
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling Amaryllidaceae alkaloids: from biosynthesis to antiviral potential – a review
Thilina U.
Jayawardena†
 a,
Natacha
Merindol†
a,
Nuwan Sameera
Liyanage
a and
Isabel
Desgagné-Penix
*ab
a,
Natacha
Merindol†
a,
Nuwan Sameera
Liyanage
a and
Isabel
Desgagné-Penix
*ab
aDepartment of Chemistry, Biochemistry, and Physics, Université du Québec à Trois-Rivières, Trois-Rivières, QC G8Z 4M3, Canada. E-mail: Isabel.Desgagne-Penix@uqtr.ca
bPlant Biology Research Group, Université du Québec à Trois-Rivières, Trois-Rivières, QC, Canada
First published on 22nd December 2023
Abstract
Covering: 2017 to 2023 (now)
Amaryllidaceae alkaloids (AAs) are a unique class of specialized metabolites containing heterocyclic nitrogen bridging that play a distinct role in higher plants. Irrespective of their diverse structures, most AAs are biosynthesized via intramolecular oxidative coupling. The complex organization of biosynthetic pathways is constantly enlightened by new insights owing to the advancement of natural product chemistry, synthetic organic chemistry, biochemistry, systems and synthetic biology tools and applications. These promote novel compound identification, trace-level metabolite quantification, synthesis, and characterization of enzymes engaged in AA catalysis, enabling the recognition of biosynthetic pathways. A complete understanding of the pathway benefits biotechnological applications in the long run. This review emphasizes the structural diversity of the AA specialized metabolites involved in biogenesis although the process is not entirely defined yet. Moreover, this work underscores the pivotal role of synthetic and enantioselective studies in justifying biosynthetic conclusions. Their prospective candidacy as lead constituents for antiviral drug discovery has also been established. However, a complete understanding of the pathway requires further interdisciplinary efforts in which antiviral studies address the structure–activity relationship. This review presents current knowledge on the topic.
 Thilina U. Jayawardena | Thilina U. Jayawardena received his bachelor's in Chemistry from the University of Colombo. He earned a master's degree and PhD degree (2022) in Life Sciences, focusing on Natural Product Chemistry at Jeju National University under Prof. You-Jin Jeon. Currently, he is continuing his postdoctoral studies at the Université du Québec à Trois-Rivières (UQTR) under the mentorship of Prof. Isabel Desgagné-Penix, having previously served as a postdoctoral fellow at the University of Calgary. His research interests include natural product chemistry, medicinal chemistry, and bioactive molecules, with a particular emphasis on specialized metabolites. |
 Natacha Merindol | Natacha Merindol obtained a PhD in microbiology and immunology from Université de Montréal in 2011. She conducted her first postdoctoral research at the INSERM Immunology unit in Lyon, working on inflammation and lymphoma and a second one at Université du Québec à Trois-Rivières (UQTR), specializing in virology. She now works as a research associate under Prof. Isabel Desgagné-Penix at UQTR, focusing on the discovery and characterization of the antiviral properties of Amaryllidaceae alkaloids and unraveling their biosynthesis. Her current projects incorporate several disciplines, such as virology, molecular biology, bioengineering, and molecular dynamics (photo credit: Isabelle Cardinal, UQTR). |
 Nuwan Sameera Liyanage | Nuwan Sameera Liyanage earned a bachelor's degree in plant science from the University of Colombo, Sri Lanka in 2013. He went on to complete his master's degree in ecology in 2020 from the Université du Québec en Abitibi-Témiscamingue. Currently, he is pursuing his PhD in Cellular and Molecular Biology at the Université du Québec à Trois-Rivières under the guidance of Prof. Isabel Desgagné-Penix. His doctoral research is focused on studying the N-methyltransferases of Amaryllidaceae alkaloid biosynthesis. |
 Isabel Desgagné-Penix | Isabel Desgagné-Penix, a professor at the University of Québec at Trois-Rivières in Canada, has always been interested in the way plants make medicines. She holds a PhD in Cell and Molecular Biology (2008) from the University of Texas-San Antonio and has become a world pioneer in the use of advanced technologies for specialized plant metabolism research. She aims to elucidate the biosynthetic pathways of selected molecules and to develop alternative sources to produce them. Strongly involved in the promotion of women and indigenous people in science, she contributes, through her work, to designing and building new biological systems for purposes useful to humanity. |
1 Introduction
1.1 Alkaloids
Alkaloids are organic compounds containing basic nitrogen. Most alkaloids are derived from amino acids, such as phenylalanine, tyrosine, tryptophan, and lysine, and represent a wide range of chemical structures. Although most alkaloids are isolated from plants, they are found in microorganisms, marine organisms,10,11 and, interestingly, terrestrial animals, such as insects.12Alkaloids are often classified based on their carbon skeleton. Indole and isoquinoline alkaloids are the two leading groups in terms of number. Tropane, pyridine, pyrrolizidine, and steroidal alkaloids are also part of this large family.15 Moreover, alkaloids are possibly categorized according to their botanical backgrounds, such as opium alkaloids, cactus alkaloids, Solanum alkaloids, and Amaryllidaceae alkaloids.16
Two tyrosine derivatives, dopamine and 4-hydroxyphenylacetaldehyde, condense and undergo cyclization to give rise to benzylisoquinoline alkaloid (BIA) precursor, (S)-norcoclaurine. The product undergoes a series of aromatic hydroxylation and methylation enzymatic reactions to reach (S)-reticuline. Most isoquinoline scaffolds dwell in (S)-reticuline as a common intermediate, leading to structural diversification. This main scaffold then undergoes functionalization reactions, such as N-, O-methylation, oxido-reduction, and oxidation bridge formation, resulting in the structural diversity of alkaloids.17 BIA is more complex than simple isoquinoline alkaloids (benzene rings fused to the isoquinoline ring) because of their third aromatic ring. Many other alkaloid groups, such as protoberberines, protopines, pavines, and aporphines, are structurally related to BIA.
Due to its structure, alkaloid crystallization is not easy although its salt counterparts are crystallizable. The water solubility of alkaloids largely depends on the amine functional group, and this is readily protonated under acidic conditions. Despite having a conjugated double bond structure in most alkaloids, they appear to be colourless in most cases, and few highly conjugated structures are found to be coloured (e.g. sanguinarine and serpentine) and otherwise exhibit robust fluorescence (e.g. quinine). N-Oxidation leaves alkaloids vulnerable to stability. Further, heat, light, and solvents interfere with their stability. Higher stabilities of alkaloids are generally obtained in ethyl acetate, toluene, and alcoholic solutions.18
In this study, our aim is to investigate Amaryllidaceae alkaloids (AAs), giving priority to their structural diversity, metabolic pathway, and antiviral potential, which are discussed in detail in later sections.
1.2 Amaryllidaceae alkaloids
As the name implies, the special group of alkaloids is primarily observed in the Amaryllidaceae plant family sensu APG II.19 Over 900 species clustered under 75 genera are distributed in multiple regions of the Amaryllidaceae plant family.20 South Africa and South America cradle the major diversity, whereas the Mediterranean region has comparatively fewer genera. Habitats from seasonally dry rainforests to ephemeral pools provide locations for Amaryllidaceae plants to thrive. Current evidence supports the idea that the origin of the family lies in Africa.21,22 The literature teaches of approximately 650 naturally occurring Amaryllidaceae alkaloids identified.23,24 Amaryllidaceae plants are of ethnopharmacological value. They have been used in traditional medicine. Nair and Staden (2014) explained the extensive use of Amaryllidaceae (Boophone disticha) in traditional medicine by various indigenous populations, such as Sotho, Zhaso, Zulu, and San in South Africa.25 Although not limited to Africa, the significant presence of Amaryllidaceae spans multiple ethnic groups worldwide. The ethnopharmacological relevance originates from its antifungal, antiparasitic, anti-inflammatory, insect antifeedant, and antibacterial effects. The deeper involvement of AAs as antibacterial agents is out of the scope of this review although a comprehensive review on the subject matter published by Nair et al. (2017) is worth referring to.26The Amaryllidaceae family is unique because of its capability to produce structurally diverse alkaloids. Although their structure appears to be different, the formation of AA is known to occur via the intramolecular oxidative coupling of norbelladine. AAs are isoquinoline alkaloids, a well-conserved group in higher plants. However, biochemical and molecular phylogenetical studies suggest an evolutionary monophyletic role in basal angiosperms.27 The group adopts the isoquinoline nucleus as the basic structural feature. The recent increase in the interest in isoquinoline alkaloids owing to their multimodal capacity has encouraged pharmacological research.28,29 In evolutionary terms, isoquinoline alkaloids are presumed to play a role in an ancient conservative and survival strategy of defense mechanism. Further, isoquinoline alkaloids and phenylpropanoid derivatives (tannin and lignin) are proposed to have intrinsic biochemical and evolutive biosynthetic relationships explaining preferential biosynthesis and accumulation concepts.30 Isoquinolines are again divided into subclasses from simple scaffolds to complex dimeric structures. The biosynthetic precursor for isoquinoline alkaloids is aromatic amino acid tyrosine. Three main groups of enzymes are involved in the process of generating AA: oxidoreductases, transferases, and lyases. Some of the oxidoreductases that catalyse the reactions are cytochromes P450 (CYP) and in most cases act in oxidative couplings and methylenedioxy bridge formation. Additionally, alkaloid biosynthesis requires stereo selective enzymes that lead to the formation of (S)-enantiomers.30,31
AA classification has been conducted using different methods by scientists according to their biogenesis, carbon skeleton and ring systems (Fig. 1). In the literature, a 9 ring-,23,24 12 ring-,32 and 11 ring-33 type classifications can be identified. However, these classifications over the years have been subjected to changes and modifications because of novel AA identified, similarities between groups and the non-specificities identified by each scientist. Therefore, it is difficult to establish a universal classification system because of its large scale and frequently updated databases of natural product chemistry research.
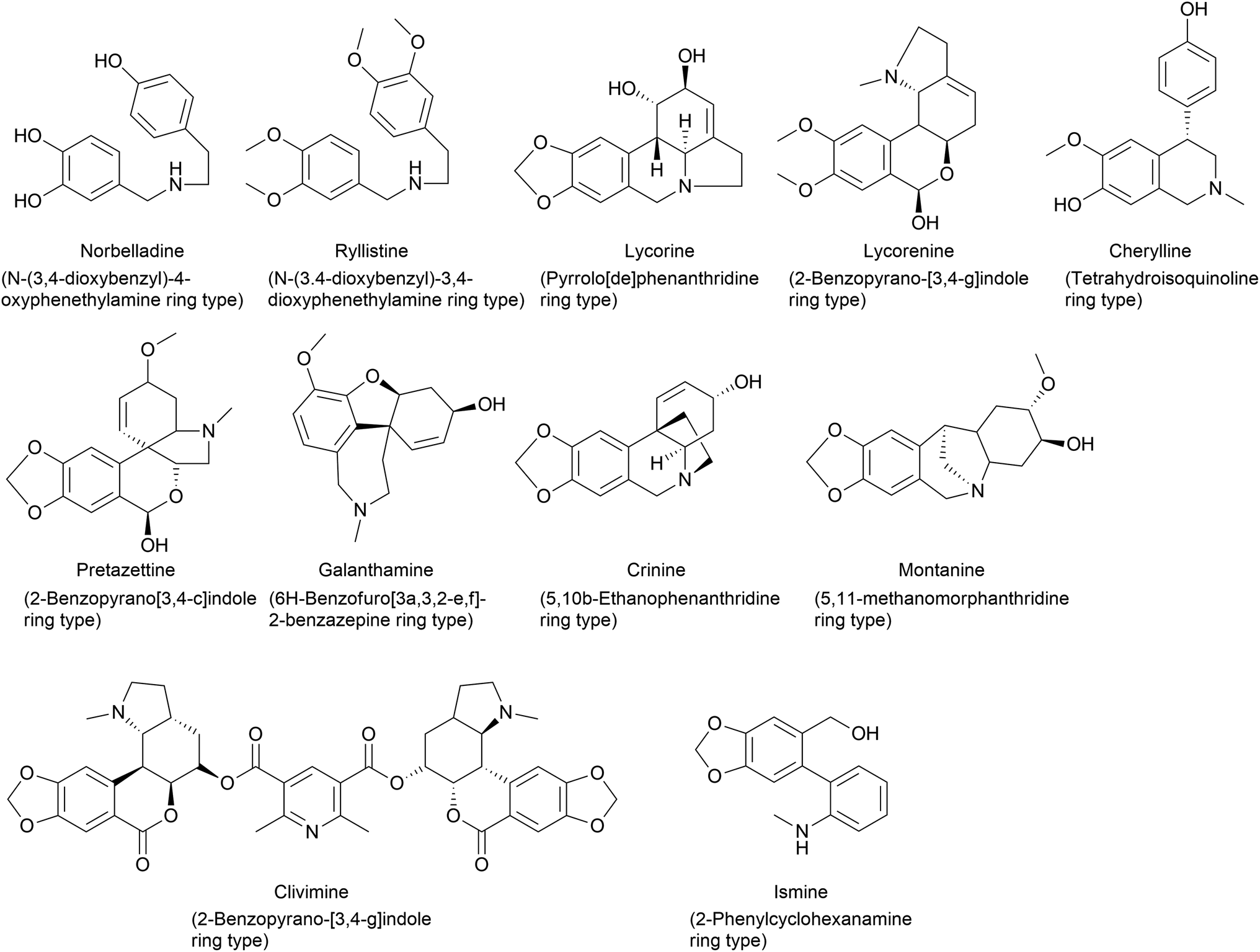 | ||
| Fig. 1 Chemical structures representing ring type(s) present in Amaryllidaceae alkaloids depending on the carbon skeleton and the ring system. | ||
The identification of novel natural products and the elucidation of their biosynthetic and degradation pathways are essential for enhancing crop characteristics, advancing drug discovery, and driving innovation in synthetic biology. To make strides in identifying compounds and genes, it is crucial to map and annotate the metabolic landscape of plants comprehensively. Although contemporary technologies can readily accomplish the initial identification, the subsequent annotation and classification of metabolites remain a significant challenge that hinders rapid advancement. Untargeted metabolomics can detect the presence of a wide array of metabolites in plants without prior knowledge of their existence or identity. However, relying solely on factors such as mass-to-charge ratio (m/z), retention time, and ion abundance does not lead to unequivocal structural annotations of metabolites and does not provide evidence of their origin. This is because numerous metabolites share similar or even identical masses. These factors can introduce additional complexities in the interpretation of the results. A targeted approach involves determining the precursor of origin for metabolites through labelling. In this method, plant tissue is exposed to a metabolite that contains a radio/heavy isotope on one or more of its atoms. When stable isotope labelling, such as 13C, is combined with mass spectrometry detection, it becomes possible to identify labelled metabolites and clarify their origin. The biochemical exploration of Amaryllidaceae alkaloid biosynthesis involves the use of labelled precursors and intermediates, leading to the formulation of biochemical models to produce different types of alkaloids. The ideas were initiated and implemented in the 1950s and 60s with radiolabelling.34 Labelling experiments provide insights into the intermediates and reaction mechanisms of biosynthetic pathways, shedding light on their interactions and interrelationships.
2 Structural diversity and biogenesis of Amaryllidaceae alkaloids
2.1 Cherylline type
Cherylline is a 4-aryltetrahydroisoquinoline alkaloid isolated from Amaryllidaceae plants. The first attempt to isolate unknowingly cherylline-type alkaloid was conducted by Boit in 1954,35 which was first named crinine. Its structure was later elucidated by Brossi and his team in 1970 (ref. 36) who named it cherylline. However, this was a racemic mixture of (+) and (−) isomers (given its C4 chiral center).37,38 Comparatively, Schwartz and Scott took a different approach to the total synthesis of racemic cherylline using a biogenetically patterned pathway.39 They yielded a quinone methide to achieve (±)-cherylline. Accordingly, in the synthetic literature, it is probable to predict that norbelladine moves to hydroxy-O,N-dimethylnorbelladine before forming the quinone methide. However, it is questionable whether methylation takes priority over hydroxylation. Despite the lack of previous records on the biosynthesis of cherylline, it has received much attention with regard to synthetic chemistry aspects and natural product isolation, even until today.40–42Few studies regarding cherylline have been published since 2017. Shi et al. (2018) examined the one-pot synthesis of two constitutional isomers: (±)-latifine and (±)-cherylline.40 Nair et al. (2020) reported the identification of cherylline and ambelline from the larvae of the Crinum moorei moth Brithys crini.43 Ambelline is a crinane alkaloid and is discussed in a different subsection. Although isoquinoline alkaloids notoriously exhibit cytotoxicity, this study identified the alkaloids in plant leaves, moth larvae, and larvae excrement material by performing HPLC and LC-MS analysis, which was further confirmed by HRMS and NMR techniques. This study lacks the tools that insects use to overcome the toxic effects of these compounds and opens up opportunities for prospective studies. Cherylline-type AA, cherylline, gigantelline, and gigantellinine were isolated from Crinum jagus.42 According to the report, cherylline derivatives were isolated and identified for the first time in a given species. Both gigantelline and gigantellinine are predicted to emerge from cherylline given their common structural features. In the case of gigantellinine, the para substituted phenol group is modified by methylation to phenyl-OH and hydroxylation at the neighbouring ortho position, whereas in gigantelline, the C7 hydroxyl group is methylated. Gigantelline was unable to inhibit the activity of acetylcholinesterase (AChE) in a dose-dependent manner, whereas cherylline and gigantellinine showed positive outcomes. Furthermore, cherylline showed weak cytotoxic effects against the breast cancer cell line MCF-7. The study further established that purified cherylline and gigantellinine had an S configuration, while gigantelline was assigned to the R configuration. However, none of the recent studies have attempted any metabolic pathway evaluation. According to our understanding, cherylline could originate through the common precursor norbelladine via the proposed route depicted in Fig. 2.
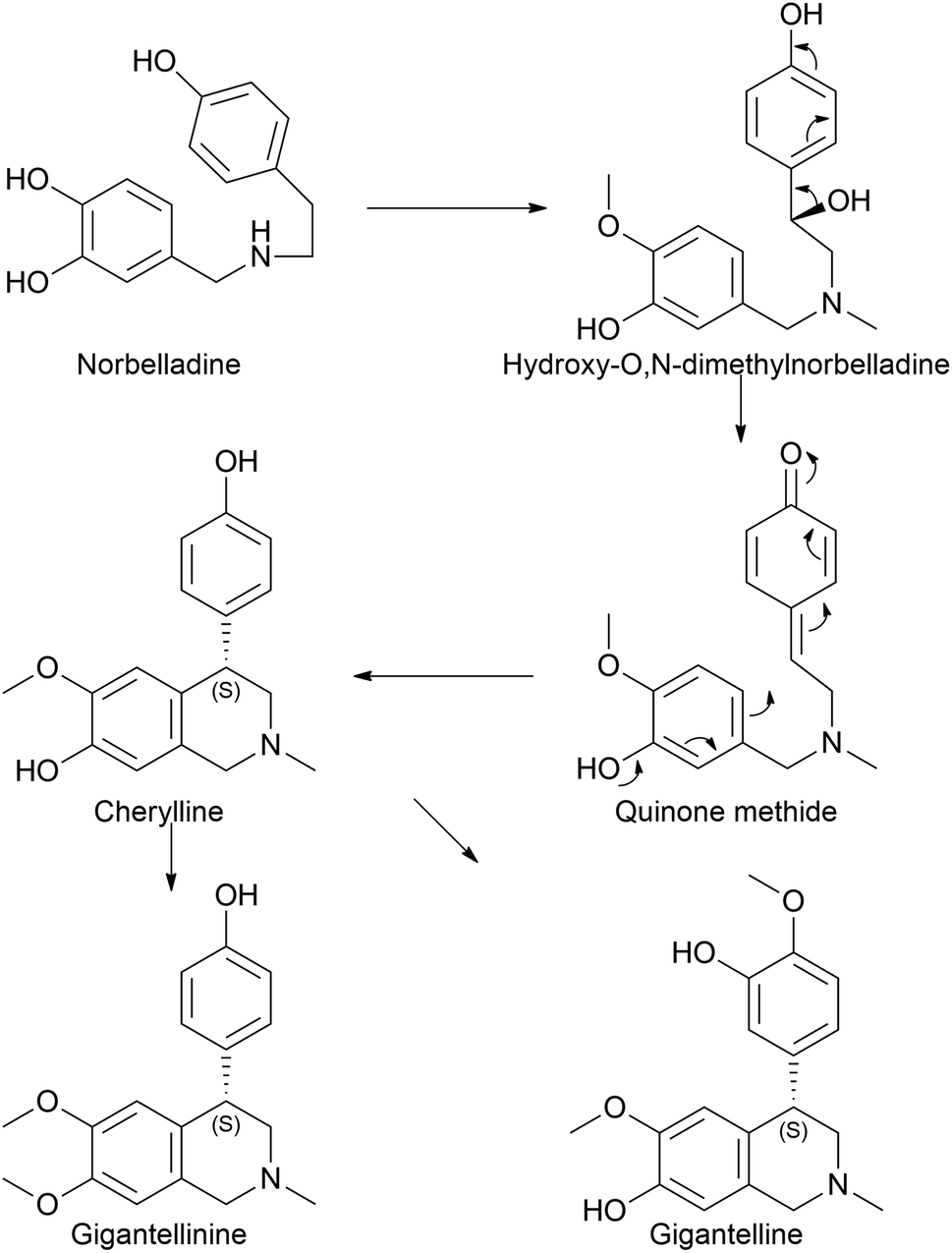 | ||
| Fig. 2 Biosynthetic pathway of cherylline-type Amaryllidaceae alkaloids. | ||
2.2 Norbelladine/belladine-type
Norbelladine/belladine-type is the originating structure of alkaloids that leads to other types of AA, such as galanthamine, lycorine, and crinine via oxidative coupling. Phenylalanine is a precursor of protocatechuic aldehyde/3,4-dihydroxybenzaldehyde (3,4-DHBA) and combines with tyramine, which is yielded from tyrosine to produce the Shiff base (norcraugsodine).44 Norbelladine/benzyl-1-phenylethylamine is formed by the reduction of the above, and subsequent methylation generates O-methylnorbelladine (Fig. 3).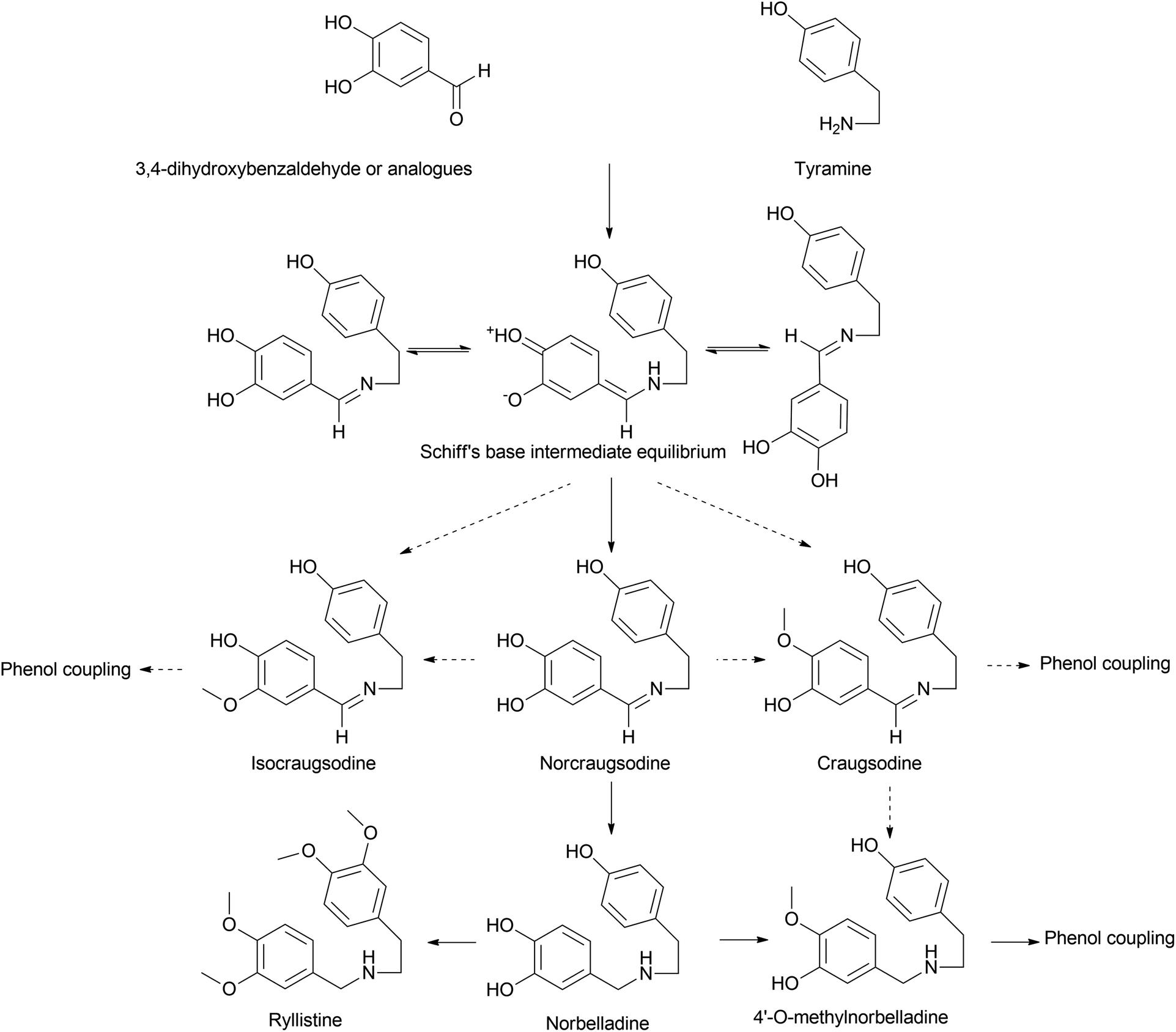 | ||
| Fig. 3 Biosynthetic pathway of belladine-type Amaryllidaceae alkaloids. Interconversion of R1 and R2 in the C6–C1 unit may yield various components, including 3,4-DHBA, vanillin, isovanillin, and corresponding Schiff base intermediates. A dashed arrow represents potential outcomes. | ||
Singh et al. (2018) argued that considering the first committed step in BIA formation via the condensation of dopamine and 4-hydroxyphenylacetaldehyde to form norcoclaurine, the same could be applied to norbelladine formation via the norcoclaurine synthase (NCS) ortholog in Amaryllidaceae identified as norbelladine synthase from Narcissus pseudonarcissus (NpNBS).48 NCS and NBS are members of the same protein family, Bet v1/PR10. This can be classified into two groups: intracellular pathogenesis-related proteins (IPR) and (S)-norcoclaurine synthases (NCS). IPR proteins indicate cytosolic localization of NCS in subcellular compartments other than the cytosol considering its putative N-terminal signal peptides.49 It was previously reported that this family, specifically the NCS group, played a pivotal role in alkaloid production.50 The very same was discussed above in the presence of NR though in lower specific activity. The study left the question of whether tyramine and 3,4-DHBA were converted to norbelladine via a single-step process under the catalysis of NBS or a two-step mechanism, as explained by Kilgore et al. (2014), involving NR.51 However, further studies by our group44 suggest that NBS and NR could cooperatively catalyze the reaction swiftly yet effectively through condensation and imine reduction to achieve norbelladine, avoiding the degradation of unstable norcraugsodine. A report on the NBS of Leucojum aestivum (LaNBS) indicates recent elucidation of the enzyme at the molecular level with data on subcellular localization (likely cytosolic), suggesting that AA biosynthesis is initiated in the cytosol.52 Phylogenetic analysis indicated that LaNBS shares a high amino acid identity with NpNBS1 and NpNBS2. The catalytic activity of LaNBS was confirmed via LC-MS/MS analysis, providing tyramine and 3,4-DHBA as substrates to produce norbelladine.
A considerable increase in publications has been reported on other Amaryllidaceae OMTs related to norbelladine. Sun et al. (2018) characterized an OMT from Lycoris aurea (LaOMT1)53 with activity on both caffeic acid and 3,4-DHBA towards both meta and para positions albeit with significant preferred methylation of the meta position. Similarly, this enzyme converted norbelladine into its successor 4′-O-methylnorbelladine but also into 3′-O-methylnorbelladine. Unlike NpN4OMT, LaOMT1 could convert caffeic acid to ferulic and isoferulic acids in the presence of methyl group donor S-adenosyl-L-methionine (SAM), and 3,4-DHBA yielded vanillin and isovanillin. However, 3,4-DHBA analogues, vanillin and isovanillin, were not methylated by LaOMT1 under their experimental conditions. This implies that once methylated, further methylation/double methylation was not a character at disposal with LaOMT1. The research further revealed that the enzyme was mainly localized in the cytoplasm and endosome.
Li et al. (2019) investigated Lycoris radiata OMT (LrOMT).54 They proposed that given the methoxy positions of different AAs, O-methylation involves different OMTs or a single OMT expressing multiple O-methylation activities. However, after successful molecular cloning, heterologous overexpression and functional characterization of LrOMT, they report it to exhibit both para- and meta-O-methylation using norbelladine as substrate to produce 4′- and 3′-O-methylnorbelladine although moderate preferences towards para-OH, respectively. Comparatively, LaOMT1 previously reported by Sun et al.53 (2018) preferred the meta position against its native substrate, caffeic acid. All three substrates, norbelladine, 3,4-DHBA, and caffeic acid, produced respective outputs with LrOMT, demonstrating its promiscuity. At least two vicinal hydroxyl groups in the aromatic substrate analogue can be recognised by OMT. Catalysis of O-methylation by LrOMT depended on the properties of the binding groups, i.e. electron donating amine groups (norbelladine, dopamine, 3,4-dihydroxybenzylamine, higenamine, (−)-epinephrine, (−)-norepinephrine, and 1,2,3,4-4H-6,7-isoquinolinediol) prefer para position, where electron withdrawing groups, such as carboxyl (caffeic acid) and aldehydes (3,4-DHBA, 5-hydroxyvanillin, 3,4,5-trihydroxybenzaldehyde, and ethyl 3,4-dihydroxybenzoate) (Fig. 4), favoured meta substitution. This summarized the difference of LrOMT from LaOMT1 (ref. 53) and NpN4OMT.51 This article further discusses the vitality of divalent cations in the reaction mixture. Magnesium (divalent, Mg2+) was shown to increase the O-methylation potential, whereas Zn2+, Cu2+ and EDTA (a divalent metal ion chelator) expressed significant inhibition. A recent study by Li et al. (2020) revealed Lycoris longituba OMT (LlOMT) to be a class 1 OMT, where it catalyses norbelladine to generate 4′-O-methylnorbelladine.55 However, they did not report any preferential methyl transfer by LlOMT; in addition, their focus was more on the action of this particular enzyme on the expression of galanthamine content. Another report by the same team suggested that methyl jasmonate treatment on L. longituba induces the expression of NBS and 4′-O-methyltransferase.56Narcissus sp. in AA biosynthesis with respect to its putative genes based on galanthamine was assessed by Aleya (2021).57 The study indicates the expression level of NpN4OMT variation between the field-grown bulb and basal tissues (significantly higher) compared to in vitro cultures (bulblet and callus). Differential expression of NpN4OMT leads to alkaloid biosynthesis in later steps and thus to cellular differentiation.
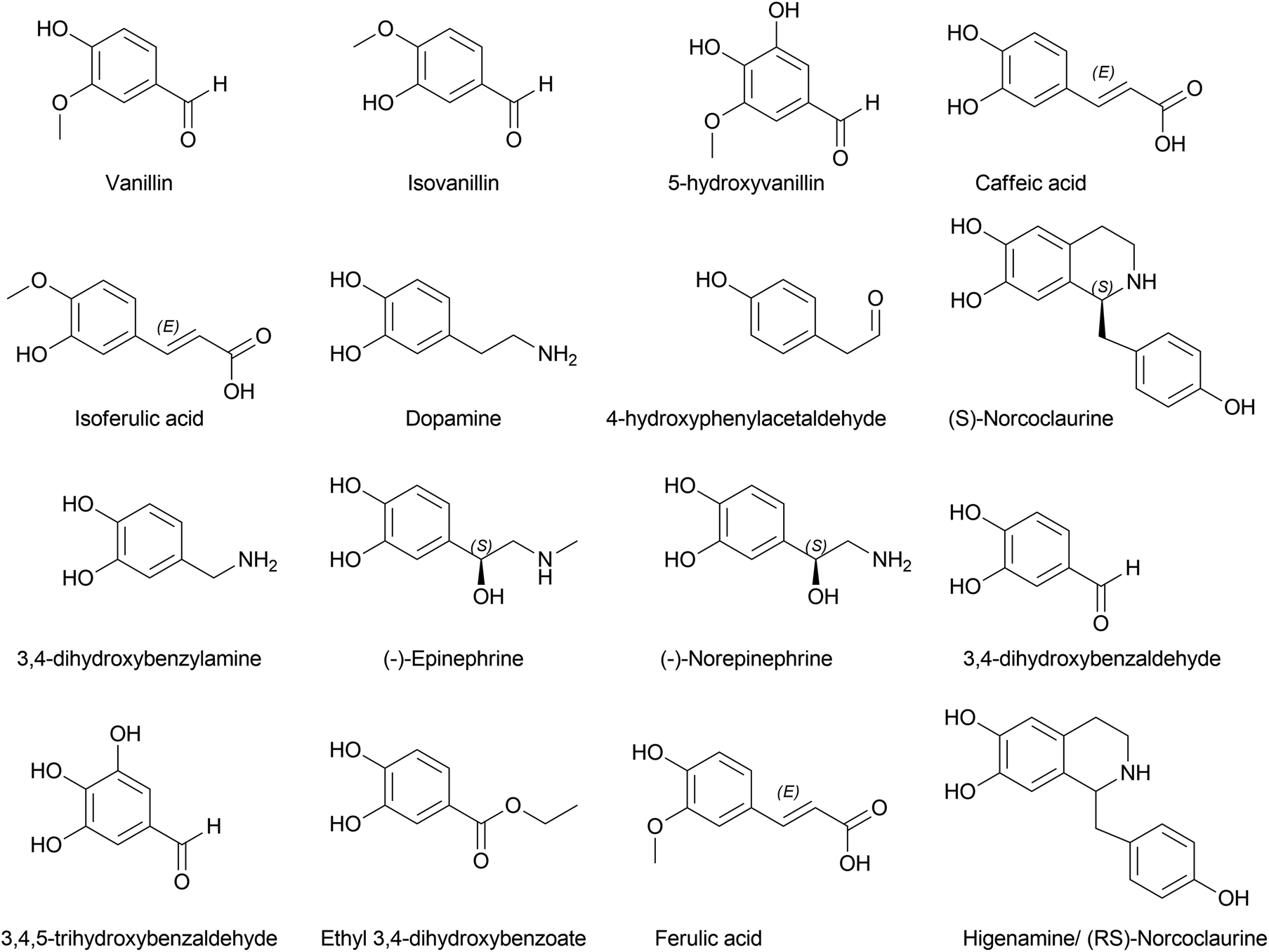 | ||
| Fig. 4 Representative structures discussed in the norbelladine/belladine-type alkaloid formation. | ||
The belladine-type alkaloids can be extended to ryllistine, a 4-oxygenated norbelladine. Ghosal et al. (1984) specially mentioned this compound as a potential candidate for 1,2-epoxy-5,10b-ethanophenanthridine alkaloids, such as (−)crinine and powelline-type alkaloid synthesis, instead of reaching them via the oxidation of (−)oxocrinine owing to its co-occurrence with 1,2-β-epoxyalkaloids.58 However, pathways reaching these compounds or their metabolism have been reported scarcely.
2.3 Downstream pathways: phenol coupling of 4′-O-methylnorbelladine
The plant kingdom possesses C–O or C–C phenol-couple reactions utilized in specialized metabolism processes, such as alkaloid, lignan, and lignin biosynthesis.59 The reactions are highly regio- and/or stereo-selective often owing to cytochrome P450 (CYP) enzymes. A 1% of total gene annotations is estimated to be covered by CYPs in plant genomes, implying their key role in versatile reactions.60 CYP is a diverse class of enzymes with multiple functions, including hydroxylation, oxide bridge formation, demethylation, carbon bond cleavage, skeleton rearrangements, and phenol coupling.61 A detailed report on CYP in plant-specialized metabolism elaborating on unusual reactions was published by Mizutani and Sato (2011).61 Multiple reports in the literature record its potential in salutaridine (CYP719A1), cyclodopeptide cyclo(1-Tyr-1-Tyr) (CYP121), and (S)-corytuberine (CYP80G2) biosynthesis, indicating CYP involvement in alkaloids.59,62,63 Internal phenol coupling is reported here given its highly efficient and selective process. Distinct types of phenol coupling reactions are believed to occur owing to the proximity of the phenolic groups in precursor molecules, allowing for the formation of multiple products from a single substrate. This increases the diversity of alkaloids produced by the plant, which may provide a survival advantage. Alternatively, distinct coupling could occur within a single enzyme, CYP96, which may increase the efficiency of the reaction. CYP96 has evolved to catalyze specific types of coupling. Additionally, internal phenol coupling can avoid the need for the substrate to cross cell membranes and interact with other enzymes and molecules. This reduces the chances of substrate loss or competing reactions, which can increase the yield of the desired product.Phenol coupling occurs in the late–late pathway of AA biosynthesis. Highly complex structures are formed owing to sequential phenolic coupling and reduction reactions. The pathway is characterised by the formation of a C–C bond between two phenolic precursors, leading to a new carbon skeleton. The common precursor 4′-O-methylnorbelladine is modified by the catalysis of CYP96 in Amaryllidaceae. For the first time, Barton and Cohen (1957)64 proposed the oxidative coupling of Amaryllidaceae based on norbelladine or related compounds.61 The condition suggested was the suitable protection of ring A phenolic group(s) via methylation. Oxidative coupling leads to distinct skeletons of AA. Secondary cyclization occurs via the oxidative coupling of O-methylnorbelladine, leading to restructuring and diversification. Three distinct pathways are suggested depending on the C–C bridge formation between ring A (C6–C1 unit) and C (C6–C2 unit): para–ortho′, ortho–para′, and para–para′ (Fig. 5). Hydrogen abstraction from phenol believed to be an action initiated by the CYP leads to the delocalization of unpaired/radical electrons via resonance structure formation, and phenol radicals are then quenched by coupling, producing the aforementioned combinations. Multiple intermediates are formed in this process, determining the end product. Biosynthesis reactions conducted using radioisotope labels have contributed much to the understanding of the mechanisms and pathways in this step of AA biosynthesis.65–68 A hypothetical pathway involving intermediates is presented in Fig. 5.
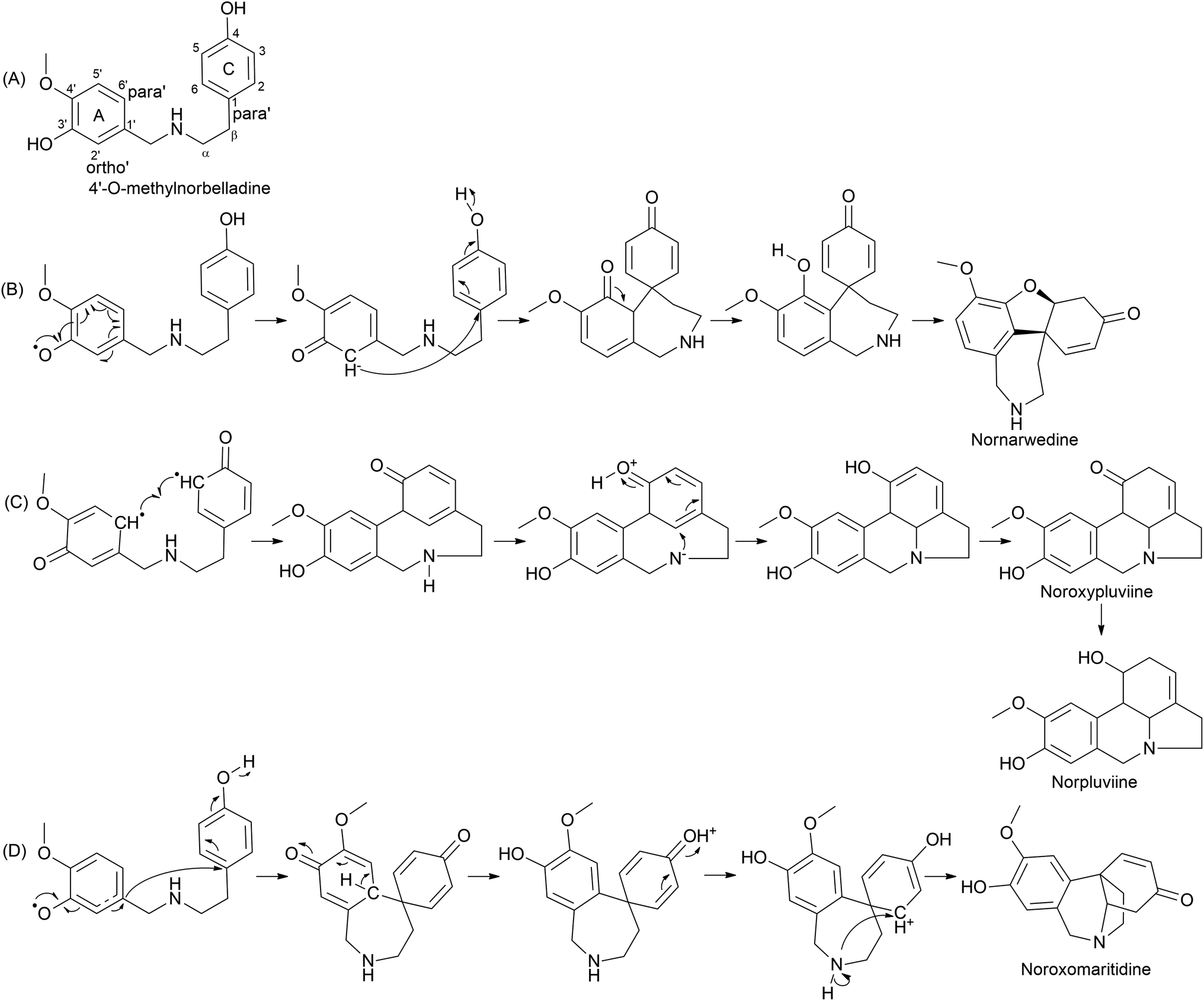 | ||
| Fig. 5 Proposed mechanism for the phenol coupling reaction. (A) The structure of 4′-O-methylnorbelladine; (B) para–ortho′ coupling leading to nornarwedine; (C) ortho–para′ coupling resulting norpluviine; (D) para–para′ coupling leads to noroxomaritidine. | ||
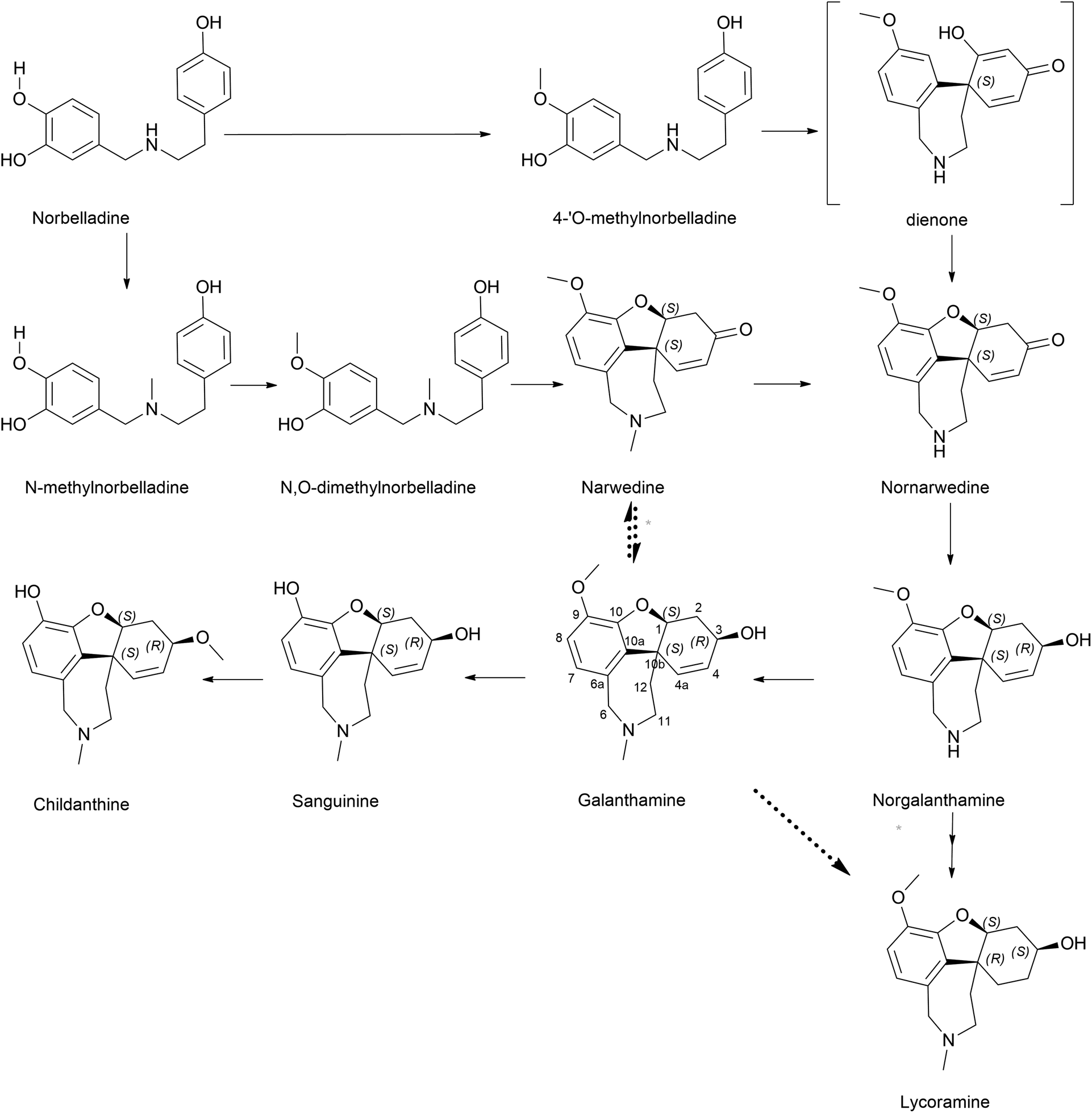 | ||
| Fig. 6 Para–ortho′ coupling pathway-related components. Galanthamine is the primary pathway illustrated. Dashed arrows indicate minor controversial steps, where more than one arrow represents multiple steps. | ||
Sanguinine and childanthine comprise the same dibenzofuran nucleus, confirming their relationship with this pathway. However, either the pathway proceedings to the above or the enzymes involved have not yet been reported or critically analysed.
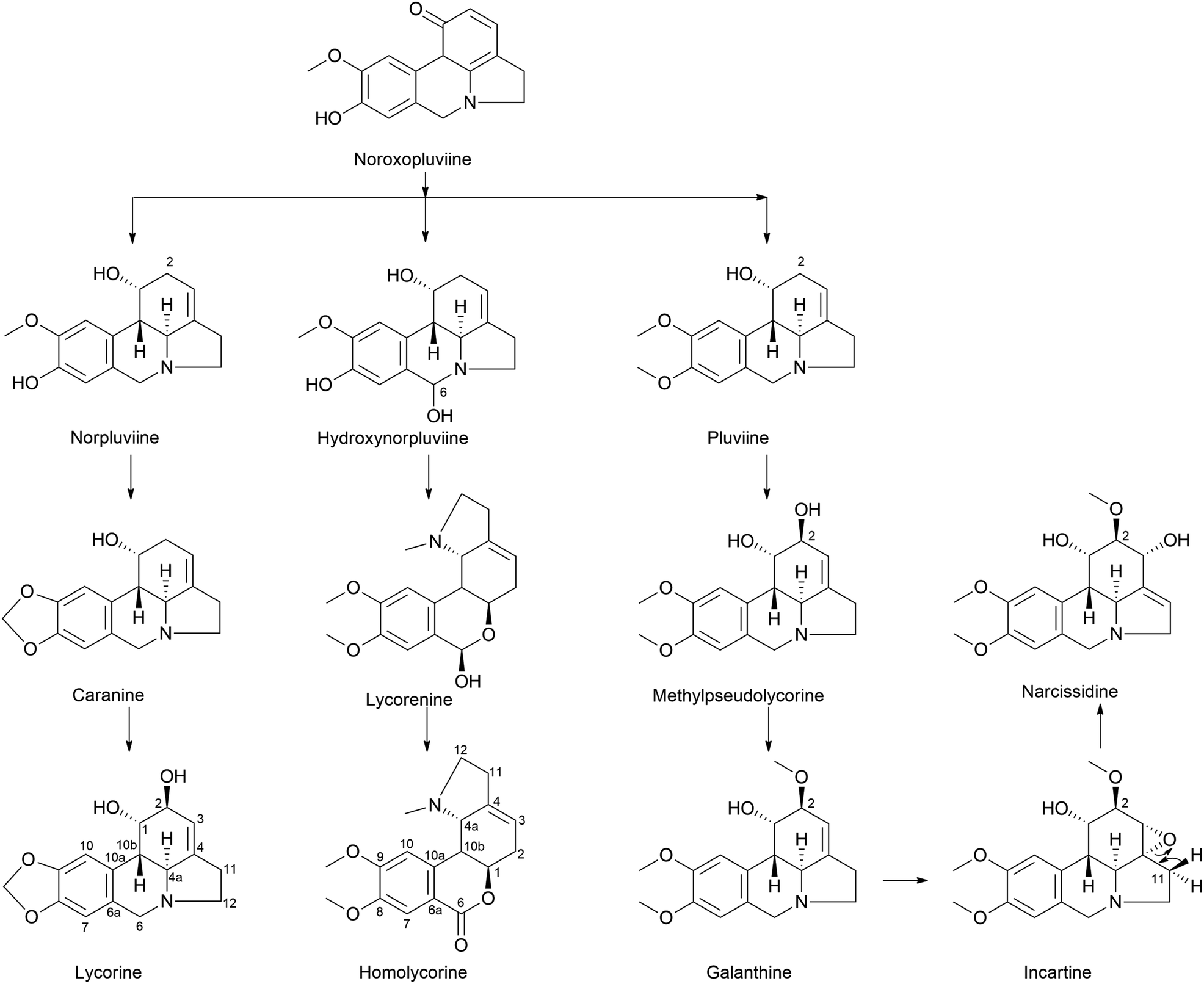 | ||
| Fig. 7 Ortho–para′ coupling pathway in lycorine and homolycorine-type alkaloid biosynthesis. Narcissidine biosynthesis through galanthine also illustrated. | ||
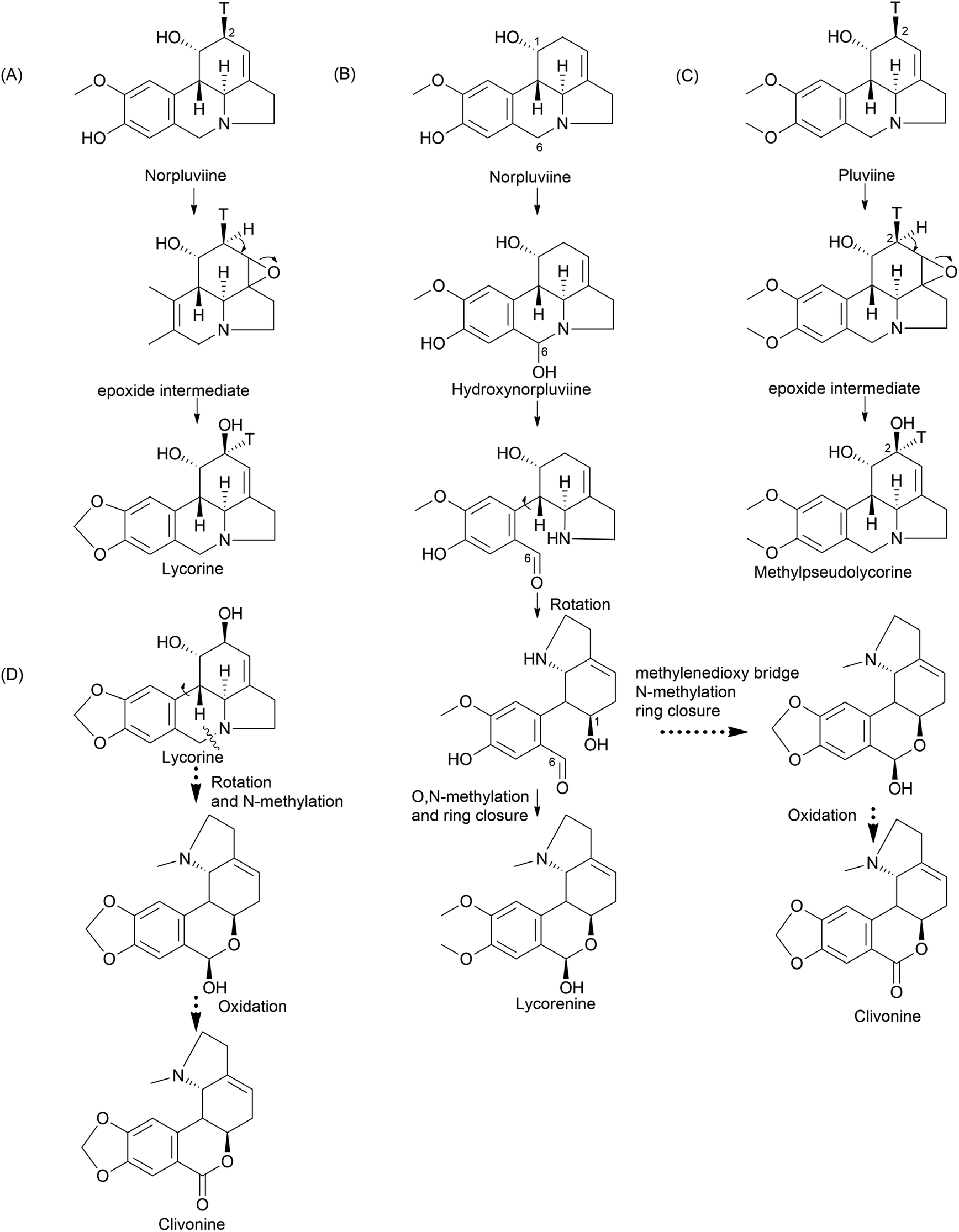 | ||
| Fig. 8 Ortho–para′ coupling pathway-related components biosynthetic mechanisms. Norpluviine proceeds to lycorine through (A) inversion and/or retention of C2 tritium elucidates hydroxylation stereospecificity; (B) bond cleavage, rotation, and hemiacetal bond establishment, and another pathway to reach clivonine; (C) suggested epoxide intermediate formation to achieve methylpseudolycorine; (D) clivonine biosynthesis via lycorine. | ||
Furthermore, the double incorporation of tritium and 14C to study norpluviine to lycorine solidified the notion that the hydroxylation process is stereospecific based on isotope label integration ratios. Wildman and Heimer reported similar observations with Zephyranthes candida.78 Interestingly, pluviine-to-galanthine conversion via methylpseudolycorine is reported to occur similarly, with inversion at C2.81 The alkaloid incartine was first identified and proposed as an intermediate of galanthine to narcissidine transformation by Kihara and a team in L. incarnate.82,83 Epoxide formation and the stereospecific C11 loss of hydrogen ultimately result in narcissidine. However, a contradicting view of the retention of configuration 2β-3H in caranine was also reported in Clivia miniata.77 Intriguingly, there was a complete loss of tritium in the original ortho position (C3) of the C6–C2 fragment, which leads to an altered stereochemical hydroxylation pathway, hence irradicating the implication of the 2-oxo-derivative. It is noteworthy that norpluviine should undergo methylenedioxy bridge formation from O-methoxyphenol at the C6–C1 unit and allylic hydroxylation at the C6–C2 unit (C2). This has led to the understanding that norpluviine yields caranine and then lycorine.80 The structure of lycorine was first determined by Nakagawa et al. in 1956, and absolute stereochemistry was assigned.84,85 Since then, lycorine and its type alkaloids have been investigated because of their structure, bioactivity and relevant relationship although the metabolic pathway involving particular enzymes is yet to be revealed.
The homolycorine pathway derives from different directions initiated from norpluviine by reducing its C6 position to obtain hydroxynorpluviine, and the oxidation of the same leads to a ring opening between C6 and nitrogen. This leaves ortho–para′ C–C coupling bond vulnerable to free rotation. Consequently, it allows the C6 ketone and C1 hydroxyl groups to create a hemiacetal linkage. However, it is questionable whether ring closure or O/N-methylation occurs primarily to produce lycorenine. The stereochemistry of lycorenine production from norpluviine was studied by Fuganti and Mazza (1973),68 where they confirmed the removal of C6 pro-R hydrogen by allowing hydroxylation in the final product lycorenine. However, the behaviour of stepwise intermediates is not reported here. Further oxidation of the C6 hydroxyl of lycorenine ultimately results in homolycorine.
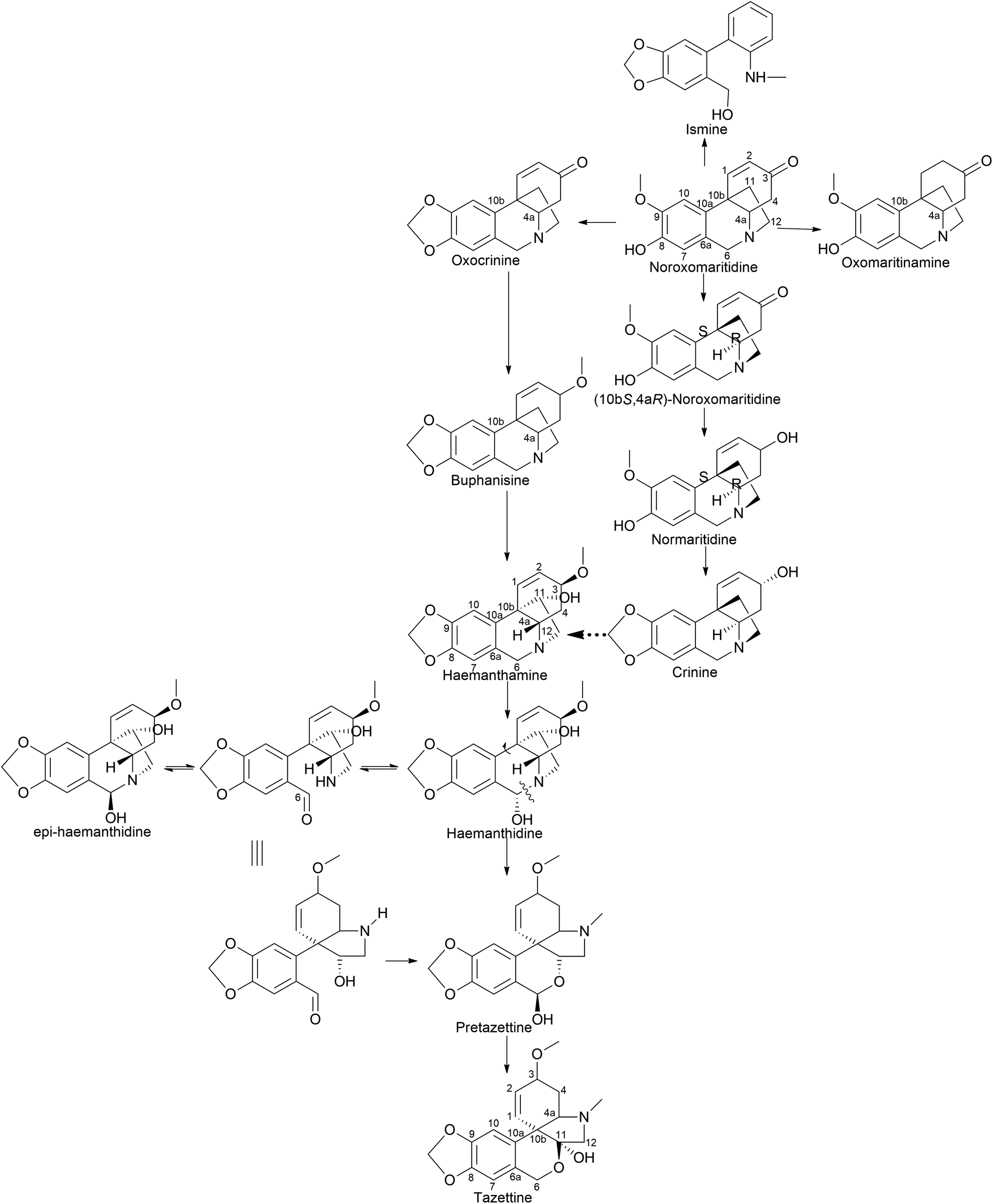 | ||
| Fig. 9 Para–para′ coupling to reach crinine and tazettine type skeletons. | ||
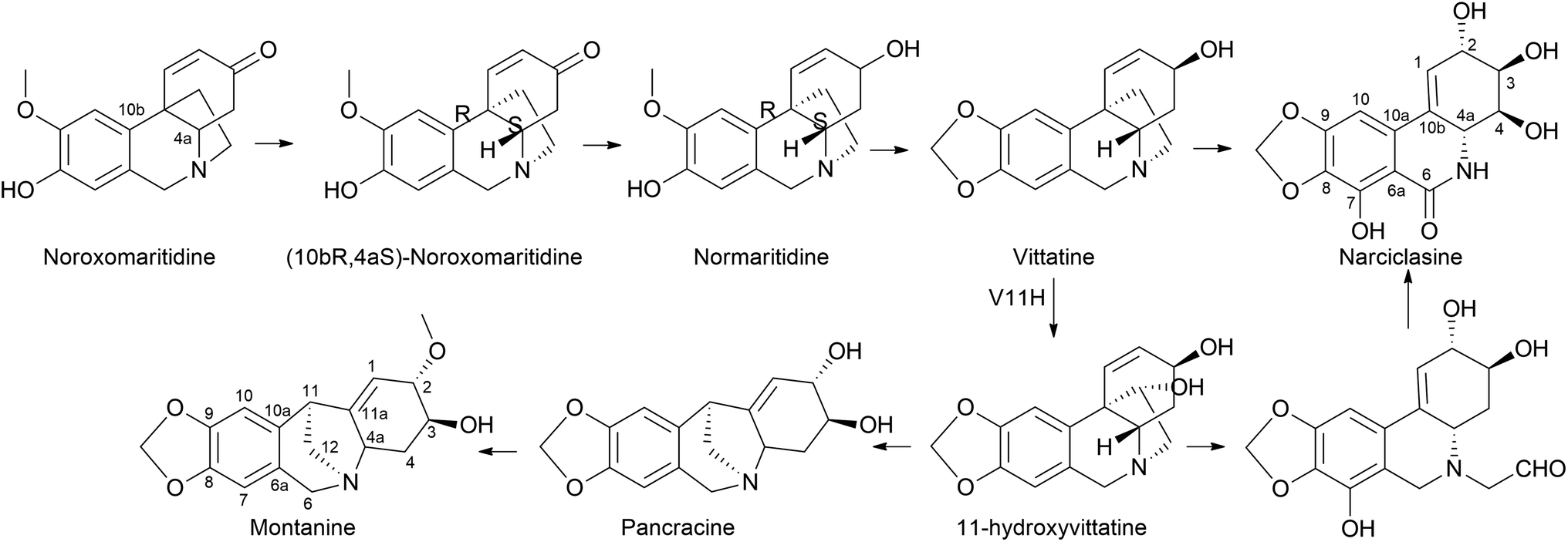 | ||
| Fig. 10 Para–para′ coupling to reach narciclasine and montanine type skeletons. | ||
It is surprising that irrespective of the widely elaborated structural diversity studies on the pathway of AA after phenol coupling reactions, only few biosynthetic evaluation attempts have been made to reveal the enzymes involved. Kilgore et al. (2016) for the first time published on CYP96T1 can produce two out of four enantiomers of noroxomaritidine [(10bR,4aS) and (10bS,4aR)] from 4′-O-methylnorbelladine, thus exhibiting its potential in para–para′ direction. However, minor amounts of nornarwedine were also observed, opening the pathway towards para–ortho′ direction. It was reported to be the first phenol coupling enzyme to be characterized from a monocot. It is noteworthy that CYP96T1 also converted 4′-O-methyl-N-methylnorbelladine to produce several C–C phenol-coupled products, with narwedine being one of them (Fig. 5). Interestingly, no product formation was observed when 3′-O-methylnorbelladine was fed as the substrate.70 The same team reported noroxomaritidine reductase (NR) (discussed in Subsubsection 2.2.1 as norcraugsodine reductase) for its involvement in the conversion of noroxomaritidine to oxomaritinamine47 (Fig. 9).
Barton proposed a novel concept that extended the scope of intermolecular phenolic coupling to form tazettine and lycorenine subclasses.69 This involved rearranging haemanthamine and lycorine-type precursors through a process referred to as “ring switching” by Giró Mañas et al. (2010).94 Wildman later confirmed these theories through tritium feeding experiments in Sprekelia formosissima, supporting the formation of tazettine, and in Narcissus ‘King Alfred’ for lycorenine considered earlier.78,95 Wildman introduced a biomimetic procedure for generating pretazettine from haemanthidine. However, challenges were encountered in developing an analogous protocol for the biomimetic conversion of lycorine into lycorenine-type ring systems. The transformation necessitates a ∼180° rotation and involves minimal strain relief unlike the ∼90° rotation with significant strain relief observed in the haemanthidine/pretazettine series.81,96 Consequently, Barton's original hypothesis remained unconfirmed from a synthetic perspective until Giró Mañas et al. (2010)94 finally corroborated experimentally the biogenetic hypothesis.
Clivimine classification in the literature is a subject of contention, with some scientists categorizing it as an independent ring structure,32 while most assert it belongs to the lycorenine subclass.94,97,98 Clivonine, a compound first isolated and characterized from Clivia miniata Regel in 1956 by Wildman,99 shares the same core ring system (2-benzopyrano[3,4-g]indole) as lycorenine-type alkaloids albeit with a slight modification involving methylenedioxy bridge formation from O-methoxyphenols at the C6–C1 unit and lactol oxidation at C7. The biosynthesis of clivonine from a lycorine-type progenitor necessitates benzylic oxidation, a ∼180° ring switch, followed by N-methylation and lactol oxidation.94
Limited information is available regarding the mechanistic aspects of the catabolic pathway that lead to ismine. Fuganti (1973)100 reported feeding experiments conducted to elucidate certain stereochemical aspects of this biosynthetic process. Sprekelia formosissima was used [6,10-3H2; 12-14 C] noroxomaritidine for labelling, which resulted in incorporations of 0.03% into ismine, 2% into haemanthamine, and 0.8% into haemanthidine. Ismine showed no 14C activity. The radioactive ismine's labelling pattern was indirectly determined by its conversion into phenanthidone, which retained approximately 25% of the tritium activity. This suggests that during the biological transformation of noroxomaritidine into ismine, there is a loss of the original tritium located at C-6 in noroxomaritidine, specifically at the benzylic position adjacent to tertiary nitrogen, a stereospecific removal of hydrogen at some point in the biosynthesis, followed by a reduction step. Furthermore, the complete absence of 14C activity indicates that the carbon atom at C-1 of [6,10-3H2; 12-14C]noroxomaritidine is not retained as an N-methyl group in ismine. Hence, the report suggests that ismine is derived through para–para coupling, followed by late elimination of the ethanol bridge from the C-15 crinane skeleton.
3 Recently reported anti-viral activities of Amaryllidaceae alkaloids and their possible molecular targets
Amaryllidaceae are recognized for their therapeutic properties24 owing to the isoquinoline alkaloids they produce.23 In addition to their probable endogenous role in the protection against plant foreign invaders,101 AA possess multiple biological properties relevant to human health. For example, galantamine is a strong inhibitor of acetylcholinesterase and has been approved for treating mild symptoms of Alzheimer's disease. Other AAs display anti-microbial properties against various animal and human threats. The first available report on the antiviral properties of alkaloids dates back to 1976 by Furusawa et al.,102 and impressively, the discovery of new antiviral activities has been unceasing. In 1989, Renard-Nozaki and colleagues showed that several types of AA target the herpes simplex virus, a DNA virus.103 In 1992, Gabrielsen et al. uncovered that many AAs also impede the replication of several families of RNA viruses in vitro, and in a mouse model.104 Lycorine has emerged as one of the most efficient antiviral compounds of this family, targeting a broad range of virus types at nM concentrations. The last 5 years have been scarred by the Coronavirus Disease 2019 (COVID-19) pandemic, orienting research towards the discovery of anti-severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) molecules. In this section, recent discoveries on AA antiviral activity against coronaviruses but also against other emerging or reemerging diseases impacting human health (such as dengue, Zika, influenza and chikungunya viruses); or animals (duck tembusu virus, DTMV); and plants (tobacco mosaic virus, TMV) are summarized (Table 1).| Virus | Alkaloid EC50, CC50 | Target (IC50) | Model | Ref. | |
|---|---|---|---|---|---|
| a SARS-CoV: severe acute respiratory syndrome – coronavirus, MERS-CoV: middle east respiratory syndrome – coronavirus, DENV: dengue virus, ZIKV: Zika virus, WNV: West Nile Virus, DTMUV: duck tembusu virus, INFV: influenza virus, CHIKV: chikungunya virus, SINV: Sindbi Virus, SFV: Semiliki Forest Virus, HSV: Herpes Simplex Virus, TMV: tobacco mosaic virus. RdRp: RNA-dependent RNA polymerase. MPro: SARS-CoV main protease. RNP: ribonucleoprotein. N.d.: not determined. | |||||
| Coronavirodeae | SARS-CoV | Lycorine (EC50 = 0.31 μM, CC50 > 40 μM) | n.d. | In cellulo (simian VeroE6 and human Huh7 cells) | 106 |
| Lycorine (EC50 = 1.02 μM; CC50 > 10 μM) | n.d. | In vitro + in cellulo (Vero) | 8 | ||
| SARS-CoV-2 | Lycorine (EC50 = 0.878 μM; CC50 > 10 μM) | In silico docking with RdRP | In vitro + in cellulo (Vero) | 8 | |
| Lycorine (EC50 = 0.439 μM, CC50 > 1000 μM in VeroE6, 0.834 μM in Huh7, 1.044 μM in HEK293T cells) | (1) Ribosome (in silico), (2) binding to frameshift stimulation element (FSE) in SARS-CoV-2 genome (in silico), (3) RdRp (in silico and in vitro), (4) viral core assembly (in vitro) | In silico, in vitro and in cellulo (VeroE6, Huh7, HEK293T) | 2 | ||
| Lycorine (EC50 = 0.01 μM, CC50 = 18.78 μM) | MPro | In vitro (cell protease assay) | 4 | ||
| Trans-dihydrolycoricidine (EC50 = 0.18 μM, CC50 = 3.48 μM), dihydronarciclasine analog with ring-A mono-substituted C7-OH (EC50 = 0.09 μM, CC50 = 1.92 μM) | n.d. | In cellulo (Calu3) | 14 | ||
| MERS-CoV | Lycorine (EC50 = 2.123 μM; CC50 > 10 μM) | RdRp (IC50= 1.406 μM) | In vitro (cell-based reporter assay), in cellulo | 8 | |
| Flaviviridae | ZIKV | Hippeastrine hydrobromide (EC50 = 3.62 μM; CC50 > 100 μM) | Post-entry | In cellulo (hNPCs, human fetal-like forebrain organoids), in vivo (adult mouse) | 109 |
| Lycorine (EC50 = 0.22–0.39 μM; CC50 = 4.4–21 μM) | Post-entry step during RNA replication, RdRp (IC50 = 25 μM) | In cellulo (A549, Huh7 and Vero), in vivo AG6 mice (10 mg kg−1) | 110 | ||
| Cherylline (EC50 = 20.3 μM; CC50 > 100 μM) | Post-entry step – RNA synthesis | In cellulo (VeroE6 and Huh7) | 111 | ||
| Lycorine (EC50 = 0.9 μM; CC50 = 4.3 and 3.4 μM in BHK-21 and Vero), pretazettine (1.9 μM; CC50 = 5.4 and 7.2 μM), narciclasine (EC50 = 0.02 μM; CC50 = 0.09 and 0.12 μM), narciclasine-4-O-β-D-xylopyranoside (EC50 = 7.9 μM; CC50 = 39.3 and 51.8 μM) | n.d. | In cellulo (BHK-21 and Vero) | 112 | ||
| DENV | Cherylline (EC50 = 8.8 μM; CC50 > 100 μM) | Post-entry step – RNA synthesis | In cellulo (VeroE6 and Huh7.5) | 111 | |
| Haemanthamine (EC50 = 337 nM; CC50 > 10 μM), pancracine (EC50 = 357 nM; CC50 > 10 μM) haemanthidine (EC50 = 476 nM; CC50 > 10 μM) | n.d. | In cellulo (Huh7) | 1 | ||
| 3,4-Dihydroxybenzaldehyde (EC50 = 24.1 μM; CC50 = 173.1 μM), 3′,4′-O-dimethylnorbelladine (EC50 = 27.5 μM; CC50 = 131.4 μM), norcraugsodine (EC50 = 37.7 μM; CC50 = 121.8 μM) | n.d. | In cellulo (Huh7) | 3 | ||
| Lycorine (EC50 = 0.9 μM), pretazettine (EC50 = 1.9 μM), narciclasine (EC50 = 0.02 μM), narciclasine-4-O-β-D-xylopyranoside (EC50 = 7.9 μM) | n.d. | In cellulo (BHK-21 and Vero) | 112 | ||
| DTMUV | Lycorine (EC50 = 2 μM) | Post-entry step | In cellulo (BHK-21) | 113 | |
| Orthomyxoviridae | INFV | Lycorine (EC50 = nd) | Nuclear–cytoplasmic transport impairment, decrease Nup93 expression | In cellulo (GD178 and MDCK) | 114 and 115 |
| Lycorine (EC50=nd) | Blockage of nuclear export of vRNP through repression of COX41 and ERK signaling pathway | In cellulo (MDCK), in vivo (in mice 5 mg kg−1 day−1) | 115 | ||
| Togaviridae | CHIKV, SINV, SFV, and VEEV | Lycorine (EC50 = 0.38–0.75 μM, CC50 > 10 μM); SINV, SFV, and VEEV (1.05, 0.83 and 0.31 μM) | Post-entry – viral translation | In cellulo (Vero, BHK21, Huh7, A549) | 9 |
| Picornaviridae | EV71 and CVA16 | LY-55 (2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl 2-((4-fluorophenyl)thio)acetate hydrochloride) (EC50 = 1.69–5.01 μM, CC50 = 106.36 μM) | Not virucidal, blocking autophagy | In cellulo (Vero) and in vivo (mice) | 6 |
| Herpesviridae | HSV | Dihydronarciclasine analog with ring-A mono-substituted C7-OH (EC50 = 0.209 to 0.504 μM, and no impact on viability <50 μM) trans-dihydrolycoricidine (EC50 = nd) | (1) Activation of ERK, ElF2 and PKR; (2) integrated stress response; (3) autophagy; (4) sirtuin signaling pathways; (5) innate antiviral response | In cellulo (in organoids with hiPSC), in vivo (mouse) | 14 |
| Tobamoviridae | TMV | Lycorine (EC50 = nd) | Uncharacterized | In planta | 116 |
| Lycoricidine derivatives (N-methyl-2,3,4-trimethoxylycoricidine), (N-methyl-2-methoxy-3,4-acetonidelycoricidine) (EC50 = nd) | Triggers defense enzyme activity | In planta | 117 | ||
3.1 Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
Lycorine was reported to exhibit anti-coronaviral activities in 2005.105 In the early days following the onset of the COVID-19 pandemic, Zhang et al. tested the antiviral properties of SARS-CoV-2 permissive VeroE6 cells (isolated from the kidney of an African green monkey) and human hepatocarcinoma-derived Huh7 cells.106 They showed that lycorine EC50 (compound concentration required to inhibit 50% of viral titer) was 0.31 μM (assessed at 72 h), with a CC50 > 40 μM (concentration required to kill 50% of the cells, assessed at 24 h in this case). In 2021, Jin and colleagues further investigated its mechanism of action. They first confirmed its anti-coronaviral effect against SARS-CoV, SARS-CoV-2 and MERS-CoV (Middle East Respiratory Syndrome – CoV) with slightly higher EC50 (1.021, 0.878, and 2.123 μM, respectively), (Table 1). Their results suggested that lycorine blocked RNA replication, acting as a non-nucleoside RNA-dependent RNA polymerase inhibitor (RdRp) of MERS-CoV with a measured IC50 of 1.406 μM.6 Their docking analysis demonstrated that lycorine also interacted with SARS-CoV-2 RdRp through hydrogen bonding with Asp623, Asn691, and Ser759 residues. In 2021, Ren et al. used a muti-targeting approach including both computational and experimental techniques to assess the potential of three alkaloids to tackle SARS-CoV-2 replication and clarify the lycorine effect on SARS-CoV-2 replication.2 First, they predicted that lycorine may target the host cell 80S ribosome subunit by applying computational methods. To reach this conclusion, the authors constructed a three-dimensional in silico model of the human ribosome in a complex with lycorine using the experimental structure of yeast ribosomes complexed with lycorine as a reference. The AA was found to fit tightly into the ribosome peptidyl transfer site and was stabilized by hydrogen bonds with several amino acids and Mg2+. Second, they suggested that lycorine attenuates SARS-CoV-2 propagation by binding to the frameshift stimulation element (FSE), which is a structured RNA motif from the SARS-CoV-2 genome with a 5′ heptanucleotide slippery site UUUAAAC and a three-stem pseudoknot involved in polyprotein processing.107 Their prediction demonstrates that lycorine binds near the slippery site of this viral RNA structure. Further computational analysis coupled with in vitro binding affinity data predicted that lycorine may also inhibit RNA synthesis by occupying the RNA growth site of RdRp but only in the presence of RNA. Lycorine was too small a molecule to sojourn in the RdRp large empty cavity in its apo state but could be stabilized following binding with RNA, as the enzyme cavity becomes more compact. In this sense, the affinity of lycorine for the enzyme might be secondary to its affinity to nucleic acids. Finally, in experimental settings of cell infection, an EC50 of 0.438 μM was measured by RT-qPCR in VeroE6 cells, and an unexpected CC50 > 1000 μM. However, lycorine notorious cytotoxicity was detected in human hepatocarcinoma Huh7 and human embryonic kidney HEK293T cells, with much lower CC50 of 0.834 and 1.044 μM, respectively. In 2022, Narayanan et al. indicated that lycorine may also target SARS-CoV-2 polyprotein processing through the inhibition of main protease (MPro) activity in a cell protease assay although no interaction with key residues in the catalytic site of the enzyme could be detected by docking analysis.4 Although many recent studies have focused on lycorine, narciclasine and its analogues have been previously shown to be strong inhibitors of coronaviruses and RNA viruses possibly through interaction with eukaryotic translation elongation factors eEF1A, nucleocapsid and/or nucleic acids, as extensively reviewed in.108 Earlier this year (2023), chemically synthetized trans-dihydrolycoricidine and dihydronarciclasine analog with ring-A mono-substituted C7-OH were also identified as inhibitors of SARS-CoV-2,14 with IC50 = 0.18 μM, CC50 = 3.48 μM, IC50 = 0.09 μM, and CC50 = 1.92 μM. Le et al. reported that 6α-hydroxyhippeastidine, 6β-hydroxyhippeastidine, 2-epi-lycorine, zephyranthine, and ungeremine isolated from Hymenocallis littoralis demonstrated weak anti-SARS-CoV-2 activity.109 Unfortunately, none of these studies tested the efficacy of lycorine or other AAs in animal models of SARS-CoV-2 infection, but they add to the evidence that some AAs are potent antiviral compounds against emerging coronavirus infections and that they probably target multiple steps of the viral replication cycle.3.2 Dengue and Zika viruses
Dengue fever, caused by the dengue virus (DENV), is the most prevalent arthropod-borne viral disease and is endemic to tropical and subtropical regions, progressively affecting temperate regions. Zika virus (ZIKV), a closely related flavivirus, can be transmitted vertically in utero and causes congenital Zika syndrome, other birth defects, and Guillain–Barré syndrome in adults. No approved antiviral drugs for these diseases are currently available. As early as 1992, Gabrielsen et al. detected that some AAs could inhibit flaviviruses, such as the Japanese encephalitis virus, the yellow fever virus, and DENV.104 In recent years, many studies have contributed to increasing our knowledge of the anti-flaviviral properties of AAs. In 2017, Zhou and colleagues reported that hippeastrine hydrobromide (HH) inhibited ZIKV infection at a post-entry step in human pluripotent stem cell-derived cortical neural progenitor cells (hNPCs) (EC50 = 3.62 μM, Table 1).110 They further showed that treatment with 25 μM of HH for 3 and 17 days rescued ZIKV-induced growth and differentiation defects in hNPCs and human fetal-like forebrain organoids and that it efficiently inhibited ZIKV infection in adult mouse brains in vivo. HH suppressed viral propagation when administered to adult mice with active ZIKV infection (at a dose of 100 mg kg−1 day−1 subcutaneously). The authors concluded that HH is a highly promising drug candidate for treating ZIKV-infected patients. In 2020, Chen et al. aimed to elucidate the anti-ZIKV properties of lycorine.111 They used RT-qPCR, immunofluorescence, western blot, and plaque forming assay to analyze viral RNA (vRNA), viral protein, and progeny virus counts in A549, Huh7 and Vero cells. The time-of-drug addition assay suggested that the AA acts at a post-entry step during RNA replication with EC50 ranging from 0.22 to 0.39 μM and CC50 ranging from 4.4 to 21 μM. The authors also showed that lycorine increased the thermal stability of NS5 (containing RdRp and methyltransferase domains) and that RdRp activity was reduced by 50% when the enzyme was treated with 25 μM of the compound. However, at this concentration, lycorine is highly cytotoxic to human cells mitigating the relevance of this interaction. Finally, the authors showed that treatment with lycorine (10 mg kg−1) protected AG6 mice against ZIKV-induced lethality (83% less death) by decreasing the viral load in the blood. These results differ from those of previous studies on the West Nile Virus, a related flavivirus in which lycorine does not target RdRp.112 Instead, its antiviral properties are specific to the 2K peptide, which plays a key role in the replication complex and polyprotein processing.In 2021, our group uncovered that extracts from Senegalese Crinum jagus inhibited DENV infection and that cherylline was a potent anti-flaviviral AA, targeting both DENV (EC50 = 8.8 μM) and ZIKV replication (EC50 = 20.3 μM).5 Further experiments revealed that cherylline and lycorine targeted a post-entry step, specifically hindering the viral RNA synthesis step but not viral translation. The cherylline ring structure is distinct from that of lycorine (Fig. 1), and their molecular targets probably differ although both inhibit a post-entry step related to RNA synthesis. In silico reverse screening predicted that human cellular proteins, such as estrogen and dopamine receptors, could be targets of cherylline.
In previous studies, we found that alkaloid extracts from another Senegalese Amaryllidaceae, Pancratium trianthum, were highly effective in blocking DENVGFP replication5 although the isolated molecules were not individually tested. We later characterized the activity of several alkaloids isolated from P. maritimum, namely lycorine, 9-O-demethyllycorine, haemanthidine, haemanthamine, 11-hydroxyvittatine, homolycorine, pancracine, obliquine, tazettine and vittatine.1 All the tested AA inhibited DENV replication (EC50 ranges from 0.34 to 73.59 μM) at low or non-cytotoxic concentrations (CC50 ranges from 6.25 μM to >100 μM), haemanthamine (EC50 = 337 nM), pancracine (EC50 = 357 nM) and haemanthidine (EC50 = 476 nM) being the most potent anti-DENV inhibitors. Haemanthamine-type alkaloids are known to target other RNA viruses and retroviruses. Although the mechanism of antiflaviviral activity has not been studied, previous studies have shown that haemanthamine targets the ribosome peptidyl transferase center A-site on the large ribosomal subunit through interactions with 25S rRNA to disrupt the protein synthesis elongation phase and cancer cell growth.113 This interaction could lead to the inhibition of viral protein synthesis and block viral replication.
In 2022, we studied the anti-DENV activity of norbelladine, its derivatives and its precursors.3 We revealed that the 3,4-dihydroxybenzaldehyde (selectivity index (SI) = 7.2), 3′,4′-O-dimethylnorbelladine (SI = 4.8), 4′-O-methylnorbelladine (SI > 4.9), 3′-O-methylnorbelladine (SI > 4.5), and norcraugsodine (SI = 3.2) reduced DENV infection with EC50 values ranging from 24.1 to 44.9 μM, and the mechanism of action is currently under investigation. Barbosa et al. searched for plant-derived antiviral agents against DENV and ZIKV.114 Hippeastrum extracts were highly antiviral. Lycorine (0.5 and 0.9 μM), pretazettine (0.8 and 1.9 μM), narciclasine (0.02 and 0.02 μM), and narciclasine-4-O-β-D-xylopyranoside (7.5 and 4.9 μM) were identified as anti-DENV-2 and -ZIKV inhibitors although they were also cytotoxic. These results agree with previous studies,104 suggesting that optimization of the AA structure could lead to new therapeutic strategies to fight flaviviral infectious diseases.
3.3 Duck tembusu virus (DTMUV)
DTMUV is a mosquito-borne flavivirus discovered in 2010 in China 114 and is responsible for fatal outbreaks in ducks115–117 and other bird species in China and Southeast Asia. Despite the availability of efficient vaccines, outbreaks continue to occur, weighing heavily on the farming economy. In 2021, Lv et al. investigated lycorine ability to inhibit the infection of this avian flavivirus.118 As viral target cells, they used hamster cell lines BHK-21 and evaluated DTMUV replication by real-time PCR, virus titer, western blot and immunofluorescence (IFA) assays. Upon treatment with 5 μM of lycorine (associated with weak cytotoxicity), the authors showed that DTMUV viral titers and envelope protein expression were reduced. Their results are consistent with lycorine targeting a post-entry step, as observed in the context of infections with other flaviviruses or with coronaviruses. The authors also concluded that lycorine is virucidal and blocks viral entry and internalization. Lycorine previously reported that interaction with RdRp, RNA and/or ribosomes could be implicated in DTMUV inhibition. Although the cell line may not be the best representative of DTMUV avian infection, their results suggest that the lycorine structure could serve as a scaffold for the development of new treatments against this disease.3.4 Influenza virus
In 2013, AA lycorine, hippeastrine, heamanthamine and 11-hydroxyvittatine were reported to display anti-influenza virus type A (A/Chicken/GuangDong/178/2004 (H5N1)) activity at a post-entry step, inhibiting nuclear-to-cytoplasmic export of ribonucleoprotein (RNP).119 Recent efforts have focused on elucidating the mechanism of action of lycorine.120,121 In 2019, Yang and colleagues performed a proteomic analysis using a multiplexed tandem mass tag approach to identify changes in protein expression in Madin–Darby Canine Kidney cells infected with H5N1 for 1 hour and treated with 0.52 μM lycorine for 12 hours. The expression levels of 71 proteins were significantly modulated when comparing virus-infected/mock with virus-infected/lycorine-treated cells. Interestingly, influenza virus infection triggered the expression of nuclear pore complex protein 93 (Nup93), while lycorine treatment decreased it. This was confirmed by western blot. Their results endorsed lycorine association with nuclear–cytoplasmic transport impairment, providing new insights into how lycorine may trap viral ribonucleoprotein (RNP) in the nucleus, an inhibitory mechanism of lycorine that appears specific to the context of influenza virus infection (Table 1). In 2022, He and colleagues showed that lycorine might have yet another target.121 They first identified that the endogenous cytochrome c oxidase subunit 4 isoform 1 (COX41) cellular protein plays an important role in H5N1 infection, promoting viral replication. Intriguingly, when this gene was knocked out, H5N1 RNPs were retained in the cell nucleus, similar to the effect of lycorine on infected cells. They further showed that treatment of cells with 0.52 μM of lycorine inhibited COX41, reducing radical oxygen species levels and phosphorylation of extracellular signal-regulated kinase (ERK), which resulted in the blockage of nuclear export of vRNP and inhibition of viral replication. Continuous treatment with lycorine did not lead to the emergence of a resistant virus. The authors then administered mice with 5 mg kg−1 day−1 lycorine or mock 1 day prior to viral challenge (2 LD50) and for the remaining 16 consecutive days. On day 5 or 14 post-infection, they observed a reduction in viral titers and inhibition of pathological changes in the lung and trachea tissues. Altogether, these results show that, as is the case for other viruses, lycorine anti-influenza properties might be the consequence of multiple targets and confirm that AA is a potential therapeutic target for the influenza virus.3.5 Chikungunya virus and other alphaviruses
Chikungunya is a critical re-emerging pathogen caused by a mosquito-borne alphavirus (CHIKV) within the Togaviridae family. Other arboviruses of this family include Sindbis virus (SINV), Semliki Forest virus (SFV), which mainly targets rodents, and Venezuelan equine encephalitis virus (VEEV), which affects horses, donkeys and zebras. Li et al. investigated whether lycorine and some derivatives could block the replication of these alphaviruses.9 They observed that lycorine impeded CHIKV with EC50 values of 0.38, 0.75, 0.41 and 0.4 μM in Vero, BHK-21, Huh7 and A549 cells, respectively (Table 1). At 10 μM (with some cytotoxicity), CHIKV viral titers were decreased by 103–105 fold. The authors later showed that lycorine inhibited the replication of SINV, SFV, and VEEV with EC50 values of 1.05, 0.83 and 0.31 μM, respectively. They investigated the alkaloid mode of action and convincingly showed that a post-entry step of the CHIKV life cycle was targeted, mainly viral translation, similar to some viruses (SARS-CoV-2), but not to others (DENV, ZIKV, and INFV). Several previously reported mechanisms of lycorine antiviral activity, such as its affinity to RNA and/or ribosome, could affect viral translation. Their results extended the antiviral spectrum of lycorine (Fig. 11).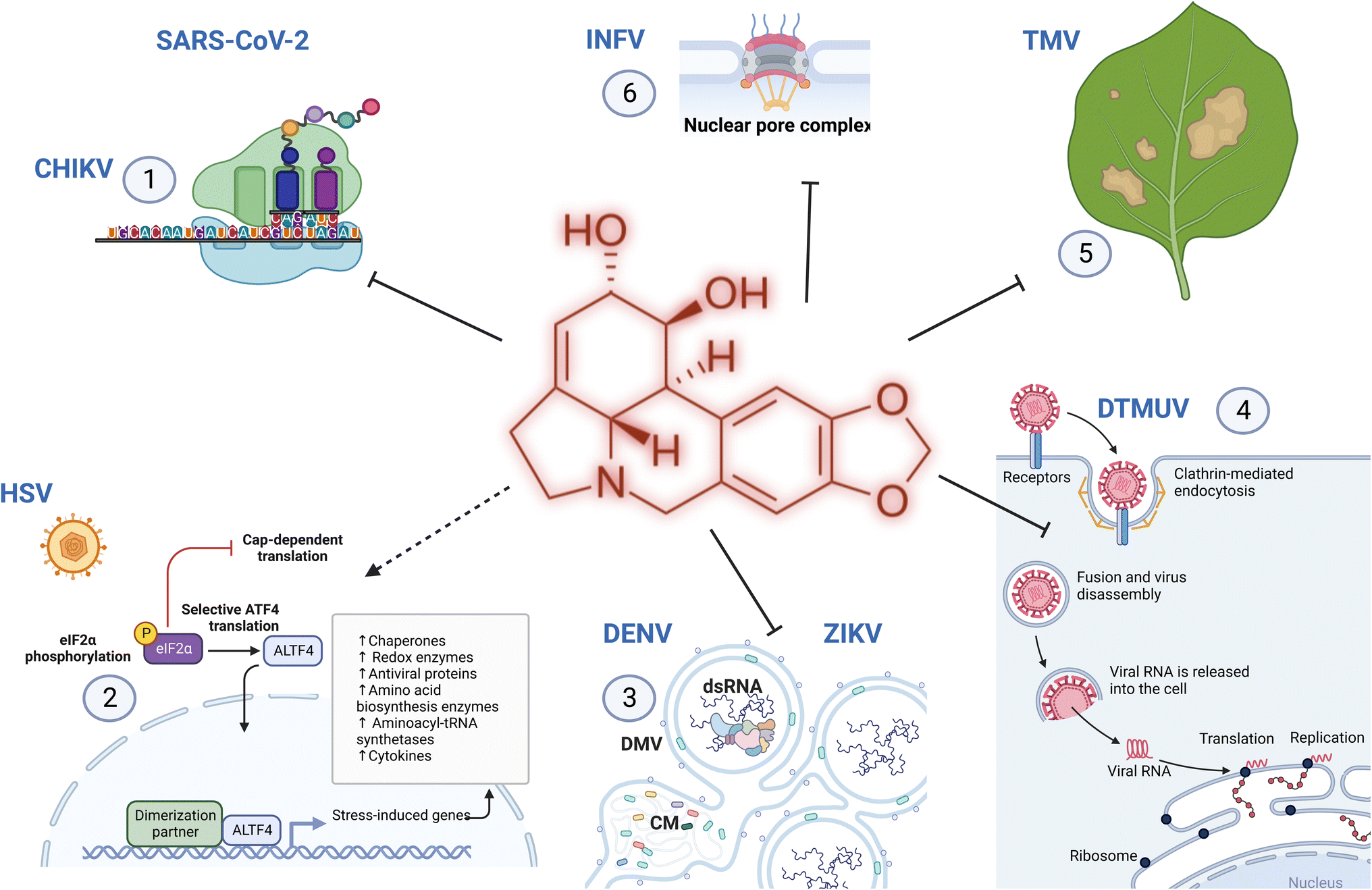 | ||
| Fig. 11 Multiple targets of antiviral Amaryllidaceae alkaloids. AAs such as lycorine target ribosomal translation during infection with chikungunya virus (CHIKV) and SARS-CoV-2 (1), trigger stress-induced and innate responses to counter herpes simplex virus (HSV) replication (2), target the RNA synthesis step of flaviviruses, such as Zika virus (ZIKV) and dengue virus (DENV) (3), inhibit Duck Tembusu Virus (DTMUV) at a post-entry step (4), decrease leaves damages caused by Tobacco Mosaic Virus (TMV) infection, triggering defense enzyme activity (5), and traps the influenza virus (INFV) at the nuclear pore complex (6). Created using Biorender. | ||
3.6 Picornaviridae
Human Enterovirus 71 (EV71) and Coxsackievirus A16 (CVA16) are RNA viruses mostly affecting children below 5 years old, causing hand, foot and mouth disease.122 Lycorine was shown to inhibit EV71 replication in cellulo and in animal models in previous studies.123 In 2019, Wang et al. showed that a lycorine derivative LY-55 (2-hydroxy-2,3a1,4,5,7,12b-hexahydro-1H-[1,3]dioxolo[4,5-j]pyrrolo[3,2,1-de]phenanthridin-1-yl 2-((4-fluorophenyl)thio)acetate hydrochloride) inhibited EV71 and CVA16 with EC50 ranging from 1.69 to 5.02 μM, and TC50 of 106.36 μM7 (Table 1). They determined that LY-55 inhibited the autophagy induced by the two viruses and was required for their replication. Alternatively, lycorine was also shown to modulate the Wnt/β-catenin pathway, which has been implicated in the replication of several RNA viruses, such as flavivirideae hepatitis C virus and enterovirus.124 Finally, the authors showed that treatment of mice with LY-55 (1.5 mg kg−1) decreased viral titers and reduced the mortality rate associated with the lethal EV71 challenge, protecting 4 out of 8 mice from death.73.7 Herpes simplex virus (HSV)
Herpes Simplex Virus (HSV) is a DNA virus. In 2018, Brown et al. developed 10b-aza-analogues of trans-hydronarciclasine and trans-hydrolycoricidine with B/C-ring fusion-modified derivatives, which completely lost their anti-HSV activity.13 In 2023, McNulty et al. confirmed that dihydronarciclasine analogs and trans-dihydrolycoricidine blocked HSV-1 replication.14 Two derivatives (at 10 μM) displayed strong inhibition of HSV-1 infection in the brain organoids. The dihydronarciclasine analog with ring-A mono-substituted C7-OH was particularly potent (EC50 = 0.504 μM in 9001 and 0.209 μM in 01SD organoids with no impact on viability < 50 μM; Table 1). Both compounds activated the eukaryotic initiation factor 2 (ElF2) signalling pathway, the eukaryotic integrated stress response (ISR) and the resulting signaling network, together with autophagy and sirtuin-1 signaling pathways (Fig. 11). Their results suggested that the compounds triggered an innate antiviral response. Previous studies have shown that narciclasine and other alkaloids interact with larger ribosome subunits, inhibiting peptide bond formation, with eukaryotic translation elongation factor 1A necessary for elongating ribosomes, and inhibiting Hsp70, which affects a broad range of viruses.125,126 The authors further tested the dihydronarciclasine analog in vivo and reported a delayed and decreased viral titer in a mouse model of HSV-1 ocular infection.3.8 Tobacco mosaic virus
Viruses target all living forms, such as the tobacco mosaic virus (TMV) from the genus Tobamovirus, which infects a wide range of plants, damaging crops of tobacco, all Solanaceae family members, ornamental plants, cucumbers, and beans. Hu et al. investigated the ethanolic extract of Lycoris aurea bulbs and their alkaloids for their anti-TMV activity.127 Lycorine and some derivatives display protective effects (15.3 and 45.2%) at high concentrations of 100 and 500 mg L−1 (0.348 mM and 1.74 mM) in contrast with galanthamine and lycoramine, which did not display any antiviral activity at 100 mg L−1. Yang et al. tested AA isolated from the bulbs of L. radiata, including sanguinine, 11-hydroxyvittatine, lycoramine, pancratinine C, homolycorine, hippeastrine, O-demethylhomolycorine N-oxide, zephyranthine, O-methyllycorenine N-oxide, lycoranine C, tazettine, lycorine, 9-O-demethylhomolycorine, homolycorine N-oxide, galanthamine, and lycoricidine derivatives owing to their anti-TMV properties in vivo by the conventional half-leaf method and on systemic infection by western blot and RT-PCR analysis.128 Some compounds, such as 11-hydroxyvittatine, zephyranthrine, and lycoranine C, displayed a moderate protective effect. Lycoricidine derivatives showed significant antiviral effects, particularly L1 (N-methyl-2,3,4-trimethoxylycoricidine) (60.8% of protective inhibitory activity at 100 μg mL−1) and L3 (N-methyl-2-methoxy-3,4-acetonidelycoricidine) (62.0%), which were almost similar to the positive control, ningnanmycin (66.4%). L1, L3, and L5 (N-allyl-2,3,4-triallyloxylycoricidine) antiviral activity was further evidenced by a reduction in TMV-coat protein expression. Finally, the authors showed that L1 also triggered defensive enzyme activities (phenylalanine ammonia lyase, peroxidase and superoxide dismutase) in systemic leaves, which is a strategy that could be used to improve disease resistance to tobacco (Fig. 11). These studies pave the way for the possible role of AAs as antiviral compounds in fighting plant diseases.4 Structure activity relationship
4.1 SARS-CoV-2 and lycorine
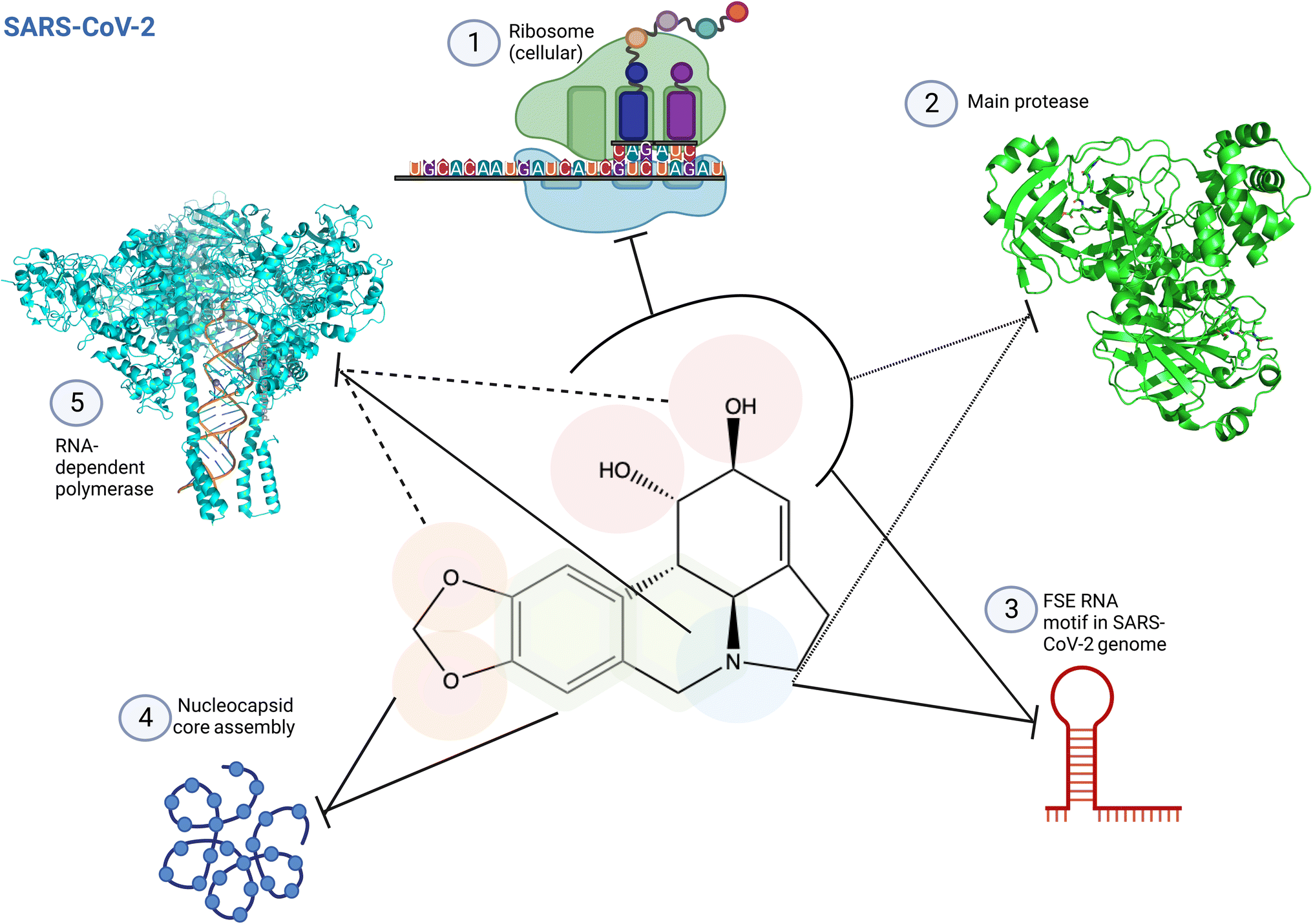 | ||
| Fig. 12 Structure activity relationship of lycorine specific to SARS-CoV-2. (1) Lycorine C1 and C2 hydroxyl groups are predicted to interact with the ribosome peptidyl transfer site, inhibiting protein translation.2 (2) C1 and C2 hydroxyl groups and the amine base are predicted to be involved in SARS-CoV-2 main protease inhibition occupying partially the enzyme active site (PDB: 7P35). This impaired correct polyprotein processing.4 (3) These pharmacophores are also involved in the interactions with the frameshifting element of SARS-CoV-2 genome, preventing correct translation.2 (4) Lycorine oxygens on methylenedioxy C8 and C9, and interactions with the adjacent ring prevent virus maturation, targeting nucleocapsid core assembly through π-stacking and hydrogen bonding.2 (5) Lycorine blocks coronavirus RNA replication through C1 and C2 hydroxyl groups, and C9 oxygen6 and/or through its amine-containing ring2 by interacting with RNA in the RNA-dependent RNA polymerase (RdRp) active site, as predicted by docking, surface plasma resonance (SPR) and structure similarity with known inhibitor (PDB: 8GWG). Small dashed lines reference to4 using antiviral assays and docking; full dash lines reference to6 using cell protease assay, in cellulo antiviral assays and docking; and full lines reference to2 using docking, SPR and antiviral in cellulo assays as main methods. Created using Biorender. | ||
4.2 Herpes-simplex virus
4.3 Zika and dengue virus
Haemanthamine specific conformational structure of C11 hydroxyl, C3 methoxy groups and ring C1–C10b and C1–C2 bonds are key to its DENV inhibition, as suggested by comparison with 11-hydroxyvittatine and with other very similar compounds being less active.1 Previous studies had shown that haemanthamine binding to the 25S rRNA A-site cleft induced a ‘flip up’ conformation of residue U2875, which was not observed upon narciclasine binding and was attributed to the absence of the phenolic hydroxyl in haemanthamine.113 In addition, haemanthamine interacted with rRNA, whereas narciclasine did not. A haemanthamine aromatic ring fused to the methylenedioxy moiety could form π-stacking interactions with the nucleotide base residues U2875 and C2821 sandwiching the AA. Haemanthamine amino group further reinforced interaction with U2875 through hydrogen bonding, stabilizing the ‘flip-up’ conformation of the uracil residue. Its C11-hydroxyl group on the ethanol bridge was implicated in two additional H-bonds with U2873. O-Acylation of C11 resulted in compromised activity, and the α-configured ethanol bridge was required for rRNA binding. Interestingly, these pharmacophores have been implicated in antiviral activities in previous studies (Fig. 13).133
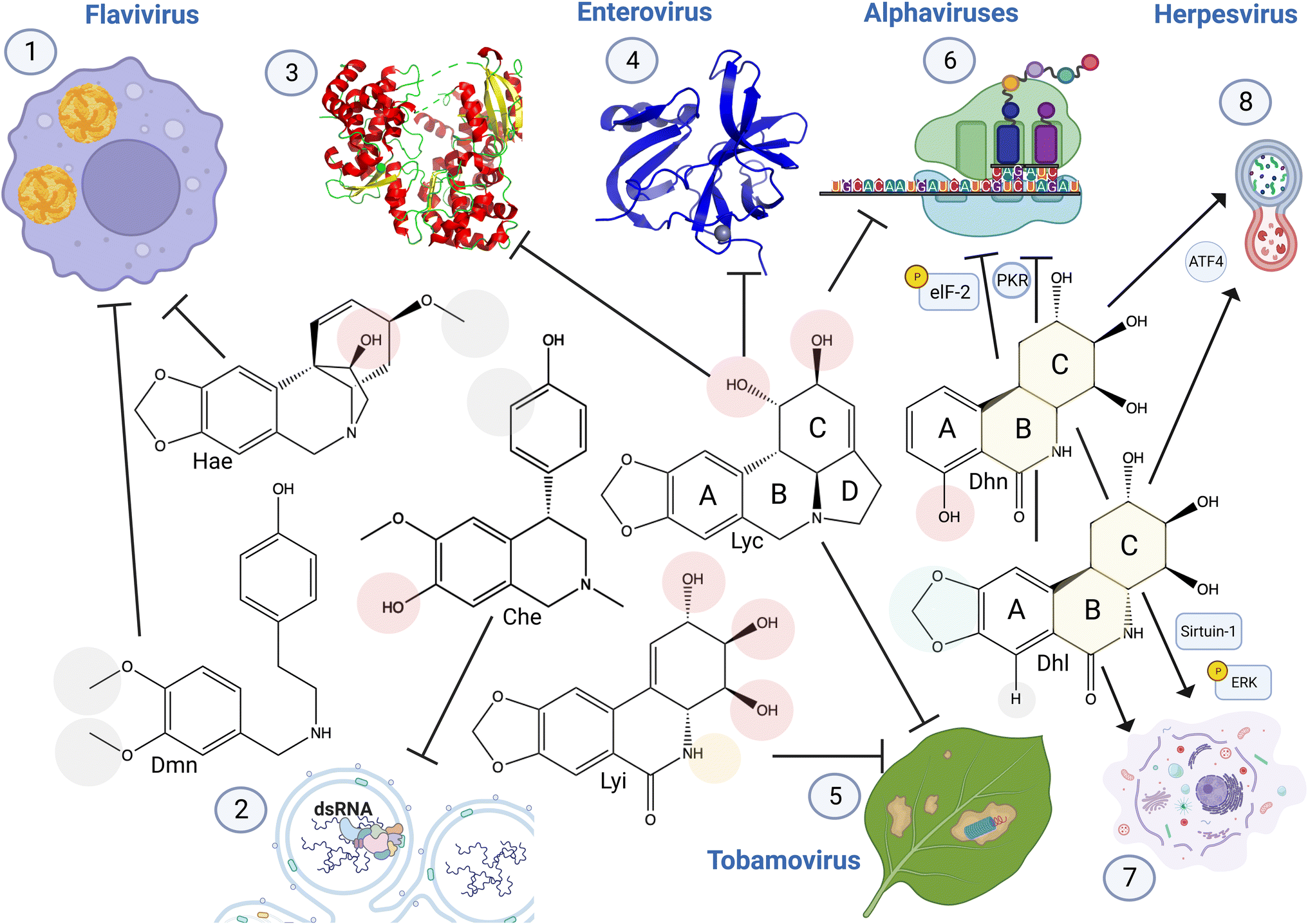 | ||
| Fig. 13 Structure activity relationship of anti-viral Amaryllidaceae alkaloids. (1) Haemanthamine structural conformation of C11 hydroxyl and C3 methoxy groups are required for DENV inhibition.1 3′,4′-O-Dimethylnorbelladine methyl groups increased anti-DENV activity.3 (2) Cherylline non-methylated 3′OH (compared to gigantelline), and the absence of hydroxyl group on the phenol cycle C3 (compared to gigantellinine) is required for its anti-viral activity specific to RNA replication (PDB: 5WZ3).5 (3) Lycorine C1 hydroxyl group interacts with Zika virus RNA-dependent RNA polymerase to block RNA synthesis, (as shown by in vitro enzyme assay, docking evidence, and interaction with His110). (4) Addition of a functional group on this hydroxyl conserves lycorine inhibition of enteroviruses 2A proteinase with reduced cytotoxicity (higher selectivity index), resulting also in a loss of RNA replication (PDB: 3W95).7,8 (5) Lycorine and lycoricidine derivatives block Tobacco Mosaic Virus. Modifications of lycorine C1 and C2 and of lycoricidine ring C and B functional groups modulate anti-TMV activity. (6) Lycorine C1 and C2 are required to block alphavirus replication through viral RNA translation inhibition.9 7-Hydroxyl derivative trans-dihydrolycoricidine, and dihydronarciclasine analog with ring-A mono-substituted and C7-OH with fully functionalized ring-C targets herpesvirus replication,13 affecting eukaryotic initiation factor 2 (elF2) pathway involved in protein translation.14 (7) These derivatives also triggered autophagy and (8), apoptosis-related signaling pathways involving ERK and Sirtuin-1. Hae: haemanthamine, Dmn, 3′4′-O-dimethylnorbelladine, Che: cherylline, Lyc: lycorine, Lyi: lycoricidine: Dhn: a dihydronarciclasine analog with ring-A mono-substituted and C7-OH, Dhl: trans-dihydrolycoricidine. Created using Biorender. | ||
In less studied ring types, 3′,4′-O-dimethylnorbelladine methyl groups increased anti-DENV activity compared to single-methylated O-norbelladine, suggesting a key role played by these two functional groups in its interaction with its target although not yet characterized.3 In contrast, norcraugsodine O-methylation abolished its anti-DENV potential and reduced its cytotoxicity, indicating the structure–activity relationship of norbelladine-type alkaloids.
4.4 Enterovirus, alphavirus and tobamovirus
Similar to flavivirus-related SAR, the addition of more complex functional groups to lycorine C1 conserved lycorine inhibition of enteroviruses 2A proteinase with reduced cytotoxicity, resulting in a loss of RNA replication.7,8 Likewise, acetylation, methylation or replacement with Cl of lycorine C1 or C2 hydroxyl and of lycoricidine C2, C3, C4 and N increased anti-TMV properties. In contrast, lycorine C1 and C2 hydroxyl groups are required for blocking alphavirus CHIKV replication through viral RNA translation inhibition,9 and acetylation at these positions results in a loss of activity.4.5 Current challenges in the AA structure–activity relationship
Although recent studies confirm and unravel key pharmacophores, the analysis also shows that modifications can yield a virus-specific impact. AAs inhibit multiple viruses and may interact with diverse targets. This complicates the analysis of the relative importance of each functional group to each of its biological properties. Future studies will need to combine powerful systematic biological methods and thorough structure–activity relationship analysis to gather key knowledge to fight viral diseases while preserving cellular processes, such as protein synthesis, proliferation and stress response, but also assessing solvability and brain blood barrier permeability.134 Moreover, AA pleiotropy might be their greatest strength because it multiplies the mechanisms required for virus escape.5 Conclusions
Recent studies have highlighted the potential of AA as a scaffold for developing new antiviral compounds for both humans and plants. In recent years, lycorine has been predominantly studied probably owing to its potency, its broad spectrum of targeted microorganisms and its relative abundance in many plants. Lycorine is also readily commercially available. However, some of its biological characteristics prevent its safe use in humans: its broad spectrum implies that it may alter non-desired cellular targets, leading to additional undesired side effects. Lycorine is cytotoxic to many human cells at low concentrations, and it is highly poisonous to humans when ingested, leading to diarrhea, vomiting, convulsions and even deaths (http://www.t3db.ca/toxins/T3D4064). Consequently, research should continue to focus on structure–activity relationships with the help of computational analysis. Finally, the discovery of the full antiviral spectrum of the other ∼650 AA should be encouraged.Natural product chemists, synthetic organic chemists, biochemists, and bioengineers have co-contributed to the research interest and understanding enrichment of Amaryllidaceae alkaloid structural diversity. All these fronts examine the interpretation and execution of AA for the benefit of mankind. Although it is dubious which method is most appropriate for the sustainable supplementation of AA in the pharmaceutical and medicinal sectors in the long run, AA structural diversity could serve as antiviral components, as emphasized in this review. Further, an improved understanding of biosynthetic pathways could support overcoming the limitations of synthetic biological approaches. However, these are achievable through interdisciplinary research in the aforementioned fields. Thus, this article serves as a tool to facilitate our objectives.
6 Author contributions
Thilina U. Jayawardena and Natacha Merindol: conceptualization, methodology, writing original draft, writing, reviewing, and editing; Nuwan Sameera Liyanage: conceptualization, methodology, writing, reviewing, and editing; Isabel Desgagné-Penix: resources, funding acquisition, supervision, writing – reviewing and editing.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
This research was funded by the Canada by the Natural Sciences and Engineering Research Council of Canada (NSERC) award number RGPIN-2021-03218 and the Canada Research Chair on plant specialized metabolism Award No. 950-232164. Many thanks are extended to the Canadian taxpayers and to the Canadian government for supporting the Canada Research Chairs Program.9 References
- M. Masi, R. Di Lecce, N. Merindol, M. P. Girard, L. Berthoux, I. Desgagne-Penix, V. Calabro and A. Evidente, Toxins, 2022, 14, 262 CrossRef CAS PubMed.
- P. X. Ren, W. J. Shang, W. C. Yin, H. Ge, L. Wang, X. L. Zhang, B. Q. Li, H. L. Li, Y. C. Xu, E. H. Xu, H. L. Jiang, L. L. Zhu, L. K. Zhang and F. Bai, Acta Pharmacol. Sin., 2022, 43, 483–493 CrossRef CAS PubMed.
- M. P. Girard, V. Karimzadegan, M. Heneault, F. Cloutier, G. Berube, L. Berthoux, N. Merindol and I. Desgagne-Penix, Molecules, 2022, 27, 5621 CrossRef CAS PubMed.
- A. Narayanan, M. Narwal, S. A. Majowicz, C. Varricchio, S. A. Toner, C. Ballatore, A. Brancale, K. S. Murakami and J. Jose, Commun. Biol., 2022, 5, 169 CrossRef CAS PubMed.
- S. Ka, N. Merindol, I. Seck, S. Ricard, A. Diop, C. S. B. Boye, K. Landelouci, B. Daoust, L. Berthoux, G. Pepin, M. Seck and I. Desgagne-Penix, Molecules, 2021, 26, 7382 CrossRef CAS PubMed.
- Y. H. Jin, J. S. Min, S. Jeon, J. Lee, S. Kim, T. Park, D. Park, M. S. Jang, C. M. Park, J. H. Song, H. R. Kim and S. Kwon, Phytomedicine, 2021, 86, 153440 CrossRef CAS PubMed.
- H. Wang, T. Guo, Y. Yang, L. Yu, X. Pan and Y. Li, Front. Cell. Infect. Microbiol., 2019, 9, 277 CrossRef CAS PubMed.
- Y. Guo, Y. Wang, L. Cao, P. Wang, J. Qing, Q. Zheng, L. Shang, Z. Yin and Y. Sun, Antimicrob. Agents Chemother., 2016, 60, 913–924 CrossRef CAS PubMed.
- N. Li, Z. Wang, R. Wang, Z. R. Zhang, Y. N. Zhang, C. L. Deng, B. Zhang, L. Q. Shang and H. Q. Ye, Virol. Sin., 2021, 36, 1465–1474 CrossRef CAS PubMed.
- V. M. Dembitsky, Bioorg. Khim., 2002, 28, 196–208 CAS.
- K. C. Guven, A. Percot and E. Sezik, Mar. Drugs, 2010, 8, 269–284 CrossRef CAS PubMed.
- J. C. Braekman, D. Daloze and J. M. Pasteels, in Alkaloids, ed. M. F. Roberts and M. Wink, Springer US, Boston, MA, 1998, ch. 15, pp. 349–378 Search PubMed.
- C. E. Brown, T. Kong, J. F. Britten, N. H. Werstiuk, J. McNulty, L. D'Aiuto, M. Demers and V. L. Nimgaonkar, ACS Omega, 2018, 3, 11469–11476 CrossRef CAS PubMed.
- J. McNulty, C. Babu-Dokuburra, J. Scattolon, C. Zepeda-Velazquez, M. A. Wesesky, J. K. Caldwell, W. Zheng, J. Milosevic, P. R. Kinchington, D. C. Bloom, V. L. Nimgaonkar and L. D'Aiuto, Sci. Rep., 2023, 13, 1639 CrossRef CAS PubMed.
- P. Dey, A. Kundu, A. Kumar, M. Gupta, B. M. Lee, T. Bhakta, S. Dash and H. S. Kim, in Recent Advances in Natural Products Analysis, ed. A. Sanches Silva, S. F. Nabavi, M. Saeedi and S. M. Nabavi, Elsevier, 2020, pp. 505–567 Search PubMed.
- R. Verpoorte, in Encyclopedia of Analytical Science, ed. P. Worsfold, A. Townshend and C. Poole, Elsevier, Oxford, 2005, pp. 56–61 Search PubMed.
- G. A. Beaudoin and P. J. Facchini, Planta, 2014, 240, 19–32 CrossRef CAS PubMed.
- R. Verpoorte, in Encyclopedia of Separation Science, ed. I. D. Wilson, Academic Press, Oxford, 2000, pp. 1949–1956 Search PubMed.
- T. A. P. Group, Bot. J. Linn. Soc., 2003, 141, 399–436 CrossRef.
- M. J. M. Christenhusz and J. W. Byng, Phytotaxa, 2016, 261, 201–217 CrossRef.
- J. C. Cedrón, M. Del Arco-Aguilar, A. Estévez-Braun and Á. G. Ravelo, in The Alkaloids: Chemistry and Biology, ed. G. A. Cordell, Academic Press, 2010, vol. 68, pp. 1–37 Search PubMed.
- A. W. Meerow, J. Francisco-Ortega, D. N. Kuhn and R. J. Schnell, Syst. Bot., 2006, 31, 42–60 CrossRef.
- I. Desgagné-Penix, Phytochem. Rev., 2020, 20, 409–431 CrossRef.
- S. Ka, M. Koirala, N. Merindol and I. Desgagne-Penix, Molecules, 2020, 25, 4901 CrossRef CAS PubMed.
- J. J. Nair and J. Van Staden, J. Ethnopharmacol., 2014, 151, 12–26 CrossRef CAS PubMed.
- J. J. Nair, A. Wilhelm, S. L. Bonnet and J. van Staden, Bioorg. Med. Chem. Lett., 2017, 27, 4943–4951 CrossRef CAS PubMed.
- D. K. Liscombe, B. P. Macleod, N. Loukanina, O. I. Nandi and P. J. Facchini, Phytochemistry, 2005, 66, 1374–1393 CrossRef CAS PubMed.
- T. Ahmed, A. U. Gilani, M. Abdollahi, M. Daglia, S. F. Nabavi and S. M. Nabavi, Pharmacol. Rep., 2015, 67, 970–979 CrossRef CAS PubMed.
- E. Plazas, S. Hagenow, M. Avila Murillo, H. Stark and L. E. Cuca, Bioorg. Chem., 2020, 98, 103722 CrossRef PubMed.
- J. D. Phillipson, M. F. Roberts and M. Zenk, The Chemistry and Biology of Isoquinoline Alkaloids, Springer Science & Business Media, 1985 Search PubMed.
- N. Samanani and P. J. Facchini, J. Biol. Chem., 2002, 277, 33878–33883 CrossRef CAS PubMed.
- A. Kornienko and A. Evidente, Chem. Rev., 2008, 108, 1982–2014 CrossRef CAS PubMed.
- M. He, C. Qu, O. Gao, X. Hu and X. Hong, RSC Adv., 2015, 5, 16562–16574 RSC.
- A. R. Battersby, H. M. Fales and W. C. Wildman, J. Am. Chem. Soc., 2002, 83, 4098–4099 CrossRef.
- H. G. Boit, Chem. Ber., 2006, 87, 1704–1707 CrossRef.
- A. Brossi, G. Grethe, S. Teitel, W. C. Wildman and D. T. Bailey, J. Org. Chem., 2002, 35, 1100–1104 CrossRef.
- A. Brossi and S. Teitel, J. Org. Chem., 2002, 35, 3559–3561 CrossRef.
- A. Brossi and S. Teitel, Tetrahedron Lett., 1970, 11, 417–419 CrossRef.
- M. A. Schwartz and S. W. Scott, J. Org. Chem., 2002, 36, 1827–1829 CrossRef.
- H. Shi, C. Du, X. Zhang, F. Xie, X. Wang, S. Cui, X. Peng, M. Cheng, B. Lin and Y. Liu, J. Org. Chem., 2018, 83, 1312–1319 CrossRef CAS PubMed.
- H. Yu, R. Lee, H. Kim and D. Lee, J. Org. Chem., 2019, 84, 3566–3578 CrossRef CAS PubMed.
- S. Ka, M. Masi, N. Merindol, R. Di Lecce, M. B. Plourde, M. Seck, M. Gorecki, G. Pescitelli, I. Desgagne-Penix and A. Evidente, Phytochemistry, 2020, 175, 112390 CrossRef CAS PubMed.
- J. J. Nair, A. L. Andresen, H. B. Papenfus, D. Staerk and J. van Staden, S. Afr. J. Bot., 2020, 131, 351–359 CrossRef CAS.
- B. B. Majhi, S. E. Gelinas, N. Merindol, S. Ricard and I. Desgagne-Penix, Front. Plant Sci., 2023, 14, 1231809 CrossRef PubMed.
- J. R. Lewis, in Second Supplements to the 2nd Edition of Rodd's Chemistry of Carbon Compounds, ed. M. Sainsbury, Elsevier, 1991, pp. 165–249 Search PubMed.
- S. Ghosal, A. Shanthy and S. K. Singh, Phytochemistry, 1988, 27, 1849–1852 CrossRef CAS.
- M. B. Kilgore, C. K. Holland, J. M. Jez and T. M. Kutchan, J. Biol. Chem., 2016, 291, 16740–16752 CrossRef CAS PubMed.
- A. Singh, M. A. Massicotte, A. Garand, L. Tousignant, V. Ouellette, G. Berube and I. Desgagne-Penix, BMC Plant Biol., 2018, 18, 338 CrossRef CAS PubMed.
- N. Samanani, D. K. Liscombe and P. J. Facchini, Plant J., 2004, 40, 302–313 CrossRef CAS PubMed.
- N. Samanani, S. U. Park and P. J. Facchini, Plant Cell, 2005, 17, 915–926 CrossRef CAS PubMed.
- M. B. Kilgore, M. M. Augustin, C. M. Starks, M. O'Neil-Johnson, G. D. May, J. A. Crow and T. M. Kutchan, PLoS One, 2014, 9, e103223 CrossRef PubMed.
- L. Tousignant, A. M. Diaz-Garza, B. B. Majhi, S. E. Gelinas, A. Singh and I. Desgagne-Penix, Planta, 2022, 255, 30 CrossRef CAS PubMed.
- B. Sun, P. Wang, R. Wang, Y. Li and S. Xu, Int. J. Mol. Sci., 2018, 19, 1911 CrossRef PubMed.
- W. Li, C. Qiao, J. Pang, G. Zhang and Y. Luo, Int. J. Biol. Macromol., 2019, 141, 680–692 CrossRef CAS PubMed.
- Q. Li, J. Xu, L. Yang, X. Zhou, Y. Cai and Y. Zhang, Front. Plant Sci., 2020, 11, 519752 CrossRef PubMed.
- Q. Li, J. Xu, Y. Zheng, Y. Zhang and Y. Cai, Front. Plant Sci., 2021, 12, 713795 CrossRef PubMed.
- F. Aleya, C. Xianmin, H. Anthony and J. Meriel, Gene, 2021, 774, 145424 CrossRef CAS PubMed.
- S. Ghosal, K. S. Saini and S. Razdan, Phytochemistry, 1985, 24, 2141–2156 CrossRef CAS.
- A. Gesell, M. Rolf, J. Ziegler, M. L. Diaz Chavez, F. C. Huang and T. M. Kutchan, J. Biol. Chem., 2009, 284, 24432–24442 CrossRef CAS PubMed.
- D. R. Nelson, R. Ming, M. Alam and M. A. Schuler, Trop. Plant Biol., 2008, 1, 216–235 CrossRef CAS.
- M. Mizutani and F. Sato, Arch. Biochem. Biophys., 2011, 507, 194–203 CrossRef CAS PubMed.
- N. Ikezawa, K. Iwasa and F. Sato, J. Biol. Chem., 2008, 283, 8810–8821 CrossRef CAS PubMed.
- P. Belin, M. H. Le Du, A. Fielding, O. Lequin, M. Jacquet, J. B. Charbonnier, A. Lecoq, R. Thai, M. Courcon, C. Masson, C. Dugave, R. Genet, J. L. Pernodet and M. Gondry, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7426–7431 CrossRef PubMed.
- D. H. R. Barton and T. Cohen, Some biogenetic aspects of phenol oxidation, Festschrift Prof. Dr. Arthur Stoll, Birkhäuser, Basel, 1957 Search PubMed.
- J. Eichhorn, T. Takada, Y. Kita and M. H. Zenk, Phytochemistry, 1998, 49, 1037–1047 CrossRef CAS.
- W. R. Bowman, I. T. Bruce and G. W. Kirby, J. Chem. Soc. D, 1969, 1075b–1077b RSC.
- C. Fuganti and M. Mazza, J. Chem. Soc. D, 1971, 1196–1197 RSC.
- C. Fuganti and M. Mazza, J. Chem. Soc., Perkin Trans. 1, 1973, 954–956 RSC.
- D. H. R. Barton, G. W. Kirby, J. B. Taylor and G. M. Thomas, J. Chem. Soc., 1963, 4545–4558 RSC.
- M. B. Kilgore, M. M. Augustin, G. D. May, J. A. Crow and T. M. Kutchan, Front. Plant Sci., 2016, 7, 225 Search PubMed.
- S. Uyeo and S. Kobayashi, Pharm. Bull., 1953, 1, 139–142 CrossRef CAS PubMed.
- N. Hazama, H. Irie, T. Mizutani, T. Shingu, M. Takada, S. Uyeo and A. Yoshitake, J. Chem. Soc., 1968, 2947–2953 CAS.
- J. Q. Chen, J. H. Xie, D. H. Bao, S. Liu and Q. L. Zhou, Org. Lett., 2012, 14, 2714–2717 CrossRef CAS PubMed.
- L. Li, Q. Yang, Y. Wang and Y. Jia, Angew Chem. Int. Ed. Engl., 2015, 54, 6255–6259 CrossRef CAS PubMed.
- S. Majumder, A. Yadav, S. Pal, A. Khatua and A. Bisai, J. Org. Chem., 2022, 87, 7786–7797 CrossRef CAS PubMed.
- I. T. Bruce and G. W. Kirby, Chem. Commun., 1968, 207–208 RSC.
- C. Fuganti and M. Mazza, J. Chem. Soc. Chem. Commun., 1972, 936–937 RSC.
- W. C. Wildman and N. E. Heimer, J. Am. Chem. Soc., 1967, 89, 5265–5269 CrossRef CAS PubMed.
- G. W. Kirby and H. P. Tiwari, J. Chem. Soc. C, 1966, 676–682 RSC.
- A. R. Battersby, R. Blinks, S. W. Breuer, H. M. Fales, W. C. Wildman and R. J. Highet, J. Chem. Soc., 1964, 1595–1609 RSC.
- R. D. Harken, C. P. Christensen and W. C. Wildman, J. Org. Chem., 2002, 41, 2450–2454 CrossRef.
- M. Kihara, L. Xu, K. Konishi, K. Kida, Y. Nagao, S. Kobayashi and T. Shingu, Chem. Pharm. Bull., 1994, 42, 289–292 CrossRef CAS.
- M. Kihara, L. XU, K. Konishi, Y. Nagao, S. Kobayashi and T. J. H. Shingu, Heterocycles, 1992, 34, 1299–1301 CrossRef CAS.
- Y. Nakagawa, S. Uyeo and H. J. C. Yajima, Chem. Ind., 1956, 1238–1239 CAS.
- K. Kotera, Y. Hamada and R. Mitsui, Tetrahedron, 1968, 24, 2463–2479 CrossRef CAS PubMed.
- C. Fuganti and M. Mazza, J. Chem. Soc. D, 1970, 1466 RSC.
- C. Fuganti, J. Staunton and A. R. Battersby, J. Chem. Soc. D, 1971, 1154–1155 RSC.
- C. Fuganti, J. Gazz. Chim. Ital., 1973, 103, 1255–1264 CAS.
- M. B. Kilgore, The Identification of alkaloid pathway genes from Non-Model Plant Species in the Amaryllidaceae, Washington University in St. Louis, 2015 Search PubMed.
- C. Fuganti and M. Mazza, J. Chem. Soc. Chem. Commun., 1972, 239 RSC.
- A. I. Feinstein and W. C. Wildman, J. Org. Chem., 2002, 41, 2447–2450 CrossRef.
- H. M. Fales and W. C. Wildman, J. Am. Chem. Soc., 2002, 86, 294–295 CrossRef.
- W. C. Wildman and D. T. Bailey, J. Am. Chem. Soc., 2002, 89, 5514–5515 CrossRef.
- C. Giró Mañas, V. L. Paddock, C. G. Bochet, A. C. Spivey, A. J. White, I. Mann and W. Oppolzer, J. Am. Chem. Soc., 2010, 132, 5176–5178 CrossRef PubMed.
- W. C. Wildman and D. T. Bailey, J. Am. Chem. Soc., 2002, 91, 150–157 CrossRef.
- S. Danishefsky, J. Morris, G. Mullen and R. Gammill, J. Am. Chem. Soc., 2002, 104, 7591–7599 CrossRef.
- H. Haning, C. Giro-Manas, V. L. Paddock, C. G. Bochet, A. J. White, G. Bernardinelli, I. Mann, W. Oppolzer and A. C. Spivey, Org. Biomol. Chem., 2011, 9, 2809–2820 RSC.
- M. Safratova, J. Kroustkova, N. Maafi, D. Suchankova, R. Vrabec, J. Chlebek, J. Kunes, L. Opletal, F. Bucar and L. Cahlikova, Plants, 2022, 11, 3034 CrossRef CAS PubMed.
- C. K. Briggs, P. F. Highet, R. J. Highet and W. C. Wildman, J. Am. Chem. Soc., 2002, 78, 2899–2904 CrossRef.
- C. Fuganti, Tetrahedron Lett., 1973, 14, 1785–1788 CrossRef.
- T. Robinson, Science, 1974, 184, 430–435 CrossRef CAS PubMed.
- E. Furusawa, S. Furusawa, J. Y. Lee and S. Patanavanich, Proc. Soc. Exp. Biol. Med., 1976, 152, 186–191 CrossRef CAS PubMed.
- J. Renard-Nozaki, T. Kim, Y. Imakura, M. Kihara and S. Kobayashi, Res. Virol., 1989, 140, 115–128 CrossRef CAS PubMed.
- B. Gabrielsen, T. P. Monath, J. W. Huggins, D. F. Kefauver, G. R. Pettit, G. Groszek, M. Hollingshead, J. J. Kirsi, W. M. Shannon and E. M. Schubert, et al. , J. Nat. Prod., 1992, 55, 1569–1581 CrossRef CAS PubMed.
- S. Y. Li, C. Chen, H. Q. Zhang, H. Y. Guo, H. Wang, L. Wang, X. Zhang, S. N. Hua, J. Yu, P. G. Xiao, R. S. Li and X. Tan, Antiviral Res., 2005, 67, 18–23 CrossRef CAS PubMed.
- Y. N. Zhang, Q. Y. Zhang, X. D. Li, J. Xiong, S. Q. Xiao, Z. Wang, Z. R. Zhang, C. L. Deng, X. L. Yang, H. P. Wei, Z. M. Yuan, H. Q. Ye and B. Zhang, Emerging Microbes Infect., 2020, 9, 1170–1173 CrossRef CAS PubMed.
- K. Zhang, I. N. Zheludev, R. J. Hagey, M. T. Wu, R. Haslecker, Y. J. Hou, R. Kretsch, G. D. Pintilie, R. Rangan, W. Kladwang, S. Li, E. A. Pham, C. Bernardin-Souibgui, R. S. Baric, T. P. Sheahan, D. S. V. D′Souza, J. S. Glenn, W. Chiu and R. Das, bioRxiv, 2020, preprint, DOI:10.1101/2020.07.18.209270.
- S. Tan, M. G. Banwell, W. C. Ye, P. Lan and L. V. White, Chem. – Asian J., 2022, 17, e202101215 CrossRef CAS PubMed.
- N. T. Le, S. De Jonghe, K. Erven, T. Vermeyen, A. M. Balde, W. A. Herrebout, J. Neyts, C. Pannecouque, L. Pieters and E. Tuenter, Molecules, 2023, 28, 3222 CrossRef CAS PubMed.
- T. Zhou, L. Tan, G. Y. Cederquist, Y. Fan, B. J. Hartley, S. Mukherjee, M. Tomishima, K. J. Brennand, Q. Zhang, R. E. Schwartz, T. Evans, L. Studer and S. Chen, Cell Stem Cell, 2017, 21, 274–283 CrossRef CAS PubMed.
- H. Chen, Z. Lao, J. Xu, Z. Li, H. Long, D. Li, L. Lin, X. Liu, L. Yu, W. Liu, G. Li and J. Wu, Virology, 2020, 546, 88–97 CrossRef CAS PubMed.
- G. Zou, F. Puig-Basagoiti, B. Zhang, M. Qing, L. Chen, K. W. Pankiewicz, K. Felczak, Z. Yuan and P. Y. Shi, Virology, 2009, 384, 242–252 CrossRef CAS PubMed.
- S. Pellegrino, M. Meyer, C. Zorbas, S. A. Bouchta, K. Saraf, S. C. Pelly, G. Yusupova, A. Evidente, V. Mathieu, A. Kornienko, D. L. J. Lafontaine and M. Yusupov, Structure, 2018, 26, 416–425 CrossRef CAS PubMed.
- E. de Castro Barbosa, T. M. A. Alves, M. Kohlhoff, S. T. G. Jangola, D. E. V. Pires, A. C. C. Figueiredo, E. A. R. Alves, C. E. Calzavara-Silva, M. Sobral, E. G. Kroon, L. H. Rosa, C. L. Zani and J. G. de Oliveira, Virol. J., 2022, 19, 31 CrossRef CAS PubMed.
- Z. Cao, C. Zhang, Y. Liu, Y. Liu, W. Ye, J. Han, G. Ma, D. Zhang, F. Xu, X. Gao, Y. Tang, S. Shi, C. Wan, C. Zhang, B. He, M. Yang, X. Lu, Y. Huang, Y. Diao, X. Ma and D. Zhang, Emerging Infect. Dis., 2011, 17, 1873–1875 CrossRef PubMed.
- J. Su, S. Li, X. Hu, X. Yu, Y. Wang, P. Liu, X. Lu, G. Zhang, X. Hu, D. Liu, X. Li, W. Su, H. Lu, N. S. Mok, P. Wang, M. Wang, K. Tian and G. F. Gao, PLoS One, 2011, 6, e18106 CrossRef CAS PubMed.
- Y. Tang, Y. Diao, X. Gao, C. Yu, L. Chen and D. Zhang, Transboundary Emerging Dis., 2012, 59, 336–343 CrossRef CAS PubMed.
- X. Lv, M. Zhang, S. Yu, C. Zhang, T. Fang, D. Liu, B. Jia, M. Zhu, B. Wang, Q. Wang, Y. Zhu and G. Wang, Poult. Sci., 2021, 100, 101404 CrossRef CAS PubMed.
- J. He, W. B. Qi, L. Wang, J. Tian, P. R. Jiao, G. Q. Liu, W. C. Ye and M. Liao, Influenza Other Respir. Viruses, 2013, 7, 922–931 CrossRef CAS PubMed.
- L. Yang, J. H. Zhang, X. L. Zhang, G. J. Lao, G. M. Su, L. Wang, Y. L. Li, W. C. Ye and J. He, PeerJ, 2019, 7, e7697 CrossRef PubMed.
- J. He, H. Huang, B. Li, H. Li, Y. Zhao, Y. Li, W. Ye, W. Qi, W. Tang and L. Wang, Front. Microbiol., 2022, 13, 862205 CrossRef PubMed.
- B. Cox and F. Levent, JAMA, 2018, 320, 2492 CrossRef PubMed.
- J. Liu, Y. Yang, Y. Xu, C. Ma, C. Qin and L. Zhang, Virol. J., 2011, 8, 483 CrossRef CAS PubMed.
- W. J. van Zuylen, W. D. Rawlinson and C. E. Ford, Rev. Med. Virol., 2016, 26, 340–355 CrossRef PubMed.
- A. Jimenez, A. Santos, G. Alonso and D. Vazquez, Biochim. Biophys. Acta, 1976, 425, 342–348 CrossRef CAS PubMed.
- G. Van Goietsenoven, V. Mathieu, F. Lefranc, A. Kornienko, A. Evidente and R. Kiss, Med. Res. Rev., 2013, 33, 439–455 CrossRef CAS PubMed.
- Z. Hu, Z. Wang, Y. Liu and Q. Wang, Pest Manage. Sci., 2018, 74, 2783–2792 CrossRef CAS PubMed.
- D. Q. Yang, Z. R. Chen, D. Z. Chen, X. J. Hao and S. L. Li, Nat. Prod. Bioprospect., 2018, 8, 189–197 CrossRef PubMed.
- C. Roman, A. Lewicka, D. Koirala, N. S. Li and J. A. Piccirilli, ACS Chem. Biol., 2021, 16, 1469–1481 CrossRef CAS PubMed.
- A. Baez and D. Vazquez, Biochim. Biophys. Acta, 1978, 518, 95–103 CrossRef CAS PubMed.
- P. Wang, L. F. Li, Q. Y. Wang, L. Q. Shang, P. Y. Shi and Z. Yin, ChemMedChem, 2014, 9, 1522–1533 CrossRef CAS PubMed.
- D. Z. Chen, C. X. Jing, J. Y. Cai, J. B. Wu, S. Wang, J. L. Yin, X. N. Li, L. Li and X. J. Hao, J. Nat. Prod., 2016, 79, 180–188 CrossRef CAS PubMed.
- N. Suzuki, S. Tani, S. Furusawa and E. Furusawa, Proc. Soc. Exp. Biol. Med., 1974, 145, 771–777 CrossRef CAS PubMed.
- E. Kohelova, R. Perinova, N. Maafi, J. Korabecny, D. Hulcova, J. Marikova, T. Kucera, L. Martinez Gonzalez, M. Hrabinova, K. Vorcakova, L. Novakova, A. De Simone, R. Havelek and L. Cahlikova, Molecules, 2019, 24, 1307 CrossRef.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |