
Parent 1,4-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole and its derivatives as precursors for the design of promising high energy density materials†
Agata N.
Kuznetsova
 ab,
Nikita E.
Leonov
a,
Oleg V.
Anikin
a,
Michael S.
Klenov
*a,
Aleksandr M.
Churakov
*a,
Yurii A.
Strelenko
a,
Roman A.
Novikov
a,
Ivan V.
Fedyanin
c,
Alla N.
Pivkina
d,
Tatiana S.
Kon’kova
d,
Valery P.
Sinditskii
e,
Anastasia D.
Smirnova
e and
Vladimir A.
Tartakovsky
a
ab,
Nikita E.
Leonov
a,
Oleg V.
Anikin
a,
Michael S.
Klenov
*a,
Aleksandr M.
Churakov
*a,
Yurii A.
Strelenko
a,
Roman A.
Novikov
a,
Ivan V.
Fedyanin
c,
Alla N.
Pivkina
d,
Tatiana S.
Kon’kova
d,
Valery P.
Sinditskii
e,
Anastasia D.
Smirnova
e and
Vladimir A.
Tartakovsky
a
aN. D. Zelinsky Institute of Organic Chemistry Russian Academy of Sciences, 47 Leninsky prospect, Moscow, 119991, Russian Federation. E-mail: klenov@ioc.ac.ru; churakov@ioc.ac.ru
bDepartment of Chemistry, Lomonosov Moscow State University, 1 Leninskie Gory, Moscow, 119991, Russian Federation
cA. N. Nesmeyanov Institute of Organoelement Compounds Russian Academy of Sciences, 28 Vavilova st., Moscow, 119991, Russian Federation
dN. N. Semenov Federal Research Center for Chemical Physics Russian Academy of Sciences, 4 Kosygin st., Moscow, 119991, Russian Federation
eMendeleev University of Chemical Technology, 9 Miusskaya Square, Moscow, 125047, Russian Federation
First published on 26th November 2024
Abstract
The design of novel high energy density materials (HEDMs) is still one of the significant challenges in the field of applied chemistry. Newly discovered scaffolds are rare, but they open up new possibilities for HEDM design. Herein, a simple and effective two-step approach to previously unknown 1,4-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole (1) is presented. This compound is an individual energetic material (d = 1.84 g cm−3,  , Tonset = 193 °C (all experimental values), D = 8.5 km s−1, P = 34 GPa (calculated ones)), but may also serve as a scaffold for the synthesis of structurally diversified related compounds. The precursors of triazolo–triazole 1 are previously unknown 5-amino-4-diazo-4H-1,2,3-triazolium chloride (4) and 5-amino-4-diazo-4H-1,2,3-triazole (5), which can also serve as starting materials for the synthesis of a variety of novel heterocycles. For example, the diazo compound 5 was used for the regioselective synthesis of a 2,5-disubstituted triazolo–triazole 10 bearing an N4-tripoid fragment. Overall, the developed approach to triazolo–triazole 1, 5-amino-4-diazo-4H-1,2,3-triazolium chloride (4) and 5-amino-4-diazo-4H-1,2,3-triazole (5) is expected to make these high nitrogen systems more accessible and affordable for the design of nitrogen containing fused heterocycles.
, Tonset = 193 °C (all experimental values), D = 8.5 km s−1, P = 34 GPa (calculated ones)), but may also serve as a scaffold for the synthesis of structurally diversified related compounds. The precursors of triazolo–triazole 1 are previously unknown 5-amino-4-diazo-4H-1,2,3-triazolium chloride (4) and 5-amino-4-diazo-4H-1,2,3-triazole (5), which can also serve as starting materials for the synthesis of a variety of novel heterocycles. For example, the diazo compound 5 was used for the regioselective synthesis of a 2,5-disubstituted triazolo–triazole 10 bearing an N4-tripoid fragment. Overall, the developed approach to triazolo–triazole 1, 5-amino-4-diazo-4H-1,2,3-triazolium chloride (4) and 5-amino-4-diazo-4H-1,2,3-triazole (5) is expected to make these high nitrogen systems more accessible and affordable for the design of nitrogen containing fused heterocycles.
Introduction
The creation of high-energy compounds with balanced properties continues to be the major focus of research in the modern chemistry of energetic materials (EMs). There is an increased demand for high energy density materials (HEDMs) with enhanced energetic performance for both military and civilian applications. A key challenge is the synthesis of novel HEDMs consisting of a nitrogen-rich heterocyclic backbone with different explosophoric functionalities combined in a single molecule.1 In the long term, structural diversity in heterocyclic cores is required. Polynitrogen fused heterocycles are promising scaffolds for constructing various HEDMs. Due to a number of structural features, such as the inherent planarity and high conjugation efficiency of heteroaromatic systems, this class of compounds is one of the most sought-after structural motifs. The planarity of the molecules leads to π–π stacking, increasing the density and lowering the mechanical sensitivity of the material, while at the same time high conjugation within the system ensures acceptable thermal stability. In addition, numerous N–N and C–N bonds in the molecules, as well as significant ring-strain energy, result in a high positive enthalpy of formation of fused nitrogen heterocycles.2–8These advantages can be used to improve the energetic performance of target molecules.2 For a long time, a limited number of available fused heterocyclic backbones for the synthesis of structurally diverse energetic compounds have hindered the progress of the area of HEDMs. Over recent years, there have been several achievements that encouraged the dynamic development of this field of energetic materials. A variety of fused-ring-based high-energy materials have been elaborated by various research teams.9–14
[5,5]-Bicyclic heterocyclic EMs are at the forefront of high-energy research due to their high enthalpy of formation, good thermal stability and high density.
Pyrazolo[4,3-c]pyrazole (type A), pyrazolo[3,4-d][1,2,3]triazole (type B), [1,2,4]triazolo[4,3-b][1,2,4]triazole (type C), pyrazolo[3,4-c][1,2,5]oxadiazole 5-oxide (type D) and [1,2,3]triazolo[4,5-c][1,2,5]oxadiazole (type E) are promising polynitrogen scaffolds for the construction of novel high-energy materials (Fig. 1). In the last few years these heterocyclic systems made up the backbone of a number of modern EMs synthesized by the research teams led by Shreeve, Klapötke and others.15–31
 | ||
| Fig. 1 Fused [5,5]-bicyclic EMs synthesized previously. | ||
One of the promising [5,5]-bicyclic heterocyclic scaffolds for perspective HEDMs is the [1,2,3]triazolo[4,5-d][1,2,3]triazole (TT) core.21 At the same time, known [1,2,3]triazolo[4,5-d]-[1,2,3]triazoles are limited to a number of examples bearing non-energetic aliphatic or aryl substituents at the 1,5- or 2,5-positions (Fig. 2).32–38 The parent unsubstituted 1,4-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole 1 has not been previously described.
 | ||
| Fig. 2 The previously known non-energetic triazolo–triazoles, triazolo–triazole TT 1 synthesized here, and the future prospects of the functionalization of the triazolo–triazole framework. | ||
Here, we report the first synthesis and properties of parent 1,4-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole (1), which is a potential precursor for novel highly functionalized energetic triazolo–triazoles. TT 1 has several positions for functionalization, and there is the possibility of introducing various substituents which could subsequently be transformed into explosophore groups (Fig. 3).
 | ||
| Fig. 3 Positions for N-functionalization of the triazolo–triazole backbone. | ||
Results and discussion
Synthesis
A two-step method for the synthesis of 5-phenyl-1,5-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole was proposed by Thiele in 1897.32 First, 4,5-diamino-2-phenyl-1H-1,2,3-triazole or its N-monoacetylated derivative was diazotized to form the corresponding diazonium salt. The latter was then treated with a base under heating to obtain a triazolo–triazole system. This synthetic approach was also applied to the pyrazolo-1,2,3-triazole series.39,40 Taking into account this synthetic protocol, we chose N-(5-amino-1H-1,2,3-triazol-4-yl)formamide (2) as a starting material for the synthesis of TT 1 (Scheme 1). Aminotriazole 2 can be easily obtained from dithioxamide in accordance with the procedure described in the literature.41 | ||
| Scheme 1 Synthesis of diazonium salt 4. | ||
Diazotization of aminotriazole 2 with NaNO2 in AcOH led to the formation of an intermediate diazonium salt, which, after dilution of the reaction mixture with ethanol, precipitated from the solution as diazo compound 3 in 35% yield. The replacement of AcOH with CF3CO2H resulted in an increase in the yield of diazotriazole 3 to 85%.
The formyl group of compound 3 was removed with concentrated HCl to afford diazonium salt 4. The latter is the first unsubstituted 1,2,3-triazole bearing an adjacent diazonium group and unsubstituted amino group.
Treatment of diazonium salt 4 with one equivalent of NaOH afforded 5-amino-4-diazo-4H-1,2,3-triazole (5) (Scheme 2a). The reaction was accompanied by a change in colour from yellowish to dark red. Compound 5 is the first heterocyclic diazo compound bearing adjacent diazo and amino groups. In the solid state it decomposed slowly at room temperature, but we were able to obtain good 13C and 14N NMR spectra in D2O solution recorded at 10 °C, which confirm its structure (for details, see the ESI†).
Treatment of diazonium salt 4 with 3 equiv. of NaOH followed by heating under reflux for 15 min was accompanied by a color change from dark-red to colorless, indicating completion of the cyclization process with the formation of sodium salt 6a. Subsequent neutralization of the solution with 3 equiv. of HCl gave target TT 1, which precipitated from the reaction mixture in 61% yield (see Scheme 2a). It was demonstrated that a three-fold excess of HCl led to the opening one of the triazole rings of TT 1, returning diazonium salt 4 in quantitative yield.
 | ||
| Scheme 2 Synthesis of TT 1 from diazonium salt 4 (a) and from diazotriazole 3 (b). | ||
The optimization of the synthetic route provided a more convenient procedure for preparing TT 1 (Scheme 2b). Heating under reflux diazo compound 3 with 3 equiv. of NaOH led to removal of the N-formyl group. Subsequent acidification of the reaction mixture with 3 equiv. of HCl gave TT 1 in 92% yield. This one-pot conversion of compound 3 into TT 1 is a convenient and efficient method for the preparation of the latter in gram scale quantities.
TT 1 on treatment with aqueous bases (NaOH, KOH or 25% aqueous solution of tetraethylammonium hydroxide) afforded the corresponding disodium (6a), dipotassium (6b) and di tetraethylammonium (6c) salts in quantitative yields (Scheme 3a). Sodium salt 6a exists in a form of dihydrate, which is confirmed by elemental analysis and DSC/TGA. Salt 6c is a stable heptahydrate and its structure and molecular composition were confirmed by single crystal X-ray diffraction analysis and elemental analysis.
 | ||
| Scheme 3 Methylation (a) and tert-butylation (b) of TT 1. | ||
To gain insight into the regioselectivity of alkylation of TT 1 and its anion, we used methylation and tert-butylation reactions (see Scheme 3). Methylation of salts 6a–c with MeI resulted in four possible isomers 7a–d. The major products were 1,5-dimethyl TT 7a (57%) and 2,5-dimethyl TT 7b (25%), while 1,6- and 1,4-isomers 7c and 7d were minor products. The highest total yield of 7a–d (96%) was achieved from salt 6c·7H2O in MeCN at room temperature for 3 days (for details, see the ESI,† Table TS1). Replacing MeI with Me2SO4 resulted in a significantly lower overall yield of methylated isomers (34%).
tert-Butylation of TT 1 with excess tBuOH in the presence of 70% aqueous HClO4 for 3 h at room temperature yielded 1,5-disubstituted TT 8 (53%) and salt 9 (26%) (Scheme 3b). Increasing the reaction time to 24 h resulted in exhaustive tert-butylation of TT 8 to form salt 9 in 63% yield.
It should be noted that 1,6-isomer 7c and 1,4-isomer 7d are representatives of a hitherto unknown class of disubstituted triazolo–triazoles. Salts of 1,3,5-trisubstituted triazolo–triazole, such as salt 9, have also not been described previously.
Among dimethyl substituted triazoles, 2,5-disubstituted TT 7b is the most thermally stable isomer (Tonset >278 °C) (for more details, see the section ‘Physicochemical and energetic properties’). This is probably due to the lack of the possibility of ring-chain tautomerism in this isomer. Accordingly, it is 2,5-disubstituted triazolo–triazoles that are the most promising isomers for obtaining energetic compounds. The search for a regioselective method for the synthesis of such compounds is a challenging task.
As a model compound for developing a new synthetic protocol, we chose TT 10 bearing methyl and morpholine substituents at the 2,5-positions (Scheme 4). One of the reasons for choosing this particular compound is that we plan to create a convenient method for the synthesis of amino-substituted triazolo–triazoles, which could be converted into energetic compounds containing a tripoid N4-fragment.22
 | ||
| Scheme 4 Synthesis of the 2,5-disubstituted triazolo–triazole 10 from diazonium salt 4. | ||
The starting compound in our synthetic protocol was diazonium salt 4 (see Scheme 4). After adding two-fold excess of morpholine to the MeCN solution of this salt, the color of the reaction mixture immediately changed to red-brown, indicating the formation of the intermediate diazo compound 5. Subsequent holding of the reaction mixture for 3 days at room temperature afforded triazole 11 in a 53% yield. Reaction of the latter with diazomethane gave methylated triazole 12 in a 65% yield (other isomers were obtained in trace amounts). Diazotization of triazole 12 followed by reaction with NaN3 resulted in azido-substituted triazole 13 (78% yield). Heating the latter under reflux in PhMe for 5 h resulted in cyclization to afford TT 10. Note that this synthetic approach has analogies in the literature.33–35
The use of the developed regioselective approach for the synthesis of energetic 2,5-disubstituted triazolotriazoles will be described in the following papers.
Crystallography
The crystal structure of TT 1 was established from powder X-ray diffraction data. The small size and rigidity of the molecule facilitates a seamless initial structure solution. However, the determination of hydrogen atom positions is challenging.The crystal (space group P21/n, Z′ = 0.5) contains only half of the molecule in the asymmetric unit, as the molecule is located at an inversion center. Thus, the molecule's inherent symmetry allows for two potential isomeric configurations: a C2h symmetric isomer with protonated N1 and N4 atoms, or a D2h symmetric isomer with protonated N2 and N5 atoms.
Analysis of H-bonding patterns is also inconclusive due to the presence of multiple potential donor–acceptor pairs with similar interatomic distances (ca. 2.9–3.0 Å), including a shortened N1⋯N2 contact.
To resolve this ambiguity, periodic DFT calculations (PBE0-D3/POB-TZVP) were performed on both potential isomers. The calculations employed experimental unit cell parameters and symmetry, with initial non-H atomic coordinates derived from the PXRD solution. Comparative analysis of the optimized structures favored the 1,4-disubstituted isomer, as evidenced by its superior fit to the experimental PXRD pattern without refinement (see the ESI,† Fig. S2–S4), higher calculated lattice and cohesive energies (32.5 and 31.3 vs. 26.0 and 24.2 kcal mol−1), and a more reasonable hydrogen bonding network. The 1,4-hydrogen atom positions are also consistent with solid-state NMR data (see below). The final refined structure was obtained through restrained refinement of the PXRD pattern using the DFT-optimized 1,4-disubstituted isomer as a starting point.
While a detailed discussion of the molecular geometry is not feasible due to the data source, some general observations can be made. Notably, the refinement with weak restraints yields reasonable bond lengths and angles, consistent with a virtually planar conformation (Fig. 4). Furthermore, the N1–N2 bond, expected to be longer due to the protonated nitrogen atom, was refined to be elongated as anticipated. However, the refined length of 1.378 Å is slightly longer than in both the dimethyl analogue 7d (1.3608(15) Å, see below) and PBE0-D3/POB-TZVP optimized structure (1.359 Å). In turn, as expected, the N2–N3 bond appears to be shortened with the length of 1.320 Å but to a greater extend as compared to the 7d analogue (1.3299(14) Å).
 | ||
| Fig. 4 (a) General view of the molecule TT 1 in a crystal; non-hydrogen atoms are represented by probability spheres of atomic displacements (p = 50%); (b) fragments of the crystal packing of TT 1, viewed along the a crystallographic axis; (c) fragments of the crystal packing of TT 1, viewed along the (1 0 −1) diagonal plane. The atomic labels in the square brackets consistently follow the common atomic numbering used throughout the text, but do not reflect crystallographic symmetry. | ||
The packing pattern of TT 1 is primarily defined by H-bonds of average strength (N1⋯N2 2.848(3), H1⋯N2 1.952 Å, N1–H1⋯N2 145° with N–H 1.020 Å), which connect molecules into nearly flat layers. As discussed previously, the molecule resides at the inversion center, which in the space group P21/n necessarily belongs to the (1 0 −1) diagonal plane. Furthermore, the molecule adopts a nearly coplanar arrangement with respect to this plane. This geometric configuration allows for the straightforward calculation of the interlayer distance based on the distance between equivalent planes, which is equal to 3.206 Å. The molecules in the layers are in turn connected by π⋯π contacts. As molecules in the adjacent layers are shifted relative to each other, the shortest interlayer contacts are characterized by slightly higher distances (C3A⋯C3A 3.312(3) and N2⋯N2 3.382 Å).
Density (d) is an important characteristic of energetic materials as their detonation performance is directly proportional to this one.42 Among triazolo–triazoles obtained in this study only TT 1 is an energetic compound itself. The density of TT 1 calculated from the PXRD data is equal to 1.842 g cm−1 (298 K) and virtually coincides with the value from the pycnometer measurement. Such a high value is supported by the presence of strong intermolecular interactions in the crystal lattice, which usually lead to an increase in the density. This value of density is comparable to that of 3,3′-azo-bis(6-amino-1,2,4,5-tetrazine) (DAAT) which is considered the most dense C,H,N molecule known to date43,44 (for details see the ESI†).
The 1,4-dimethyl substituted TT 7d also crystallizes in non-centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with half of the molecule in the independent part of the unit cell (Fig. 5). Bond lengths and angles are comparable with those obtained for TT 1via a restrained refinement, and with the values characteristic of similar heterocyclic compounds containing an annulated triazole cycle, as proven by Mogul geometry check.45 The N1–N2 bond with the substituted nitrogen atom (1.3608(15) Å) is expectedly longer than the N2–N3 bond (1.3299(14)), while C3A–N1 and C3A–N3 bond lengths are nearly equal (1.3561(15) and 1.3590(16) Å).
, with half of the molecule in the independent part of the unit cell (Fig. 5). Bond lengths and angles are comparable with those obtained for TT 1via a restrained refinement, and with the values characteristic of similar heterocyclic compounds containing an annulated triazole cycle, as proven by Mogul geometry check.45 The N1–N2 bond with the substituted nitrogen atom (1.3608(15) Å) is expectedly longer than the N2–N3 bond (1.3299(14)), while C3A–N1 and C3A–N3 bond lengths are nearly equal (1.3561(15) and 1.3590(16) Å).
 | ||
| Fig. 5 (a) General view of the molecule of compound 7d in crystal form; non-hydrogen atoms are represented by probability ellipsoids of atomic displacements (p = 50%); (b) fragments of crystal packing of 7d, viewed along the crystallographic b axis. The atomic labels in the square brackets consistently follow the common atomic numbering used throughout the text, but do not reflect crystallographic symmetry. | ||
Despite the absence of the classical H-bonds as in TT 1, the crystal packing of the molecules in 7d is also layered. The layers are co-planar to the (1 0 1) and equivalent crystallographic planes, with the calculated interlayer distance of 3.169 Å. The molecule itself is tilted with respect to this plane by 9.4°, thus forming a step-like arrangement. The molecules within the layers are connected by non-classical C–H⋯N bonds (C⋯N 3.5798(18), H⋯N 2.497 Å, C–H⋯N 171° with C–H 1.092 Å), forming a pattern quite similar to that observed in TT 1, despite the apparent weakness of the intermolecular bonds. The layers are connected by π⋯π stacking interactions, with the shortest interatomic distance C⋯N of 3.3338(15) Å. In addition, due to the presence of methyl groups, there are also C–H⋯N contacts between the layers (C⋯N 3.4768(16), H⋯N 2.742 Å), which are not possible in the case of TT 1.
| Compd | N-1 | N-2 | N-3 | N-4 | N-5 | N-6 |
|---|---|---|---|---|---|---|
| TT 1 | −105.9 | −36.0 | −105.9 | −105.9 | −36.0 | −105.9 |
| TT 1 (solid state) | −168.3 | −7.8 | −77.6 | −168.3 | −7.8 | −77.6 |
| 7a | −178.1 | 14.2 | −57.7 | −91.3 | −116.4 | −74.1 |
| 7b | −79.9 | −107.8 | −79.9 | −79.9 | −107.8 | −79.9 |
| 7c | −179.0 | 3.3 | −54.6 | −54.6 | 3.3 | −179.0 |
| 7d | −172.2 | 7.4 | −71.9 | −172.2 | 7.4 | −71.9 |
NMR spectroscopy
A powerful tool for establishing the structure of polynitrogen compounds is 15N NMR spectroscopy, which allows identifying all nitrogen atoms of the molecule. Prior to this work, 15N NMR spectra had not been recorded for [1,2,3]triazolo[4,5-d][1,2,3]triazoles. In this study, we fully characterized triazolo–triazoles using 1H, 13C, 14N, and 15N NMR spectroscopy for the most important representatives of this class. The relative positions of hydrogen atoms and methyl substituents were confirmed by a set of 1H–13C, and 1H–15N heteronuclear correlations and INEPT 15N experiments. The obtained data have been thoroughly organized and can be used to identify other members representative of the [1,2,3]triazolo[4,5-d][1,2,3]triazole family in the future. As is known from the literature, the hydrogen atom in the 1,2,3-triazole ring appears as a broad signal in 1H NMR spectra due to delocalization between all three nitrogen atoms. In a solution of TT 1 rapid proton exchange is observed and the position of hydrogens is not fixed. However, in the solid state their positions can be clearly determined by solid-state NMR spectroscopy (CP/MAS). For this reason, we recorded solid-state 1H, 13C, and 15N NMR spectra for TT 1 because solvation and tautomerization effects were excluded in this case. Only one signal was observed in 1H MAS NMR at δ = 15.4 ppm, which corresponds to the NH-fragment of TT 1. A single carbon signal was also observed at δ = 153.3 ppm in 13C CP/MAS, allowing us to rule out structures F and H (Fig. 6). In the 15N CP/MAS spectrum of TT 1, three signals of varying intensities were observed (δ = −7.8 (N-2 and N-5), −77.6 (N-3 and N-6), −168.3 (N-1 and N-4) ppm) (Fig. 7a, Table 1). This fact allows us to exclude structure G. Thus, unambiguously the only one suitable option is structure I in solid form. Interestingly, the arrangement of nitrogen atom signals in TT 1 clearly coincides with the arrangement of nitrogen atom signals in 1,4-dimethyl triazolo–triazole 7d (δ = 7.4 (N-2 and N-5), −71.9 (N-3 and N-6), −172.2 (N-1 and N-4) ppm) (Fig. 7b, Table 1).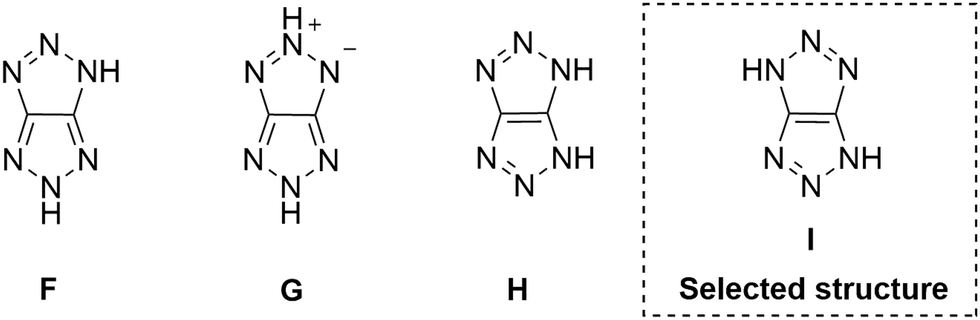 | ||
| Fig. 6 Plausible structures of TT 1. | ||
 | ||
| Fig. 7 15N NMR spectra for (a) TT 1 (solid-state, 9.5 kHz MAS, 18 msec CP) and (b) its 1,4-dimethyl derivative 7d (solution). | ||
Physicochemical and energetic properties
The thermal stability of the obtained triazolo–triazoles TT 1, 6a, 7a–d, and 8–10 was determined with differential scanning calorimetry (DSC, Table 2). For details, see the ESI.† Triazolo–triazoles 1, 6a, 7a–d, and 8–10 exhibited relatively high thermal stability. DSC analysis revealed that the decomposition of these compounds begins at temperatures above 167 °C. TT 1 is rather a stable compound that starts to decompose at 193 °C. Disodium salt 6a·2H2O shows similar thermal stability as an H-form 1 (Tonset = 207 °C). 2,5-Dimethyl derivative 7b was found to be the most stable (Tonset is not detected, this compound is vaporized at 278 °C even at elevated pressure (2 MPa)), while 1,6-dimethyl derivative 7c turned out to be the least stable (Tonset = 167 °C). At the same time the first representative of a previously unknown class of 1,3,5-trisubstituted triazolo–triazoles 9, decomposes at high temperature without melting (Tonset = 210 °C). Triazolo–triazole 10 (Tonset = 245 °C) is somewhat less stable, compared to the 2,5-dimethyl analog 7b.The combination of data on the thermal stability obtained from our work and literature allows us to conclude that 2,5-substituted derivatives exhibit the highest thermal stability among triazolo–triazoles.
The thermal stability of TT 1 was investigated under both non-isothermal (DSC analysis for solid TT 1 and its solution in 2,4,6-trinitrotoluene) and isothermal conditions (decomposition study using Bourdon manometers for solid TT 1). The decomposition of TT 1 in the solid state can be described by the Arrhenius equation: k1 = 6.18 × 1020·e(−25215/T) which characterizes the decomposition in a defect-free crystal lattice, while k2 = 1.20 × 1020·e(−23890/T) characterizes the decomposition at crystal defects. The activation energies Ea of these two processes are 210 and 199 kJ mol−1, respectively (see the ESI†). Additional experiments investigating the decomposition of TT 1 in a 2,4,6-trinitrotoluene solution have showed that the decomposition rate was significantly higher than that in the solid state. The decomposition of TT 1 in solution is described by the following equation: kliq = 9.14 × 1014·e(−16930/T) (Ea = 141 kJ mol−1). Based on the kinetic data, we proposed a mechanism for the thermal decomposition of TT 1 (Scheme 5). Apparently, the rate-limiting stage of decomposition is the opening of the TT ring.
 | ||
| Scheme 5 A plausible mechanism for the thermal decomposition of TT 1. | ||
Besides the thermal stability another important property of energetic materials is the standard enthalpy of formation  .42 The standard enthalpy of combustion
.42 The standard enthalpy of combustion  of TT 1 was experimentally determined by the combustion calorimetry (bomb calorimetry) method. The standard enthalpy of formation
of TT 1 was experimentally determined by the combustion calorimetry (bomb calorimetry) method. The standard enthalpy of formation  was calculated from
was calculated from 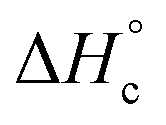 (for details see the ESI†). TT 1 has
(for details see the ESI†). TT 1 has  , which is much higher than those of the most common energetic materials such as 1,3,5-trinitro-1,3,5-triazinane (RDX) and N-methyl-N-(2,4,6-trinitrophenyl)nitramide (tetryl) (Table 3). Preliminary safety testing and detonation performance evaluation of TT 1 were carried out. As can be seen in Table 3, TT 1 has a calculated detonation velocity of 8.5 km s−1 and detonation pressure of 34 GPa. Compared to DAAT and common energetic materials (RDX, N-methyl-N-(2,4,6-trinitrophenyl)-nitramide (tetryl)), the studied compound demonstrated superior detonation performance to DAAT (8.4 km s−1 and 33.0 GPa), and tetryl (7.6 km s−1 and 26.0 GPa). However, the performance of TT 1 was somewhat lower than that of RDX (8.9 km s−1 and 36.0 GPa).46 The impact sensitivity of TT 1 (8 J) was slightly higher than that of RDX (10 J). TT 1 has a friction sensitivity value close to the primary explosives (FS = 15 N).
, which is much higher than those of the most common energetic materials such as 1,3,5-trinitro-1,3,5-triazinane (RDX) and N-methyl-N-(2,4,6-trinitrophenyl)nitramide (tetryl) (Table 3). Preliminary safety testing and detonation performance evaluation of TT 1 were carried out. As can be seen in Table 3, TT 1 has a calculated detonation velocity of 8.5 km s−1 and detonation pressure of 34 GPa. Compared to DAAT and common energetic materials (RDX, N-methyl-N-(2,4,6-trinitrophenyl)-nitramide (tetryl)), the studied compound demonstrated superior detonation performance to DAAT (8.4 km s−1 and 33.0 GPa), and tetryl (7.6 km s−1 and 26.0 GPa). However, the performance of TT 1 was somewhat lower than that of RDX (8.9 km s−1 and 36.0 GPa).46 The impact sensitivity of TT 1 (8 J) was slightly higher than that of RDX (10 J). TT 1 has a friction sensitivity value close to the primary explosives (FS = 15 N).
| Compounds | TT 1 | DAATo | RDXr | Tetrylu |
|---|---|---|---|---|
| a Molecular weight. b Nitrogen content. c Oxygen balance toward CO2. d Oxygen coefficient for a compound with the molecular formula of CxHyNwOz, α = z/(2x + y/2). e Density. f Melting point. g Decomposition temperature measured at a heating rate of 5 °C min−1. h Experimentally measured standard enthalpy of formation. i Calculated detonation velocity. j Calculated detonation pressure. k Impact sensitivity. l Friction sensitivity. m Density measured by powder diffraction at 298 K. n Calculated from empirical equations implemented in the PILEM application. o 3,3′-Azo-bis(6-amino-1,2,4,5-tetrazine). p Ref. 43. q Ref. 44. r 1,3,5-Trinitro-1,3,5-triazinane. s Ref. 47. t Ref. 48. u N-Methyl-N-(2,4,6-trinitrophenyl)-nitramide. | ||||
| Formula | C2H2N6 | C4H4N12 | C3H6N6O6 | C7H5N5O8 |
| M w | 110.03 | 220.07 | 222.03 | 287.01 |
| Nb (%) | 76.35 | 76.35 | 37.84 | 24.39 |
| Ω CO2 (%) | −72.7 | −72.7 | −21.6 | −47.4 |
| α | 0 | 0 | 0.67 | 0.49 |
| d (g cm−3) | 1.84m | 1.84p | 1.82s | 1.73s |
| 1.76q | ||||
| T m (°C) | — | — | 204s | 129s |
| T onset (°C) | 193 | 252p | 204s | 188t |
| 301q | ||||
ΔHf°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h (kcal kg−1) h (kcal kg−1) |
+1005 | +940p | +72s | +17s |
| +1123q | ||||
| D (km s−1) | 8.5n | 8.4n | 8.9n | 7.60n |
| 8.1n | 8.75s | 7.57s | ||
| P C–J (GPa) | 34.0n | 33.0n | 36.0n | 26.0n |
| 30.0n | 35.0s | |||
| ISk (J) | 8 | — | 10t | 14t |
| IFl (N) | 15 | 324q | 130t | 204t |
Conclusions
In summary, we have developed a facile two-step method for the synthesis of a hitherto unknown parent unsubstituted 1,4-dihydro-[1,2,3]triazolo[4,5-d][1,2,3]triazole (TT 1). This compound showed high thermal stability (Tonset = 193 °C), and can be considered as an energetic material with high density (1.84 g cm−3) and excellent experimental enthalpy of formation (+1005 kcal kg−1). TT 1 opens up access to a hitherto unknown and diverse series of N-functionalized energetic [1,2,3]triazolo[4,5-d][1,2,3]triazoles which may be of interest as high energy density materials (HEDMs). A previously unknown ring-opened form of TT 1, namely 5-amino-4-diazo-4H-1,2,3-triazole (5), as well as its salt, namely 5-amino-4-diazo-4H-1,2,3-triazolium chloride (4), have also been described. The latter two compounds are valuable starting materials for new HEDMs as well as for new heterocyclic bioactive compounds. They were key precursors for the regioselective synthesis of a previously unknown unsymmetrically 2,5-disubstituted triazolo–triazole, namely 5-methyl-2-morpholino-2H-[1,2,3]triazolo[4,5-d][1,2,3]triazole (10). The latter contains an energetically promising N4-tripoid fragment. We believe that the developed TT 10 synthesis protocol can be used to obtain a whole range of new HEDMs with this fragment. Due to our ongoing studies9,10,14,18–24 in the field of fused heterocyclic compounds, there is space to design novel HEDMs with improved performance, optimized characteristics, and environmental compatibility.Author contributions
All authors have given approval to the final version of the manuscript. CRediT: Agata N. Kuznetsova: investigation, methodology, writing – original draft; Nikita E. Leonov: investigation, methodology, writing – original draft; Oleg V. Anikin: investigation, methodology, writing – original draft; Michael S. Klenov: conceptualization, methodology, supervision, writing – review & editing; Alexander M. Churakov: conceptualization, methodology, supervision, writing – review & editing; Vladimir A. Tartakovsky: conceptualization, methodology, supervision, writing – review & editing; Yurii A. Strelenko: data curation, formal analysis, writing – original draft; Roman A. Novikov: data curation, formal analysis, writing – original draft; Ivan V. Fedyanin: data curation, formal analysis, writing – original draft; Alla N. Pivkina: data curation, formal analysis, writing – original draft; Tatiana S. Kon'kova: data curation, formal analysis, writing – original draft; Valery P. Sinditskii: data curation, formal analysis, writing – original draft; Anastasia D. Smirnova: data curation, formal analysis, writing – original draft.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (Project No. 22-13-00089) for the exception of X-ray structural studies, solid state NMR studies, thermochemical studies, thermal stability and sensitivity studies. This work was performed using the solid-state NMR facility equipment in the Shared Research Center (Department of Structural Studies) of N.D. Zelinsky Institute of Organic Chemistry RAS, Moscow (Dr R. A. Novikov). The X-ray structural studies of compounds 1, 3, 4, 6c, 7b, 7d and 9 were supported by the Ministry of Science and Higher Education of the Russian Federation (Contract No. 075-00277-24-00). The thermochemical studies were performed within the State Program of Fundamental Researches supported by the Ministry of Science and Higher Education of Russia, registration number 122040500073-4. The thermal stability and sensitivity studies were performed within the State Program of Fundamental Researches supported by the Ministry of Science and Higher Education of Russia, registration number 122040500070-3. The crystal structure determination of compounds 7c and 12 was performed in the Department of Structural Studies of Zelinsky Institute of Organic Chemistry, Moscow.Notes and references
- Y. Qu and S. P. Babailov, Azo-linked high-nitrogen energetic materials, J. Mater. Chem. A, 2018, 6, 1915–1940 RSC.
- H. Gao, J. Zhang and J. M. Shreeve, Fused heterocycle-based energetic materials (2012–2019), J. Mater. Chem. A, 2020, 8, 4193–4216 RSC.
- W. Li, J. Tian, X. Qi, K. Wang, Y. Jin, B. Wang and Q. Zhang, Synthesis of 4,8-Dinitraminodifurazano[3,4-b,e]pyrazine Derived Nitrogen-Rich Salts as Potential Energetic Materials, ChemistrySelect, 2018, 3, 849–854 CrossRef CAS.
- D. E. Chavez, J. C. Bottaro, M. Petrie and D. A. Parrish, Synthesis and Thermal Behavior of a Fused, Tricyclic 1,2,3,4-Tetrazine Ring System, Angew. Chem., Int. Ed., 2015, 54, 12973–12975 CrossRef CAS PubMed.
- Y. Tang, D. Kumar and J. M. Shreeve, Balancing Excellent Performance and High Thermal Stability in a Dinitropyrazole Fused 1,2,3,4-Tetrazine, J. Am. Chem. Soc., 2017, 139, 13684–13687 CrossRef CAS.
- C. Bian, X. Dong, X. Zhang, Z. Zhou, M. Zhang and C. Li, The unique synthesis and energetic properties of a novel fused heterocycle: 7-nitro-4-oxo-4,8-dihydro-[1,2,4]triazolo[5,1-d]-[1,2,3,5]tetrazine 2-oxide and its energetic salts, J. Mater. Chem. A, 2015, 3, 3594–3601 RSC.
- B. J. Zhao, P. Wang, W. Fu, C. Li and Z. M. Zhou, High Density of a New Fused Heterocycle: 7,8-Dinitro-4-oxo-4,6-dihydropyrazolo[5,1-d][1,2,3,5]tetrazine 2-oxide and Its Energetic Salts, ChemistrySelect, 2018, 3, 4797–4803 CrossRef CAS.
- D. E. Chavez, D. A. Parrish, L. Mitchell and G. H. Imler, Azido and Tetrazolo 1,2,4,5-Tetrazine N-Oxides, Angew. Chem., Int. Ed., 2017, 56, 3575–3578 CrossRef CAS PubMed.
- M. S. Klenov, A. A. Guskov, O. V. Anikin, A. M. Churakov, Yu. A. Strelenko, I. V. Fedyanin, K. A. Lysenko and V. A. Tartakovsky, Synthesis of Tetrazino-tetrazine 1,3,6,8-Tetraoxide (TTTO), Angew. Chem., Int. Ed., 2016, 55, 11472–11475 CrossRef CAS PubMed.
- O. V. Anikin, N. E. Leonov, M. S. Klenov, A. M. Churakov, A. A. Voronin, A. A. Guskov, Yu. A. Strelenko, N. V. Muravyev, I. V. Fedyanin and V. A. Tartakovsky, An Energetic (Nitro-NNO-azoxy)triazolo-1,2,4-triazine, Eur. J. Org. Chem., 2019, 4189–4195 CrossRef CAS.
- D. Kumar, G. H. Imler, D. A. Parrish and J. M. Shreeve, A Highly Stable and Insensitive Fused Triazolo-Triazine Explosive (TTX), Chem. – Eur. J., 2016, 23, 1743–1747 CrossRef PubMed.
- D. G. Piercey, D. E. Chavez, B. L. Scott, G. H. Imler and D. A. Parrish, An Energetic Triazolo-1,2,4-Triazine and its N-Oxide, Angew. Chem., Int. Ed., 2016, 55, 15315–15318 CrossRef CAS PubMed.
- M. C. Schulze, B. L. Scott and D. E. Chavez, A high density pyrazolo-triazine explosive (PTX), J. Mater. Chem. A, 2015, 3, 17963–17965 RSC.
- A. M. Churakov, S. L. Loffe and V. A. Tartakovsky, Synthesis of [1,2,5]Oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-Di-N-oxide, Mendeleev Commun., 1995, 5, 227–228 CrossRef.
- S. A. Shevelev, I. L. Dalinger, T. K. Shkineva, B. I. Ugrak, V. I. Gulevskaya and M. I. Kanishchev, Synthesis, transformations, and physicochemical properties of nitro derivatives of 1H,4H-pyrazolo[4,3-c]pyrazole, Russ. Chem. Bull., 1993, 42, 1063–1068 CrossRef.
- T. M. Klapötke, P. C. Schmid, S. Schnell and J. Stierstorfer, 3,6,7-Triamino-[1,2,4]triazolo[4,3-b][1,2,4]triazole: A Non-toxic, High-Performance Energetic Building Block with Excellent Stability, Chem. – Eur. J., 2015, 21, 9219–9228 CrossRef.
- A. Matsumoto, M. Yoshida and O. Simamura, Preparations and Properties of 5-Phenyl-5H[1,2,3]-triazolo[4,5-c][1,2,5]-oxadiazole, -thiadiazole, and -selenadiazole, Bull. Chem. Soc. Jpn., 1974, 47, 1493–1495 CrossRef CAS.
- A. A. Voronin, I. V. Fedyanin, A. M. Churakov, A. N. Pivkina, N. V. Muravyev, Y. A. Strelenko, M. S. Klenov, D. Lempert and V. A. Tartakovsky, 4H-[1,2,3]Triazolo[4,5-c][1,2,5]oxadiazole 5-oxide and Its Salts: Promising Multipurpose Energetic Materials, ACS Appl. Energy Mater., 2020, 3, 9401–9407 CrossRef CAS.
- V. P. Zelenov, A. A. Lobanova, N. I. Lyukshenko, S. V. Sysolyatin and A. I. Kalashnikov, Behavior of [1,2,5]oxadiazolo[3,4-e][1,2,3,4]tetrazine 4,6-dioxide in various media, Russ. Chem. Bull., 2008, 57, 1384–1389 CrossRef CAS.
- A. Gunasekaran, M. L. Trudell and J. H. Boyer, Dense energetic compounds of C, H, N, and O atoms IV nitro and azidofurazan derivatives, Heteroat. Chem., 1994, 5, 441–446 Search PubMed.
- A. M. Churakov, S. L. Ioffe, Y. A. Strelenko and V. A. Tartakovsky, Synthesis of 4H-[1,2,3]triazolo[4,5-c][1,2,5]oxadiazole 5-oxide and its N-and O-alkyl derivatives, Tetrahedron Lett., 1996, 37, 8577–8580 CrossRef CAS.
- A. A. Voronin, S. P. Balabanova, I. V. Fedyanin, A. M. Churakov, A. N. Pivkina, Yu. A. Strelenko, M. S. Klenov and V. A. Tartakovsky, Anions Containing Tripoid Conjugated N4− System: Salts of 5-(Substituted Amino)-[1,2,3]triazolo[4,5-c][1,2,5]oxadiazol-5-ium-4-ides, as well as Their Synthesis, Structure, and Thermal Stability, Molecules, 2022, 27, 6287 CrossRef CAS.
- A. Gunasekaran and J. H. Boyer, Dense energetic compounds of C, H, N, and O atoms. III. 5-[4-nitro-(1,2,5)oxadiazolyl]-5H-[1,2,3]triazolo[4,5-c][1,2,5]oxadiazole, Heteroat. Chem., 1993, 4, 521–524 Search PubMed.
- S. P. Balabanova, A. A. Voronin, A. M. Churakov, M. S. Klenov, I. V. Fedyanin, A. N. Pivkina, D. B. Meerov, T. S. Konkova, Yu. N. Matyushin, Yu. A. Strelenko, K. S. Erokhin, V. P. Zelenov and V. A. Tartakovsky, Insight into structural and energetic features of substituted triazolofurazans, CrystEngComm, 2024, 26, 3349–3362 Search PubMed.
- J. Zhang, D. A. Parrish and J. M. Shreeve, 3,6-dinitropyrazolo[4,3-c]pyrazole-based energetic materials, Chem. – Asian J., 2014, 9, 2953–2960 CrossRef CAS PubMed.
- P. Yin, C. He and J. M. Shreeve, Fused heterocycle-based energetic salts: alliance of pyrazole and 1,2,3-triazole, J. Mater. Chem. A, 2016, 4, 1514–1519 RSC.
- T. M. Klapötke, M. Leroux, P. C. Schmid and J. Stierstorfer, Energetic Materials Based on 5, 5′-Diamino-4, 4′-dinitramino-3,3′-bi-1, 2, 4-triazole, Chem. – Asian J., 2016, 11, 844–851 CrossRef.
- C. He, J. Zhang, D. A. Parrish and J. M. Shreeve, 4-Chloro-3,5-dinitropyrazole: a precursor for promising insensitive energetic compounds, J. Mater. Chem. A, 2013, 1, 2863–2868 RSC.
- J. Zhang, D. A. Parrish and J. M. Shreeve, Curious cases of 3,6-dinitropyrazolo[4,3-c]pyrazole-based energetic cocrystals with high nitrogen content: an alternative to salt formation, Chem. Commun., 2015, 51, 7337–7340 RSC.
- J. Zhang, D. A. Parrish and J. M. Shreeve, Thermally Stable 3,6-Dinitropyrazolo[4,3-c]pyrazole-based Energetic Materials, Chem. – Asian J., 2014, 9, 2953–2960 CrossRef CAS.
- T. M. Klapötke, Chemistry of High-Energy Materials, 6th edn, De Gruyter, Berlin/Boston, 2022 Search PubMed.
- J. Thiele and K. Schleussner, Ueber Diamidophenylosotriazol, Liebigs Ann. Chem., 1897, 295, 129–172 CrossRef.
- A. Matsumoto, M. Yoshida and O. Simamura, 2H,5H-Triazolo[4,5-d]triazoles. A New Heteroaromatic System, Bull. Chem. Soc. Jpn., 1970, 43, 3587–3590 CrossRef.
- L. A. Henderson, US Pat., 3541107A, 1970 Search PubMed.
- R. A. Carboni, US Pat., 3190886, 1965 Search PubMed.
- A. Matsumoto, J. H. Lee, M. Yoshida and O. Simamura, A Polarographic Study of Aryl-substituted Tetra-aza- and Hexa-aza-pentalenes in Dimethylformamide, Bull. Chem. Soc. Jpn., 1974, 47, 1490–1492 CrossRef CAS.
- T. Ohta, M. Yoshida and H. Kuroda, Electronic Structures of Meso-ionic Azapentalenes Studied by X-Ray Photoelectron Spectroscopy, Bull. Chem. Soc. Jpn., 1977, 50, 1917–1919 Search PubMed.
- T. Kaihoh, T. Itoh, K. Yamaguchi and A. Ohsawa, First Synthesis of a 1,2,3,4-Tetrazine, J. Chem. Soc., Perkin Trans. 1, 1991, 2045–2048 Search PubMed.
- F. De Sio, L. Cecchi and F. Melani, Chemical and photochemical behavior of 5-benzoylamido-4-diazol-methyl-3-phenyl-pyrazole, Heterocycles, 1984, 22, 2309–2311 Search PubMed.
- G. Daidone, M. L. Bajardi, D. Raffa and B. Maggio, Facile Synthesis of 5-Benzamido-4-Diazopyrazole Derivatives, a Class of Biologically Active Agents and Key Intermediates, Synth. Commun., 1995, 25, 1441–1449 Search PubMed.
- V. G. Andrianov, V. G. Semenikhina and A. V. Eremeev, Rearrangements of 1-oxa-2-azoles 8. Synthesis and rearrangement of amidrazones of 1,2,4-oxadiazole-3-carboxylic acid, Chem. Heterocycl. Compd., 1992, 28, 803–807 Search PubMed.
- M. H. Keshavarz and T. M. Klapötke, The properties of energetic materials: sensitivity, physical and thermodynamic properties, Walter de Gruyter GmbH & Co KG, 2021 Search PubMed.
- D. E. Chavez, M. A. Hiskey and R. D. Gilardi, 3,3-azobis (6-amino-1,2,4,5-trerazine): A novel high nitrogen energetic material, Angew. Chem., Int. Ed., 2000, 39, 1791 CrossRef CAS.
- J. Kerth and S. Löbbecke, Synthesis and Characterization of 3,3′-Azobis(6-Amino-1,2,4,5-Tetrazine) DAAT – A New Promising Nitrogen-Rich Compound, Propellants, Explos., Pyrotech., 2002, 27, 111 Search PubMed.
- I. J. Bruno, J. C. Cole, M. Kessler, J. Luo, W. D. S. Motherwell, L. H. Purkis, B. R. Smith, R. Taylor, R. I. Cooper, S. E. Harris and A. G. Orpen, Retrieval of crystallographically-derived molecular geometry information, J. Chem. Inf. Model., 2004, 44, 2133–2144 Search PubMed.
- N. V. Muravyev, D. R. Wozniak and D. G. Piercey, Progress and performance of energetic materials: open dataset, tool, and implications for synthesis, J. Mater. Chem. A, 2022, 10, 11054–11073 Search PubMed.
- R. Meyer, J. Kohler and A. Homburg, Explosives, Wiley-VCH, Weinheim, 7th edn, 2016 Search PubMed.
- N. V. Muravyev, D. B. Meerov, K. A. Monogarov, I. N. Melnikov, E. K. Kosareva, L. L. Fershtat, A. B. Sheremetev, I. L. Dalinger, I. V. Fomenkov and A. N. Pivkina, Sensitivity of energetic materials: Evidence of thermodynamic factor on a large array of CHNOFCl compounds, Chem. Eng. J., 2021, 421, 129804 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds including NMR spectra and X-ray. CCDC 2382714 and 2382722–2382729. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj04427d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |