
Huperzine alkaloids: forty years of total syntheses
Bichu
Cheng
 *ac,
Lili
Song
a and
Fener
Chen
*abc
*ac,
Lili
Song
a and
Fener
Chen
*abc
aEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: rfchen@fudan.edu.cn
bCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang, Jiangxi 330022, China
cSchool of Science, Green Pharmaceutical Engineering Research Center, Harbin Institute of Technology, Shenzhen 518055, China. E-mail: chengbichu@hit.edu.cn
First published on 11th October 2023
Abstract
Covering: up to 2023
Huperzine alkaloids are a group of natural products belonging to the Lycopodium alkaloids family. The representative member huperzine A has a unique structure and exhibits potent inhibitory activity against acetylcholine esterase (AChE). This subfamily of alkaloids provides a great opportunity for developing synthetic methodologies and asymmetric synthesis. The efforts towards the synthesis of huperzine A have cultivated dozens of total syntheses and a rich body of new chemistry. Impressive progress has also been made in the synthesis of other huperzine alkaloids. The total syntheses of huperzines B, U, O, Q and R, structure reassignment and total syntheses of huperzines K, M and N have been reported in the past decade. This review focuses on the synthetic organic chemistry and the biosynthesis and medicinal chemistry of huperzines are also covered briefly.
1. Introduction
Alzheimer's disease (AD) is a progressive neurodegenerative disease that slowly destroys memory, thinking, behavior and social skills. The disease is named after Dr Alois Alzheimer, who reported changes in the brain tissue of a woman who had died of an unusual mental illness in 1906. There have been many hypotheses on the cause of the disease, with one of the major deficits in AD patients involving the cholinergic system.Acetylcholine (1) is a neurotransmitter in the cholinergic system. It was first isolated in 1914, and its role as a neurotransmitter was identified by the work of Otto Loewi. During cholinergic neurotransmission, acetylcholine stored in the synaptic vesicles is released to the synaptic cleft, where it binds to a postsynaptic receptor and causes depolarization. Since acetylcholine (1) is rapidly degraded to choline and acetate ions by the enzyme acetylcholinesterase (AChE), it only has a brief duration of action. Cholinergic neurotransmission plays an important role in memory and learning, but it has impacts on patients with AD. Since one of the major deficits in AD patients involves the cholinergic system, the use of reversible inhibitors of AChE to increase the level of the neurotransmitter in the central nervous system (CNS) is one viable therapeutic approach to the disease.1,2
Experimental evidence has shown that inhibitors of AChE might elevate the levels of acetylcholine (1) in the brains of these patients and reverse the cognitive decline. Several reversible inhibitors of AChE, such as tacrine (2), donepezil (3), rivastigmine (4) and galantamine (5), have been approved in the USA for the treatment of patients suffering from mild to moderate Alzheimer's disease (Fig. 1). Although certain treatments may help relieve some physical or mental symptoms associated with neurodegenerative diseases, slowing their progression is not currently possible, and no cures exist.
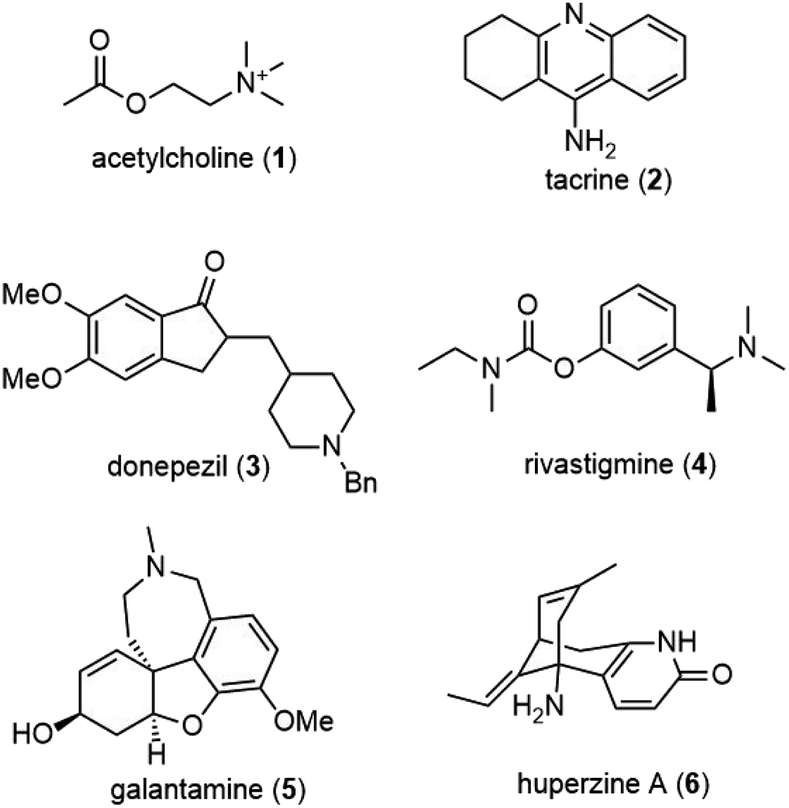 | ||
| Fig. 1 Structure of acetylcholine and inhibitors of AChE. | ||
Huperzine A (6) is a Lycopodium alkaloid isolated by a Chinese team in 1986 from the club moss Huperzia serrata (Thunb.) Trev. Lycopodium serratum Thunb.3 This club moss has been used in traditional Chinese medicine for the treatment of contusion, strain, swelling, and schizophrenia. The total alkaloids from this plant could alleviate the symptom of myasthenia gravis. It is a potent and selective reversible inhibitor of AChE, which appears to be superior to other AChE inhibitors, such as tacrine (2) or galanthamine (5), because of its longer duration of action and higher therapeutic index. Huperzine A (6) has already been marketed as a new drug in the treatment of AD in China and as a dietary supplement in the USA.
2. Biosynthesis of huperzines
Earlier extensive isotope labeling studies by Spenser and coworkers revealed a plausible metabolic route for these alkaloids.4–6 More recently, through combined metabolomic profiling and transcriptomics, Sattely and coworkers identified a developmentally controlled set of biosynthetic genes for the Lycopodium alkaloids.7In the early stage of the proposed biosynthesis, the decarboxylation of lysine (7) by lysine decarboxylase (LDC) produced cadaverine (8) (Scheme 1).7 Subsequent oxidative deamination of cadaverine (8) by copper amine oxidase (CAO) formed 5-aminopentanal, which could cyclize to Δ1-piperideine (9). However, type III PKS enzymes piperidyl-ketide synthase (PIKS) PtPIKS-1 and PtPIKS-2 catalyzed the decarboxylative condensation of two molecules of malonyl-CoA (10) to form acetonedicarboxylic acid (11, or its bisCoA ester). A decarboxylative Mannich reaction of 11 with Δ1-piperideine (9) generated 4-(2-piperidyl) acetoacetate (4PAA, 12). Further decarboxylation by an unknown decarboxylase formed pelletierine (13), the first general intermediate to Lycopodium alkaloids.
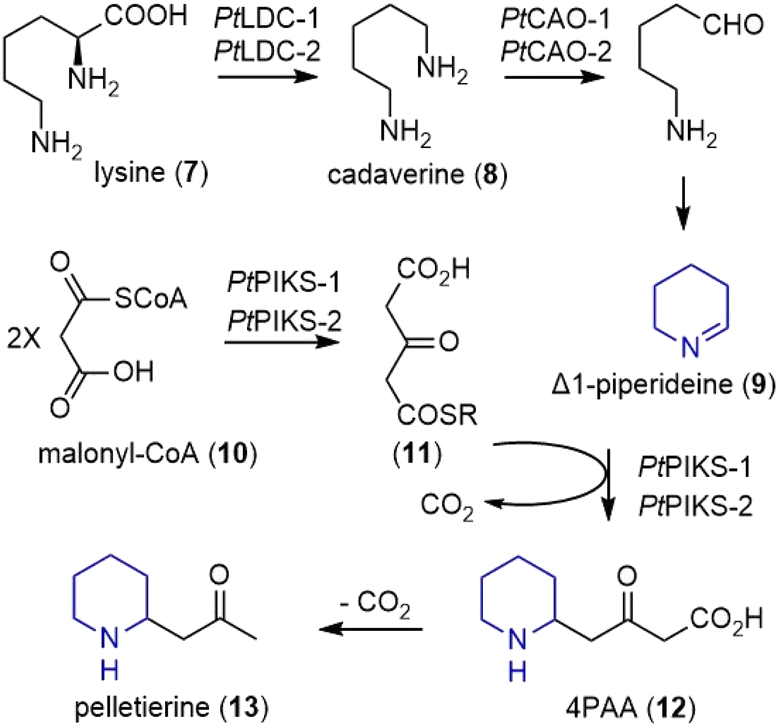 | ||
| Scheme 1 Possible biosynthesis of huperzines (part 1). | ||
Pelletierine (13) and 4PAA, or some derivatives thereof underwent a decarboxylative coupling reaction by an unknown enzyme(s) to form phlegmarine (14), the second general intermediate to all Lycopodium alkaloids (Scheme 2). The late stage of the proposed biosynthesis of huperzine A was also studied. Three Fe(II)/2-oxoglutarate-dependent dioxygenases (OGDs) catalyze transformations within downstream huperzine A biosynthesis. The desaturation of des-N-methyl-α-obscurine (15) was catalyzed by Pt2OGD-3, and resulted in the formation of the aromatic pyridone ring to produce des-N-methyl-β-obscurine (16). Another enzyme, Pt2OGD-1, transformed huperzine B (17) to huperzine C (18). A C–N bond and a C–C bond were both cleaved during this process, resulting in the opening of the piperidine ring and the loss of a carbon atom. The third enzyme Pt2OGD-2 catalyzed the redox-neutral double bond isomerization of huperzine C (18) to yield huperzine A (6).
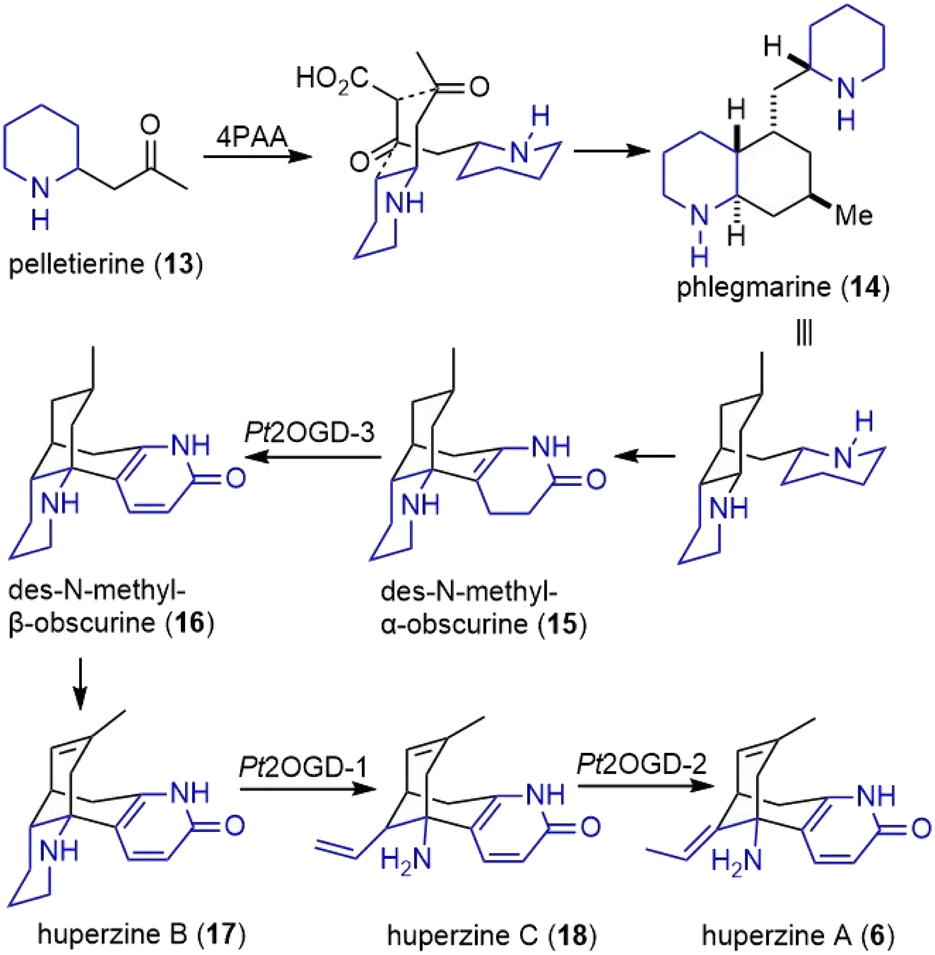 | ||
| Scheme 2 Possible biosynthesis of huperzines (part 2). | ||
Among the hitherto reported Lycopodium alkaloids, huperzine A is a typical representative with efficient, reversible, and highly selective inhibitory activities against AChE. It is therefore used as a promising therapeutic agent for the treatment of Alzheimer's disease. Given their unique and intriguing structures, biogenesis, and wide-ranging biological activities, Lycopodium alkaloids have gained immense interest worldwide. The chemical strategies for the construction of Lycopodium alkaloids have been extensively reported.5,8
In this review, we highlight the progress in the total synthesis of natural products in this group including huperzine A (6), huperzine B (17), 8,15-dihydrohuperzine A (19),9 huperzine U (20),10 huperzine K (21),11,12 huperzine M (22),13 huperzine N (23),13 huerzine O (24),14 huperzine Q (25)15 and huperzine R (26)16 (Fig. 2).
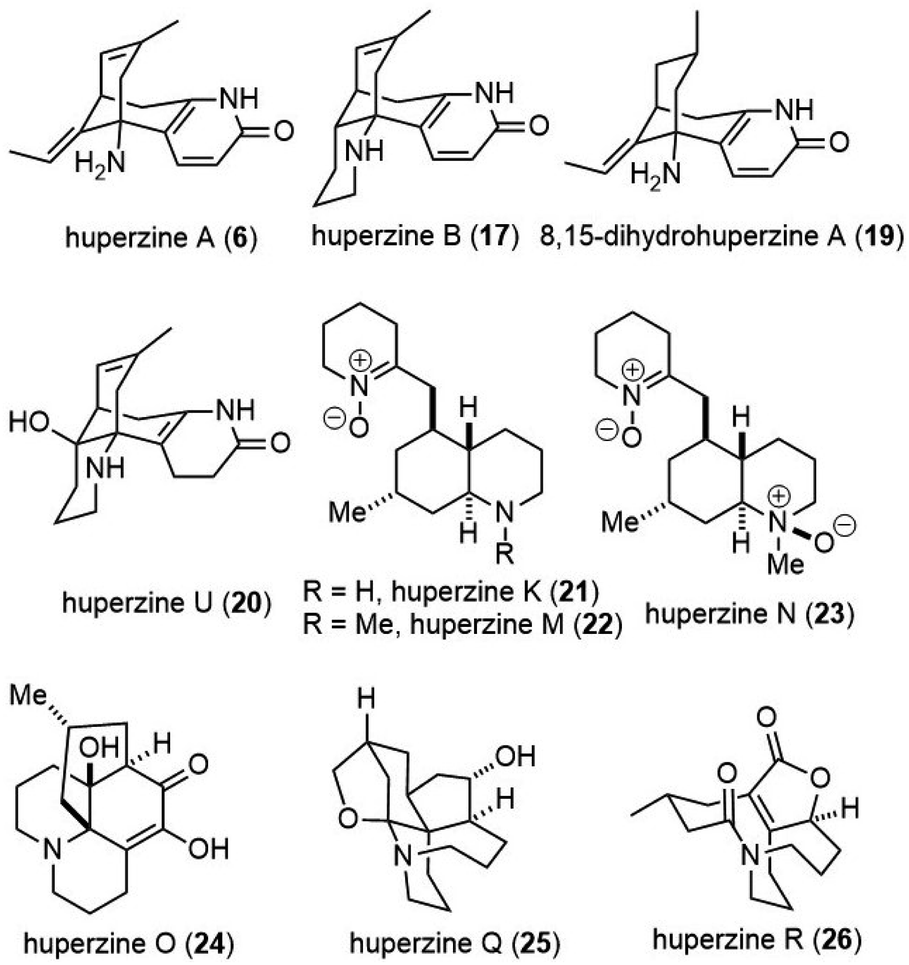 | ||
| Fig. 2 Huperzine alkaloids discussed in this review. | ||
3. Huperzine A
In 1986, Liu and coworkers reported the isolation of huperzines A (6) and B (17) from Huperzia serrata (Thunb.) Trev., a Chinese folk medicine Qian Ceng Ta (Fig. 3).3 Their structures were elucidated by spectroscopic analysis and chemical derivatization. Huperzine A (6) has a quite unique structure, which contains a bicyclo[3.3.1] skeleton fused with a pyridone moiety, as well as two stereocenters and two trisubstituted olefins. They were also reported to exhibit strong anticholinesterase activity in pharmacological studies and markedly increase the efficiency for learning and memory in animals.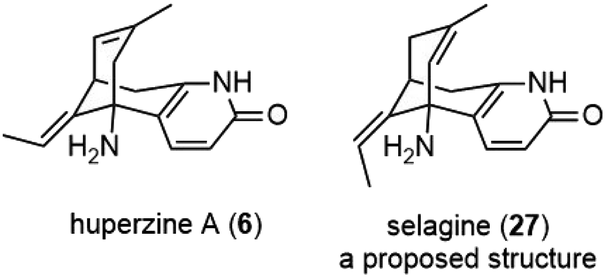 | ||
| Fig. 3 Structure of huperzine A and selagine. | ||
A structurally similar alkaloid selagine (27) was isolated by the Wiesner group in 1960 from Lycopodium selago.17 The original proposed structure for selagine (27) was an olefin isomer of huperzine A (Fig. 3). After Liu's report on the isolation of huperzine A, Ayer and coworkers reinvestigated Wiesner's work and authentical samples left behind. In 1989, they reported that selagine (27) had the same identity as huperzine A (6).18
The unique chemical structure, in combination with the potent inhibitory activity of AChE, has made huperzine A (6) a subject of intensive investigation in chemical synthesis and pharmacology.5,8,19–21 Due to the slow growth of the producing species and the low extraction yield of huperzine A (6) from the natural sources, overharvesting has caused a rapid decline in the abundance of Huperziaceae. A sustainable way for the large-scale production of huperzine A (6) is required to meet the current medical demand and for further development. After decades of research and investigation worldwide, dozens of chemical syntheses of huperzine A (6) have been reported, including a kilogram scale synthesis.
Before the structure reassignment of selagine (27) was made, two groups have already reported their synthetic efforts on the originally reported structure, and these early efforts will be briefly discussed first.
3.1. Synthetic studies of selagine
The Kende group first reported the synthesis of a model system of selagine (27) in 1979 (Scheme 3).22 The readily available 3-chloro-2-methyl-1-thiophenylpropene (28) was used as an equivalent of methacrolein. The alkylation of the dianion of 30 with 28, followed by the intramolecular cyclization of the vinyl thioether with β-keto ester under acidic conditions generated 31, and further oxidative elimination gave the bridged bicyclic structure 32. The Gravel group reported a similar strategy later, but their methacrolein equivalent compound 3-chloro-2-methyl-1-selenophenylpropene (29) was much less readily available.23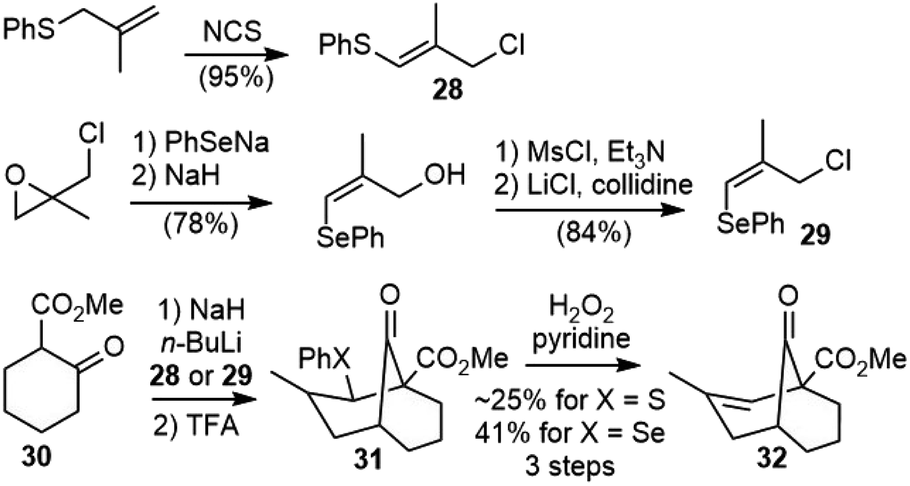 | ||
| Scheme 3 Kende's and Gravel's synthetic studies of selagine. | ||
The Kende group later reported a second strategy to the total synthesis of selagine (Scheme 4).24 Birch reduction of benzoic acid 33, followed by C-allylation and esterification, afforded 34. A palladium-catalyzed cyclization reaction of 34 gave the bridged bicyclic structure 35. Protection of the ketone and selective hydroboration of the less hindered olefin followed by oxidation gave ketone 37, which could be a key intermediate for the synthesis of the proposed structure of selagine.
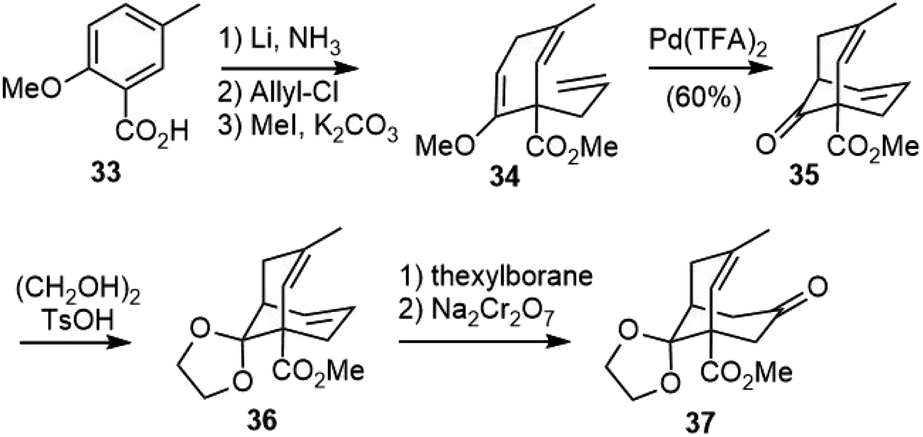 | ||
| Scheme 4 Kende's synthetic studies of selagine. | ||
3.2. Kozikowski's total synthesis
Kozikowski and coworkers reported the first total synthesis of (±)-huperzine A in 1989.25,26 They envisioned that the core structure of huperzine A could be generated by a cascade Michael addition/aldol reaction of bicyclic ketoester 39 with methacrolein 40, followed by dehydration (Scheme 5).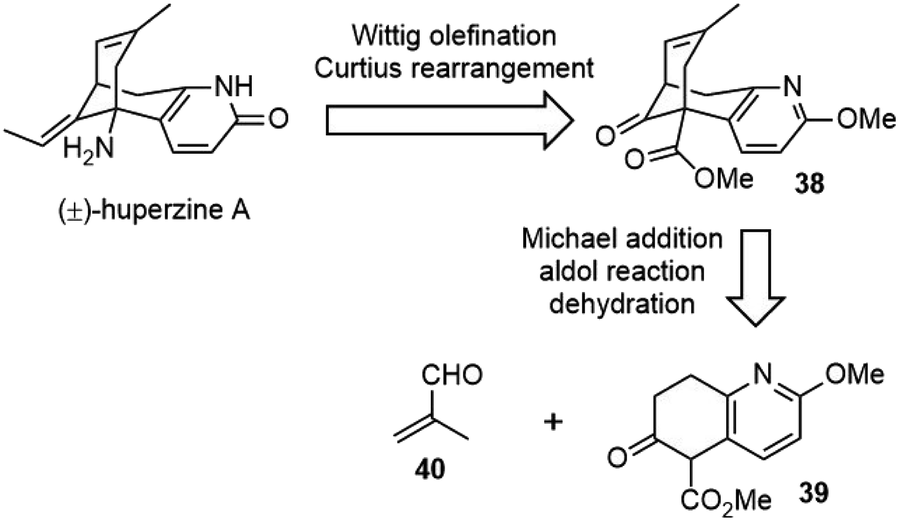 | ||
| Scheme 5 Kozikowski's retrosynthetic analysis of (±)-huperzine A. | ||
To prepare the ring-fused pyridone 43, they chose Stork's “aza-annelation” method (Scheme 6).27 Conjugate addition of the pyrrolidine enamine of cyclohexanone derivative 41 to acrylamide, followed by hydrolysis and cyclization, gave product 42 as a mixture of two olefin isomers. N-benzylation of ring nitrogen and direct dehydrogenation with DDQ could afford the desired product, but only in 40% yield. Alternatively, stepwise selenenylation followed by oxidative elimination afforded the pyridone system 43 in higher yield. Due to the poor stability of the N-protected pyridone during the later transformations, it was debenzylated by hydrogenation and O-methylated with silver carbonate and methyl iodide to afford methoxypyridine. Hydrolysis of the ketal group and α-carbomethoxylation reaction afforded the desired β-keto ester fragment 39.
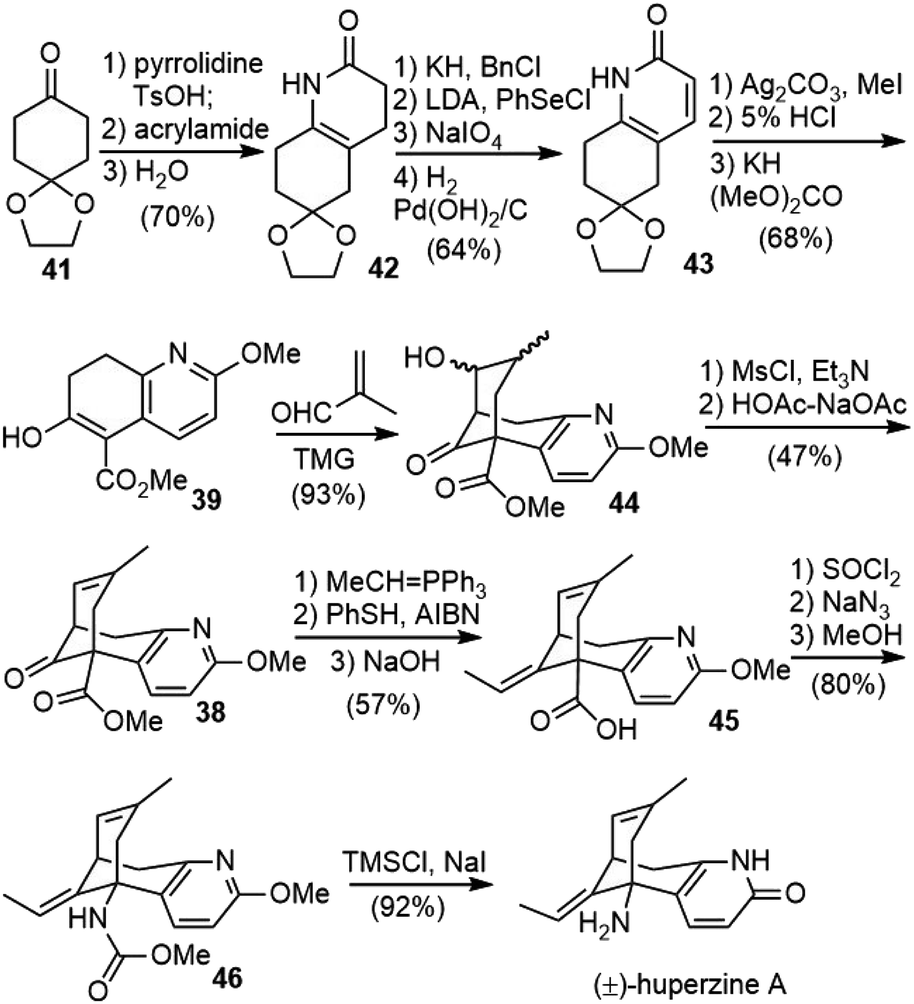 | ||
| Scheme 6 Kozikowski's total synthesis of (±)-huperzine A. | ||
The key cascade Michael addition–aldol reaction of β-keto ester 39 with methacrolein (10 equiv.) was realized with the catalysis of 20 mol% 1,1,3,3-tetramethylguanidine (TMG) to generate the bridged structure 44 in 93% yield as a mixture of inconsequential diastereomers. The ketol mixture 44 was then dehydrated to alkene 38 by the elimination of the derived mesylate in acetic acid with sodium acetate in modest yield. A Wittig olefination of the hindered ketone group afforded a trisubstituted olefin as a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Z/E mixture. The mixture of olefins was isomerized by heating with thiophenol (PhSH) and azobisisobutyronitrile (AIBN) at 170 °C to a mixture comprised predominantly of the desired E-olefin (95:5 E/Z). Hydrolysis of the E/Z mixture of esters provided solely the acid 45 of E stereochemistry, with the hindered Z-olefinic ester intact. The acid 45 was converted to carbamate 46via a three-step, one-pot process, including acid chloride synthesis, acyl azide formation with concomitant Curtius rearrangement, and methanolysis of the resulting isocyanate. Lastly, global deprotection with trimethylsilyl iodide (TMSI) finished the first total synthesis of (±)-huperzine A (±-6).
:1 Z/E mixture. The mixture of olefins was isomerized by heating with thiophenol (PhSH) and azobisisobutyronitrile (AIBN) at 170 °C to a mixture comprised predominantly of the desired E-olefin (95:5 E/Z). Hydrolysis of the E/Z mixture of esters provided solely the acid 45 of E stereochemistry, with the hindered Z-olefinic ester intact. The acid 45 was converted to carbamate 46via a three-step, one-pot process, including acid chloride synthesis, acyl azide formation with concomitant Curtius rearrangement, and methanolysis of the resulting isocyanate. Lastly, global deprotection with trimethylsilyl iodide (TMSI) finished the first total synthesis of (±)-huperzine A (±-6).
Since the first-generation synthesis of the fused pyridone intermediate 43 was lengthy and required the use of several expensive reagents, Kozikowski and coworkers examined several alternate routes (Table 1), and discovered a highly efficient three-component, one-pot process to make this key intermediate.28 Heating a mixture of ketone 41 with ammonia and methyl propiolate afforded the desired product 43 in 70% yield on a gram scale (entry 4).
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yield |
| 1 | (1) TMSCl, Et3N; (2) (Me2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)+Cl− (3) (PyCH2CONH2) +Cl−; (4) HOAc CH2)+Cl− (3) (PyCH2CONH2) +Cl−; (4) HOAc |
9% |
| 2 | (1) Pyrrolidine, TsOH; (2) CH2CClCONH2 |
11% |
| 3 | (1) LDA; ClCHCHCO2Me; (2) NH4OH |
22% |
| 4 | NH3, methyl propiolate | 70% |
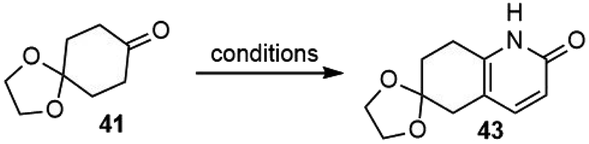
3.3. Ji's total synthesis
Shortly after Kozikowski's report, Ji and coworkers published their total synthesis of (±)-huperzine A in the same year.29 Coincidently, they used the same strategy to make the bridged bicyclic structure, but the keto ester intermediate 39 was prepared in a different way (Scheme 7). The starting material 47 was transformed into diol 48 in three steps, including O-methylation, ester reduction and side chain homologation. The diol was then transformed into a diester, which underwent a Dieckmann condensation to generate the desired fragment 39. The reaction of 39 with methacrolein under basic conditions underwent a cascade Michael addition and aldol reaction to generate the bridged system 44. A further two-step dehydration afforded olefin 38. Wittig olefination afforded the exocyclic olefin 49, favoring the undesired Z-isomer. Selective hydrolysis of the less hindered E-isomeric ester to the acid and Curtius rearrangement with diphenylphosphoryl azide (DPPA) afforded the product 50 directly.30 Final deprotection afforded (±)-huperzine A.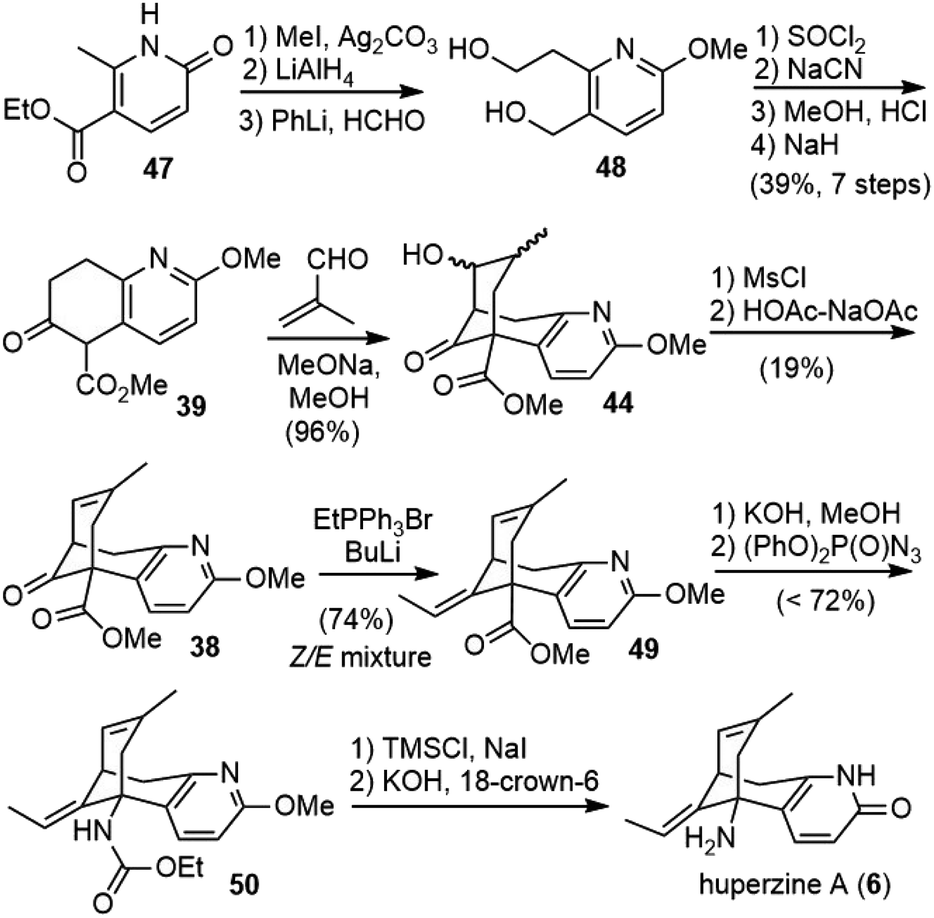 | ||
| Scheme 7 Ji's total synthesis of (±)-huperzine A. | ||
The lengthy synthesis of 39 was optimized later by Ji's group.31 A four-step approach from the readily available dimethyl 4-oxopimelate 51 was developed (Scheme 8). The enamine 52 was directly condensed with propiolamide to give pyridone 53. O-Methylation of pyridone and the Dieckmann condensation of diester 53 afforded 39 in 39% overall yield.
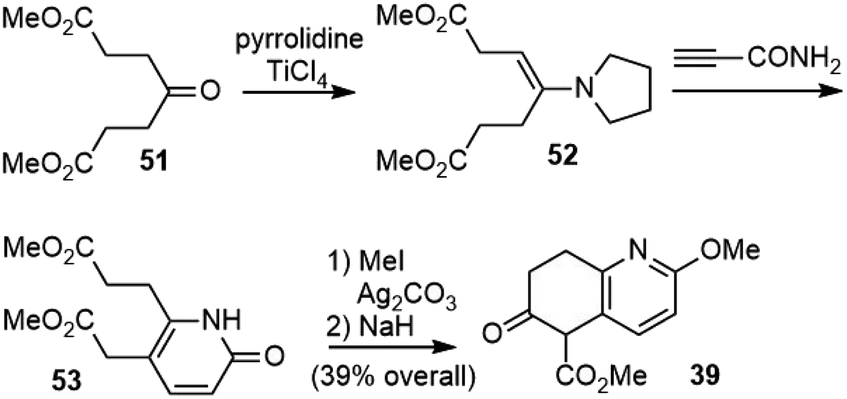 | ||
| Scheme 8 Improved synthesis of intermediate 39. | ||
3.4. Kozikowski's asymmetric total synthesis
With (±)-huperzine A successfully synthesized, Kozikowski and coworkers tested the biological activity of their synthetic sample. Both in vitro and in vivo pharmacological assays show that the racemate is half as potent as natural (−)-huperzine A, which would suggest that the (+)-isomer is poorly active as an inhibitor of AChE. Therefore, they developed a chiral auxiliary-based route to the asymmetric synthesis of both (−)-huperzine A and (+)-huperzine A.32Transesterification of 39 with (−)-8-phenylmenthol afforded chiral ester 54, which underwent the cascade Michael addition–aldol reaction with methacrolein in the presence of TMG to give a 90% yield of the product (Scheme 9). Further dehydration afforded the product 55 in a 9:1 diastereomeric ratio, which was chromatographically separable. A Wittig olefination generated the second trisubstituted olefin. The PhSH/AIBN-mediated olefin isomerization was less efficient in this case; a 1:1.4 Z/E mixture of olefin was obtained after two cycles. After chromatographic separation, the desired E isomer was reduced with lithium aluminum hydride (LiAlH4) to give alcohol 56. The alcohol was oxidized to the acid by the Jones reagent, and the subsequent Curtius rearrangement afforded 46. The final global deprotection then gave (−)-huperzine A (6).
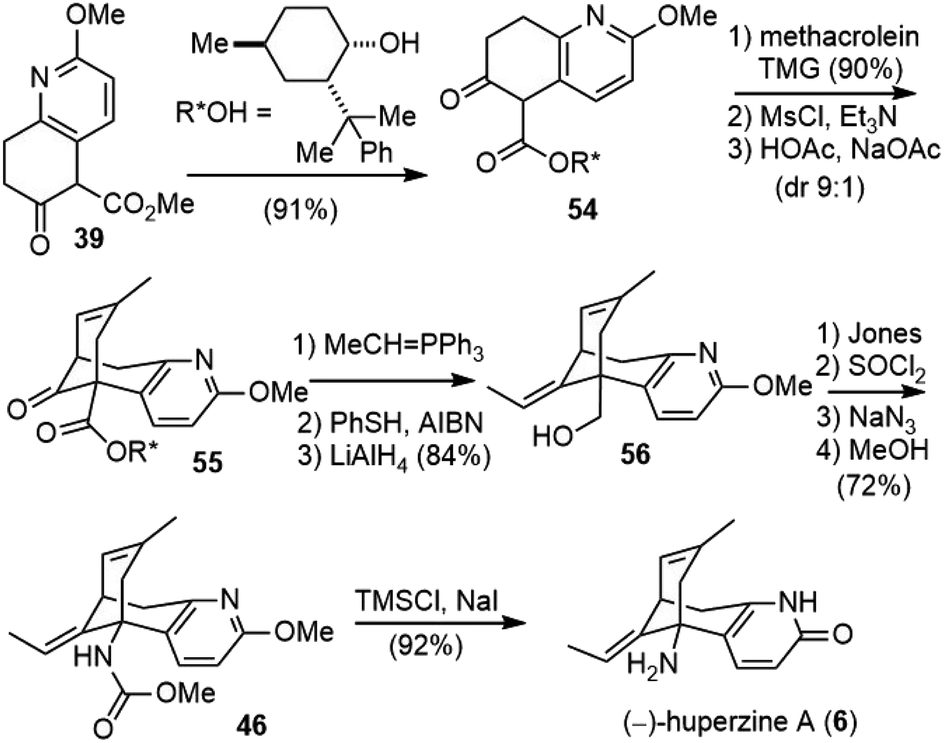 | ||
| Scheme 9 Kozikowski's total synthesis of (−)-huperzine A. | ||
They also developed a synthetic route to (+)-huperzine A at the same time (Scheme 10). Since the desired diastereomer for (+)-huperzine A synthesis was only a minor product in the above synthesis, it was more efficient to react racemic alcohol (±)-Z-56 with (S)-MTPA-Cl and the resulting diastereomers were separated by column chromatography. The optically pure ester was reduced with LiAlH4 to provide alcohol (+)-Z-56. After functional group manipulation and olefin isomerization, the acid (+)-45 was obtained, which was transformed into (+)-huperzine A following the same sequence as above.
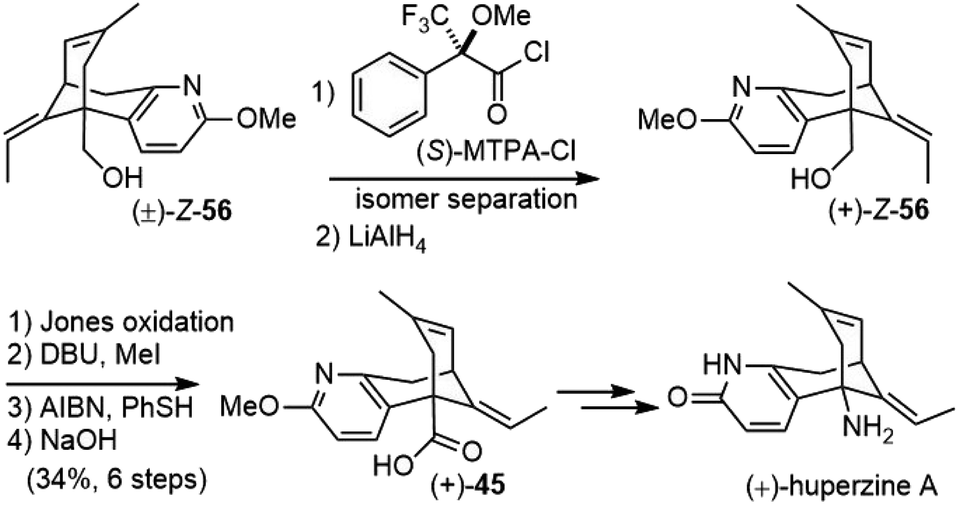 | ||
| Scheme 10 Kozikowski's total synthesis of (+)-huperzine A. | ||
With both enantiomers of huperzine A successfully prepared, Kozikowski and coworkers tested their ability to inhibit AChE from rat cortex. The IC50 value for (+)-huperzine A was found to be 1448 ± 62.4 nM, which is 33-fold larger than that of (−)-huperzine A (IC50 = 44.5 ± 2.9 nM). Racemic huperzine A has an IC50 of 71.5 ± 2.4 nM. The difference in IC50's of the two optically pure enantiomers demonstrates a reasonably large stereoselectivity of action for huperzine A.
3.5. Kozikowski's second generation total synthesis
In their first-generation total synthesis of (±)-huperzine A, dehydration of the key annulation product 44 to olefin 38 was the lowest yielding step. In order to improve the synthesis, the Kozikowski group developed a second-generation synthesis of (±)-huperzine A in 1993.33,34 To circumvent the problematic dehydration step, they chose to investigate an alternative bicycloannulation methodology reported by Huang and Lu (Scheme 11).35 The readily available 2-methylene-1,3-propanediol diacetate 57 served as an a1, a3 synthon, which reacted with dicarbanions to give methylenecyclohexane derivatives under the catalysis of palladium. The application of the bicyclic keto ester 39 as a bis-nucleophile in this transformation would make the bicyclic bridged structure 58 directly.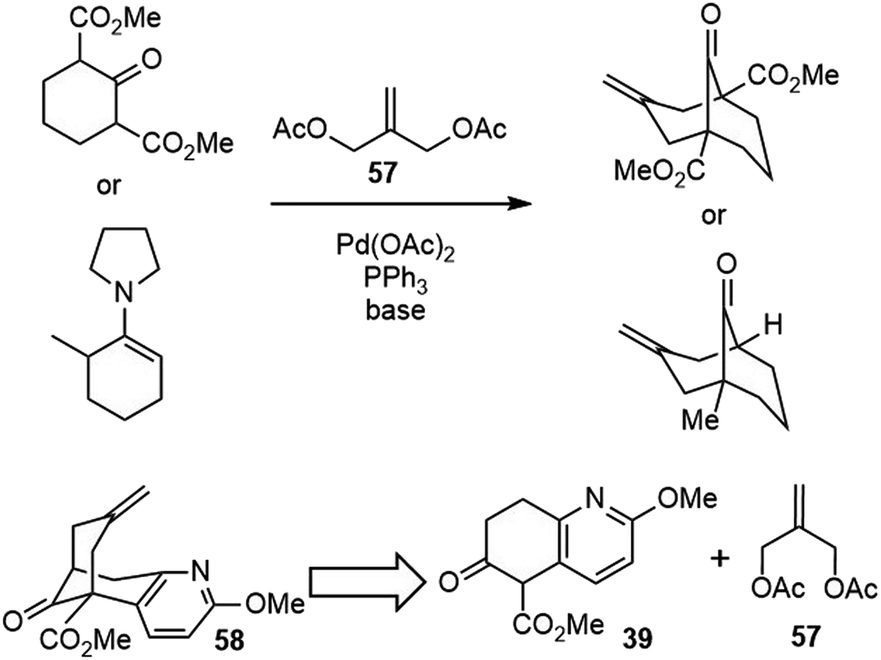 | ||
| Scheme 11 Lu's palladium-catalyzed bicycloannulation reaction and Kozikowski's retrosynthetic analysis. | ||
After the screening and optimization of the reaction conditions, it was found that the palladium-catalyzed bicycloannulation reaction of β-keto ester 39 with 2-methylene-1,3-propanediol diacetate 57 using TMG as a base afforded the methylene bridged structure 58 in 92% yield (Scheme 12). A Wittig olefination, followed by PhSH/AIBN-mediated isomerization, led to a 95:5 mixture of (E)- and (Z)-alkenes. Further hydrolysis of the ester gave the acid 59 cleanly in 63% overall yield. The Curtius rearrangement of the acid with DPPA, followed by methanolysis of the resulting isocyanate, provided the carbamate 60. Global deprotection with TMSI afforded product 61. Finally, isomerization of the exocyclic olefin with triflic acid (TfOH) afforded (±)-huperzine A as the sole product in 84% yield. The palladium-catalyzed bicycloannulation methodology effectively replaced the low-yielding, two-step elimination protocol used in their first-generation total synthesis. The overall yield of (±)-huperzine A from compound 39 was raised to 40%.
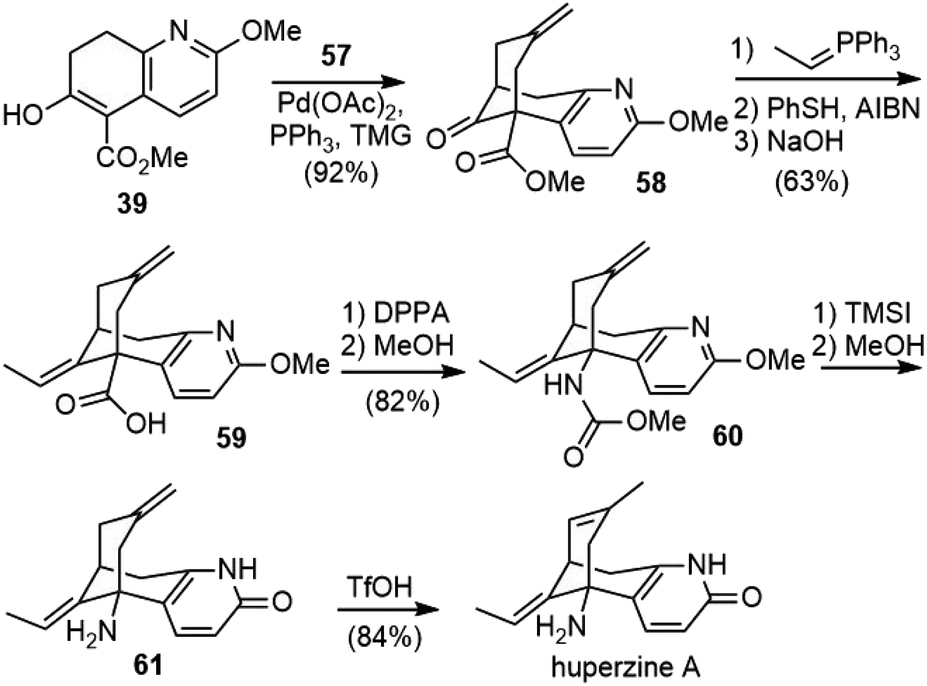 | ||
| Scheme 12 Kozikowski's second-generation total synthesis of (±)-huperzine A. | ||
3.6. Azadi-Ardakani's total synthesis
To meet the medical demand of (−)-huperzine A (6), a safe, practical and scalable process for the manufacture of (−)-huperzine A (6) was reported in 2012 by Azadi-Ardakani based on a multi-team effort (Scheme 13).36 Several hundred grams of (−)-huperzine A (6) with a chemical and optical purity of >99% had been obtained. The process was based on the previous synthetic efforts from the community, including the improved procedure developed by Kozikowski,34 the chiral bicycloannulation methodology of Terashima (vide infra)37 and Ji's modified Curtius rearrangement reaction and ether cleavage methodology.29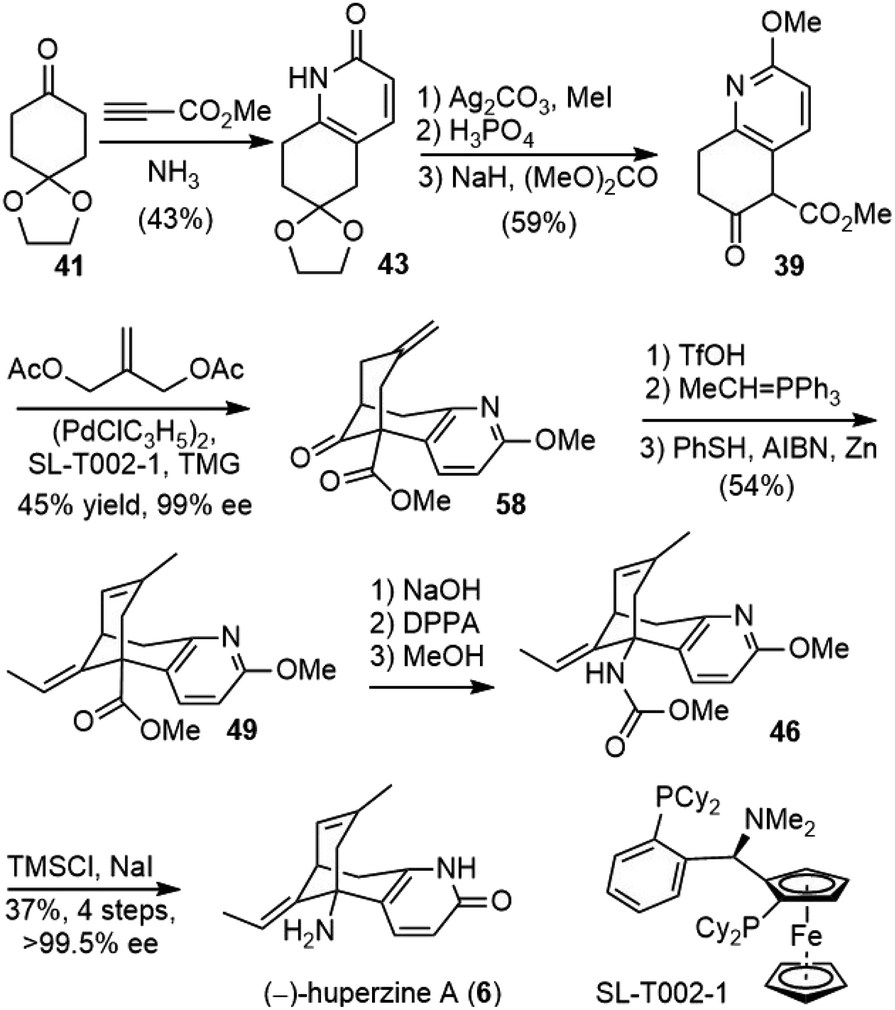 | ||
| Scheme 13 Azadi-Ardakani's total synthesis of (−)-huperzine A. | ||
Bicyclic ketoester 39 was prepared on a kilogram scale from 1,4-cyclohexanedione monoethylene ketal 41 in 43% yield, with minor modifications of Kozikowski's simplified procedure.28O-methylation of 43 was best performed under phase-transfer conditions with 1 equiv. of Ag2CO3, 3 equiv. of MeI, and 0.5 equiv. of benzyltriethylammonium chloride as the phase transfer catalyst. The ethylene ketal was hydrolyzed to a ketone in dilute phosphoric acid in 98% yield. The reaction of the ketone with dimethyl carbonate (30 volumes) as both the solvent and the reactant in the presence of sodium hydride (1.2 equiv.) gave the β-keto ester 39 in 65% yield. All the above-mentioned reactions were performed on a kilogram scale, and more than 9 kilograms of 39 had been obtained.
The palladium-catalyzed asymmetric bicycloannulation reaction of β-keto ester 39 with compound 57 was based on the work of Terashima.37 After the screening of a variety of ligands, the Taniaphos ligand SL-T002-1 was identified as the best lead ligand. Further optimization of reaction parameters led to the desired annulation product 58 with 99% conversion and 84% ee, under the catalysis of 2 mol% palladium and ligand each. Recrystallisation from isopropyl alcohol upgraded 58 to 99% ee in 45% overall yield.
The exocyclic olefin was isomerized to an endocyclic olefin with TfOH in ethylene dichloride at room temperature over just 1 h. A Wittig olefination afforded the product as a mixture of Z/E olefin isomers, and the phosphine byproduct was removed by acid/base wash. The isomerization of olefin to enhance the E-isomer ratio was conducted with PhSH and AIBN in the presence of a catalytic amount of fresh zinc dust. The role of zinc may be responsible for removing trace amounts of disulfide, which would inhibit alkene isomerization. Crystallization from methanol furnished the pure product 49 in 54% yield from intermediate 58 with an E-to-Z-isomer ratio of greater than 100 and a purity (E + Z isomers) of 99.4%. The ester of 49 was hydrolyzed with NaOH to give an acid. The Curtius rearrangement of the acid with DPPA gave the isocyanate, which was converted into carbamate 46 after refluxing with methanol. The carbamate and O-methyl groups were removed with TMSI, which was generated in situ by the reaction of trimethylsilyl chloride and sodium iodide. Repeated crystallization furnished (−)-huperzine A in a modest yield with high purity.
The whole process consisted of 11 chemical stages starting from commercially available materials with only nine isolation steps and no chromatography purification. A total of 275 g (−)-huperzine A of high chemical and optical purity has been manufactured in a cGMP environment.
3.7. Fukuyama's total synthesis
In 2009, the Fukuyama group reported a new strategy to the total synthesis of (−)-huperzine A.38 Their synthesis featured a desymmetrization reaction to set up the chiral centers, a serendipitously discovered cation-olefin cyclization to build the bicyclic bridged structure, and a new protocol for 2-pyridone synthesis.The desymmetrization of commercially available anhydride 62 with a stoichiometric amount of quinine and excess benzyl alcohol gave carboxylic acid 63 in 93% ee (Scheme 14).39 Selective reduction of the carboxylic acid to alcohol via a mixed anhydride method afforded a lactone, which was hydrogenated to provide 64. Treatment of 64 with 2 equiv. of potassium hexamethyldisilazide (KHMDS) induced the cleavage of the tetrahydrofuran ring and the formation of a dienolate, which regio- and diastereoselectively reacted with methallyl bromide at the α-position of the lactone from the convex face. Homoallylic alcohol 65 was isolated after treatment with TBAF. Directed epoxidation of homoallylic alcohol 65 using a catalytic amount of VO(OEt)3 afforded the epoxide with complete diastereoselectivity. The Swern oxidation of the alcohol caused simultaneous cleavage of the epoxide to furnish hydroxyenone 66 in good yield. Attempted protection of the hydroxy group in 66 with TBSOTf led to a cation-olefin cyclization product, and further optimization found that treatment of 66 with a catalytic amount of TfOH caused a cation-olefin cyclization and afforded the tricyclic compound 67 in 61% yield. The protection of the alcohol and ring opening of the lactone with phenylthiolate generated carboxylic acid 68. The Curtius rearrangement of acid 68 with DPPA produced a methyl carbamate. The oxidation of the sulfide and subsequent elimination gave unsaturated ketone 69.
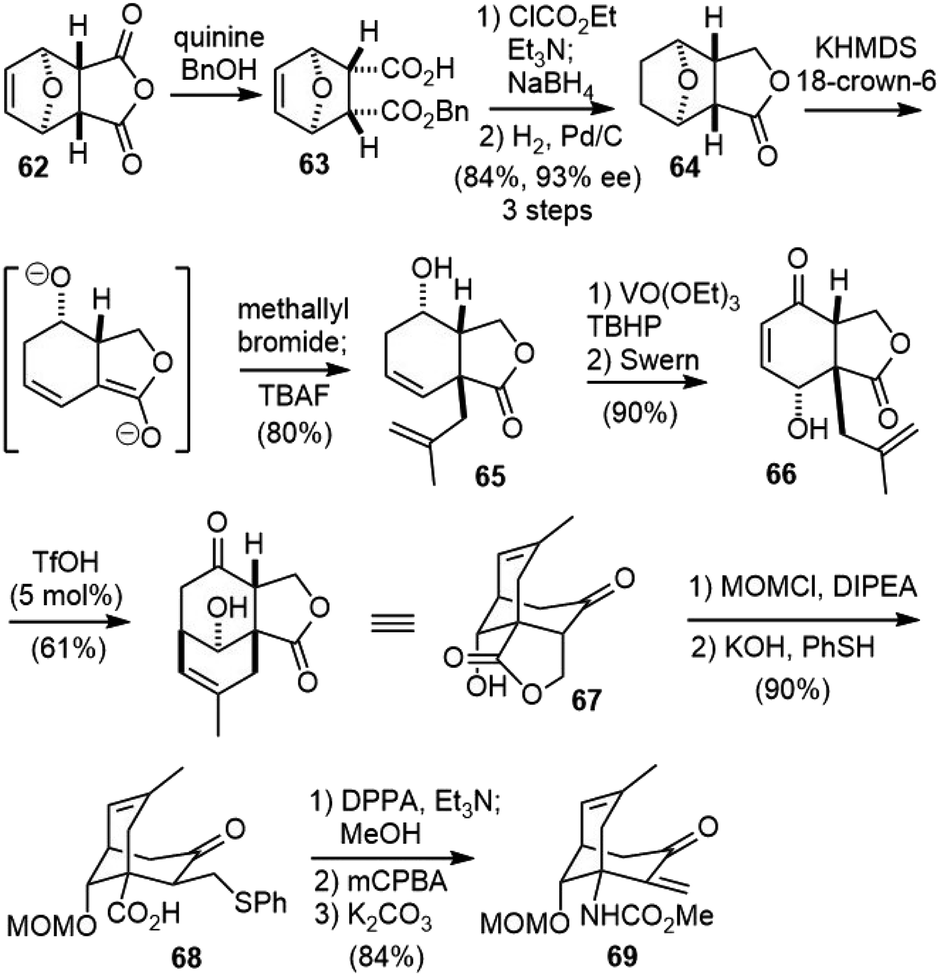 | ||
| Scheme 14 Fukuyama's total synthesis of (−)-huperzine A (part 1). | ||
Conjugate addition of sulfinylamide 70 to 69 followed by cyclization and desulfination afforded pyrone 71 (Scheme 15). Reaction with NH3 converted pyrone 71 into the corresponding pyridone, which was then protected as 2-methoxypyridine 72. Deprotection and oxidation of the resulting alcohol afforded ketone 73. Installation of the ethylidene moiety with Wittig olefination of ketone was unsuccessful, and a stepwise process was developed instead. The addition of vinyllithium to ketone 73, followed by treatment with SOCl2 gave the rearranged allyl chloride 74 as the sole isomer. The reduction of the allyl chloride 74 with LiBHEt3 furnished the exocyclic olefin, and final deprotection with TMSI finished the total synthesis of (−)-huperzine A (6). The overall process was accomplished in 23 steps and 1.8% yield from commercially available anhydride 62.
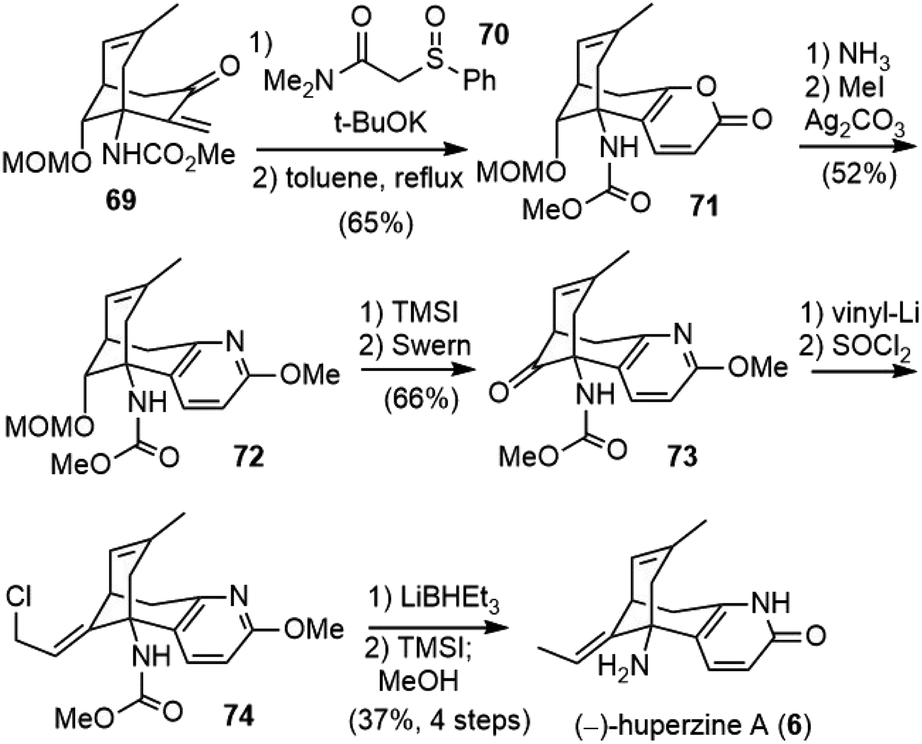 | ||
| Scheme 15 Fukuyama's total synthesis of (−)-huperzine A (part 2). | ||
The protocol for substituted 2-pyridone synthesis was further optimized to a one-pot process by the Fukuyama group.40 The conjugate addition of 2-(phenylsulfinyl)acetamide 75 to α,β-unsaturated ketones, followed by cyclization and sulfoxide elimination afforded 2-pyridones in good yields. This protocol was later applied in the total synthesis of several other natural products with 2-pyridones (Scheme 16).41–43
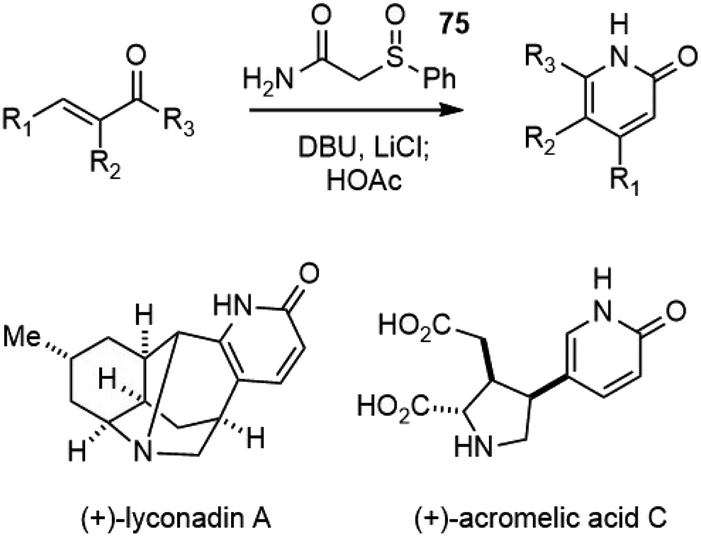 | ||
| Scheme 16 Fukuyama's synthesis of 2-pyridones. | ||
3.8. Herzon's total synthesis
Herzon and coworkers reported a robust and scalable total synthesis strategy of (−)-huperzine A (6) from a chiral pool compound (R)-pulegone (76) in 2011.44 They first transformed (R)-pulegone (76) to (R)-(+)-4-methylcyclohex-2-ene-1-one (78) following a procedure reported in the literature (Scheme 17a).45 This sequence was reproducibly executed on multigram scales, but required several steps. A more straightforward route was developed later (Scheme 17b).46 The deprotonation of (R)-(+)-pulegone and trapping of the resulting enolate with N-phenyl bis(trifluoromethanesulfonimide) (PhNTf2) provided a vinyl triflate. Selective ozonolysis of the electron-rich olefin formed ketone 79. A palladium-catalyzed reduction reaction removed the α-triflate group and provided the target enone 78 (44%, >99% ee from 76). Overall, this sequence requires three manipulations and one chromatographic purification, and employs inexpensive reagents and starting materials, which provides a convenient method to access multigram quantities of 78.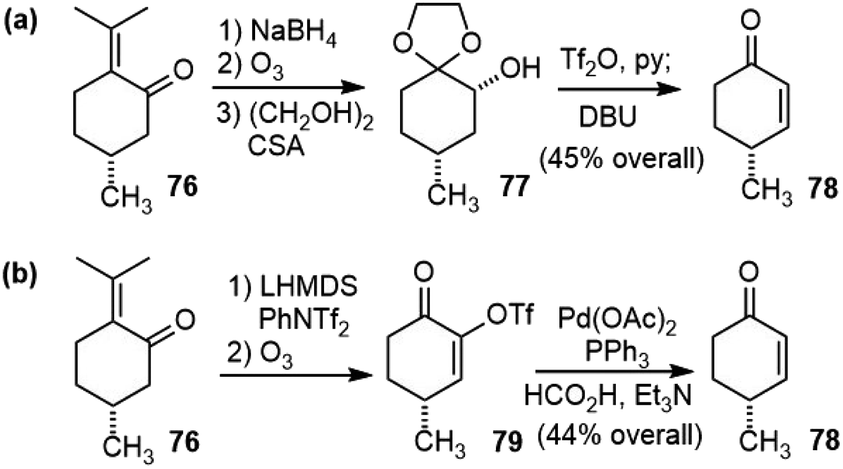 | ||
| Scheme 17 Synthesis of compound 78: (a) Lee's original procedure; (b) Herzon's modified procedure. | ||
With enough amount of 78 available, they went on to finish the total synthesis of (−)-huperzine A (Scheme 18). Conjugate addition of the silylcuprate (PhMe2SiLiCuI) to 78, followed by alkylation of the incipient enolate with bromide 80 afforded the product 81 as a single diastereomer in 84–91% yield. Kinetically controlled deprotonation of 81 and trapping of the resulting enolate with p-toluenesulfonyl cyanide, followed by immediate work up, formed a cyanoketone. The cyanoketone was then subjected to an intramolecular enolate heteroarylation reaction to generate the bicyclic bridged structure 82. The optimal conditions employed bis(tri-tert-butylphosphine) palladium (0) (Pd(Pt-Bu3)2) as the catalyst and sodium tert-butoxide (NaOt-Bu) as the base. Wittig olefination of 82 at a low concentration (0.01 M) afforded the olefin in good yield as a 5:1 mixture of E/Z isomers on a gram scale. The Fleming-Tamao oxidation of the silyl group provided the alcohol 83. The dehydration of the alcohol by the Burgess reagent, and the hydration of the nitrile group by a platinum phosphinito catalyst afforded amide 84.47 Finally, the Hofmann rearrangement reaction with iodosobenzene bis(trifluoroacetate) (PIFA), and global deprotection with TMSI afforded (−)-huperzine A. It took eight steps from cyclohexenone 78 and three chromatographic purifications to deliver over 3.5 g of (−)-huperzine A (6).
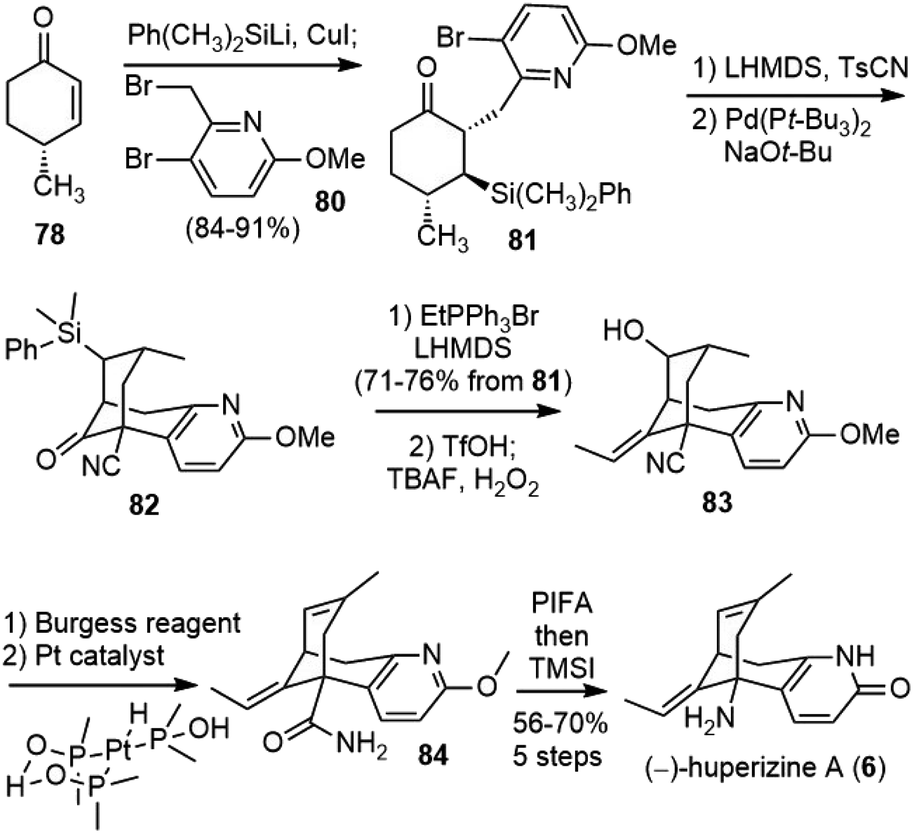 | ||
| Scheme 18 Herzon's total synthesis of (−)-huperzine A. | ||
3.9. Lin and Sun's total synthesis
Lin and Sun reported a total synthesis of (−)-huperzine A (6) in 2012 (Scheme 19).48,49 Their approach shared some common features with Herzon's route. Both syntheses used (R)-pulegone (76) as their chiral starting material and formed the same C–C bond to assemble the bridged bicyclic structure.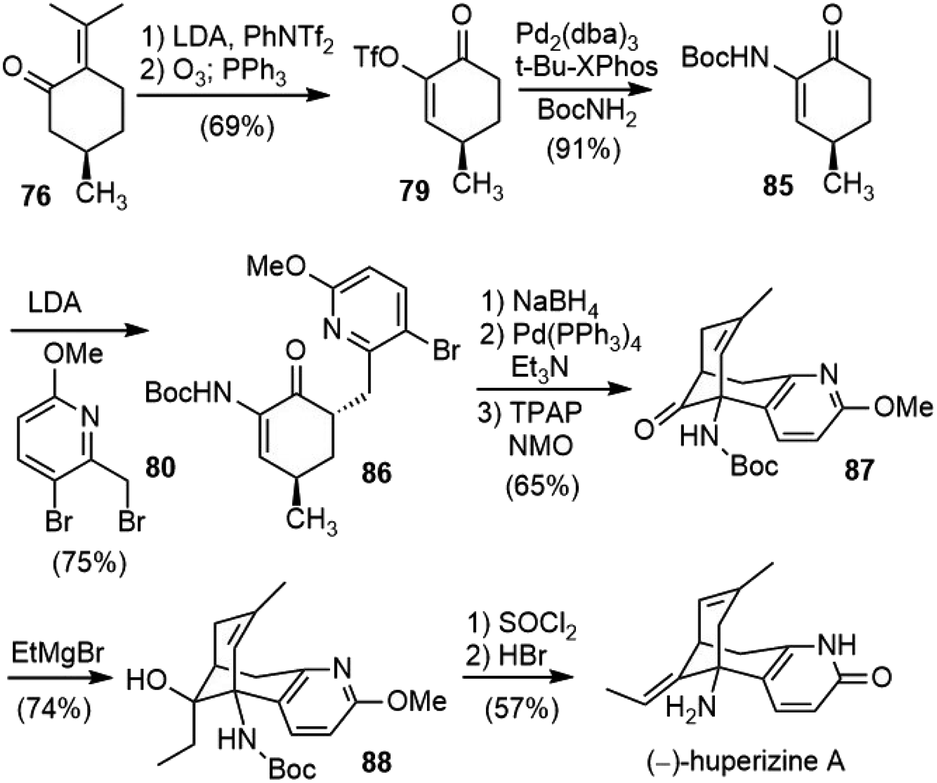 | ||
| Scheme 19 Lin and Sun's total synthesis of (−)-huperzine A. | ||
(R)-Pulegone (76) was transformed into an enol triflate with LDA/PhNTf2, followed by ozonolysis to generate 79 in 69% overall yield. The Buchwald–Hartwig coupling reaction of 79 with BocNH2 catalyzed by Pd2(dba)3/t-Bu-XPhos in toluene established the key C–N bond to give fragment 85 on a decagram scale. The kinetic deprotonation of 85 with 2.5 equiv of LDA generated a dianion, trapping of which with bromide 80 afforded product 86 in 75% yield with >20/1 dr and >94% ee. Inspired by Mann's work (vide infra),50 they found that the intramolecular Heck reaction worked well after reduction of the ketone to an allylic alcohol, and further oxidation of the Heck product provided ketone 87 in good yield over three steps from 86. The construction of the exocyclic olefin proved nontrivial. Attempted Wittig and Julia olefination reactions failed, and an addition–elimination protocol was thus developed. The reaction of 87 with ethylmagnesium bromide furnished the tertiary alcohol 88 in 74% yield. The exposure of 88 to SOCl2 at room temperature led to the slow dehydration of the alcohol. Further treatment with aqueous HBr effected demethylation and concomitant double bond transposition, providing (−)-huperzine A (6) in 57% yield.
The total synthesis of (−)-huperzine A was finished in 10 steps with a 17% overall yield from commercially abundant (R)-pulegone. Most of the steps have been operated on multigram scales, which provided an option for the supply of (−)-huperzine A.
3.10. White's total synthesis
In 2013, White and coworkers reported the total synthesis of (−)-huperzine A from (S)-4-hydroxycyclohex-2-enone (89) in 17 steps.51,52 Their strategy was distinct from previous syntheses, in which two strained cyclobutanes were synthesized first, and then fragmented sequentially in controlled ways for the synthesis of the pyridone ring and the bridged bicyclic structure.53,54The starting material (S)-(−)-4-hydroxycyclohex-2-enone (89) was obtained from (−)-quinic acid (90) in six steps following a known procedure (Scheme 20).55 The etherification of 89 with trans-crotyl bromide in the presence of silver(I) oxide (Ag2O) gave a diene, which was irradiated through Pyrex glass to form the intramolecular [2 + 2]-adduct 92. Ketone 92 was transformed into a silyl enol ether, followed by an allylic oxidative azidation reaction with iodosobenzene (PhIO) and trimethylsilyl azide (TMSN3) to give an allylic azide as a single epimer.56 The reduction of the azide to amine, followed by acylation with α-phenylselenylacrylic acid generated amide 93. Upon treatment with trimethylaluminum (AlMe3), 93 underwent an intramolecular formal [2 + 2] cyclization to generate the highly strained cycloadduct 94 in good yield.
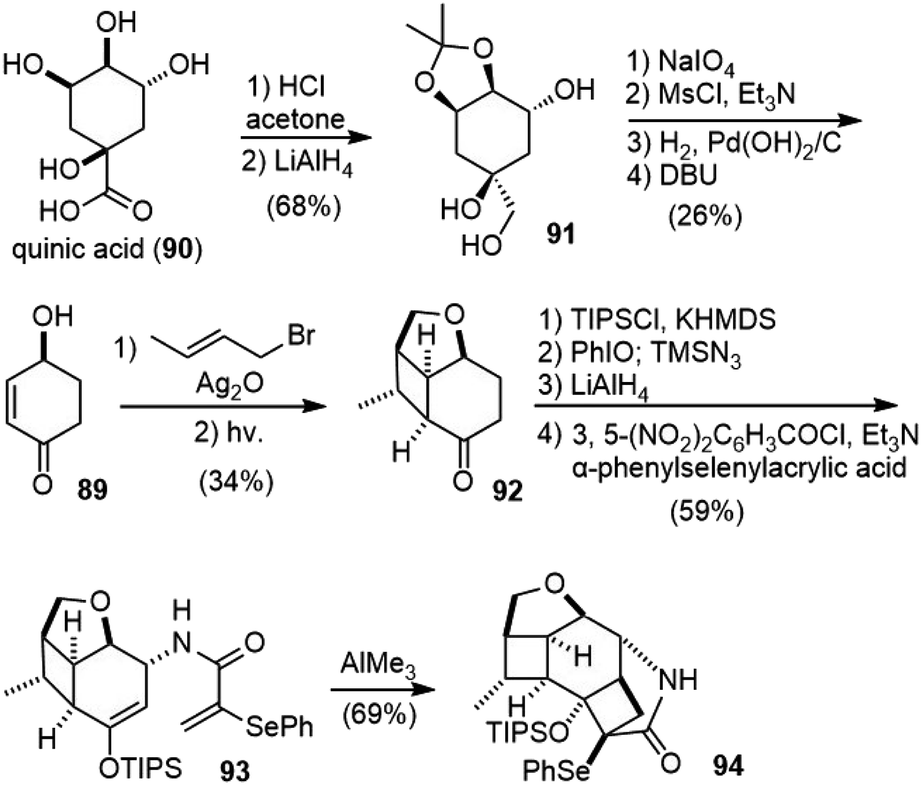 | ||
| Scheme 20 White's total synthesis of (−)-huperzine A (part 1). | ||
Cleavage of the silyl ether in 94 with aqueous fluoride caused a concomitant retro-aldol fragmentation to release the strain and furnished α-selenyl δ-lactams 95. The oxidation of 95 with sodium periodate (NaIO4) led directly to pyridone 96 in good yield (Scheme 21). O-Methylation of 96, followed by reductive cleavage of the C–O bond with activated zinc dust, generated hydroxy ketone 97. The hydroxy group was transformed into an olefin in 5 steps and gave ketone 98. Initial efforts to use the oxime or imine derivatives of ketone 98 for the skeletal rearrangement were unsuccessful. Instead, when ketone 98 was treated with methyl carbamate and anhydrous p-toluenesulfonic acid, an immediate reaction took place to deliver product 46 in good yield. Presumably, an intramolecular aza-Prins reaction triggered a stereocontrolled scission of a cyclobutylcarbinyl cation to create the bridged framework of the natural alkaloid. While alternative fragmentation modes of the cyclobutane in imine 99 are conceivable, a stereoelectronic bias should favor the desired fragmentation pathway. Treatment of 46 with TMSI completed the total synthesis of (−)-huperzine A.
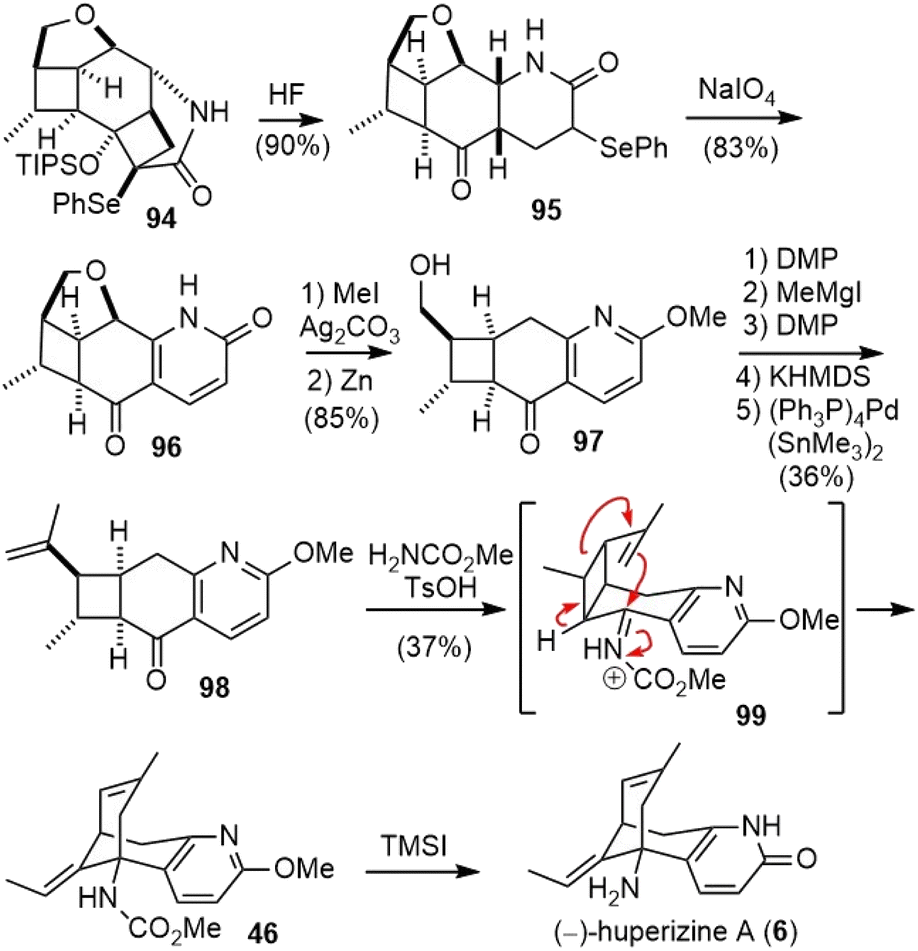 | ||
| Scheme 21 White's total synthesis of (−)-huperzine A (part 2). | ||
3.11. Langlois' formal total synthesis
In 1999, Langlois and coworkers reported a formal total synthesis of (+)-huperzine A (Scheme 22).57,58 Similar to Kozikowski's second-generation total synthesis of (±)-huperzine A, they also utilized Lu's palladium-catalyzed bicycloannulation reaction as the key step to build the bridged structure.35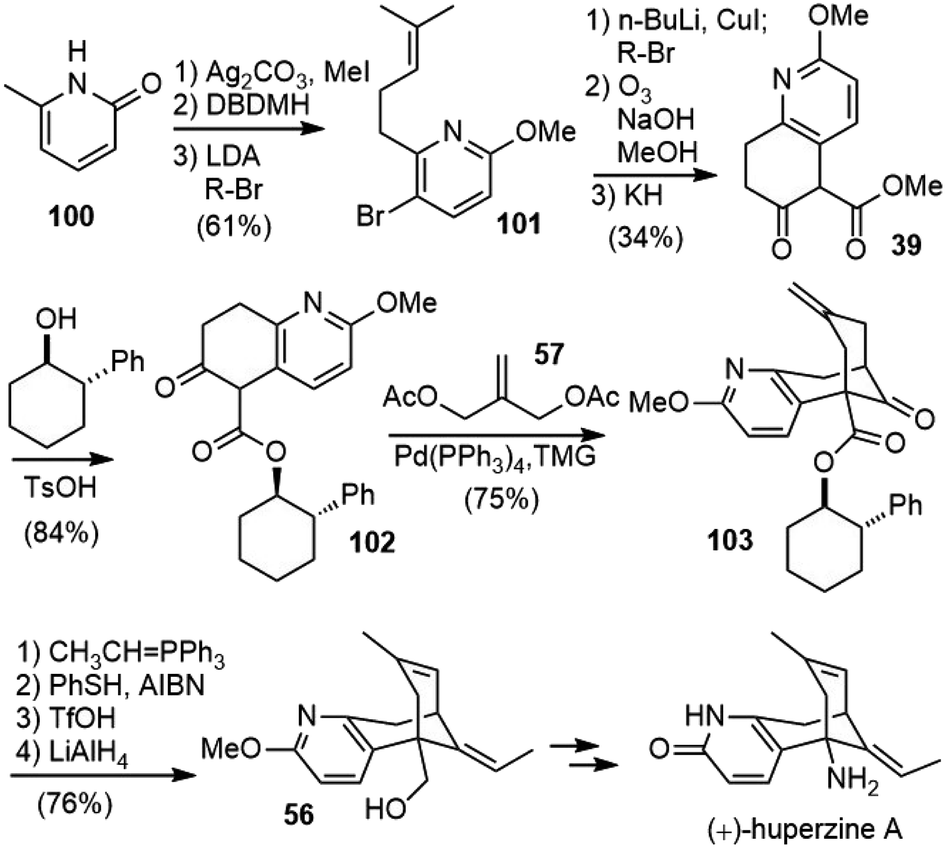 | ||
| Scheme 22 Langlois's formal total synthesis of (+)-huperzine A. | ||
They prepared β-keto ester 39 in a different way. O-Methylation of 2-hydroxy-6-methylpyridine (100) followed by regioselective bromination with dibromodimethylhydantoin (DBDMH) afforded 5-bromo-2-methoxy-6-methylpyridine. Deprotonation of the methyl side chain with LDA followed by alkylation with 4-bromo-2-methyl-2-butene (R-Br) gave the expected pyridine derivative 101 in 74% yield. The bromide was then transformed into an alkyl side chain, ozonolysis of the olefin in an alkaline medium afforded the diester directly, and further Dieckmann cyclisation of the diester with potassium hydride (KH) afforded the desired β-keto ester 39. Transesterification of 39 with (1R,2S)-2-phenylcyclohexanol afforded the corresponding chiral ester 102. The key palladium-catalyzed annulation reaction with 1.1 equiv. each of 57 and TMG afforded the expected annulated compound 103 in 75% yield. A diastereomeric excess of 92% for this reaction was obtained by the Mosher ester analysis of an alcohol derivative. Wittig olefination of 103 followed by radical isomerization with PhSH/AIBN gave a 15/85 mixture of Z- and E-isomers. It was subsequently found that Z/E and exo-endo isomerization of two double bonds could be conducted in one step by treatment with TfOH to afford the desired product in good yield. The reduction of the ester gave the known primary alcohol 56, which was a direct synthetic precursor of (+)-huperzine A in Kozikowski's total synthesis.32
3.12. Camps' formal total synthesis
In 2000, the Camps group reported their work on the formal synthesis of (±)-huperzine A (Scheme 23).59,60 A Michael addition–aldol reaction of β-keto ester 104 with methacrolein generated the bridged structure. The formation of its derived thiocarbonate, followed by pyrolytic syn-elimination, generated the olefin product 105. Following Kozikowski's and Ji's procedure, 105 was transformed to carbamate 107, via acid 106. The formation of the pyridone ring using Stork's “aza-annelation” reaction gave the known compound 46, which was transformed into (±)-huperzine A after deprotection.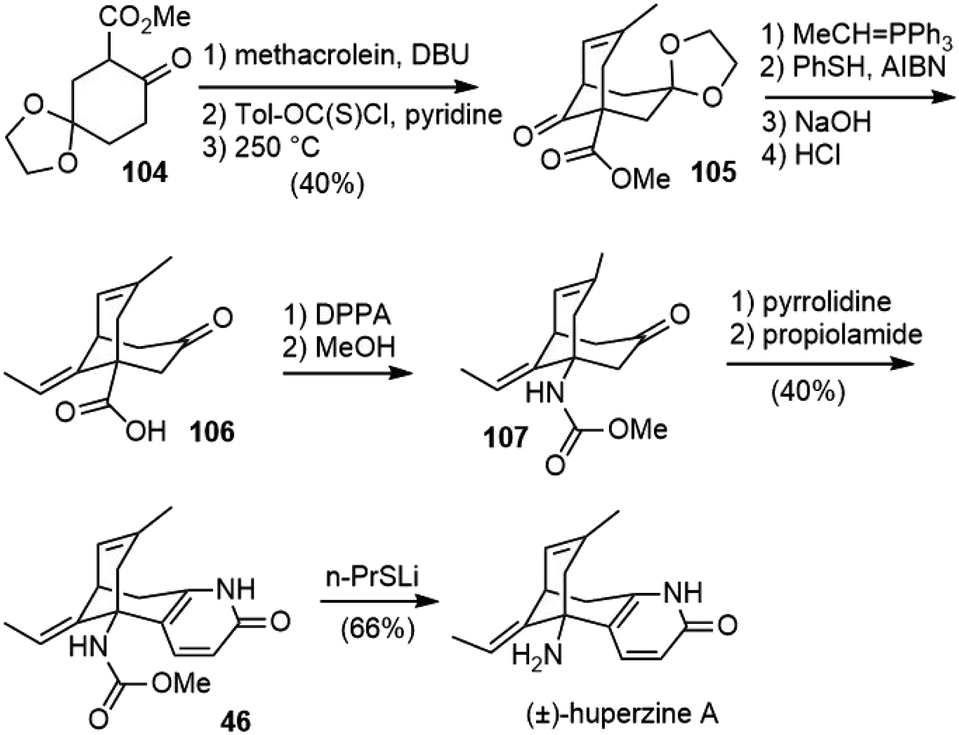 | ||
| Scheme 23 Camps' formal total synthesis of (±)-huperzine A. | ||
3.13. Lee's formal total synthesis
The Lee group reported a formal total synthesis of (±)-huperzine A in 2002 (Scheme 24).61 Starting from the same β-keto ester 39 as previous syntheses, the allylation reaction generated the quaternary center to afford compound 108, which underwent a Mn(III)-mediated intramolecular oxidative radical cyclization to the corresponding bicyclic compounds 109 as a mixture of olefin isomers; both were intermediates in Kozikowski's second-generation total synthesis.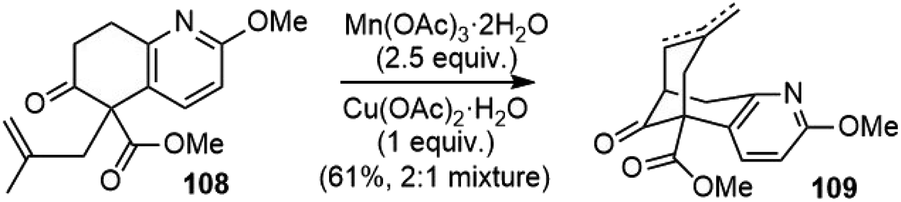 | ||
| Scheme 24 Lee's formal total synthesis of (±)-huperzine A. | ||
3.14. Mann's formal total synthesis
In 2007, Mann and coworkers reported a concise and convergent synthesis of the tricyclic skeleton of (±)-huperzine A (Scheme 25).50 Regioselective α-alkylation of the readily available 2-substituted cyclohexanone 110 was accomplished using NaHMDS as the base and trapping the resulting enolate with iodide 111. The attempted Heck cyclization of the alkylation product 112 was unsuccessful. Alternatively, the Luche reduction of the ketone to alcohol followed by the intramolecular Heck reaction generated the tricyclic product 113 in good yield. Similar reactions to form the same C–C bond were employed later in Herzon's and Lin's total syntheses (vide supra).44,48,49 The disubstituted olefin was then transformed into the trisubstituted olefin 114 in 4 steps. Desilylation and alcohol oxidation followed by esterification afforded the known intermediate 38 in Kozikowski and Ji's total synthesis.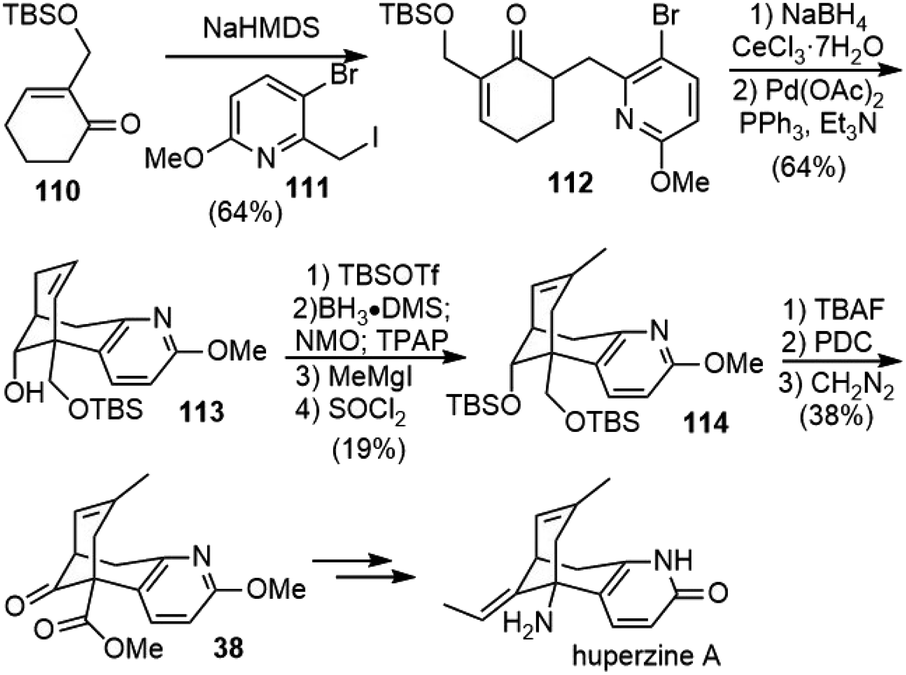 | ||
| Scheme 25 Mann's formal total synthesis of (±)-huperzine A. | ||
3.15. Asymmetric Michael addition–aldol reaction
In Kozikowski's25 and Ji's29 total syntheses of (±)-huperzine A, the same cascade Michael addition and aldol reaction of intermediate 39 with methacrolein were used to generate the bridged core structure 38. Developing an asymmetric version of this key reaction would result in an asymmetric synthesis of huperzine A. Therefore, several groups reinvestigated this reaction and reported highly efficient systems for this cascade reaction (Scheme 26).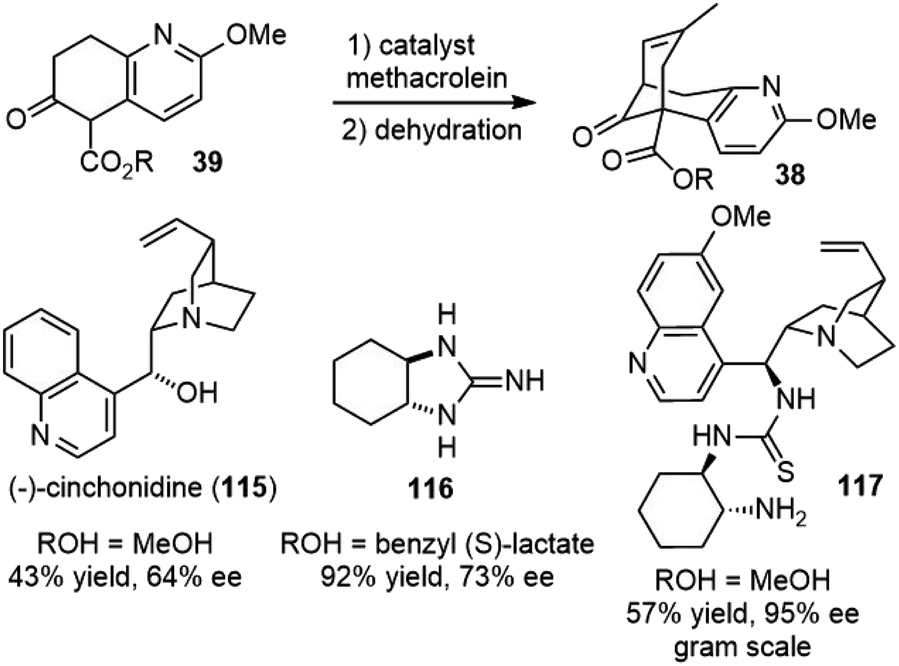 | ||
| Scheme 26 Asymmetric Michael-aldol reaction. | ||
In 1997, Terashima and coworkers reported that the reaction of compound 39 with large excess of methacrolein (10 equiv.) mediated by stoichiometric amount of (−)-cinchonidine (115) led to the bridged product in 43% yield and 64% ee after dehydration. (−)-Cinchonidine (115) could be recovered by concentration and subsequent filtration. An enantiomerically pure product (>99% ee) was readily obtained by recrystallization of the partially optically active compound from hexane.62,63
In 2003, Ma and coworkers reported chiral guanidines as efficient catalysts for this transformation. The best result was obtained when trans-1,2-diaminocyclohexane-derived guanidine 116 was employed as a catalyst and lactate ester was used as the substrate.64
In 2012, Yao and coworkers reported a multifunctional organocatalyst 117 for the cascade reaction. The bicyclic core of (−)-huperzine A was obtained with up to 95% ee on a gram scale. The methodology was also eligible to synthesize a variety of bicyclo[3.3.1]nona-2,6-dien-9-ones in high enantiopurities, and thus, it is useful for the future development of novel huperzine A analogs with medicinal interests.65
3.16. Asymmetric palladium-catalyzed annulation
Both Kozikowski's34 second-generation total synthesis and Langlois'57 formal total synthesis of (±)-huperzine A utilized Lu's palladium-catalyzed bicycloannunaltion reaction to build the bridged bicyclic structure. Developing an asymmetric version of this key reaction would also lead to enantioselective syntheses of huperzine A (Scheme 27).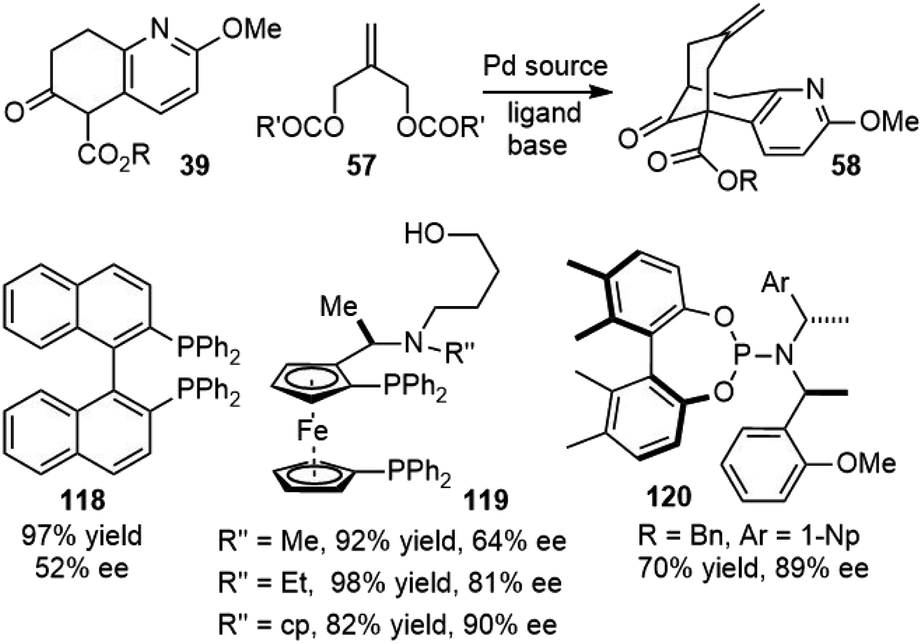 | ||
| Scheme 27 Asymmetric palladium-catalyzed annulation. | ||
In 1997, Terashima and coworkers reported the first catalytic asymmetric variant of this reaction using chiral ferrocenylphosphine ligands 119.37,63 They found that the substituents in nitrogen played important roles for the enantioselectivity. This work formed the basis of a scalable process for the manufacture of (−)-huperzine A reported by Azadi-Ardakani in 2012 (vide supra).36
Bai and coworkers also screened a library of bisphosphine ligands for this transformation, modest ee was obtained with BINAP 118 as the ligand, but up to 90% ee could be obtained using a slightly modified ferrocenylphosphine ligand 119.66,67
In 2014, Ojima and coworkers screened a small library of fine-tunable monodentate phosphoramidite (MPN) ligands 120, and the allylic substrates for the enantioselective Pd-catalyzed tandem allylic alkylation reaction.68 The reaction generated the key tricyclic intermediate with high enantiopurity for the formal total synthesis of (−)-huperzine A.
3.17. Synthesis of the core structure
Other than the successful total synthesis and formal synthesis of huperzine A, there are also several reports on the synthesis of the bridged bicyclic core structures.Mulzer and coworkers developed a synthesis strategy of 5-desamino huperzine A in 2001 (Scheme 28).69 A double Michael addition of dimethyl 1,3-acetone dicarboxylate to benzoquinone monoketal 121, followed by selective monodecarboxylation with LiOH in DMF gave keto ester 122. A Wittig olefination of the non-enolized carbonyl group gave an exocyclic olefin, which was isomerized by Pd/C to the thermodynamically favored endocyclic olefin. A further Michael reaction with acrylonitrile gave 123. The decarboxylation of 123 with LiBr led to a diastereomeric mixture of keto nitriles, and further reaction with ammonia in methanol gave dihydropyridone, which was desaturated with sulfuryl chloride to afford 124. The ketal was hydrolyzed and the resulting ketone was subjected to a Wittig olefination to give an E/Z mixture of desamino huperzine A (125). It was found that the two isomers were inseparable, and that radical isomerization was less effective than the previous synthesis of huperzine A. Therefore, 125 was transformed into pyridone O-acetates, and isomers were separated at this stage by semi-preparative HPLC. Deprotection with ammonia furnished the desired desamino huperzine 126.
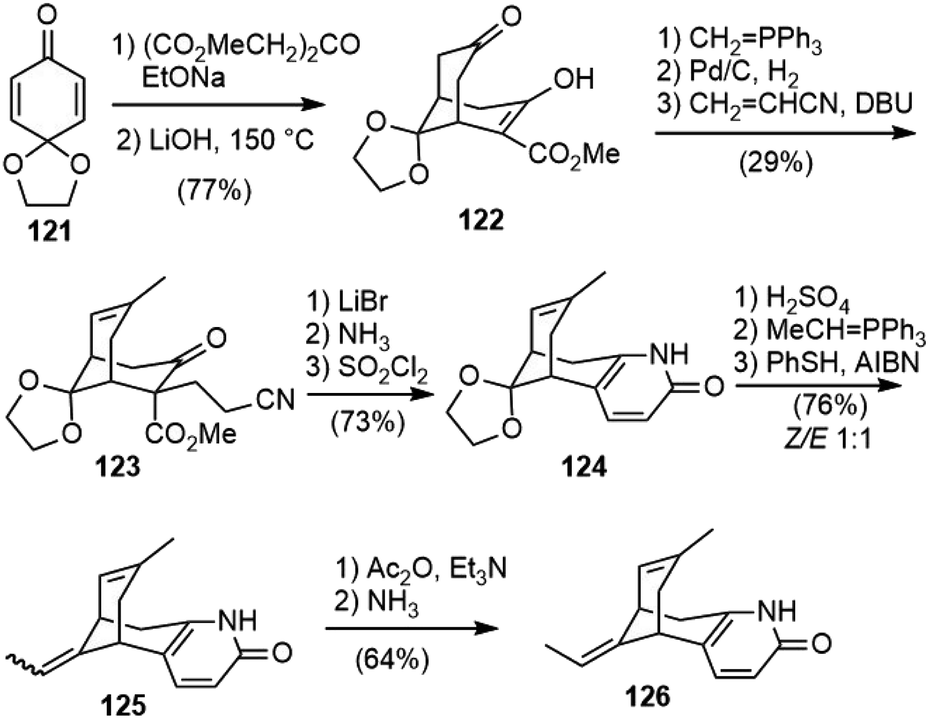 | ||
| Scheme 28 Mulzer's synthesis of 5-deamino-huperzine A. | ||
Caprio and coworkers reported a radical approach to the core structure of huperzine A (Scheme 29).70,71 The formylation of bromopyridine 127 and reaction with but-3-enylmagnesium bromide gave an alcohol. Dess–Martin oxidation and treatment of the resulting ketone with allylmagnesium bromide gave diene 128. Ring closing olefin metathesis of 128 with Grubb's first-generation catalyst gave a cyclohexenol in excellent yield. Deprotonation with 2.2 equiv. of tert-butyllithium, followed by the addition of 1.2 equiv. of phenylselenium chloride afforded a selenide. The protection of the alcohol as a triethylsilyl ether gave compound 129. Under the radical reaction conditions, 129 underwent the desired cyclization reaction to generate the bicyclic structure 130 in good yield.
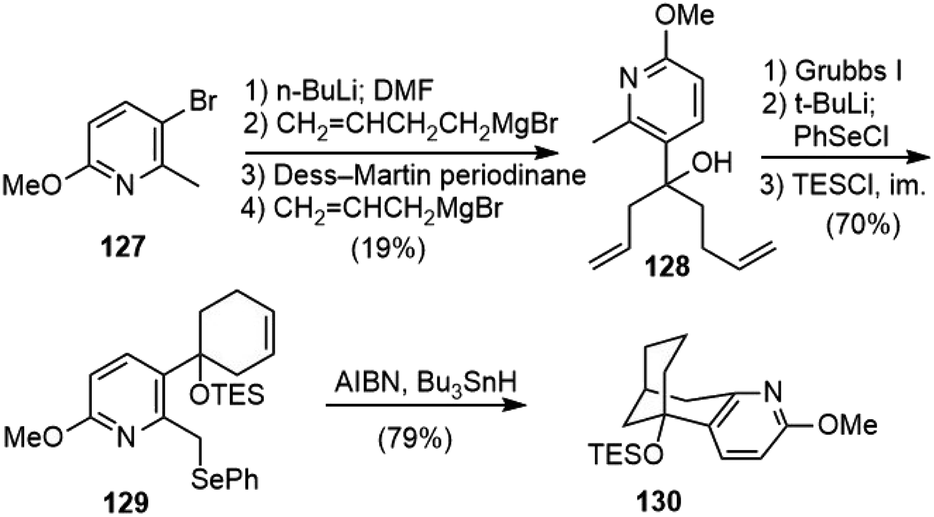 | ||
| Scheme 29 Caprio's synthetic studies on huperzine A. | ||
3.18. Summary of huperzine A's total syntheses
Several different strategies have been developed for the synthesis of the bridged core structure of huperzine A.(a) A cascade Michael addition and aldol reaction of intermediate 39 with methacrolein was used to generate the bridged core structure in Kozikowski's25 and Ji's29 (1989) total syntheses of (±)-huperzine A.
(b) Lu's palladium-catalyzed bicycloannunaltion reaction was used to build the bridged bicyclic structure in both Kozikowski's34 second-generation total synthesis and Langlois'57 formal total synthesis of (±)-huperzine A.
(c) In Fukuyama's total synthesis, a serendipitously discovered cation-olefin cyclization reaction was employed in the synthesis of the bridged structure.
(d) In Herzon's and Lin's total syntheses, palladium-catalyzed heteroaryl alkylation reactions were used.
(e) In White's total synthesis, a strain-promoted intramolecular aza-Prins reaction and a stereocontrolled scission reaction of a cyclobutylcarbinyl cation were designed and executed.
Since the bioactivity of (−)-huperzine A is much potent than its enantiomer, a variety of strategies have been developed for the asymmetric total synthesis of (−)-huperzine A, and significant progress has been achieved.
(a) Kozikowski finished the first asymmetric total synthesis of (−)-huperzine A, using a chiral auxiliary-controlled cascade Michael addition–aldol reaction to afford the product with high diastereoselectivity.
(b) A classical chiral resolution approach was used in Kozikowski's total synthesis of (+)-huperzine A.
(c) Langlois and coworkers reported a formal total synthesis of (+)-huperzine A, using a palladium-catalyzed, chiral auxiliary controlled diastereoselective bicycloannunaltion reaction.
(d) Herzon, Lin and White all used chiral pool compounds as the source of chirality in their total syntheses of (−)-huperzine A.
(e) Fukuyama utilized a desymmetrization reaction to introduce the chirality in their total synthesis.
(f) The cascade Michael addition–aldol reaction and palladium-catalyzed bicycloannunaltion reaction became the playground for the development of catalytic asymmetric synthesis. A variety of organocatalyzed systems were identified for the highly enantioselective cascade Michael addition–aldol reactions. The screening of chiral ligands in the palladium-catalyzed bicycloannunaltion reaction also led to highly efficient systems, and was applied in Azadi-Ardakani's scalable synthesis of (−)-huperzine A.
The innovation in the synthesis of pyridone fragment is also noteworthy. The strategies in Kozikowski's synthesis evolved from a low-yielding and costly multistep sequence to a highly efficient three-component, one-pot reaction (Table 1). This result inspired the improvement of Ji's synthesis of a similar fragment, and was further applied in Azadi-Ardakani's scalable synthesis of (−)-huperzine A. Fukuyama and coworkers used a conjugate addition reaction of sulfinylamide to α,β-unsaturated ketone, followed by cyclization and desulfination to afford a pyrone. Further reaction with NH3 converted the pyrone into the corresponding pyridone. The strategy was later improved into a one-pot process by using α,β-unsaturated ketones and 2-substituted acetamides as substrates for substituted 2-pyridone synthesis (Schemes 15 and 16).
4. Huperzines B and U
Huperzine B (17) was first isolated together with huperzine A by Liu in 1986.3 Huperzine B (17) displays weaker AChE inhibitory activity than huperzine A, but has a higher therapeutic index. The biological activities of huperzine U (20) have not been reported, possibly as a result of the scarceness of this natural product.4.1. Bai's total synthesis
Bai and coworkers reported the first total synthesis of (±)-huperzine B in 1997 (Scheme 30).72 Racemic huperzine B was obtained via a reaction sequence of 12 steps in 6.6% overall yield. The tetracyclic intermediate 137 was constructed by the tandem Michael addition and intramolecular Mannich cyclization reaction, using 135 and 136 as two components.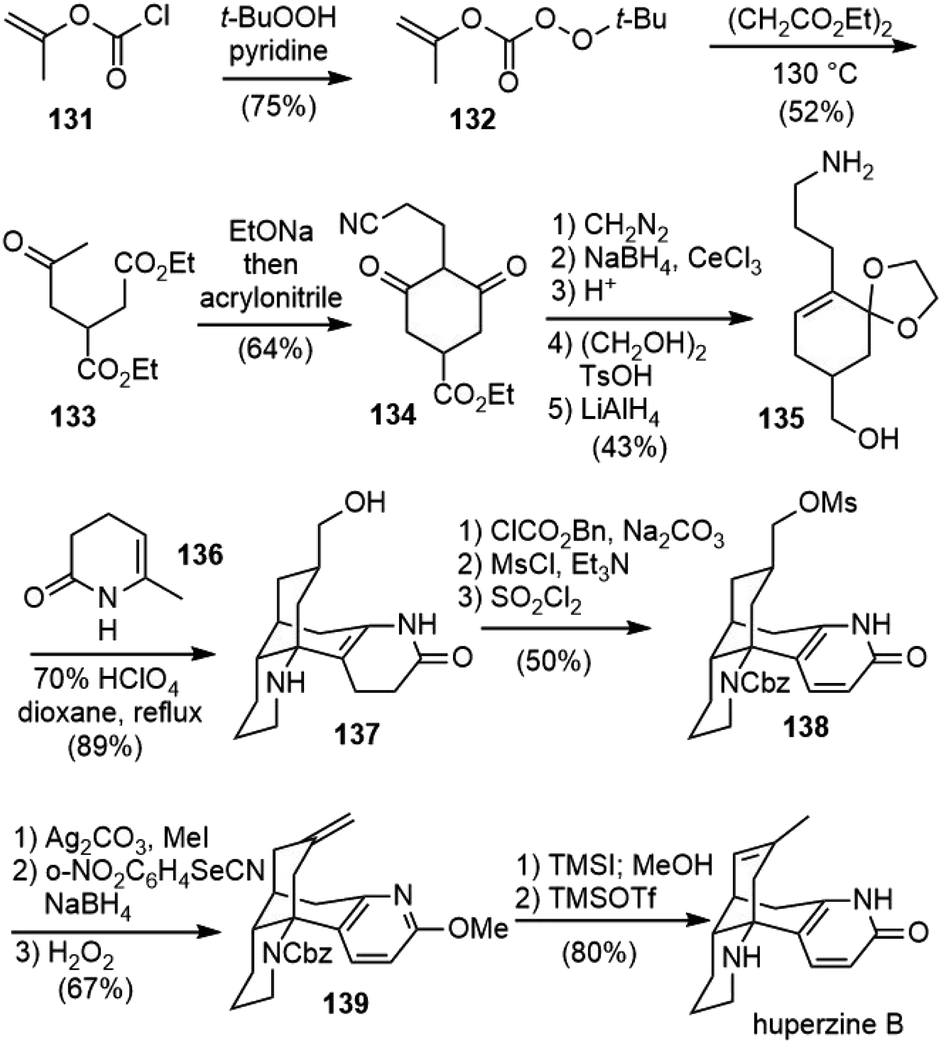 | ||
| Scheme 30 Bai's total synthesis of (±)-huperzine B. | ||
The keto ester 133 was prepared by a thermal free radical acetonylation of diethyl succinate with peroxycarbonate 132 following a method reported in the literature.73 Under basic conditions, 133 underwent an intramolecular Dieckmann condensation, followed by a one-pot conjugate addition reaction with acrylonitrile to give cyanide 134. After functional group manipulation and oxidation state adjustment, 134 was transformed into amino alcohol 135 in 5 steps. The treatment of 135 with perchloric acid (HClO4) in the presence of 136 initiated a cascade reaction to form the desired tetracyclic product 137 directly in 89% yield. Presumably 135 was first transformed into a cyclic imine and then underwent the tandem Michael addition and intramolecular Mannich reaction with 136 to generate amino alcohol 137 (vide infra). This annulation strategy was developed by Schumann and coworkers and had been successfully used in the total syntheses of other Lycopodium alkaloids.74–76
Compound 137 was transformed to 138 by the manipulation of the amino and alcohol groups, followed by the conversion of the dihydropyridone moiety into pyridone. O-Methylation of 138 with Ag2CO3-MeI, substitution of the mesylate with o-nitrophenyl selenocyanate and sodium borohydride (NaBH4), followed by oxidative elimination afforded olefin 139. Deprotection of 139 with TMSI and the shift of exocyclic double bond into endocyclic with TMSOTf afforded (±)-huperzine B (17).
4.2. Lee's semisynthesis
The Lee group reported a semisynthesis of huperzine B in 2004 (Scheme 31).77 They started from acid 45, an intermediate in Kozikowski's total synthesis of huperzine A.26 A Curtius rearrangement of 45, followed by acidic hydrolysis, afforded amine 140. N-Alkylation of the amine with 4-iodo-1-butene, and a subsequent ring closing metathesis reaction with the Grubbs second generation catalyst, gave product 141. Selective hydrogenation of double bonds in 141 with Pd/C and final deprotection with TMSI furnished (±)-huperzine B.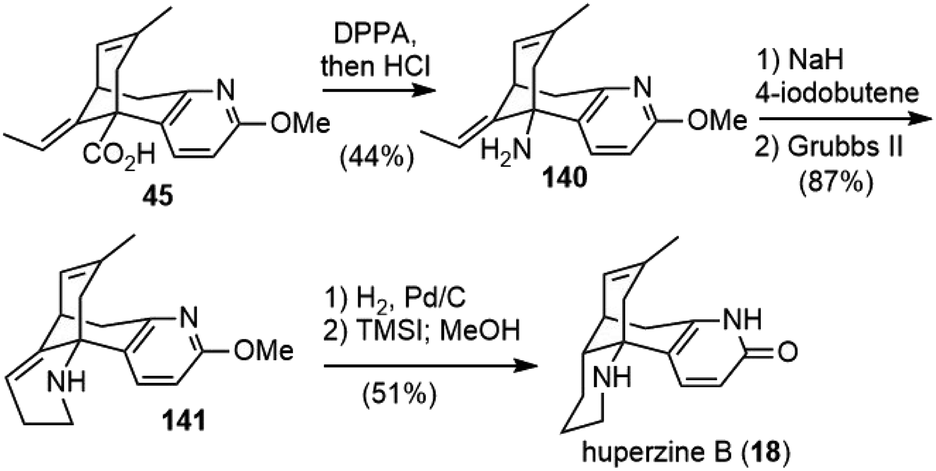 | ||
| Scheme 31 Lee's semisynthesis of (±)-huperzine B. | ||
4.3. Lin and Sun's total synthesis
After finishing the total synthesis of (−)-huperzine A, the Lin group reported the first asymmetric total synthesis of (−)-huperzines B and U in 2014 (Scheme 32).49 Starting from 87, the key intermediate in their huperzine A synthesis,48 allylation with allylmagnesium chloride delivered a tertiary alcohol. Selective hydroboration/oxidation of the terminal olefin produced a primary alcohol. The alcohol was converted into its mesylate and cyclized with nitrogen to afford the piperidine product 142 as a mixture of isomers. Dehydration of 142 with SOCl2 furnished the olefin 141. Both olefin isomerization and O-demethylation were achieved by HBr. Subsequent selective olefin hydrogenation with Pd/C furnished (−)-huperzine B (17). Meanwhile, isomerization of the olefin in 142 could be achieved with Crabtree's catalyst in a hydrogen atmosphere. Finally, O-demethylation with LiI and excess Et3N, followed by semi-reduction of the pyridone ring gave huperzine U (20).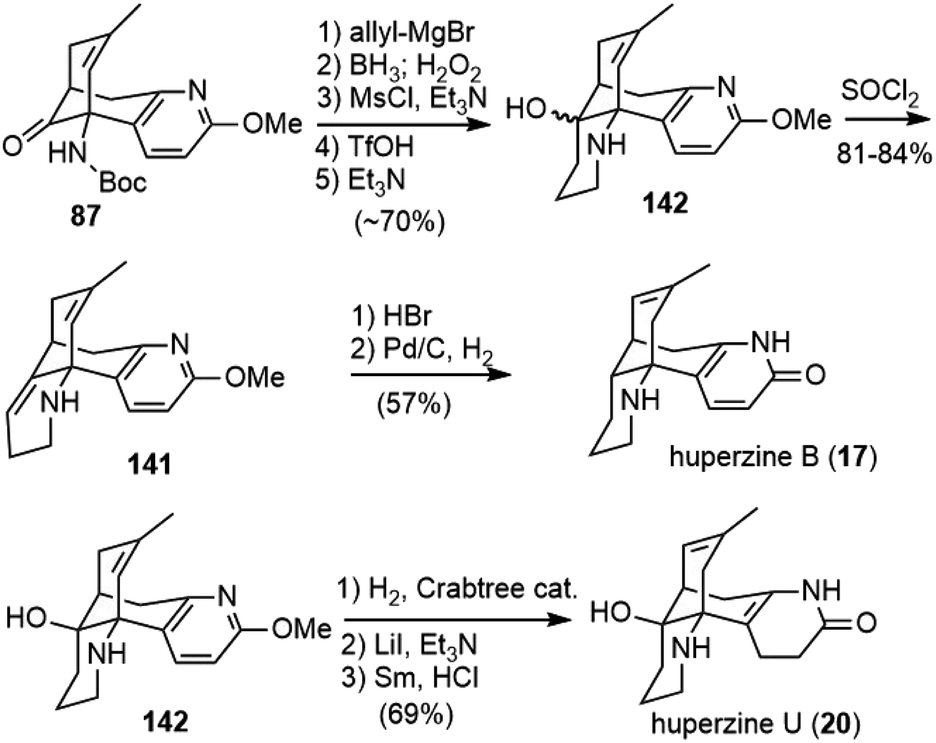 | ||
| Scheme 32 Lin and Sun's syntheses of (−)-huperzines B and U. | ||
5. 8,15-Dihydrohuperzine A
(−)-8,15-Dihydrohuperzine A (19) was isolated in 2014 by Thasana and coworkers from club moss Huperzia carinata (Desv. Ex. Poir.) Trevis collected in Thailand.9 The isolation yield from the dry plant was extremely low (0.000028%), and it exhibited weaker AChE inhibitory activity (IC50 2.63 μM) than huperzine A (IC50 0.016 μM), which revealed the importance of the double bond between C8 and C15 for the biological activity of huperzine A.In 2021, Sarpong and coworkers reported a total synthesis of 8,15-dihydrohuperzine A (19) through bioinspired late-stage diversification of a readily accessible common precursor, N-desmethyl-β-obscurine (Scheme 33).76 Dihydropyridone 136 was prepared from β-ketoester 143via a Michael addition into acrylonitrile followed by decarboxylation. Subsequent nitrile hydration and cyclization in vacuo delivered 136 in 18% overall yield. The primary amine 146 was prepared from (+)-pulegone (76) following the procedures reported in the literature with modifications.78,79 The sequence was initiated by nucleophilic epoxidation of the exocyclic olefin, and subsequent nucleophilic opening of the epoxide with sodium thiophenolate (PhSNa) and concomitant retro-aldol reaction delivered the phenylthioether, which was selectively oxidized to sulfoxide 145 with sodium perborate (NaBO3). α-Alkylation of 145 with acrylonitrile, followed by thermal syn-elimination of phenylsulfenic acid, gave an enone, which was protected as the ethylene glycol ketal and reduced with LiAlH4 to deliver primary amine 146.
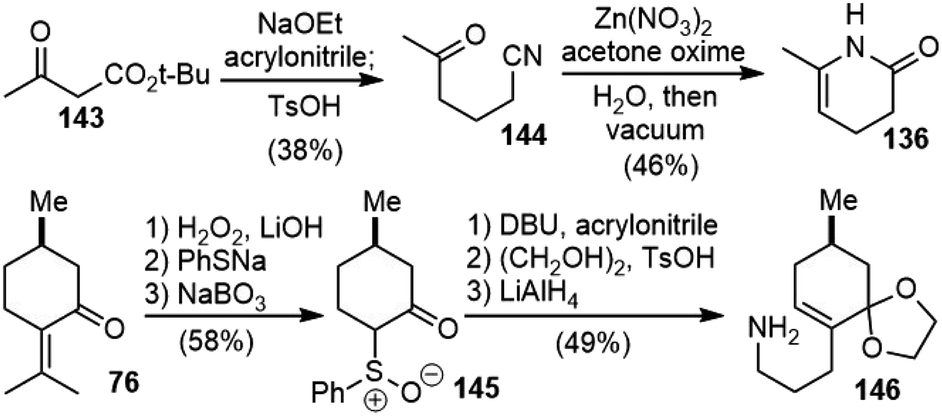 | ||
| Scheme 33 Sarpong's total synthesis of (−)-8,15-dihydrohuperzine A (part 1). | ||
Upon heating with HClO4, the two building blocks 136 and 146 were coupled via a diastereoselective formal (3 + 3)-cycloaddition reaction to afford N-desmethyl-α-obscurine (15) (Scheme 34).72,74 An α,β-unsaturated iminium ion 146a and the open-chain enolamide 136a were presumably formed in situ and underwent a tandem Michael addition and intramolecular Mannich reaction (vide supra) to furnish 15. Protection of piperidine nitrogen with Boc anhydride, dehydrogenation of the dihydropyridone ring with lead tetraacetate (Pb(OAc)4), O-triflation of pyridone, and piperidine oxidation with RuO2/NaIO4 yielded imide 147. Hydrolysis of both the imide and triflate, followed by O-methylation of pyridone and ester hydrolysis, generated the acid 148. A Pd(0)-catalyzed decarbonylative elimination of an in situ-generated mixed anhydride of 148 afforded the desired terminal olefin 149.80 Further isomerization of the terminal olefin with an in situ-generated palladium hydride catalyst led to an internal (E)-alkene.81 Finally, a TMSI-mediated deprotection of both carbamate and methyl ether delivered (−)-8,15-dihydrohuperzine A (19).
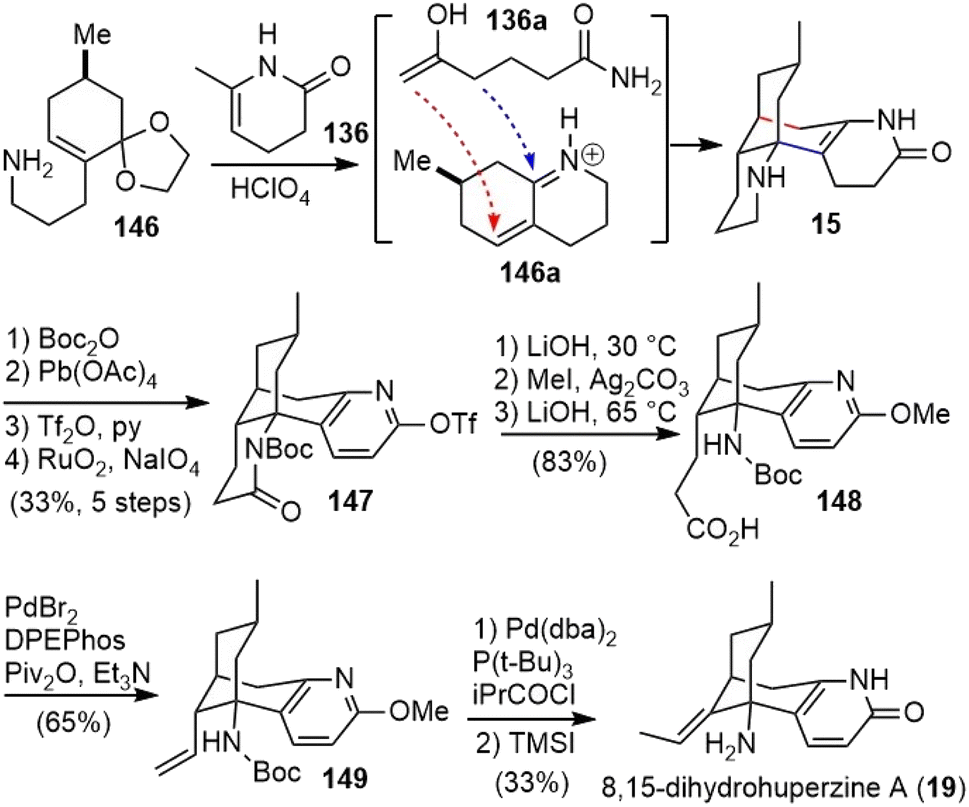 | ||
| Scheme 34 Sarpong's total synthesis of (−)-8,15-dihydrohuperzine A (part 2). | ||
6. Huperzines K, M and N
Huperzines K, M and N were also isolated from Huperzia serrata (thub.) trev. From the structural point of view, they belong to phlegmarine alkaloids, which contain the decahydroquinoline ring system and 4–5 stereocenters. The density of stereochemistry made their structure assignment difficult. In fact, the structures of all three natural products were reassigned later, and confirmed by total syntheses (Fig. 4).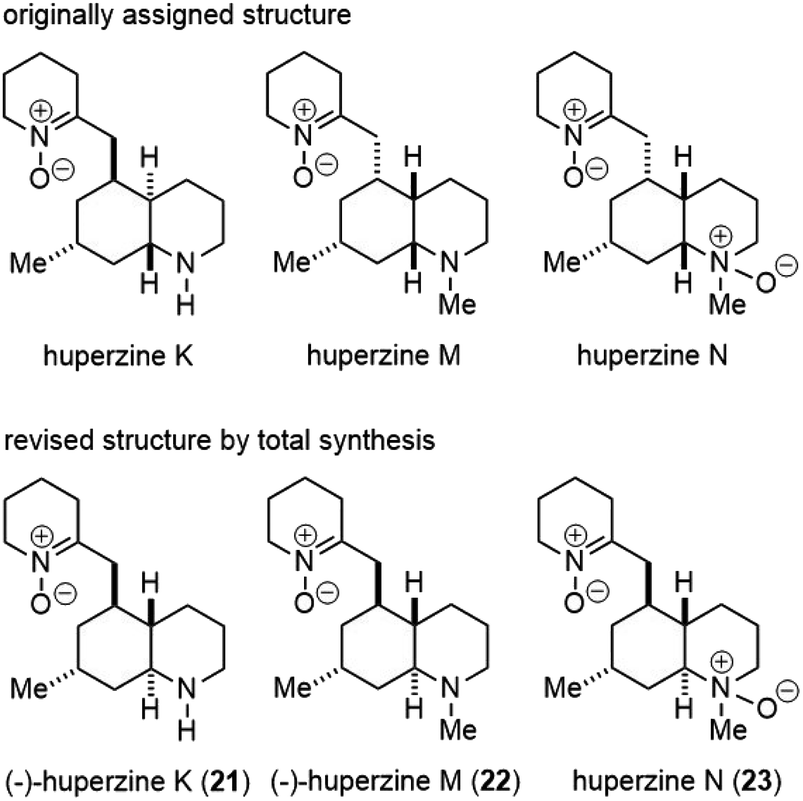 | ||
| Fig. 4 Originally assigned and revised structures of huperzines K, M and N. | ||
The Bonjoch group developed highly efficient approaches toward the synthesis of cis-decahydroquinolines (Scheme 35).82 The substrate β-keto ester 152 was prepared by N-tosylation of the commercially available 5-aminopentanoic acid 150, followed by a homologation reaction with mono-tert-butylmalonate under Masamune conditions.83 Subjecting the β-keto ester 152 and crotonaldehyde to LiOH (1 equiv.) in i-PrOH and water resulted in a cascade Robinson annulation/intramolecular aza-Michael cyclization process. The cis-decahydroquinoline 153 was delivered in only one step and as a single diastereoisomer. Two C–C bonds, one C–N bond, three stereogenic centers and two rings were formed in this process. Water was essential to drive the aza-Michael reaction to completion, and in its absence, significant amounts of the ring-opened Robinson annulation product were obtained.
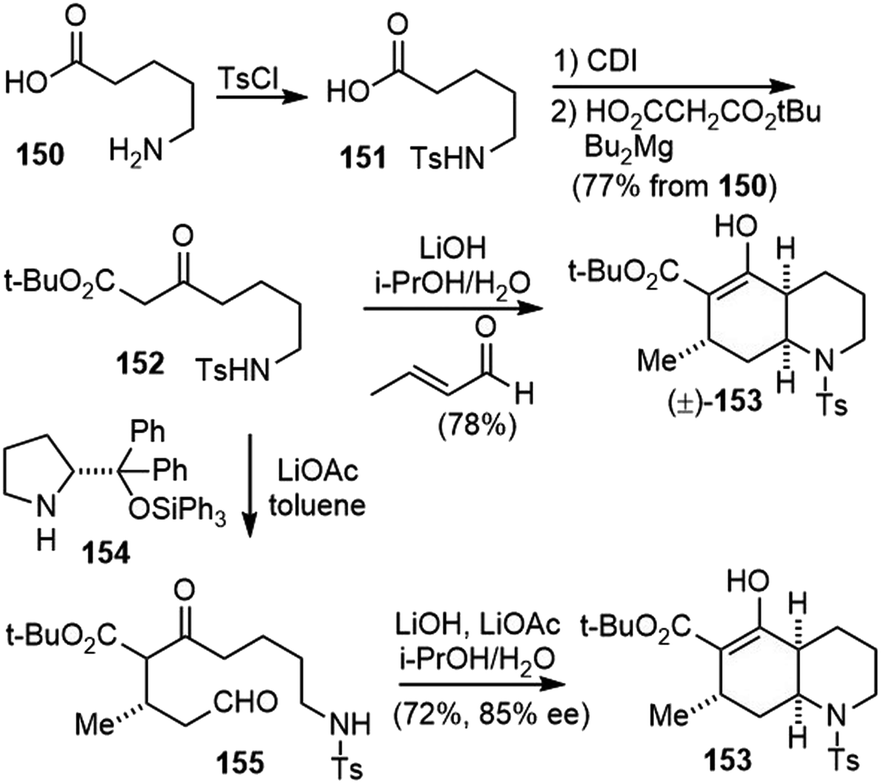 | ||
| Scheme 35 Racemic and asymmetric synthesis of cis-decahydroquinolines. | ||
Asymmetric version of this transformation was also developed. The stereochemistry was set up by a Michael addition reaction catalyzed by a triphenylsilyl-modified Hayashi catalyst (154), with LiOAc as an additive in toluene. The removal of the solvent and treatment of the mixture with LiOH in the presence of i-PrOH and water led to the tandem aldol condensation–intramolecular aza-Michael reaction, which delivered cis-decahydroquinoline 153. One recrystallization from MeOH provided the product 153 in >99% ee.
cis-Decahydroquinoline 153 was a versatile intermediate for the total syntheses of several cis-phlegmarine alkaloids. In order to synthesize the putative structure of huperzine N, epimerization of the stereocenters at the ring junction was required (Scheme 36).84,85 Treatment of 153 with TFA, followed by decarboxylation, gave ketone 156. Under basic conditions, an equilibrium process occurred, in which 156 underwent a retro aza-Michael ring opening and subsequent closure to the more stable decahydroquinoline 158. Direct addition of phosphonate 159 led to a chemoselective reaction with 158, and shifted the equilibrium to the desired direction. Vinylpyridine 160 was formed exclusively as an inconsequential mixture of Z/E isomers in 89% yield over the entire sequence from 153.
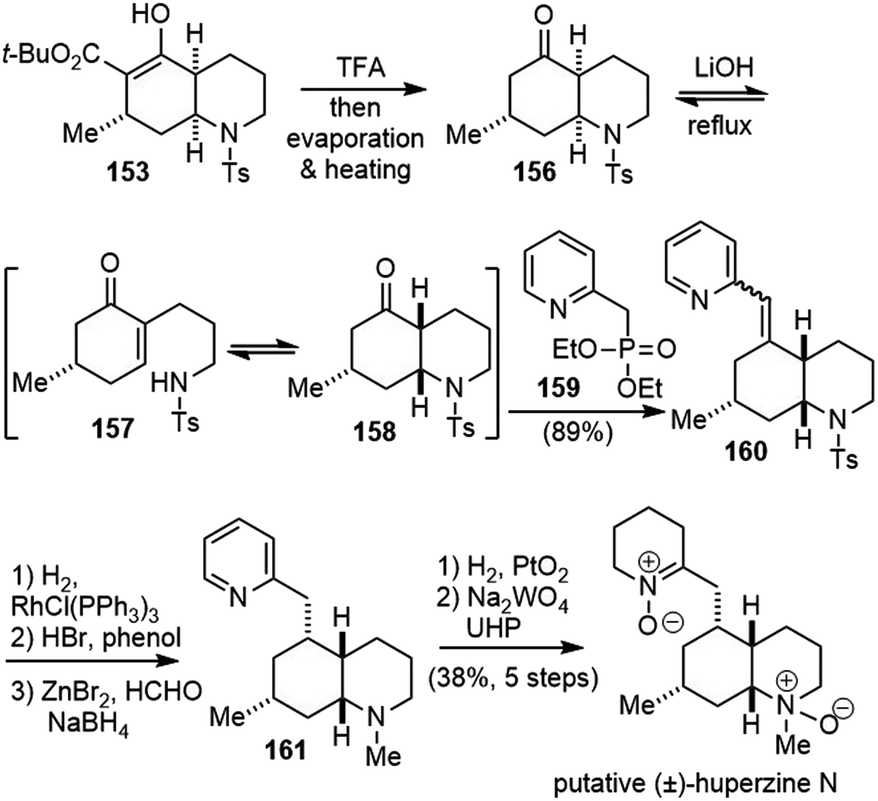 | ||
| Scheme 36 Synthesis of putative huperzine N. | ||
Hydrogenation of 160 with Wilkinson's catalyst gave the desired epimer in a 9:1 ratio.86 The removal of the tosyl group and N-methylation via reductive amination with formaldehyde generated 161. Hydrogenation of the pyridine and oxidation of amine gave the reported structure of huperzine N. Unfortunately, the NMR spectra of the synthetic sample did not match with those reported for the natural product.
By analyzing the 13C NMR spectroscopic data of 20 phlegmarines reported in the literature, the Bonjoch group developed a pattern recognition method for the stereochemical arrangement of the four stereogenic carbons in phlegmarine alkaloids. Based on this method, they made the structure reassignment of huperzines N, K and M, the latter being identical to the isolated alkaloid named lycoposerramine Y. The revised structures were validated by their first total synthesis using an enantioselective construction of the cis-decahydroquinoline skeleton (Scheme 37).87
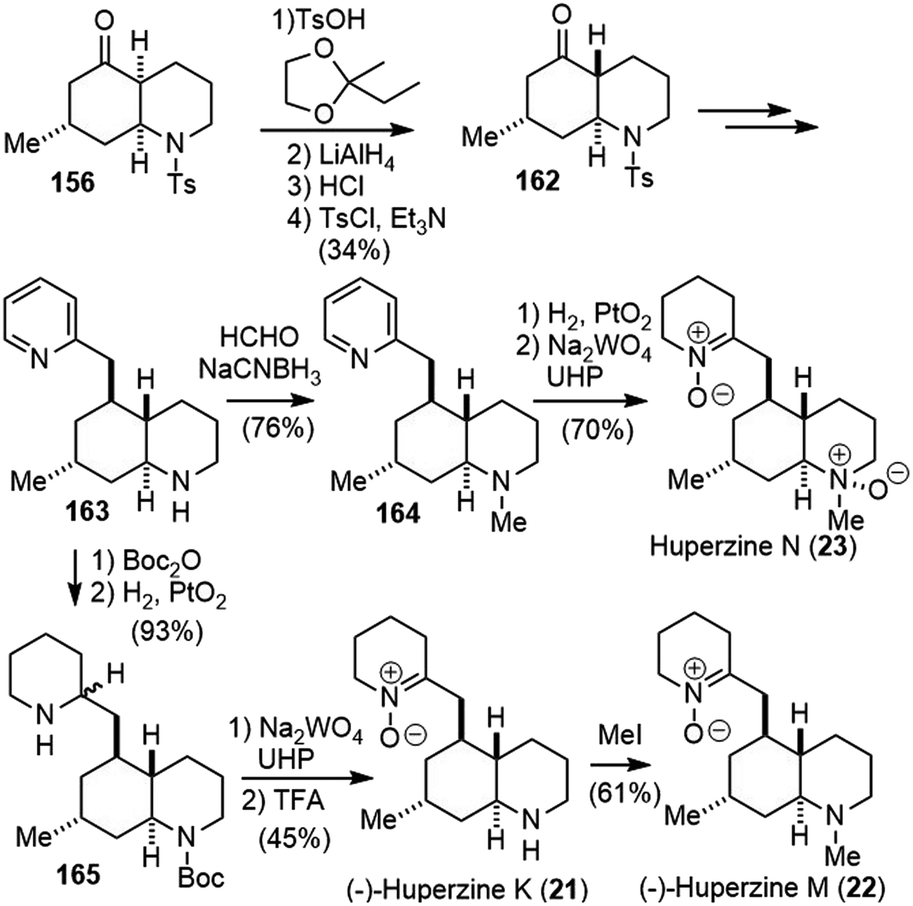 | ||
| Scheme 37 Syntheses of reassigned huperzines K, M, and N. | ||
The easily available ketone 156 was transformed into a ketal, and reductive removal of the N-tosyl group and acid-induced epimerization led to a 2:1 mixture of ketone and its C4-epimer. Tosylation of the mixture furnished the required trans-decahydroquinoline 162 as a single isomer after chromatographic separation. Following a process similar to Scheme 36, ketone 162 was transformed to piperidine 163. N-methylation via reductive amination afforded amine 164. Hydrogenation of the pyridine ring and oxidation of both amines afforded the revised structure of huperzine N (23).88 Similarly, optical pure 163 was obtained from optical pure 156. The protection of amine and hydrogenation of the pyridine ring afforded compound 165. Amine oxidation and deprotection generated the revised structure of (−)-huperzine K (21), and further N-methylation gave the revised structure of (−)-huperzine M (22). The data for all three synthetic compounds matched with those reported literature values, which supported their structure reassignment.
7. Huperzine O
Huperzine O (24) was also isolated from Huperzia serrata (thub.) trev. by Zhu and coworkers in 2000.14 Huperzine O has a tetracyclic core structure with six stereogenic centers and an unusual internal hydrogen bond. It does not possess any activity on the inhibition of AChE.Fan and coworkers reported a total synthesis of huperzine O in 2017 (Scheme 38).89 They developed a novel tandem palladium-mediated oxidative dehydrogenation/hetero-Michael addition reaction of bridged bicyclic ketones to introduce hetero substituents at the conformationally rigid bridgehead carbons. Following the literature procedure, a cascade Michael addition–aldol reaction of acrolein with enamine of 4-methylcyclohexone 166 generated the bridged structure, and a two-step dehydration process gave the known ketone 167 on 80 g scale. Selective allylation of 167 with the allylic Grignard reagent afforded the homoallylic alcohol 168 in 98% yield and 13:1 dr. Subsequent hydroboration−oxidation of both olefins, followed by alcohol oxidation with 2-iodoxybenzoic acid (IBX), furnished hemiacetal ketone 169.
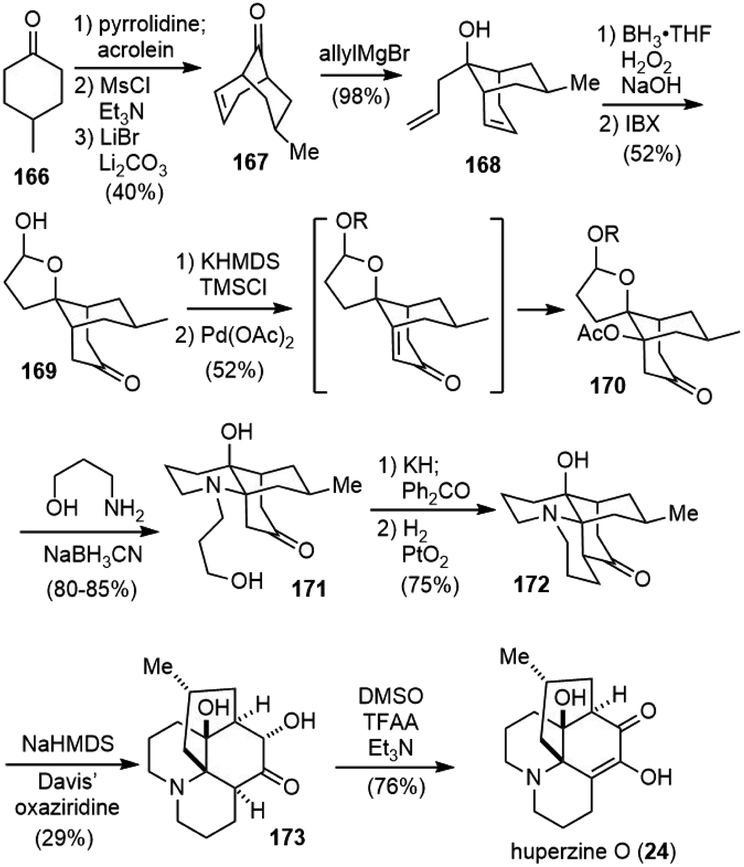 | ||
| Scheme 38 Fan's total synthesis of huperzine O. | ||
The bridgehead C–H heterofunctionalization reaction was then explored. Saegusa–Ito oxidation of the silyl enol ether of ketone 169 with a stochiometric amount of freshly prepared Pd(OAc)2 afforded the desired bridgehead acetate-functionalized product 170 directly. The reaction presumably involved a highly strained bicyclo[3.3.1] bridgehead enone intermediate, followed by a strain-driven conjugate addition reaction. With a less strained bicyclic system, the reaction stopped at the enone stage. Attempts to use catalytic amounts of palladium catalyst were unsuccessful. Reductive amination of hemiacetal 170 with 3-aminopropanol afforded an unexpected but desired piperidine-containing tricyclic product 171. After installation of the crucial bridgehead aza-quaternary center, the tetracyclic structure was formed by adopting Heathcock's protocol (Oppenauer oxidation, aldol condensation, and reductive hydrogenation)90 to afford (±)-lycodoline (172) in 75% yield on a gram scale. Kinetically controlled α-hydroxylation of the ketone using sterically bulky Davis' oxaziridine and subsequent alcohol oxidation gave 76% yield of (±)-huperzine O (24).
8. Huperzine Q
Huperzine Q (25) was isolated from the CHCl3 fraction of the basic material of the whole plant of Huperzia serrata by Zhu and coworkers in 2002.15 Its structure was determined by spectroscopic studies and X-ray crystallographic analysis. A spiroaminal moiety and six stereogenic centers, including a quaternary carbon center, were embedded in the unique pentacyclic skeleton. No bioactivity of huperzine Q was reported.8.1. Takayama's total synthesis
Takayama and coworkers reported the first total synthesis of (−)-huperzine Q in 2011 (Scheme 39).91 Ketone 176 was prepared by a coupling reaction between alkyne 174 and acyl chloride 175. The Noyori reduction of the ketone and in situ lactonization generated the optically active lactone 177. α-Allylation of the lactone, followed by epimerization and lactone reduction provided diol 178. Submitting the diol 178 directly to the Pauson–Khand reaction conditions generated the cyclized product 180’ with the wrong stereochemistry. Conformational analysis indicated that the substituents are in the equatorial position of the chair-like transition state, which led to the product with the undesired stereochemistry. Converting the diol 178 to a cyclic silyl ether 179 altered the conformation of the reaction intermediate and forced the substituents to the axial position. After optimization, a one-pot diol silylation, Pauson–Khand reaction and deprotection delivered product 180 having the desired C7 stereochemistry in 92% yield.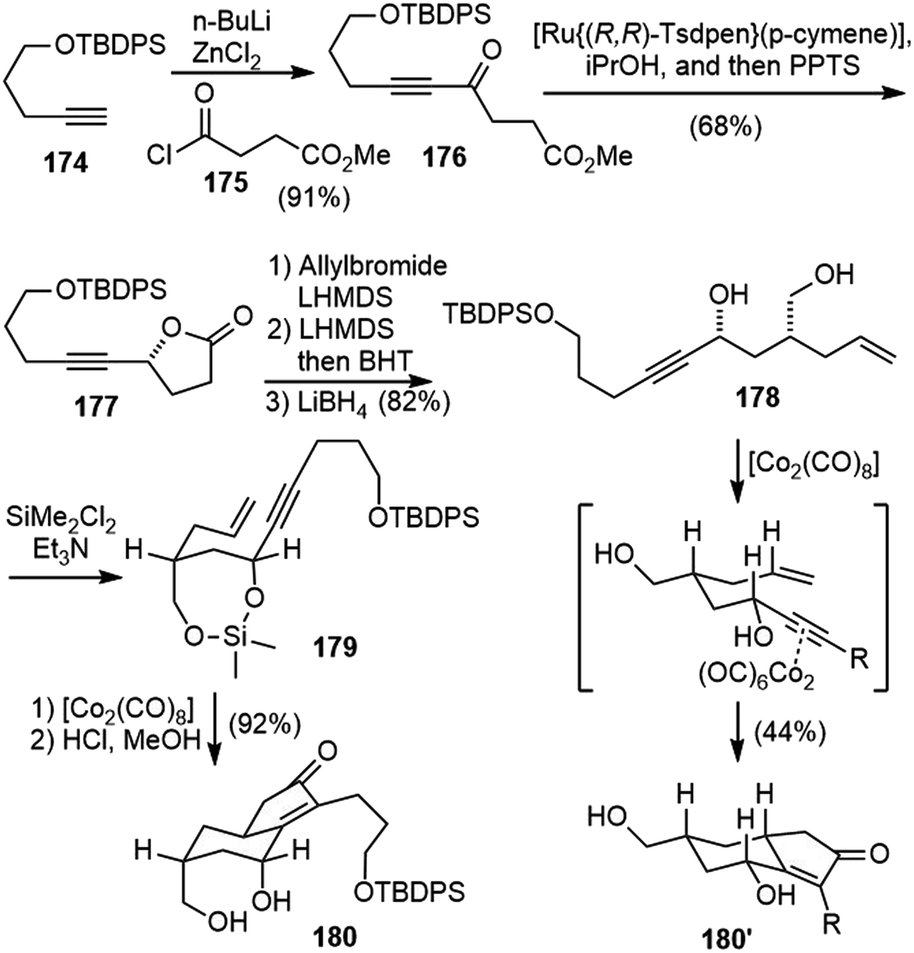 | ||
| Scheme 39 Takayama's total synthesis of (−)-huperzine Q (part 1). | ||
Protection of the diol in 180 and asymmetric reduction of the enone with (R)-Me-CBS reagent furnished an allyl alcohol with good diastereoselectivity (Scheme 40). Conjugate addition of the allylic alcohol to phenyl vinyl sulfoxide gave 181, which underwent a vinyl Claisen rearrangement to set up the quaternary center and delivered product 182.92 A Wittig olefination, followed by hydroboration/oxidation of both olefins generated a diol, which was further transformed into the nosylamide 183. Deprotection of the primary silyl ether and an intramolecular Mitsunobu reaction afforded the azonane ring 184. X-ray crystallographic analysis of 184 confirmed the absolute configuration of all the chiral centers. The removal of the two MOM groups generated a diol, and the primary alcohol was selectively acetylated and the secondary alcohol was oxidized. Successive removal of the Ns group and diacetyl groups was achieved in a one-pot fashion to afford a carbinolamine, which underwent spiroaminal formation after refluxing in toluene with anhydrous (+)-camphorsulfonic acid (CSA) to furnish (−)-huperzine Q (25).
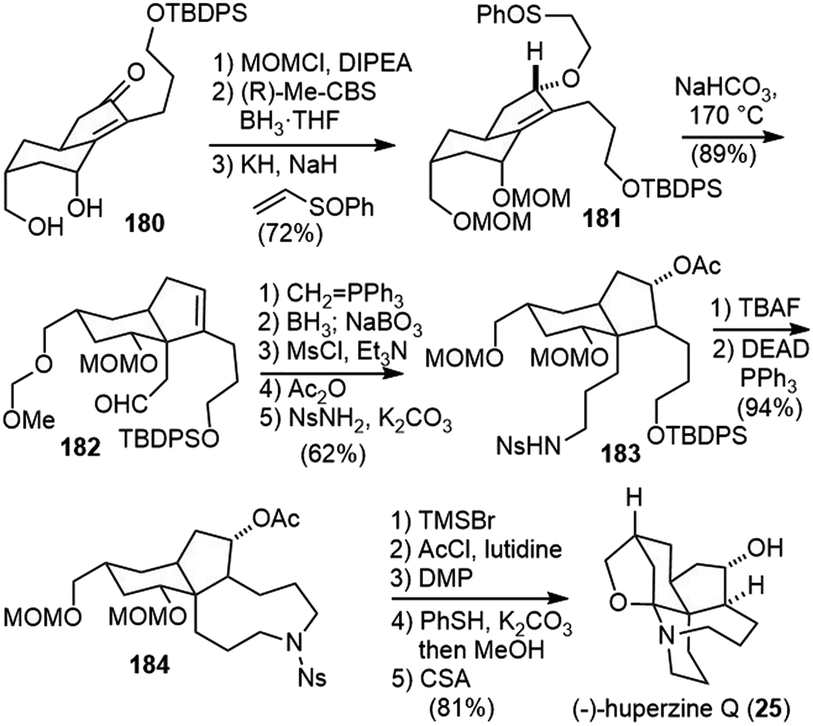 | ||
| Scheme 40 Takayama's total synthesis of (−)-huperzine Q (part 2). | ||
8.2. Lei's total synthesis
In 2015 Lei and coworkers reported a 12-step total synthesis of (−)-huperzine Q (Scheme 41).93 The commercially available acid 185 was transformed to the chiral enone 187 in 4 steps, including lipase-catalyzed kinetic resolution.94 A Noyori-type three-component Michael addition/aldol reaction of enone 187 formed adduct 188.95 After alcohol oxidation and intramolecular C-alkylation sequence, the desired bicyclic diketone 189 was obtained with the correct stereochemistry. A regioselective carbonyl–olefin metathesis reaction mediated with stoichiometric amounts of Schrock's molybdenum alkylidene complex in an ethylene atmosphere afforded the desired cyclopentene product 190. A one-pot olefin hydroboration/oxidation process generated a diketone as a mixture of diastereoisomers. The removal of the Boc group with (+)-CSA caused concomitant epimerization and enamine formation, and further hydrogenolysis afforded 191 as the sole product. The key late-stage bromofunctionalization of enamine 191 with NBS in the presence of (+)-CSA provided bromide 192 in 96% yield. An α-bromoiminium ion was proposed to be the key intermediate. The structure of 192 was determined by two-dimensional NMR spectroscopy and further confirmed by single-crystal X-ray diffraction analysis. The SmI2-mediated concurrent debromination and ketone reduction of 192 delivered huperzine Q (25) in 95% yield.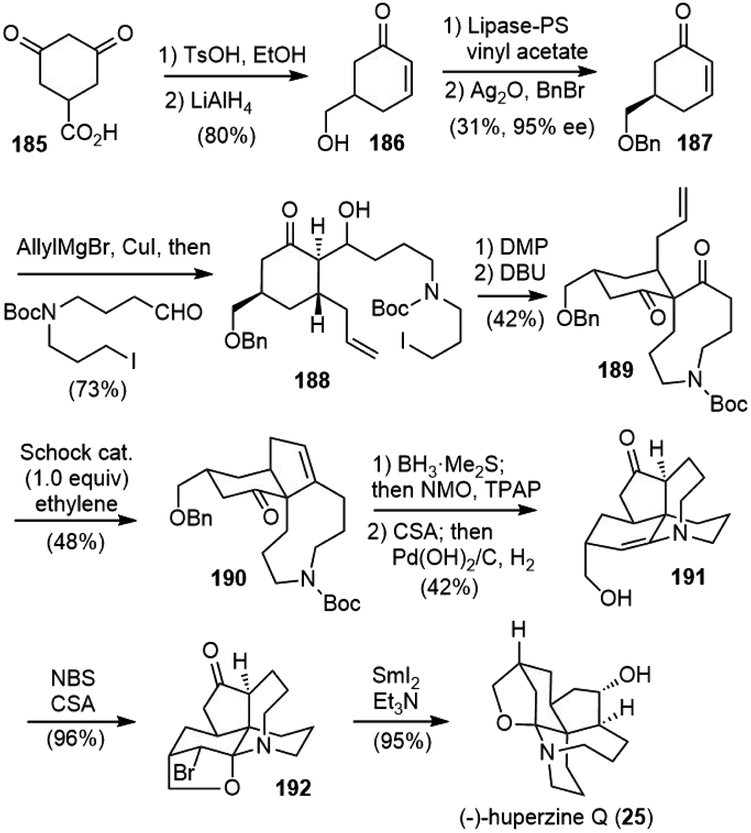 | ||
| Scheme 41 Lei's total synthesis of (−)-huperzine Q. | ||
8.3. Zhao's total synthesis
Zhao and coworkers also reported their total synthesis of (−)-huperzine Q in 2015 (Scheme 42).96 Hajos-Parrish-type ketone 195 was prepared from 1,3-cyclopentanedione 193. Selective reduction of the unconjugated carbonyl group with NaBH4 afforded an alcohol with the desired selectivity. A hydroxyl-directed stereocontrolled Birch reduction gave the cis-fused isomer 196 in 8:1 diastereoselectivity. Alcohol 196 was then transformed into enol triflate 197 with KHMDS/PhNTf2, which underwent a B-alkyl Suzuki coupling to furnish the product 198 in high yield. Successive hydroboration/oxidation of two olefins generated a diol. The diol was selectively functionalized and underwent an intramolecular substitution reaction to afford azolane 199. Dehydrogenation of ketone with IBS afforded an enone, which was transposed to another enone 200. Redox transformation and epimerization of 200 generated the hydroxylated product 201. Reductive removal of the tosylate group and subsequent cyclization under acidic conditions provided the final natural product (−)-huperzine Q.
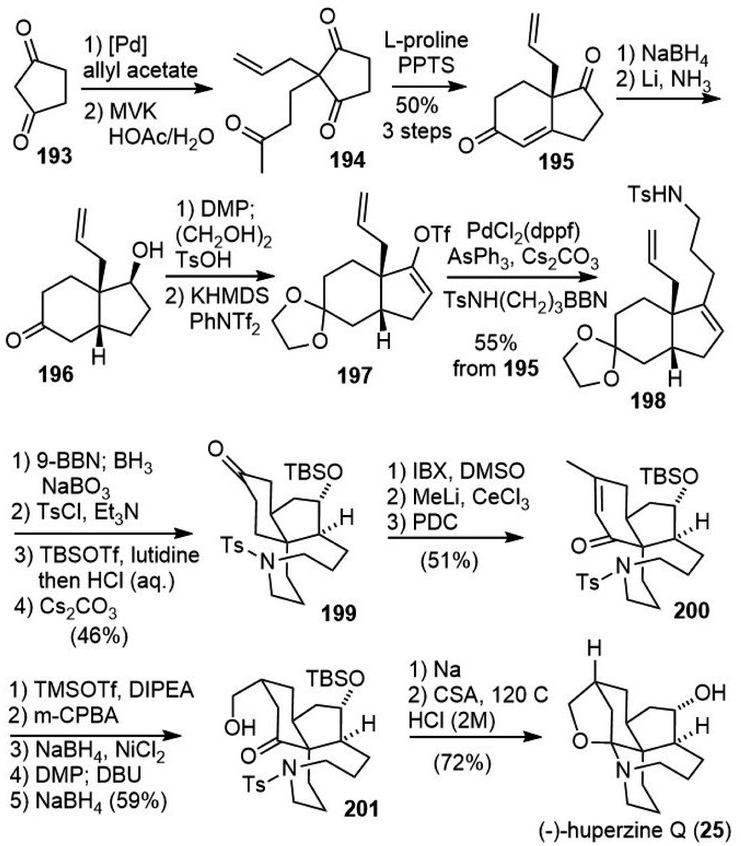 | ||
| Scheme 42 Zhao's total synthesis of (−)-huperzine Q. | ||
8.4. Fukuyama and Yokoshima's total synthesis
Fukuyama and Yokoshima reported the total synthesis of (±)-huperzine Q in 2017 (Scheme 43).97 Their synthesis commenced with ozonolysis of olefin 202 and HWE olefination of the resulting aldehyde to give an enone, which was transformed to the diene 203. The other fragment 207 was prepared by O-benzylation and α-iodination of enone 204, followed by a B-alkyl Suzuki−Miyaura coupling reaction. The crucial Diels–Alder reaction of 207 with diene 203 in the presence of zinc chloride (ZnCl2) occurred from the opposite side of the benzyloxymethyl group on the cyclohexanone ring to furnish the cis-decaline ring, which was oxidized with DDQ to afford enone 208. Sequential cleavage of the Boc and TBDPS groups gave a hydroxy nosylamide, which underwent a Mitsunobu cyclization to produce the tricyclic compound 209. The cis-decaline system was converted into the cis-hydrindane core in a two-step process. Nucleophilic epoxidation of the enone moiety in 209 afforded an epoxyketone. Upon activation with TMSOTf, the epoxide was opened to form tertiary carbocation, followed by a 1,2-shift of the carbonyl group to give a ring-contracted product 210 in 91% yield. The nosyl group and formyl group were successively removed with thiophenol to form a hemiaminal. Debenzylation and stereoselective reduction of the ketone moiety were simultaneously achieved via the Birch reduction, leaving the hemiaminal moiety intact. Finally, the formation of the N,O-acetal moiety was carried out according to Takayama's procedure to furnish (±)-huperzine Q.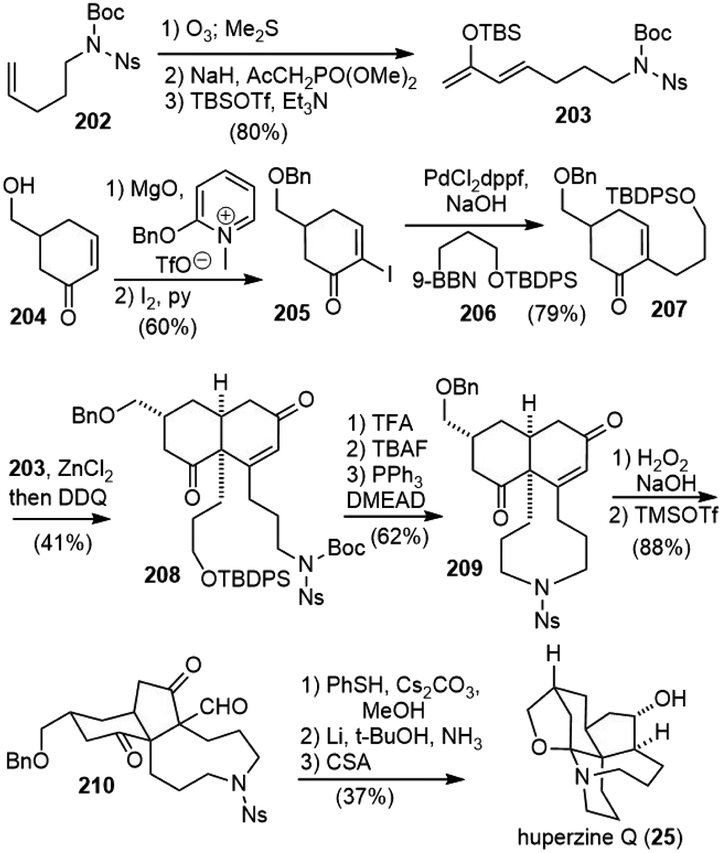 | ||
| Scheme 43 Fukuyama and Yokoshima's total synthesis of (±)-huperzine Q. | ||
8.5. Summary of huperzine Q's total syntheses
Huperzine Q (25) is a highly intricate structure within this family, which poses significant challenges to synthetic organic chemistry. The four finished total syntheses of huperzine Q share some similarities, but also some differences between them (Fig. 5).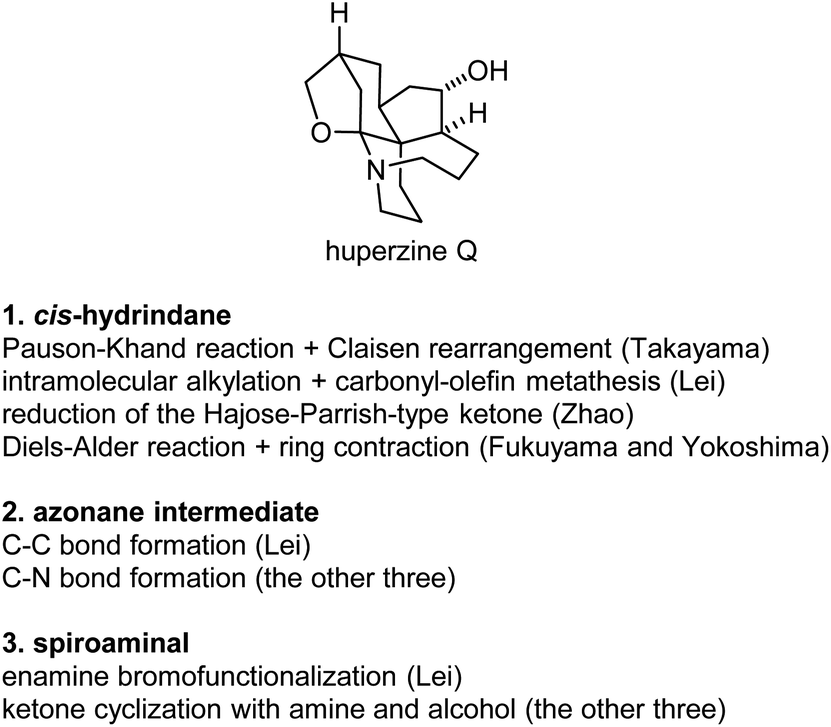 | ||
| Fig. 5 Summary and comparison of huperzine Q syntheses. | ||
(a) The cis-hydrindane structure. Each group developed a distinct method to construct the key cis-hydrindane structure. In Takayama's synthesis, a tethered diastereoselective Pauson-Khand rection formed the bicyclic structure, followed by the vinyl Claisen rearrangement to set up the quaternary center. In Lei's synthesis, a diastereoselective intramolecular alkylation reaction set up the quaternary center, followed by a carbonyl-olefin metathesis reaction to form the bicyclic structure. In Zhao's synthesis, a hydroxy directed reduction of the enone derived from the Hajos-Parrish ketone afforded the desired structure. In Fukuyama and Yokoshima's total synthesis, the cis-hydrindane structure was made by ring contraction of the corresponding cis-decalin structure, which was readily available from a diastereoselective Diels–Alder reaction.
(b) All syntheses went through an azonane intermediate, and were constructed by intramolecular substitution reactions. Three of them formed the azonane by an N-alkylation reaction of sulfonamides, while a 1,3-diketone alkylation reaction was used in Lei's synthesis.
(c) The spiroaminal structure. Takayama made this structure by intramolecular double cyclization of a ketone with an amine and an alcohol under acidic conditions; similar conditions were applied in Zhao's and Fukuyama's synthesis. An enamine bromofunctionalization, followed by reductive debromination reaction, was used in Lei's synthesis.
(d) Source of chirality. Three different methods were used in the three asymmetric syntheses. Transition metal-catalyzed asymmetric hydrogenation reaction was used in Takayama's synthesis, a lipase-catalyzed kinetic resolution reaction was applied in Lei's synthesis, and organocatalyzed desymmetrization reaction was used in Zhao's synthesis.
9. Huperzine R
Huperzine R (26) was isolated from the whole plant of Huperzia serrata by Zhu and co-workers in 2002.16 It is a novel 15-carbon Lycopodium alkaloid, characterized by a unique bicyclic lactam core. Its relative configuration was established using spectroscopic and X-ray crystallographic techniques. It shows only weak in vitro inhibition on the activity of AChE.9.1. Fukuyama and Yokoshima's synthetic studies
In 2015, Fukuyama and Yokoshima disclosed their synthetic studies on huperzine R (Scheme 44).98 The cyclopropanolactone 212 was readily prepared from a CsF-promoted annulation reaction of malonate with glycidyl nosylate 211.99 Ring opening of the cyclopropane with cuprate resulted in a lactone ester 213. A stereoselective Michael addition to methyl acrylate and protective group replacement afforded compound 214. Hydrogenolysis of the benzyl ester group followed by a Curtius rearrangement gave the desired spirocyclic lactam 215 directly. Reduction of the lactone to a lactol, and treatment with allylmagnesium chloride gave a diastereomeric mixture of diols 216. Alcohol oxidation and transformation of the silyl ether into a mesylate and the terminal olefin into a protected primary alcohol via hydroboration afforded compound 218. The secondary lactam was then reduced to an amine and stimulated the intramolecular cyclization to form the seven-membered ring, and the protected alcohol was transformed into another mesylate 219.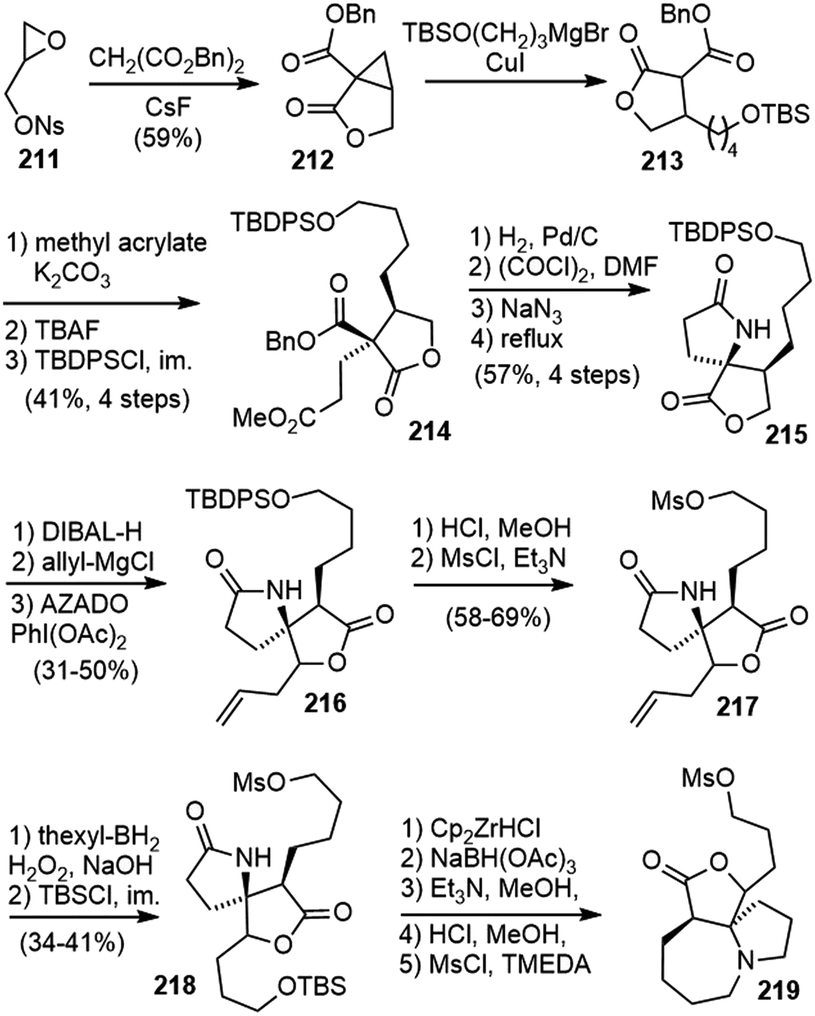 | ||
| Scheme 44 Fukuyama and Yokoshima's synthetic studies on huperzine R (part 1). | ||
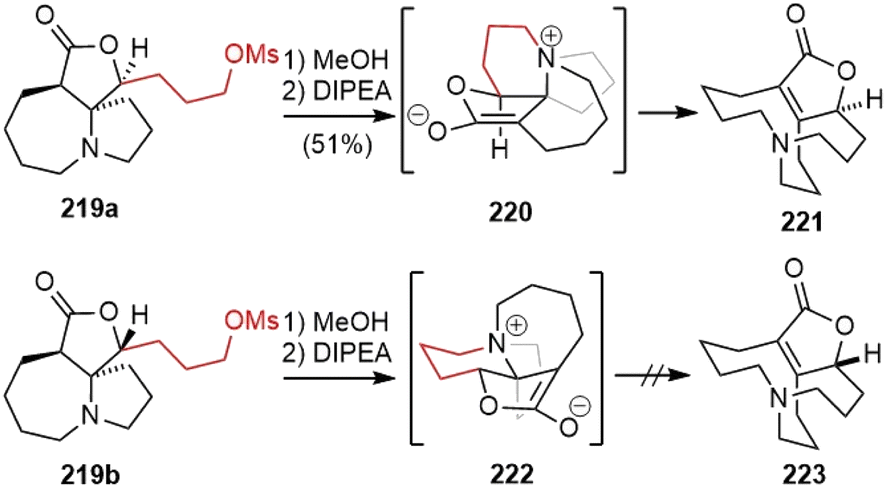
The two diastereomeric mesylates 219 were both subjected to the key fragmentation reaction (Scheme 45). The heating of mesylate 219a in methanol resulted in the formation of an ammonium salt. Subsequent treatment with N,N-diisopropylethylamine (DIPEA) led to the elimination of the ammonium moiety via enolate 220, and generated the α,β-unsaturated lactone 221, which contains the skeleton of huperzine R. In this transformation, the central chirality of the spirocyclic system is efficiently transferred to the planar chirality of the bicyclic system. Heating the isomeric mesylate 219b in refluxing methanol also gave an ammonium salt. However, upon treatment with DIPEA, it underwent extensive decomposition. The authors proposed that the stereoelectronic effect plays a key role in the outcome of the reactions. The π-orbital of the enolate and the adjacent C–N bond in 220 are parallel, resulting in the prompt elimination of the ammonium moiety. The misalignment of C–N bond with the π-orbital of the enolate in 222 caused a poor reaction.
 | ||
| Scheme 45 Fukuyama and Yokoshima's synthetic studies on huperzine R (Part 2). | ||
9.2. Fukuyama and Yokoshima's total synthesis
Three years later, Fukuyama and Yokoshima disclosed a total synthesis of huperzine R in 2018 (Scheme 46).100 The synthesis featured an intramolecular 1,3-dipolar cycloaddition of a nitrile oxide and reductive cleavage of the resulting isoxazoline to induce a retro-aldol-type reaction, forming the bicyclic lactam core.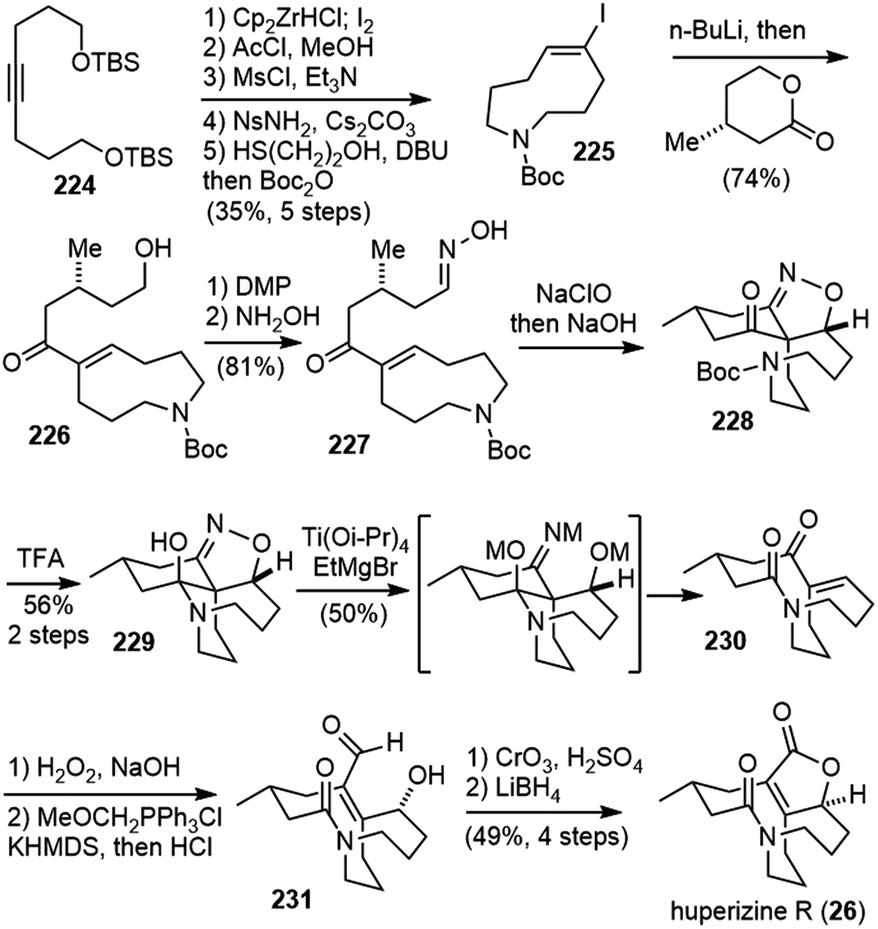 | ||
| Scheme 46 Fukuyama and Yokoshima's total synthesis of huperzine R. | ||
Symmetrical alkyne 224 was transformed into a macrocyclic vinyl iodide 225 in 5 steps, including a hydrozirconation-iodination process and a Fukuyama macrocyclization reaction. Halogen/lithium exchange of 225 generated an alkenyllithium species, which reacted with a chiral lactone to provide the hydroxy ketone 226. The oxidation of the alcohol and condensation with NH2OH afforded oxime 227. Chlorination of the oxime with NaClO and subsequent treatment with NaOH generated a nitrile oxide, which underwent the key 1,3-dipolar cycloaddition reaction to give an isoxazoline 228 with a diastereomeric ratio of 3.7:1. Cleavage of the Boc group with TFA afforded hemiaminal 229 in 56% yield. The diastereomers could be separated at this stage. Treatment of 229 with a titanium(III) reagent resulted in the reductive cleavage of the N–O bond and a retro-aldol-type reaction to afford lactam 230. Epoxidation of the enone, and a Wittig olefin of the ketone gave aldehyde 231. Final redox transformation installed the lactone ring and gave huperzine R.
10. Medicinal chemistry of huperzines
The importance of the cholinergic system and the potential role of AChE in neurodegenerative diseases have made the development of AChE inhibitors an attractive treatment for neurodegenerative disease. Due to their potent AChE inhibitory activities, huperzines A and B have gained a diverse range of interests from the medicinal chemistry community. Derivatives of huperzines A and B were investigated to search for optimal AChE inhibitors (Table 2).| Entry | modification | structure | activity | Ref. |
|---|---|---|---|---|
| 1 | (±)-10,10-Dimethylhuperzine A |
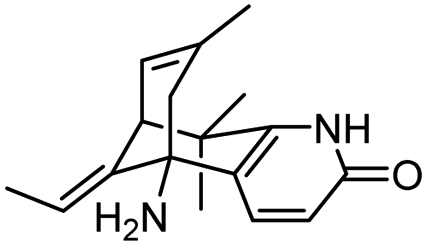
|
In vitro IC50: AChE (16.7 nM) | 102 |
| 2 | Fluorinated analogues of huperzine A |
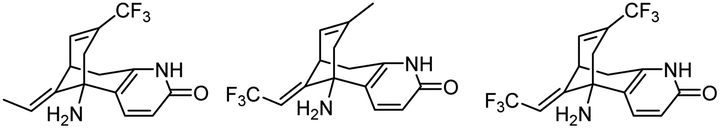
|
In vitro IC50: AChE (0.4–3 μM) | 106 |
| 3 | (±)-14-Fluorohuperzine A |
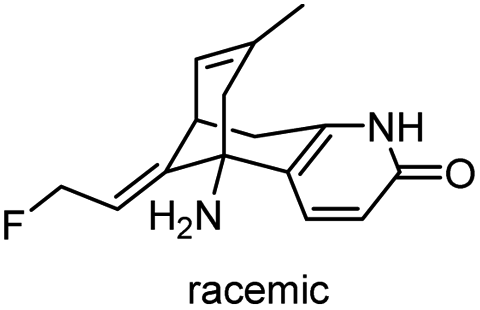
|
In vitro IC50: AChE (10 μM) | 107 |
| 4 | Analogs of huperzine A |
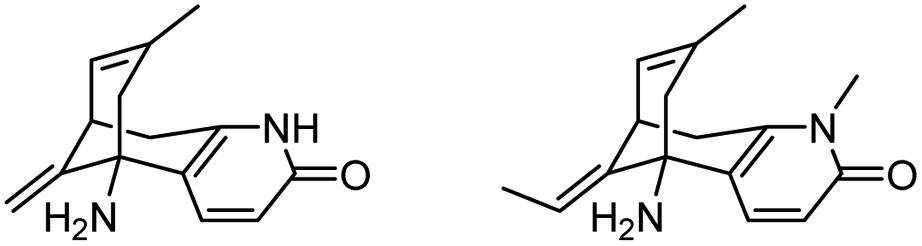
|
Improved survival of guinea pigs exposed to soman | 108 |
| 5 | Analogues of huperzine B |
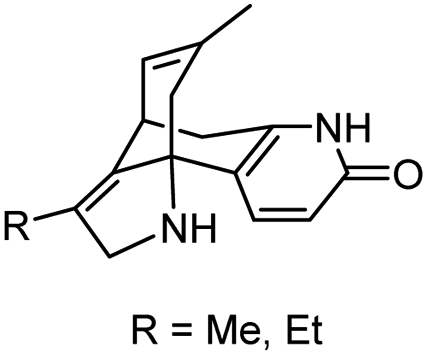
|
In vitro IC50: AChE (0.22–0.40 μM) | 109 |
| 6 | 5-Desamino huperzine A |
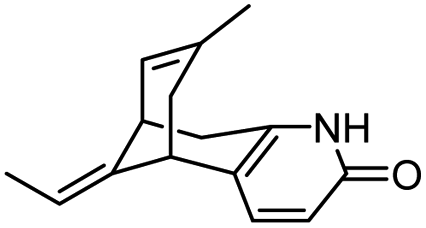
|
In vitro IC50: rAChE (12.8 μM) | 69 |
| 7 | Schiff base of huperzine A |
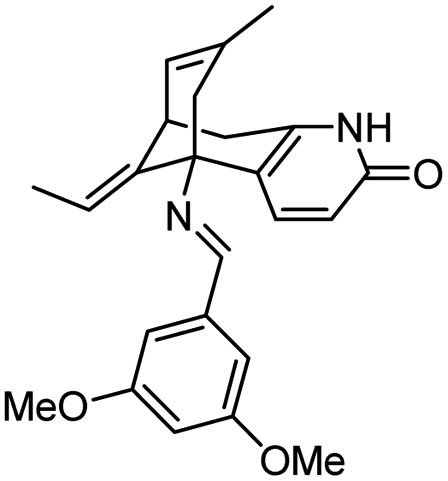
|
In vitro IC50: TcAChE (0.02 nM) hAChE (29 nM) | 110 |
| 8 | C(2)-Functionalized huperzine A |
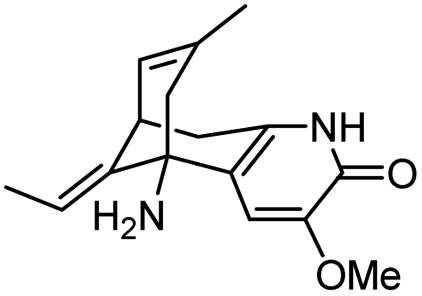
|
In vitro IC50: AChE (0.16 μM) | 111 |
| 9 | N-(Hetero)aryl analogue of huperzine A |
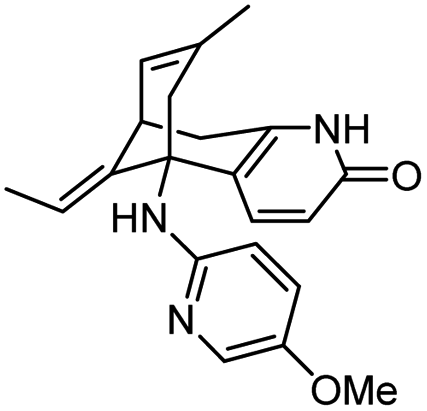
|
In vitro IC50: AChE (1.5 μM) | 112 |
| 10 | Huperzine A-tacrine hybrid |
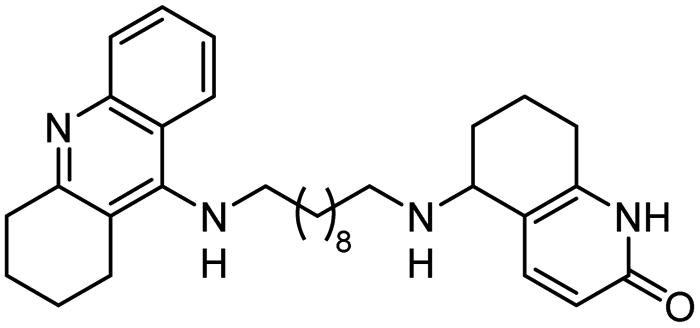
|
In vitro IC50: AChE (8.8 nM) | 113 |
| 11 | Tacrine–huperzine A hybrids |
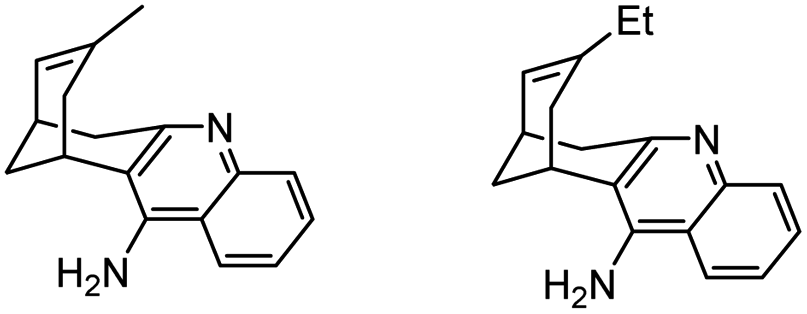
|
In vitro IC50: AChE (65 nM and 38.5 nM) | 114 |
| 12 | Tacrine–huperzine A hybrids |
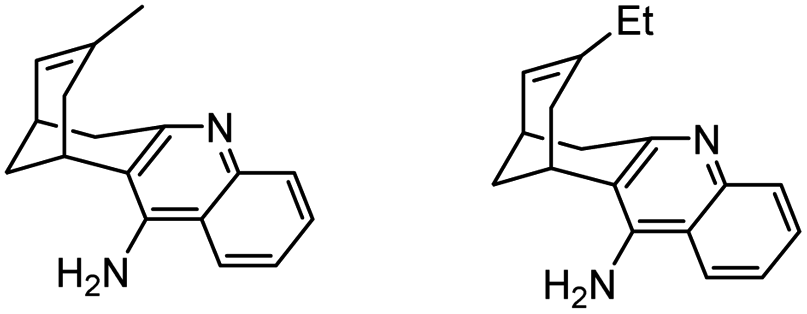
|
In vitro IC50: AChE (4.5 nM), BChE (347 nM) | 115 |
| 13 | Tacrine–huperzine A hybrids (huprines) |
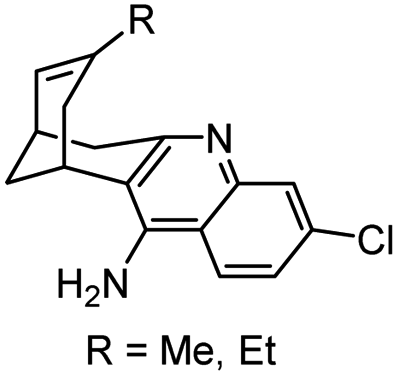
|
In vitro IC50: hAChE (0.32 nM), hBChE (159 nM) | 116 |
| 14 | syn-Huprines |
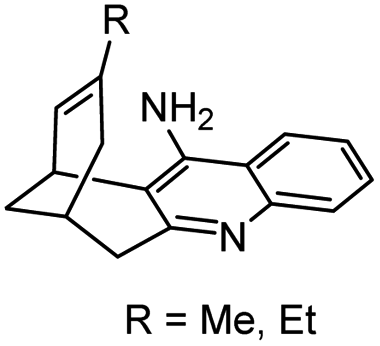
|
In vitro IC50: hAChE (77–436 nM), hBChE (63–148 nM) | 117 |
| 15 | Fragment dimer |
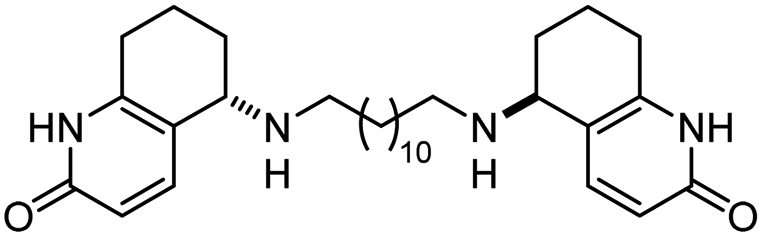
|
In vitro IC50: AChE (52 nM), BChE (9600 nM) | 104 |
| 16 | Bis-huperzine B analogue |
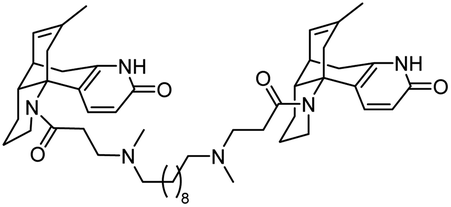
|
In vitro IC50: AChE (4.93 nM), BChE (54300 nM) |
118 |
| 17 | Huprine-tacrine heterodimer |
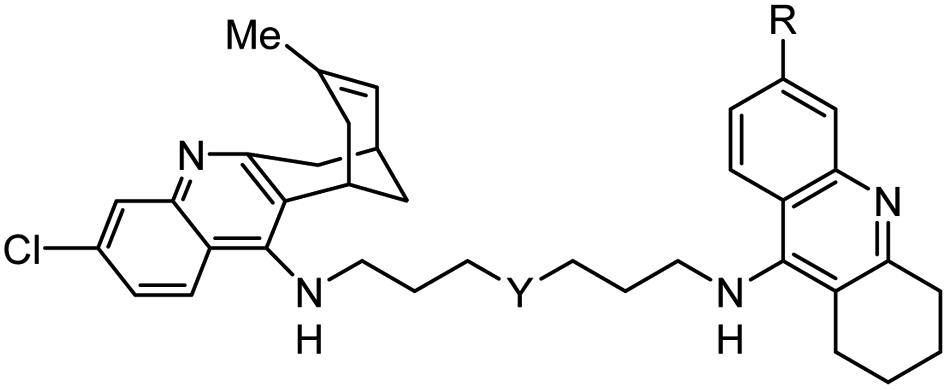
|
In vitro IC50: hAChE (0.29–0.8 nM), hBChE (4.74–31 nM) | 119 |
| 18 | Huperzine A-tacrine heterodimer |
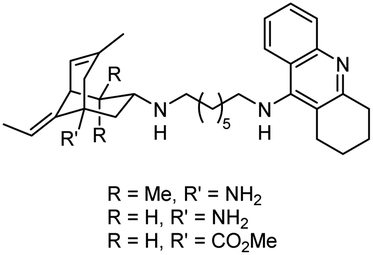
|
In vitro IC50: hAChE (6.4–16.5 nM), hBChE (19.5–30.8 nM) | 120 |
| 19 | Heterodimer of donepezil and huperzine A fragment |
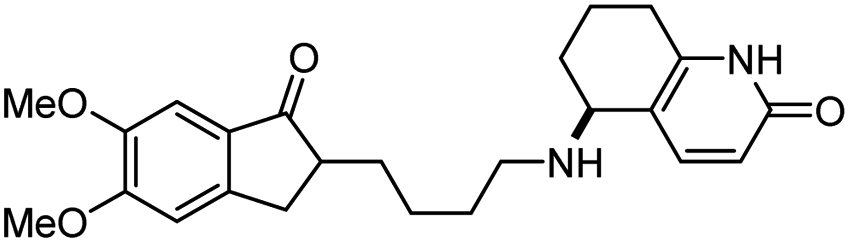
|
In vitro IC50: hAChE (9 nM), hBChE (>1000 nM) | 121 |
| 20 | Huprine–tacrine heterodimer |
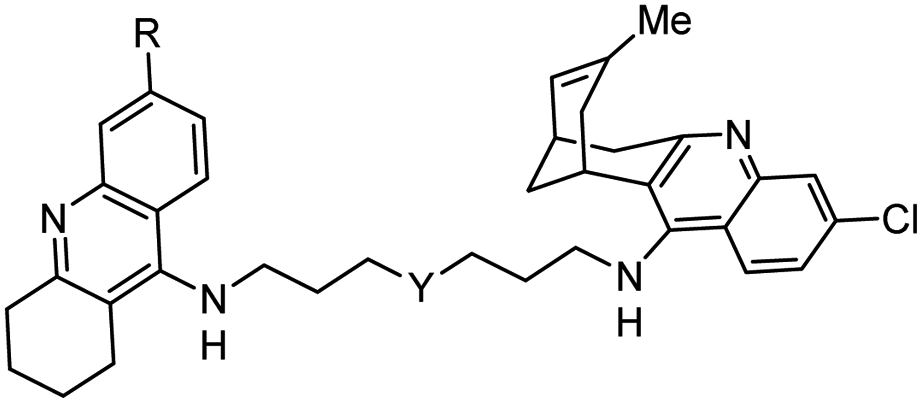
|
In vitro IC50: hAChE (0.31 nM), hBChE (25 nM) | 122 |
| 21 | Rhein–huprine heterodimer |
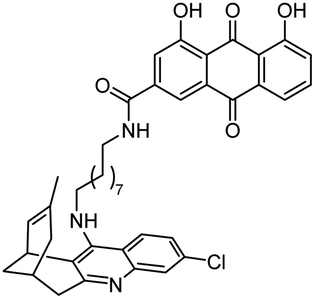
|
In vitro IC50: hAChE (2.39 nM), hBChE (513 nM) and central Aβ lowering effect in vivo | 123 |
| 22 | Levetiracetam huprine heterodimer |
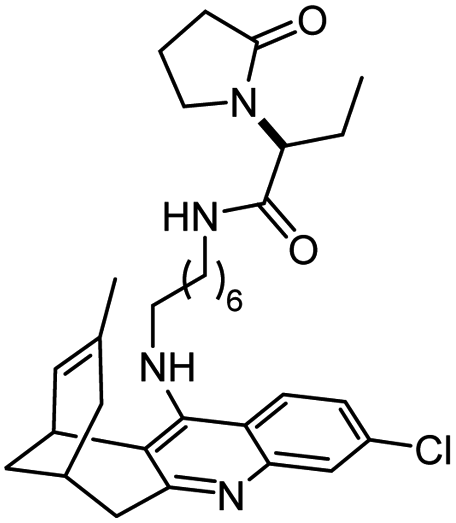
|
In vitro IC50: hAChE (4.2 nM), hBChE (232 nM) and chronic and acute effects in vivo | 124 |
| 23 | Huprine Y and capsaicin heterodimer |
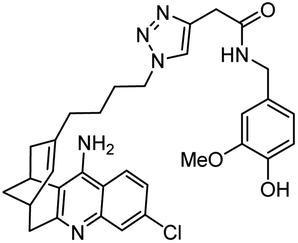
|
In vitro IC50: hAChE (1.1 nM), hBChE (7.3 nM) and antioxidant activity | 125 |
Early study on the structure–activity relationship of huperzine A focused on the modification of its core structure. Kozikowski and coworkers prepared a variety of derivatives of huperzine A, following their successful total synthesis of the natural product. Their work revealed the importance of the unsaturation and the three-carbon bridge to its AChE inhibitory activity.101 Several more potent analogues of huperzine A, including the 10-axial methyl analogue 232 (IC50 = 3 nM for FBS AChE), were developed (Fig. 6).102 They went on to collaborate with the Sussman group and reported the structure of AChE in complex with huperzine A in 1997.103 The high affinity of the natural product to AChE was mostly due to the hydrogen bonding and hydrophobic interactions between huperzine A and the aromatic residues in the active-site gorge of AChE.
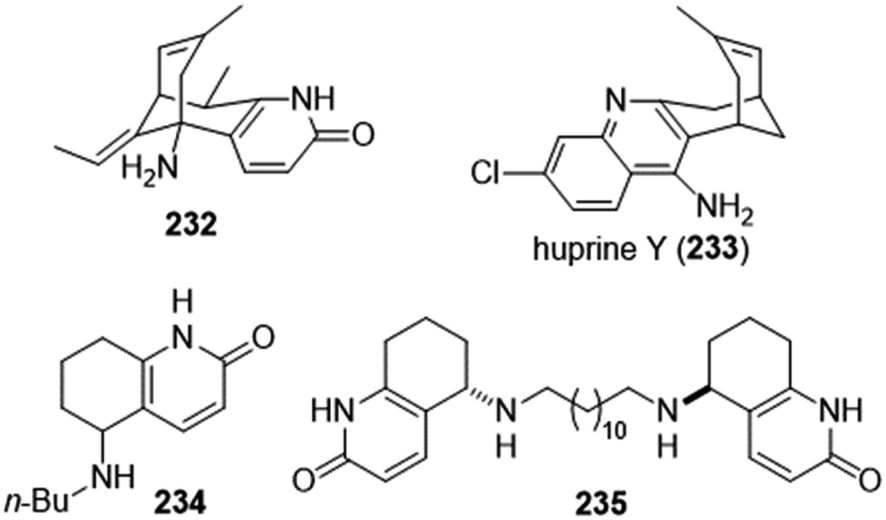 | ||
| Fig. 6 Analogues of huperzine A. | ||
A series of hybrid structures of tacrine and huperzine A, represented by huprine Y (233), possess potent inhibitory activities against AChE. In 2000, Carlier found that dimerization of an inactive fragment (234) of huperzine A produced a bivalent compound 235 with twice the potency of the natural product.104 The structure of AChE in the complex with the bivalent ligand was also solved.105 The findings revealed that the enhanced affinity of the dimer analogues for AChE was attributed to their binding to the two “anionic” sites of the enzyme. Since then, numerous homo- or hetero-bivalent ligands were developed (Table 2, entries 15–23).
11. Conclusions
Since the discovery of huperzines A and B and their potent inhibitory activity on AChE, huperzine alkaloids have been intensively investigated by the scientists from different disciplines.Their unique structure has been targeted by synthetic organic chemists for more than 40 years since the early efforts on the synthesis of seganine, a proposed wrong structure of huperzine A. These four-decade-long synthetic efforts have yielded dozens of total syntheses, including several routes that can be scaled up. The scalable synthesis of (−)-huperzine A has demonstrated that highly efficient chemical synthesis can be a viable approach to address the demand for high-value products. However, given the modest overall yield of the kilogram scale synthesis, there is still room for further improvement to reduce the cost of larger scale synthesis. The development of highly efficient catalytic asymmetric reactions and synthetic strategies, coupled with the advancements in related fields, such as flow chemistry126 and synthetic biology,127 will be beneficial for the production of these valuable alkaloids. (−)-Huperzine A has been approved for the treatment of AD only in China. To further improve its efficacy and gain wider application, it would require more efforts on the medicinal chemistry of (−)-huperzine A and its derivatives.
Other alkaloids in this family received less attention, but impressive progress has also been made in the past decade. The total syntheses of huperzines B, O, Q and R have all been reported in the past decade. The work on the structure reassignment and total syntheses of huperzines K, M and N showcased the efficiency of cascade reactions in the building of complex structures and the power of total synthesis in the structure determination of natural products. The challenges and opportunities provided by this family of alkaloids will undoubtedly continue to stimulate the development of new chemistry and pharmaceuticals.
12. Conflicts of interest
There are no conflicts to declare.13. Acknowledgements
Financially support from Shenzhen Fundamental Research Program (GXWD20201230155427003-20200822151253001) and Shenzhen Peacock Plan (HA11409033) was acknowledged.14. Notes and references
- E. J. Mufson, S. E. Counts, S. E. Perez and S. D. Ginsberg, Expert Rev. Neurother., 2008, 8, 1703–1718 CrossRef CAS.
- T. H. Ferreira-Vieira, I. M. Guimaraes, F. R. Silva and F. M. Ribeiro, Curr. Neuropharmacol., 2016, 14, 101–115 CrossRef CAS.
- J.-S. Liu, Y.-L. Zhu, C.-M. Yu, Y.-Z. Zhou, Y.-Y. Han, F.-W. Wu and B.-F. Qi, Can. J. Chem., 1986, 64, 837–839 CrossRef CAS.
- T. Hemscheidt and I. D. Spenser, J. Am. Chem. Soc., 1993, 115, 3020–3021 CrossRef CAS.
- X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752–772 RSC.
- X. Li, W. Li, P. Tian and T. Tan, Biotechnol. Adv., 2022, 60, 108026 CrossRef CAS PubMed.
- R. S. Nett, Y. Dho, Y.-Y. Low and E. S. Sattely, Proc. Natl. Acad. Sci., 2021, 118, e2102949118 CrossRef CAS.
- P. Siengalewicz, J. Mulzer and U. Rinner, in The Alkaloids: Chemistry and Biology, ed. H.-J. Knölker, Academic Press, 2013, vol. 72, pp. 1–151 Search PubMed.
- S. Thorroad, P. Worawittayanont, N. Khunnawutmanotham, N. Chimnoi, A. Jumruksa, S. Ruchirawat and N. Thasana, Tetrahedron, 2014, 70, 8017–8022 CrossRef CAS.
- C.-H. Tan, X.-Q. Ma, G.-F. Chen and D.-Y. Zhu, Can. J. Chem., 2003, 81, 315–318 CrossRef CAS.
- W. Gao, Y. Li, S. Jiang and D. Zhu, Planta Med., 2000, 66, 664–667 CrossRef CAS.
- K. Katakawa, M. Kitajima, K. Yamaguchi and H. Takayama, Heterocycles, 2006, 69, 223 CrossRef CAS.
- W.-Y. Gao, Y.-M. Li, S.-H. Jiang and D.-Y. Zhu, Helv. Chim. Acta, 2008, 91, 1031–1035 CrossRef CAS.
- B. Wang, N. Teng and D. Zhu, Chin. J. Org. Chem., 2000, 20, 812 CAS.
- C.-H. Tan, X.-Q. Ma, G.-F. Chen and D.-Y. Zhu, Helv. Chim. Acta, 2002, 85, 1058–1061 CrossRef CAS.
- C.-H. Tan, G.-F. Chen, X.-Q. Ma, S.-H. Jiang and D.-Y. Zhu, J. Nat. Prod., 2002, 65, 1021–1022 CrossRef CAS.
- Z. Valenta, H. Yoshimura, E. F. Rogers, M. Ternbah and K. Wiesner, Tetrahedron Lett., 1960, 1, 26–33 CrossRef.
- W. A. Ayer, L. M. Browne, H. Orszanska, Z. Valenta and J.-S. Liu, Can. J. Chem., 1989, 67, 1538–1540 CrossRef CAS.
- A. P. Kozikowski and W. Tückmantel, Acc. Chem. Res., 1999, 32, 641–650 CrossRef CAS.
- S. Zheng, C. Yu and Z. Shen, Chin. J. Org. Chem., 2013, 33, 2261 CrossRef CAS.
- S. Md, A. Sohel and T. Opatz, Arkivoc, 2013, 2014, 92–108 Search PubMed.
- A. S. Kende and J. A. Schneider, Synth. Commun., 1979, 9, 419–425 CrossRef CAS.
- D. Gravel, R. Déziel and L. Bordeleau, Tetrahedron Lett., 1983, 24, 699–702 CrossRef CAS.
- A. S. Kende, F. H. Ebetino, R. Battista, R. J. Boatman, D. P. Lorah and E. Lodge, Heterocycles, 1984, 21, 91 CrossRef CAS.
- Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116–4117 CrossRef CAS.
- A. P. Kozikowski, Y. Xia, E. R. Reddy, W. Tuckmantel, I. Hanin and X. C. Tang, J. Org. Chem., 1991, 56, 4636–4645 CrossRef CAS.
- G. Stork, Pure Appl. Chem., 1968, 17, 383–402 CrossRef CAS.
- A. P. Kozikowski, E. R. Reddy and C. P. Miller, J. Chem. Soc. Perkin, 1990, 1, 195–197 RSC.
- L. Qian and R. Ji, Tetrahedron Lett., 1989, 30, 2089–2090 CrossRef CAS.
- T. Shioiri, K. Ninomiya and S. Yamada, J. Am. Chem. Soc., 1972, 94, 6203–6205 CrossRef CAS PubMed.
- D. Bai, Pure Appl. Chem., 2007, 79, 469–479 CrossRef CAS.
- F. Yamada, A. P. Kozikowski, E. R. Reddy, Y. P. Pang, J. H. Miller and M. McKinney, J. Am. Chem. Soc., 1991, 113, 4695–4696 CrossRef CAS.
- A. P. Kozikowski, G. Campiani, P. Aagaard and M. McKinney, J. Chem. Soc. Chem. Commun., 1993, 860–862 RSC.
- G. Campiani, L. Q. Sun, A. P. Kozikowski, P. Aagaard and M. McKinney, J. Org. Chem., 1993, 58, 7660–7669 CrossRef CAS.
- Y. Huang and X. Lu, Tetrahedron Lett., 1988, 29, 5663–5664 CrossRef CAS.
- S. R. Tudhope, J. A. Bellamy, A. Ball, D. Rajasekar, M. Azadi-Ardakani, H. S. Meera, J. M. Gnanadeepam, R. Saiganesh, F. Gibson, L. He, C. H. Behrens, G. Underiner, J. Marfurt and N. Favre, Org. Process Res. Dev., 2012, 16, 635–642 CrossRef CAS.
- S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Tetrahedron: Asymmetry, 1997, 8, 829–832 CrossRef CAS.
- T. Koshiba, S. Yokoshima and T. Fukuyama, Org. Lett., 2009, 11, 5354–5356 CrossRef CAS.
- C. Bolm, I. Schiffers, C. L. Dinter and A. Gerlach, J. Org. Chem., 2000, 65, 6984–6991 CrossRef CAS.
- M. Fujii, T. Nishimura, T. Koshiba, S. Yokoshima and T. Fukuyama, Org. Lett., 2013, 15, 232–234 CrossRef CAS.
- T. Nishimura, A. K. Unni, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2011, 133, 418–419 CrossRef CAS.
- T. Nishimura, A. K. Unni, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2013, 135, 3243–3247 CrossRef CAS.
- H. Lei, S. Xin, Y. Qiu and X. Zhang, Chem. Commun., 2018, 54, 727–730 RSC.
- M. K. M. Tun, D.-J. Wüstmann and S. B. Herzon, Chem. Sci., 2011, 2, 2251–2253 RSC.
- H. W. Lee, S. K. Ji, I.-Y. C. Lee and J. H. Lee, J. Org. Chem., 1996, 61, 2542–2543 CrossRef CAS.
- M. K. M. Tun and S. B. Herzon, J. Org. Chem., 2012, 77, 9422–9425 CrossRef CAS PubMed.
- T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249–261 CrossRef CAS.
- R. Ding, B.-F. Sun and G.-Q. Lin, Org. Lett., 2012, 14, 4446–4449 CrossRef CAS PubMed.
- R. Ding, J.-G. Fu, G.-Q. Xu, B.-F. Sun and G.-Q. Lin, J. Org. Chem., 2014, 79, 240–250 CrossRef CAS PubMed.
- C. Lucey, S. A. Kelly and J. Mann, Org. Biomol. Chem., 2007, 5, 301–306 RSC.
- J. D. White, Y. Li, J. Kim and M. Terinek, Org. Lett., 2013, 15, 882–885 CrossRef CAS.
- J. D. White, Y. Li, J. Kim and M. Terinek, J. Org. Chem., 2015, 80, 11806–11817 CrossRef CAS.
- J. D. White, J. Kim and N. E. Drapela, J. Am. Chem. Soc., 2000, 122, 8665–8671 CrossRef CAS.
- H. Yan, G. S. Smith and F.-E. Chen, Green Synth. Catal., 2022, 3, 219–226 CrossRef.
- J. E. Audia, L. Boisvert, A. D. Patten, A. Villalobos and S. J. Danishefsky, J. Org. Chem., 1989, 54, 3738–3740 CrossRef CAS.
- P. Magnus and J. Lacour, J. Am. Chem. Soc., 1992, 114, 767–769 CrossRef CAS.
- C. Chassaing, A. Haudrechy and Y. Langlois, Tetrahedron Lett., 1999, 40, 8805–8809 CrossRef CAS.
- A. Haudrechy, C. Chassaing, C. Riche and Y. Langlois, Tetrahedron, 2000, 56, 3181–3187 CrossRef CAS.
- P. Camps, J. Contreras, R. El Achab, J. Morral, D. Muñoz-Torrero, M. Font-Bardia, X. Solans, A. Badia and N. M. Vivas, Tetrahedron, 2000, 56, 4541–4553 CrossRef CAS.
- P. Camps, J. Contreras, M. Font-Bardia and X. Solans, Synth. Commun., 1996, 26, 9–18 CrossRef CAS.
- I. Y. C. Lee, M. H. Jung, H. W. Lee and J. Y. Yang, Tetrahedron Lett., 2002, 43, 2407–2409 CrossRef CAS.
- S. Terashima, S. Kaneko, T. Yoshino and T. Katoh, Heterocycles, 1997, 46, 27 CrossRef.
- S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Tetrahedron, 1998, 54, 5471–5484 CrossRef CAS.
- Q.-B. Pan and D.-W. Ma, Chin. J. Chem., 2003, 21, 793–796 CrossRef CAS.
- X.-H. Ding, X. Li, D. Liu, W.-C. Cui, X. Ju, S. Wang and Z.-J. Yao, Tetrahedron, 2012, 68, 6240–6248 CrossRef CAS.
- X.-C. He, B. Wang and D. Bai, Tetrahedron Lett., 1998, 39, 411–414 CrossRef CAS.
- X.-C. He, B. Wang, G. Yu and D. Bai, Tetrahedron: Asymmetry, 2001, 12, 3213–3216 CrossRef CAS.
- C.-F. Lin, C.-W. Chien and I. Ojima, Org. Chem. Front., 2014, 1, 1062–1066 RSC.
- K. Högenauer, K. Baumann, A. Enz and J. Mulzer, Bioorg. Med. Chem. Lett., 2001, 11, 2627–2630 CrossRef PubMed.
- J. Ward and V. Caprio, Tetrahedron Lett., 2006, 47, 553–556 CrossRef CAS.
- V. Caprio and J. Ward, Heterocycles, 2009, 79, 791 CrossRef.
- B. Wu and D. Bai, J. Org. Chem., 1997, 62, 5978–5981 CrossRef CAS.
- R. Jaouhari, B. Maillard, C. Filliatre and J.-J. Villenave, Synthesis, 1982, 1982, 760–763 CrossRef.
- D. Schumann and A. Naumann, Liebigs Ann. Chem., 1983, 1983, 220–225 CrossRef.
- D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS.
- H. M. S. Haley, S. E. Payer, S. M. Papidocha, S. Clemens, J. Nyenhuis and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 4732–4740 CrossRef CAS.
- I. Y. Choi Lee, J. Y. Hong, M. H. Jung and H. W. Lee, Tetrahedron Lett., 2004, 45, 285–287 CrossRef.
- D. Caine, K. Procter and R. A. Cassell, J. Org. Chem., 1984, 49, 2647–2648 CrossRef CAS.
- E. Schuster, G. Jas and D. Schumann, Org. Prep. Proced. Int., 1992, 24, 670–672 CrossRef CAS.
- L. J. Gooßen and N. Rodríguez, Chem. Commun., 2004, 724–725 RSC.
- D. Gauthier, A. T. Lindhardt, E. P. K. Olsen, J. Overgaard and T. Skrydstrup, J. Am. Chem. Soc., 2010, 132, 7998–8009 CrossRef CAS PubMed.
- B. Bradshaw, C. Luque-Corredera and J. Bonjoch, Org. Lett., 2013, 15, 326–329 CrossRef CAS PubMed.
- D. W. Brooks, L. D.-L. Lu and S. Masamune, Angew. Chem., Int. Ed. Engl., 1979, 18, 72–74 CrossRef.
- B. Bradshaw, C. Luque-Corredera and J. Bonjoch, Chem. Commun., 2014, 50, 7099–7102 RSC.
- B. Bradshaw, C. Luque-Corredera, G. Saborit, C. Cativiela, R. Dorel, C. Bo and J. Bonjoch, Chem.–Eur. J., 2013, 19, 13881–13892 CrossRef CAS.
- C. Bosch, B. Fiser, E. Gómez-Bengoa, B. Bradshaw and J. Bonjoch, Org. Lett., 2015, 17, 5084–5087 CrossRef CAS.
- C. Bosch, B. Bradshaw and J. Bonjoch, J. Nat. Prod., 2019, 82, 1576–1586 CrossRef CAS.
- G. V. Saborit, C. Bosch, T. Parella, B. Bradshaw and J. Bonjoch, J. Org. Chem., 2016, 81, 2629–2634 CrossRef CAS PubMed.
- X.-H. Zhao, Q. Zhang, J.-Y. Du, X.-Y. Lu, Y.-X. Cao, Y.-H. Deng and C.-A. Fan, J. Am. Chem. Soc., 2017, 139, 7095–7103 CrossRef CAS.
- C. H. Heathcock, E. F. Kleinman and E. S. Binkley, J. Am. Chem. Soc., 1982, 104, 1054–1068 CrossRef CAS.
- A. Nakayama, N. Kogure, M. Kitajima and H. Takayama, Angew. Chem., Int. Ed., 2011, 50, 8025–8028 CrossRef CAS PubMed.
- T. Mandai, M. Ueda, S. Hasegawa, M. Kawada, J. Tsuji and S. Saito, Tetrahedron Lett., 1990, 31, 4041–4044 CrossRef CAS.
- B. Hong, H. Li, J. Wu, J. Zhang and X. Lei, Angew. Chem., Int. Ed., 2015, 54, 1011–1015 CrossRef CAS.
- M. Carda, J. Van der Eycken and M. Vandewalle, Tetrahedron: Asymmetry, 1990, 1, 17–20 CrossRef CAS.
- R. Noyori and M. Suzuki, Angew. Chem., Int. Ed. Engl., 1984, 23, 847–876 CrossRef.
- C. Zeng, J. Zhao and G. Zhao, Tetrahedron, 2015, 71, 64–69 CrossRef CAS.
- S. Tanimura, S. Yokoshima and T. Fukuyama, Org. Lett., 2017, 19, 3684–3686 CrossRef CAS.
- H. Kumazaki, R. Nakajima, Y. Bessho, S. Yokoshima and T. Fukuyama, Synlett, 2015, 26, 2131–2134 CrossRef CAS.
- K. Kitaori, M. Mikami, Y. Furukawa, H. Yoshimoto and J. Otera, Synlett, 1998, 1998, 499–500 CrossRef.
- T. Nomura, S. Yokoshima and T. Fukuyama, Org. Lett., 2018, 20, 119–121 CrossRef PubMed.
- A. P. Kozikowski, C. P. Miller, F. Yamada, Y. P. Pang, J. H. Miller, M. McKinney and R. G. Ball, J. Med. Chem., 1991, 34, 3399–3402 CrossRef CAS.
- A. P. Kozikowski, G. Campiani, L.-Q. Sun, S. Wang, A. Saxena and B. P. Doctor, J. Am. Chem. Soc., 1996, 118, 11357–11362 CrossRef CAS.
- M. L. Raves, M. Harel, Y.-P. Pang, I. Silman, A. P. Kozikowski and J. L. Sussman, Nat. Struct. Biol., 1997, 4, 57–63 CrossRef CAS.
- P. R. Carlier, D.-M. Du, Y.-F. Han, J. Liu, E. Perola, I. D. Williams and Y.-P. Pang, Angew. Chem., Int. Ed., 2000, 39, 1775–1777 CrossRef CAS.
- D. M. Wong, H. M. Greenblatt, H. Dvir, P. R. Carlier, Y.-F. Han, Y.-P. Pang, I. Silman and J. L. Sussman, J. Am. Chem. Soc., 2003, 125, 363–373 CrossRef CAS.
- S. Kaneko, N. Nakajima, M. Shikano, T. Katoh and S. Terashima, Bioorg. Med. Chem. Lett., 1996, 6, 1927–1930 CrossRef CAS.
- Z. Fanxing, J. Hualiang, T. Xican, C. Kaixian and J. Ruyun, Bioorg. Med. Chem. Lett., 1998, 8, 1661–1664 CrossRef CAS.
- H. Gunosewoyo, S. K. Tipparaju, M. Pieroni, Y. Wang, B. P. Doctor, M. P. Nambiar and A. P. Kozikowski, Bioorg. Med. Chem. Lett., 2013, 23, 1544–1547 CrossRef CAS.
- V. Rajendran, A. Saxena, B. P. Doctor and A. P. Kozikowski, Bioorg. Med. Chem. Lett., 2002, 12, 1521–1523 CrossRef CAS.
- J. Yan, L. Sun, G. Wu, P. Yi, F. Yang, L. Zhou, X. Zhang, Z. Li, X. Yang, H. Luo and M. Qiu, Bioorg. Med. Chem., 2009, 17, 6937–6941 CrossRef CAS PubMed.
- S. Anukanon, P. Pongpamorn, W. Tiyabhorn, J. Chatwichien, W. Niwetmarin, R. B. Sessions, S. Ruchirawat and N. Thasana, ACS Omega, 2021, 6, 19924–19939 CrossRef CAS PubMed.
- S.-X. Miao, L.-X. Wan, Z.-X. He, X.-L. Zhou, X. Li and F. Gao, J. Nat. Prod., 2021, 84, 2374–2379 CrossRef CAS.
- P. R. Carlier, D.-M. Du, Y. Han, J. Liu and Y.-P. Pang, Bioorg. Med. Chem. Lett., 1999, 9, 2335–2338 CrossRef CAS.
- A. Badia, J. E. Baños, P. Camps, J. Contreras, D. M. Görbig, D. Muñoz-Torrero, M. Simón and N. M. Vivas, Bioorg. Med. Chem., 1998, 6, 427–440 CrossRef CAS.
- P. Camps, R. El Achab, D. M. Görbig, J. Morral, D. Muñoz-Torrero, A. Badia, J. E. Baños, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 1999, 42, 3227–3242 CrossRef CAS PubMed.
- P. Camps, R. El Achab, J. Morral, D. Muñoz-Torrero, A. Badia, J. E. Baños, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 2000, 43, 4657–4666 CrossRef CAS.
- P. Camps, E. Gómez, D. Muñoz-Torrero, A. Badia, N. M. Vivas, X. Barril, M. Orozco and F. J. Luque, J. Med. Chem., 2001, 44, 4733–4736 CrossRef CAS PubMed.
- S. Feng, Z. Wang, X. He, S. Zheng, Y. Xia, H. Jiang, X. Tang and D. Bai, J. Med. Chem., 2005, 48, 655–657 CrossRef CAS.
- P. Camps, X. Formosa, D. Muñoz-Torrero, J. Petrignet, A. Badia and M. V. Clos, J. Med. Chem., 2005, 48, 1701–1704 CrossRef CAS PubMed.
- S. Gemma, E. Gabellieri, P. Huleatt, C. Fattorusso, M. Borriello, B. Catalanotti, S. Butini, M. De Angelis, E. Novellino, V. Nacci, T. Belinskaya, A. Saxena and G. Campiani, J. Med. Chem., 2006, 49, 3421–3425 CrossRef CAS PubMed.
- Y. Hu, J. Zhang, O. Chandrashankra, F. C. F. Ip and N. Y. Ip, Bioorg. Med. Chem., 2013, 21, 676–683 CrossRef CAS.
- C. Galdeano, E. Viayna, I. Sola, X. Formosa, P. Camps, A. Badia, M. V. Clos, J. Relat, M. Ratia, M. Bartolini, F. Mancini, V. Andrisano, M. Salmona, C. Minguillón, G. C. González-Muñoz, M. I. Rodríguez-Franco, A. Bidon-Chanal, F. J. Luque and D. Muñoz-Torrero, J. Med. Chem., 2012, 55, 661–669 CrossRef CAS.
- E. Viayna, I. Sola, M. Bartolini, A. De Simone, C. Tapia-Rojas, F. G. Serrano, R. Sabaté, J. Juárez-Jiménez, B. Pérez, F. J. Luque, V. Andrisano, M. V. Clos, N. C. Inestrosa and D. Muñoz-Torrero, J. Med. Chem., 2014, 57, 2549–2567 CrossRef CAS PubMed.
- I. Sola, E. Aso, D. Frattini, I. López-González, A. Espargaró, R. Sabaté, O. Di Pietro, F. J. Luque, M. V. Clos, I. Ferrer and D. Muñoz-Torrero, J. Med. Chem., 2015, 58, 6018–6032 CrossRef CAS.
- E. Viayna, N. Coquelle, M. Cieslikiewicz-Bouet, P. Cisternas, C. A. Oliva, E. Sánchez-López, M. Ettcheto, M. Bartolini, A. De Simone, M. Ricchini, M. Rendina, M. Pons, O. Firuzi, B. Pérez, L. Saso, V. Andrisano, F. Nachon, X. Brazzolotto, M. L. García, A. Camins, I. Silman, L. Jean, N. C. Inestrosa, J.-P. Colletier, P.-Y. Renard and D. Muñoz-Torrero, J. Med. Chem., 2021, 64, 812–839 CrossRef CAS PubMed.
- L. Wan, G. Kong, M. Liu, M. Jiang, D. Cheng and F. Chen, Green Synth. Catal., 2022, 3, 243–258 CrossRef.
- C. A. Voigt, Nat. Commun., 2020, 11, 6379 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |