
Structural insights into the diverse prenylating capabilities of DMATS prenyltransferases
Evan T.
Miller
 ,
Oleg V.
Tsodikov
and
Sylvie
Garneau-Tsodikova
*
,
Oleg V.
Tsodikov
and
Sylvie
Garneau-Tsodikova
*
Department of Pharmaceutical Sciences, College of Pharmacy, University of Kentucky, 789 South Limestone Street, Lexington, KY 40536-0596, USA. E-mail: sylviegtsodikova@uky.edu
First published on 6th November 2023
Abstract
Covering: 2009 up to August 2023
Prenyltransferases (PTs) are involved in the primary and the secondary metabolism of plants, bacteria, and fungi, and they are key enzymes in the biosynthesis of many clinically relevant natural products (NPs). The continued biochemical and structural characterization of the soluble dimethylallyl tryptophan synthase (DMATS) PTs over the past two decades have revealed the significant promise that these enzymes hold as biocatalysts for the chemoenzymatic synthesis of novel drug leads. This is a comprehensive review of DMATSs describing the structure–function relationships that have shaped the mechanistic underpinnings of these enzymes, as well as the application of this knowledge to the engineering of DMATSs. We summarize the key findings and lessons learned from these studies over the past 14 years (2009–2023). In addition, we identify current gaps in our understanding of these fascinating enzymes.
1. Introduction
1.1. Prevalence of natural products in drugs and drug leads
An examination of the current pharmacopeia available today reveals that many therapeutically relevant and essential medicinal compounds have come or are derived from natural products (NPs). NPs include secondary metabolites found in plants, bacteria, and fungi that perform specific functions outside of an organism's energy and nutrient production. Such NPs have been evolutionarily optimized to interact with the environment of the producing organism, including competing, symbiotic or host organisms, and, for this reason, they are the most common source of biologically active compounds in Nature.1 To say that NPs have been influential in drug discovery and design efforts would be an understatement. With 49.2% of all drugs approved worldwide from January 1, 1981 to September 30, 2019 owing either their structure or pharmacophore to NPs, the importance of NPs in the treatment of human diseases is quite clear.2 Historically, the most popular NPs in the development of medicines are divided into four primary groups based on their scaffold structure: nonribosomal peptides (NRPs), polyketides (PKs), alkaloids, and terpenoids. Additionally, research into the biosynthesis of a fifth group, ribosomally-synthesized and posttranslationally-modified peptides (RiPPs), has gathered attention in the past decade.3–6 These NP families can be found all over the world in both terrestrial and marine environments, representing a vast natural resource of potential therapeutic agents waiting to be discovered.The appeal of investigating NPs as therapeutic agents comes in part from their unrivaled structural diversity within biologically relevant chemical spaces.7 This structural diversity arises both from the use of distinct building blocks in the scaffold of an NP, such as different proteinogenic and nonproteinogenic amino acids and amino acid-like compounds in the biosynthesis of an NRP, and from different chemical modifications of the scaffold catalyzed by tailoring enzymes. These modifications include, but are not limited to: halogenation (by a halogenase “Hal”),8 epimerization (by an epimerase “E”), oxidation (by an oxidase “Ox”), methylation (by a methyltransferase “M”),9 and prenylation (by a prenyltransferase “PT”).10 The structural diversity created by these modifications in the course of evolution increased or altered biological activities and mechanisms of action.1,11–18 In this review, we focus on the PT class of tailoring enzymes called dimethylallyl tryptophan synthases (DMATSs), which modify many clinically relevant NPs, and which have gained popularity in the literature over the last 20 years (Fig. 1).19–23 The last review on DMATSs was published in 201524 with little reference to protein structure. Herein, we cover not only the new findings on DMATSs published in 2015–2023, but also rationalize the mechanistic observations based on the body of the structural work on DMATSs.
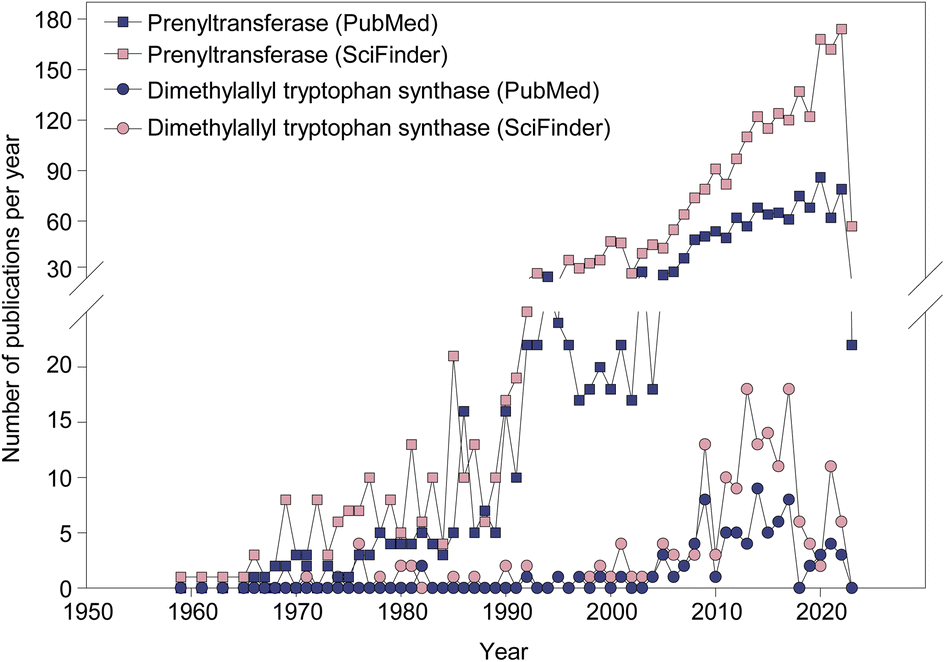 | ||
| Fig. 1 The rate of publishing on PTs (squares) and dimethylallyl tryptophan synthases (DMATSs; circles) from 1959 to 2023. | ||
1.2. Dimethylallyl tryptophan synthase (DMATS) prenyltransferases
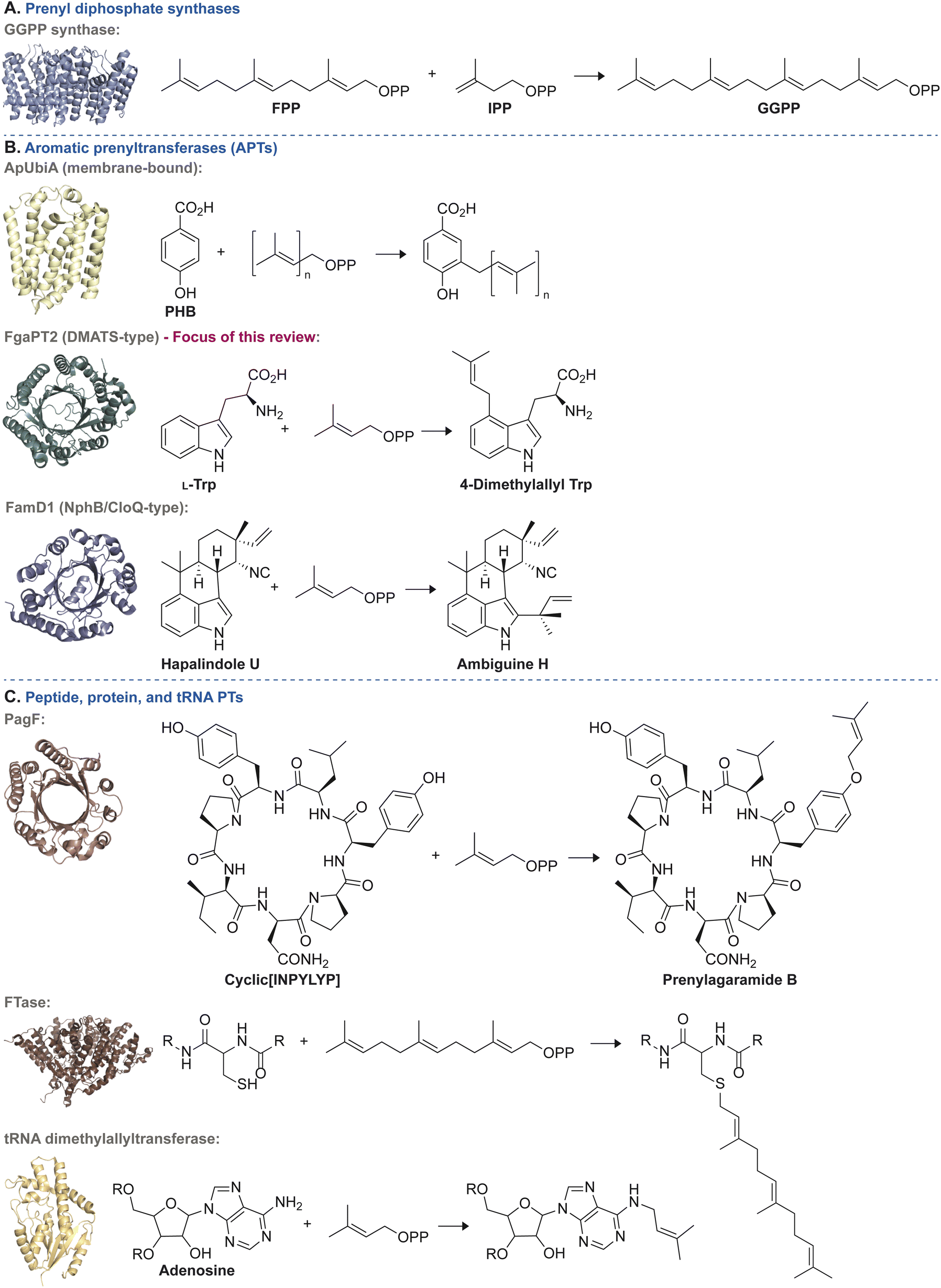 | ||
| Fig. 2 Representative examples of each of the three general categories of PT enzymes along with a common reaction they catalyze. (A) Reactions catalyzed by prenyl diphosphate synthases: condensation of farnesyl diphosphate (FPP) and isopentenyl diphosphate (IPP) to yield geranylgeranyl diphosphate (GGPP) catalyzed by the prenyl diphosphate synthase, GGPP synthase (PDB ID: 7MY7).27 (B) Reactions catalyzed by APTs: top = prenylation of p-hydroxybenzoic acid (PHB) by ApUbiA (PDB ID: 4OD4).28 Middle = prenylation of L-Trp to form 4-dimethylallyl tryptophan by FgaPT2 (PDB ID: 3I4Z).29 Bottom = prenylation of hapalindole U to form ambiguine H by FamD1 (PDB ID: 5YNT).30 (C) Reactions catalyzed by peptide, protein, and tRNA PTs: top = prenylation of cyclic[INPYLYP] by PagF to form prenylagaramide B (PDB ID: 5TTY).31 Middle = farnesylation of a C-terminal cysteine residue from a CaaX motif by human protein farnesyltransferase (PDB ID: 1JCS).32 Bottom = prenylation of adenosine by tRNA dimethylallyltransferase (PDB ID: 3CRQ).33 | ||
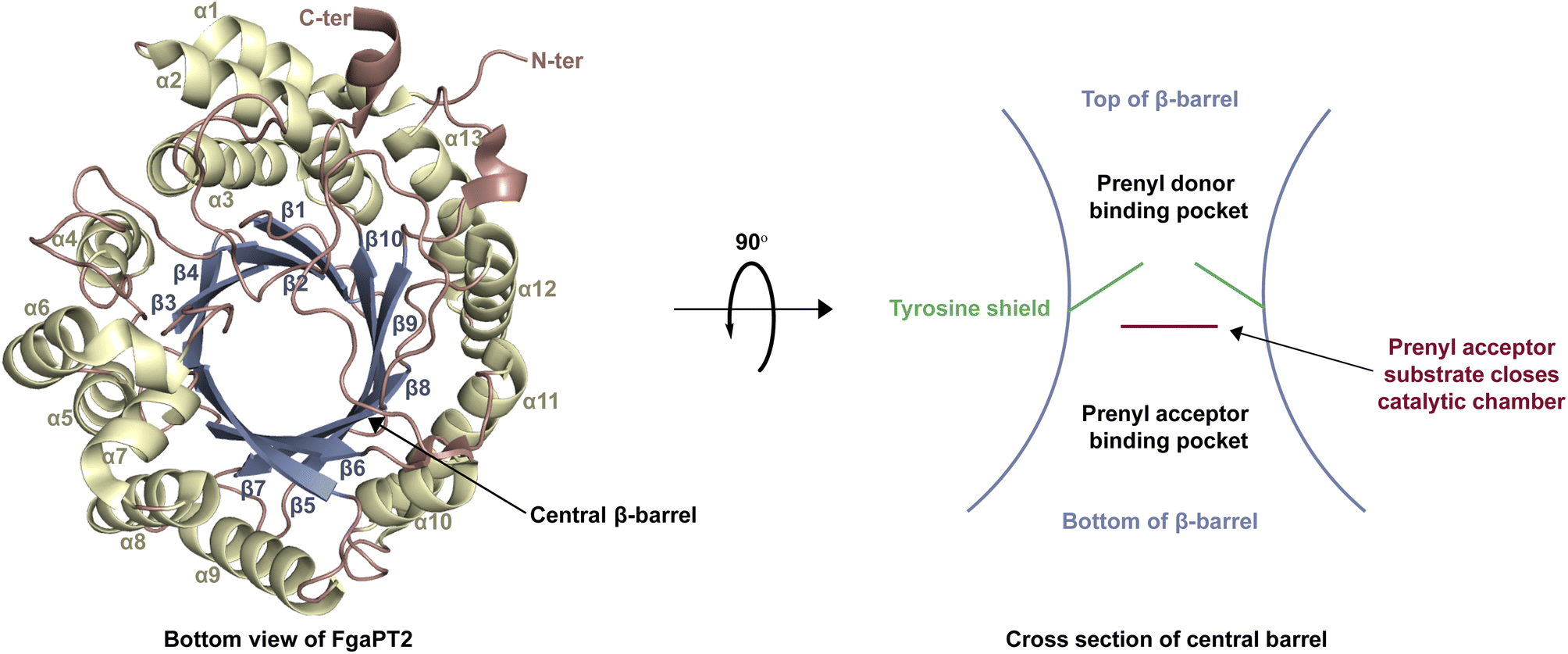 | ||
| Fig. 3 The structure of FgaPT2 (PDB ID 3I4Z)29 containing a prototypical ABBA fold and a central β-barrel. | ||
| Enzyme | Variant(s) | Organism | Proposed natural substrate for WT enzyme and best accepted substrate(s) for variant(s) | Other substrate(s) | Natural cosubstrate(s) | Other cosubstrate(s) (see Fig. 4) | Regiospecificity (see Fig. 5) | Citation(s) |
|---|---|---|---|---|---|---|---|---|
| a Abbreviations: DKP = diketopiperazine; DMAPP = dimethylallyl diphosphate; FPP = farnesyl diphosphate; GGPP = geranylgeranyl diphosphate; GPP = geranyl diphosphate; PP = diphosphate; reg = regular; rev = reverse; WT = wild-type. b N/D = not described. | ||||||||
| DKP DMATSs (Section 2.1.1.) | ||||||||
| FtmPT1 | WT | A. fumigatus | Brevianamide F | Indole DKPs, Trp derivatives, naphthalenes, ardeemin fumiquinazoline, linear indole dipeptides | DMAPP | N/D | C2-reg | 34–40 |
| Y205N, Y205L | Brevianamide F | Naphthalenes | DMAPP | N/D | C3-rev | |||
| G115T | Brevianamide F | N/D | DMAPP | N/D | C3-rev | |||
| M364G | Brevianamide F | L-Trp-L-Trp | GPP | N/D | C2-reg | |||
| NotF | WT | Aspergillus sp. MF297-2 | Brevianamide F | Indole DKPs | DMAPP | N/D | C2-rev | 41 and 42 |
| Y266A | Indole DKPs | N/D | DMAPP | N/D | N/D | |||
| Y266V | Indole DKPs | N/D | DMAPP | N/D | N/D | |||
| L193S | Indole DKPs | N/D | DMAPP | N/D | N/D | |||
| P352A | Indole DKPs | N/D | DMAPP | N/D | N/D | |||
| CdpNPT | WT | A. fumigatus | N/Db | Indole DKPs, Trp derivatives, benzodiazepinediones, daptomycin, ardeemin fumiquinazoline, linear indole dipeptides, β-carbolines | DMAPP | 6–9, 14, 15, 17, 18, 22, 23, 69, 71–74, 79, 84, 98, 101, 102, 104–106 | C3-rev | 38, 40 and 43–45 |
| M349G | Indole DKPs | N/D | GPP | N/D | C6-reg, C3-rev | |||
| AnaPT | WT | N. fischeri | (R)-Benzodiazepinedione | Aszonalenins, cyclo-Trp-Ala, cyclo-Trp-Pro, ardeemin fumiquinazoline, chalcones, flavonoids, acylphloroglucinols | DMAPP, GPP | N/D | C3-rev (DMAPP), C6-reg, C7-reg (GPP) | 40 and 46–50 |
| AtaPT | WT | A. terreus | N/D | Lignanoids, indole DKPs, quinoline alkaloids, xanthones, benzophenones, flavonoids, glycosides, hydroxynaphthalenes, phenylethylchromone, curcuminoid, stilbene, coumarins, p-hydroxybenzaldehyde, chalcones, acylphloroglucinols | DMAPP, GPP, FPP | N/D | C4-reg, C7-reg (indole DKPs), C-reg, O-reg (other substrates) | 25, 46 and 51–53 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||||||
| Trp DMATSs (Section 2.1.2.) | ||||||||
| FgaPT2 | WT | A. fumigatus | L-Trp | Trp derivatives, L-Tyr, daptomycin, select indole DKPs | DMAPP | GPP, 3, 6, 7, 9, 10, 17, 73, 76, 77, 78, 80, 81, 82, 85, 88, 89–92, 96–98 | C4-reg | 54–60 |
| K174F | L-Tyr | L-Trp | DMAPP | N/D | C3-reg | |||
| R244L | Indole DKPs | N/D | DMAPP | N/D | C4-reg | |||
| K174F, R244N | Indole DKPs | N/D | DMAPP | N/D | C3-rev | |||
| K174F, R244L | Indole DKPs | N/D | DMAPP | N/D | C3-rev | |||
| M328C | L-Trp | N/D | GPP | DMAPP, FPP | C4-reg | |||
| M328A | L-Trp | N/D | GPP | DMAPP, FPP | C4-reg | |||
| M328T | L-Trp | N/D | DMAPP | GPP | C4-reg | |||
| M328S | L-Trp | N/D | GPP | DMAPP, FPP | C4-reg | |||
| M328G | L-Trp | N/D | GPP | DMAPP, FPP | C4-reg | |||
| M328V | L-Trp | N/D | DMAPP | GPP | C4-reg | |||
| M328N | L-Trp | N/D | DMAPP | GPP | C4-reg | |||
| 5-DMATSSc | WT | S. coelicor | L-Trp | Trp derivatives | DMAPP | 2, 78, 2-pentenyl-PP | C5-reg, C5/6-reg | 61–63 |
| Y326H, Q255V | L-Trp | N/D | DMAPP | N/D | C5-reg, C5/6-reg | |||
| Y326H, Q255N | L-Trp | N/D | DMAPP | N/D | C5-reg, C5/6-reg | |||
| L81A | L-Trp | N/D | GPP | N/D | C5-reg | |||
| 6-DMATSMo | WT | M. olivasterospora | L-Trp | Trp derivatives | DMAPP | GPP | C6-reg | 61 and 63 |
| L78A, F197W | L-Trp | N/D | DMAPP | N/D | C5-reg, C6-reg | |||
| V259Q, H329Y | L-Trp | N/D | DMAPP | N/D | C5-reg | |||
| L78A | L-Trp | N/D | GPP | N/D | C6-reg | |||
| IptA | WT | Streptomyces sp. SN-593 | L-Trp | Trp derivatives | DMAPP | N/D | C6-reg | 64 |
| W154A | L-Trp | N/D | GPP, FPP | N/D | C6-reg | |||
| PriB | WT | Streptomyces sp. RM-5-8 | L-Trp | Trp derivatives, indole derivatives, naphthalene derivatives, anthranilic acid, pindolol, daptomycin | DMAPP, GPP, FPP, GGPP | 30–65 | C6-reg | 65 |
| CymD | WT | S. arenicola CNS-205 | L-Trp | N/D | DMAPP | N/D | N1-rev | 66 |
| DMATS1Ff | WT | F. fujikoroi | L-Trp | L-Tyr | DMAPP | GPP | N1-rev | 67 and 68 |
Enzymes of the DMATS family primarily catalyze the prenylation of indole derivatives, including diketopiperazines (DKPs) containing a Trp or a Trp-like moiety.20 The substrate structure is the basis for further stratification of the DMATS family into four different groups based on their natural substrates: (i) Tyr DMATSs, (ii) Trp DMATSs, (iii) DKP DMATSs, and (iv) xanthone DMATSs.67,69 Even though certain enzymes have been observed to prenylate substrates from more than one group, such as 7-DMATS with both L-Tyr and L-Trp as substrates, these enzymes typically prefer one substrate over another, as shown by different rates of conversion, which determines which groups they belong to.67
Prenylation reactions can be divided into two categories: (i) regular prenylation, where the primary carbon of the prenyl donor is added to the acceptor and (ii) reverse prenylation, where the tertiary carbon of the prenyl donor is added to the acceptor (Fig. 5A and B).67 The majority of reactions catalyzed by DMATSs are regular prenylations, which correspond to the formation of more thermodynamically stable regioisomers. Additionally, the site of prenylation in nearly 75% of all prenylation reactions, including reverse prenylations, is a member of a ring system. DMATSs also catalyze prenylations, albeit rarely, on exocyclic oxygen and nitrogen atoms (e.g., benzylic oxygen of Tyr or nitrogen of aniline). Reverse prenylations only occur on nitrogen or carbon atoms (Fig. 5C).70
 | ||
| Fig. 4 (A) A list of possible natural prenyl donor cosubstrates of DMATSs. (B) Collection of unnatural alkyl donor cosubstrates tested against selected DMATSs.43,45,54,65,72 | ||
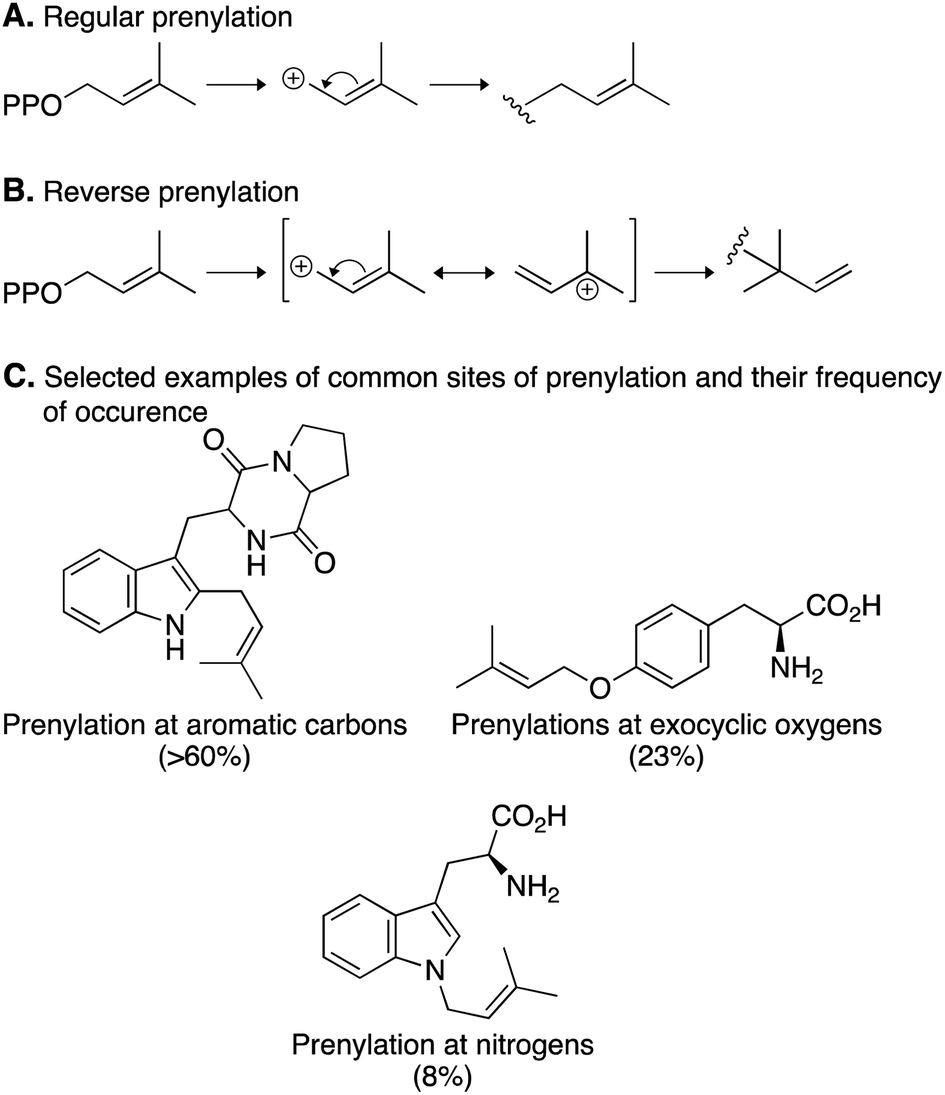 | ||
| Fig. 5 Prenylation reactions catalyzed by DMATSs. (A) A regular prenylation with DMAPP. (B) A reverse prenylation with DMAPP. (C) Common sites of prenylation and their frequency of occurrence. | ||
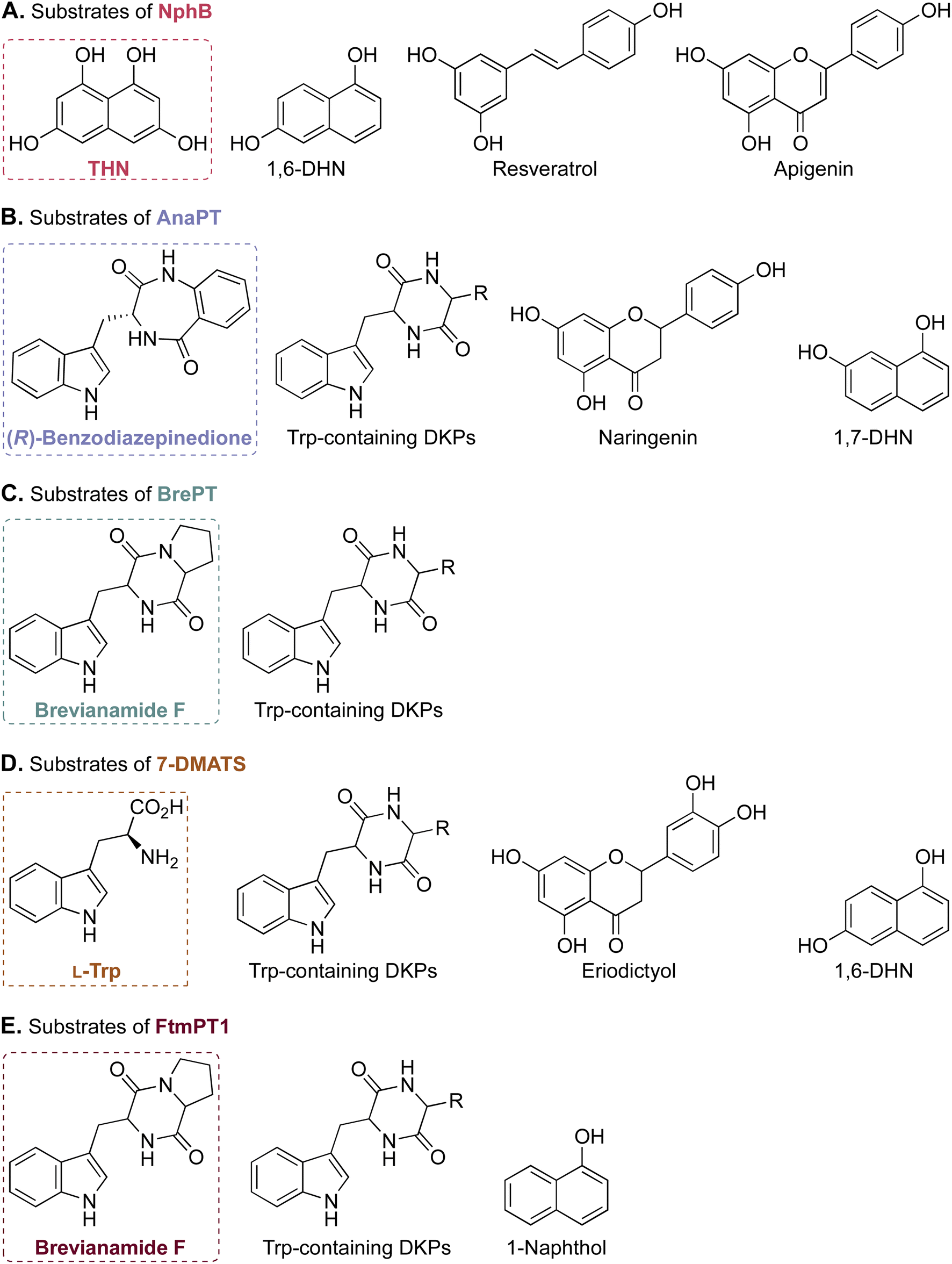 | ||
| Fig. 6 Selected examples of the prenyl acceptor substrate promiscuity of ABBA-PTs. Substrates of NphB, AnaPT, BrePT, 7-DMATS, and FtmPT1 are presented in panels (A)–(E). The natural substrate of each enzyme is in a box. | ||
The molecular basis of the promiscuity as well as the regio- and stereospecificity of reactions catalyzed by DMATSs are popular topics of investigation in the research community.64,67,73 Considering that only ∼50 enzymes out of more than 200 putative members of the DMATS family have been characterized, much work remains to be done before the mechanistic puzzle of DMATSs can be solved in its totality.20
Despite many developments, an exact chemical mechanism for DMATSs is still not known with certainty. There are two overarching mechanisms that are thought to occur in DMATSs: (i) direct prenylation (i.e., nucleophilic attack from the indole ring from the site incorporating the prenyl moiety) (Fig. 7A and C), or (ii) a multistep process in which the prenyl moiety is incorporated at one position on the indole ring, then transferred to another via a rearrangement (Fig. 7B and D). There are conflicting data regarding which mechanism is utilized in each DMATS, and the exact answer is yet to be definitively found. The results of many mechanistic studies performed, as expertly reviewed by Tanner,73 contain a level of ambiguity that makes their interpretation difficult. The chemical and biochemical rationale for both mechanisms are plausible in many situations, and the utilization of different substrates and/or mutants calls into question whether the results can be attributed to different binding modes.67,73–84 Although there has been recent evidence to suggest that the particular mechanism may depend on the individual PT or the substrate,61,65,85 any mechanistic explanation at present remains speculative. The continued effort of determining and examining the atomic structures of DMATSs will be essential for resolving this uncertainty.
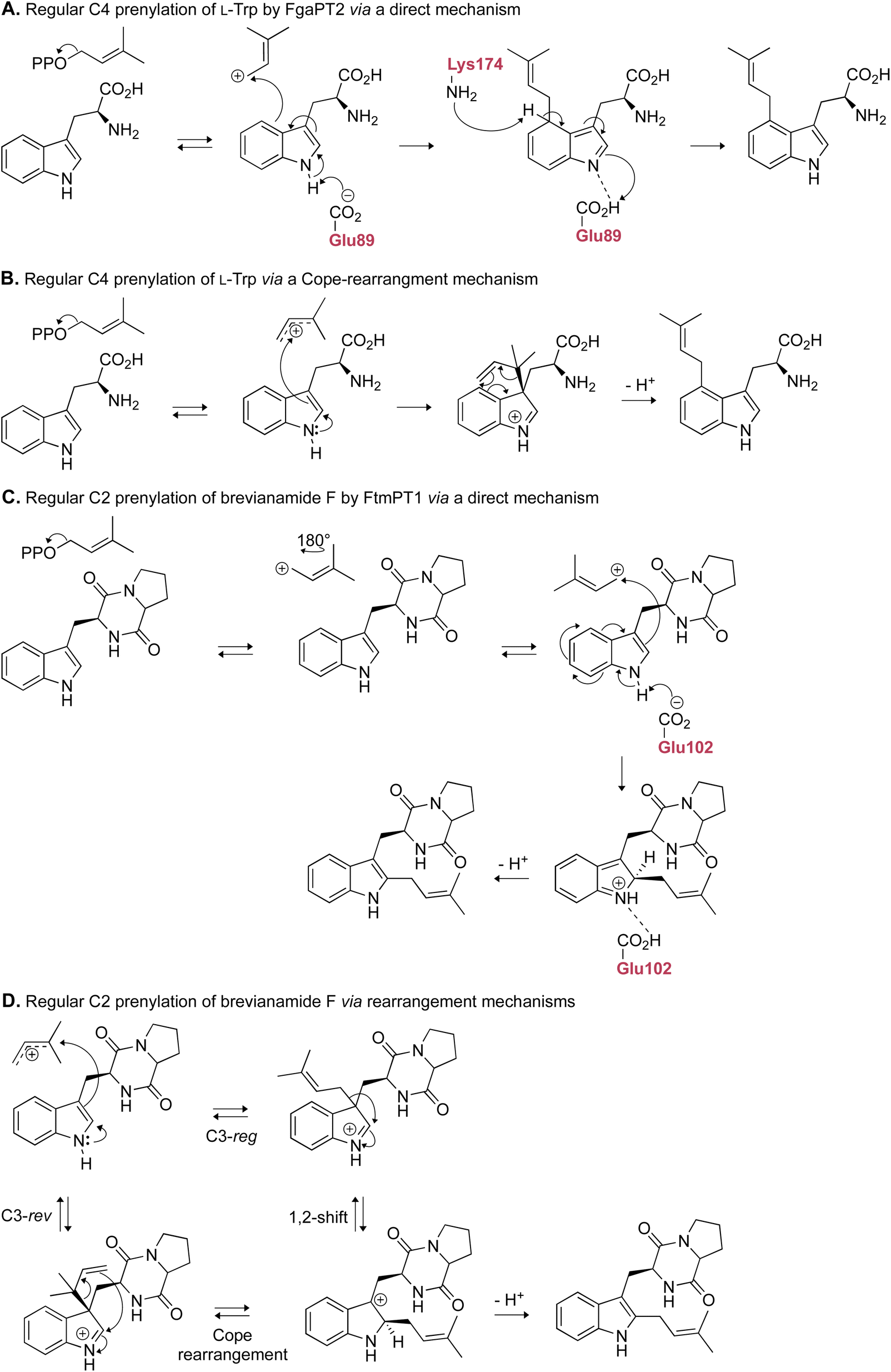 | ||
| Fig. 7 Representative examples of the proposed mechanistic routes that FgaPT2 and FtmPT1 may use to arrive at their respective final products. (A) Direct prenylation of the indole ring at C4. (B) Indirect prenylation of C4 via a Cope rearrangement. (C) Direct prenylation of the indole ring at C2. (D) Indirect prenylation of C2 via either a Cope rearrangement or a 1,2-shift. | ||
The utilization of DMATSs to modify aromatic scaffolds can be hindered due to their low turnover or catalytic efficiency in the presence of unnatural substrates.36,37,57 Therefore, to increase the utility of DMATSs for chemoenzymatic synthesis, two goals must be accomplished: (i) engineering methods must be established to efficiently confer the ability of the substrate binding pocket to incorporate substrates of varying sizes and conformations, and (ii) the kinetic parameters of these engineered enzymes in the presence of a selected unnatural donor or acceptor substrate must be comparable to the respective wild-type (WT) enzyme in the presence of its natural substrate. The next section summarizes the structure–function relationships that have been elucidated from the available crystal structures of DMATSs. We also describe the principles gleaned from active-site engineering efforts towards providing guidance for future efforts in accomplishing the two goals stated above.
2. Structure, mechanism, and engineering of DMATSs
2.1. Analysis of the prenyl acceptor binding pocket
The ability of DMATSs to prenylate a stunningly wide variety of substrates has been the subject of many studies. With the goal of elucidating the molecular basis for their promiscuity, the prenyl acceptor binding pocket of DMATSs has been a subject of fascination for enzymologists over the past twenty years. Nevertheless, even though approximately 50 DMATSs have been characterized biochemically,20 there have been comparatively few structural studies performed on this family of enzymes. To the best of our knowledge, of the known biochemically characterized DMATSs, there are only 12 DMATSs with crystal structures released in the Protein Data Bank (PDB) from 2009 to 2023 (Tables 2 and 3). Of these published structures, the vast majority are of WT enzymes in either their apo form or in ternary complex with their natural substrates and an unreactive diphosphate analogue, such as dimethylallyl S-thiolodiphosphate (DMSPP) or geranyl S-thiolodiphosphate (GSPP). While the number of structures of DMATS mutants and those in complex with unnatural substrates and cosubstrates has increased in recent years, a continued effort to produce these structures is needed to further the engineering and biotechnological application of DMATSs.| Enzyme | PDB ID | Resolution (Å) | Complexed prenyl acceptor | Complexed prenyl donor | Citation(s) |
|---|---|---|---|---|---|
| a Abbreviations: DMSPP = dimethylallyl S-thiolodiphosphate; GSPP = geranyl S-thiolodiphosphate; SPP = thiolodiphosphate. b N/P = not present in the structure. | |||||
| FtmPT1 | 3O24 | 2.50 | N/Pb | N/P | 34 |
| 3O2K | 2.40 | Brevianamide F | DMSPP | ||
| NotF | 6VY9 | 3.19 | N/P | N/P | 41 |
| 6VYA | 3.0 | Brevianamide F | DMSPP | ||
| CdpNPT | 4E0T | 2.25 | N/P | N/P | 44, 85 and 97 |
| 4E0U | 2.60 | (S)-Benzodiazepinedione | SPP | ||
| 7XVJ | 2.40 | Harmol | N/P | ||
| 7Y3V | 2.43 | Harmane | N/P | ||
| AnaPT | 4LD7 | 2.83 | N/P | SPP | 48 |
| AtaPT | 5KCG | 1.90 | N/P | N/P | 25 |
| 5KCL | 2.10 | N/P | DMSPP | ||
| 5KCQ | 2.0 | N/P | GSPP | ||
| 5KCY | 2.30 | (+)-Butyrolactone II | GSPP | ||
| 5KD6 | 1.84 | (−)-Butyrolactone II | GSPP | ||
| 5KDA | 2.0 | Genistein | DMSPP | ||
| 5KD0 | 2.82 | (+)-Butyrolactone II | GSPP | ||
| AtaPTE91A | 5KD0 | 2.82 | (+)-Butyrolactone II | GSPP | 25 |
| Enzyme | PDB ID | Resolution (Å) | Complexed prenyl acceptor | Complexed prenyl donor | Citation |
|---|---|---|---|---|---|
| a Abbreviations: DMSPP = dimethylallyl S-thiolodiphosphate; GSPP = geranyl S-thiolodiphosphate; PPi = diphosphate. b N/P = not present in the structure. | |||||
| FgaPT2 | 3I4Z | 1.76 | N/Pb | N/P | 29 |
| 3I4X | 2.10 | L-Trp | DMSPP | ||
| 5-DMATSSc | 6ZS0 | 1.50 | N/P | N/P | 61 |
| 6ZRZ | 1.70 | L-Trp | DMSPP | ||
| 6-DMATSMo | 6ZRY | 1.65 | N/P | N/P | 61 |
| 6ZRX | 1.70 | L-Trp | DMSPP | ||
| IptA | 7W8U | 2.25 | N/P | N/P | 64 |
| 7W8V | 1.61 | L-Trp | DMSPP | ||
| 7W8W | 1.80 | 5-Methyl-L-Trp | N/P | ||
| 7W8X | 1.45 | 6-Methyl-L-Trp | DMSPP | ||
| 7W8Y | 1.39 | Nα-Methyl-L-Trp | DMSPP | ||
| PriB | 5JXM | 1.15 | N/P | N/P | 65 |
| 5K9M | 1.50 | N/P | PPi | ||
| 5INJ | 1.40 | L-Trp | DMSPP | ||
| DMATS1Ff | 8DB1 | 2.72 | N/P | N/P | 68 |
| 8DB0 | 2.26 | L-Trp | DMSPP | ||
| 8DAZ | 2.49 | L-Trp | GSPP | ||
| 8DAY | 2.55 | L-Tyr | DMSPP | ||
| CymD | 6OS3 | 1.70 | N/P | N/P | 66 |
| 6OS5 | 1.66 | L-Trp | N/P | ||
| 6OS6 | 1.33 | L-Trp | DMSPP | ||
DMATSs have two primary binding sites: (i) the prenyl acceptor (substrate) binding site and (ii) the prenyl donor (cosubstrate) binding site. This section will detail the features of the prenyl acceptor binding site of the DKP-converting and free Trp-converting groups of the DMATSs. While DMATSs that convert free Tyr and xanthones exist, these enzymes are yet to be structurally characterized. We also acknowledge that the structures of the indolactam-specific PTs TleC and MpnD have been published.71 However, conflicting information exists in the literature as to whether they belong to the CloQ/NphB-type or DMATS PT families.61,67,78,98,99 Therefore, we opted not to include them in this review.
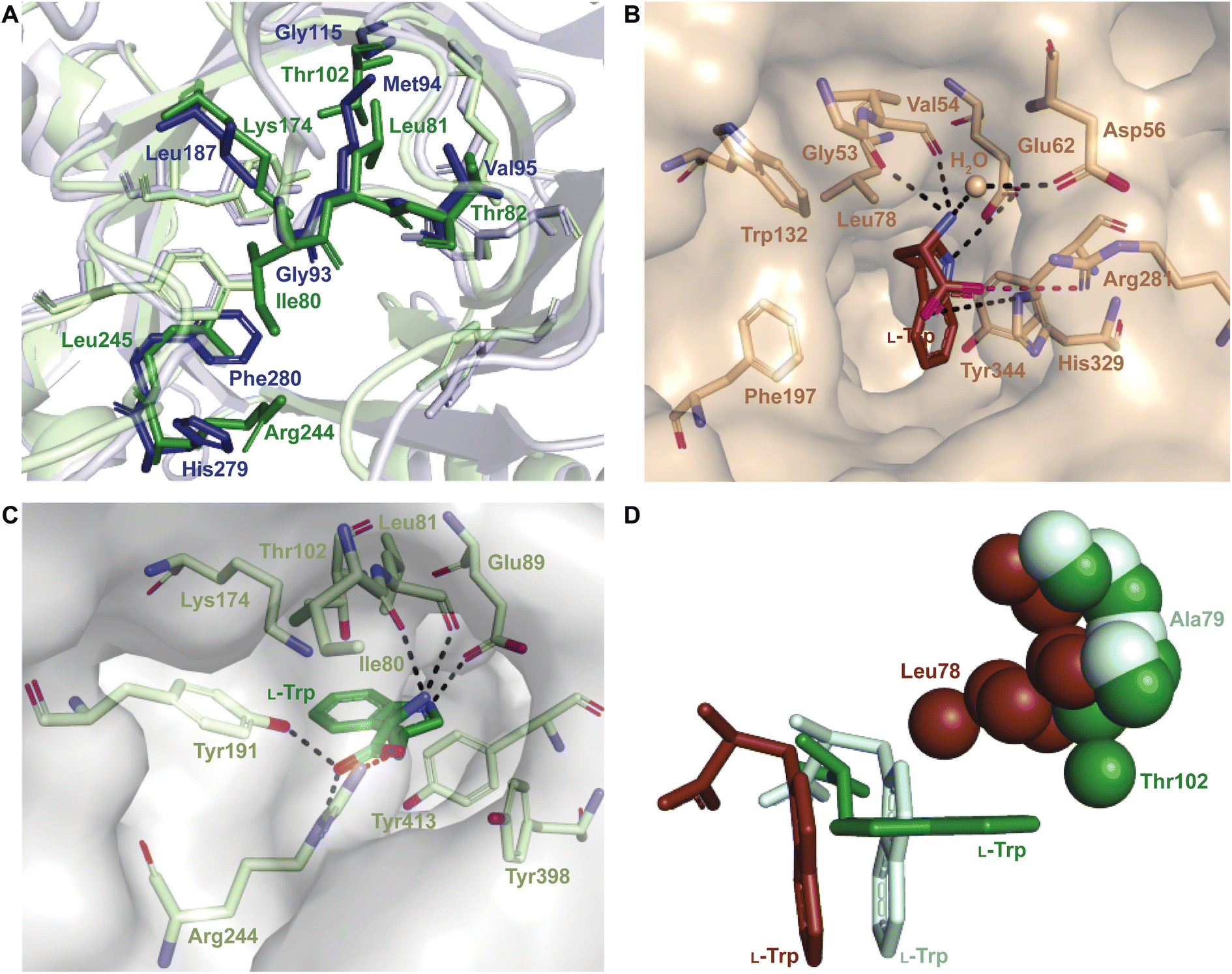
While each DMATS enzyme has a unique architecture of its prenyl acceptor binding site, X-ray crystallographic studies have established common physiochemical and structural characteristics of DMATSs (Fig. 8). A conserved feature of the catalytic chamber of DMATSs is its hydrophobic nature, as dictated by the chemical process. The prenylation reaction proceeds via a carbocation intermediate (i.e., the prenyl cation; Fig. 7) that must be shielded from the solvent for the reaction to occur. The bottom of the β-barrel becomes closed upon binding, with the acceptor substrate serving as a lid over the active site, shielding it from the solvent and protecting the carbocation intermediate during catalysis (Fig. 3).67 As such, aromaticity and hydrophobicity are mandatory factors involved in the substrate preference of DMATSs, as a lack of either could compromise the active site. The prenyl acceptor binding pocket (Fig. 3) is isolated from the solvent-accessible prenyl donor binding pocket by a rigid ring of four tyrosine residues that has been termed the tyrosine (Tyr) shield (Fig. 8A). These four Tyr residues play an essential role in catalysis by protecting the carbocation intermediate from solvent and stabilizing the intermediate via cation–π interactions.34,65 The fact that the four residues making up the Tyr shield are conserved in all PTs is consistent with their utility in reducing the energy barrier of carbocation formation and in shielding the carbocation.34 Furthermore, all structurally characterized DMATSs contain a conserved Glu residue within their active site, which is shown to be essential to catalysis in most cases, although some exceptions have been noted, as in AtaPT and 5-DMATSSc.25,29,34,61,67,100 This Glu is implicated in the hydrogen bonding with the hydrogen of the indole amine to increase the electron density of the ring while also stabilizing the positive charge of the σ-complex intermediate formed after nucleophilic attack on the prenyl cation (Fig. 8B).29,34 Other conserved elements in DMATSs are residues controlling regio- and stereoselectivity. In many DMATSs, a catalytic base (i.e., Lys or His) deprotonates the arenium intermediate and aids in the regioselectivity of the prenylation reaction (Fig. 8C).29,61 The catalytic base acts in conjunction with another residue that serves to control the position of the indole moiety relative to the prenyl donor; we refer to this residue here as the angle-determining residue (ADR) (Fig. 8D). This residue also controls stereoselectivity, as incorporation of a prenyl/alkyl moiety is only possible from the re-face of the ring. The exact position of the indole determines the stereochemistry of the prenyl/alkyl substituent.97 Additionally, the proximity of the primary or the tertiary carbon of the prenyl donor to the reactive atom of the indole ring (activated and positioned by the above residues) would result in regular or reverse prenylation, respectively. Even though the indole PT MpnD71 is not definitively classified as a DMATS and, therefore, not covered in detail in this review, it serves as an excellent illustration of this structure-based principle. A structure of this enzyme in complex with GPP (modeled in place of the DMSPP observed in the crystal structure) and its substrate, (−)-indolactam V, revealed similar distances from the C5 of the substrate indole and the C1 of GPP and from the C7 of the indole and the C3 of GPP. The similarity of these distances explained why MpnD catalyzed regular prenylation at C5 and reverse prenylation at C7. To the best of our knowledge, a specific catalytic base has been identified only in structures of Trp DMATSs. DKP DMATSs appear to rely principally on an ADR for regioselectivity control, and no obvious catalytic base has been observed in their crystal structures.34,97 Several studies have suggested a conserved Glu residue, in addition to the secondary amine on the DKP ring, as potential candidates for catalytic bases (Fig. 8C).34,97,101 The L-Tyr DMATS SirD (UniProt: Q6Q874) and the xanthone DMATS XptB (UniProt: P0DP82) have not been structurally characterized yet. Due to their unique substrate preferences, we retrieved predicted structures of these enzymes from the Alphafold Protein Structure Database.69,102–105 We compared these structural models with the structure of the prototypical DMATS FgaPT2 (Fig. 9). Like FgaPT2, SirD accepts both L-Trp derivatives and L-Tyr. Both DMATSs contain the conserved Glu residue interacting with the indole NH in other DMATS enzymes (Glu111 in SirD and Glu89 in FgaPT2), as well as the Tyr shield residues, suggesting a conserved substrate recognition and reaction mechanism.57 SirD lacks a corresponding catalytic base to the one observed in FgaPT2 (Lys174) (Fig. 9A). As SirD can generate N1- and C7-prenylated indole-containing products, the model suggests that its catalytic base is the conserved Glu residue or a water molecule. The role of the Glu as a catalytic base is in agreement with studies on other N1- and C7-prenylating DMATSs, which have suggested that the conserved Glu is oriented to deprotonate the arenium ion intermediate following prenylation at these positions.64,101,105 The side chains of the residues of the substrate binding pocket of SirD are generally the same size as those of FgaPT2, consistent with the overlapping substrate preferences, but the identities of these residues differ in several cases (Fig. 9A). The residues of the substrate binding pocket of XptB contain smaller side chains than those of FgaPT2. Superimposition of the two binding pockets shows that Arg83, Lys174, Tyr191, Arg244, Leu245, Met328, and Tyr398 in FgaPT2 are replaced by Leu96, Val189, His206, Phe254, Cys255, Val350, and Phe422 in XptB, respectively. The result is a less sterically hindered XptB binding pocket, presumably evolved to accommodate the larger xanthone scaffold (Fig. 9B). The exact function of the conserved Glu residue (Glu102) of this enzyme is not known, as its natural substrate does not contain an indole moiety.69
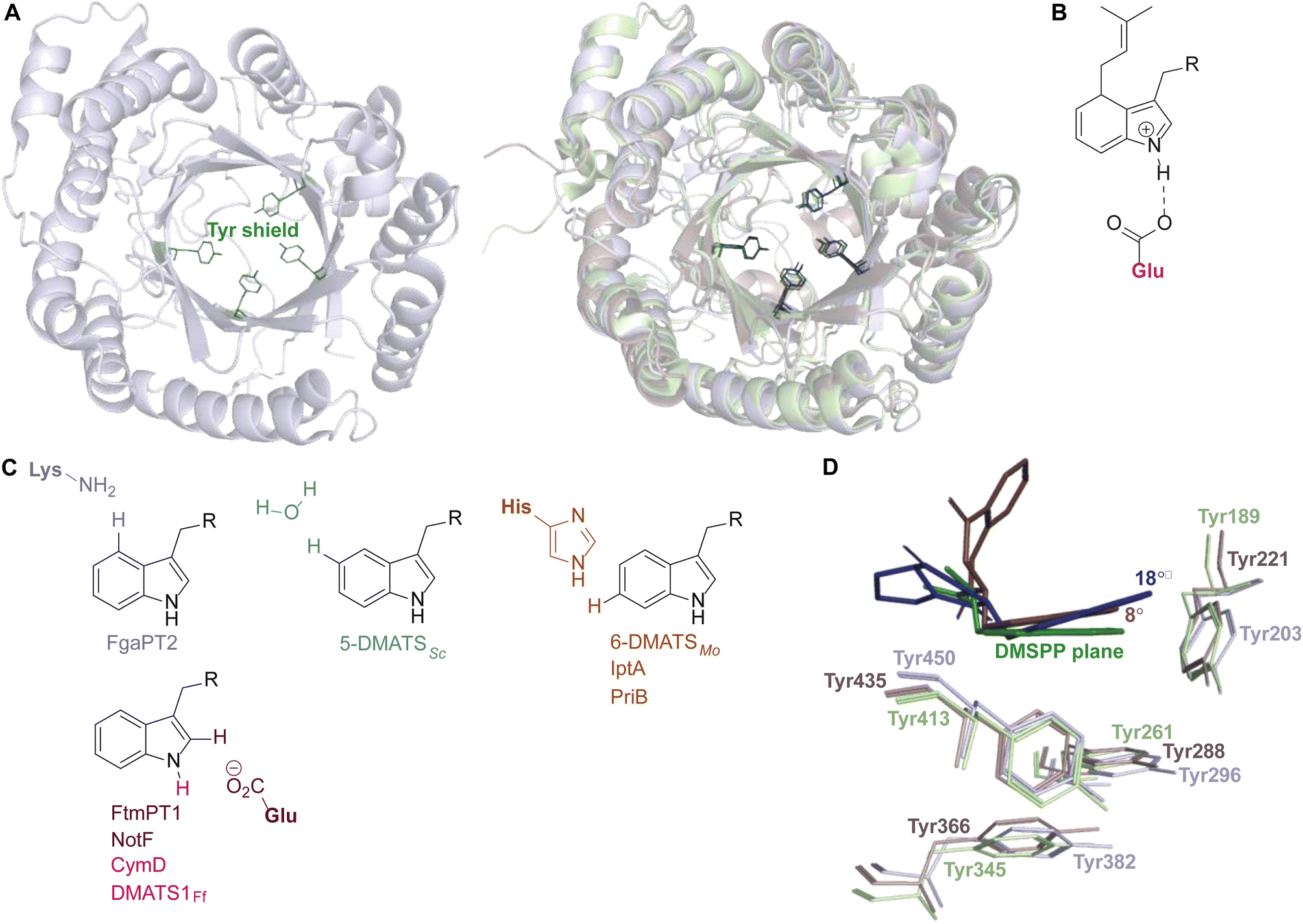 | ||
| Fig. 8 Conserved elements of the prenyl acceptor binding pocket. (A) FtmPT1 with the Tyr shield in green (left structure). Superimposition of FtmPT1 (light blue) with FgaPT2 (light green) and CdpNPT (light brown), showing the conserved nature of the Tyr shield in DMATSs (right overlay). (B) The charge stabilization by a conserved Glu residue in the presence of the arenium ion intermediate formed from a direct prenylation at C4. (C) Catalytic bases identified at or near the site of prenylation. (D) Superimposition of the indole moieties of substrates in complexes with FtmPT1 (brevianamide F, navy), CdpNPT ((S)-benzodiazepinedione, brown), and FgaPT2 (L-Trp, green) demonstrating the tilt of the indole moiety resulting from the steric effects of the ADR. | ||
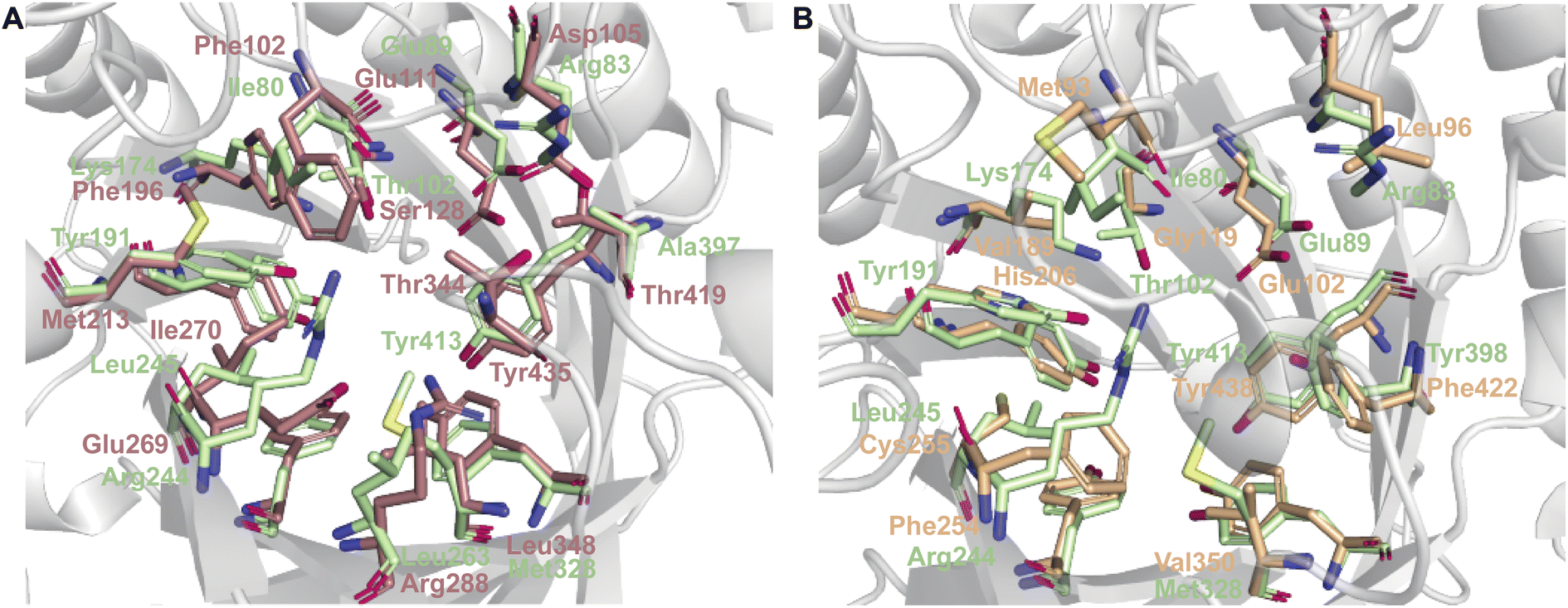 | ||
| Fig. 9 Comparison of SirD and XptB model predictions with FgaPT2. (A) The substrate binding pocket of SirD (light brown) and FgaPT2 (light green). (B) The substrate binding pocket of XptB (light orange) and FgaPT2 (light green). Some of the residues lining the substrate binding pocket and the Tyr shield are shown as sticks. | ||
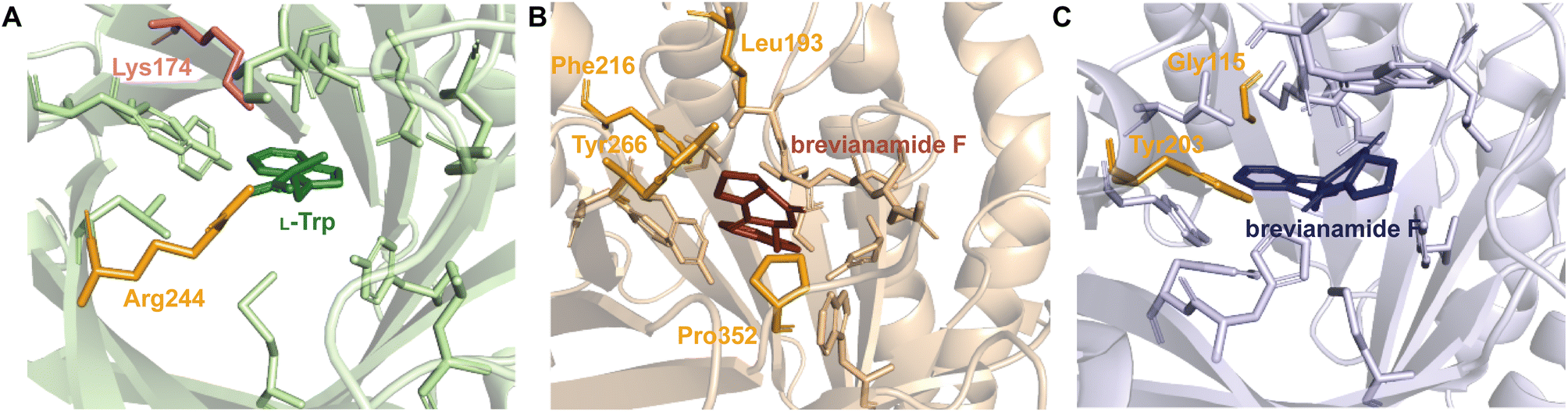
2.1.1.1. Active site architecture. Structurally characterized prenyl acceptor binding pockets of DKP DMATSs have been shown to be predominantly hydrophobic and more spacious than those of Trp DMATSs.25,34,48,97 While an exact explanation for the molecular basis of the unusual substrate promiscuity seen in DMATSs has remained elusive, it has been postulated that the hydrophobic nature of the binding pocket plays a role. Because the pocket is mostly hydrophobic, it allows for potentially favorable interactions with unnatural substrates via surface complementarity.97 In order for efficient catalysis to occur in the presence of a given substrate, polar (i.e., hydrogen bonds (H-bonds) and salt bridges) and nonpolar (i.e., hydrophobic effect and π–π) interactions must act in concert to properly position the substrate.97 Reduced rates of conversion of unnatural substrates have been observed for DMATSs most likely for this reason, as these substrates likely cannot be optimally positioned for the prenyl transfer. Structural characterization of DMATSs (Tables 2 and 3) has revealed that the interactions employed in substrate binding are generally conserved throughout DKP DMATSs, mostly in the form of hydrophobic interactions and H-bonds to the heteroatoms of the substrate, although the degree to which each type of an interaction was utilized differed from enzyme to enzyme (Fig. 10).25,34,41,48,97 Despite this general conserved nature, DKP DMATSs have been documented as displaying different regioselectivities, prenylation types, and substrate scopes.25,34,41,48,97 These differences have been attributed to the overall architecture of their respective active site, which has been shown to vary in three primary ways: (i) the overall size of the binding pocket, (ii) the angle of the indole moiety, and (iii) the rigidity of the binding pocket.25,34,41,48,97
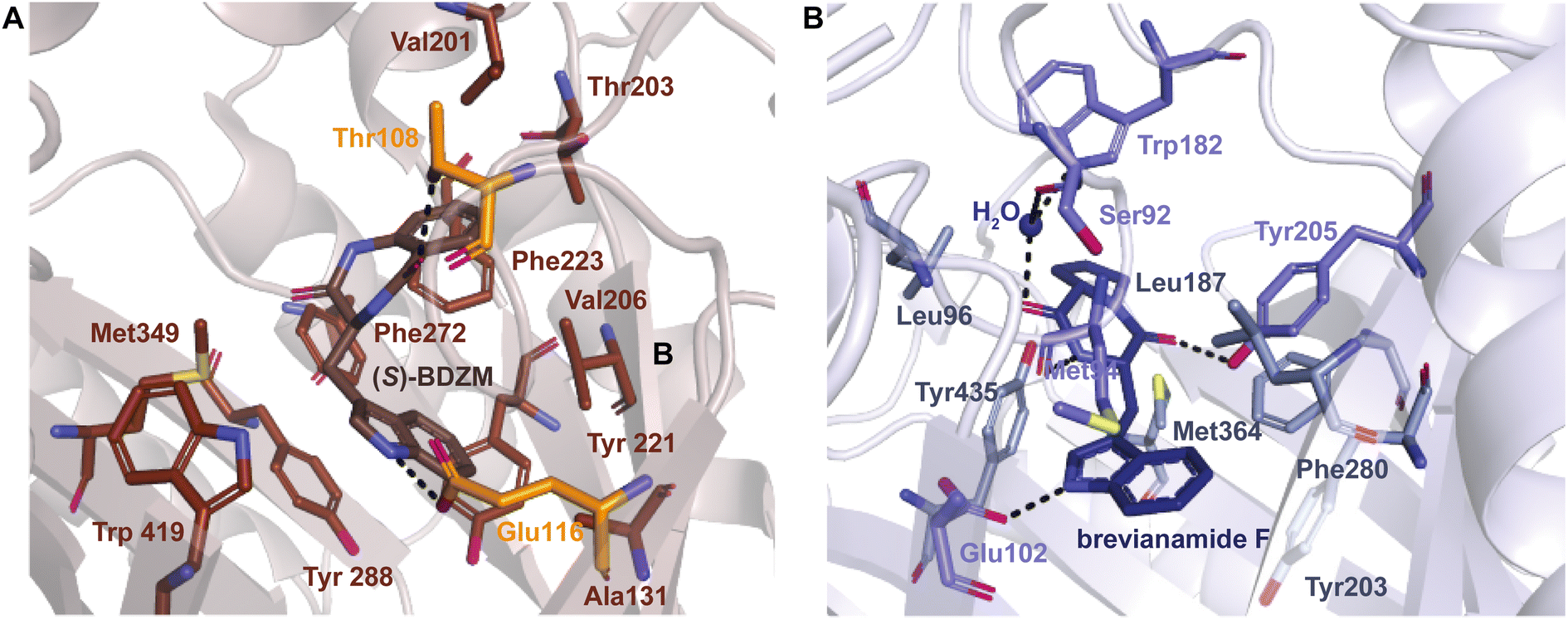 | ||
| Fig. 10 (A) Residues in brown and orange are implicated in hydrophobic contacts and H-bonds in CdpNPT, respectively. (B) Residues in gray and blue are implicated in hydrophobic contacts and H-bonds in FtmPT1, respectively. H-bonds are shown as dotted black lines. | ||
As a whole, DKP DMATSs possess less sterically constrained binding pockets than Trp DMATSs to accommodate their larger substrates.25 Structural studies have determined that the sizes of these binding pockets vary widely from one DKP DMATS to another due to: (i) smaller side chains within the binding pocket, and (ii) different orientation of the α3–β1 and α6–β3 binding loops (Fig. 11B and C).25,34,48,97 Mutations to smaller residues to increase the volume of the active site can have a dramatic impact on the substrate scope. For instance, the binding pocket of FtmPT1 has a solvent accessible volume of approximately 1600 Å3. Among the structurally characterized DKP DMATSs, this volume is one of the smallest.25 Nevertheless, the binding pocket of FtmPT1 can accommodate a wide array of substrates, such as simple indole derivatives, indole DKPs, and hydroxynaphthalenes.37,39,106 When smaller side chains are present in the active site of an enzyme (e.g., Leu252, Cys175, Ser170, and Ser192 in AtaPT) in place of the larger side chains (e.g., Phe280, Leu187, Trp182, and Tyr205, among others in FtmPT1), the binding pocket volume nearly doubles (Fig. 11A).25 Consequently, the spacious binding pocket of AtaPT was observed to prenylate an unprecedented variety of structurally diverse substrates including lignanoids, indole DKPs, glycosides, flavonoids, p-hydroxybenzaldehyde, as well as several diverse examples of PKs.25,46,51–53,107 This dichotomy between FtmPT1 and AtaPT demonstrates the importance of the binding pocket size as a determining factor of substrate scope. The same effect has been shown in DKP DMATSs in which the difference in binding pocket sizes is not as drastic. The binding pocket of CdpNPT, for example, was shown to only be ∼500 Å3 larger than that of FtmPT1. Despite this difference in size, CdpNPT was shown to prenylate indole DKPs, aszonalenins, β-carbolines, and even the indole moiety of the macrocyclic antibiotic daptomycin.43,45,85,89,97
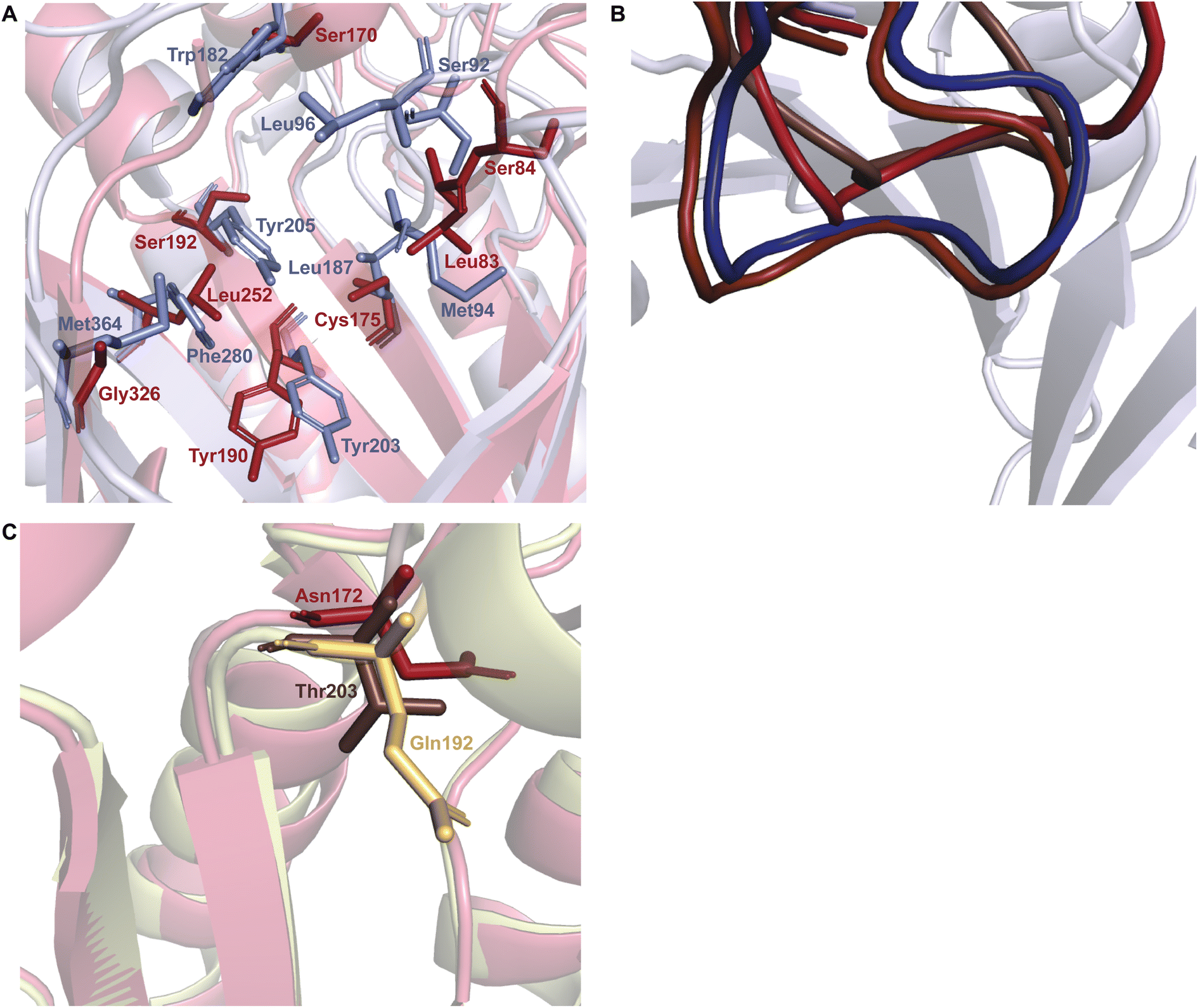 | ||
| Fig. 11 Important structural factors determining substrate scope. (A) The binding pocket residues of FtmPT1 (navy) and AtaPT (brick red) demonstrating the less sterically hindered binding pocket of AtaPT. (B) Superimposition of the α3–β1 binding loops of CdpNPT (brown), FtmPT1 (navy), NotF (chocolate brown), and AtaPT (brick red). (C) Superimposition of the α6–β3 binding loops of CdpNPT (brown), AnaPT (dark beige), and AtaPT (brick red). | ||
In addition to the more spacious binding pocket of CdpNPT, other structural features of DMATSs also serve as determinants of the substrate scope, specifically the α3–β1 and α6–β3 loop regions, termed the binding loops (Fig. 11B and C).48,97 The α3–β1 binding loop has been implicated in providing polar interactions to the substrate in FtmPT1 and in the Trp DMATS FgaPT2.29,34 The binding loop points inward toward the center of the inner barrel, constricting both the entrance to and the size of the binding pocket (Fig. 11B).48,97 In contract, this binding loop in CdpNPT does not stabilize the substrate.97 The α3–β1 binding loop in CdpNPT is oriented differently from that of FtmPT1, pointing outward, towards the side of the barrel.97 Therefore, the increased space provided by this structural difference likely allows larger substrates to gain access to the binding pocket. Interestingly, in DKP DMATSs with larger binding pocket volumes (i.e., AnaPT and AtaPT) the α3–β1 binding loop adopted an inward orientation, though to a lesser degree than in FtmPT1 and NotF (Fig. 11B). Furthermore, the α6–β3 binding loop also showed different orientations in these enzymes. In CdpNPT, this loop contained less bulky side chains, which further opened the binding pocket. AtaPT and AnaPT, in contrast, displayed more sterically constrained loops, closing the corresponding space in the binding pocket.25,34,48,97 These observations indicate that while the access to the binding pocket is important in determining the substrate scope, it is not as impactful as the size of the binding pocket itself.
How DMATSs control regio- and stereoselectivity of prenylation has been a long-standing question in the PT research community.64,67,73 While the chemical basis for these properties has remained a subject of debate,73 the available crystal structures of DKP DMATSs have revealed that the angle of the indole moiety relative to the prenyl donor may be a critical determinant (Fig. 12A and B). Even though the indole ring is held by many steric interactions within the binding pocket, its angle was shown to be controlled primarily by the ADR. For DKP DMATSs, we define an ADR as a residue within the indole binding cleft corresponding to position Ala131 (CdpNPT) or Gly115 (FtmPT1) that dictates the deviation from co-planarity of the indole moiety and the prenyl donor, measured as a tilt angle between the indole ring plane and the plane of the dimethylallyl moiety of DMSPP (x − z) plane (Fig. 12A).34,97 This angle has been proposed to determine the prenylation site of the indole ring. For example, in the crystal structure of the ternary complex of FtmPT1 with brevianamide F and DMSPP, the indole moiety of brevianamide F had an 18° tilt relative to the DMSPP plane, so that the C4, C5, C6, and C7 positions of the indole ring were too far from the cosubstrate, while the C2 faced the prenyl cation, resulting in a regularly C2-prenylated product.34,39,97 In CdpNPT, the indole ring was tilted by 8° relative to the DMSPP plane (Fig. 12B; the dimethylallyl moiety was modeled based on the structure of FtmPT1),97 positioning the indole C3 for prenylation and resulting in a reversely C3-prenylated product.97 These studies have demonstrated that the magnitude of the indole tilt is inversely proportional to the steric bulk of the ADR side chains (e.g., the ADR Gly115 in FtmPT1 led to a tilt of 18°, the largest tilt among structurally characterized DMATSs, whereas the ADR Ala131 in CdpNPT only allowed the indole ring to tilt by 8°). Extrapolation of this dependence to the Trp DMATS FgaPT2 identified the bulkier Thr102 as an ADR that placed the indole ring in a co-planar orientation with the DMSPP plane (Fig. 12B).29,34
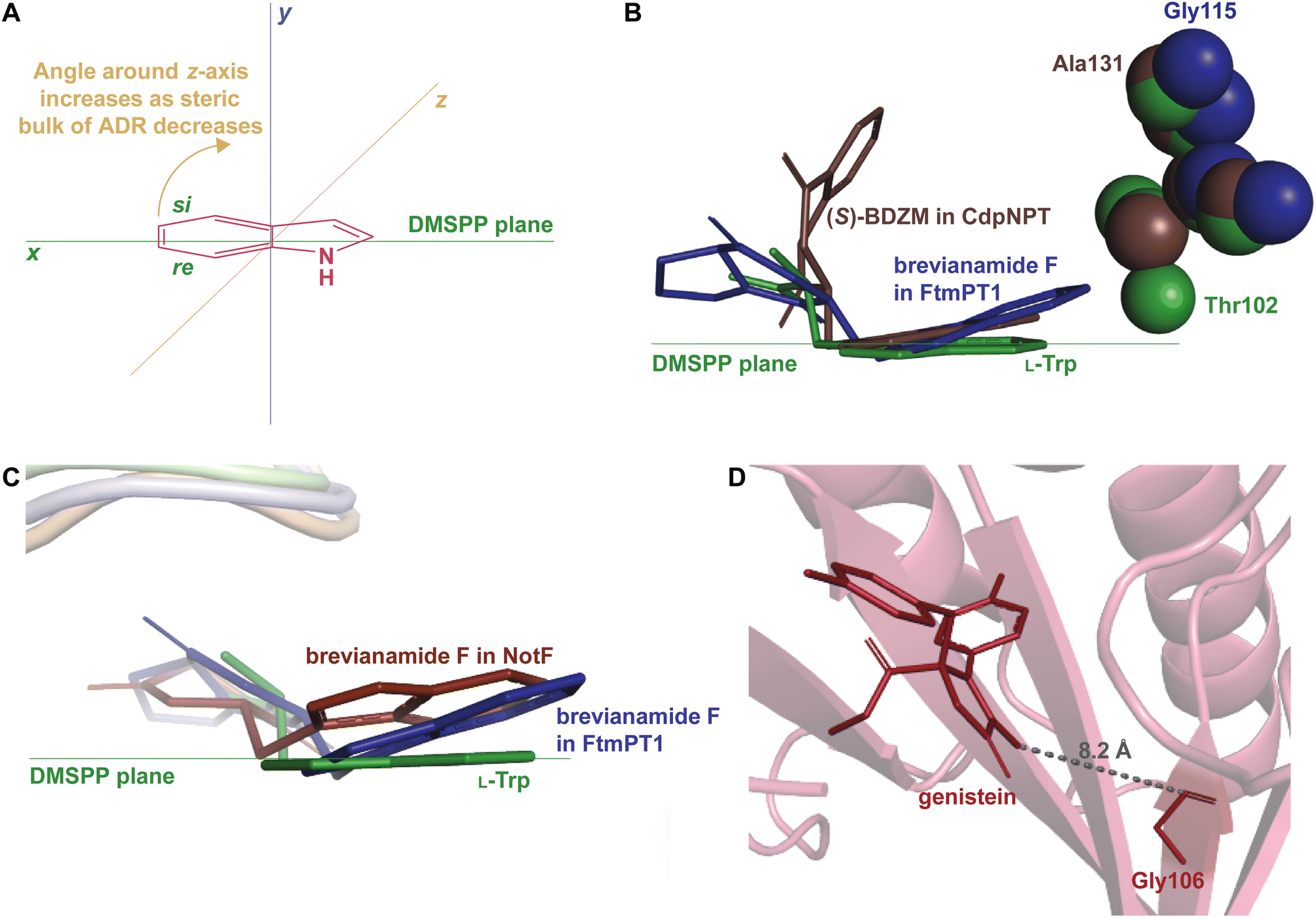 | ||
| Fig. 12 The ADR and its role in determining regioselectivity. (A) Representation of the tilt angle relative to the DMSPP plane, controlled by the steric bulk of the ADR. (B) Superimposition of the bound substrates of FgaPT2 (green), FtmPT1 (navy), and CdpNPT (brown) showing the relationship between the steric bulk of the ADR and the tilt of the indole moiety. (C) Superimposition of the bound substrates of FtmPT1 (navy) and NotF (chocolate brown). The FgaPT2 substrate (green) is superimposed as a reference to the DMSPP plane. (D) AtaPT in complex with genistein (brick red) showing the distance between the ADR and the genistein. | ||
The effect of the ADR in controlling the regioselectivity through the control of the positioning of the indole ring is applicable to indole-containing substrates of DKP DMATSs, but it does not appear to be determined only by the identity of the ADR, even among DKP DMATSs. The corresponding structurally analogous ADRs for the DKP DMATSs NotF, AnaPT, and AtaPT are Gly124, Gly126, and Gly106, respectively.25,41,48 Based solely on the ADR identity in NotF, the binding mode of the indole moiety of brevianamide F in this enzyme should display a similar tilt angle to that in FtmPT1.34,41 In contrast, in NotF the indole ring was rotated differently, where a tilt from the DMSPP plane was relatively minor, but the ring was moved away from this plane due to a rotation of the indole moiety around the x-axis (Fig. 12C). This rotation apparently resulted from interactions with other structural features of the enzyme.41 While this difference was likely partially due to additional steric constraints from the α3–β1 binding loop, the indole was positioned so that the C3 of the dimethyallyl moiety of the DMSPP was at 3.5 Å and at 4.2 Å from the C3 and the C2 atoms of the indole ring, respectively, consistent with reverse prenylation at one or both of these two positions. The authors suggested that the reaction occurred at the C2 based on the higher intrinsic reactivity of this carbon atom. In another example, both AnaPT and CdpNPT have the same regio- and chemoselectivities, suggesting that the binding mode of the indole ring in AnaPT should be similar to that in CdpNPT. On the other hand, the ADR in AnaPT is a Gly and not Ala, as in CdpNPT. A similar discrepancy exists for AtaPT, which catalyzes regular C4- or C7-prenylation of the indole ring (Table 1), while the ADR is a Gly. Therefore, other structural features must be involved in indole ring positioning in AnaPT and AtaPT. Without crystal structures of these two enzymes in complexes with an indole DKP, the roles of their respective Gly126 and Gly106 in substrate positioning cannot be elucidated. In addition, the impact of the ADR in the catalysis of non-indole containing compounds likely depends on the structure of a particular DKP. In the crystal structure of AtaPT in complex with the non-indole compound genistein, the genistein is bound ∼8 Å away from Gly106 (Fig. 12D). It is therefore unlikely that this residue plays a major role in determining the regioselectivity of the prenylation reaction. This survey leads us to a conclusion that the ADR, as is defined here, should only be utilized with caution in describing the tilt of an indole moiety bound to FtmPT1, CdpNPT, and other highly similar enzymes that are yet to be studied.
Stereospecificity has also been proposed to be impacted by the position at which the indole ring is held within the binding pocket.97 Because the si-face of the indole moiety shields the active site from solvent, prenylation must take place on the re-face side of the indole ring.97 This is indeed the case for the reverse C3-prenylated hexahydropyrroloindoline products generated as major products from CdpNPT (Fig. 13), and the regular C3-prenylated hexahydropyrrolindoline compounds found as minor side products in FtmPT1.34,37,97 The products were prenylated in a syn-cis (2S, 3R, 11S) configuration. That is, the prenyl moiety at C3 was incorporated on the same side as the carbonyl at C11 of the DKP substrate (Fig. 13).48 CdpNPT was also shown to prenylate in an anti-cis manner, but at a much lower frequency.48
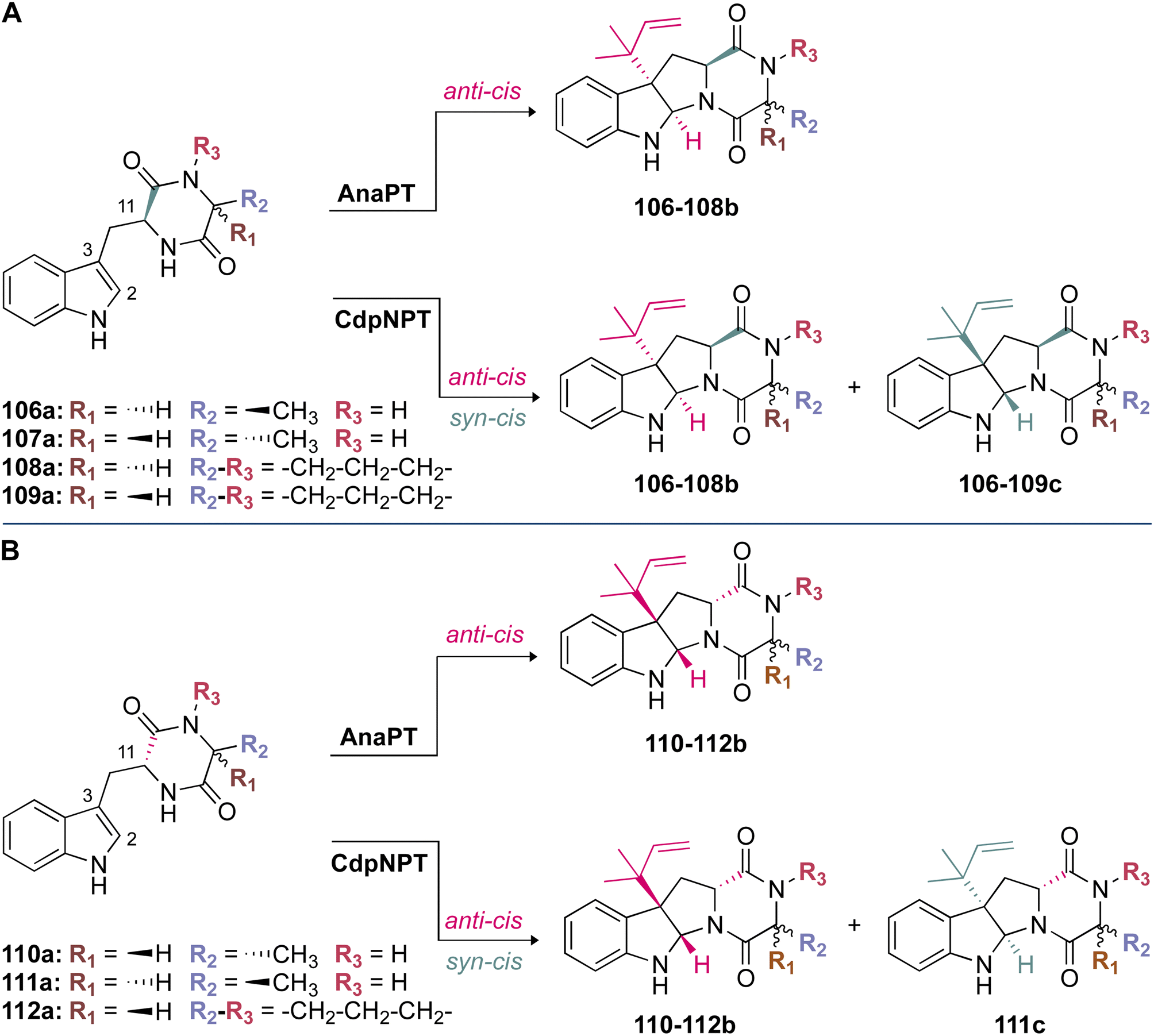 | ||
| Fig. 13 Stereospecificity of prenylation among DKP DMATSs. (A) anti-cis and syn-cis hexahydropyrroloindoline products generated from compounds 106a–109a (with L-Trp side chain) by AnaPT and CdpNPT. Stereochemistry of 106–108b: (2R, 3S, 11S). Stereochemistry of 106–109c: (2S, 3R, 11S). (B) anti-cis and syn-cis hexahydropyrroloindoline products generated from compounds 110a–112a (with D-Trp side chain) by AnaPT and CdpNPT. Stereochemistry of 110–112b: (2S, 3R, 11R). Stereochemistry of 111c: (2R, 3S, 11R). | ||
The crystal structures of FtmPT1 and CdpNPT revealed a basis for the formation of syn-cis (2S, 3R, 11S)-prenylated hexahydropyrroloindolines in that both prenyl moieties were oriented in the same half-space.48 However, at least in the case of CdpNPT, this diastereoselectivity was shown to be largely substrate-dependent. For instance, when the Trp moiety of the DKP substrate was in an L-configuration, syn-cis (2S, 3R, 11S)-configured compounds (106–109c) were shown to be the major products (Fig. 13A). This was in contrast to D-configured Trp moieties, in which anti-cis (2S, 3R, 11R)-configured compounds (110–112b) were shown to be the major products (Fig. 13B). A loss of stereoselective control occurred when the second amino acid residue of the DKP was replaced with smaller residues (i.e., substitution of Pro by Ala). While the major products still followed the above trend, a larger product ratio of anti-cis to syn-cis was observed. These results formed the basis of a proposal that the size of the second residue plays a role in the stereoselectivity of CdpNPT.48 This proposal echoed the conclusions of kinetic experiments performed on CdpNPT in the presence of different stereoisomers of benzodiazepinedione (BDZM). CdpNPT behaved differently towards (R)- and (S)-BDZM, with the (S) isomer being preferred, most likely because the (R) isomer had fewer polar interactions with the enzyme.97 For the smaller DKPs that were likely not efficiently stabilized within the binding pocket, the incorporation of the prenyl group occurred from different faces of the ring.48
Unlike CdpNPT, AnaPT generated anti-cis (2S, 3R, 11R)- and (2R, 3S, 11S)-prenylated hexahydropyrroloindolines as major products (110–112b in Fig. 13B and 106–108b in Fig. 13A, respectively). Similarly to CdpNPT, the stereoselectivity was dependent on the configuration of the Trp moiety, but it was not impacted by the size of the second amino acid residue (Fig. 13).48 This was a profound observation, because the prenylation occurred on the opposite face of the indole ring from what would have been expected based on the CdpNPT prenylation.48,73 The stereoselectivity of AnaPT was determined not to be substrate-dependent. In the presence of (R)- and (S)-BDZM AnaPT generated prenylated products with configurations that were opposite to those of the products of CdpNPT.48 These results taken together formed the current basis of understanding for the behavior of AnaPT, which was thought to exhibit an entirely different binding mode of the indole moiety from those of CdpNPT or FtmPT1, to facilitate the incorporation of the prenyl moiety in the observed configuration.48 This binding mode was suggested to require a 90° rotation of both substrates from the orientation observed in the CdpNPT binding mode.48 However, until a crystal structure of AnaPT in complex with an indole-containing substrate can be obtained, the proposed AnaPT binding mode will remain only a hypothesis.
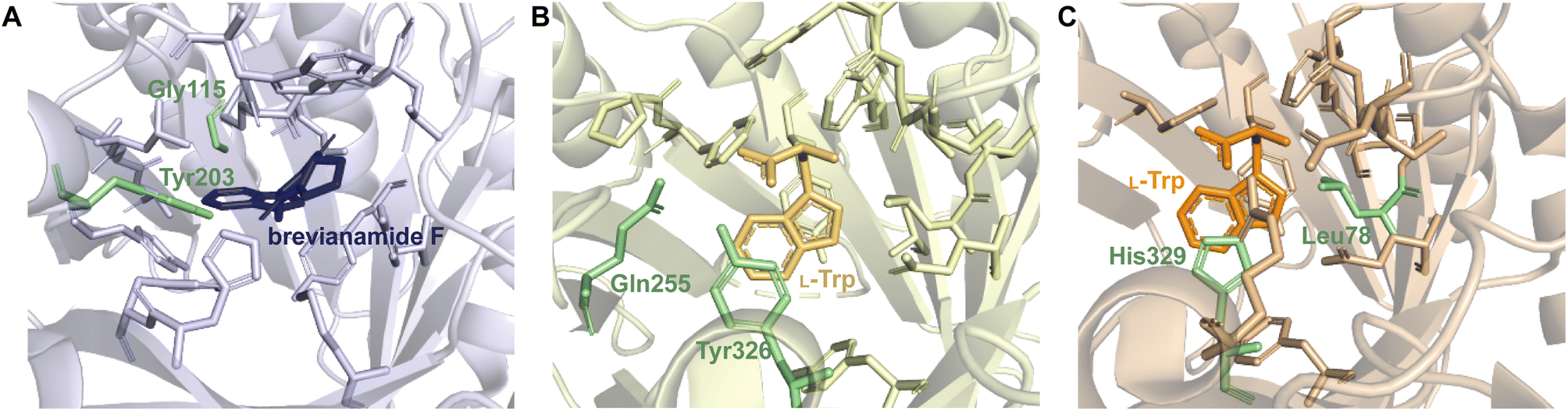
Despite the structural variation among the prenyl acceptor binding pockets of DKP DMATSs, the structures of ternary complexes of these enzymes with a substrate and a cosubstrate have indicated generally minimal conformational changes upon substrate binding.25,34,41,48 Only relatively minor changes were observed in the Tyr shield regions of CdpNPT, NotF, and FtmPT1. This was not the case for AtaPT. AtaPT was shown to perform catalysis via an ordered sequential mechanism, where binding of the preferred prenyl donor, GPP, was required for binding of the prenyl acceptor substrate.25 Furthermore, closed and open conformations were observed to be localized to the Tyr shield residues Tyr190 and Tyr268 (Fig. 14B). The closed conformation of the substrate binding pocket observed in apo AtaPT and in AtaPT in complex with the unreactive GPP analogue, GSPP, where it appeared to serve the purpose of constricting the size of the binding pocket. The open conformation was observed for AtaPT bound to the prenyl acceptor (PDB ID: 5KCY).25 The open conformation showed both Tyr residues orienting themselves away from the prenyl acceptor binding pocket and toward GSPP, which accommodated binding of the acceptor substrate (Fig. 14B).25 Due to its large size, the substrate binding channel of AtaPT was observed to bind different substrates at different sites. Interestingly, the conformations of Tyr190 and Tyr268 were dependent on which site certain substrates were bound in. The genistein binding site was further away from the Tyr shield and, therefore, binding to this site apparently did not require remodeling of the shield to open the binding pocket. On the other hand, the (+)-butyrolactone II binding site was close to the Tyr shield, and binding of this substrate was coupled to conformational remodeling of the two Tyr residues (Fig. 14A and B).25 Because the natural substrate(s) of AtaPT is/are not known with certainty,108,109 we do not know if their binding requires the opening of the binding site pocket. This question was answered in part by the structural analysis of CdpNPT complexed with the unnatural substrate harmol (Fig. 14D).85 In this structure, Trp419, a residue previously shown to be stationary upon (S)-BDZM binding (Fig. 14C), was reported to be shifted by 2.20 Å to accommodate the substrate (Fig. 14D). Other crystal structures of DKP DMATSs in complexes with diverse substrates are needed to improve our understanding of the enzyme dynamics.
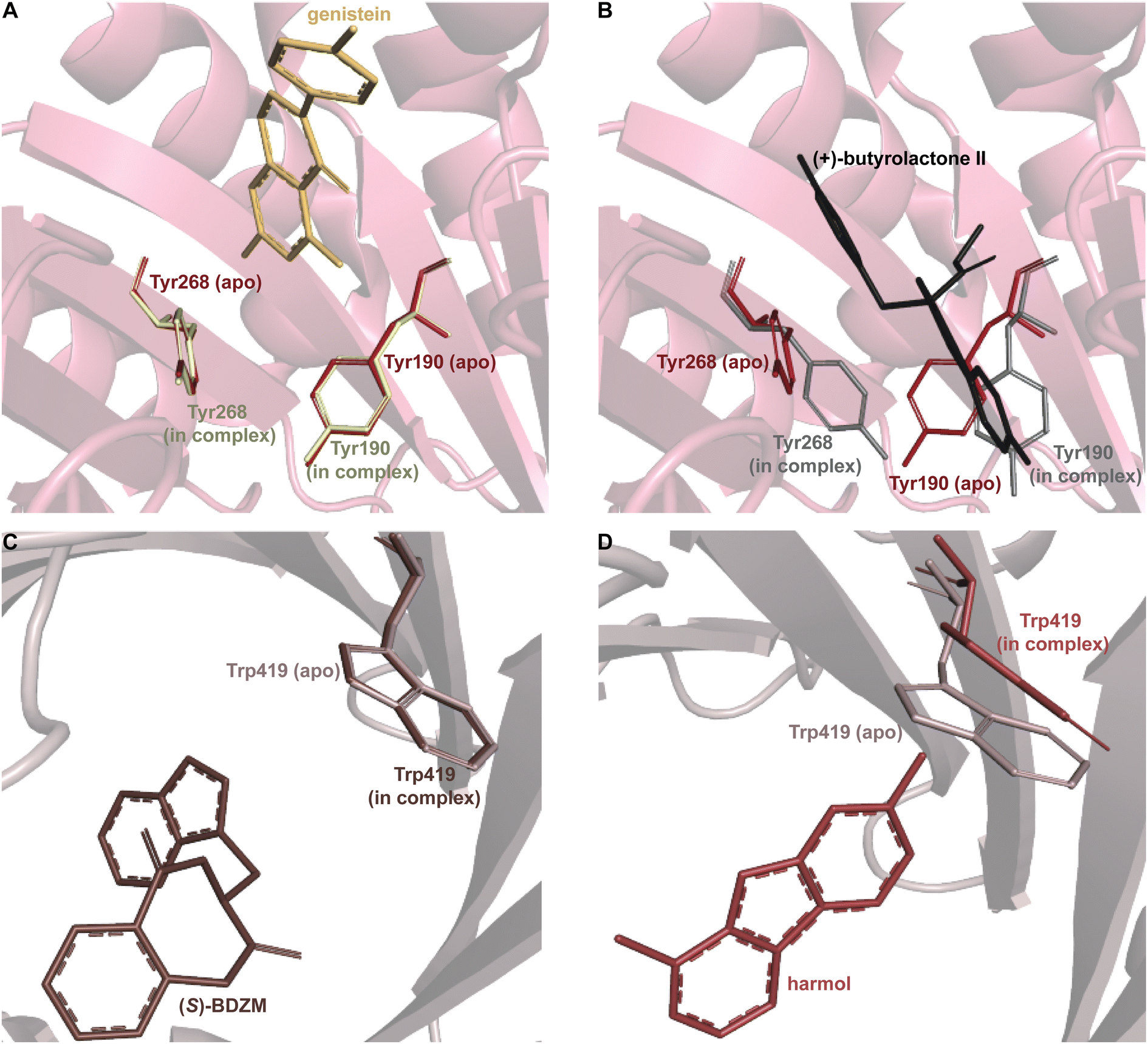 | ||
| Fig. 14 Substrate-dependent binding pocket dynamics. (A) Superimposition of the Tyr shield region of apo AtaPT (brick red) in the closed state and AtaPT in complex with genistein (light yellow) showing minimal differences from the closed state. (B) Superimposition of the Tyr shield region of apo AtaPT (brick red) and AtaPT in complex with (+)-butyrolactone II (gray) showing the open state of Tyr190 and Tyr268. (C) Superimposition of the rigid conformation of Trp419 in both apo CdpNPT (gray) and CdpNPT in complex with (S)-BDZM (brown). (D) An analogous superimposition showing a shift in the conformation of the CdpNPT Trp419 residue when in complex with harmol (chocolate brown). | ||
2.1.1.2. Mechanism. Among the DKP DMATSs, only FtmPT1 has been the subject of multiple mechanistic studies. FtmPT1 is an intriguing case for the Cope rearrangement. In that structure, the atoms C2 and C3 of the indole moiety of the substrate were within appropriate distances from the C3 atom of DMSPP (3.8 Å and 3.5 Å, respectively),34 whereas the C1 atom of DMSPP was too far, making the observed regularly prenylated product difficult to explain. The authors proposed that the prenyl group or its carbocation changed its orientation from that observed in the crystal structure during catalysis. In the same study, the mutant of Gly115, a residue close to the C7 of the indole, to a Thr, changed the dominant reaction to the reverse prenylation at the C3 of the indole. This result and other studies observing reverse prenylation at C3 and other positions among products suggested the possibility of an initial reverse prenylation at the C3 or neighboring positions followed by a Cope rearrangement or a 1,2-alkyl shift to the final product (Fig. 7D).73,110 An alternative mechanism, termed the cation-flip mechanism (Fig. 7C), invoked a rotation of the dimethylallyl cation by 180° to facilitate direct regular C2 prenylation via nucleophilic attack at the primary carbocation, to explain the positioning of the indole moiety relative to the prenyl donor.34,110 The cation-flip mechanism has been disputed, because a drastic re-orientation of such a highly reactive species in an active site is not the most efficient method of catalysis. Much of the evidence that has been used to argue the case of rearrangement-based mechanisms has come from analysis of products of unnatural substrates produced by FtmPT1 or by the FtmPT1G115T mutant, where the reversely C3-prenylated product dominated.73 Therefore, it is possible that these observations are a result of a sub-optimal spatial control by the enzyme, or they are due to unnatural binding modes. Nevertheless, the continued effort to elucidate the mechanistic basis of prenylation is essential for future engineering efforts, as such knowledge provides insight into strategic alterations of the binding pocket while preserving catalytic activity.61 These efforts could be aided by quantum mechanical and molecular mechanical (QM/MM) approaches, in addition to more intricate crystallographic experiments such as time-resolved serial femtosecond crystallography (TR-SFX),111–113 and traditional mechanistic enzymology techniques like rapid quenching and intermediate trapping, to establish the chemical mechanism.
While structural characterization and mutagenesis experiments have outlined the important factors and the key players of the catalytic cycles of DKP DMATSs, there is still an uncertainty regarding the final deprotonation step to restore aromaticity to the arenium intermediate.34,97,101 For FtmPT1 and NotF, the only reported residue that could act as a catalytic base in this step is the conserved Glu residue responsible for coordinating the indole N–H (Fig. 8C).34,101 A mutation of Glu102 to Gln in FtmPT1 revealed that catalysis required a negatively-charged side chain, not just H-bond accepting capabilities, making Glu102 a strong catalytic base candidate.34 Alternatively, the amide nitrogen in DKP or DKP-like rings could perform an intramolecular deprotonation at the indole C2. The hexahydropyrroloindole product generated by FtmPT1G115T demonstrated its capability to perform a nucleophilic attack, which made it a plausible catalytic base as well.34 At present, the proton at C2 of the arenium intermediate is thought be abstracted by Glu102.34 Reverse and regular C3 prenylations of indole DKPs have been shown to result in ring closure to form prenylated hexahydropyrroloindole ring systems, as evidenced by the products of CdpNPT and AnaPT (Fig. 13).48,97 This process was proposed to occur via a nucleophilic attack of the DKP ring amide nitrogen at C2 of the indole ring, followed by deprotonation by a water molecule, which would therefore exclude the need for a catalytic base.97 These mechanistic details are yet to be experimentally determined.
2.1.2.1. Active site architecture. Trp DMATSs share conserved characteristics with DKP DMATSs in their structures and the intermolecular interactions involved in substrate binding.29,61,64–66,68 As with DKP DMATSs, the details of polar and nonpolar contacts differ from among Trp DMATSs.66 The active site architecture observed in structurally characterized Trp DMATSs has been shown to differ from that of DKP DMATSs in three ways: (i) the steric confines of the prenyl acceptor binding pocket, (ii) the binding mode of the prenyl acceptor, and (iii) the extent of conformational changes of the active site upon substrate binding.
Trp DMATSs generally have smaller and more sterically constrained substrate binding pockets than DKP DMATSs (Fig. 15A), resulting in the respective difference in the substrate sizes. Whereas DKP DMATSs can accommodate relatively large substrates (i.e., DKPs, naphthalenes, and aszonalenins), Trp DMATSs primarily prenylate simple indole derivatives, although FgaPT2 and PriB are notable exceptions (Table 1).54,58,59,61–63,65,114–119 These two Trp DMATSs prenylated the indole moiety of the large antibiotic daptomycin, the structural basis of which has not yet been fully defined.65 The ability of PriB, FgaPT2, and CdpNPT (the only three DMATSs shown to modify daptomycin) to generate prenylated daptomycin analogues provides a convenient starting point for the discussion of the steric constraints of the binding pockets of Trp DMATSs relative to those of DKP DMATSs. Of the three PTs, PriB had the least percentage of substrate converted to product in the presence of daptomycin (11%), while FgaPT2 and CdpNPT appeared to be more efficient, with 65% and 75% conversion, respectively.65
 | ||
| Fig. 15 Differences in binding pocket architecture among Trp DMATSs. (A) Superimposition of the α3–β1 binding loops of FgaPT2 (light green) and FtmPT1 (light blue) showing the tighter binding pocket of FgaPT2. (B) The surface representation of the 6-DMATSMo binding pocket showing the residues lining the restrictive BM2-adopting binding pocket. (C) The surface representation of the FgaPT2 binding pocket showing the steric and polar interactions that make up the BM1-adopting binding pocket. (D) Superimposition of the residues corresponding to the FgaPT2 ADR and binding modes of L-Trp observed in FgaPT2 (green), 6-DMATSMo (brown), and CymD (light teal). | ||
As with other enzymes, access to and the size of the prenyl donor binding pocket are important determinants of the ability of a Trp DMATS to accept large substrates, controlled, as in DKP DMATSs, by the orientations of the α3–β1 and α6–β3 loops and the sizes of the residues lining the binding pocket.
Superimposition of structures of PriB and FgaPT2 showed that their α3–β1 loops adopt an inward orientation (Fig. 16A), restricting their substrate binding pockets, as opposed to the outward orientation observed in CdpNPT (Fig. 16A). The regions of the α3–β1 loop in contact with L-Trp in the two structures differ (Ile80, Leu81, Thr82, and Arg83 in FgaPT2 and Phe85, Leu86, Ser87, and Asp88 in PriB. Note: these residues are not shown in Fig. 16A). The residues of FgaPT2 were shown to be more flexible upon substrate binding than the corresponding residues of PriB.120 Furthermore, FgaPT2 appeared to adopt a similar conformation of its α6–β3 loop to that of CdpNPT. In contrast, the α6–β3 loop of PriB protruded further into the barrel (Fig. 16B). The α6–β3 loops in FgaPT2 and CdpNPT contain two residues that point towards the center of the barrel. In FgaPT2 these residues are Ile169 and Thr171, and in CdpNPT, they are Val201 and Thr203. PriB contains three residues whose side chains are directed towards the center of the barrel: Pro161, Leu162, and Trp165 (Fig. 16B). These residues serve to close the binding pocket on one side. The relationship between the daptomycin turnover rates of PriB and FgaPT2 as well as the structural analysis argue that the binding loop orientation and the size of the side chains in the substrate binding pocket influence the substrate scope of Trp DMATSs.
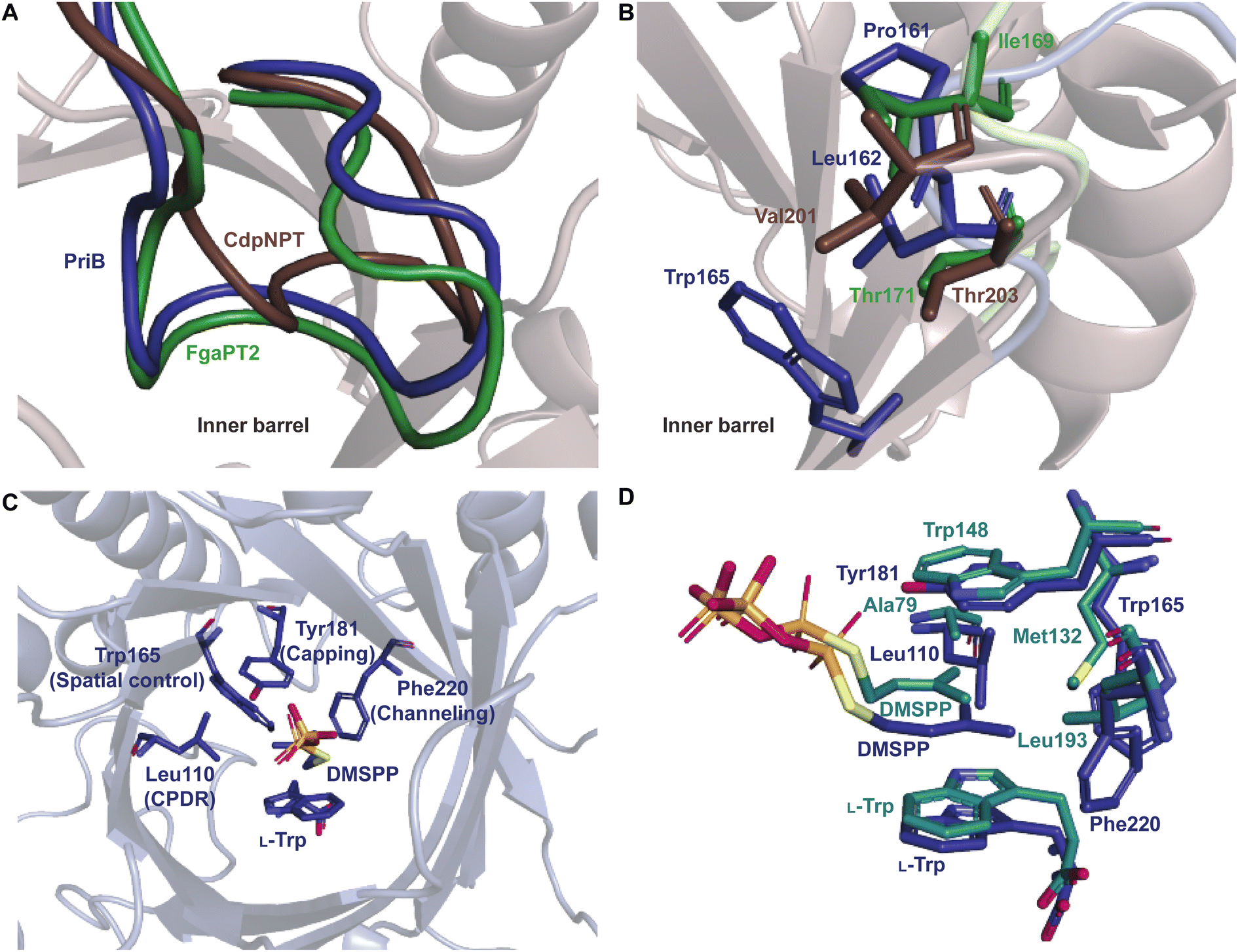 | ||
| Fig. 16 (A) Superimposition of the α3–β1 binding loop orientations observed in FgaPT2 (green), PriB (navy), and CdpNPT (brown). (B) Inward-oriented residues in the α6–β3 binding loop in FgaPT2 (green), PriB (navy), and CdpNPT (brown). (C) The residues of PriB implicated in regioselectivity control for DMATSs that utilize binding mode 2 (BM2). (D) Superimposition of the residues controlling regioselectivity of PriB (navy) and CymD (teal). | ||
The L-Trp was shown to be bound to PriB similarly to all structurally characterized Trp DMATSs, except FgaPT2.61,64–66,68 The indole ring bound to PriB is coplanar with the dimethylallyl moiety of DMSPP, but this plane is rotated by 90° relative to the L-Trp-DMSPP plane in FgaPT2 (Fig. 16B–D).61 The proximity of the prenylation site C6 of the indole ring to the primary carbon of the dimethylallyl moiety of the bound DMSPP (the C–C distance of 3.6 Å) and to the likely base His312 in PriB is in excellent agreement with the regular prenylation at the C6. The C7 is also at an appropriate distance from the primary carbon of the DMSSP, but it is much further from His312. Herein, we refer to the indole binding mode observed in FgaPT2 and the DKP DMATSs as binding mode 1 (BM1), and the binding mode observed in PriB and other Trp DMATSs as binding mode 2 (BM2). The ADRs of BM2-adopting Trp DMATSs do not appear to control the orientation of the indole ring (Fig. 15D). Nevertheless, these residues have been implicated in determining the regioselectivity of the enzyme. In all the structurally characterized C6-prenylating Trp DMATSs, the residue corresponding to the FgaPT2 ADR is a Leu, while in the N1-prenylating Trp DMATSs the analogous residue is an Ala (Fig. 15D). Instead of impacting the angle of the indole ring of L-Trp relative to the prenyl donor, the residue positioned in place of the ADR was shown to influence the proximity of the dimethylallyl group of the prenyl donor to the site of prenylation, thereby aiding in regioselectivity control.66 For a large side chain at this position (e.g., Leu), the prenyl donor binds close to the benzene part of the indole ring, while for small side chains (e.g., Ala) the prenyl donor is bound in proximity to the indole nitrogen.66 Because the residue corresponding to the ADR in BM2 DMATSs does not impact the angle of the indole, we herein refer to the residue at this position as the cosubstrate proximity determining residue (CPDR). The CPDR has been shown to be only a part of a network of residues implicated in directing regioselectivity. In addition to the CPDR, three to four other residues have been identified: (i) the capping group, (ii) the channeling residue, and (iii) the spatial control residue(s) (Table 4, Fig. 16C and D). Four to five residues form a channel that has been proposed to direct the prenyl donor to the site of prenylation. The capping group is an aromatic residue that stabilizes the dimethylallyl carbocation. The size of the capping group was thought to impact regioselectivity: large residues (e.g., Trp148 in CymD) would facilitate carbocation movement to the pyrrole ring of the indole. In turn, somewhat smaller residues (e.g., Tyr181 in PriB) were proposed to localize carbocation movement to the benzene ring of the indole.66 However, this regioselectivity control mechanism is dubious. For instance, the capping group of the reverse N1-prenylating Trp DMATS from the fungus Fusarium fujikoroi, DMATS1Ff, is Tyr189. As both CymD, whose capping group is a Trp, and DMATS1Ff, whose capping group is a Tyr, catalyze the same reaction, the size of the capping group does not appear to be a definitive factor in regioselectivity control. The channeling residue exists on the side opposite the CPDRs and sterically guides the carbocation towards the prenylation site. The spatial control residue(s) form favorable interactions with the geminal methyl groups of the prenyl donor, and hold the prenyl donor near L-Trp.66 The identities of these residues vary slightly from enzyme to enzyme, controlling regioselectivity by positioning the prenyl donor in an optimal orientation for nucleophilic attack by the prenyl acceptor substrate. This was demonstrated in the structure of CymD in a complex with L-Trp and DMSPP, where the indole nitrogen was oriented at an angle of 104° relative to the plane of the tertiary carbon of DMSPP (Fig. 17A). This orientation was close to the optimal angle of 107° for a nucleophilic attack.66,121 For both DMATS1Ff and CymD, the N1 of the indole group was at an appropriate distance from the tertiary carbon of DMSPP (3.3 Å in DMATS1Ff and 3.4 Å in CymD) and the catalytic Glu (the N–Oε distance of 2.8 Å in both enzymes) for reverse prenylation at the N1. The structures of both enzymes also contained a water molecule at 3.3 Å from the N1, which could, in principle, also serve as a catalytic base.
| Enzyme | Capping residue | CPDR | Channeling residue | Spatial control residue | Citation |
|---|---|---|---|---|---|
| 5-DMATSSc | Tyr151 | Leu81 | Phe190 | Trp135 | 61 |
| PriB | Tyr181 | Leu110 | Phe220 | Trp165 | 65 |
| 6-DMATSMo | Tyr148 | Leu78 | Phe197 | Trp132 | 61 |
| IptA | Tyr170 | Leu100 | Phe209 | Trp154 | 64 |
| CymD | Trp148 | Ala79 | Leu193 | Met132 | 66 |
| DMATS1Ff | Tyr189 | Ala107 | Met239 | Phe174 | 68 |
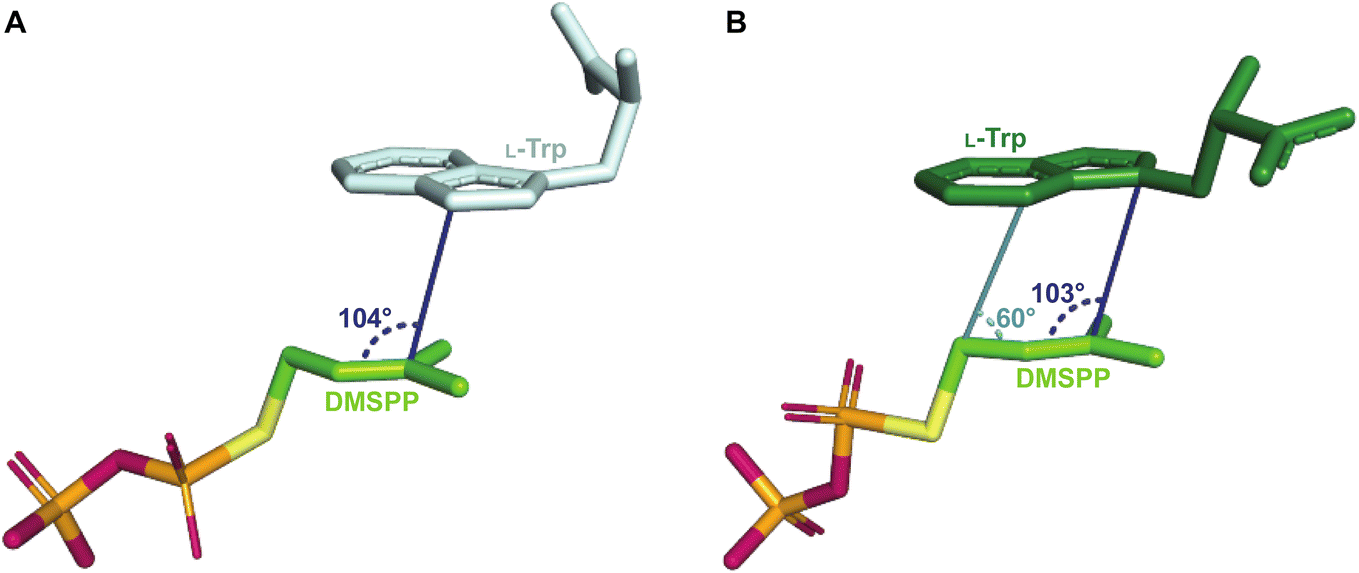 | ||
| Fig. 17 (A) The disposition of DMSPP and L-Trp in ternary complexes with Trp DMATS. (A) The DMSPP and L-Trp in the ternary complex with CymD. The orientation of the tertiary carbon of DMSPP relative to the site of prenylation on the indole ring (N1) of L-Trp is close to the optimal angle of attack. (B) The DMSPP and L-Trp in the ternary complex with FgaPT2. The angle in light blue indicates the orientation of the putative reactive carbon of the prenyl moiety in the regular prenylation of the indole C4 relative to the site of prenylation. The angle in navy shows the orientation of the tertiary carbon of the prenyl moiety relative to the hypothesized prenylation site in the proposed Cope rearrangement mechanism. | ||
The binding mode was also shown to be important for catalytic activity with unnatural prenyl acceptor substrates. Screening of Trp derivatives for prenylation by IptA demonstrated that a racemic mixture of 5-methyl-Trp was prenylated with a relative activity nearly twice that of L-Trp.64 Substrates containing electron donating substituents were shown to increase kcat values of DMATSs, but the activity measurements on Trp derivatives containing hydroxyl and methoxy substituents were not able to replicate the levels of conversion observed with the 5-methyl group.64,67,122 Additionally, a racemic mixture of 6-methyl-Trp showed a drastic reduction in turnover. The structures of the respective complexes showed that 5-methyl-L-Trp adopted a binding mode consistent with regular prenylation at the C6 position of the indole, in which the C6 was 0.3 Å closer to the C1 of the prenyl donor than that of L-Trp. In this binding mode the indole N–H of 5-methyl-L-Trp was positioned by 0.4 Å closer to a conserved Glu (Glu84) than that of L-Trp (Fig. 18B). The increased proximity of the C6-prenylation site and of the N-H of 5-methyl-L-Trp to the prenyl donor and Glu84, respectively, compared to those of L-Trp, was proposed to facilitate more efficient catalysis of 5-methyl-L-Trp compared to L-Trp.64 In contrast, 6-methyl-L-Trp was bound with the indole N–H positioned 0.3 Å further from Glu84 than that in bound L-Trp (Fig. 18A), which could explain the decrease in turnover rate (Fig. 18C).64 This orientation of 6-methyl-L-Trp also showed that the indole C7 was positioned closer to Glu84.64 In this state, Glu84 was proposed to act as the catalytic base, which was consistent with the observed C7-prenylation of 6-methyl-Trp by IptA. This structural explanation was further supported by the similarities of the observed binding mode of 6-methyl-L-Trp to the proposed binding of L-Trp to 7-DMATSNeo.64,101 Taken together, these observations demonstrate how relatively small differences in binding can have large impacts on both the kinetic parameters and the regioselectivity of DMATSs. This large effect of small positional changes calls into question the use of unnatural substrates to elucidate the chemical mechanism(s) of DMATSs. This structural analysis can inform future engineering strategies. If efficient methods of increasing proximity of a substrate to either the cosubstrate or Glu84 could be established, the enzyme kinetics would be possible to control.
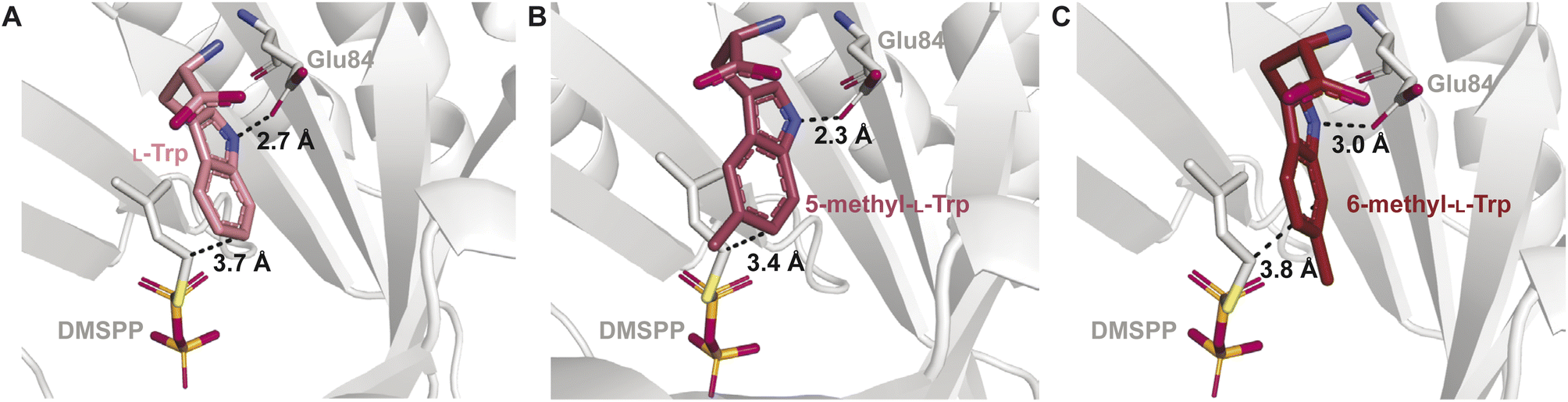 | ||
| Fig. 18 The effect of indole modifications on the indole binding mode in the binding pocket of IptA for (A) L-Trp, (B) 5-methyl-L-Trp, and (C) 6-methyl-L-Trp. Distances from the prenylation site on the indole ring to the DMSPP carbon corresponding to dimethylallyl carbocation formation and Glu84 are shown by dashed lines. | ||
The extent of conformational remodeling of the substrate binding pocket has been shown to be different in Trp DMATSs than in DKP DMATSs. While the apo and complexed states of FgaPT2, 5-DMATSSc, and DMATS1Ff have not been described as open or closed states, as they were in AtaPT, these three Trp DMATSs were reported as having notable degrees of conformational change upon binding of L-Trp.29,61,68 A relationship between significant conformational remodeling and an ordered sequential kinetic mechanism appears to exist for FgaPT2, as it does for AtaPT. Evidence for this association came from an FgaPT2 homolog from Claviceps sp. that was found to undergo catalysis via an ordered sequential mechanism.100 NphB was also found to be prenylated in an ordered sequential manner,123,124 which raised a possibility that an ordered sequential mechanism was conserved within the ABBA-PT superfamily. Additional enzymes need to be studied mechanistically to test this possibility. For instance, CymD did not display any significant conformational changes upon binding L-Trp. Isothermal titration calorimetry (ITC) experiments on CymD suggested a synergistic effect between the prenyl acceptor and donor,66 but whether they must bind in a strict order or not, is yet to be ascertained.
2.1.2.2. Mechanism. The catalytic mechanism has been most extensively studied for FgaPT2 among Trp DMATSs. As with FtmPT1, this enzyme was in the focus of the debate trying to establish a direct or a rearrangement-based prenylation mechanism (Fig. 7). Both mechanisms have been supported by experimental data.61,73,77,84 Despite this evidence, arguments based on the reverse prenylation at the C3 by the FgaPT2 mutant Lys174Ala have been made for a rearrangement-based mechanism,84 which have not been conclusively discounted.29,73,84 In addition to these arguments, the orientation of the dimethylallyl group relative to L-Trp in the binding pocket also favors the rearrangement mechanism. The C3 of the indole ring is located at an angle of 103° relative to the DMSPP plane, an orientation close to the Bürgi–Dunitz angle (107°) that is more consistent with a nucleophilic attack on C3 (Fig. 17B). Furthermore, the distance from the C3 of the indole to the C3 of the dimethylallyl moiety is 3.5 Å, somewhat shorter than the distance from the C4 of the indole to the C1 of the dimethylallyl (3.8 Å), favoring an initial reverse prenylation at the intrinsically more reactive C3 followed by a Cope rearrangement to the regularly installed prenyl at the C4. While these observations support the existence of a rearrangement-based prenylation mechanism, direct experimental evidence using the WT enzyme with L-Trp as a substrate is still lacking.
Mechanisms of other Trp DMATS have also been investigated. The reverse N1-prenylating Trp DMATSs CymD and DMATS1Ff have also been subjects of debate regarding direct or rearrangement-based mechanisms.66,67,125 The idea that the CymD reaction proceeded via a direct prenylation mechanism was questioned, as the lone pair of the indole nitrogen exists in a p-orbital and contributes to the aromaticity of the ring system, making it unlikely to act as a nucleophile during the incorporation of an alkyl group.125 Therefore, an initial regular C3 prenylation followed by an aza-Cope rearrangement to yield the reverse N1-prenylated product was considered. While such a mechanism was not ruled out completely, the observation that the cleavage of the indole N–H bond was rate-limiting pointed to a mechanism resembling direct prenylation at N1. Deprotonation of the heteroatom could increase the nucleophilicity of the N1 position, and, with the methylene carbon of DMSPP being ∼5 Å from the C3 position of the indole, a direct prenylation mechanism for the reaction catalyzed by CymD appeared highly plausible.66 A similar conclusion was reached for DMATS1Ff upon its biochemical and structural characterizations, suggesting that reverse N1 prenylation occurred via a direct mechanism in both bacteria and fungi.66–68
Another mechanistic puzzle described recently was the reaction catalyzed by 5-DMATSSc.61 Even though the active site of this enzyme was highly homologous to that of 6-DMATSMo, the reason for the regioselectivity at C5 and not C6 was not obvious from the structural comparison. L-Trp is bound to both PTs nearly identically, with the only differences being Gln255 and Tyr326 in 5-DMATSSc and Val259 and His329 (the catalytic base) in 6-DMATSMo, respectively. Mutagenesis of Gln255 and Tyr326 of 5-DMATSSc (the best candidates for a catalytic base) showed that both residues were important, but not essential, for catalysis. A logical explanation for this observation and for the regioselectivity control of 5-DMATSSc is that an active site water coordinated by Gln255 serves a catalytic function and aids in the re-establishment of the aromaticity of the arenium intermediate. Because the tertiary carbon of DMSPP bound to 5-DMATSSc is closer to the C7 (a C–C distance of 3.8 Å) and C6 (4.1 Å) of the indole ring than to the C5 (4.3 Å), and the primary carbon of DMSPP is relatively far from the C5 (5.2 Å), it is likely that a reverse prenylation occurs at one of these alternative positions, followed by one or more rearrangement steps to yield the regularly C5-prenylated product.61 In 6-DMATSMo, the regular 6-prenylation of the indole moiety was consistent with the proximity of the C6 atom of the indole to the primary carbon of DMSPP and to the imidazole moiety of His329.61
 | ||
| Fig. 19 Target residues for substrate scope expansion (orange) and specificity alteration (pink). (A) The FgaPT2 binding pocket. (B) The NotF binding pocket. (C) The FtmPT1 binding pocket. | ||
 | ||
| Fig. 20 Target residues for regioselectivity switching (pale green). (A) The FtmPT1 binding pocket. (B) The 5-DMATSSc binding pocket. (C) The 6-DMATSMo binding pocket. | ||
| Enzyme | Variant(s) | Compound(s) | Product score | Activity score | Overall weighted score | Citation(s) |
|---|---|---|---|---|---|---|
| a Calculated using reported reaction rate values. b N/C = not calculated due to the lack of required quantitative data. c Calculated using reported WT kinetic parameters.56 | ||||||
| FtmPT1 | WT | 1-Naphthol | 0.06 | 1.0 | 0.81 | 126 |
| 1,6-Dihydroxynaphthalene | 0.02 | 1.0 | 0.80 | |||
| 1,7-Dihydroxynaphthalene | 0.06 | 1.0 | 0.81 | |||
| 2,7-Dihydroxynaphthalene | 0.06 | 1.0 | 0.81 | |||
| 1-Amino-7-hydroxynaphthalene | 0.13 | 1.0 | 0.83 | |||
| Y205M | 1-Naphthol | 0.25 | 12.05 | 9.70 | ||
| 1,6-Dihydroxynaphthalene | 0.09 | 4.82 | 3.90 | |||
| Y205L | 1,7-Dihydroxynaphthalene | 0.13 | 5.80 | 4.70 | ||
| 2,7-Dihydroxynaphthalene | 0.06 | 1.24 | 1.0 | |||
| Y205F | 1-Amino-7-hydroxynaphthalene | 0.16 | 19.1 | 15.31 | ||
| NotFa | WT | Brevianamide F | 1.0 | 1.0 | 1.0 | 41 |
| cyclo-L-Trp-L-phenylglycine | 0.21 | 1.0 | 0.84 | |||
| cyclo-L-Trp-L-homophenylalanine | 0.15 | 1.0 | 0.83 | |||
| cyclo-L-Trp-L-Trp | 0.11 | 1.0 | 0.82 | |||
| cyclo-L-Trp-L-cyclo-methylTrp | 0.55 | 1.0 | 0.91 | |||
| Y266A | Brevianamide F | 0.11 | 0.05 | 0.06 | ||
| cyclo-L-Trp-L-phenylglycine | 0.22 | 0.85 | 0.72 | |||
| cyclo-L-Trp-L-homophenylalanine | 0.22 | 1.10 | 0.92 | |||
| cyclo-L-Trp-L-Trp | 0.13 | 1.53 | 1.25 | |||
| cyclo-L-Trp-L-cyclo-methylTrp | 0.38 | 1.10 | 0.96 | |||
| Y266V | Brevianamide F | 0.05 | 0.03 | 0.03 | ||
| cyclo-L-Trp-L-phenylglycine | 0.12 | 0.40 | 0.34 | |||
| cyclo-L-Trp-L-homophenylalanine | 0.15 | 0.80 | 0.70 | |||
| cyclo-L-Trp-L-Trp | 0.08 | 1.70 | 1.40 | |||
| cyclo-L-Trp-L-cyclo-methylTrp | 0.32 | 1.30 | 1.10 | |||
| L193S | Brevianamide F | 0.20 | 0.10 | 0.12 | ||
| cyclo-L-Trp-L-phenylglycine | 0.18 | 0.73 | 0.62 | |||
| cyclo-L-Trp-L-homophenylalanine | 0.26 | 1.11 | 0.93 | |||
| cyclo-L-Trp-L-Trp | 0.13 | 1.60 | 1.31 | |||
| cyclo-L-Trp-L-cyclo-methylTrp | 0.38 | 0.90 | 0.80 | |||
| P352A | Brevianamide F | 0.03 | 0.01 | 0.01 | ||
| cyclo-L-Trp-L-phenylglycine | 0.05 | 0.15 | 0.13 | |||
| cyclo-L-Trp-L-homophenylalanine | 0.19 | 0.73 | 0.62 | |||
| cyclo-L-Trp-L-Trp | 0.03 | 0.41 | 0.33 | |||
| cyclo-L-Trp-L-cyclo-methylTrp | 0.35 | 1.03 | 0.90 | |||
| FgaPT2 | WT | L-Trp | 1.0 | 1.0 | 1.0 | 55–57 |
| L-Tyr | 0.18 | 1.0 | 0.83 | |||
| cyclo-L-Trp-L-Tyr | 0.21 | 1.0 | 0.84 | |||
| Brevianamide F | 0.15 | 1.0 | 0.83 | |||
| cyclo-L-Trp-D-Pro | N/Cb | 1.0 | N/C | |||
| cyclo-L-Trp-L-Leu | N/C | 1.0 | N/C | |||
| cyclo-L-Trp-Gly | 0.15 | 1.0 | 0.83 | |||
| cyclo-L-Trp-L-Trp | 0.21 | 1.0 | 0.84 | |||
| cyclo-L-Trp-L-Phe | 0.07 | 1.0 | 0.81 | |||
| cyclo-L-Trp-L-Ala | 0.10 | 1.0 | 0.82 | |||
| K174F | L-Trp | 0.002 | N/C | N/C | ||
| L-Tyr | 0.50 | 4.9 | 4.0 | |||
| R244L | L-Trp | N/C | N/C | N/C | ||
| cyclo-L-Trp-L-Tyr | 0.28 | 20 | N/C | |||
| Brevianamide F | 0.33 | 147 | 117.7 | |||
| cyclo-L-Trp-D-Pro | 0.50 | N/C | N/C | |||
| cyclo-L-Trp-L-Leu | N/C | N/C | N/C | |||
| cyclo-L-Trp-Gly | N/C | 27 | N/C | |||
| cyclo-L-Trp-L-Trp | N/C | N/C | N/C | |||
| cyclo-L-Trp-L-Phe | N/C | N/C | N/C | |||
| K174F, R244N | cyclo-L-Trp-L-Ala | 0.20 | N/C | N/C | ||
| cyclo-L-Trp-L-Trp | 0.08 | 25.0c | 20.0 | |||
| cyclo-L-Trp-Gly | 0.37 | 25.5c | 20.5 | |||
| cyclo-L-Trp-L-Phe | 0.37 | N/C | N/C | |||
| Brevianamide F | N/C | N/C | N/C | |||
| cyclo-L-Trp-L-Tyr | N/C | N/C | N/C | |||
| K174F, R244L | cyclo-L-Trp-L-Ala | N/C | N/C | N/C | ||
| cyclo-L-Trp-L-Trp | N/C | N/C | N/C | |||
| cyclo-L-Trp-Gly | N/C | N/C | N/C | |||
| cyclo-L-Trp-L-Phe | N/C | N/C | N/C | |||
| Brevianamide F | 0.36 | 174.0 | 139.3 | |||
| cyclo-L-Trp-L-Tyr | 0.41 | 84.0 | 67.3 | |||
| Enzyme | Variant(s) | Compound | Product score | Activity score | Overall weighted score | Citation(s) |
|---|---|---|---|---|---|---|
| a The regioselectivity of each variant can be found in Table 1. b Calculated using relative activity data. | ||||||
| FtmPT1 | WT | Brevianamide F | 0.96 | 1.0 | 0.8 | 34 and 126 |
| G115Tb | 0.95 | 0.17 | 0.33 | |||
| Y205N | −0.17 | 0.08 | 0.03 | |||
| Y205L | −0.26 | 0.14 | 0.06 | |||
| 5-DMATSSc | WT | L-Trp | 0.74 | 1.0 | 1.74 | 61 |
| Q255V, Y326H | −0.17 | 0.41 | 0.29 | |||
| Q255N, Y326H | −0.18 | 0.59 | 0.44 | |||
| 6-DMATSMo | WT | L-Trp | 0.86 | 1.0 | 0.97 | 61 |
| L78A | 0.24 | 1.24 | 1.04 | |||
| V259Q, H329Y | 0.77 | 0.87 | 0.85 | |||
Rational binding pocket engineering requires knowledge of key residues involved in catalysis and substrate binding. This information is obtained from structure–function studies of the enzyme of interest or a homologous enzyme. Residues in the active site can be either essential or nonessential to catalysis. Based on observations from the literature, we define essential residues as those associated with a significant (>90%) loss of catalytic activity when substituted with other amino acid residues, such as the conserved Glu residue or the catalytic base. These residues often serve as a catalytic acid or base or make key contacts with the bound substrate. Nonessential residues (NERs), in contrast, are associated with a reasonable retention of activity (∼15% or greater) when substituted with another amino acid residue (e.g., Gly115 in FtmPT1 and Arg244 in FgaPT2). As expected, nearly all binding pocket engineering attempts described have involved NER mutagenesis (Fig. 19 and 20).34,36,56,57,126 One of the main inherent problems in substrate binding pocket engineering to bind unnatural substrate is that the position and the orientation of the unnatural substrate can significantly differ from those of the natural substrate. One strategy to deal with this problem is to perform in silico docking with an unnatural substrate, by using AUTODOCK127 and GLIDE128 programs. A combination of computational and experimental approaches can be useful for planning enzyme engineering studies.41,57
Engineering to alter the substrate scope of an enzyme can be accomplished either by altering (usually enlarging) the substrate binding pocket to accept different scaffolds, or by redesigning the binding pocket specifically for one compound. To do either, past results have shown that the most successful attempts for DMATSs achieve both the complementarity of the enzyme–substrate interface and a set of other favorable enzyme–substrate interactions. Steric factors have been established as important in these engineering criteria, as they control the amount of space available within the binding pocket to accommodate substrates while excluding solvent. Exchanging bulkier NERs for smaller ones has been a central tenet in expanding substrate scope. A downside of this strategy is that expanding the binding pocket space can open the access to the active site for solvent, with deleterious effects on activity and a loss of spatial control, while constricting the pocket risks making it too small to bind the substrate. Therefore, an engineered binding pocket needs to be optimized to the size of the desired substrate(s).
An illustration of these principles was described in studies of FgaPT2 by the Li group in 2015.57 FgaPT2 was shown to prenylate both L-Trp and L-Tyr, but with a 5-fold lower activity for L-Tyr than for L-Trp (Table 5). Mutagenesis of identified NERs to accelerate catalysis with L-Tyr demonstrated that expanding the binding pocket (i.e., substitution of Thr102 by a Gly), drastically decreased enzymatic activity. Significantly constricting the binding pocket by substituting Lys174 by a Trp resulted in similar loss of activity. Ultimately, the replacement of Lys174 by a Phe, a residue smaller than Trp and available for π–π interactions with L-Tyr resulted in increased activity towards L-Tyr compared to the WT enzyme and switched the substrate preference to L-Tyr. The design process which culminated in the FgaPT2K174F construct was an excellent case study for the application of the principle of balancing steric and other interactions. This study also underscored the complexity and a trial–error nature of the enzyme engineering efforts. Studies of NotF yielded decreased turnover rates for indole DKP substrates containing unsaturated or aromatic moieties (Table 5). This observation was proposed to be a result of inhibited product dissociation due to noncovalent π–π interactions with aromatic residues in the binding pocket.41 Mutations of a residue engaged in π–π stacking with substrate (e.g., Tyr266) to aliphatic residues resulted in increased turnover rates for large sp2-hybridized substrates, in agreement with this hypothesis.
While the altering of substrate scope has been successful in some studies, alteration of regioselectivity has proven to be extremely challenging. Several attempts have been made to accomplish this goal (Table 6). Engineering methods for reprogramming regioselectivity of DMATSs have used two main strategies: (i) mutating NERs and (ii) changing locations of catalytic residues. Mutations of NERs are meant to change the position of the bound substrate relative to the catalytic residues for prenylation at a different position. Therefore, this method is generally applicable and dependent on the availability of structural and biochemical data on NERs. This method was utilized in the in the generation of FtmPT1G115T, FtmPT1Y205N, FtmPT1Y205L, and 6-DMATSMoL78A.34,61,126 These studies yielded enzyme variants that had reduced catalytic activities and generated a significant fraction of side products.34,61 Method (ii) focuses on changing positions of the main catalytic residues to those of another DMATS that has the desired regioselectivity. This method was used to generate 5-DMATSScQ255V,Y326H, 5-DMATSScQ255N,Y326H, and 6-DMATSMoV259Q,H329Y.61
This approach has been used with success, particularly in 6-DMATSMo.61 However, in contrast to method (i), it is a less universal approach to accomplish regioselectivity switching. Moving main catalytic residues to another position relies both on the mechanistic information and on significant homology between the enzyme of interest and the characterized enzyme with the desired regioselectivity. For instance, the template enzyme used for the switching of 6-DMATSMo from a C6- to a C5-prenylating enzyme was 5-DMATSSc. The two enzymes were highly homologous, and the L-Trp substrate was bound to them in an almost identical manner.61 On the other hand, attempts to use enzymes such as FgaPT2 to prenylated at C6 would require extensive active site remodeling to create a similar binding mode to 6-DMATSMo or PriB, which would have a low likelihood of success. Similar attempts to switch regioselectivity were performed on 5-DMATSSc, but with little success. As the mechanistic knowledge of 5-DMATSSc is still incomplete, it may have been difficult to determine which residues to use to generate the mutants.61 However, as with method (i), method (ii) has only been used a few times. Therefore, it is possible that this approach will find more applications as mechanistic information on DMATSs accumulates.
2.2. The prenyl donor binding pocket
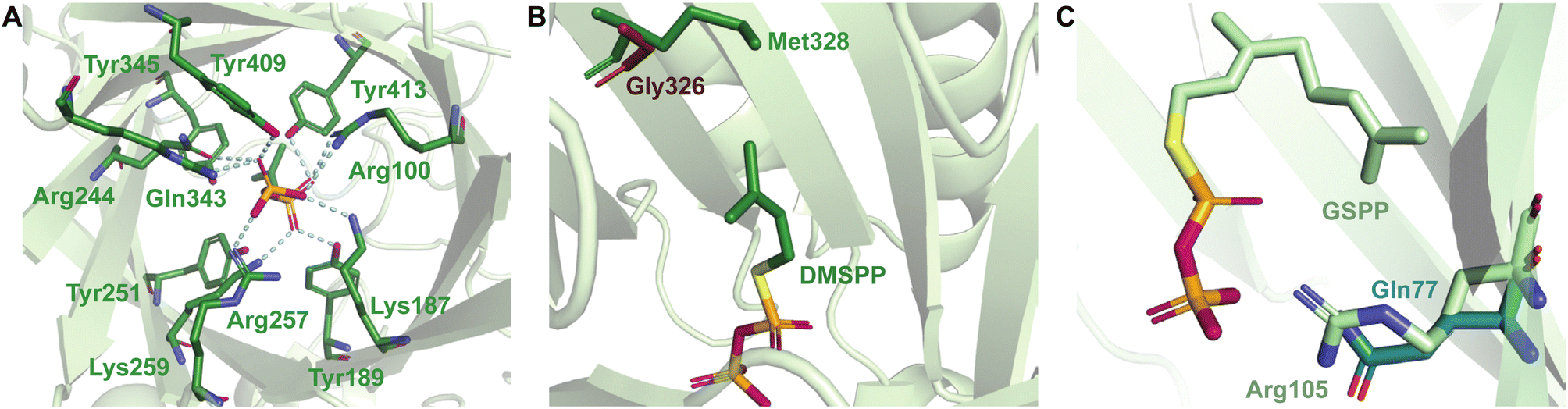 | ||
| Fig. 21 Key structural features of the prenyl donor binding pocket. (A) The diphosphate binding site of FgaPT2. Salt bridges and H-bonds are denoted by blue dashed lines. (B) Superimposition of the gatekeeping residues of FgaPT2 (green) and AtaPT (brick red). (C) The diphosphate binding site of DMATS1Ff in complex with GSPP (light green) showing the Arg105 (light green) conformation reported to allow GPP binding and the more rigid Gln77 of CymD (green) thought to prohibit GPP binding. | ||
The structural determinants for the cosubstrate scope include primarily residues collectively known as gatekeeping residues.35,60 These residues act as steric limiters of the cosubstrate size and prevent binding of prenyl diphosphates with carbon chains longer than a specific length. The gatekeeping residues of DKP DMATSs and the Trp DMATS FgaPT2 have been more extensively characterized than those of other Trp DMATSs. In DKP DMATSs and FgaPT2, the gatekeeping residue consists of one amino acid side chain that protrudes from β7 into the substrate binding pocket, where it applies steric pressure to the prenyl moiety of the cosubstrate (Fig. 21B). The most restrictive gatekeeping residue was shown to be a Met, present in FgaPT2, FtmPT1, and CdpNPT.60 The Met gatekeeping residue typically restricts the preferred cosubstrate to DMAPP. Despite this, DMATSs with this gatekeeping residue have demonstrated the ability to incorporate several unnatural prenyl and alkyl donors (Table 1).35,43,54,60 The most permissive characterized gatekeeping residue was shown to be a Gly, for which prenyl donors with side chains up to 15 carbons (FPP) in AtaPT were accepted.25,35 This enzyme with the Gly gatekeeper was tested with DMAPP, GPP, and FPP.25,35,60 Screening of unnatural prenyl and alkyl donor libraries would provide further valuable information on the limits of cosubstrate promiscuity of Gly as a gatekeeping residue.
The gatekeeping residues of Trp DMATSs utilizing BM2 have not been sufficiently described. The existing information suggests that multiple structural factors exist that determine the cosubstrate scope. Even these multiple factors do not fully explain the observed cosubstrate scopes of these enzymes. Proposed candidates for the gatekeeping residue in 5-DMATSSc and 6-DMATSMo were their respective CPDRs, Leu81 and Leu78.61 Another candidate, Trp154 in IptA, was a spatial control residue.64 While these residues were shown to have an impact on cosubstrate scope, they were insufficient to explain the cosubstrate scope of PriB, which also contains Leu (Leu110) and Trp (Trp165) at the same positions. IptA, 5-DMATSSc, and 6-DMATSMo preferred DMAPP as a cosubstrate.61,63,64,115,122 Of the three, only 6-DMATSMo accepted GPP as a cosubstrate, but with a low turnover rate.64 In contrast, PriB was reported to incorporate GPP with a turnover rate near 100%, demonstrating that the CPDR was not the only structural factor determining the cosubstrate scope, if at all.65,66 Structural studies comparing the active sites of DMATS1Ff and CymD provided insight into this problem. Like PriB and 6-DMATSMo, DMATS1Ff accepted GPP as a cosubstrate, while CymD did not. This difference in the cosubstrate scopes was thought to be caused by Arg105, which resulted in the presence of a cavity in the active site of DMATS1Ff. The corresponding residue in CymD, Gln77, was unable to generate a similar cavity due to its rigidity (Fig. 21C).68 Comparison of the structures of PriB, 6-DMATSMo, and DMATS1Ff revealed that this Arg residue is conserved in all three enzymes, making it an intriguing structural aspect to investigate in the future. Notably, this conserved Arg residue is also present in 5-DMATSSc, even though this enzyme does not accept GPP. As the final deprotonation step in the catalytic cycle of 5-DMATSSc is thought to require an ordered water as a catalytic base, it is possible that a large cosubstrate like GPP could displace the water and abolish activity. Because IptA, which does not generate detectable products in the presence of GPP, also contains the conserved Arg and does not utilize an ordered water in catalysis, the above explanation is unlikely.64 An alternative explanation involves the size of the active site. For instance, DMATS1Ff has a larger active site than CymD by 27 Å3, which could aid in accepting larger cosubstrates.68 The active site sizes of other Trp DMATSs remain as yet unreported, making this explanation inconclusive at present. To summarize, the active site volume as a factor in cosubstrate scope determination of Trp DMATSs could prove to be an interesting subject for future computational studies.
Unfortunately, the breadth of information on the structural basis of cosubstrate scope determination does not extend to the differences in activity observed between GPP-accepting DMATSs. DMATS1Ff, 6-DMATSMo, and PriB use GPP with turnover rates of 2.4%, 10%, and 97%, respectively.61,65,68 At present, there is no explanation for these drastic differences in activity. Potential answers may lie in the identity and orientation of the residues stabilizing the prenyl carbocation. Further studies are needed to uncover the structural and chemical bases of these differences.
| Enzyme | Construct(s) | Donor cosubstrateb | Product score | Activity score | Overall weighted score | Citation |
|---|---|---|---|---|---|---|
| a Indicates calculations were made using relative activity data. b Abbreviations: DMAPP = dimethylallyl diphosphate; GPP = geranyl diphosphate. c N/C = not calculated due to the lack of required quantitative data. | ||||||
| FtmPT1a | WT | DMAPP | 0.87 | 1.0 | 0.97 | 35 |
| GPP | 0 | 0 | 0 | |||
| M364G | GPP | 0.34 | 54.8 | 43.9 | ||
| CdpNPTa | WT | DMAPP | 0.52 | 1.0 | 0.90 | |
| GPP | 0 | 0 | 0 | |||
| M349G | GPP | 0.13 | 23.5 | 18.83 | ||
| 5-DMATSSc | WT | DMAPP | 0.82 | 1.0 | 0.96 | 61 |
| GPP | 0 | 0 | 0 | |||
| L81A | GPP | 0.28 | 0.052 | 0.09 | ||
| 6-DMATSMo | WT | DMAPP | 0.88 | 1.0 | 0.98 | 61 |
| GPP | 0.10 | 1.0 | 0.82 | |||
| L78A | GPP | 1.0 | 10.0 | 8.2 | ||
| FgaPT2 | WT | DMAPP | 1.0 | 1.0 | 1.0 | 60 |
| GPP | 0.04 | 1.0 | 0.81 | |||
| M328C | GPP | N/Cc | 229 | N/C | ||
| M328A | GPP | N/C | 250 | N/C | ||
| M328T | GPP | N/C | 181 | N/C | ||
| M328S | GPP | N/C | 317 | N/C | ||
| M328G | GPP | N/C | 267 | N/C | ||
| M328V | GPP | N/C | 73 | N/C | ||
| M328N | GPP | N/C | 47 | N/C | ||
The results of the studies summarized in Table 7 point to three properties that should be noted when altering the gatekeeping residue: (i) steric effects, (ii) polarity, and (iii) polarizability. Removal of steric pressure of larger gatekeeping residues (e.g., Met) by mutations to smaller side chains (e.g., Gly, Ala) allowed the active sites of engineered enzymes to accept larger cosubstrate like GPP and FPP.60 The decreased steric pressure was also associated with a loss of the spatial control of the dimethylallyl moiety.60 These effects should be considered when attempting to engineer DMATSs with high specificity for smaller cosubstrates such as DMAPP and 1–21 (Fig. 4B). Engineering of more robust DMATSs with the ability to incorporate prenyl diphosphates of varying sizes was accomplished via the utilization of polar side chains.60 FgaPT2 constructs FgaPT2M328V and FgaPT2M328T mutants retained high turnover rates with DMAPP as a cosubstrate (∼80%), and FgaPT2M328T converted GPP at a higher rate (∼44%) than FgaPT2M328V did (∼26%), demonstrating that polarity was an important factor. Remarkably, FgaPT2M328C demonstrated comparable turnover rates for both DMAPP and GPP, with conversion yields of approximately 40 and 50%, respectively. This result was attributed to the properties of the thiol group of Cys328, which compensated for the lack of hydrophobic contacts due to its larger atomic radius and more polarizable electron shell than the alkyl and hydroxyl side chains of Val328 and Thr328, respectively.60
To the best of our knowledge, gatekeeping residue engineering has been utilized only to achieve the use of large cosubstrates, like GPP and FPP. More studies are needed to understand engineering strategies for cosubstrate scope expansion. Specifically, future research could focus on the engineering strategies for the incorporation of unnatural prenyl and alkyl donor cosubstrates. Recently developed libraries of prenyl and alkyl diphosphates contain compounds with diverse steric and electronic properties.43,65 It would be invaluable to determine how to engineer the prenyl donor binding pockets of DMATSs for the use of these molecules as cosubstrates.
2.3. Oligomerization of DMATSs
Several DMATS structures were reported as homodimers based on their crystal structures (Fig. 22A).68 Dimerization appears to be limited primarily to fungal DMATSs. Structural and biochemical studies of these enzymes have revealed that protomer–protomer interfaces contain conserved protein–protein interactions.68 In almost all cases, the dimer interface consists of a four-helix bundle made up of the first ααββ motif near the N-terminus of fungal DMATSs. In this interface, a conserved Tyr residue has been proposed to be a key mediator for dimerization (Fig. 22B). The existence of this residue supports earlier findings that suggested that the N-terminal domains of fungal DMATSs were required for dimerization.68,129 This Tyr residue corresponds to Tyr53 in DMATS1Ff, Tyr52 in FgaPT2, Tyr59 in FtmPT1, Tyr80 in CdpNPT, Tyr73 in AnaPT, and Tyr53 in AtaPT.25,68 The Tyr in the protomer–protomer interface of each dimer is proposed to engage in stabilizing homodimer π–π interactions with its counterpart of the adjacent protomer.68 Notably, the dimerization interface in AtaPT is different from that of other fungal DMATSs, so that Tyr53 is not a part of this interface. Instead, the dimer interface in AtaPT consists primarily of polar residues (Fig. 22C). AtaPT was proposed to exist as a homotetramer (a dimer of dimers) stabilized by salt bridges in the interface between the two dimers (Fig. 22C and D).25,130 At present, it is unknown what biological role oligomerization plays in fungal DMATSs. Activity was maintained when these enzymes were purified as monomers, and the residues in the dimer interfaces did not appear to impact substrate binding. However, it has been speculated that dimerization/oligomerization could play some allosteric role in catalysis.25 | ||
| Fig. 22 Dimerization of DMATSs. (A) The dimer of DMATS1Ff. (B) A part of the dimeric interface of DMATS1Ff showing the conserved Tyr residue. (C) The tetramer of AtaPT. (D) The dimer interface of AtaPT showing the predominately a polar nature of the interface. | ||
3. Conclusions
In this review, we have described the current state of our understanding of the structure of DMATSs and its relationship to the catalytic mechanism, the substrate and cosubstrate scopes, and the regioselectivity of these enzymes. In addition, we have outlined the principles and key steps involved in engineering of the active sites of DMATSs to alter their functional properties. Furthermore, we have also identified current gaps in knowledge in this field.The amount of structural information on DMATSs has grown significantly since the first crystal structure of FgaPT2 was published in 2009.65 As a result, a solid foundation has been laid for the use of DMATSs in chemoenzymatic synthesis and industrial biotechnology. However, many unknowns remain to be addressed before this can occur. These include, but are not limited to: (i) the elucidation of exact chemical mechanisms of DMATSs, (ii) the structural and/or chemical basis of conversion rate discrepancies in GPP-accepting DMATSs, (iii) the structural basis for the activity of DMATSs with unnatural substrates, and (iv) engineering strategies to confer favorable substrate scope(s) and kinetic properties to DMATSs in a predictable fashion.
The majority of attempts to determine the substrate scope of DMATSs have made use of different libraries of substrates and cosubstrates from one study to the next, which makes it difficult to compare the substrate promiscuities of different enzymes. Crystal structures and homology models are not always accurate indicators of substrate scope, as demonstrated in the study of FgaPT2 with daptomycin.65 In our opinion, it would be useful to develop a library of substrates and cosubstrates containing known natural and unnatural substrates and cosubstrates, such as those listed in Table 1. The use of such a standard library would provide a better understanding of the substrate scope and the limitations of each enzyme. This information will also be invaluable for rational enzyme engineering.
4. Abbreviations
| ABBA | Protein superfamily with a unique αββα fold |
| ADR | Angle-determining residue |
| APT | Aromatic prenyltransferase |
| BM1 | Binding mode one |
| BM2 | Binding mode two |
| BDZM | Benzodiazepinedione |
| CPDR | Cosubstrate proximity determining residue |
| DKP | Diketopiperazine |
| DMAPP | Dimethylallyl diphosphate |
| DMATS | Dimethylallyl tryptophan synthase |
| DMSPP | Dimethylallyl S-thiolodiphosphate |
| E | Epimerase |
| FPP | Farnesyl diphosphate |
| GGPP | Geranylgeranyl diphosphate |
| GPP | Geranyl diphosphate |
| GSPP | Geranyl S-thiolodiphosphate |
| Hal | Halogenase |
| ITC | Isothermal titration calorimetry |
| M | Methyltransferase |
| N/C | Not calculated |
| N/D | Not described |
| NER | Nonessential residue |
| NP | Natural product |
| N/P | Not present |
| NRP | Nonribosomal peptide |
| Ox | Oxidase |
| PDB | Protein data bank |
| PHB | p-Hydroxybenzoic acid |
| PK | Polyketide |
| PP or PPi | Diphosphate |
| PT | Prenyltransferase |
| QM/MM | Quantum mechanical and molecular mechanical |
| RiPP | Ribosomally-synthesized and posttranslationally-modified peptide |
| SPP | Thiolodiphosphate |
| tRNA | Transfer ribonucleic acid |
| TR-SFX | Time-resolved serial femtosecond crystallography |
| WT | Wild-type |
5. Conflicts of interest
There are no conflicts to declare.6. Acknowledgements
This work was supported by fellowship #2239063 from the Graduate Research Fellowship Program (GRFP) from the National Science Foundation (NSF) Division of Graduate Education (to E. T. M.).7. References
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Engineering bifunctional enzymes capable of adenylating and selectively methylating the side chain or core of amino acids, ACS Synth. Biol., 2018, 7, 399–404 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Mechanistic understanding of lanthipeptide biosynthetic enzymes, Chem. Rev., 2017, 117, 5457–5520 CrossRef CAS.
- C. Wu and W. A. van der Donk, Engineering of new-to-nature ribosomally synthesized and post-translationally modified peptide natural products, Curr. Opin. Biotechnol., 2021, 69, 221–231 CrossRef CAS PubMed.
- G. A. Hudson and D. A. Mitchell, RiPP antibiotics: biosynthesis and engineering potential, Curr. Opin. Microbiol., 2018, 45, 61–69 CrossRef CAS PubMed.
- Y. Chen, M. Garcia de Lomana, N. O. Friedrich and J. Kirchmair, Characterization of the chemical space of known and readily obtainable natural products, J. Chem. Inf. Model., 2018, 58, 1518–1532 CrossRef CAS PubMed.
- D. Cantillo and C. O. Kappe, Halogenation of organic compounds using continuous flow and microreactor technology, React. Chem. Eng., 2017, 2, 7–19 RSC.
- C. Dufes, Chapter 6 – Brain delivery of peptides and proteins, in Peptide and Protein Delivery, ed. C. V. D. Walle, Academic Press, 2011, pp. 105–122 Search PubMed.
- H. D. Jain, C. Zhang, S. Zhou, H. Zhou, J. Ma, X. Liu, X. Liao, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, K. S. Smith, T. L. Macdonald, H. Kakeya, H. Osada and J. M. Cook, Synthesis and structure-activity relationship studies on tryprostatin A, an inhibitor of breast cancer resistance protein, Bioorg. Med. Chem., 2008, 16, 4626–4651 CrossRef CAS PubMed.
- C. S. Neumann, D. G. Fujimori and C. T. Walsh, Halogenation strategies in natural product biosynthesis, Chem. Biol., 2008, 15, 99–109 CrossRef CAS.
- N. Molchanova, J. E. Nielsen, K. B. Sorensen, B. K. Prabhala, P. R. Hansen, R. Lund, A. E. Barron and H. Jenssen, Halogenation as a tool to tune antimicrobial activity of peptoids, Sci. Rep., 2020, 10, 14805 CrossRef CAS PubMed.
- G. Capranico, E. Butelli and F. Zunino, Change of the sequence specificity of daunorubicin-stimulated topoisomerase II DNA cleavage by epimerization of the amino group of the sugar moiety, Cancer Res., 1995, 55, 312–317 CAS.
- J. Chatterjee, C. Gilon, A. Hoffman and H. Kessler, N-methylation of peptides: a new perspective in medicinal chemistry, Acc. Chem. Res., 2008, 41, 1331–1342 CrossRef CAS PubMed.
- S. K. Shrestha and S. Garneau-Tsodikova, Expanding substrate promiscuity by engineering a novel adenylating-methylating NRPS bifunctional enzyme, ChemBioChem, 2016, 17, 1328–1332 CrossRef CAS.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, Lessons learned in engineering interrupted adenylation domains when attempting to create trifunctional enzymes from three independent monofunctional ones, RSC Adv., 2020, 10, 34299–34397 RSC.
- T. A. Lundy, S. Mori, N. Thamban Chandrika and S. Garneau-Tsodikova, Characterization of a unique interrupted adenylation domain that can catalyze three reactions, ACS Chem. Biol., 2020, 15, 282–289 CrossRef CAS PubMed.
- R. Fasan, Tuning P450 enzymes as oxidation catalysts, ACS Catal., 2012, 2, 647–666 CrossRef CAS.
- K. Yazaki, K. Sasaki and Y. Tsurumaru, Prenylation of aromatic compounds, a key diversification of plant secondary metabolites, Phytochemistry, 2009, 70, 1739–1745 CrossRef CAS PubMed.
- J. Winkelblech, A. Fan and S. M. Li, Prenyltransferases as key enzymes in primary and secondary metabolism, Appl. Microbiol. Biotechnol., 2015, 99, 7379–7397 CrossRef CAS PubMed.
- P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, South Sussex, United Kingdom, 3rd edn, 2009 Search PubMed.
- A. Kralj, S. Kehraus, A. Krick, E. Eguereva, G. Kelter, M. Maurer, A. Wortmann, H. H. Fiebig and G. M. Konig, Arugosins G and H: prenylated polyketides from the marine-derived fungus Emericellanidulans var. acristata, J. Nat. Prod., 2006, 69, 995–1000 CrossRef CAS PubMed.
- A. Fan, J. Winkelblech and S. M. Li, Xanthones are privileged scaffolds in medicinal chemistry – but are they over-privileged?, in Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation, ed. S. Brase, The Royal Society of Chemistry, Cambridge, UK, 2016, pp. 312–347 Search PubMed.
- A. Fan, J. Winkelblech and S. M. Li, Impacts and perspectives of prenyltransferases of the DMATS superfamily for use in biotechnology, Appl. Microbiol. Biotechnol., 2015, 99, 7399–7415 CrossRef CAS.
- R. Chen, B. Gao, X. Liu, F. Ruan, Y. Zhang, J. Lou, K. Feng, C. Wunsch, S. M. Li, J. Dai and F. Sun, Molecular insights into the enzyme promiscuity of an aromatic prenyltransferase, Nat. Chem. Biol., 2017, 13, 226–234 CrossRef CAS.
- T. Bonitz, V. Alva, O. Saleh, A. N. Lupas and L. Heide, Evolutionary relationships of microbial aromatic prenyltransferases, PLoS One, 2011, 6, e27336 CrossRef CAS.
- A. Satta, L. Esquirol, B. E. Ebert, J. Newman, T. S. Peat, M. Plan, G. Schenk and C. E. Vickers, Molecular characterization of cyanobacterial short-chain prenyltransferases and discovery of a novel GGPP phosphatase, FEBS J., 2022, 289, 6672–6693 CrossRef CAS.
- W. Cheng and W. Li, Structural insights into ubiquinone biosynthesis in membranes, Science, 2014, 343, 878–881 CrossRef CAS PubMed.
- U. Metzger, C. Schall, G. Zocher, I. Unsold, E. Stec, S. M. Li, L. Heide and T. Stehle, The structure of dimethylallyl tryptophan synthase reveals a common architecture of aromatic prenyltransferases in fungi and bacteria, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS.
- J. Wang, C. C. Chen, Y. Yang, W. Liu, T. P. Ko, N. Shang, X. Hu, Y. Xie, J. W. Huang, Y. Zhang and R. T. Guo, Structural insight into a novel indole prenyltransferase in hapalindole-type alkaloid biosynthesis, Biochem. Biophys. Res. Commun., 2018, 495, 1782–1788 CrossRef CAS.
- Y. Hao, E. Pierce, D. Roe, M. Morita, J. A. McIntosh, V. Agarwal, T. E. Cheatham 3rd, E. W. Schmidt and S. K. Nair, Molecular basis for the broad substrate selectivity of a peptide prenyltransferase, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 14037–14042 CrossRef CAS.
- S. B. Long, P. J. Hancock, A. M. Kral, H. W. Hellinga and L. S. Beese, The crystal structure of human protein farnesyltransferase reveals the basis for inhibition by CaaX tetrapeptides and their mimetics, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12948–12953 CrossRef CAS PubMed.
- W. Xie, C. Zhou and R. H. Huang, Structure of tRNA dimethylallyltransferase: RNA modification through a channel, J. Mol. Biol., 2007, 367, 872–881 CrossRef CAS PubMed.
- M. Jost, G. Zocher, S. Tarcz, M. Matuschek, X. Xie, S. M. Li and T. Stehle, Structure-function analysis of an enzymatic prenyl transfer reaction identifies a reaction chamber with modifiable specificity, J. Am. Chem. Soc., 2010, 132, 17849–17858 CrossRef CAS.
- G. Liao, P. Mai, J. Fan, G. Zocher, T. Stehle and S. M. Li, Complete decoration of the indolyl residue in cyclo-L-Trp-L-Trp with geranyl moieties by using engineered dimethylallyl transferases, Org. Lett., 2018, 20, 7201–7205 CrossRef CAS.
- W. Zhao, A. Fan, S. Tarcz, K. Zhou, W. B. Yin, X. Q. Liu and S. M. Li, Mutation on Gly115 and Tyr205 of the cyclic dipeptide C2-prenyltransferase FtmPT1 increases its catalytic activity toward hydroxynaphthalenes, Appl. Microbiol. Biotechnol., 2017, 101, 1989–1998 CrossRef CAS.
- B. Wollinsky, L. Ludwig, A. Hamacher, X. Yu, M. U. Kassack and S. M. Li, Prenylation at the indole ring leads to a significant increase of cytotoxicity of tryptophan-containing cyclic dipeptides, Bioorg. Med. Chem. Lett., 2012, 22, 3866–3869 CrossRef CAS.
- A. Kremer and S. M. Li, Tryptophan aminopeptidase activity of several indole prenyltransferases from Aspergillus fumigatus, Chem. Biol., 2008, 15, 729–738 CrossRef CAS.
- A. Grundmann and S. M. Li, Overproduction, purification and characterization of FtmPT1, a brevianamide F prenyltransferase from Aspergillus fumigatus, Microbiology, 2005, 151, 2199–2207 CrossRef CAS PubMed.
- P. Mai, L. Coby and S. M. Li, Different behaviors of cyclic dipeptide prenyltransferases toward the tripeptide derivative ardeemin fumiquinazoline and its enantiomer, Appl. Microbiol. Biotechnol., 2019, 103, 3773–3781 CrossRef CAS PubMed.
- S. P. Kelly, V. V. Shende, A. R. Flynn, Q. Dan, Y. Ye, J. L. Smith, S. Tsukamoto, M. S. Sigman and D. H. Sherman, Data science-driven analysis of substrate-permissive diketopiperazine reverse prenyltransferase NotF: applications in protein engineering and cascade biocatalytic synthesis of (−)-eurotiumin A, J. Am. Chem. Soc., 2022, 144, 19326–19336 CrossRef CAS.
- K. Yang, S. M. Li, X. Liu and A. Fan, Reinvestigation of the substrate specificity of a reverse prenyltransferase NotF from Aspergillus sp. MF297-2, Arch. Microbiol., 2020, 202, 1419–1424 CrossRef CAS.
- E. D. Gardner, D. A. Dimas, M. C. Finneran, S. M. Brown, A. W. Burgett and S. Singh, Indole C6 functionalization of tryprostatin B using prenyltransferase CdpNPT, Catalysts, 2020, 10, 1247–1259 CrossRef CAS.
- S. A. Hamdy, T. Kodama, Y. Nakashima, X. Han and H. Morita, Catalytic potential of a fungal indole prenyltransferase toward beta-carbolines, harmine and harman, and their prenylation effects on antibacterial activity, J. Biosci. Bioeng., 2022, 134, 311–317 CrossRef CAS.
- E. M. Scull, C. Bandari, B. P. Johnson, E. D. Gardner, M. Tonelli, J. You, R. H. Cichewicz and S. Singh, Chemoenzymatic synthesis of daptomycin analogs active against daptomycin-resistant strains, Appl. Microbiol. Biotechnol., 2020, 104, 7853–7865 CrossRef CAS PubMed.
- Y. Li, X. Zhou, S. M. Li, Y. Zhang, C. M. Yuan, S. He, Z. Yang, S. Yang and K. Zhou, Increasing structural diversity of prenylated chalcones by two fungal prenyltransferases, J. Agric. Food Chem., 2022, 70, 1610–1617 CrossRef CAS PubMed.
- S. Isogai, N. Okahashi, R. Asama, T. Nakamura, T. Hasunuma, F. Matsuda, J. Ishii and A. Kondo, Synthetic production of prenylated naringenins in yeast using promiscuous microbial prenyltransferases, Metab. Eng. Commun., 2021, 12, e00169 CrossRef CAS PubMed.
- X. Yu, G. Zocher, X. Xie, M. Liebhold, S. Schutz, T. Stehle and S. M. Li, Catalytic mechanism of stereospecific formation of cis-configured prenylated pyrroloindoline diketopiperazines by indole prenyltransferases, Chem. Biol., 2013, 20, 1492–1501 CrossRef CAS PubMed.
- K. Zhou, L. Ludwig and S. M. Li, Friedel-crafts alkylation of acylphloroglucinols catalyzed by a fungal indole prenyltransferase, J. Nat. Prod., 2015, 78, 929–933 CrossRef CAS.
- D. Pockrandt, C. Sack, T. Kosiol and S. M. Li, A promiscuous prenyltransferase from Aspergillus oryzae catalyses C-prenylations of hydroxynaphthalenes in the presence of different prenyl donors, Appl. Microbiol. Biotechnol., 2014, 98, 4987–4994 CrossRef CAS.
- Q. Ran, L. Tao, X. Zhou, S. M. Li, C. M. Yuan, S. Yang and K. Zhou, Geranylation of chalcones by a fungal aromatic prenyltransferase, J. Agric. Food Chem., 2023, 71, 4675–4682 CrossRef CAS.
- K. Xu, C. Yang, Y. Xu, D. Li, S. Bao, Z. Zou, F. Kang, G. Tan, S. M. Li and X. Yu, Selective geranylation of biflavonoids by Aspergillus terreus aromatic prenyltransferase (AtaPT), Org. Biomol. Chem., 2019, 18, 28–31 RSC.
- K. Zhou, C. Wunsch, J. Dai and S. M. Li, gem-Diprenylation of acylphloroglucinols by a fungal prenyltransferase of the dimethylallyltryptophan synthase superfamily, Org. Lett., 2017, 19, 388–391 CrossRef CAS PubMed.
- C. Bandari, E. M. Scull, T. Bavineni, S. L. Nimmo, E. D. Gardner, R. C. Bensen, A. W. Burgett and S. Singh, FgaPT2, a biocatalytic tool for alkyl-diversification of indole natural products, MedChemComm, 2019, 10, 1465–1475 RSC.
- L. Zheng, P. Mai, A. Fan and S. M. Li, Switching a regular tryptophan C4-prenyltransferase to a reverse tryptophan-containing cyclic dipeptide C3-prenyltransferase by sequential site-directed mutagenesis, Org. Biomol. Chem., 2018, 16, 6688–6694 RSC.
- A. Fan and S. M. Li, Saturation mutagenesis on Arg244 of the tryptophan C4-prenyltransferase FgaPT2 leads to enhanced catalytic ability and different preferences for tryptophan-containing cyclic dipeptides, Appl. Microbiol. Biotechnol., 2016, 100, 5389–5399 CrossRef CAS PubMed.
- A. Fan, G. Zocher, E. Stec, T. Stehle and S. M. Li, Site-directed mutagenesis switching a dimethylallyl tryptophan synthase to a specific tyrosine C3-prenylating enzyme, J. Biol. Chem., 2015, 290, 1364–1373 CrossRef.
- M. Liebhold and S. M. Li, Regiospecific benzylation of tryptophan and derivatives catalyzed by a fungal dimethylallyl transferase, Org. Lett., 2013, 15, 5834–5837 CrossRef CAS PubMed.
- N. Steffan and S. M. Li, Increasing structure diversity of prenylated diketopiperazine derivatives by using a 4-dimethylallyltryptophan synthase, Arch. Microbiol., 2009, 191, 461–466 CrossRef CAS PubMed.
- P. Mai, G. Zocher, T. Stehle and S. M. Li, Structure-based protein engineering enables prenyl donor switching of a fungal aromatic prenyltransferase, Org. Biomol. Chem., 2018, 16, 7461–7469 RSC.
- E. Ostertag, L. Zheng, K. Broger, T. Stehle, S. M. Li and G. Zocher, Reprogramming substrate and catalytic promiscuity of tryptophan prenyltransferases, J. Mol. Biol., 2021, 433, 166726–166739 CrossRef CAS PubMed.
- J. Winkelblech, M. Liebhold, J. Gunera, X. Xie, P. Kolb and S. M. Li, Tryptophan C5-, C6-, and C7-Prenylating enzymes displaying a preference for C6 of the indole ring in the presence of unnatural dimethylallyl diphosphate analogues, Adv. Synth. Catal., 2015, 357, 975–986 CrossRef CAS.
- J. Winkelblech, X. Xie and S. M. Li, Characterisation of 6-DMATS(Mo) from Micromonospora olivasterospora leading to identification of the divergence in enantioselectivity, regioselectivity and multiple prenylation of tryptophan prenyltransferases, Org. Biomol. Chem., 2016, 14, 9883–9895 RSC.
- H. Suemune, D. Nishimura, K. Mizutani, Y. Sato, T. Hino, H. Takagi, Y. Shiozaki-Sato, S. Takahashi and S. Nagano, Crystal structures of a 6-dimethylallyltryptophan synthase, IptA: insights into substrate tolerance and enhancement of prenyltransferase activity, Biochem. Biophys. Res. Commun., 2022, 593, 144–150 CrossRef CAS PubMed.
- S. I. Elshahawi, H. Cao, K. A. Shaaban, L. V. Ponomareva, T. Subramanian, M. L. Farman, H. P. Spielmann, G. N. Phillips Jr., J. S. Thorson and S. Singh, Structure and specificity of a permissive bacterial C-prenyltransferase, Nat. Chem. Biol., 2017, 13, 366–368 CrossRef CAS.
- B. W. Roose and D. W. Christianson, Structural basis of tryptophan reverse N-prenylation catalyzed by CymD, Biochemistry, 2019, 58, 3232–3242 CrossRef CAS.
- I. Burkhardt, Z. Ye, S. Janevska, B. Tudzynski and J. S. Dickschat, Biochemical and mechanistic characterization of the fungal reverse N-1-dimethylallyltryptophan synthase DMATS1Ff, ACS Chem. Biol., 2019, 14, 2922–2931 CrossRef CAS PubMed.
- S. A. Eaton, T. A. Ronnebaum, B. W. Roose and D. W. Christianson, Structural basis of substrate promiscuity and catalysis by the reverse prenyltransferase N-dimethylallyl-L-tryptophan synthase from Fusarium fujikuroi, Biochemistry, 2022, 61, 2025–2035 CrossRef CAS.
- D. Pockrandt, L. Ludwig, A. Fan, G. M. Konig and S. M. Li, New insights into the biosynthesis of prenylated xanthones: XptB from Aspergillus nidulans catalyses an O-prenylation of xanthones, ChemBioChem, 2012, 13, 2764–2771 CrossRef CAS PubMed.
- J. Gunera, F. Kindinger, S. M. Li and P. Kolb, PrenDB, a substrate prediction database to enable biocatalytic use of prenyltransferases, J. Biol. Chem., 2017, 292, 4003–4021 CrossRef CAS.
- T. Mori, L. Zhang, T. Awakawa, S. Hoshino, M. Okada, H. Morita and I. Abe, Manipulation of prenylation reactions by structure-based engineering of bacterial indolactam prenyltransferases, Nat. Commun., 2016, 7, 10849 CrossRef CAS PubMed.
- C. Bandari, E. M. Scull, J. M. Masterson, R. H. Q. Tran, S. B. Foster, K. M. Nicholas and S. Singh, Determination of alkyl-donor promiscuity of tyrosine-O-prenyltransferase SirD from Leptosphaeria maculans, ChemBioChem, 2017, 18, 2323–2327 CrossRef CAS PubMed.
- M. E. Tanner, Mechanistic studies on the indole prenyltransferases, Nat. Prod. Rep., 2015, 32, 88–101 RSC.
- E. Wenkert and H. Sliwa, A model study of ergot alkaloid biosynthesis, Bioorg. Chem., 1977, 6, 443–452 CrossRef CAS.
- M. P. Siler, PhD dissertation No. 4574, ETH Zurich, 1970, https://www.research-collection.ethz.ch/bitstream/handle/20.500.11850/132629/1/eth-32691-01.pdf.
- H. G. Floss, Biosynthesis of ergot alkoids and related compounds, Tetrahedron, 1976, 32, 873–912 CrossRef CAS.
- J. D. Rudolf, H. Wang and C. D. Poulter, Multisite prenylation of 4-substituted tryptophans by dimethylallyltryptophan synthase, J. Am. Chem. Soc., 2013, 135, 1895–1902 CrossRef CAS.
- H. P. Chen and I. Abe, Microbial soluble aromatic prenyltransferases for engineered biosynthesis, Synth. Syst. Biotechnol., 2021, 6, 51–62 CrossRef CAS PubMed.
- L. L. Pan, L. F. Song, Y. Miao, Y. Yang and K. M. Merz Jr., Mechanism of formation of the nonstandard product in the prenyltransferase reaction of the G115T mutant of FtmPT1: a case of reaction dynamics calling the shots?, Biochemistry, 2017, 56, 2995–3007 CrossRef CAS PubMed.
- J. Chen, H. Morita, T. Wakimoto, T. Mori, H. Noguchi and I. Abe, Prenylation of a nonaromatic carbon of indolylbutenone by a fungal indole prenyltransferase, Org. Lett., 2012, 14, 3080–3083 CrossRef CAS.
- C. C. Loh, G. Raabe and D. Enders, Enantioselective synthesis of tetrahydrocarbazoles through a Michael addition/Ciamician-Plancher rearrangement sequence: asymmetric synthesis of a potent constrained analogue of MS-245, Chemistry, 2012, 18, 13250–13254 CrossRef CAS.
- M. Nakazaki, K. Yamamoto and K. Yamagami, Mechanism of Plancher's rearrangement. I. Tworfold Wagner-Meerwein type rearrangement of indolenines, Bull. Chem. Soc. Jpn., 1960, 33, 466–472 CrossRef CAS.
- R. J. Sundberg, Electrophilic substitution reactions of indoles, Heterocycl. Chem., 2010, 26, 47–115 CAS.
- L. Y. Luk, Q. Qian and M. E. Tanner, A cope rearrangement in the reaction catalyzed by dimethylallyltryptophan synthase?, J. Am. Chem. Soc., 2011, 133, 12342–12345 CrossRef CAS.
- S. A. Hamdy, T. Kodama, Y. Nakashima, X. Han, T. Matsui and H. Morita, Enzymatic formation of a prenyl beta-carboline by a fungal indole prenyltransferase, J. Nat. Med., 2022, 76, 873–879 CrossRef CAS PubMed.
- R. Liu, H. Zhang, W. Wu, H. Li, Z. An and F. Zhou, C7-Prenylation of tryptophan-containing cyclic dipeptides by 7-dimethylallyl tryptophan synthase significantly increases the anticancer and antimicrobial activities, Molecules, 2020, 25, 3676 CrossRef CAS PubMed.
- T. M. Khopade, K. Ajayan, S. S. Joshi, A. L. Lane and R. Viswanathan, Bioinspired Bronsted acid-promoted regioselective tryptophan isoprenylations, ACS Omega, 2021, 6, 10840–10858 CrossRef CAS PubMed.
- W. Li, L. Coby, J. Zhou and S. M. Li, Diprenylated cyclodipeptide production by changing the prenylation sequence of the Nature's synthetic machinery, Appl. Microbiol. Biotechnol., 2023, 107, 261–271 CrossRef CAS.
- W. B. Yin, J. Cheng and S. M. Li, Stereospecific synthesis of aszonalenins by using two recombinant prenyltransferases, Org. Biomol. Chem., 2009, 7, 2202–2207 RSC.
- A. E. Fraley, K. Caddell Haatveit, Y. Ye, S. P. Kelly, S. A. Newmister, F. Yu, R. M. Williams, J. L. Smith, K. N. Houk and D. H. Sherman, Molecular basis for spirocycle formation in the paraherquamide biosynthetic pathway, J. Am. Chem. Soc., 2020, 142, 2244–2252 CrossRef CAS.
- A. E. Fraley, H. T. Tran, S. P. Kelly, S. A. Newmister, A. Tripathi, H. Kato, S. Tsukamoto, L. Du, S. Li, R. M. Williams and D. H. Sherman, Flavin-dependent monooxygenases NotI and NotI' mediate spiro-oxindole formation in biosynthesis of the notoamides, ChemBioChem, 2020, 21, 2449–2454 CrossRef CAS PubMed.
- Y. Ye, L. Du, X. Zhang, S. A. Newmister, M. McCauley, J. V. Alegre-Requena, W. Zhang, S. Mu, A. Minami, A. E. Fraley, M. L. Adrover-Castellano, N. A. Carney, V. V. Shende, F. Qi, H. Oikawa, H. Kato, S. Tsukamoto, R. S. Paton, R. M. Williams, D. H. Sherman and S. Li, Fungal-derived brevianamide assembly by a stereoselective semipinacolase, Nat. Catal., 2020, 3, 497–506 CrossRef CAS PubMed.
- X. Chen, E. Mukwaya, M. S. Wong and Y. Zhang, A systematic review on biological activities of prenylated flavonoids, Pharm. Biol., 2014, 52, 655–660 CrossRef CAS.
- S. Preciado, L. Mendive-Tapia, G. Torres-Garcia, R. Zamudio-Vazquez, V. Soto-Cerrato, T. R. Perez, F. Albericio, E. Nicolas and R. Lavilla, Synthesis and biological evaluation of a post-synthetically modified Trp-based diketopiperazine, Med. Chem. Commun., 2013, 4, 1171–1174 RSC.
- T. Awakawa and I. Abe, Biosynthesis of the teleocidin-type terpenoid indole alkaloids, Org. Biomol. Chem., 2018, 16, 4746–4752 RSC.
- P. Zhao, Y. Xue, J. Li, X. Li, X. Zu, Z. Zhao, C. Quan, W. Gao and S. Feng, Non-lipopeptide fungi-derived peptide antibiotics developed since 2000, Biotechnol. Lett., 2019, 41, 651–673 CrossRef CAS PubMed.
- J. M. Schuller, G. Zocher, M. Liebhold, X. Xie, M. Stahl, S. M. Li and T. Stehle, Structure and catalytic mechanism of a cyclic dipeptide prenyltransferase with broad substrate promiscuity, J. Mol. Biol., 2012, 422, 87–99 CrossRef CAS PubMed.
- T. Mori, Enzymatic studies on aromatic prenyltransferases, J. Nat. Med., 2020, 74, 501–512 CrossRef CAS PubMed.
- T. An, X. Feng and C. Li, Prenylation: a critical step for biomanufacturing of prenylated aromatic natural products, J. Agric. Food Chem., 2023, 71, 2211–2233 CrossRef CAS PubMed.
- S. L. Lee, H. G. Floss and P. Heinstein, Purification and properties of dimethylallylpyrophosphate: tryptophan dimethylallyl transferase, the first enzyme of ergot alkaloid biosynthesis in Claviceps sp. SD 58, Arch. Biochem. Biophys., 1976, 177, 87–94 CrossRef.
- K. Miyamoto, F. Ishikawa, S. Nakamura, Y. Hayashi, I. Nakanishi and H. Kakeya, A 7-dimethylallyl tryptophan synthase from a fungal Neosartorya sp.: biochemical characterization and structural insight into the regioselective prenylation, Bioorg. Med. Chem., 2014, 22, 2517–2528 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- M. Varadi, S. Anyango, M. Deshpande, S. Nair, C. Natassia, G. Yordanova, D. Yuan, O. Stroe, G. Wood, A. Laydon, A. Zidek, T. Green, K. Tunyasuvunakool, S. Petersen, J. Jumper, E. Clancy, R. Green, A. Vora, M. Lutfi, M. Figurnov, A. Cowie, N. Hobbs, P. Kohli, G. Kleywegt, E. Birney, D. Hassabis and S. Velankar, AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models, Nucleic Acids Res., 2022, 50, D439–D444 CrossRef CAS.
- D. M. Gardiner, A. J. Cozijnsen, L. M. Wilson, M. S. Pedras and B. J. Howlett, The sirodesmin biosynthetic gene cluster of the plant pathogenic fungus Leptosphaeria maculans, Mol. Microbiol., 2004, 53, 1307–1318 CrossRef CAS.
- J. D. Rudolf and C. D. Poulter, Tyrosine O-prenyltransferase SirD catalyzes S-, C-, and N-prenylations on tyrosine and tryptophan derivatives, ACS Chem. Biol., 2013, 8, 2707–2714 CrossRef CAS PubMed.
- H. Zou, X. Zheng and S. M. Li, Substrate promiscuity of the cyclic dipeptide prenyltransferases from Aspergillus fumigatus, J. Nat. Prod., 2009, 72, 44–52 CrossRef CAS.
- A. Klamrak, J. Nabnueangsap, P. Puthongking and N. Nualkaew, Synthesis of ferulenol by engineered Escherichia coli: structural elucidation by using the in silico tools, Molecules, 2021, 26, 6264–6284 CrossRef CAS PubMed.
- C. J. Guo, W. W. Sun, K. S. Bruno, B. R. Oakley, N. P. Keller and C. C. C. Wang, Spatial regulation of a common precursor from two distinct genes generates metabolite diversity, Chem. Sci., 2015, 6, 5913–5921 RSC.
- Y. Shen, J. Zou, D. Xie, H. Ge, X. Cao and J. Dai, Butyrolactone and cycloheptanetrione from mangrove-associated fungus Aspergillus terreus, Chem. Pharm. Bull., 2012, 60, 1437–1441 CrossRef CAS PubMed.
- N. Mahmoodi and M. E. Tanner, Potential rearrangements in the reaction catalyzed by the indole prenyltransferase FtmPT1, ChemBioChem, 2013, 14, 2029–2037 CrossRef CAS PubMed.
- J. M. Martin-Garcia, C. E. Conrad, J. Coe, S. Roy-Chowdhury and P. Fromme, Serial femtosecond crystallography: a revolution in structural biology, Arch. Biochem. Biophys., 2016, 602, 32–47 CrossRef CAS PubMed.
- R. K. Y. Cheng, Towards an optimal sample delivery method for serial crystallography at XFEL, Crystals, 2020, 10, 215 CrossRef CAS.
- C. Kupitz, I. Grotjohann, C. E. Conrad, S. Roy-Chowdhury, R. Fromme and P. Fromme, Microcrystallization techniques for serial femtosecond crystallography using photosystem II from Thermosynechococcus elongatus as a model system, Philos. Trans. R. Soc. London, Ser. B, 2014, 369, 20130316–20130324 CrossRef.
- A. Fan, X. Xie and S. M. Li, Tryptophan prenyltransferases showing higher catalytic activities for Friedel-Crafts alkylation of o- and m-tyrosines than tyrosine prenyltransferases, Org. Biomol. Chem., 2015, 13, 7551–7557 RSC.
- T. Ozaki, M. Nishiyama and T. Kuzuyama, Novel tryptophan metabolism by a potential gene cluster that is widely distributed among actinomycetes, J. Biol. Chem., 2013, 288, 9946–9956 CrossRef CAS PubMed.
- H. L. Ruan, E. Stec and S. M. Li, Production of diprenylated indole derivatives by tandem incubation of two recombinant dimethylallyltryptophan synthases, Arch. Microbiol., 2009, 191, 791–795 CrossRef CAS PubMed.
- N. Steffan, I. A. Unsold and S. M. Li, Chemoenzymatic synthesis of prenylated indole derivatives by using a 4-dimethylallyltryptophan synthase from Aspergillus fumigatus, ChemBioChem, 2007, 8, 1298–1307 CrossRef CAS.
- S. Subramanian, X. Shen, Q. Yuan and Y. Yan, Identification and biochemical characterization of a 5-dimethylallyltryptophan synthase in Streptomyces coelicor A3(2), Process Biochem., 2012, 47, 1419–1422 CrossRef CAS.
- I. A. Unsold and S. M. Li, Overproduction, purification and characterization of FgaPT2, a dimethylallyltryptophan synthase from Aspergillus fumigatus, Microbiol., 2005, 151, 1499–1505 CrossRef PubMed.
- J. J. Clark, M. L. Benson, R. D. Smith and H. A. Carlson, Inherent versus induced protein flexibility: comparisons within and between apo and holo structures, PLoS Comput. Biol., 2019, 15, e1006705 CrossRef.
- H. B. Bürgi, J. D. Dunitz, J. M. Lehn and G. Wipff, Stereochemistry of reaction paths at carbonyl centres, Tetrahedron, 1974, 30, 1563–1572 CrossRef.
- S. Takahashi, H. Takagi, A. Toyoda, M. Uramoto, T. Nogawa, M. Ueki, Y. Sakaki and H. Osada, Biochemical characterization of a novel indole prenyltransferase from Streptomyces sp. SN-593, J. Bacteriol., 2010, 192, 2839–2851 CrossRef CAS.
- T. Kumano, S. B. Richard, J. P. Noel, M. Nishiyama and T. Kuzuyama, Chemoenzymatic syntheses of prenylated aromatic small molecules using Streptomyces prenyltransferases with relaxed substrate specificities, Bioorg. Med. Chem., 2008, 16, 8117–8126 CrossRef CAS.
- G. Zocher, O. Saleh, J. B. Heim, D. A. Herbst, L. Heide and T. Stehle, Structure-based engineering increased the catalytic turnover rate of a novel phenazine prenyltransferase, PLoS One, 2012, 7, e48427 CrossRef CAS PubMed.
- Q. Qian, A. W. Schultz, B. S. Moore and M. E. Tanner, Mechanistic studies on CymD: a tryptophan reverse N-prenyltransferase, Biochemistry, 2012, 51, 7733–7739 CrossRef CAS PubMed.
- K. Zhou, W. Zhao, X. Q. Liu and S. M. Li, Saturation mutagenesis on Tyr205 of the cyclic dipeptide C2-prenyltransferase FtmPT1 results in mutants with strongly increased C3-prenylating activity, Appl. Microbiol. Biotechnol., 2016, 100, 9943–9953 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- Induced Fit Docking Protocol 2015-2, Glide Version 6.4, Prime Version 3.7, Schrödinger: LLC New York, NY, 2015 Search PubMed.
- X. Liu and C. T. Walsh, Characterization of cyclo-acetoacetyl-L-tryptophan dimethylallyltransferase in cyclopiazonic acid biosynthesis: substrate promiscuity and site directed mutagenesis studies, Biochemistry, 2009, 48, 11032–11044 CrossRef CAS PubMed.
- B. Gao, R. Chen, X. Liu, J. Dai and F. Sun, Expression, purification, crystallization and crystallographic study of the Aspergillus terreus aromatic prenyltransferase AtaPT, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2015, 71, 889–894 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |