
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rate constants for H-atom abstraction by HOO˙ from H-donor compounds of antioxidant relevance†
Mario C.
Foti
 *a,
Concetta
Rocco
a,
Zongxin
Jin
b and
Riccardo
Amorati
b
*a,
Concetta
Rocco
a,
Zongxin
Jin
b and
Riccardo
Amorati
b
aConsiglio Nazionale delle Ricerche, Istituto di Chimica Biomolecolare, via Paolo Gaifami 18, 95126 Catania, Italy. E-mail: mario.foti@cnr.it; Tel: +390957338343
bDepartment of Chemistry “G. Ciamician”, University of Bologna, Via Gobetti 83, 40129 Bologna, Italy
First published on 22nd August 2024
Abstract
The rate constants for the reaction of hydroperoxyl (or perhydroxyl) radical HOO˙ with fifteen phenols and ascorbyl palmitate were measured in acetonitrile at 37 °C by evaluating the effect that the antioxidants had on the rate of autoxidation of γ-terpinene. The HOO˙ radical represents an important reactive species that can be formed by protonation of superoxide anions (O2˙−) or by fragmentation of alkylperoxyl radicals (ROO˙) formed during the autoxidation of pro-aromatic derivatives like γ-terpinene. The phenols investigated in this study include natural compounds like phenolic acids (protocatechuic, caffeic and dihydrocaffeic acids), flavonoids (3-hydroxyflavone, pinobanksin, galangin, catechin, luteolin, quercetin, 6-methoxyluteolin), 4-methylcatechol and antioxidant additives ascorbyl palmitate and the a-tocopherol analogue 2,2,5,7,8-pentamethyl-6-chromanol. The rate constants for the reaction of HOO˙ with the above compounds (kinh) spanned from 1 × 103 M−1 s−1 for the unsubstituted phenol, to 7 × 104 and 9 × 104 M−1 s−1 for 4-methylcatechol and ascorbyl palmitate, respectively. As in a typical Evans–Polanyi plot, the log(kinh) was found to be inversely proportional to the bond dissociation enthalpy of the reactive OH. The comparison of the results with the data reported in the literature shows an unusual kinetic solvent effect that enlightens the unique behavior of HOO˙ and provides a rationale for the superior radical trapping ability of catechols and ascorbyl palmitate.
1. Introduction
In aerobic organisms, the superoxide radical anion O2˙− and its conjugate acid, the hydroperoxyl radical, HOO˙ (pKa ≈ 4.7), are largely present.1,2 It has been estimated that in healthy humans, 1.7 to 17 kg year−1 of inhaled oxygen are converted to O2˙− as a side product of mitochondrial respiration.1 Furthermore, enzymes like NADPH oxidase are deputed to produce superoxide for signaling or immune response.3 The superoxide anion O2˙− is not a strong oxidizing radical and is unable to abstract H-atoms from most common oxidizable substrates like polyunsaturated fatty acids.4 On the other hand, the protonated form HOO˙ has a well-documented H-atom abstraction activity that is similar (but not identical, vide infra) to that of alkylperoxyl radicals (ROO˙).5Apart from the reactivity, there is another important difference that characterizes HOO˙ relative to ROO˙. While the latter is usually little influenced by pH, the concentration of HOO˙ in aqueous media is pH dependent. At physiological pH, the conjugate base O2˙− is the predominant species and as such it may act as a reservoir of HOO˙. Many detrimental effects to human health can therefore be caused by HOO˙ because this radical is able to initiate radical reactions.4 The chemical diversity between ROO˙ and HOO˙/O2˙− has an influence on the strategies that Nature implements in order to keep their concentration under control. While the ROO˙ radicals are almost exclusively scavenged by H-atom donors,6 like α-tocopherol and ubiquinol,7 the removal of HOO˙/O2˙− can also occur by disproportionation to H2O2 and O2 catalyzed by super oxide dismutase enzymes (SOD).8
The importance and peculiarities of HOO˙ have not surprisingly led to a great research interest, but only a few works on its reactivity with phenolic antioxidants are present in the literature. The only rate constants available have, in fact, been obtained some decades ago in pulse-radiolysis studies with ascorbic acid and a few phenols in acidic aqueous solutions.9–11 More recently, the finding that the autoxidation of cyclohexadiene derivatives occurs through a radical-chain reaction mediated exclusively by HOO˙ radicals (see Scheme 1)12,13 has paved the way to studies on the reactivity of some phenolic and non-phenolic antioxidants with HOO˙, including α-tocopherol,5 catechol,14 dialkyl nitroxides,15 and “nanoantioxidants” like poly-1,8-dihydroxynaphthalene16 and CeO2 nanoparticles.17
 | ||
| Scheme 1 Radical-chain mechanism for liquid-phase autoxidation of γTH2 to Cy. The process can be slowed down by blocking the propagation step with a donor of H-atoms (AOH) to the chain carrier HOO˙. | ||
The reactivity of phenolic antioxidants of biological relevance remains, however, largely unknown. For this reason, we decided to determine the rate at which HOO˙ is quenched by the antioxidants 1–16 (see Scheme 2) including simple phenols, flavonoids and ascorbyl palmitate. We studied these reactions in acetonitrile and not in water or alcohols in order to avoid the mechanism of Sequential Proton Loss Electron Transfer (SPLET) which is active in protic solvents.18 In acetonitrile, ionization of phenols is poor, and the reaction proceeds by Hydrogen Atom Transfer (HAT) rather than by electron transfer from the anions.19 The absence of SPLET makes easier the kinetic treatment of our system. Among the different approaches that could be used to determine the rate constants of HOO˙ + 1–16, we used the method of inhibited autoxidation of γ-terpinene (γTH2), a well-known natural compound found in the essential oils of aromatic plants and featuring a 1,4-cyclohexadiene moiety (see Scheme 1).13 The formation of p-cymene (Cy) from γTH2 aromatization was induced by azo-bisisobutyronitrile (AIBN) decomposition and was determined via the GC-MS techniques. NMR experiments showed that Cy was the sole organic product formed in equimolar concentrations with H2O2 (see below). The rate constant of HOO˙ with 1–16, kinh, can be easily obtained from the rate of Cy formation in the presence and in the absence of antioxidants.
 | ||
| Scheme 2 Structure of the investigated antioxidants. | ||
The log(kinh) values were found to be inversely dependent on the calculated bond dissociation enthalpy (BDE) of the AO–H groups.6 This work enlightens for the first time some remarkable differences between HOO˙ and alkylperoxyl (ROO˙) as H-atom abstracting radicals from catechols and related poly-hydroxylated antioxidants.
2. Results
2.1. Mechanism of autoxidation of γTH2 and NMR of the products
The 2,2′-azobis(isobutyronitrile) (AIBN)-induced autoxidation of γTH2 at 37 °C produces Cy and hydrogen peroxide in a radical-chain reaction having HOO˙ as a chain-carrying radical, see Scheme 1. This mechanism was studied by Foti and Ingold following the observation that γTH2 could delay the peroxidation of linoleic acid. In this context, they observed that the autoxidation of γTH2 pertains, as for other hydrocarbons (1,4-cyclohexadiene, styrene, cumene, lipid chains, etc.), to a radical-chain process, involving initiation, propagation and termination steps. The conversion sets in with the thermal decomposition of the radical initiator AIBN, which produces a small and constant flux of carbon radicals. Subsequently, these initial radicals In˙ react rapidly with oxygen20 and abstract a hydrogen atom from γTH2, see Scheme 1.12,13 The formation of the chain carrier, HOO˙, occurs by a relatively fast unimolecular elimination of HOO˙ from γTHOO˙ via intramolecular 1,4-hydrogen-atom transfer (1,4-HAT) with a rate constant kf of ∼5 × 104 s−1 (see Scheme 1).20 The rate law of γTH2 autoxidation was found to depend on the concentration of γTH2,13,21 presumably because of a competition between fragmentation of γTHOO˙ and its reaction with another γTH2. The presence of the HOO˙ radical was also suggested by the NMR spectra of the reaction products, see Fig. 1. | ||
Fig. 1
1H NMR of a solution of γTH2 (0.898 M), AIBN (0.06 M) and mesitylene (0.022 M) in acetonitrile after 24 hours of reaction at 37 °C. The acetonitrile solution was then diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 with CDCl3 and the NMR spectrum was recorded. The singlet at 8.41 ppm was assigned to H2O2 while the aromatic moiety of p-cymene gave rise to a multiplet at 6.91 ppm. The broad signal at 5.25 ppm was assigned to the protons of the residual γTH2. The peak at 6.6 ppm was due to mesitylene (the internal standard). The area ratio of the peaks of Cy to HOOH was 4:2 and this corresponds to a molar ratio of 1:1. :10 with CDCl3 and the NMR spectrum was recorded. The singlet at 8.41 ppm was assigned to H2O2 while the aromatic moiety of p-cymene gave rise to a multiplet at 6.91 ppm. The broad signal at 5.25 ppm was assigned to the protons of the residual γTH2. The peak at 6.6 ppm was due to mesitylene (the internal standard). The area ratio of the peaks of Cy to HOOH was 4:2 and this corresponds to a molar ratio of 1:1. | ||
The Cy was found to be in equimolar concentration with H2O2, as required by the mechanism shown in Scheme 1. Moreover, these compounds were the only products formed and so they confirm that the hydroperoxyl radical was quantitatively generated by γTH2 autoxidation.
The large amount of H2O2 present in the solution made us wonder whether H2O2 could have some oxidizing effect on γTH2 or Cy. If H2O2 reacted with γTH2 or Cy, its concentration would decrease over time. By 1H-NMR in deuterated chloroform, we observed instead that, once O2 was depleted, the concentration of H2O2 did not change in a time interval of 48 hours and remained essentially equal to that of Cy. Apparently, these findings demonstrate that H2O2, under our conditions, did not react at an appreciable rate with γTH2 or Cy. The foregoing supports the use of γTH2 autoxidation as a convenient source of HOO˙ radicals in acetonitrile solution.
2.2. Determination of the rate constant kinh
The progress of γTH2 autoxidation was followed by determining via GC-MS the concentration of Cy overtime (see Material and methods). The autoxidation of γTH2 could be inhibited by the antioxidants 1–16 shown in Scheme 2. These antioxidants (AOH) capture and quench the chain carrier HOO˙ as shown in Scheme 1 and reaction (1). | (1) |
 | (2) |
 | (3) |
The efficiency of inhibition is expressed by the rate constant of the H-atom abstraction, nkinh, where n is the stoichiometric factor, i.e. the number of HOO˙ radicals quenched by one molecule of antioxidant. While with alkylperoxyl radicals (ROO˙), the stoichiometry is usually close to 2,22 in the case of HOO˙, higher values of n may be found because of the regeneration reaction of AOH (reaction (3)), which competes with the irreversible radical termination (reaction (2)).5,23 Moreover, in the case of catechols, an ortho-quinone may be generated. These ortho-quinones can be reduced back to the corresponding semiquinone radicals by HOO˙, increasing the duration of the inhibition period.14,24,25 However, in acetonitrile, HOO˙ is largely H-bonded to the solvent (˙OO–H–NCCH3) and as such H-atom donation is relatively unimportant compared to reactions (1) and (2).14 So, phenol regeneration, especially at the beginning of the reaction where the concentration of quinones is low, becomes secondary and n → 2. The rate constants nkinh were measured through the kinetic effects that compounds 1–16 had on the initial rate of γTH2 autoxidation, see Fig. 2. The presence of an antioxidant in a solution of γTH2 slows down the process of oxidation. Curiously, diluted and “aged” solutions of ascorbyl palmitate (16) accelerated the oxidation process, thus exhibiting a pro-oxidant behavior, presumably by the effect of dehydroascorbyl palmitate formed by spontaneous autoxidation of 16. To avoid these inconveniences, we measured the kinh of ascorbyl palmitate shortly after the preparation of the solutions.
 | ||
| Fig. 2 Formation of Cy measured by GC-MS during the autoxidation of γTH2 (0.95 M) initiated by AIBN (0.02 M) in the absence (black circles) and in the presence (open circles) of 4-methylcatechol (8.50 × 10−5 M) in acetonitrile at 37 °C. The initial rates of the uninhibited and inhibited reactions were R0 = 1.16 × 10−5 M s−1 and Rinh = 0.24 × 10−5 M s−1, respectively. The plateau visible in the black circle graph shows the moment in which oxygen was depleted from the vial and the reaction was over. | ||
For determining nkinh, we used a standard solution of acetonitrile at 37 °C containing γTH2, AIBN and mesitylene (1,3,5-trimethylbenzene) used as an internal standard. The initial rate of autoxidation in the absence of antioxidants R0 was (10.1 ± 4.0) × 10−6 M s−1. Then varying amounts of antioxidants were added to the blank solution and the initial rate, Rinh, was redetermined. The presence of inhibitors caused a reduction of Rinh relative to R0. These initial rates are related to the rate constant kinh through eqn 4, which can be solved to give the rate constant nkinh, eqn 5 (see also the ESI† for kinetic details).26
 | (4) |
 | (5) |
In eqn 5, Γinh = R0/Rinh − Rinh/R0; Ri is the rate of initiation; 2kt is the rate constant for the termination reaction HOO˙ + HOO˙ (see Scheme 1). In acetonitrile at 50 °C, 2kt ∼ 8.2 × 107 M−1 s−113 and thus the value at 37 °C can be estimated (by assuming that reaction rates double for every 10 degrees rise in temperature) to be ∼4.1 × 107 M−1 s−1. Ri can be calculated from the known decomposition rate of AIBN at 37 °C, Ri = 2kd × [AIBN] – 1 × 10−6 × 6.0 × 10−2 = 6.0 × 10−8 M s−1.13,21 Therefore, the term  and eqn 5 becomes eqn 6, where [AOH]0 is the initial concentration of antioxidant.
and eqn 5 becomes eqn 6, where [AOH]0 is the initial concentration of antioxidant.
 | (6) |
Table 1 gathers the values of nkinh for 1–16. These values range from 1.1 × 103 M−1 s−1 for phenol to 92 × 103 M−1 s−1 for ascorbyl palmitate. Table 1 also contains the rate constants of the reaction with ROO˙ radicals and a few of the compounds 1–16. These rates were determined by means of the autoxidation of styrene at 30 °C in MeCN.26–29 The nkinh value found in the present study for 2,2,5,7,8-pentamethyl-6-chromanol (PMHC) (4.0 × 104 M−1 s−1, see Table 1) is almost identical to the value previously obtained by O2 uptake at 30 °C in the same solvent (kHOO˙ = 6.8 × 104 M−1 s−1),5 indicating substantial agreement between the different techniques used to measure the rates.
| Compound | nk inh × 10−4 | Concentration range × 104/M | Relative rate constants kinh,rela | k ROO˙ × 10−4 |
|---|---|---|---|---|
a Rate constants relative to phenol. These values are independent of the value of  , see text.
b The rate constant nkinh for pinobanksin is too low to be determined. The value does not exceed 100 M−1 s−1.
c Under some circumstances, see text, ascorbyl palmitate behaved as a pro-oxidant, i.e. Rinh > R0.
d For caffeic acid phenethyl ester, ref. 26.
e From ref. 27,
f From ref. 6.
g Value for 3,5-di-tert-butylcatechol, from ref. 28.
h From ref. 29. , see text.
b The rate constant nkinh for pinobanksin is too low to be determined. The value does not exceed 100 M−1 s−1.
c Under some circumstances, see text, ascorbyl palmitate behaved as a pro-oxidant, i.e. Rinh > R0.
d For caffeic acid phenethyl ester, ref. 26.
e From ref. 27,
f From ref. 6.
g Value for 3,5-di-tert-butylcatechol, from ref. 28.
h From ref. 29.
|
||||
| Pinobanksin (9) | ≤ 0.01b | 1.113–2.23 | ≤ 0.1 | |
| Phenol (1) | 0.11 ± 0.05 | 7.20–10.77 | 1.00 | |
| Galangin (10) | 0.13 ± 0.05 | 1.15–6.04 | 1.18 | |
| Protocatechuic acid (5) | 0.28 ± 0.08 | 1.15–6.45 | 2.55 | |
| 3-Hydroxyflavone (8) | 0.29 ± 0.10 | 3.54–6.37 | 2.64 | |
| 4-Methoxyphenol (2) | 1.1 ± 0.6 | 0.958–3.83 | 10.0 | |
| Caffeic acid (7) | 1.15 ± 0.13 | 1.19–7.16 | 10.5 | 1.3d |
| Luteolin (12) | 1.4 ± 0.2 | 0.92–5.21 | 12.7 | |
| Di-hydrocaffeic acid (6) | 1.5 ± 0.8 | 1.20–7.21 | 13.6 | |
| Catechol (3) | 1.7 ± 0.4 | 1.06–5.80 | 15.5 | |
| 6-Methoxyluteolin (13) | 2.6 ± 0.3 | 0.56–4.87 | 23.6 | |
| Catechin (11) | 2.7 ± 0.4 | 0.66–4.00 | 24.6 | |
| Quercetin (14) | 3.5 ± 2.0 | 0.89–5.14 | 31.8 | 1.2e |
| PMHC (15) | 3.6 ± 1.2 | 0.97–5.85 | 32.7 | 68f |
| 4-Methylcatechol (4) | 7 ± 2 | 0.962–3.26 | 63.6 | 2.0g |
| Ascorbyl palmitate (16) | 9.2 ± 2.5c | 0.735–5.96 | 83.6 | 8.3h |
2.3. Calculation of the AO–H BDE
To explain the kinetic results reported in Table 1, we calculated the bond dissociation enthalpies (BDE) of the O–H groups by theoretical methods.25,30 These calculations were aimed at finding the most stable conformation and the weakest OH bonds in the compounds. To minimize errors, we used phenol as a reference compound, and the Δ(OH BDE) was calculated in acetonitrile as the difference between the investigated compounds and the simple phenol, whose OH BDE is reported in benzene as 86.7 ± 0.7 kcal mol−1.31 The most stable conformations in molecules and radicals and the Δ(OH BDE) values for compounds 1–16 are reported in Table 2.| Name | Parent molecule | Radical | ΔBDE (kcal mol−1) | |
|---|---|---|---|---|
| 1 | Phenol |

|

|
0 |
| 2 | 4-Methoxyphenol |

|

|
−6.32 |
| 3 | Catechol |
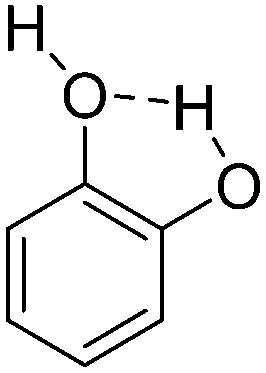
|

|
−7.11 |
| 4 | 4-Methylcatechol |
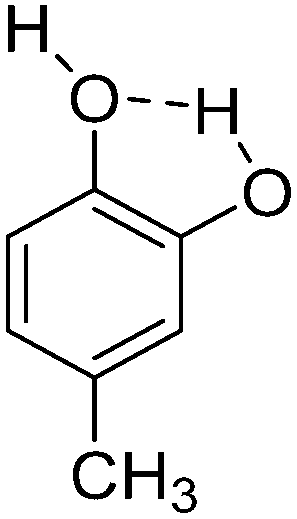
|
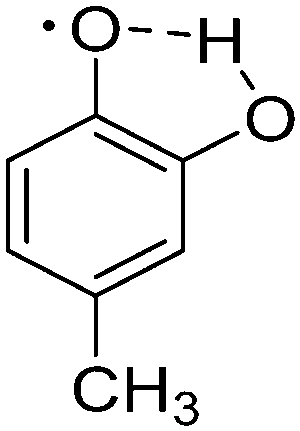
|
−9.72 |
| 5 | Protocatechuic acid |

|

|
−3.81 |
| 6 | Dihydrocaffeic acid |
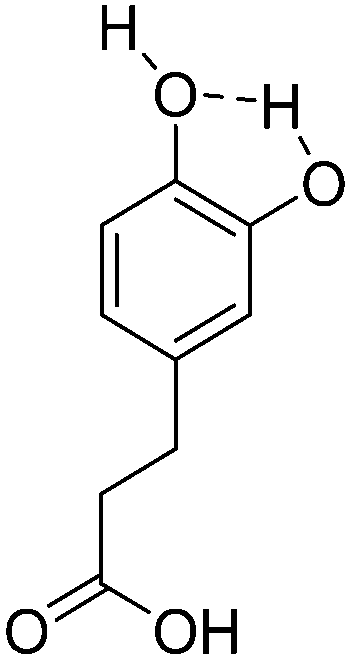
|

|
−8.41 |
| 7 | Caffeic acid |

|

|
−7.25 |
| 8 | 3-Hydroxyflavone |

|

|
−1.75 |
| 9 | Pinobanksin |

|

|
7.70 |
| 10 | Galangin |

|

|
−2.80 |
| 11 | (+)-Catechin |
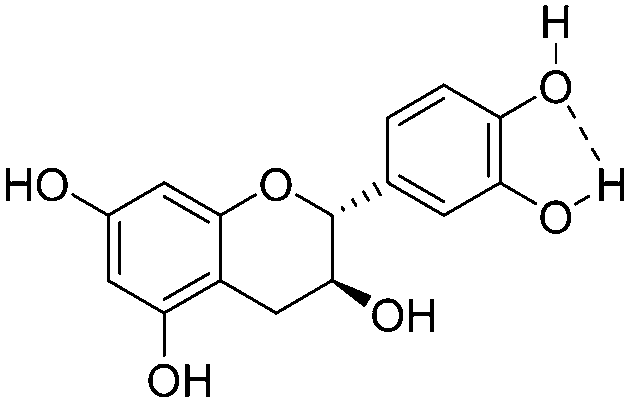
|

|
−7.29 |
| 12 | Luteolin |

|
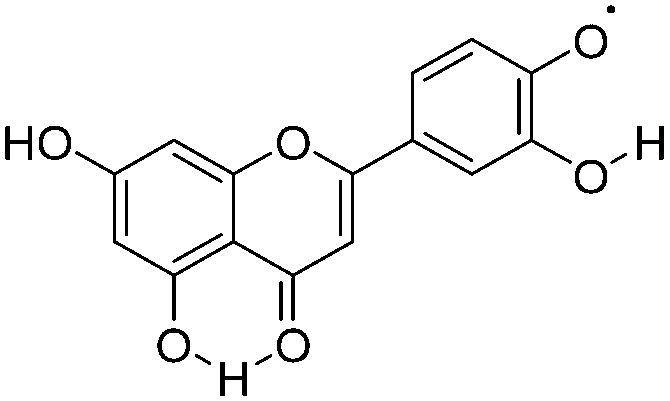
|
−5.90 |
| 13 | 6-Methoxyluteolin (nepetin) |
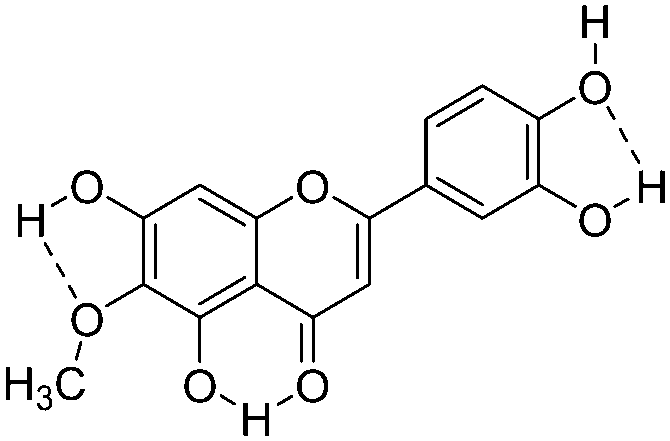
|

|
−6.07 |
| 14 | Quercetin |

|

|
−7.67 |
| 15 | PMHC |

|

|
−11.93 |
| 16 | Ascorbyl palmitate |

|
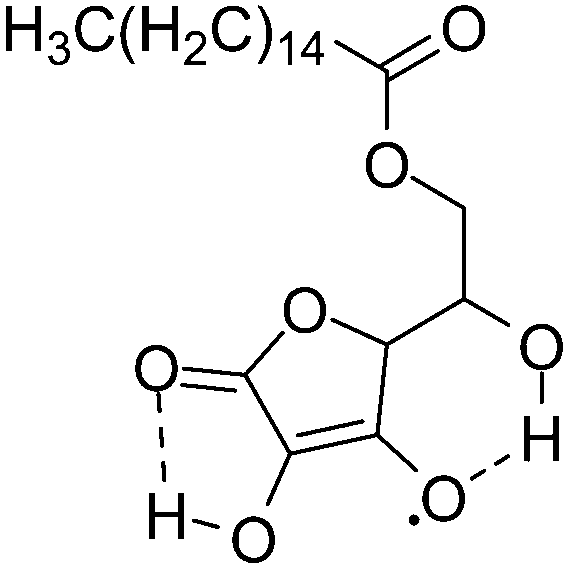
|
−7.95 |
As expected, the results show that phenols with multiple electron-donating groups, such as PMHC, or with a catechol moiety have a low O–H BDE.6 Generally, the reactive OH group in polyphenols is the weakest O–H.32 There are cases where the reactive group is a non-phenolic O–H group with low BDE as in the case of 3-hydroxyflavone and galangin, see Table 2, in which the reactive center is the enolic 3-OH. In the case of pinobanksin, the 3-OH is an alcoholic group and as such its OH BDE is high, see Table 2. On the other hand, the BDE of the 7-OH is slightly lower than that of the other OHs and this could justify its minimal reactivity (see Table 2). In conclusion, the most powerful antioxidants among 1–16 appear to be PMHC, 4-methylcatechol and ascorbyl palmitate. In the latter, two weak enolic OHs are present, the weakest being the 4-OH.29
Our thermodynamic calculations agree well with the kinetic data, see Fig. 3. The log(kinh) vs. Δ(OH BDEs) were summarized in the form of an Evans–Polanyi plot.6,33 The regression coefficient of the plot (R2 = 0.878, see Fig. 3) was reasonably good and showed that the OH BDE is a good descriptor to predict the rate of reaction for HOO˙ + H-donor, similar to what has been reported for ROO˙,34 and other radicals, e.g. DPPH˙.6
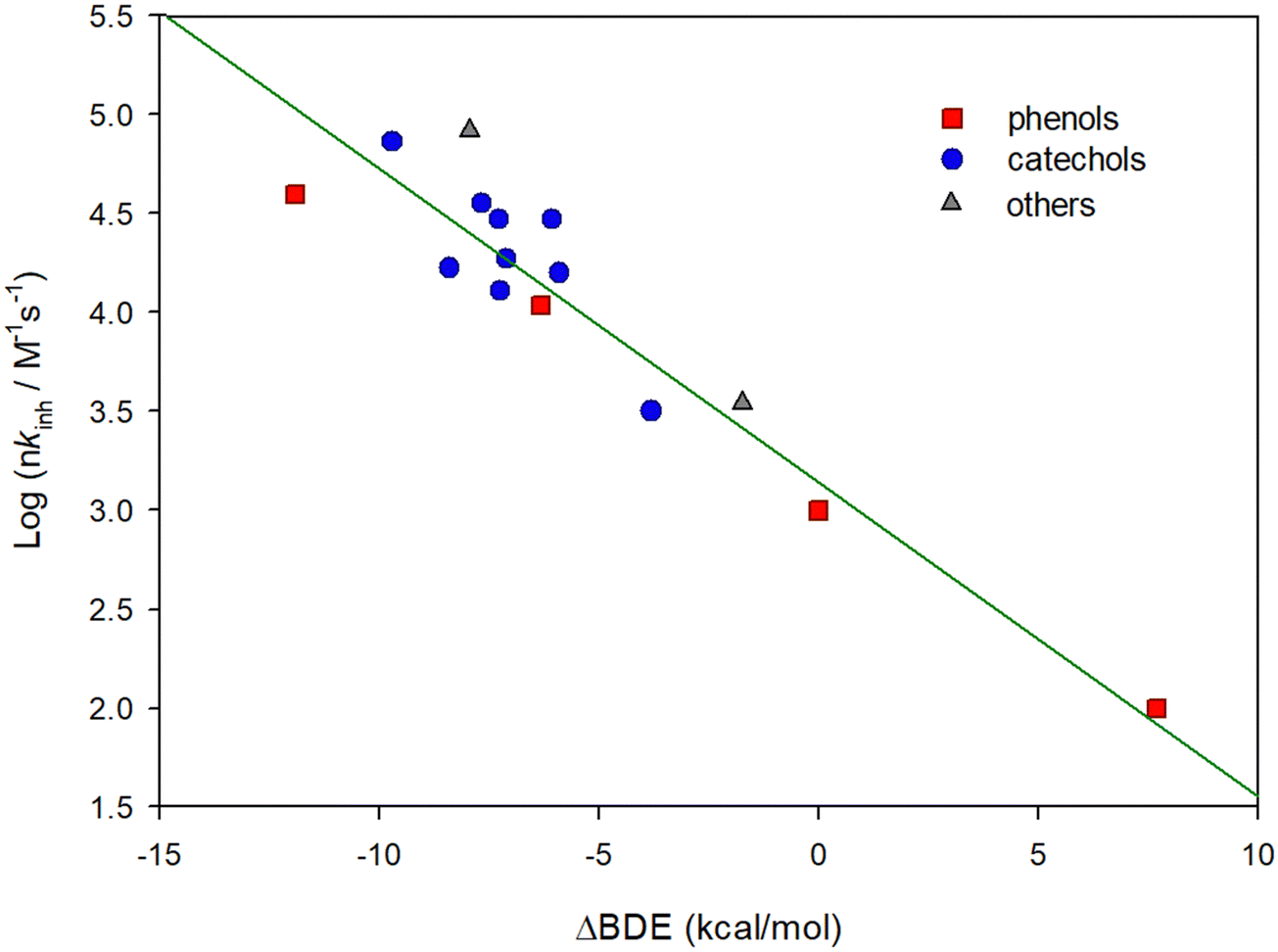 | ||
| Fig. 3 Linear relationship between the logarithm of the rate constant for the reaction with HOO˙, nkinh, and the calculated O–H BDE. The linear regression line has R2 = 0.878 and slope of −0.16. | ||
3. Discussion
The rate constants collected in this work show that the reactivity of phenolic antioxidants with HOO˙ obeys the same rules as for ROO˙, that is, the rate constants increase as the bond dissociation enthalpies of the reactive OH decrease, see Fig. 3.33 In many cases, the rate constants for HOO˙ and ROO˙ in acetonitrile are similar, although for PMHC + HOO˙/ROO˙ the rate constants are largely different, see Table 1. This calls for further studies particularly on the reactivity of the HOO˙ radical. The kinetic solvent effect (KSE) observed in the reactions PMHC + HOO˙/ROO˙ on the passage from chlorobenzene to acetonitrile must be considered.22,35 While in chlorobenzene the rate constants reported in the literature for the reaction between PMHC and HOO˙ or ROO˙ are similar (kHOO˙ = 1.6 × 106 M−1 s−1 and kROO˙ = 3.2 × 106 M−1 s−1 respectively, at 30 °C), in acetonitrile these are distinctly different, being 3.6 × 104 and 68 × 104 M−1 s−1 for kHOO˙ and kROO˙, respectively (see Table 1). The passage from chlorobenzene to acetonitrile determines a reduction of the rate constant of PMHC by a factor of 4.7 when the attacking radical is ROO˙, and by 44.4 when the radical is HOO˙. The reactivity reduction observed with ROO˙ radicals was explained on the basis of the mechanism reported in Scheme 3A, which implies that OH groups donating an H-bond to the solvent are not reactive toward ROO˙. | ||
| Scheme 3 Kinetic schemes that can be used to explain the KSEs with alkylperoxyl radicals (A) and with hydroperoxyl radicals (B). Proposed transition states for the H-atom transfer from catechol H-bonded to MeCN by ROO˙ (C) or HOO˙ (D). | ||
In polar solvents, phenols with ortho-alkyl substituents (e.g. PMHC) or ortho-methoxy groups are weak H-bond donors because they are sterically protected against the formation of a H-bond complex. Thus, a significant part of ortho-alkylphenols remains “free” in polar solvents and can react with radicals. On the other hand, antioxidants that are efficient H-bond donors, like catechols, show in acetonitrile a marked decrease in kROO˙ and thus a reduction of their antioxidant efficiency.34
The noticeable difference between ROO˙ and HOO˙'s reactivity in polar solvents originates from the fact that HOO˙ (but not ROO˙) can establish an equilibrium with acetonitrile strongly shifted to the right
| HOO˙ + MeCN ⇌ MeCN–HOO˙ |
As shown in Scheme 3B, the new H-bonded radical is less reactive than free HOO˙ and ROO˙ because the H-bond in the radical (MeCN–HOO˙) becomes weaker as the reaction proceeds from the reactants to the products (i.e. MeCN–HOOH), and H2O2 is a weaker H-bond donor than HOO˙.6
The foregoing suggests that in acetonitrile nkinh can be much smaller than kROO˙ as a general rule, i.e., for any H-atom donor. However, the results reported in Table 1 show that this is not the case. If we exclude PMHC, in all other cases where a comparison can be made, nkinh is similar to kROO˙. This suggests that ROO˙ and HOO˙ behave differently in the H-atom abstraction from H-bonded catechols or ascorbyl palmitate, as tentatively shown in Scheme 3C and D. In catechols, HAT may only occur from the intramolecularly H-bonded OH group. In the transition state (TS) with HOO˙, see Scheme 3D, two H-bonds with catechol might be present similarly to the TS previously identified in the reaction of HOO˙ with ortho-methoxy phenols.36 This “doubly bound” TS allows for a closer proximity of HOO˙ to catechol. Of course, this is not possible with ROO˙, see Scheme 3C, and thus the TS in Scheme 3C would be less stable. It has been demonstrated that the proximity of the O–atoms of the radicals to the phenol ring favors the transfer of electrons through orbital overlap with the H-atom being transferred as a proton.37,38 Therefore, the “doubly bound” TS would allow a better proton-coupled electron transfer (PCET) relative to the transition state formed by ROO˙. However, the binding of HOO˙ to catechol with the help of two H-bonds requires that HOO˙ breaks its H-bond with MeCN. Therefore, the feasibility of this process certainly depends on the relative strength of the two competing interactions.
4. Conclusions
A new method for generating HOO˙ radicals has been presented in this paper. This method yields HOO˙ via the autoxidation of γ-terpinene to p-cymene induced by AIBN in acetonitrile at 37 °C. The autoxidation process of γ-terpinene described in Scheme 1 is a radical-chain reaction carried exclusively by HOO˙ radicals. The reaction can be followed by measuring via GS-MS the quantity of Cy produced over time. From these data, the initial rates, in the presence and in the absence of antioxidant, were obtained. Then, the rate constants kinh for the quenching of HOO˙ by phenols and ascorbyl palmitate were calculated with good approximation. The rate constants so obtained vary from 1.1 × 103 M−1 s−1 for phenol to 92 × 103 M−1 s−1 for ascorbyl palmitate. These numbers represent the first examples of rate constants obtained for the quenching of HOO˙ by flavonoids, catechols and ascorbyl palmitate. The log of the rate constants kinh was inversely proportional to the OH BDEs of these antioxidants as in the typical Evans–Polanyi plots. The stronger antioxidants were those in which electron-donating groups were present in the ring since these phenols have low OH BDEs. Also, two different TS were hypothesized by us in order to explain the different reactivity of HOO˙ relative to ROO˙ in the HAT from catechol. The TS of HOO˙ would be stabilized by two intramolecular H-bonds. This “doubly bound” TS would allow a better proton-coupled electron transfer (PCET) process relative to the transition state formed by ROO˙. Additional kinetic experiments and theoretical computations are therefore required to validate these intriguing conjectures.5. Materials and methods
5.1 Materials
Solvents were HPLC grade. All reagents were commercially available. AIBN was purchased from Merck, recrystallized from methanol and its purity checked by NMR (≥98%). Mesitylene, from Aldrich, was used without further purification. Acetonitrile (HPLC grade) was purchased from Sigma-Aldrich and was used without further purification. γ-Terpinene (Aldrich) was percolated through a column of activated silica gel to remove the stabilizer present in the oil (α-tocopherol 0.08% w/w). Purified samples were then kept in the freezer at −20 °C until use. After one month, however, the sample became opaque for the formation of hydroperoxides and moisture. Therefore, it was centrifuged, and the upper layer was checked by 1H NMR which confirmed that the sample was constituted by γ-TH2 with a relatively small amount of Cy (<5%). Therefore, we collected the organic layer and continued to use it until the sample ran out.5.2 GC-MS experiments
Screw-capped glass vials with a perforable septum and a volume of 5.2 to 40.0 ml, from Supelco, were used in all experiments. They were washed in sequence with a solution of 0.001 M HCl, 0.001 M NaOH, water, acetone and finally acetonitrile. The acid/base washing was done in order to remove possible impurities released from the glass of the vials. The septum of the vials was changed at the end of the experiments. Stock solutions of AIBN (7.93 × 10−2 M) and mesitylene (1.438 M), employed as an internal standard, in acetonitrile were kept in the freezer at −20 °C until use. Two final solutions were obtained by mixing the following volumes or multiples of them: 500 µL of the AIBN solution with 100 µL of 95% pure γTH2, 10 or 30 µL of stock solution of mesitylene and finally 50 or 30 µL of acetonitrile. The solutions were therefore 0.898 M in γTH2, 6.00 × 10−2 M in AIBN, and 2.18 × 10−2 or 6.54 × 10−2 M in mesitylene. As expected, the internal standard did not influence the kinetics, therefore, we used in most experiments the concentration [mesitylene] = 6.54 × 10−2 M. Finally, the antioxidants 1–16 were added to a final concentration in the range 1 × 10−5–1 × 10−3 M.In a typical experiment, equal volumes (200–800 µL) of solution were loaded in three identical vials of 5.2 mL volume so that all experiments were run in triplicate. The vials were left open to the air for 20 min and then were capped and put in a shaker at 37 °C with or without shaking (250 rpm). At given time intervals, aliquots of solution (2–3 µL) were withdrawn with a syringe from the vials and immediately diluted with acetonitrile 1:50 v/v, then, 1 µL of the solution was injected in a HP5890A GC-MS instrument. The carrier gas was helium and the capillary column a Zebron ZB-5, 30 m × 0.25 mm i.d. × 0.25 µm. The GC program was, initial temp of 80 °C, increase 3 deg min−1 to 150 °C, increase 10 deg min−1 to 230 °C (hold 1.0 min).
The peaks of Cy and mesitylene in the gas-chromatogram were identified, and the Cy concentration was calculated by the equation  where ACy, and Amesitylene were the areas of the corresponding peaks and ACy0 the area due to the amount of pre-existing pCy in solution. The initial rate
where ACy, and Amesitylene were the areas of the corresponding peaks and ACy0 the area due to the amount of pre-existing pCy in solution. The initial rate 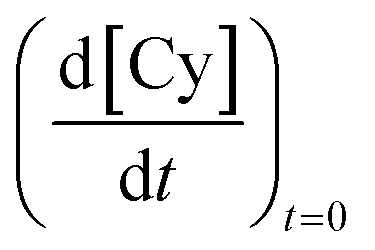 was calculated after a reaction time Δt of 60–100 min, therefore when γTH2 consumption was negligible.
was calculated after a reaction time Δt of 60–100 min, therefore when γTH2 consumption was negligible.
 | (7) |
Eqn 7 was used to calculate the initial rate for inhibited (with antioxidant) and uninhibited (no antioxidant) reactions (see ESI†).
5.3 1H-NMR experiments
A solution of γTH2 (0.898 M) + AIBN (6.00 × 10−2 M) + mesitylene (2.18 × 10−2 M) in acetonitrile was reacted for 24 hours at 37 °C. The 1H NMR of spectrum was done by mixing 50 µL of the final acetonitrile solution with 500 µL of CDCl3. The singlet at 8.41 ppm was assigned to HOOH while the aromatic moiety of p-cymene gave rise to the multiplet at 6.91 ppm. The broad signal at 5.25 ppm was assigned to the protons of the residual γTH2. The aliphatic upfield signals (not shown in this spectrum) at δ < 3 ppm were due to acetonitrile, CH3 and (CH3)2CH of Cy and γTH2. Finally, the peak at 6.6 ppm was due to mesitylene (the internal standard). The peaks of Cy and HOOH were in the area ratio of 4:2 corresponding to a molar ratio of 1:1 (as required by the reaction reported in Scheme 1).
When using acetone-d6 as the NMR solvent, besides the presence of γTH2, H2O2 and Cy, two more peaks at 10.25 and at 5.28 ppm were observed. 13C-NMR and DEPT spectra showed the presence of a quaternary carbon at 101 ppm. These signals were assigned to the condensation product between one molecule of H2O2 and one of (CD3)2CO.39
5.4 Theoretical calculations
Geometry optimizations and frequency calculations were carried out at the B3LYP/6-31+G(d,p) level with implicit solvent acetonitrile (polarized continuum model) using the Gaussian 16 package.30 Preliminary optimization calculations have been performed to identify the most stable conformers and the most stable radical. Stationary points were confirmed by checking the absence of imaginary frequencies. The ΔBDE values were determined from the difference of the enthalpy-corrected energies of the studied molecule and phenol. The Cartesian coordinates of the structures used to calculate the ΔBDE values are reported in the ESI.†Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. J. acknowledges a fellowship from China Scholarship Council (CSC: 202209120002). R. A. acknowledges funding by the European Union – NextGenerationEU under the National Recovery and Resilience Plan (PNRR) – Mission 4 Education and research – Component 2 From research to business – Investment 1.1 Notice Prin 2022 – DD N. 104 del 2/2/2022, from PRIN20227XZKBY – Superoxide responsive redox-active systems and nano smart materials to target ferroptosis – FEROX – CUP J53D23008550006”. We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support.References
- B. Halliwell and J. M. Gutteridge, Free radicals in biology and medicine, Oxford university press, USA, 2015 Search PubMed.
- B. H. Bielski, Photochem. Photobiol., 1978, 28, 645–649 CrossRef CAS.
- A. Cipriano, M. Viviano, A. Feoli, C. Milite, G. Sarno, S. Castellano and G. Sbardella, J. Med. Chem., 2023, 66, 11632–11655 CrossRef CAS PubMed.
- J. Aikens and T. A. Dix, Arch. Biochem. Biophys., 1993, 305, 516–525 CrossRef CAS.
- J. Cedrowski, G. Litwinienko, A. Baschieri and R. Amorati, Chem. – Eur. J., 2016, 22, 16441–16445 CrossRef CAS.
- M. C. Foti, C. Daquino, I. D. Mackie, G. A. DiLabio and K. Ingold, J. Org. Chem, 2008, 73, 9270–9282 CrossRef CAS.
- J. Helberg and D. A. Pratt, Chem. Soc. Rev., 2021, 50, 7343–7358 RSC.
- Y. Sheng, I. A. Abreu, D. E. Cabelli, M. J. Maroney, A.-F. Miller, M. Teixeira and J. S. Valentine, Chem. Rev., 2014, 114, 3854–3918 CrossRef CAS PubMed.
- D. E. Cabelli and B. H. J. Bielski, J. Phys. Chem., 1983, 87, 1809–1812 CrossRef CAS.
- Z. Kozmér, E. Arany, T. Alapi, E. Takács, L. Wojnárovits and A. Dombi, Radiat. Phys. Chem., 2014, 102, 135–138 CrossRef.
- B. Bielski and D. Cabelli, Int. J. Radiat., 1991, 59, 291–319 CrossRef CAS PubMed.
- D. G. Hendry and D. Schuetzle, J. Am. Chem. Soc., 1975, 97, 7123–7127 CrossRef CAS.
- M. C. Foti and K. Ingold, J. Agric. Food Chem., 2003, 51, 2758–2765 CrossRef CAS PubMed.
- Y. Guo, A. Baschieri, F. Mollica, L. Valgimigli, J. Cedrowski, G. Litwinienko and R. Amorati, Angew. Chem., Int. Ed., 2021, 60, 15220–15224 CrossRef CAS.
- A. Baschieri, L. Valgimigli, S. Gabbanini, G. A. DiLabio, E. Romero-Montalvo and R. Amorati, J. Am. Chem. Soc., 2018, 140, 10354–10362 CrossRef CAS PubMed.
- A. Mavridi-Printezi, F. Mollica, R. Lucernati, M. Montalti and R. Amorati, Antioxidants, 2023, 12, 1511 CrossRef CAS.
- R. Amorati, Y. Guo, B. M. Budhlall, C. F. Barry, D. Cao and S. S. R. K. Challa, ACS Omega, 2023, 8, 40174–40183 CrossRef CAS.
- G. Litwinienko and K. U. Ingold, J. Org. Chem., 2004, 69, 5888–5896 CrossRef CAS.
- R. Amorati, S. Menichetti, C. Viglianisi and M. C. Foti, Chem. Commun., 2012, 48, 11904–11906 RSC.
- S. Sortino, S. Petralia and M. C. Foti, New J. Chem., 2003, 27, 1563–1567 RSC.
- M. C. Foti, S. Sortino and K. U. Ingold, Chem. – Eur. J., 2005, 11, 1942–1948 CrossRef CAS.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022–9046 CrossRef CAS.
- J.-F. Poon, O. Zilka and D. A. Pratt, J. Am. Chem. Soc., 2020, 142, 14331–14342 CrossRef CAS.
- Y. Guo, A. Baschieri, R. Amorati and L. Valgimigli, Food Chem., 2021, 345, 128468 CrossRef CAS PubMed.
- N. Cardullo, F. Monti, V. Muccilli, R. Amorati and A. Baschieri, Molecules, 2023, 28, 735 CrossRef CAS.
- C. Spatafora, C. Daquino, C. Tringali and R. Amorati, Org. Biomol. Chem., 2013, 11, 4291–4294 RSC.
- R. Amorati, A. Baschieri, A. Cowden and L. Valgimigli, Biomimetics, 2017, 2, 9 CrossRef.
- L. Valgimigli, R. Amorati, S. Petrucci, G. F. Pedulli, D. Hu, J. J. Hanthorn and D. A. Pratt, Angew. Chem., 2009, 121, 8498–8501 CrossRef.
- R. Amorati, G. F. Pedulli and L. Valgimigli, Org. Biomol. Chem., 2011, 9, 3792–3800 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- P. Mulder, H.-G. Korth, D. A. Pratt, G. A. DiLabio, L. Valgimigli, G. Pedulli and K. Ingold, J. Phys. Chem. A, 2005, 109, 2647–2655 CrossRef CAS.
- M. Musialik, R. Kuzmicz, T. S. Pawłowski and G. Litwinienko, J. Org. Chem, 2009, 74, 2699–2709 CrossRef CAS.
- M. C. Foti, E. R. Johnson, M. R. Vinqvist, J. S. Wright, L. R. C. Barclay and K. U. Ingold, J. Org. Chem., 2002, 67, 5190–5196 CrossRef CAS PubMed.
- M. Lucarini and G. F. Pedulli, Chem. Soc. Rev., 2010, 39, 2106–2119 RSC.
- M. C. Foti, J. Pharm. Pharmacol., 2007, 59, 1673–1685 CrossRef CAS.
- A. Galano, J. R. Leon-Carmona and J. R. L. Alvarez-Idaboy, J. Phys. Chem. B, 2012, 116, 7129–7137 CrossRef CAS PubMed.
- L. D. Mena and M. T. Baumgartner, J. Am. Chem. Soc., 2022, 144, 15922–15927 CrossRef CAS PubMed.
- G. A. DiLabio and E. R. Johnson, J. Am. Chem. Soc., 2007, 129, 6199–6203 CrossRef CAS.
- E. A. Espinosa-Fuentes, L. C. Pacheco-Londoño, M. Hidalgo-Santiago, M. Moreno, R. Vivas-Reyes and S. P. Hernández-Rivera, J. Phys. Chem. A, 2013, 117, 10753–10763 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj03030c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |