DOI:
10.1039/D4NJ04175E
(Paper)
New J. Chem., 2024,
48, 20140-20148
Structures and properties of the mono- and diprotonated species of acetylenedicarbonyl fluoride†
Received
24th September 2024
, Accepted 17th November 2024
First published on 22nd November 2024
Abstract
In this study, the first crystal structure of acetylenedicarbonyl fluoride is reported. Furthermore, the title compound was investigated in the superacidic media XF/SbF5 (X = H, D) which led to the formation of the monoprotonated species. Sur-prisingly, the diprotonated species was only formed by employing the monoprotonated species in 1,1,1,2-tetrafluoroethane whereas attempts at its synthesis in anhydrous hydrogen fluoride were unsuccessful. The vibrational spectroscopy is discussed together with quantum chemical calculations on the DFT/aug-cc-pVTZ level of theory. The stability of the Z-conformation of protonated acyl fluorides is calculated on the MP2/aug-cc-pVTZ level of theory.
Introduction
Acid fluorides are often prepared from their chlorides. Via the synthetic route of metathesis, a chlorine–fluorine exchange is often explored with antimony fluorides, less commonly with aHF.1 Acetylenedicarbonyl chloride can be obtained by the decomposition of an anthracene derivative as the starting material.2 The metathesis for acetylenedicarbonyl fluoride is not ecologically or economically viable due to the low yield of acetylenedicarbonyl chloride.2 The direct synthesis of acetylenedicarbonyl chloride starting with acetylenedicarboxylic acid with classical chlorinating agents reacts after removal of the solvent to the addition product with added HCl (PCl5, POCl3) or Cl2 (SOCl2) at the triple bond.3,4 The acetylenedicarbonyl fluoride is the best-known structure compared to the corresponding acetylenedicarbonyl halides. One of the most efficient reaction pathways to obtain acetylenedicarbonyl fluoride is the use of sulfur tetrafluoride.5 The first synthesis was reported in 1960 by Hasek.5 In 1973, the synthesis of acetylenedicarbonyl fluoride was optimised and supplemented by the synthesis using the potassium salt of the carboxylic acid as starting material.5,6 Although its synthesis is reported in the literature, many properties are still unknown. Acetylenedicarbonyl fluoride was investigated in CCl4 solution in Raman and 19F NMR spectroscopy (49.2 ppm) in 1973.6 The Raman spectrum is only described by the stretching oscillation of C–F (1822 cm−1) and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (2262 cm−1).6 In 1975, the pure substance was only characterized by its mass spectrum, vapor pressure, boiling (46 °C), and melting point (−51 °C).7 In summary, compared to other acid fluorides, acetylenedicarbonyl fluoride is rarely described in the literature, and its basicity remains unknown. Additionally, the investigations of protonated acyl fluorides are very rare. Only five crystal structures are known from protonated acyl fluorides the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 This prompted us to investigate acetylenedicarbonyl fluoride in superacidic media.
C (2262 cm−1).6 In 1975, the pure substance was only characterized by its mass spectrum, vapor pressure, boiling (46 °C), and melting point (−51 °C).7 In summary, compared to other acid fluorides, acetylenedicarbonyl fluoride is rarely described in the literature, and its basicity remains unknown. Additionally, the investigations of protonated acyl fluorides are very rare. Only five crystal structures are known from protonated acyl fluorides the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 This prompted us to investigate acetylenedicarbonyl fluoride in superacidic media.
Results and discussion
Synthesis and properties of [C4(OH)OF2][SbF6] (2) and [C4(OD)OF2][SbF6] (3)
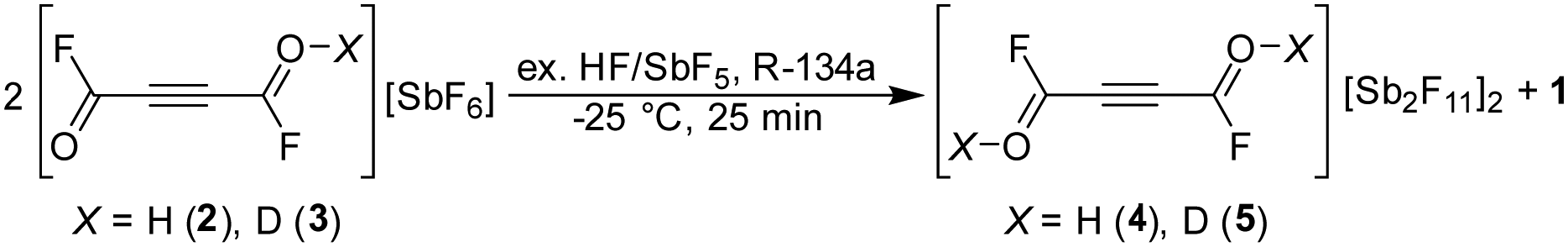
The monoprotonated species of acetylenedicarbonyl fluoride is obtained in the superacidic system HF/SbF5, using anhydrous hydrogen fluoride (aHF), or aDF for deuterated salts, as reagent and solvent (eqn (1)). The salts are soluble in aHF and grew as colourless crystals in solution. The salts of 2 and 3 are stable up to −43 °C. The solvent was removed overnight at −78 °C.| |  | (1) |
Synthesis and properties of [C4(OH)2F2][Sb2F11]2·2HF (4) [C4(OD)2F2][Sb2F11]2·2HF (5)
The salts of diprotonated species 4 and 5 are prepared from the corresponding salts of the monoprotonated species (2 resp. 3). The salts (2 and 3) decompose when mixed with an excess of SbF5 in the aprotic solvent 1,1,1,2-tetrafluoroethane (R-134a) at −25 °C over a period of 25 minutes. The decomposition results in the formation of the starting material (1) and salts of the diprotonated species are formed which is described in eqn (2). Salt containing the diprotonated species could not be obtained in anhydrous HF or deuterium fluoride even in the presence of 15 equivalents of SbF5. Although the diprotonation can only occur by decomposition, the salts are stable in aHF and can be recrystallized. The products were obtained as yellowish salts stable up to 0 °C.| |  | (2) |
Vibrational spectroscopy
Vibrational spectroscopy of C4O2F2 (1).
The low (l.t.) and room temperature (r.t.) Raman and IR of C4O2F2 (1) are illustrated in Fig. 1. In Table 1 selected observed Raman and IR frequencies of 1 are summarized together with the quantum chemically calculated frequencies of the [C4(OH)OF2]+·HF cation as well as their assignments. The complete analysis of the vibrational frequencies is provided in the ESI† in Table S1. The C4O2F2 molecule possesses C2 symmetry, whereby 18 fundamental vibrational modes are expected.
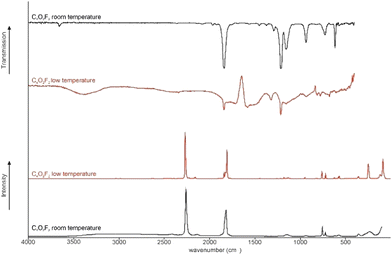 |
| | Fig. 1 Low-temperature Raman and IR spectra of C4O2F2 (1). | |
Table 1 Selected observed vibrational frequencies [cm−1] of C4O2F2 (1) and calculated vibrational frequencies [cm−1] of C4O2F2
|
1 at room temperaturea |
1 at low temperaturea |
ν (IR/Ra)bc |
Assignment |
| IR |
Ra |
IR |
Ra |
|
Abbreviations for IR intensities: vs = very strong, s = strong, m = medium, w = weak, sh = shoulder, br = broad. Experimental Raman intensities are relative to a scale of 1 to 100.
Calculated on the B3LYP/aug-cc-pVTZ level of theory. Scaling factor: 0.968.
IR intensities in km mol−1; Raman intensities in Å4/u.
|
|
|
2263 (100) |
|
2271 (100) |
2257 (0/515) |
ν1 |
A |
ν
s(CC) |
| 1842 (s) |
|
1842 (s) |
1843 (12) |
1812 (578/19) |
ν11 |
B |
ν
as(CO) |
|
|
1819 (57) |
|
1810 (60) |
1810 (190/172) |
ν2 |
A |
ν
s(CO) |
| 1215 (s) |
|
1215 (vs) |
1179 (2) |
1183 (696/1) |
ν12 |
B |
ν
as(CF) |
| 1159 (s) |
1150 (4) |
1155 (s) |
1133 (3) |
1102 (174/14) |
ν3 |
A |
ν
s(CF) |
| 939 (m) |
938 (2) |
935 (m) |
950 (3) |
908 (122/3) |
ν13 |
B |
ν
as(C–C) |
|
|
757 (21) |
781 (m) |
763 (16) |
729 (12/8) |
ν4 |
A |
ν
s(C–C) |
In fair agreement with the values reported in the literature, the CC stretching vibration is detected at 2263 cm−1 (r.t. Ra) and 2271 cm−1 (l.t. Ra,), respectively.6 The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations are observed at 1842 cm−1 (IR) at both temperatures and. 1843 cm−1 (Ra) at low temperatures, respectively.6 The symmetric CO stretching vibration is detected at 1819 cm−1 (r.t. Ra) and at 1810 cm−1 (l.t. Ra), respectively. In comparison to FC(O)NCS (1854 cm−1) the CO stretching vibration is in the same range.11 Compared to FC(O)SCN (1867 cm−1) and FC(O)SOC(O)CF3 (trans ap–sp: 1876 cm−1) the CO stretching vibration is red shifted.11,12 The antisymmetric C–F oscillation is identified at 1215 cm−1 (IR) at both temperatures and 1179 cm−1 (l.t. Raman). The C–F stretching vibration is in the same range compared to FC(O)NCS (1245 cm−1).11 The symmetric C–F stretching vibration is detected at 1159 cm−1 (r.t. IR), 1150 cm−1 (l.t. Raman), 1155 cm−1 (r.t. IR) resp. 1133 cm−1 in Raman. Compared to the C–F stretching vibration of FC(O)NCS (1108 cm−1) the oscillation is blue-shifted.11 The antisymmetric C–C vibration is observed at room temperature in IR at 939 cm−1 (IR) and. 938 cm−1 (Ra) and at low at 935 cm−1 (IR) and. 950 cm−1 in (Ra). The antisymmetric C–C oscillation is identified at room temperature in Raman at 757 cm−1 and low temperature at 763 cm−1 (Ra) and 781 cm−1 (IR).
O stretching vibrations are observed at 1842 cm−1 (IR) at both temperatures and. 1843 cm−1 (Ra) at low temperatures, respectively.6 The symmetric CO stretching vibration is detected at 1819 cm−1 (r.t. Ra) and at 1810 cm−1 (l.t. Ra), respectively. In comparison to FC(O)NCS (1854 cm−1) the CO stretching vibration is in the same range.11 Compared to FC(O)SCN (1867 cm−1) and FC(O)SOC(O)CF3 (trans ap–sp: 1876 cm−1) the CO stretching vibration is red shifted.11,12 The antisymmetric C–F oscillation is identified at 1215 cm−1 (IR) at both temperatures and 1179 cm−1 (l.t. Raman). The C–F stretching vibration is in the same range compared to FC(O)NCS (1245 cm−1).11 The symmetric C–F stretching vibration is detected at 1159 cm−1 (r.t. IR), 1150 cm−1 (l.t. Raman), 1155 cm−1 (r.t. IR) resp. 1133 cm−1 in Raman. Compared to the C–F stretching vibration of FC(O)NCS (1108 cm−1) the oscillation is blue-shifted.11 The antisymmetric C–C vibration is observed at room temperature in IR at 939 cm−1 (IR) and. 938 cm−1 (Ra) and at low at 935 cm−1 (IR) and. 950 cm−1 in (Ra). The antisymmetric C–C oscillation is identified at room temperature in Raman at 757 cm−1 and low temperature at 763 cm−1 (Ra) and 781 cm−1 (IR).
Vibrational spectroscopy of [C4(OH)OF2][SbF6] (2) and [C4(OD)OF2][SbF6] (3).
The low-temperature Raman and IR spectra of [C4(OH)OXF2][SbF6] (2, 3) (X = H, D) are illustrated in Fig. 2. In Table 2 selected observed Raman and IR frequencies of 2, and 3 are listed together with the quantum chemically calculated frequencies of [C4(OH)OF2]+·HF cation as well as their assignments. The complete analysis of the vibrational frequencies is summarized in the ESI† in Table S2.
 |
| | Fig. 2 Low-temperature Raman and IR spectra of C4O2F2, 2, and 3. | |
Table 2 Selected observed vibrational frequencies [cm−1] of [C4(OH)OF2][SbF6], [C4(OD)OF2][SbF6], and calculated vibrational frequencies [cm−1] of [C4(OH)OF2]+·HF
|
2 exp.a |
3 exp.a |
[C4(OH)OF2]+·HF calc.bc |
Assignment |
| IR |
Ra |
IR |
Ra |
IR/Ra |
|
Abbreviations for IR intensities: vs = very strong, s = strong, m = medium, w = weak, sh = shoulder, br = broad. Experimental Raman intensities are relative to a scale of 1 to 100.
Calculated on the B3LYP/aug-cc-pVTZ level of theory. Scaling factor: 0.968.
IR intensities in km mol−1; Raman intensities in Å4/u.
|
| 3084 (m) |
|
|
|
2878 (2945/159) |
ν1 |
A |
ν(OH) |
| 2257 (m) |
2269 (14) |
2264 (m) |
2264 (56) |
2237 (490/425) |
ν2 |
A |
ν(CC) |
| 1840 (s) |
1846 (9) |
1838 (s) |
1848 (20) |
1823 (213/194) |
ν3 |
A |
ν(CO) |
| 1653 (m) |
1659 (5) |
1647 (m) |
1654 (13) |
1597 (634/37) |
ν4 |
A |
ν(CO) |
| 1458 (m) |
1445 (2) |
1443 (m) |
1418 (6) |
1422 (376/1) |
ν5 |
A |
ν(CF) |
|
|
|
1194 (m) |
|
1164 (324/26) |
ν7 |
A |
ν(CF) |
| 986 (m) |
986 (2) |
989 (m) |
1000 (7) |
963 (34/39) |
ν8 |
A |
ν
as(C–C) |
| 768 (w) |
766 (4) |
|
764 (6) |
737 (12/7) |
ν10 |
A |
ν
s(C–C) |
The monoprotonated species [C4(OH)OF2]+ possesses C1 symmetry with 21 expected fundamental vibrational modes, showing both Raman and IR activity. In comparison with the starting material, the monoprotonation has no influence on the CC bond, the unprotonated CO bond, and the symmetric C–C stretching vibration. Clear evidence of a monoprotonation is the stretching vibration of the CO bond, which is red-shifted in IR to 1653 cm−1 and in Ra at 151 cm−1 to 1659 cm−1. Also, in IR the OH oscillation is identified at 3084 cm−1. Furthermore, the protonation is detected by a blue shift of the C–F bond by 243 cm−1 in IR at 1458 cm−1 and in Ra at 1445 cm−1. The antisymmetric stretching vibration is blue-shifted in IR and Ra to 986 cm−1.
Vibrational spectroscopy of [C4(OH)2F2][Sb2F11]·2HF (4) and [C4(OD)2F2][Sb2F11]·2HF (5).
The low-temperature Raman and IR spectra of [C4(OX)2OF2][Sb2F11] (X = H, D) (4, 5) are illustrated in Fig. 3. In Table 3 are selected observed Raman and IR frequencies of 4 and 5 are listed together with the quantum chemically calculated frequencies of [C4(OH)OF2]+·HF as well as their assignments. The complete analysis of the vibrational frequencies can be seen in the ESI† in Table S3. The diprotonated species [C4(OH)2F2]+ possesses C2h symmetry with 24 expected fundamental vibrational modes. The diprotonation causes a slight effect on the CC by a blue shift of 37 cm−1 to 2300 cm−1. Furthermore, after protonation the CC bond leads to a change of dipole moment which is not present in the starting material and is detected at 2334 cm−1 (IR). The CO stretching vibrations are red-shifted and can be observed at 1626 cm−1 in IR and 1688 cm−1 in Ra. In IR the antisymmetric C–F bond is blue-shifted to 1445 cm−1 and the symmetric C–F bond to 1418 cm−1. The effect on the C–C vibration is within the standard deviation.
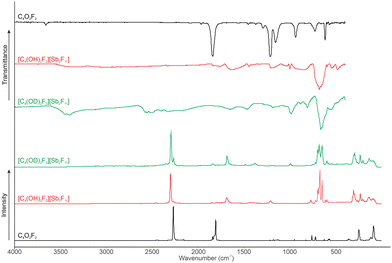 |
| | Fig. 3 Low-temperature Raman and IR spectra of C4O2F2, 4, and 5. | |
Table 3 Selected observed vibrational frequencies [cm−1] of [C4(OH)2F2][Sb2F11]2, [C4(OD)2F2][Sb2F11]2, and calculated vibrational frequencies [cm−1] of [C4(HO)2F2]2+·2HF
|
4 exp.a |
5 exp.a |
[C4(OH)2F2]+ ·HF calc.bc |
Assignment |
| IR |
Ra |
IR |
Ra |
IR/Ra |
|
Abbreviations for IR intensities: vs = very strong, s = strong, m = medium, w = weak, sh = shoulder, br = broad. Experimental Raman intensities are relative to a scale of 1 to 100.
Calculated on the B3LYP/aug-cc-pVTZ level of theory. Scaling factor: 0.968.
IR intensities in km mol−1; Raman intensities in Å4/u.
|
|
|
|
3446 (m) |
|
|
ν1 |
A |
ν
s(OH) |
| 2334 (s) |
2300 (88) |
2328 (m) |
2296 (100) |
2266 (24/843) |
ν2 |
A |
ν(CC) |
| 1626 (s) |
1688 (18) |
1649 (m) |
1686 (32) |
1659 (174/68) |
ν4 |
A |
ν
s(CO) |
|
|
|
1635 (m) |
|
1651 (1262/6) |
ν15 |
B |
ν
as(CO) |
| 1445 (s) |
|
1472 (w) |
1488 (7) |
1422 (405/1) |
ν16 |
B |
ν
as(CF) |
| 1418 (s) |
|
|
1382 (10) |
1387 (298/35) |
ν5 |
A |
ν
s(CF) |
| 957 (s) |
|
986 (m) |
994 (9) |
984 (163/1) |
ν19 |
B |
ν
as(CC) |
| 770 (vw) |
772 (11) |
|
|
762 (20/14) |
ν8 |
A |
ν
s(CC) |
Crystal structure
Crystal structure of C4O2F2 (1).
The starting material 1 crystallizes in the tetragonal space group P41212 with four formula units per unit cell. Selected bond lengths, bond angles, dihedral angles and interatomic contacts are listed in Table 4. Further crystallographic data can be found in the ESI† (Chapter S2). The formula unit is displayed in Fig. 4 the CC bond length of 1.186(2) Å is in the expected range for an average triple bond (1.183 Å).13 The C1–C2 bond of 1.445(2) Å and is slightly shortened compared to an average single carbon bond (1.466 Å) with a similar bonding situation. The CO bond length of 1.178(2) Å is comparable to the corresponding bond lengths of malonyl difluoride (1.177(3) Å) and fumaryl fluoride (1.177(4) Å), respectively.14,15 The C–F bond of 1 (1.332(1) Å) is significantly shortened compared to the C–F bond reported for malonyl difluoride (1.349(4) Å) and fumaryl fluoride (1.334(2) Å).14,15 In 1 the molecules are connected via C⋯O donor–acceptor interactions. The molecules within one layer are connected by 3.078(2) Å interatomic contacts and the next layer is connected by 2.896 Å C⋯O contacts. The interatomic contacs are 90% and 96% below the radii of the van-der-Waals radii. The distance is comparable to the contact of FC(O)⋯C(O)CF3 (3.037 Å) and FC(O)⋯CF3 (3.217 Å).16
Table 4 Selected bond lengths, interionic distances [Å] and bond angels [°] of 1 and symmetry codes: i = −x, −y, −z; ii = −0.5 − x, 0.5 + y, −0.25 + z; iii = −1.5 − x, 0.5 + y, −0.25 + z
| Bond lengths [Å] |
| C1–C2 |
1.445(2) |
C1–O1 |
1.178(2) |
| C2–C2i |
1.186(2) |
C1–F1 |
1.332(1) |
| Bond angles [°] |
| C1–C2–C2i |
177.9(1) |
F1–C1–C2 |
111.3(1) |
| O1–C1–C2 |
126.7(1) |
O1–C1–F1 |
121.9(1) |
| Intermolecular contacts [Å] |
| O1⋯C1ii |
3.078(2) |
O1⋯C1iii |
2.896(2) |
 |
| | Fig. 4 Formula unit of C4O2F2 (displacement ellipsoids with 50% probability). Symmetry operation i = −x, −y, −z. | |
Crystal structure of [C4(OH)OF2][SbF6] (2).
The monoprotonated species of C4O2F2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two formula units per unit cell. Selected bond lengths, bond angles, dihedral angles, and interatomic contacts are listed in Table 5. Further crystallographic data can be found in the ESI† (Chapter S2). Two formula units are displayed in Fig. 5 together with the intermolecular contacts.
with two formula units per unit cell. Selected bond lengths, bond angles, dihedral angles, and interatomic contacts are listed in Table 5. Further crystallographic data can be found in the ESI† (Chapter S2). Two formula units are displayed in Fig. 5 together with the intermolecular contacts.
Table 5 Selected bond lengths, interionic distances [Å] and bond angels [°] of 2 and symmetry codes: i = 1 − x, 1 − y, −z; ii = 1 − x, 1 − y, 1 − z; iii = x, −1 + y, z; iv = 2 − x, −1 − y,1 − z
| Bond lengths [Å] |
| C1–C2 |
1.423(9) |
C4–O2 |
1.174(8) |
| C2–C3 |
1.184(9) |
C1–F1 |
1.289(5) |
| C3–C4 |
1.457(9) |
C4–F2 |
1.321(7) |
| C1–O1 |
1.218(6) |
|
|
| Bond angles [°] |
| C1–C2–C3 |
176.5(6) |
F1–C1–O1 |
120.6(5) |
| C2–C3–C4 |
176.5(6) |
O2–C4–C3 |
125.1(5) |
| O1–C1–C2 |
122.9(5) |
F2–C4–C3 |
111.8(5) |
| F1–C1–C2 |
116.4(5) |
F2–C4–O2 |
123.1(5) |
| Intermolecular contacts [Å] |
| O1–(H1)…F3 |
2.384(6) |
C2–F8iii |
3.057(6) |
| C1–F7i |
2.711(7) |
C3–F5iv |
3.064(5) |
| C1–F4ii |
2.942(6) |
C4–F5iv |
2.754(4) |
 |
| | Fig. 5 Two formula units with intermolecular contacts and hydrogen bonds in the crystal structure of 2 (displacement ellipsoids with 50% probability). Symmetry codes: i = 2 − x, 1 − y, 1 − z. | |
In comparison with the starting material, the monoprotonation does not significantly affect the bond lengths of the unprotonated acyl fluoride moiety, the C3–C4 and the CC bond. The C1![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O1 bond is significantly elongated by approximately 0.040 Å to 1.218(6) Å compared to the neutral compound. The C1–F1 bond (1.289(5) Å) is shortened by 0.043 Å compared to 1. The Sb–F bond lengths of anion are in the range from 1.853(3) Å to 1.961(3) Å and in agreement with those reported in the literature.17,18 Distorting the ideal Oh symmetry is caused by strong hydrogen bonds of O1–(H1)⋯F3 with a length of 2.384(6) Å. Also, the C–F interaction leads to a distortion of the ideal symmetry. The layer structure is built via the hydrogen bonds and the C1–F7 interatomic interaction. The layers are linked by four additional C–F interactions.
O1 bond is significantly elongated by approximately 0.040 Å to 1.218(6) Å compared to the neutral compound. The C1–F1 bond (1.289(5) Å) is shortened by 0.043 Å compared to 1. The Sb–F bond lengths of anion are in the range from 1.853(3) Å to 1.961(3) Å and in agreement with those reported in the literature.17,18 Distorting the ideal Oh symmetry is caused by strong hydrogen bonds of O1–(H1)⋯F3 with a length of 2.384(6) Å. Also, the C–F interaction leads to a distortion of the ideal symmetry. The layer structure is built via the hydrogen bonds and the C1–F7 interatomic interaction. The layers are linked by four additional C–F interactions.
Crystal structure of [C4(OH)2F2][Sb2F11]·2HF (4).
The diprotonated species of C4O2F2 crystallizes in the triclinic space group P with two formula units per unit cell. Selected bond lengths, bond angles, dihedral angles and interatomic contacts are listed in Table 6. Further crystallographic data can be found in the ESI† (Chapter S2). The formula unit of 3 is displayed in Fig. 6.
Table 6 Selected bond lengths, interionic distances [Å] and bond angels [°] of 4 and symmetry codes: i = 1 − x, 2 − y, −z, ii = 1 − x, 2 − y, 1 − z, iii = x, 1 + y, −1 + z
| Bond lengths [Å] |
|
|
|
| C1–C2/C1i–C2i |
1.430(6) |
C1–O1/C1i–O1i |
1.226(5) |
| C2–C2i |
1.190(6) |
C1–F1/C1i–F1i |
1.281(4) |
| Bond angles [°] |
|
|
|
| C1–C2–C2i |
179.5(5) |
F1–C1–C2/F1i–C1i–C2i |
116.5(4) |
| O1–C1–C2/O1i–C1i–C2i |
122.1(4) |
F1–C1–O1/F1i–C1i–O1i |
121.5(4) |
| Intermolecular contacts [Å] |
|
|
|
| O1–(H1)⋯F2i |
2.413(4) |
C2–F10ii |
2.794(6) |
| F2–(H2)⋯F5 |
2.481(5) |
C1i–F6iii |
2.871(6) |
| C1–F10ii |
2.582(6) |
|
|
 |
| | Fig. 6 Formula unit of 4 (displacement ellipsoids with 50% probability). Symmetry codes: i = 1 − x, 1 − y, 1 − z. | |
Compared to the neutral compound 1, the CC is not affected by the protonation. The shortening of the C–C bond is within the 3 σ range. The C1O1 bond is significantly elongated by 0.048 Å to 1.226(5) Å compared to the starting material. The diprotonation shortens the C1–F1 bond by approximately 0.051 Å to 1.281(4) Å. The bond length of the [Sb2F11]− anion is in the range from 1.847(2) Å to 2.046(2) Å and agrees with those reported in the literature.19,20 The anion is connected to the cation with hydrogen bonds via a co-crystallized HF molecule. According to Jeffrey, the hydrogen bonds can be classified as strong.21 Surprisingly, in contrast to 3 and 2, the diprotonation has a planar molecular geometry.
NMR spectroscopy
The 1H, 13C, and 19F NMR spectra of 1 and its protonated species were measured at −50 °C in anhydrous HF (aHF) and SO2. In Table 7 selected observed NMR shifts and couplings constants in aHF are listed. The complete data and the measured NMR spectra are given in the ESI† as well as SO2 data. In the 1H NMR spectrum, the peaks at 9.92 ppm for 2 and 9.72 ppm for 4 are the first evidence of mono- respectively diprotonation. The 13C{1H} NMR spectra show a trend of the shifts by protonation steps. The higher the level of protonation, the more the peak of the acid fluoride moiety is shifted downfield, and the coupling constant increases. The observation is consistent with the values reported in the literature by the protonated species of haloacetyl fluorides, CCl2HCOF and fumaryl fluoride.8–10 The shift downfield of cations compared to the neutral compound is reported in the literature.8–10,22–24 The triple bond carbon atoms possess the opposite effect. The higher the protonation level, the more they are shielded, and the coupling constants decrease. In 19F NMR the acyl fluoride moiety is shifted also slightly downfield by each protonation step. The change in 19F NMR shift is also known from the protonation of haloacetyl fluorides, CCl2HCOF, however is much smaller in the protonated species of C4O2F2.8,9 Compared to fumaryl fluoride the shifts in aHF are in the same range.10
Table 7 Observed 1H and 19F NMR chemical shifts [ppm] at −50 °Cab
|
|
C4O2F2 (1) |
[C4(HO)OF2][SbF6] (2) |
[C4(HO)2F2][Sb2F11]2·(4) |
|
The observed nuclei in the assigned species are highlighted in bold characters.
The multiplicity of the signal is given in parentheses.
|
|
1H NMR |
|
9.92(OH, s) |
9.72(OH, s) |
|
13C{1H} NMR |
140.2(COF,d, J = 322.7 Hz) |
141.9(COF, d, J = 324.0 Hz) |
143.7(COF, d, J = 326.1 Hz) |
| 72.9(CC, dd, J = 111.9 Hz, 14.4 Hz) |
72.8(CC, dd, J = 108.8 Hz, 14.3 Hz) |
74.0(CC, dd, J = 104.0 Hz, 13.1 Hz) |
|
19F NMR |
45.22(COF, s) |
45.48(COF, s) |
45.91(COF, s) |
| −120.99(s) |
−120.68(s) |
| −128.00(s) |
−126.69(s) |
Theoretical calculations
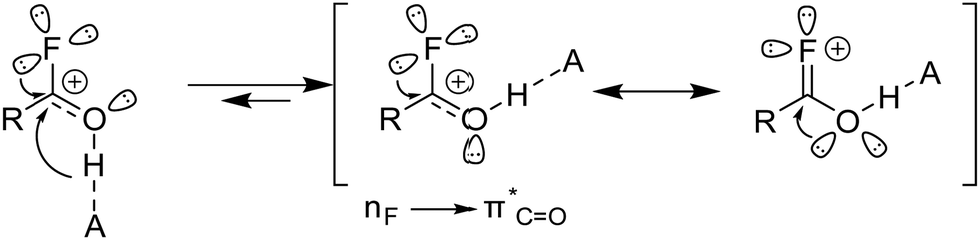
The quantum chemical calculations were performed on the MP2/aug-cc-pVTZ level of theory at 298 K using the Gaussian16 program package.25 Further details on a comparison of calculated and measured bond lengths are shown in ESI.† The stereoelectronic features of oxygen and the supramolecular stereoelectronic effect (SSE) in carboxylic acids are well-documented in the literature.26–28 Studies indicate that the conformation of carboxylic acids is strongly influenced by the  electron donation from the oxygen atom into the carbonyl group.26 Consideration of the isolated molecule shows that the E-conformation is energetically less favorable compared to the Z-conformation.26 As soon as hydrogen bonds can form this energetic disadvantage is reversed, making the E-conformation more stable than the Z-conformation.26,28 Protonated carboxylic acids also show two different conformers, with the syn–syn and the syn–anti conformation.24,29 Protonated acyl fluorides have a similar geometry compared to carboxylic acids. A few protonated acid fluorides have been published in recent years, the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 All five crystal structures are in the Z-conformation. The monoprotonated and diprotonated species of acetylenedicarbonyl fluoride also adopt the Z conformation. The C–F bond is shortened due to the protonation of an acyl fluoride. This shortening of the C–F bond is reported in the literature by the +R effect.10 However, the +R effect alone is no explanation for the favoured Z conformation in protonated acyl fluorides. The classic Z effect is stronger than the SSE effect in protonated acyl fluorides (Fig. 7). The +R effect additionally stabilizes the classic Z effect.
electron donation from the oxygen atom into the carbonyl group.26 Consideration of the isolated molecule shows that the E-conformation is energetically less favorable compared to the Z-conformation.26 As soon as hydrogen bonds can form this energetic disadvantage is reversed, making the E-conformation more stable than the Z-conformation.26,28 Protonated carboxylic acids also show two different conformers, with the syn–syn and the syn–anti conformation.24,29 Protonated acyl fluorides have a similar geometry compared to carboxylic acids. A few protonated acid fluorides have been published in recent years, the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 All five crystal structures are in the Z-conformation. The monoprotonated and diprotonated species of acetylenedicarbonyl fluoride also adopt the Z conformation. The C–F bond is shortened due to the protonation of an acyl fluoride. This shortening of the C–F bond is reported in the literature by the +R effect.10 However, the +R effect alone is no explanation for the favoured Z conformation in protonated acyl fluorides. The classic Z effect is stronger than the SSE effect in protonated acyl fluorides (Fig. 7). The +R effect additionally stabilizes the classic Z effect.
 |
| | Fig. 7 The illustration of the classic Z effect (middle) is stronger than the supramolecular stereoelectronic effect (left). The +R effect stabilizes additionally due to the C–F bond shortening the Z conformation (right). | |
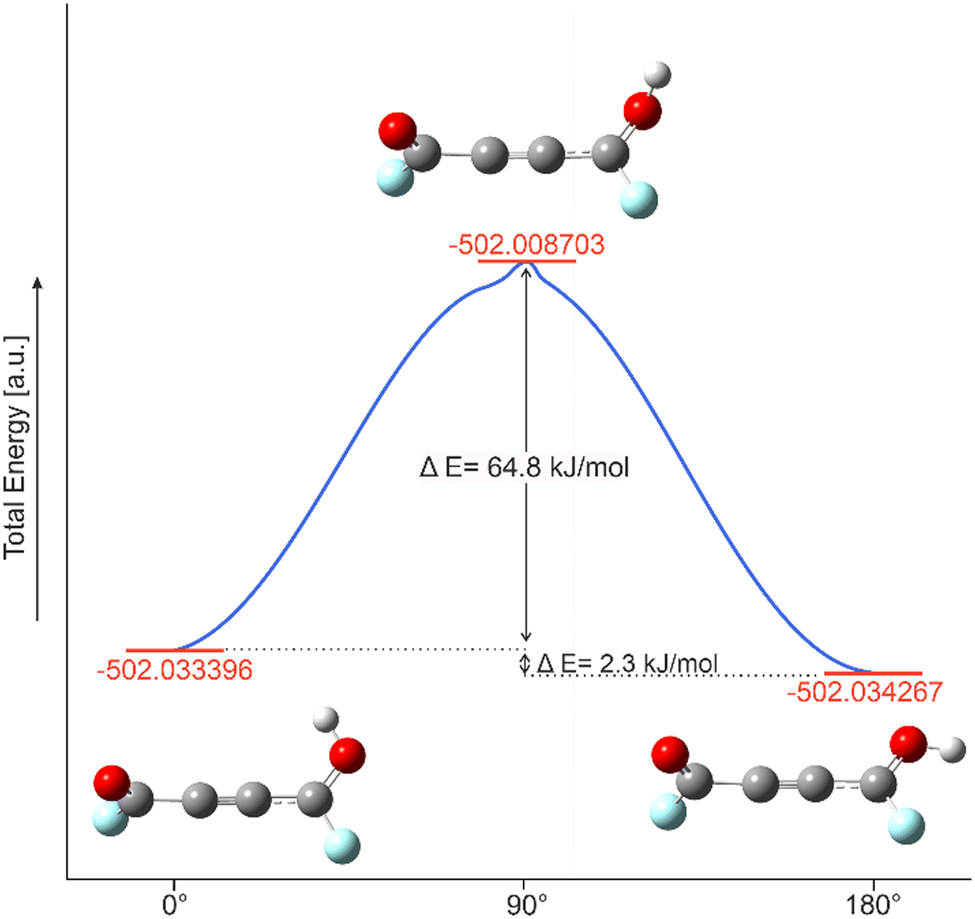
Due to the stabilization by the classic Z effect and the +R effect the rotational barrier of the monoprotonated species of acetylenedicarbonyl fluoride is calculated. Fig. 8 displays the internal reaction coordinate (IRC) of the E and Z conformation with the transition state. The energetic accuracy for the method MP2/aug-cc-pVTZ is between 0.64 kJ mol−1 (mean) and 1.64 kJ mol−1 (maximum).30
 |
| | Fig. 8 IRC of the rotational barrier of the monoprotonated species of C4O2F2 from Z to E conformation. | |
The E- and Z-conformers have only a small energy gap of 2.3 kJ mol−1 between each other. This is in the same range as the calculated energy differences for protonated propiolic acid of 1.4 kJ mol−1.24 The energy barrier (64.8 kJ mol−1) is in the same area compared to the protonated carboxylic acids formic (15.3 kcal mol−1 ≙ 64.0 kJ mol−1), acetic (11.2 kcal mol−1 ≙ 49.8 kJ mol−1), and propiolic acids (18.2 kJ mol−1).24,29 The not observed E-conformer in protonated acyl fluoride is a result of the highly temperature unstable cations. Another example for the stabilisation of conformation is the syn conformation of FC(O)SCl which is 0.31 kcal mol−1 (≙1.30 kJ mol−1) more stable than the anti-conformer by an lpπ → π*(CO) orbital interaction.31 The system of protonated acyl fluorides would have to be exposed to very high temperatures for the thermal energy to be sufficient to enable the conformational change. The thermal decomposition of the protonated acyl fluorides is an obstacle in this process.
Conclusions
In this work, the crystal structure of C4O2F2 is reported for the first time. Furthermore, the structure and properties of its mono- and diprotonated species are reported.2 The diprotonated species is the first example, which is formed by the decomposition of the monoprotonation in R-134a. All compounds were characterized by single-crystal X-ray diffraction, low-temperature NMR-spectroscopy, and low-temperature vibrational spectroscopy. The experimental data are discussed together with the DFT/aug-cc-pVTZ level of theory.32 The quantum chemical calculations in the theoretical calculations part were performed on the MP2/aug-cc-pVTZ level of theory. The theoretical calculation part explains the favoured Z conformation of protonated acyl fluorides. The rotational barrier is notable higher compared to the carboxylic acids.24,29
Experimental section
Caution! The hydrolysis of SF4, SbF5 C4F2O2, and the prepared salts (2–5) might form HF which burns skin and causes irreparable damage. Safety precautions must be taken while using and handling these materials.
Apparatus and materials
All experiments were executed on an electropolished stainless-steel vacuum line. For the synthesis of acetylenedicarbonyl fluoride, 1-L-autoclaves were employed. The autoclave was dried over 1 h at 350 °C. Room- and low-temperature Raman spectroscopic studies were performed using a Bruker MultiRAM FT-Raman spectrometer with Nd: YAG laser excitation (λ = 1064 cm−1) under vacuum at −196 °C. For a measurement, the synthesized compounds were transferred into a cooled glass cell. At low temperatures. IR spectra were recorded in a vacuum using a Bruker Vertex −80 V FTIR spectrometer. A small amount of the synthesized samples was placed on a CsBr single crystal plate in a cooled cell for measurement.33 The low-temperature single-crystal X-ray diffraction of C4O2F2 (1), [C4(OH)OF2][SbF6] (2), was performed on an Oxford XCalibur 3 diffractometer equipped with a Kappa CCD detector, operating with Mo-Kα (0.71073 Å) radiation (3 errors) and a Spellman generator (voltage 50 kV, current 40 mA). The program CrysAlisPro 1.171.38.46 (Rigaku OD, 2015)34 was employed for the data collection and reduction. The structures were solved utilizing SHELXT35 and SHELXL-2018/336 of the WINGX software package.37 The structures were checked using the software PLATON.38 The absorption correction was performed using the SCALE3 ABPSACK multiscan method.39 Selected data and parameters of the measured single-crystal X-ray structures analyses are summarized in Table S3 (see ESI†). NMR samples were prepared by adding the HF solution to a small FEP tube under a nitrogen stream. The tube was sealed under vacuum and inserted into a standard NMR tube. For 1H, 19F, and 13C{1H} NMR measurements a Bruker AV400TR and a JEOL ECX 400 NMR spectrometer were used. For evaluation, MNOVA by Mestrelab was used.40 The quantum chemical calculations in the experimental section were performed on the B3LYP/aug-cc-pVTZ level of theory with the Gaussian16 program package.25 The quantum chemical calculations were performed on the MP2/aug-cc-pVTZ level of theory at 298 K with the Gaussian16 program package.25 For visualization and illustration of the MEP calculations GaussView 6.0 was used.32,41
Synthesis of acetylenedicarbonyl fluoride (1).
The preparation procedure previously reported by Hasek et al. was optimized.5 A dry 1-L-autoclave was filled with dried C4H2O4 (93 mmol) in a nitrogen atmosphere. The autoclave was evacuated and SF4 (185 mmol) was condensed in the autoclave at −196 °C. The reaction mixture was allowed to warm up to room temperature for 24 h. The product was purified by trap to trap-distillation (−78 °C and −196 °C). A colorless liquid was formed in all the experiments with a yield of around 73%. The reaction with sulfur tetrafluoride and acetylene dicarboxylic acid leads to a very pure product of acetylenedicarbonyl fluoride.
Synthesis of [C4(OH)OF2][SbF6] (2).
Antimony pentafluoride (200 mg, 0.923 mmol,1.5 eq.) was condensed in an FEP tube reactor in a static vacuum at −196 °C. Afterward, 0.75 mL anhydrous hydrogen fluoride (aHF) was added, and the mixture homogenized at −40 °C. At −196 °C. Acetylenedicarbonyl fluoride (72.6 mg, 0.615 mmol, 1.0 eq.) was added at −196 °C to the superacidic system and warmed up to −40 °C. The solvent was removed at −78 °C overnight. The product was a white salt.
Synthesis of [C4(OD)OF2][SbF6] (3).
Antimony pentafluoride (60.0 mg, 0.277 mmol, 2.2 eq.) was condensed in a static vacuum in an FEP tube reactor at −196 °C. Afterwards, 0.75 mL anhydrous deuterated hydrogen fluoride (aDF) was added, and the mixture homogenized at −40 °C. At −196 °C Acetylenedicarbonyl fluoride (14.8 mg, 0.125 mmol, 1.0 eq.) was condensed into the FEB tube reaction vessel. The mixture was warmed up to −40 °C. Finally, the excess solvent was removed at −78 °C overnight. The product was observed as a colorless salt.
Synthesis of [C4(OH)2F2][Sb2F11]2·2HF (4).
At first (2) was synthesized. Afterward, R134a was condensed at −78 °C and after 20 min, the reaction mixture was warmed up to −25 °C for 20 min and mixed. The solvent was removed very quickly at −25 °C. The product occurs as a colorless to yellow salt.
Synthesis of [C4(OD)2F2][Sb2F11]2·2DF (5).
For the synthesis of (5), R134a was added to synthesized salt (3) at −78 °C and after 20 min, the reaction mixture was warmed up to −25 °C for 20 min and mixed. The solvent was removed very quickly at −25 °C. The product was a colorless yellow salt.
Data availability
The data supporting this article have been included as part of the ESI.† For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details. Crystallographic data has been deposited at the CCDC under 2371787, 2371788, and 2371790.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the Department of Chemistry at the Ludwig Maximilian University of Munich, the Deutsche Forschungsgemeinschaft (DFG), the F-Select GmbH, and Prof. Dr. Karaghiosoff for their support.
Notes and references
-
T. Schirmeister, C. Schmuck and P. R. Wich, Beyer/Walter Organische Chemie, Hirzel Verlag, Stuttgart, 25th edn, 2016 Search PubMed.
- O. Diels and W. E. Thiele, Zur Kenntnis der Dien-Synthesen, XXX. Mitteil.: Über das Chlorid der Acetylendicarbonsäure, Ber. Dtsch. Chem. Ges., 1938, 71, 1173–1178 CrossRef.
- G. Maier and W. A. Jung, Aktivierung von acetylendicarbonsäure durch überführung in gemischte anhydride [1], Tetrahedron Lett., 1980, 21, 3875–3878 CrossRef CAS.
- R. N. McDonald and R. A. Krueger, The Catalyzed Reaction of Acetylenedicarboxylic Acid and Thionyl Chloride, J. Org. Chem., 1963, 28, 2542–2544 CrossRef CAS.
- W. R. Hasek, W. C. Smith and V. A. Engelhardt, The Chemistry of Sulfur Tetrafluoride. II. The Fluorination of Organic Carbonyl Compounds 1, J. Am. Chem. Soc., 1960, 82, 543–551 CrossRef CAS.
- F. E. Herkes and H. E. Simmons, Acetylenedicarbonyl Fluoride, Synthesis, 1973, 166 CrossRef CAS.
- F. E. Herkes and H. E. Simmons, Acetylenedicarbonyl fluoride. I. Its physical properties and reaction with nucleophilic reagents, J. Org. Chem., 1975, 40, 420–423 CrossRef CAS.
- S. Steiner, A. Nitzer, C. Jessen and A. J. Kornath, Reactions of Dichloroacetyl Fluoride in Superacidic Media, Z. Anorg. Allg. Chem., 2024, 650 DOI:10.1002/zaac.202400013.
- S. Steiner, C. Jessen and A. J. Kornath, Synthesis and Structural Investigation of Protonated Haloacetyl Fluorides, Z. Anorg. Allg. Chem., 2022, 648 DOI:10.1002/zaac.202200060.
- M. C. Bayer, C. Kremser, C. Jessen, A. Nitzer and A. J. Kornath, Strengthening of the C-F Bond in Fumaryl Fluoride with Superacids, Chemistry, 2022, 28, e202104422 CrossRef CAS PubMed.
- L. A. Ramos, S. E. Ulic, R. M. Romano, M. F. Erben, C. W. Lehmann, E. Bernhardt, H. Beckers, H. Willner and C. O. Della Védova, Vibrational spectra, crystal structures, constitutional and rotational isomerism of FC(O)SCN and FC(O)NCS, Inorg. Chem., 2010, 49, 11142–11157 CrossRef CAS PubMed.
- S. E. Ulic, A. Kosma, C. O. Della Vedova, H. Willner and H. Oberhammer, S-(fluoroformyl)O-(trifluoroacetyl) thioperoxide, FC(O)S-OC(O)CF3: gas-phase structure and conformational properties, J. Phys. Chem. A, 2006, 110, 10201–10205 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- M. C. Bayer, C. Jessen and A. J. Kornath, Structure and Properties of Fumaryl Fluoride, Z. Anorg. Allg. Chem., 2021, 647, 258–265 CrossRef CAS.
- A. Jin, H. G. Mack, A. Waterfeld and H. Oberhammer, Gas-phase structure and conformations of malonyl difluoride (COF-CH2-COF) and difluoromalonyl difluoride (COF-CF2-COF). An electron diffraction and ab initio study, J. Am. Chem. Soc., 1991, 113, 7847–7852 CrossRef CAS.
- M. F. Erben, C. O. Della Védova, H. Willner, F. Trautner, H. Oberhammer and R. Boese, Fluoroformyl trifluoroacetyl disulfide, FC(O)SSC(O)CF3: synthesis, structure in solid and gaseous states, and conformational properties, Inorg. Chem., 2005, 44, 7070–7077 CrossRef CAS PubMed.
- R. Minkwitz, C. Hirsch and T. Berends, Synthesis and Characterisation of New Arsonium Salts and Crystal Structures of Trimethylarsonium Undecafluorodiarsenate (CH3)3AsH + As2F11− and Trimethylarsonium Hexafluoroantimonate (CH3)3AsH + SbF6−, Eur. J. Inorg. Chem., 1999, 2249–2254 CrossRef CAS.
- R. Minkwitz and S. Schneider, Synthesis and Characterization of the Tetrahydroxyphosphonium Hexafluorometalates P(OH)4 + MF6− (M = As, Sb), Angew. Chem., Int. Ed., 1999, 38, 210–212 CrossRef CAS.
- J. F. Lehmann, G. J. Schrobilgen, K. O. Christe, A. Kornath and R. J. Suontamo, X-ray crystal structures of XF(6)Sb(2)F(11) (X = Cl, Br, I); (35,37)Cl, (79,81)Br, and (127)I NMR studies and electronic structure calculations of the XF(6)(+) cations, Inorg. Chem., 2004, 43, 6905–6921 CrossRef CAS PubMed.
- T. Drews, W. Koch and K. Seppelt, The Cl 2 O 2+ Cation: Preparation and Structural Investigation of Cl 2 O 2+ SbF 6- and Cl 2 O 2+ Sb 2 F 11-, J. Am. Chem. Soc., 1999, 121, 4379–4384 CrossRef CAS.
-
G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, Oxford, 1997 Search PubMed.
- D. Hollenwäger, Y. Morgenstern, L. Daumer, V. Bockmair and A. J. Kornath, Structural Investigation of Diprotonated Glycine, Diprotonated Glycine Methyl Ester, and Monoprotonated Glycinoyl Fluoride, ACS Earth Space Chem., 2024, 8, 2101–2109 CrossRef.
- G. A. Olah, A. L. Berrier and G. K. Prakash, C NMR chemical shift correlations in application of “tool of increasing electron demand” to stable long-lived carbocations: Comprehensive evaluation, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 1998–2002 CrossRef CAS PubMed.
- D. Hollenwäger, S. Thamm, V. Bockmair, A. Nitzer and A. J. Kornath, Synthesis and Structure of Protonated Propiolic Acid, J. Org. Chem., 2024, 89, 11421–11428 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- I. V. Alabugin, L. Kuhn, M. G. Medvedev, N. V. Krivoshchapov, V. A. Vil, I. A. Yaremenko, P. Mehaffy, M. Yarie, A. O. Terent'ev and M. A. Zolfigol, Stereoelectronic power of oxygen in control of chemical reactivity: the anomeric effect is not alone, Chem. Soc. Rev., 2021, 50, 10253–10345 RSC.
- S. Z. Vatsadze, A. V. Medved’ko, M. K. Mirakbarov, M. E. Minyaev, V. N. Khrustalev, D. U. Zaripov, M. G. Medvedev and I. V. Alabugin, Not all carbon—carbon bonds are equivalent: anomeric effect of sp-hybridized carbon atom, Russ. Chem. Bull., 2024, 73, 363–371 CrossRef CAS.
- M. G. Medvedev, I. S. Bushmarinov and K. A. Lyssenko, Z-effect reversal in carboxylic acid associates, Chem. Commun., 2016, 52, 6593–6596 RSC.
- H. Hogeveen, Chemistry and spectroscopy in strongly acidic solutions. Part XX: Configurational equilibration in protonated formic acid and protonated acetic acid (dihydroxycarbonium ions): (Short communication), Recl. Trav. Chim. Pays-Bas, 1968, 87, 1313–1317 CrossRef CAS.
- J. Kaminsky, R. A. Mata, H.-J. Werner and F. Jensen, The accuracy of local MP2 methods for conformational energies, Mol. Phys., 2008, 106, 1899–1906 CrossRef CAS.
- M. F. Erben, C. O. Della Védova, R. M. Romano, R. Boese, H. Oberhammer, H. Willner and O. Sala, Anomeric and mesomeric effects in methoxycarbonylsulfenyl chloride, CH(3)OC(O)SCl: an experimental and theoretical study, Inorg. Chem., 2002, 41, 1064–1071 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- L. Bayersdorfer, R. Minkwitz and J. Jander, Eine Infrarot-Tieftemperaturkvette fr die Vermessung temperaturempfindlicher Gase, Flssigkeiten und Feststoffe, Z. Anorg. Allg. Chem., 1972, 392, 137–142 CrossRef CAS.
-
Rigaku Oxford Diffraction, CrysAlisPro Software System, Version 1.171.38.46, Rigaku Oxford Diffraction, Rigaku Corporation, Oxford, UK, 2015 Search PubMed.
- G. M. Sheldrick, SHELXT – integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
-
SCALE3 ABSPACK, An Oxford Diffraction Program, Oxford Diffraction Ltd, UK, 2005 Search PubMed.
-
MestReNova 14.0, Mestrelab Research, 2019 Search PubMed.
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6.0, Shawnee Mission, KS, 2016 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Alexander
Nitzer
,
Dominik
Leitz
and
Andreas J.
Kornath‡
*,
Alexander
Nitzer
,
Dominik
Leitz
and
Andreas J.
Kornath‡







 electron donation from the oxygen atom into the carbonyl group.26 Consideration of the isolated molecule shows that the E-conformation is energetically less favorable compared to the Z-conformation.26 As soon as hydrogen bonds can form this energetic disadvantage is reversed, making the E-conformation more stable than the Z-conformation.26,28 Protonated carboxylic acids also show two different conformers, with the syn–syn and the syn–anti conformation.24,29 Protonated acyl fluorides have a similar geometry compared to carboxylic acids. A few protonated acid fluorides have been published in recent years, the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 All five crystal structures are in the Z-conformation. The monoprotonated and diprotonated species of acetylenedicarbonyl fluoride also adopt the Z conformation. The C–F bond is shortened due to the protonation of an acyl fluoride. This shortening of the C–F bond is reported in the literature by the +R effect.10 However, the +R effect alone is no explanation for the favoured Z conformation in protonated acyl fluorides. The classic Z effect is stronger than the SSE effect in protonated acyl fluorides (Fig. 7). The +R effect additionally stabilizes the classic Z effect.
electron donation from the oxygen atom into the carbonyl group.26 Consideration of the isolated molecule shows that the E-conformation is energetically less favorable compared to the Z-conformation.26 As soon as hydrogen bonds can form this energetic disadvantage is reversed, making the E-conformation more stable than the Z-conformation.26,28 Protonated carboxylic acids also show two different conformers, with the syn–syn and the syn–anti conformation.24,29 Protonated acyl fluorides have a similar geometry compared to carboxylic acids. A few protonated acid fluorides have been published in recent years, the haloacetyl fluorides, dichloroacetyl fluoride, and fumaryl fluoride.8–10 All five crystal structures are in the Z-conformation. The monoprotonated and diprotonated species of acetylenedicarbonyl fluoride also adopt the Z conformation. The C–F bond is shortened due to the protonation of an acyl fluoride. This shortening of the C–F bond is reported in the literature by the +R effect.10 However, the +R effect alone is no explanation for the favoured Z conformation in protonated acyl fluorides. The classic Z effect is stronger than the SSE effect in protonated acyl fluorides (Fig. 7). The +R effect additionally stabilizes the classic Z effect.