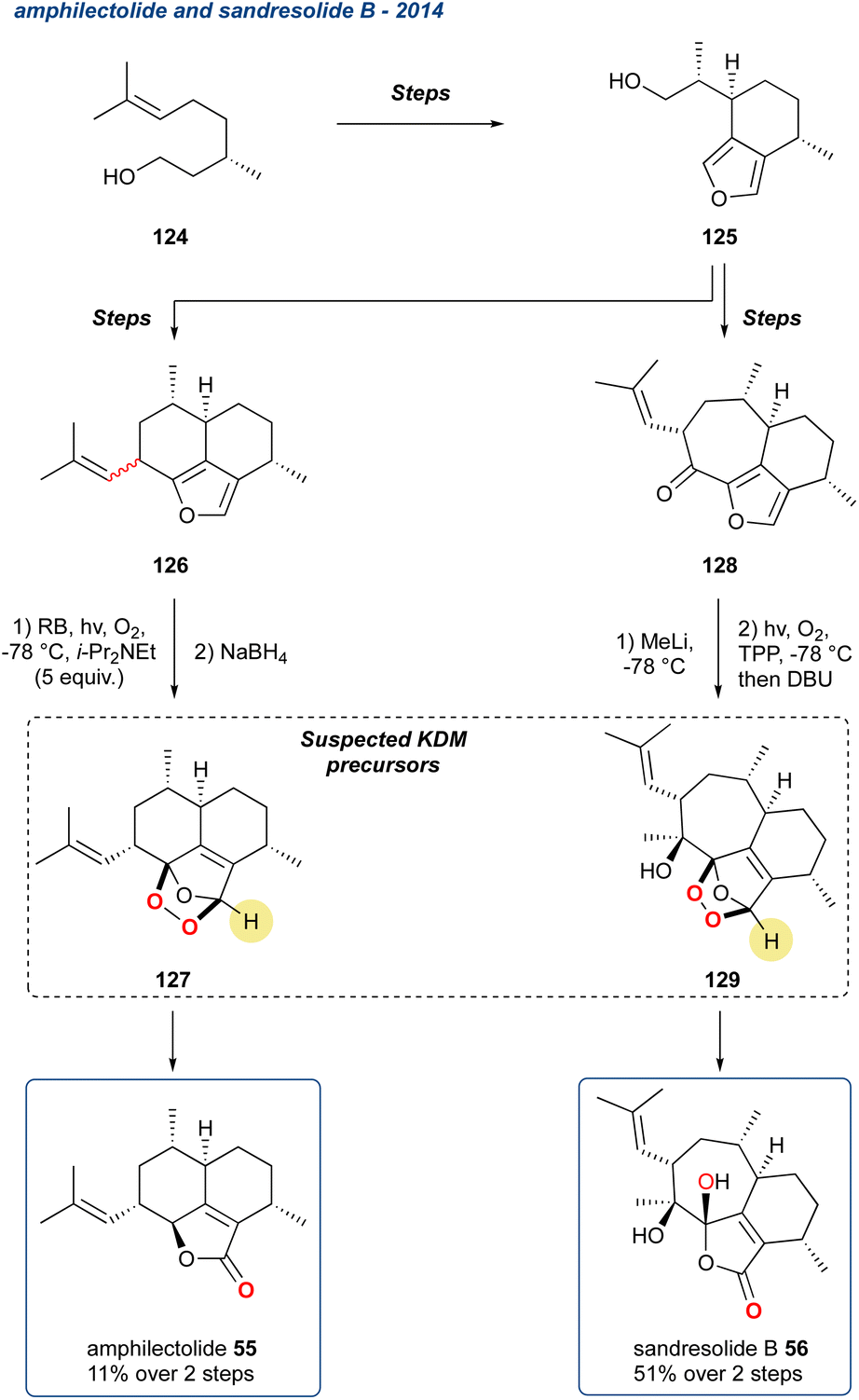
5.1.13 Amphilectolide and sandresolide B, 2014. Two related natural products, amphilectolide (55) and sandresolide B (56) were synthesised by Chen et al. from a common intermediate where a regioselective KDM rearrangement was employed in the final step of the synthesis (Scheme 19).94
Scheme 19 The synthesis of amphilectolide (55) and sandresolide B (56) via a suspected KDM rearrangement from endoperoxide precursors 127 and 129, respectively.
Belonging to a large family of marine metabolites, originally isolated from Pseudopterogorgia elisabethae, a Caribbean coral, natural products of this type demonstrate a broad range of biological activity, i.e., inflammation, tuberculosis, anti-cancer, and antiplasmodial.95 To complete their synthesis of amphilectolide (55) the researchers treated the diastereomeric mixture of 126 with singlet oxygen in the presence of Hünigs base (i-Pr2NEt), and then immediately reduced with NaBH4 yielding 55 in 11%, from a complex mixture of products, over the 2-steps (Scheme 19). It is unclear from the report whether the reaction proceeded by a KDM rearrangement, but given the conditions, the presence of a carbonyl at the 5-membered ring, it is highly likely the KDM precursors was 127. The other target, sandresolide B (56), was obtained in 51% yield over 2-steps, where methylated 128 was subjected to, after some optimization, photo-oxidation with tetraphenylporphyrin (TPP) followed by treatment with DBU to trigger the regioselective KDM rearrangement. During optimisation, the authors found that DBU was the only base to give efficient transformation of the peroxide intermediate.
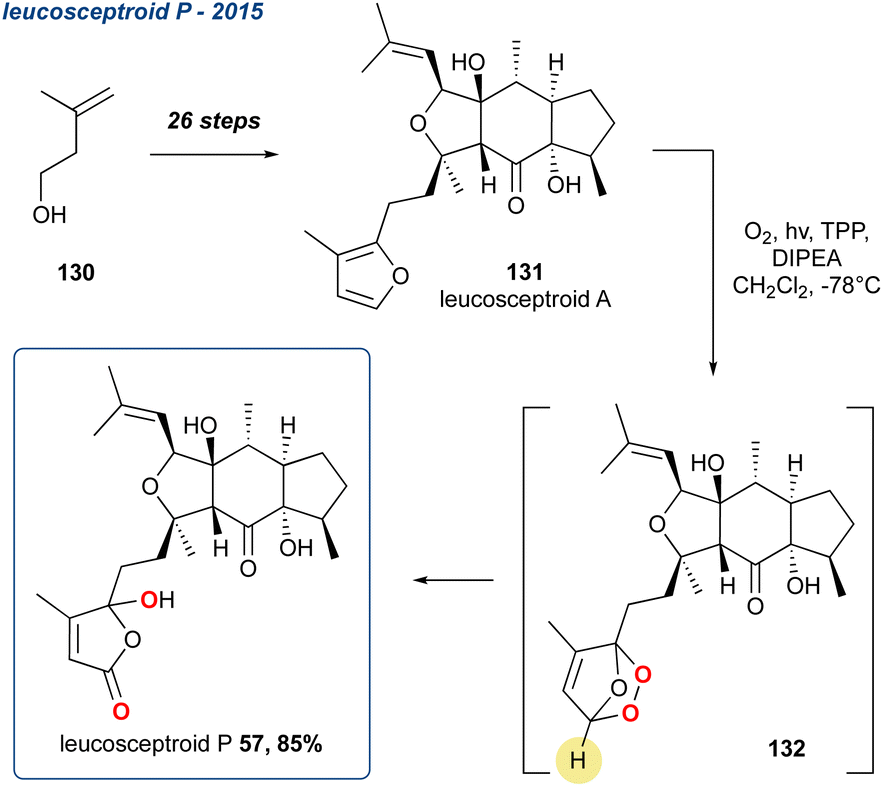
5.1.14 Leucosceptroid P, 2015. Hugelshofer et al. reported the synthesis of a range of leucosceptroids, including a leucosceptroid P (57) which was proposed via oxidation and KDM rearrangement of leucosceptroid A (131) (Scheme 20).96,97
Scheme 20 Hugelshofer et al. use of the KDM rearrangement in the synthesis of leucosceptroid P. Singlet oxidation of the furan is unselective, therefore leucosceptroid is obtained as a 1:1 mixture.
They are a family of terpenes isolated from Leucosceptrum canum that exhibit antifeedant activities. Building upon their related synthesis of norleucosceptroids A–H, the researchers synthesised leucosceptroid A (131) in 26-steps from alcohol 130.98 Oxidation with singlet oxygen yielded the corresponding endoperoxide (132) which was then converted in situ by Hünigs base forming the γ-hydroxyenone regioselectively in 85% yield. Due to a lack of substrate control, the oxidation of the furan ring by singlet oxygen provides a mixture of endoperoxides. This was predicted by the authors given their previous disclosure on the biomimetic studies on this family of natural products.97 The KDM rearrangement therefore gives a 1:1 mixture of diastereoisomers in the furan ring, endoperoxide was formed racemically, therefore leading to leucosceptroid P (57) to be obtained as a 1:1 mixture of diastereoisomers at the carbon containing the newly formed hydroxyl group. This is inconsequential as the natural product is originally obtained as a 1:1 mixture.
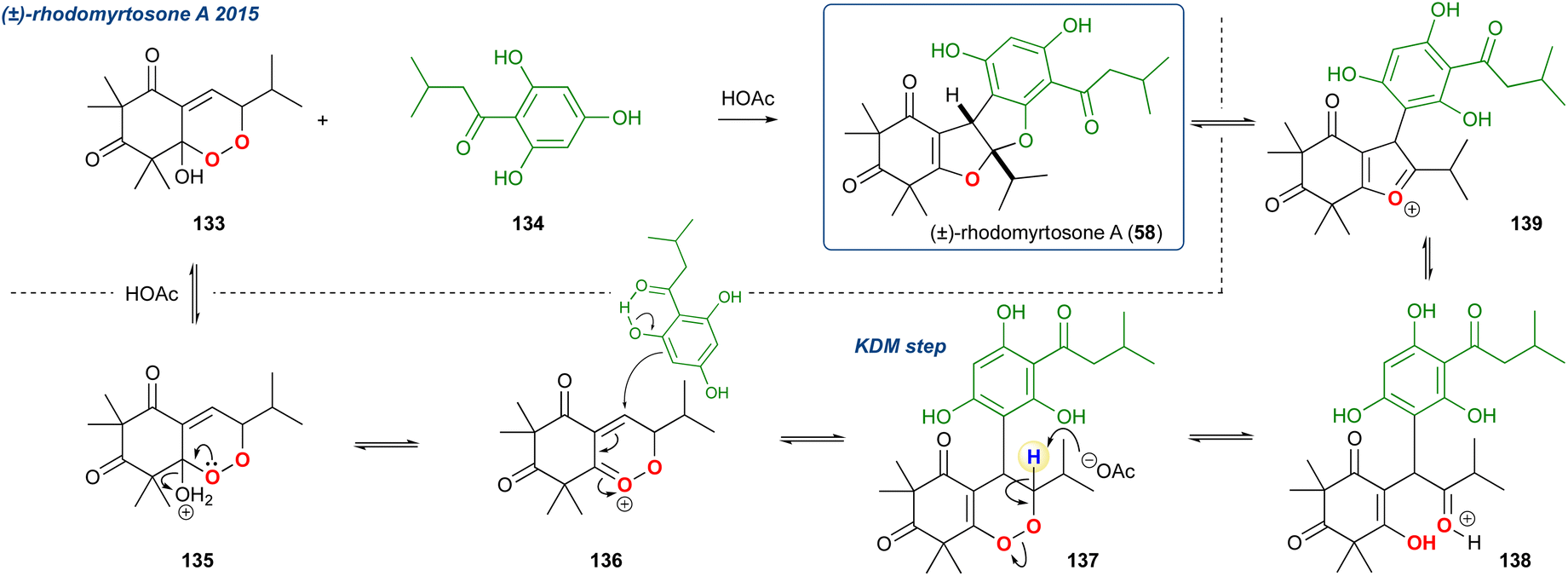
5.1.15 Rhodomyrtosone A and related, 2015. Gervais et al. reported the total synthesis of rhodomyrtosone A (58), a bis-furan β-triketone natural product that was isolated from Rhodomyrtus tomentosa.99 In their approach they synthesised endoperoxide 133 in 4-steps and then heated it in acetic acid at 100 °C in the presence of acylphloroglucinol 134 to give 58 in 60% yield (Scheme 21). Although no mechanistic studies were performed, it is proposed that the KDM rearrangement is part of a cascading sequence of steps starting with protonation of the peroxy acetal 135. Subsequent elimination of water forms peroxycarbenium intermediate 136 which then undergoes Michael-type addition by 134. Removal of the acidic proton adjacent to the peroxide of addition product 137 in KDM fashion yields diketone 138 which then cyclises twice under the acidic conditions with elimination of water yielding the desired natural product 58.
Scheme 21 Gervias et al. approach to (±)-rhodomyrtosone A. A unique KDM rearrangement occurs due to an initial conjugate addition to the peroxide oxycation 136.
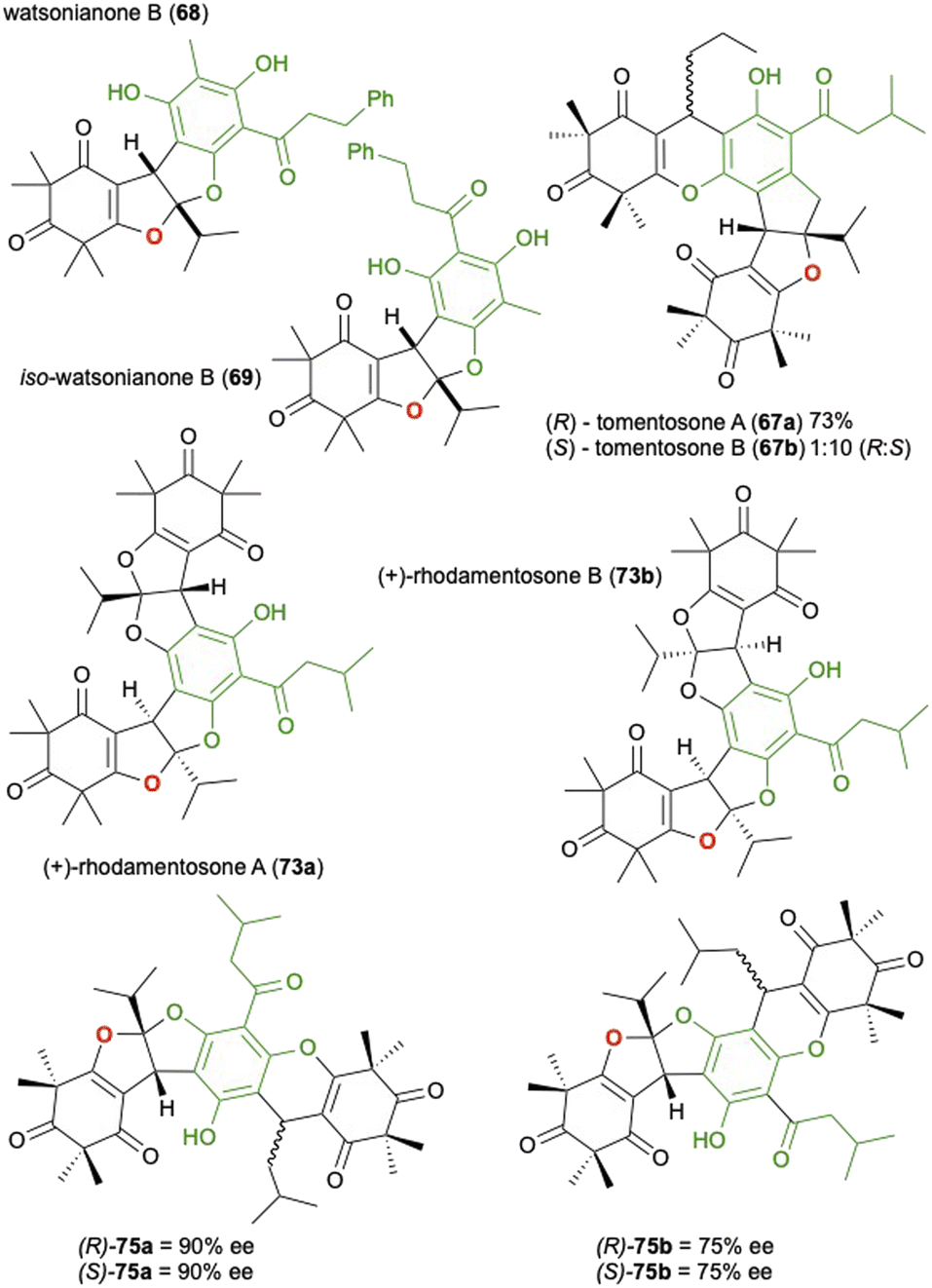
Following the initial report of this synthesis by Gervais et al. there have been several reports utilizing the same approach, occasionally with minor modification to the conditions, to realise a variety of related natural products isolated from Rhodomyrtus tomentosa or other related plants in the same genus. Watsonianone B (68) and its isomer (69) were synthesised by Zhang et al. in a 2:1 ratio and 27% isolated yield for the desired 68 (Fig. 2).100 Tomentosones A (67a) and B (67b) were also reported by the same group and synthesised in a similar manner, only this time using pyridinium p-toluenesulfonate (PPTS) in toluene which yielded a 1:10 mixture of 67a and 67b in 73% yield.101 They also reported that the two isomers could be interconverted in the presence of p-toluenesulfonic acid. The following year Deng et al. reported the asymmetric synthesis of rhodomentosones A (73a) and B (73b), a pair of phloroglucinol trimers that contain an unusual 6/5/5/6/5/5/6 fused ring system.102 In this report the group described an asymmetric approach to the domino reaction first reported by Gervais et al. by employing a catalytic amount of a chiral phosphoric acid in the presence of AlF3 as a Lewis acid they could access the desired bis-furan in high ee. After separation of the isomers, the second bis-furan unit was installed giving rhodomentosone A (73a) and B (73b) in a ratio of 1:2 with 90% ee and a combined yield of 45%. This asymmetric approach was applied again by Deng et al. in the synthesis of four unnamed phloroglucinol trimers (75a and 75b) isolated again from Rhodomyrtus tomentosa.103
Fig. 2 The synthesis of related natural products from Rhodomyrtus tomentosa or related plants in the same genus using the cascade approach described in Scheme 20.
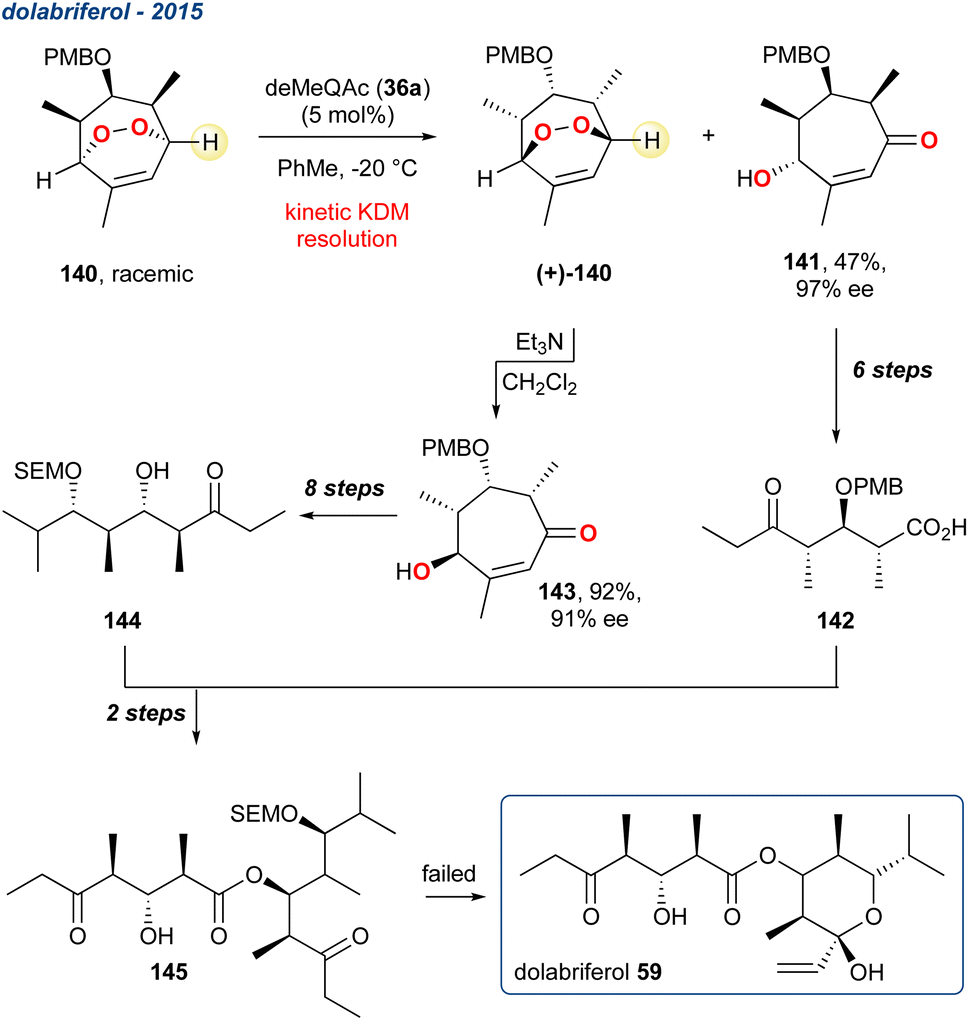
5.1.16 Dolabriferol, 2015, enantioselective. Dolabriferol (58) belonging to the family of noncontiguous polypropionates, is a marine natural product isolated from Dolabrifera dolabrifera and was isolated from the acetone extracts of sea hare (Anaspidean mollusk) that was collected off the Cuban coast.104 Gesinski et al. reported the attempted total synthesis of 58 in 2015, which centred around a divergent/convergent strategy that utilised a KDM kinetic resolution as the key step (Scheme 22).105
Scheme 22 The use of an organocatalyst in a KDM rearrangement and kinetic resolution in the attempted total synthesis of dolabriferol 59.
The two enantioenriched fragments of the target were constructed from a common racemic peroxide 140. The authors used the established organocatalysts deMeQAc (36a), but unlike the centrosymmetric desymmetrisation described by Stanben et al.,43 the authors used a racemic endoperoxide to achieve a kinetic resolution. The regioselectivity of the KDM rearrangement is central to this approach, given the highlighted proton in 140 is less sterically encumbered, and therefore more likely to be deprotonated in the key KDM rearrangement step. After some optimisation, they found that in toluene and when 140 was treated with deMeQAc (36a) a selective deprotonation occurred providing enone 141 as a single enantiomer in 47% yield and 97% ee. The remaining enantioenriched peroxide (+)-140 could then be cleanly converted using a subsequent KDM rearrangement but using Et3N, providing enantioenriched enone 143 in 92% yield and 91% ee. Each fragment was then taken forward in several steps and recombined, but unfortunately the synthesis of 59 failed on the last step.
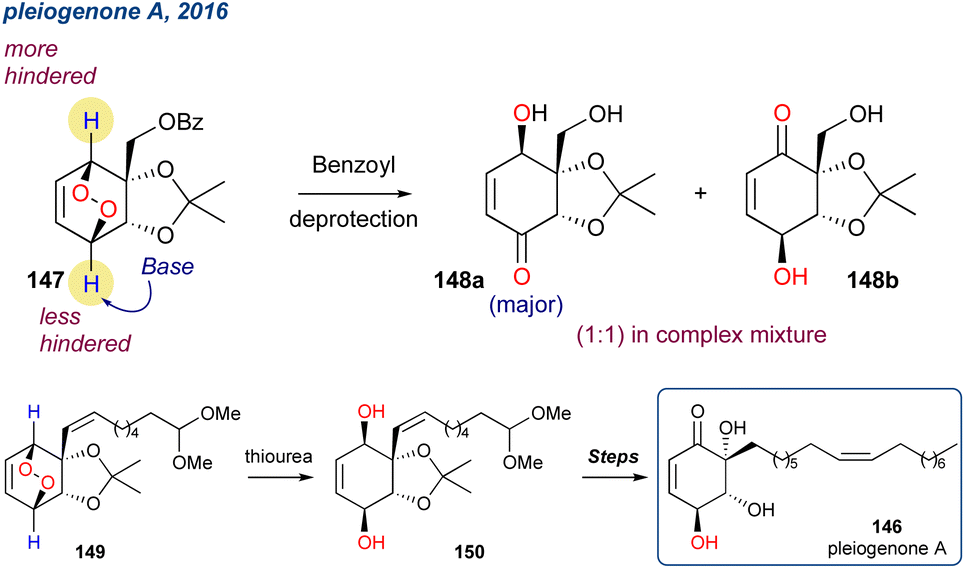
5.1.17 Pleiogenone A, 2016. Froese et al. reported the first total synthesis of pleiogenone A (146),106 a hydroxylated cyclohexenone that was isolated from the bark of Pleiogynium timorense with demonstrated potent biological activity (Scheme 23). In their approach they used acetal protected ipso-diol, synthesised with an enzymatic dihydroxylation, as the starting point. The subsequent endoperoxide 146 was then subjected to base catalysed KDM rearrangement conditions but suffered from undesired regioselectivity from abstraction of the least hindered proton as reported by Lewis and co-workers.107 The group attempted to direct the proton abstraction with benzoyl deprotection (conditions not given in the original report) to yield an oxo-anion to serve as the base for the KDM rearrangement step, however this approach yielded equal amounts of 147a and 147b as part of a complex mixture and their isolation proved too laborious to be viable moving forward. Ultimately, they completed the synthesis using an alternative approach and installed the γ-hydroxyenone core by reduction of a peroxide with thiourea to yield a diol and then a selective TIPS protection and IBX oxidation (148 to 149 to 150).
Scheme 23 Attempted synthesis of a key precursor (147) of pleiogenone A via a KDM rearrangement.
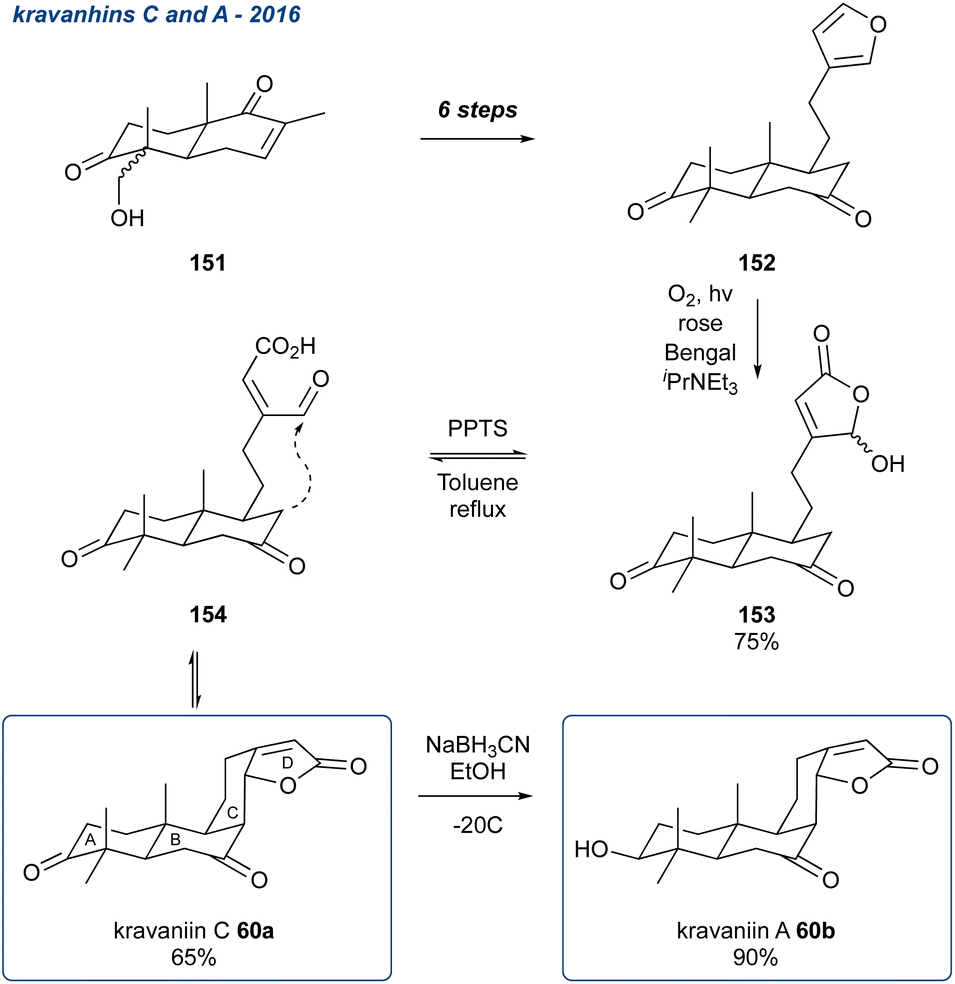
5.1.18 Kravanhins C and A, 2016. Zhong and co-workers reported an innovative ‘bioinspired’ synthesis of kravanhins A and C (Scheme 24; 60a and 60b).108 These natural products were isolated from the fruits of the Amomum kravanh; which are used in traditional Chinese medicine to treat digestive disorders.109 The synthesis began with 151, obtained in 4-steps from (−)-carvone, which was then carried forward in a further 6-steps to the furan 152. The authors then unmasked the furan using the Faulkner method (see Section 5.2)110 to reveal the γ-hydroxy butenolide 153 in 75%. The authors postulated that the ring opened form 153 would undergo an aldol cyclisation to form ring C; this was ultimately realised using PPTS in refluxing toluene, to give kravanhin C (60b) in 65%. Subsequent stereoselective reduction of the carbonyl in ring A then gave kravanhin A (60a) in 90% yield.
Scheme 24 Synthesis of kravanhins A (60b) and C (60a) using an oxidative ring opening of a 3-substituted furan and subsequent aldol condensation.
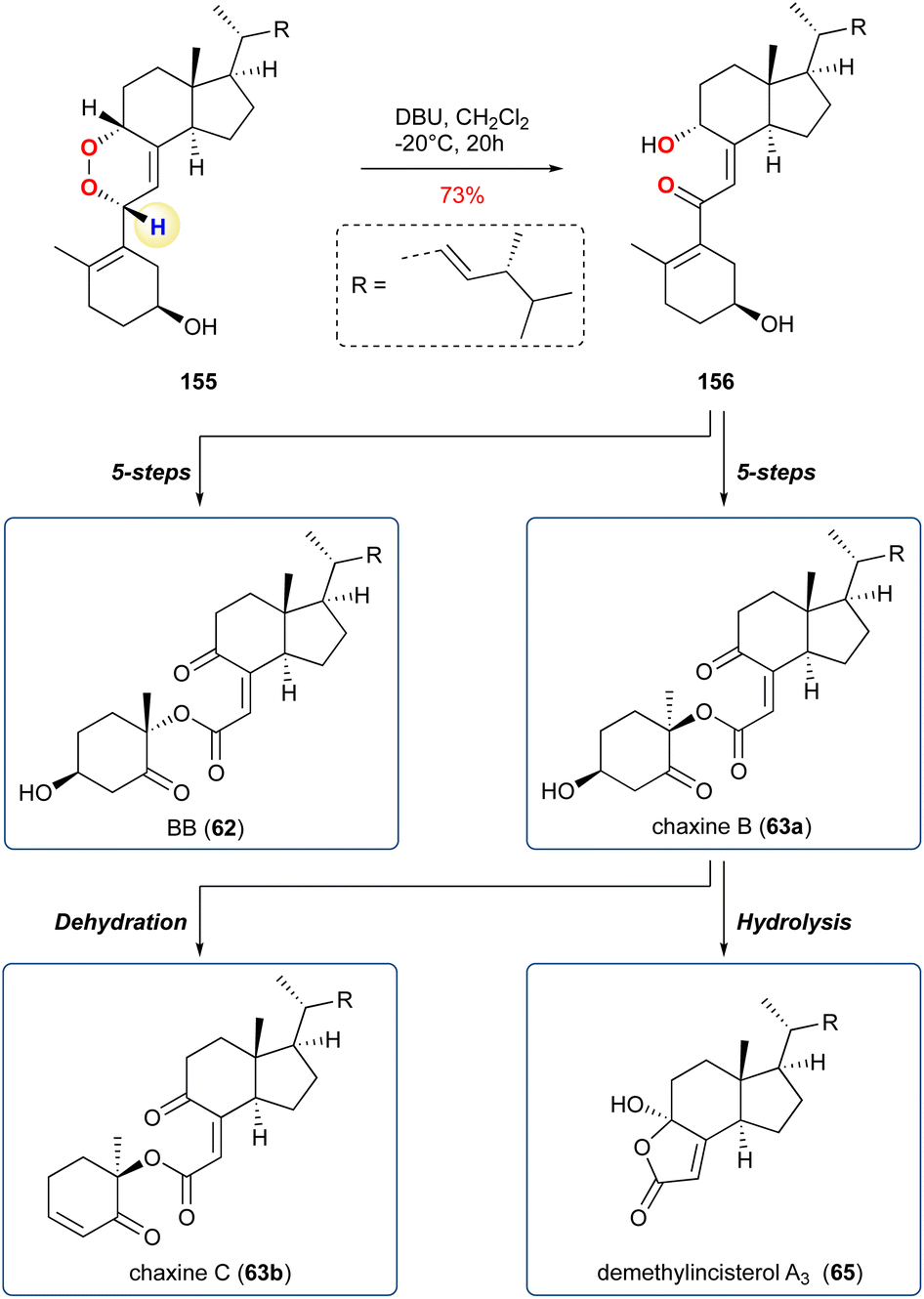
5.1.19 Chaxine B, C and related analogues, 2017, 2020. Hirata et al. reported the synthesis and structural revision of chaxine B and related natural products, which have been isolated from fungi and marine animals, with ergosterol as the starting point (Scheme 25).111 Following their proposed biosynthetic route they planned a KDM rearrangement, which would lay the path for the synthesis of a key 1,4-diketone intermediate for a Baeyer–Villiger oxidation. From the endoperoxide 155 they used DBU as the base and obtained 156 in 73% yield and as a single product, with regioselectivity likely arising from the increased acidity of the proton in the bis-allyl position (Scheme 24 – in blue/yellow). Following the synthesis chaxine B (63a) and its epimer 62 (so called BB) which were achieved in 5-steps each and chaxine C (63b) could be obtained through dehydration. In a subsequent report, the group applied the chemistry to a range of unnatural analogues and in addition demonstrated that demethylincisterol A3 (65) could be obtained through hydrolysis of 63a.112
Scheme 25 The KDM rearrangement was employed by Hirata and co-workers to deliver a key cis γ-hydroxyenone.
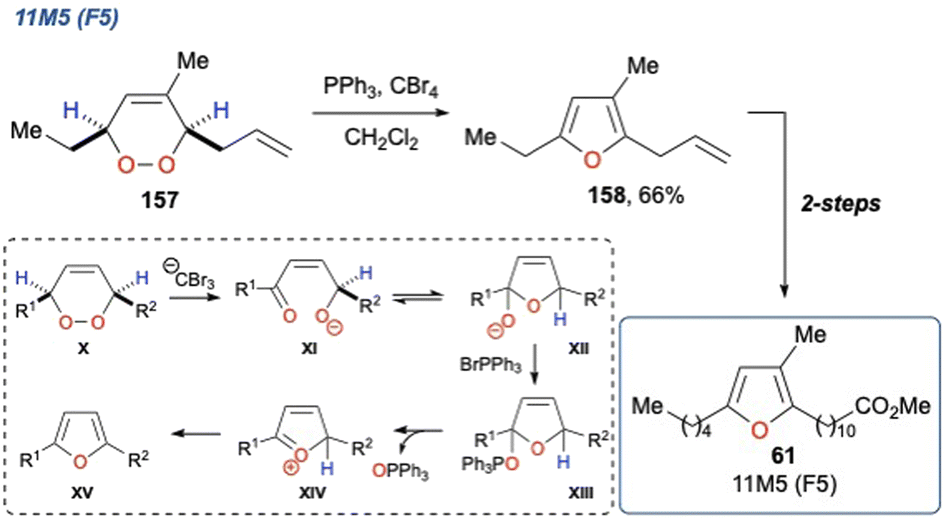
5.1.20 11M5 (F5), 2017. Lee et al. reported the total synthesis of the furan fatty acid 11M5 (F5) (61), a class of natural products and metabolites that have demonstrated activity toward a range of inflammatory diseases (Scheme 26).35
Scheme 26 Lee et al. use of the Appel reagent in a KDM rearrangement, and its application to the synthesis of 11M5.
In their synthesis, they employed Appel reaction conditions (PPh3, CBr4) on endoperoxide 157 (which was synthesised in 4-steps) to perform a dehydrative cyclisation to yield furan 158 in 66% yield. In this case the in situ generated CBr3− was used as a base to initiate the KDM rearrangement step yielding enone XI, and upon cyclisation to XII and subsequent dehydration with PPh3Br+ the desired furan XV could be realised. It is not known which proton is deprotonated in the initial KDM step (X to XI); however, this is immaterial given that final product destination is the fully dehydrated furan. The synthesis of 61 was completed with a cross metathesis and hydrogenation to install the ester functionality. The researchers also demonstrated the scope and versatility of the reaction on a range of substrates, yielding a variety of multi-substituted furans in 50–98% yield. The authors framed this as a biosynthetically inspired route to these important natural products, given the biosynthesis of furan fatty acids in marine bacteria.113
5.1.21
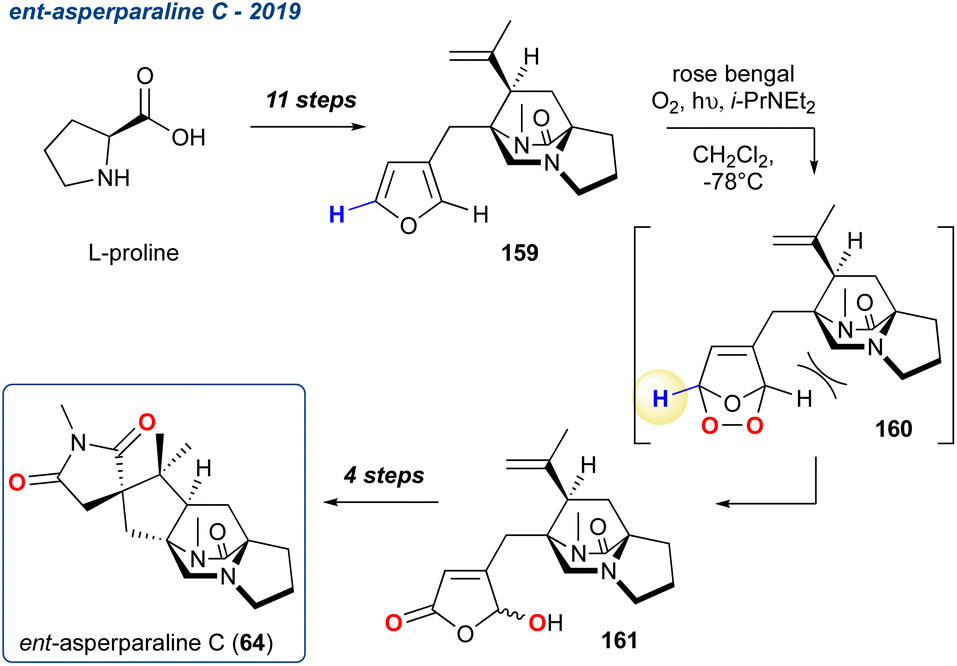
Ent-asperparaline C, 2019. Recently, Dokli, et al. reported the total synthesis of ent-asperparaline C (64), a natural product from a wider family of alkaloids containing a diketopiperazine unit, and a smaller subfamily that contains a bridged diazabicyclo-[2.2.2]-octanone core (Scheme 27).114 Asperparalines A–C were isolated from Aspergillus japonicus and have shown strong paralytic effects on silkworms.115 In their reported synthesis of 64, the authors accessed furan 159 in 11-steps from L-proline, which was then converted to the γ-hydroxyenone 161 in 85% yield using singlet oxygen and Hünigs base in one-pot. This reaction showed high regioselectivity for the desired product, likely due to the bulky group on one side of the furan ring, in addition, the authors noted that 161 was isolated as an inseparable mixture of diastereoisomers in which the ratio could not be determined. It was also noted that no oxidation or ring fragmentation occurred under the conditions of oxidation-KDM rearrangement. Another 4-steps took 161 to the completed synthesis of 64.
Scheme 27 The use of a regioselective KDM rearrangement by Dokli et al. in their synthesis of ent-asperparaline C (64).
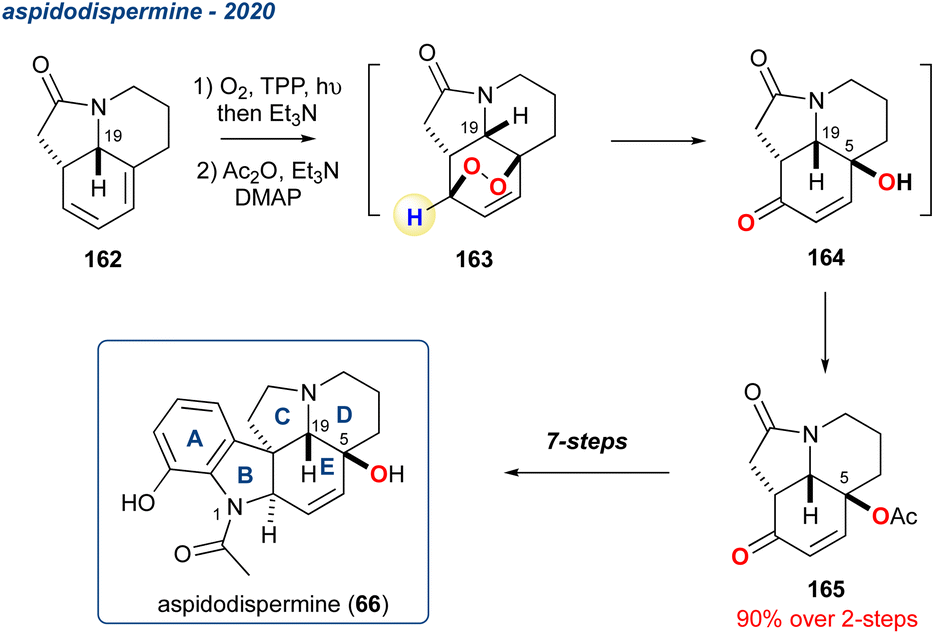
5.1.22 Aspidodispermine, 2020. Reuß and Heretsch reported the use of a KDM rearrangement in a route to aspidodispermine (66), a member of the pyrroloquinoline alkaloids (Scheme 28).116 Diene 162 was accessed in 10-steps and then subjected to photo-oxidation conditions to yield the endoperoxide which was reacted in situ with Et3N with regioselectivity coming from the presence of the quaternary carbon in the diene. The KDM rearrangement product was then acetyl protected without isolation, yielding 165 in 90% over the 2-steps. The stereochemistry of the hydroxyl group at C5 on 165 is installed via the initial photooxidation, which occurs on the convex face (syn to the H at C19). The subsequent regioselective KDM then provides the desired stereochemistry at C5. The synthesis of the target 66 was completed in an additional 7-steps.
Scheme 28 A regioselective KDM rearrangement in the synthesis of aspidodispermine (66).
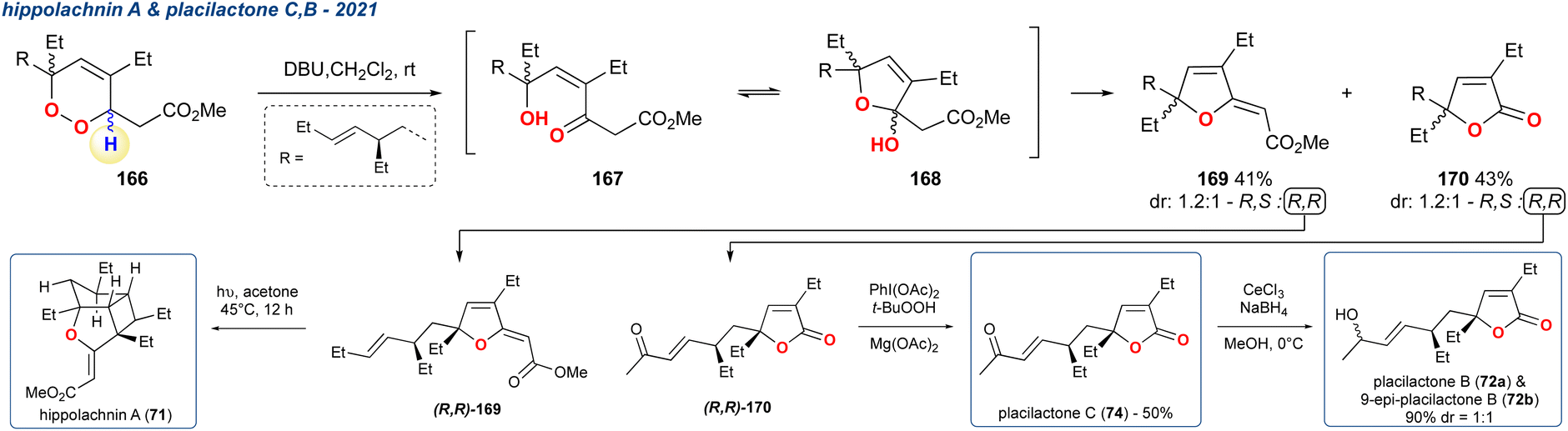
5.1.23 (+)-Hippolachnin A and plakilactone C, B, 2021. Several Plakortin polyketides were synthesised by Li et al. and among them was (+)-hippolachnin A (71), a tricyclic marine natural product isolated from a marine sponge Plakinastrella mamillaris (Scheme 29).117–119 In this report the group employed the KDM rearrangement as a key strategic step in their synthesis. After accessing racemic peroxide 166 in 14-steps it was treated it with DBU as the base, forming the KDM rearrangement product (167) which upon cyclisation formed hemiketal 168. Hemiketal 168 was found to undergo elimination yielding two major products 169 and 170 as a 1.2:1 mixture of diastereoisomers, of which the R,R-isomer (R,R-169) underwent UV-induced [2 + 2]-cyclisation to complete the formal synthesis of (+)-hippolachnin A (71). The group also demonstrated that the R,R-isomer of the undesired product (R,R-170) obtained from the KDM rearrangement step could be taken forward to access another series of natural products plakilactone C (74) and B (72) in 1- and 2-steps, respectively.
Scheme 29 Synthesis of hippolachnin A and placilactone C and B.
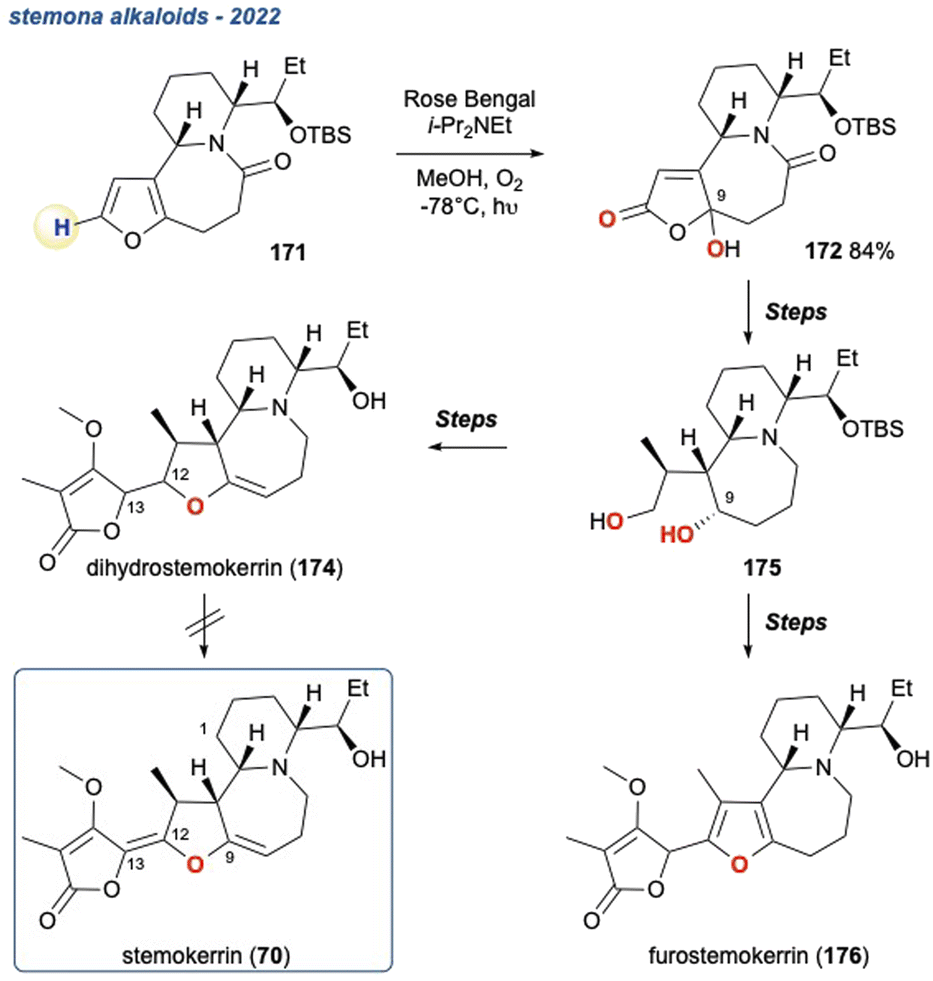
5.1.24 Toward stemokerrin – pyrido[1,2-a]azepine stemona alkaloid skeleton, 2022. In an attempt to synthesise stemokerrin, a natural product of the pyrido[1,2-a]azepine subset of Stemona alkaloids, Morgenstern et al. employed a KDM rearrangement in the latter half of their synthesis (Scheme 30).120 The peroxide intermediate was generated from furan 171 and quenched in situ by i-Pr2NEt, in this case the regioselectively of the KDM rearrangement was due to the availability of a single H-atom. The γ-hydroxybutenolide 172 was then taken forward in their attempts to synthesise 70 and ultimately resulted in 2 non-natural products; dihydrostemokerrin 175 and furostemokerrin 176, when attempts to construct the C12–C13 double bond of 174 proved unsuccessful.
Scheme 30 Morgenstern et al. use of the KDM rearrangement to install a γ-hydroxybutenolide in their unsuccessful synthesis of stemokerrin. However, two non-natural products dihydrostemokerren and furostemokerrin could be accessed.
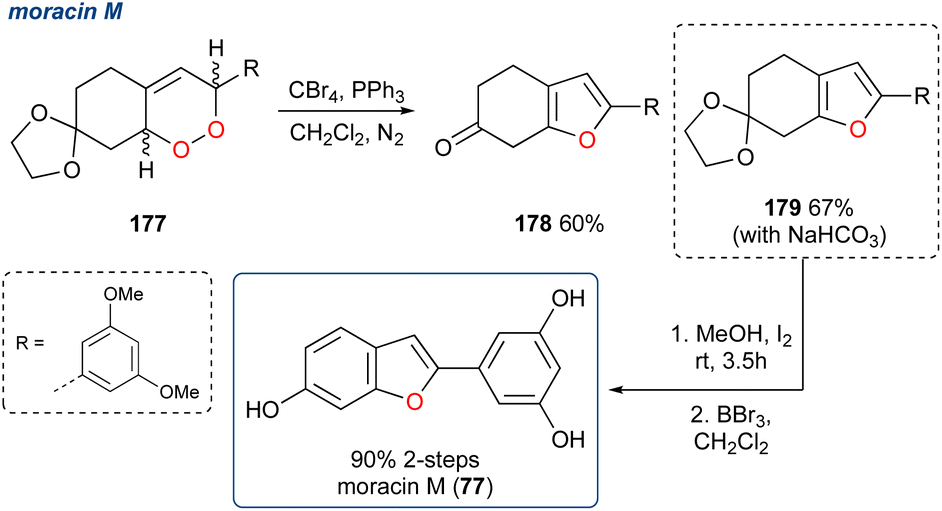
5.1.25 Moracin M, 2023. Using the same Appel approach to synthesise functionalized furans (Scheme 31), Al-Jawaheri et al. completed the synthesis of the benzofuran natural product moracin M (77).121
Scheme 31 Synthesis of the benzofuran, moracin M (77) using an Appel reagent mediated KDM rearrangement.
Using their reported route to 1,3-dienes122,123 they synthesised endoperoxide 177 and subjected it to Appel conditions, which unexpectedly yielded ketone 178 in 60% yield. Here the furan was successfully constructed but due to the build-up of HBr during the reaction the acetal deprotection occurred and attempts to aromatise 178 failed. A minor modification to the procedure saw the inclusion of NaHCO3 to quench the HBr in situ and prevent deprotection and under these conditions acetal 179 was obtained in 67% yield. The synthesis of moracin M (77) was completed in 90% yield over two deprotection steps.
5.2 Unmasking 3-substituted furans to reveal 4-hydroxybutenolides using the KDM rearrangement
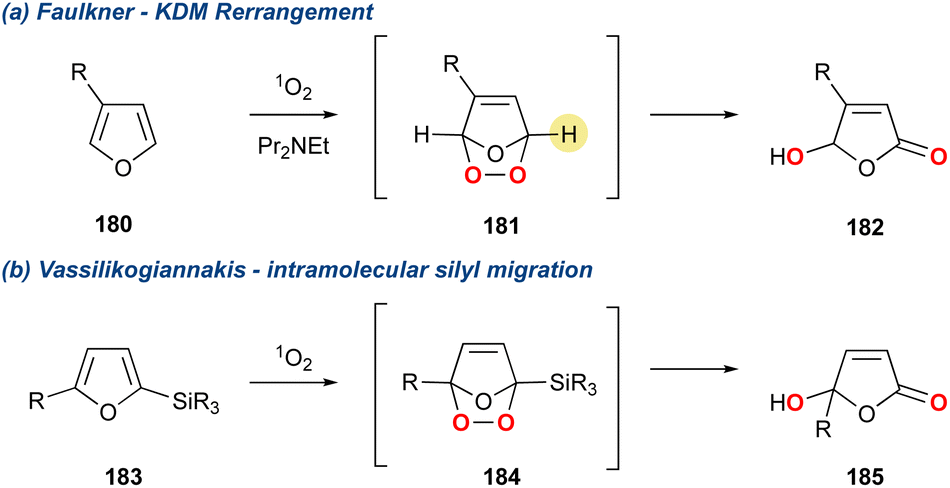
The butenolide motif is found over several natural product families whose synthesis has been recently reviewed.124,125 In 1988 Faulkner and co-workers110 reported a convenient method for the direct conversion of 3-alkyl furans to 4-hydroxybutenolides through the action of singlet oxygen and a hindered base (Scheme 32a).126 This method makes use of a hindered base to direct the deprotonation, thereby proving 182. This methodology was further modified by Vassilikogiannakis and co-workers127 who utilised 2-trialkylsilylfurans which could be directly converted to the corresponding butanolide using singlet oxygen, but in the absence of base (Scheme 32b). This latter method goes via an intramolecular silyl migration as described by Adam and Rodriguez.128 Both methods have found application in the total synthesis of natural products over the past 25 years. However, this section will only centre on those syntheses that convert 3-alkylfurans instead of 2-trialkylsilylfurans, as the former proceeds solely by a base mediated KDM rearrangement.
Scheme 32 Conversion of furans into butenolides via the KDM rearrangement.
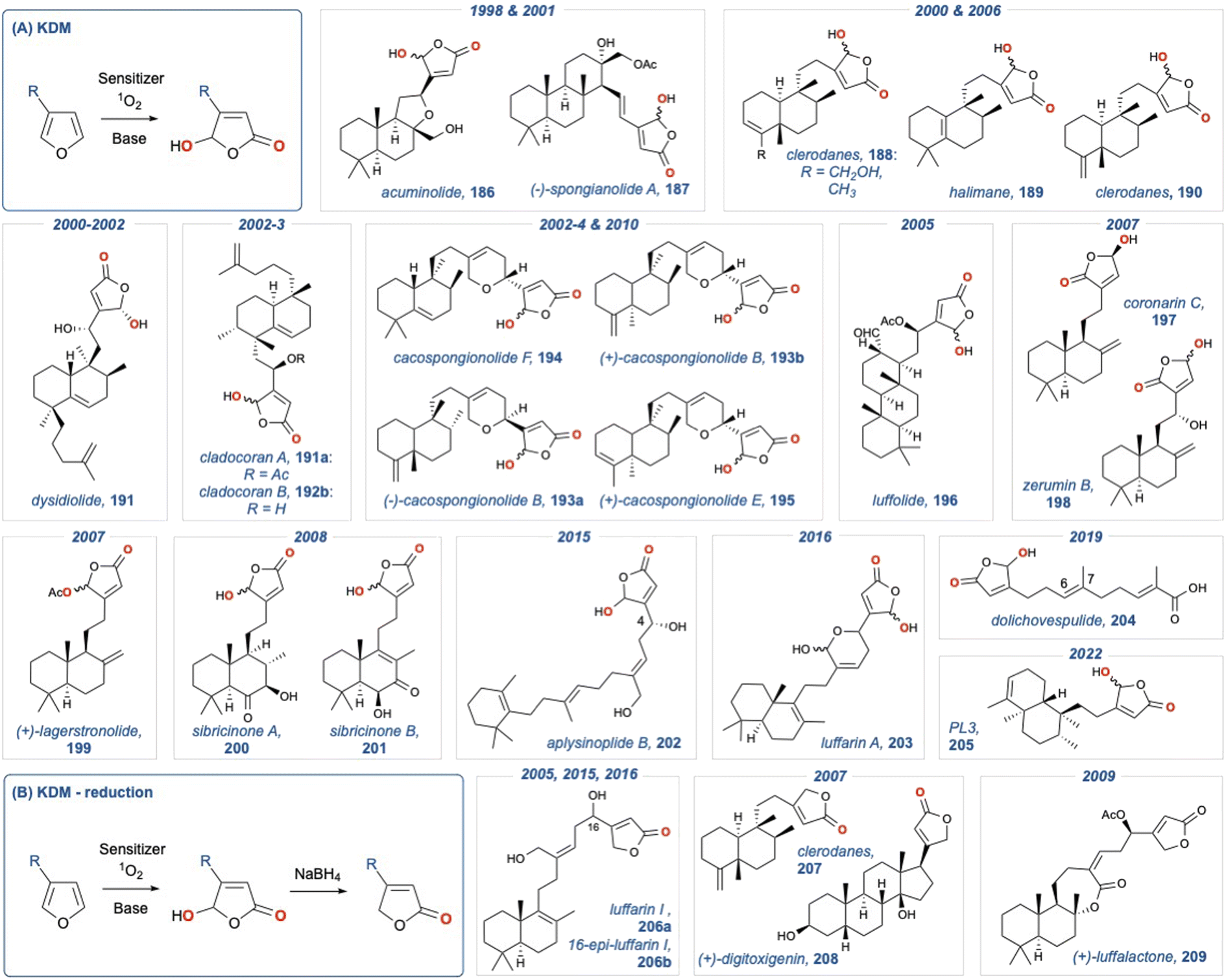
We have summarised all the natural products that have been synthesised using this approach in Fig. 3. Fig. 3A demonstrates natural products giving a γ-hydroxybutenolide in the last step, while Fig. 3B, gives natural products that possess a butenolide, which is installed through a subsequent reduction. Given the uniform approach across all these natural products in unmasking of the 3-substituted furan in the last step using the Faulkner method, detailed analysis has not been undertaken; however, a summary is given below.
Fig. 3 Natural products that have been synthesised using (A) unmasking of a 3-substituted furan in the final step; (b) unmasking of a 3-substituted furan in the final step and then reduction.
Dubay and co-workers synthesised acuminolide (186) in 1998 (and its 17-O-acetyl analogue, 187) using a semisynthetic approach.129 Acuminolide (186) is a labdane diterpene with cytotoxic activity, and their syntheses was achieved from commercially available (+)-sclareolide, with the key butenolide being revealed using a KDM rearrangement. Soetjipto and co-workers disclosed a synthesis of acuminolide (186) in 2001,130 and again, the key butenolide was revealed using a last step KDM rearrangement. The clerodanes and halimanes (188–190) were synthesised by Costa and co-workers. The authors were able to assign the absolute configuration through a successful semi-synthesis from methyl (+)-hardwickiate; again, they utilised the KDM rearrangement to reveal the key butenolide.131,132 Dysiodiolide (191) is a natural product isolated form the marine sponge Dysidera etheris and is a potent inhibitor of the human cdc25A protein phosphatase.133 This natural product has received considerable attention since is identification in 1996. Consequently, several total syntheses134–139 have been reported, with the key butenolide being revealed in the final step. Cladocoran A and B (191a,b), isolated form the coral Cladocora cespitosa140 show structural homology with dysidiolide (191), consequently several groups use a final KDM rearrangement to reveal the butenolide. Marcos et al.141,142 used a semisynthetic approach from ent-halimic acid to give a synthesis of their purported structures. Miyaoka et al.143 then used a de novo synthesis and established clardocoran B (192b) to be an olefinic isomer of dysodiolide (191), with clardocoran A (191a) being its acetate. The cacospongionolides (193–195) are sesterterpenes isolated from sponges of the Thorectidae family and are inhibitors of phospholipase A2 (PLA2) and therefore show potential in treating inflammation. First to be synthesised was (−)- and (+)-cacospongionolides B (193a,b) by Chueng and Snapper,144 who used a KDM rearrangement in the final step to reveal the butenolide, this was followed by a synthesis of (−)-cacospongionolides F (194) by Demeke and Forsyth,145 and a follow up synthesis of cacospongionolides E (195) again by Snapper,146 as well as latter synthesis of (−)-cacospongionolides B (193a) by Oshida et al.147 Luffoilide (196) is a metabolite from the sponge Luffariella, and its semi-synthesis148 was completed using a KDM rearrangement in the final step from methyl isoanticopalate, itself obtained from sclareol. The same group then reported a synthesis of (+)-laffalactone (209);149 however, the key γ-hydroxybutenolide from the final KDM rearrangement was subsequently reduced to provide the desired natural product. Laffarin I (206a) was first synthesised by Urosa and co-workers from commercially available sclareol.150 Luffarin A (203), a related metabolite, was synthesised using a similar last step KDM rearrangement approach, along with luffarin I (206a) and 16-epi-luffarin I (206b); the last two natural products were obtained by a subsequent reduction of the γ-hydroxybutenolide.151 The labdane diterpenes, coronarin C (197) and zerumin B (198) underwent oxidation using rose Bengal or TPP as the photosensitiser. Oxidation using rose Bengal was undertaken in the absence of base, giving the required regioisomer of the butenolide; whereas, in the presence of base the undesired product was isolated.152 Basabe and co-workers completed a semi-synthesis of the diterpene (+)-lagerstronolide (199) from (+)-sclareol.153 (+)-Lagerstronolide (199) is a metabolite isolated from the Lagerstreomia lancasteria shrub in south-eastern Australia.154 The key butenolide was introduced via a KDM rearrangement in the penultimate step, with the resultant γ-hydroxybutenolide being acetylated to give the desired natural product. A de novo synthesis of (+)-digitoxigenin (208) was reported by Homma and Nakata in 2007.155 (+)-Digitoxigenin (208) has clinical value as a treatment for congestive heart failure and as well as displaying anticancer activity. The butenoilide was introduced by a two-step sequence involving a KDM rearrangement with subsequent reduction of the γ-hydroxybutenolide. Marcos and co-workers completed syntheses of both sibiricinone A and B (200 and 201), again from (+)-sclareol.156 Kutsumura and co-workers completed a total synthesis of aplysinoplide B (202),157 a cytotoxic sesterterpeniod isolated from the marine sponge Aplysinopsis digitata.158 The purpose of the synthesis was the promising activity of these natural products against P388 mouse leukemia cells, as well as confirming the C4 hydroxyl group. Again, the butenolide was introduced in the last step via a KDM rearrangement. Dolichovespulide (204) is a sesquiterpene isolated from the body surface extracts of hornet queens (Dolichovespula maculata). This pheromone is a new member of relatively small class of terpenoids isolated from the Vespidae insect family. The key butenolide was unveiled using a KDM rearrangement, and the total synthesis confirmed the C6–C7 double bond geometry.159 Technically, revealing of butenolide was the penultimate step in the synthesis, as the free acid needed to be revealed in the last step. In 2022, Zhu and co-workers provided a modular synthetic approach, using a metal mediated tail-to-head cyclisation strategy, to synthesise the central diterpene ring system of several trans-clerodane analogues. In their report they provided an effective synthesis of annonene, which was oxidised using a KDM rearrangement condition to provide the butenolide PL3 (205).160
6 Conclusions
This review provides valuable insight into the versatility of the KDM rearrangement in natural product synthesis. It is clear from the number of reports that this transformation is increasingly recognised as a key biomimetic step. There are several reports that utilise the enantioselective KDM, but here the methodology is limited to larger rings (≥6-membered) that contain some pre-existing steric bias. New developments in this area would open-up a raft of possibilities for installing chiral alcohols on a wider variety of substrates. Presently, there are limited reports of the reaction taking place under acidic conditions, apart from the report by Bach and co-workers.40 This may provide scope to improve on the existing enantioselective desymmetrisation of prochiral endoperoxides described by Toste.43 This method uses a chiral base to affect the KDM, has substrate limitations. The successful development of an enantioselective acid catalysed process could have significant impact and improve upon the already impressive range of natural products that can be synthesised using the KDM rearrangement.
7 Author contributions
MCK and DSL contributed equally to the conceptualisation and investigation of this research. They both contributed equally to the writing and preparation of the manuscript.
8 Conflicts of interest
There are no conflicts to declare.
9 Acknowledgements
MCK would like to thank Loughborough University. DSL would like to thank the Center for Green Chemistry and Green Engineering for their support and the JA team for their advice and fruitful discussions.
10 Notes and references
N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880–881 CrossRefCAS.
N. A. Milas and D. M. Surgenor, J. Am. Chem. Soc., 1946, 68, 205–208 CrossRefCAS.
W. Baker and D. M. Easty, Nature, 1950, 166, 156 CrossRefPubMed.
M. C. Kimber and D. K. Taylor, Trends Org. Chem., 2001, 9, 53 CAS.
M. Oda and Y. Kitahara, Tetrahedron Lett., 1969, 10, 3295–3296 CrossRef.
A. Daştan and M. Balci, Tetrahedron, 2006, 62, 4003–4010 CrossRef.
F. Koc, E. Cadirci, A. Albayrak, Z. Halici, A. Hacimuftuoglu and H. Suleyman, Med. Chem. Res., 2010, 19, 84–93 CrossRefCAS.
Y. Morita, E. Matsumura, T. Okabe, M. Shibata, M. Sugiura, T. Ohe, H. Tsujibo, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2003, 26, 1487–1490 CrossRefCASPubMed.
M. Balci and B. Atasoy, Tetrahedron Lett., 1984, 25, 4033–4036 CrossRefCAS.
M. E. Sengül, Z. Ceylan and M. Balci, Tetrahedron, 1997, 53, 10401–10408 CrossRef.
M. Güney, Z. Ç. Ceylan, A. Daştan and M. Balci, Can. J. Chem., 2005, 83, 227–235 CrossRef.
M. G. Zagorski and R. G. Salomon, J. Am. Chem. Soc., 1980, 102, 2501–2503 CrossRefCAS.
M. G. Zagorski and R. G. Salomon, J. Am. Chem. Soc., 1984, 106, 1750–1759 CrossRefCAS.
I. A. Yaremenko, V. A. Vil', D. V Demchuk and A. O. Terent'ev, Beilstein J. Org. Chem., 2016, 12, 1647–1748 CrossRefCASPubMed.
A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRefCASPubMed.
R. D. Bach and H. B. Schlegel, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRefCASPubMed.
E. Mete, R. Altundas, H. Secen and M. Balci, Turk. J. Chem., 2003, 27, 145 CAS.
D. R. Kelly, H. Bansal and J. J. G. Morgan, Tetrahedron Lett., 2002, 43, 9331–9333 CrossRefCAS.
T. D. Avery, J. A. Culbert and D. K. Taylor, Org. Biomol. Chem., 2006, 4, 323–330 RSC.
N. Akbulut and M. Balci, J. Org. Chem., 1988, 53, 3338–3342 CrossRefCAS.
T. Fujishima, F. Kitoh, T. Yano and R. Irie, Synlett, 2010, 2010, 2279–2282 CrossRef.
V. L. Paddock, R. J. Phipps, A. Conde-Angulo, A. Blanco-Martin, C. Giró-Mañas, L. J. Martin, A. J. P. White and A. C. Spivey, J. Org. Chem., 2011, 76, 1483–1486 CrossRefCASPubMed.
C. Liu, E. Shi, F. Xu, Q. Luo, H. Wang, J. Chen and X. Wan, Chem. Commun., 2015, 51, 1214–1217 RSC.
J. Jiang, J. Liu, L. Yang, Y. Shao, J. Cheng, X. Bao and X. Wan, Chem. Commun., 2015, 51, 14728–14731 RSC.
I. Erden, N. Öcal, J. Song, C. Gleason and C. Gärtner, Tetrahedron, 2006, 62, 10676–10682 CrossRefCASPubMed.
X. Zheng, S. Lu and Z. Li, Org. Lett., 2013, 15, 5432–5435 CrossRefCASPubMed.
F. Zhang, P. Du, J. Chen, H. Wang, Q. Luo and X. Wan, Org. Lett., 2014, 16, 1932–1935 CrossRefCASPubMed.
R. A. Kumar, C. U. Maheswari, S. Ghantasala, C. Jyothi and K. R. Reddy, Adv. Synth. Catal., 2011, 353, 401–410 CrossRefCAS.
T. D. Avery, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2000, 65, 5531–5546 CrossRefCASPubMed.
B. W. Greatrex, M. C. Kimber, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2003, 68, 4239–4246 CrossRefCASPubMed.
M. C. Kimber and D. K. Taylor, J. Org. Chem., 2002, 67, 3142–3144 CrossRefCASPubMed.
B. W. Greatrex, M. C. Kimber, D. K. Taylor, G. Fallon and E. R. T. Tiekink, J. Org. Chem., 2002, 67, 5307–5314 CrossRefCASPubMed.
X. Zhang, S. I. Khan and C. S. Foote, J. Org. Chem., 1993, 58, 7839–7847 CrossRefCAS.
R. J. Lee, M. R. Lindley, G. J. Pritchard and M. C. Kimber, Chem. Commun., 2017, 53, 6327–6330 RSC.
P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Org. Lett., 2012, 14, 4537–4539 CrossRefCASPubMed.
M. Rössle, T. Werner, A. Baro, W. Frey and J. Christoffers, Angew. Chem., Int. Ed., 2004, 43, 6547–6549 CrossRefPubMed.
G. S. Buchanan, K. P. Cole, Y. Tang and R. P. Hsung, J. Org. Chem., 2011, 76, 7027–7039 CrossRefCASPubMed.
M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Chem.–Eur. J., 2012, 18, 4766–4774 CrossRefCASPubMed.
C. Wiegand, E. Herdtweck and T. Bach, Chem. Commun., 2012, 48, 10195 RSC.
L. J. Roberts, R. G. Salomon, J. D. Morrow and C. J. Brame, FASEB J., 1999, 13, 1157–1168 CrossRefCASPubMed.
R. G. Salomon, Acc. Chem. Res., 1985, 18, 294–301 CrossRefCAS.
S. T. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658–12659 CrossRefCASPubMed.
J. Priest, M. R. Longland, M. R. J. Elsegood and M. C. Kimber, J. Org. Chem., 2013, 78, 3476–3481 CrossRefCASPubMed.
T. D. Avery, N. F. Jenkins, M. C. Kimber, D. W. Lupton and D. K. Taylor, Chem. Commun., 2002, 28–29 RSC.
S. V. A.-M. Legendre, C. J. Sumby, A. Karton and B. W. Greatrex, J. Org. Chem., 2023, 88, 11444–11449 CrossRefCASPubMed.
J. M. Aubry, J. Am. Chem. Soc., 1985, 107, 5844–5849 CrossRefCAS.
J. M. Aubry and B. Cazin, Inorg. Chem., 1988, 27, 2013–2014 CrossRefCAS.
M. Elsherbini, R. K. Allemann and T. Wirth, Chem.–Eur. J., 2019, 25, 12486–12490 CrossRefCASPubMed.
D. F. Evans and M. W. Upton, J. Chem. Soc., Dalton Trans., 1985, 1151 RSC.
W. Adam, D. V. Kazakov and V. P. Kazakov, Chem. Rev., 2005, 105, 3371–3387 CrossRefCASPubMed.
M. C. Carreño, M. González-López and A. Urbano, Angew. Chem., Int. Ed., 2006, 45, 2737–2741 CrossRefPubMed.
C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRefCASPubMed.
A. Burgard, T. Gieshoff, A. Peschl, D. Hörstermann, C. Keleschovsky, R. Villa, S. Michelis and M. P. Feth, Chem. Eng. J., 2016, 294, 83–96 CrossRefCAS.
H. F. Grantham, R. J. Lee, G. M. Wardas, J.-R. Mistry, M. R. J. Elsegood, I. A. Wright, G. J. Pritchard and M. C. Kimber, J. Org. Chem., 2024, 89, 484–497 CrossRefCASPubMed.
F. Lévesque and P. H. Seeberger, Org. Lett., 2011, 13, 5008–5011 CrossRefPubMed.
K. N. Loponov, J. Lopes, M. Barlog, E. V. Astrova, A. V. Malkov and A. A. Lapkin, Org. Process Res. Dev., 2014, 18, 1443–1454 CrossRefCAS.
L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752–2906 CrossRefCASPubMed.
G. I. Ioannou, T. Montagnon, D. Kalaitzakis, S. A. Pergantis and G. Vassilikogiannakis, ChemPhotoChem, 2017, 1, 173–177 CrossRefCAS.
C. A. Clark, D. S. Lee, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2016, 20, 1792–1798 CrossRefCAS.
D. S. Lee, Z. Amara, C. A. Clark, Z. Xu, B. Kakimpa, H. P. Morvan, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2017, 21, 1042–1050 CrossRefCASPubMed.
D. S. Lee, M. Sharabi, R. Jefferson-Loveday, S. J. Pickering, M. Poliakoff and M. W. George, Org. Process Res. Dev., 2020, 24, 201–206 CrossRefCAS.
R. A. Bourne, X. Han, M. Poliakoff and M. W. George, Angew. Chem., Int. Ed., 2009, 48, 5322–5325 CrossRefCASPubMed.
A. Chaudhuri, S. D. A. Zondag, J. H. A. Schuurmans, J. van der Schaaf and T. Noël, Org. Process Res. Dev., 2022, 26, 1279–1288 CrossRefCASPubMed.
H. Tan, X. Chen, Z. Liu and D. Z. Wang, Tetrahedron, 2012, 68, 3952–3955 CrossRefCAS.
G. Zhou, X. Gao, W. Z. Li and Y. Li, Tetrahedron Lett., 2001, 42, 3101–3103 CrossRefCAS.
W.-D. Z. Li, G. Zhou, X. Gao and Y. Li, Tetrahedron Lett., 2001, 42, 4649–4651 CrossRefCAS.
R. C. Brown, D. K. Taylor and G. M. Elsey, Org. Lett., 2006, 8, 463–466 CrossRefCASPubMed.
N. Sitachitta and W. H. Gerwick, J. Nat. Prod., 1998, 61, 681–684 CrossRefCASPubMed.
T. D. Avery, B. W. Greatrex, D. K. Taylor and E. R. T. Tiekink, J. Chem. Soc., Perkin Trans. 1, 2000, 1319–1321 RSC.
T. D. Avery, G. Fallon, B. W. Greatrex, S. M. Pyke, D. K. Taylor and E. R. T. Tiekink, J. Org. Chem., 2001, 66, 7955–7966 CrossRefCASPubMed.
J. S. Clark, J. M. Northall, F. Marlin, B. Nay, C. Wilson, A. J. Blake and M. J. Waring, Org. Biomol. Chem., 2008, 6, 4012 RSC.
S. C. Fields, L. Mireles-Lo and B. C. Gerwick, J. Nat. Prod., 1996, 59, 698–700 CrossRefCAS.
T. Amagasa, R. N. Paul, J. J. Heitholt and S. O. Duke, Pestic. Biochem. Physiol., 1994, 49, 37–52 CrossRefCAS.
I. S. Marcos, L. Castañeda, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2009, 65, 9256–9263 CrossRefCAS.
Y. Tang, K. P. Cole, G. S. Buchanan, G. Li and R. P. Hsung, Org. Lett., 2009, 11, 1591–1594 CrossRefCASPubMed.
M. Sugano, A. Sato, Y. Iijima, T. Oshima, K. Furuya, H. Kuwano, T. Hata and H. Hanzawa, J. Am. Chem. Soc., 1991, 113, 5463–5464 CrossRefCAS.
K. P. Cole and R. P. Hsung, Org. Lett., 2003, 5, 4843–4846 CrossRefCASPubMed.
K. P. Cole and R. P. Hsung, Chem. Commun., 2005, 5784 RSC.
M. Kawasumi, N. Kanoh and Y. Iwabuchi, Org. Lett., 2011, 13, 3620–3623 CrossRefCASPubMed.
S. Ohno, K. Tomita-Yokotani, S. Kosemura, M. Node, T. Suzuki, M. Amano, K. Yasui, T. Goto, S. Yamamura and K. Hasegawa, Phytochemistry, 2001, 56, 577–581 CrossRefCASPubMed.
H. Yokoe, H. Sasaki, T. Yoshimura, M. Shindo, M. Yoshida and K. Shishido, Org. Lett., 2007, 9, 969–971 CrossRefCASPubMed.
K. C. Nicolaou, S. Totokotsopoulos, D. Giguère, Y.-P. Sun and D. Sarlah, J. Am. Chem. Soc., 2011, 133, 8150–8153 CrossRefCASPubMed.
K. C. Nicolaou, M. Lu, S. Totokotsopoulos, P. Heretsch, D. Giguère, Y.-P. Sun, D. Sarlah, T. H. Nguyen, I. C. Wolf, D. F. Smee, C. W. Day, S. Bopp and E. A. Winzeler, J. Am. Chem. Soc., 2012, 134, 17320–17332 CrossRefCASPubMed.
G. R. Schulte and B. Ganem, Tetrahedron Lett., 1982, 23, 4299–4302 CrossRefCAS.
S. D. Jolad, J. J. Hoffmann, K. H. Schram, J. R. Cole, M. S. Tempesta and R. B. Bates, J. Org. Chem., 1981, 46, 4267–4272 CrossRefCAS.
C.-R. Zhang, S.-P. Yang, S.-G. Liao, Y. Wu and J.-M. Yue, Helv. Chim. Acta, 2006, 89, 1408–1416 CrossRefCAS.
Y. Takeuchi, Q. Cheng, Q.-W. Shi, T. Sugiyama and T. Oritani, Biosci., Biotechnol., Biochem., 2001, 65, 1395–1398 CrossRefCASPubMed.
D. A. Mulholland, A. Langlois, M. Randrianarivelojosia, E. Derat and J.-M. Nuzillard, Phytochem. Anal., 2006, 17, 87–90 CrossRefCASPubMed.
W.-D. Xuan, H.-S. Chen, J.-L. Du, S. Liang, T.-Z. Li and D.-G. Cai, J. Asian Nat. Prod. Res., 2006, 8, 719–722 CrossRefCASPubMed.
L. Mao, L. Xin and Y. Dequan, Planta Med., 1984, 50, 459–461 CrossRefCASPubMed.
J. G. Buta, J. L. Flippen and W. R. Lusby, J. Org. Chem., 1978, 43, 1002–1003 CrossRefCAS.
M. Zhang, N. Liu and W. Tang, J. Am. Chem. Soc., 2013, 135, 12434–12438 CrossRefCASPubMed.
I. T. Chen, I. Baitinger, L. Schreyer and D. Trauner, Org. Lett., 2014, 16, 166–169 CrossRefCASPubMed.
T. J. Heckrodt and J. Mulzer, in Natural Products Synthesis II, ed. J. Mulzer, Spinger, Berlin, Heidelber, 2005, pp. 1–41 Search PubMed.
C. L. Hugelshofer and T. Magauer, J. Am. Chem. Soc., 2015, 137, 3807–3810 CrossRefCASPubMed.
S.-H. Luo, C. L. Hugelshofer, J. Hua, S.-X. Jing, C.-H. Li, Y. Liu, X.-N. Li, X. Zhao, T. Magauer and S.-H. Li, Org. Lett., 2014, 16, 6416–6419 CrossRefCASPubMed.
C. L. Hugelshofer and T. Magauer, Angew. Chem., Int. Ed., 2014, 53, 11351–11355 CrossRefCASPubMed.
A. Gervais, K. E. Lazarski and J. A. Porco, J. Org. Chem., 2015, 80, 9584–9591 CrossRefCASPubMed.
X. Zhang, G. Wu, L. Huo, X. Guo, S. Qiu, H. Liu, H. Tan and Y. Hu, J. Nat. Prod., 2020, 83, 3–7 CrossRefCASPubMed.
X. Zhang, C. Dong, G. Wu, L. Huo, Y. Yuan, Y. Hu, H. Liu and H. Tan, Org. Lett., 2020, 22, 8007–8011 CrossRefCASPubMed.
L.-M. Deng, L.-J. Hu, Y.-T.-Z. Bai, J. Wang, G.-Q. Qin, Q.-Y. Song, J.-C. Su, X.-J. Huang, R.-W. Jiang, W. Tang, Y.-L. Li, C.-C. Li, W.-C. Ye and Y. Wang, Org. Lett., 2021, 23, 4499–4504 CrossRefCASPubMed.
L.-M. Deng, W. Tang, S.-Q. Wang, J.-G. Song, X.-J. Huang, H.-Y. Zhu, Y.-L. Li, W.-C. Ye, L.-J. Hu and Y. Wang, J. Org. Chem., 2022, 87, 4788–4800 CrossRefCASPubMed.
M. L. Ciavatta, M. Gavagnin, R. Puliti, G. Cimino, E. Martinez, J. Ortea and C. A. Mattia, Tetrahedron, 1996, 52, 12831–12838 CrossRefCAS.
M. R. Gesinski, W. E. Brenzovich, S. T. Staben, D. J. Srinilta and F. D. Toste, Tetrahedron Lett., 2015, 56, 3643–3646 CrossRefCAS.
J. Froese, C. Overbeeke and T. Hudlicky, Chem.–Eur. J., 2016, 22, 6180–6184 CrossRefCASPubMed.
M. J. Palframan, G. Kociok-Köhn and S. E. Lewis, Chem.–Eur. J., 2012, 18, 4766–4774 CrossRefCASPubMed.
Z. Zhong, G. Zhao, D. Xu, B. Dong, D. Song, X. Xie and X. She, Chem.–Asian J., 2016, 11, 1542–1547 CrossRefCASPubMed.
H. Yin, J.-G. Luo and L.-Y. Kong, J. Nat. Prod., 2013, 76, 237–242 CrossRefCASPubMed.
M. R. Kernan and D. J. Faulkner, J. Org. Chem., 1988, 53, 2773–2776 CrossRefCAS.
Y. Hirata, A. Nakazaki, H. Kawagishi and T. Nishikawa, Org. Lett., 2017, 19, 560–563 CrossRefCASPubMed.
M. Niki, Y. Hirata, A. Nakazaki, J. Wu, H. Kawagishi and T. Nishikawa, J. Org. Chem., 2020, 85, 4848–4860 CrossRefCASPubMed.
N. Shirasaka, K. Nishi and S. Shimizu, Biochim. Biophys. Acta, Lipids Lipid Metab., 1997, 1346, 253–260 CrossRefCASPubMed.
I. Dokli, R. Pohl, B. Klepetářová and U. Jahn, Chem. Commun., 2019, 55, 3931–3934 RSC.
H. Hayahsi, Y. Nishimoto, K. Akiyama and H. Nozaki, Biosci., Biotechnol., Biochem., 2000, 64, 111–115 CrossRefPubMed.
F. Reuß and P. Heretsch, Org. Lett., 2020, 22, 3956–3959 CrossRefPubMed.
Q. Li, K. Zhao, A. Peuronen, K. Rissanen, D. Enders and Y. Tang, J. Am. Chem. Soc., 2018, 140, 1937–1944 CrossRefCASPubMed.
C. Festa, S. De Marino, M. V. D'Auria, E. Deharo, G. Gonzalez, C. Deyssard, S. Petek, G. Bifulco and A. Zampella, Tetrahedron, 2012, 68, 10157–10163 CrossRefCAS.
Z. Xue, Q. Li, J. Zhang and Y. Tang, Org. Lett., 2021, 23, 8783–8788 CrossRefCASPubMed.
M. Morgenstern, C. Mayer, A. Pöthig and T. Bach, Synthesis, 2023, 55, 1671–1689 CrossRefCAS.
Y. Al-Jawaheri, M. R. J. Elsegood, J.-R. Mistry and M. C. Kimber, Tetrahedron Lett., 2023, 114, 154273 CrossRefCAS.
Y. Al-Jawaheri and M. C. Kimber, Org. Lett., 2016, 18, 3502–3505 CrossRefCASPubMed.
Y. Al-Jawaheri, M. Turner and M. Kimber, Synthesis, 2018, 50, 2329–2336 CrossRefCAS.
K. Tadiparthi and S. Venkatesh, J. Heterocycl. Chem., 2022, 59, 1285–1307 CrossRefCAS.
S. Chatterjee, R. Sahoo and S. Nanda, Org. Biomol. Chem., 2021, 19, 7298–7332 RSC.
B. L. Feringa, Recl. Trav. Chim. Pays-Bas, 2010, 106, 469–488 CrossRef.
T. Montagnon, M. Tofi and G. Vassilikogiannakis, Acc. Chem. Res., 2008, 41, 1001–1011 CrossRefCASPubMed.
W. Adam and A. Rodriguez, Tetrahedron Lett., 1981, 22, 3505–3508 CrossRefCAS.
P. A. Zoretic, H. Fang, A. A. Ribeiro and G. Dubay, J. Org. Chem., 1998, 63, 1156–1161 CrossRefCAS.
N. Furuichi, T. Hata, H. Soetjipto, M. Kato and S. Katsumura, Tetrahedron, 2001, 57, 8425–8442 CrossRefCAS.
P. M. Imamura and M. Costa, J. Nat. Prod., 2000, 63, 1623–1625 CrossRefCASPubMed.
R. M. Ide, M. Costa and P. M. Imamura, J. Braz. Chem. Soc., 2006, 17, 417–420 CrossRefCAS.
S. P. Gunasekera, P. J. McCarthy, M. Kelly-Borges, E. Lobkovsky and J. Clardy, J. Am. Chem. Soc., 1996, 118, 8759–8760 CrossRefCAS.
S. R. Magnuson, L. Sepp-Lorenzino, N. Rosen and S. J. Danishefsky, J. Am. Chem. Soc., 1998, 120, 1615–1616 CrossRefCAS.
D. Demeke and C. J. Forsyth, Tetrahedron, 2002, 58, 6531–6544 CrossRefCAS.
H. Miyaoka and Y. Yamada, Bull. Chem. Soc. Jpn., 2002, 75, 203–222 CrossRefCAS.
H. Miyaoka, Y. Kajiwara, Y. Hara and Y. Yamada, J. Org. Chem., 2001, 66, 1429–1435 CrossRefCASPubMed.
M. Takahashi, K. Dodo, Y. Hashimoto and R. Shirai, Tetrahedron Lett., 2000, 41, 2111–2114 CrossRefCAS.
D. Demeke and C. J. Forsyth, Org. Lett., 2000, 2, 3177–3179 CrossRefCASPubMed.
A. Fontana, M. L. Ciavatta and G. Cimino, J. Org. Chem., 1998, 63, 2845–2849 CrossRefCAS.
I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, P. Basabe, F. A. Hernández, H. B. Broughton and J. G. Urones, Synlett, 2002, 2002, 0105–0109 CrossRef.
I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, P. Basabe, N. García, R. F. Moro, H. B. Broughton, F. Mollinedo and J. G. Urones, J. Org. Chem., 2003, 68, 7496–7504 CrossRefCASPubMed.
H. Miyaoka, M. Yamanishi, Y. Kajiwara and Y. Yamada, J. Org. Chem., 2003, 68, 3476–3479 CrossRefCASPubMed.
A. K. Cheung and M. L. Snapper, J. Am. Chem. Soc., 2002, 124, 11584–11585 CrossRefCASPubMed.
D. Demeke and C. J. Forsyth, Org. Lett., 2003, 5, 991–994 CrossRefCASPubMed.
A. K. Cheung, R. Murelli and M. L. Snapper, J. Org. Chem., 2004, 69, 5712–5719 CrossRefCASPubMed.
S. Kobayashi, M. Oshida, M. Ono and A. Nakazaki, Heterocycles, 2010, 80, 313 CrossRefPubMed.
P. Basabe, S. Delgado, I. S. Marcos, D. Diez, A. Diego, M. De Román and J. G. Urones, J. Org. Chem., 2005, 70, 9480–9485 CrossRefCASPubMed.
P. Basabe, O. Bodero, I. S. Marcos, D. Díez, A. Blanco, M. de Román and J. G. Urones, J. Org. Chem., 2009, 74, 7750–7754 CrossRefCASPubMed.
A. Urosa, I. Marcos, D. Díez, A. Lithgow, G. Plata, J. Padrón and P. Basabe, Mar. Drugs, 2015, 13, 2407–2423 CrossRefCASPubMed.
A. Urosa, I. S. Marcos, D. Díez, G. B. Plata, J. M. Padrón and P. Basabe, Mol. Diversity, 2016, 20, 369–377 CrossRefCASPubMed.
J. E. Villamizar, J. Juncosa, J. Pittelaud, M. Hernández, N. Canudas, E. Tropper, F. Salazar and J. Fuentes, J. Chem. Res., 2007, 2007, 342–346 CrossRef.
P. Basabe, O. Bodero, I. S. Marcos, D. Diez, M. de Román, A. Blanco and J. G. Urones, Tetrahedron, 2007, 63, 11838–11843 CrossRefCAS.
P. K. Chaudhuri, Phytochemistry, 1987, 26, 3361–3362 CrossRefCAS.
M. Honma and M. Nakada, Tetrahedron Lett., 2007, 48, 1541–1544 CrossRefCAS.
I. S. Marcos, L. Castañeda, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2008, 64, 10860–10866 CrossRefCAS.
N. Kutsumura, K. Matsuo and T. Saito, Tetrahedron Lett., 2015, 56, 2602–2604 CrossRefCAS.
R. Ueoka, Y. Nakao, S. Fujii, R. W. M. van Soest and S. Matsunaga, J. Nat. Prod., 2008, 71, 1089–1091 CrossRefCASPubMed.
W. Ren, R. Gries, K. L. Kurita, C. S. McCaughey, S. K. Alamsetti, R. G. Linington, G. Gries and R. Britton, J. Nat. Prod., 2019, 82, 2009–2012 CrossRefCASPubMed.
W. Zhu, Q. Yin, Z. Lou and M. Yang, Nat. Commun., 2022, 13, 6633 CrossRefCASPubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Darren S.
Lee
*a and
Darren S.
Lee


![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: