
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
HF-addition to haloacetyl fluorides in superacidic media†
Sebastian
Steiner
 *,
Zurwa M.
Shafiq
,
Alexander
Nitzer
,
Dirk
Hollenwäger
and
Andreas J.
Kornath‡
*,
Zurwa M.
Shafiq
,
Alexander
Nitzer
,
Dirk
Hollenwäger
and
Andreas J.
Kornath‡
Department Chemie, Ludwig-Maximilians-Universität München, Butenandtstr. 5-13, 81377 Munich, Germany. E-mail: sebastian.steiner@cup.uni-muenchen.de
First published on 16th July 2024
Abstract
The reactions of difluoroacetyl fluoride and trifluoroacetyl fluoride were investigated in the binary superacid HF/SbF5 by low-temperature NMR spectroscopy. Whereas both haloacetyl fluorides form oxonium species after the addition of HF, the protonated acyl fluorides were not observed. Protonated 1,1,2,2-tetrafluoroethanol was isolated as a solid and represents an example of a protonated α-fluoroalcohol. The salt was characterized by low-temperature vibrational spectroscopy and single-crystal X-ray diffraction. [CHF2CF2OH2][SbF6] crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with two formula units per unit cell. Protonated perfluoroethanol is only stable in solution. The reactivity of both haloacyl fluorides is discussed based on quantum chemical calculations at the MP2/aug-cc-pVTZ-level of theory.
with two formula units per unit cell. Protonated perfluoroethanol is only stable in solution. The reactivity of both haloacyl fluorides is discussed based on quantum chemical calculations at the MP2/aug-cc-pVTZ-level of theory.
Introduction
Perfluorinated alcohols are of special interest in chemical syntheses. Due to their low nucleophilicity and high hydrogen bond donor strength, they are used as versatile applicable solvents and as precursors for the syntheses of weakly coordinating anions (WCAs).1–6 As primary and secondary perfluoroalcohols are thermally unstable due to facile HF-elimination, only a limited number of isolated compounds are reported in the literature.7,8 The simplest representative of this compound class, trifluoromethanol, was synthesized first by Seppelt from trifluoromethyl hypochlorite and hydrogen chloride at −110 °C.9 CF3OH is thermally unstable and decomposes rapidly to COF2 and HF.10 On a laboratory scale, perfluoroalcohols are synthesized by reacting either perfluorinated ketones or acyl fluorides in aHF.7 By this method, heptafluorocyclobutanol was prepared by Baxter, and the first crystal structure of an α-fluoroalcohol was elucidated by single-crystal X-ray diffraction.11,12 In addition, perfluorinated methanol, ethanol, and n-propanol were synthesized from the respective acyl fluorides, and the temperature dependency of the HF-addition to these compounds was investigated by NMR spectroscopy.13 Thus, the respective acyl fluoride is the predominant species at room temperature, but the addition of strong Lewis acids to HF enables the shift of the equilibrium to the side of the perfluorinated alcohols and the stabilization of these compounds as oxonium salts.13In previous studies of our group, the properties of acetyl fluoride and haloacetyl fluorides were investigated in the binary superacidic systems HF/AsF5 and HF/SbF5. While acetyl fluoride reacts under the formation of stable acetylium salts, the substitution of the CH3 moiety for electron-withdrawing groups leads to the formation of protonated acyl fluorides, as in the case of chloroacetyl fluoride and fluoroacetyl fluoride.14 When even more electron-withdrawing groups such as the CCl2H moiety were introduced, dichloroacetyl fluoride showed a different reactivity. Thus, oxonium salts were formed when the Lewis acid SbF5 was applied in excess. Thereby, the COF moiety is protonated and then HF is added due to an increased electrophilicity of the carbonyl carbon.15 Nevertheless, the intermediate protonated dichloroacetyl fluoride was isolated as a solid and characterized by single-crystal X-ray diffraction.15 This prompted us to perform investigations on the properties of further haloacetyl fluorides with even more electron-withdrawing groups in superacidic media. It was the aim to investigate their reactivity regarding the addition of HF to their carbonyl bonds and to find out if there is a limit to isolating even more electron-deficient protonated haloacetyl fluorides. We herein report the reactions of difluoroacetyl fluoride and trifluoroacetyl fluoride in the binary superacidic systems HF/SbF5 and DF/SbF5.
Results and discussion
Syntheses and properties
Difluoroacetyl fluoride (F2AcF) was reacted at −60 °C in the binary superacidic system HF/SbF5. To trace the reactivity of the haloacetyl fluoride in superacidic media, NMR spectroscopy was performed at −60 °C, whereas one equivalent of the Lewis acid was applied. Accordingly, the measured 1H, 19F, and 13C NMR spectra indicate the presence of [CHF2CF2OH2][SbF6] (1) in the solution, as presented in eqn (1). The oxonium species 1 is formed after the addition of HF to the carbonyl bond of F2AcF.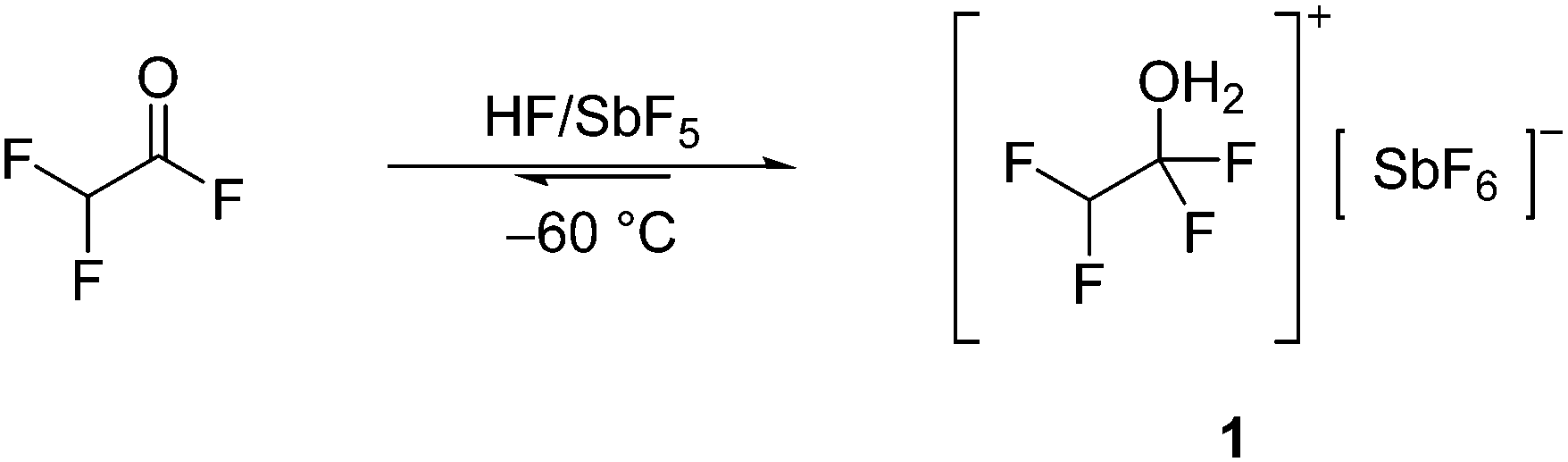 | (1) |
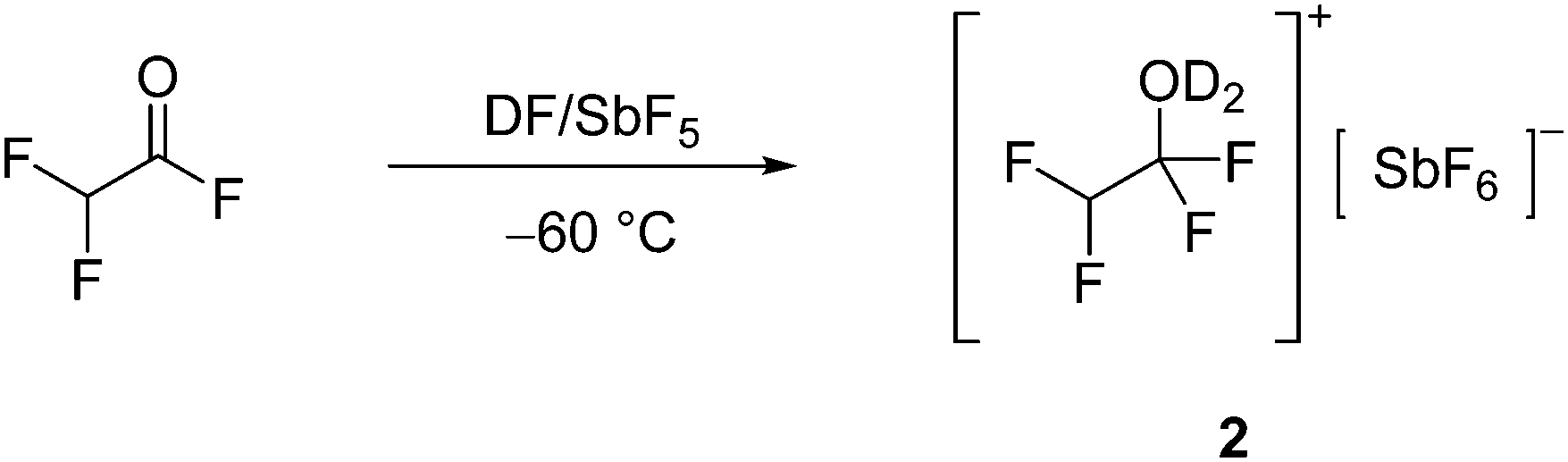 | (2) |
To investigate the reactivity of trifluoroacetyl fluoride (F3AcF) in HF/SbF5, NMR spectroscopy was performed at −70 °C with one equivalent of SbF5 being applied. Analog to the observed reactivity of F2AcF in superacidic media, the measured NMR spectra indicate the presence of [CF3CF2OH2][SbF6] (3) in the solution (see eqn (3)), but also the formation of an equilibrium between F3AcF and the oxonium species 3. As previously reported by Christe,13 the equilibrium is strongly shifted to the side of 3. Here, as in the case of F2AcF, protonated F3AcF is also not observed under these conditions.
 | (3) |
NMR spectroscopy
To trace the reactivity of difluoroacetyl fluoride and trifluoroacetyl fluoride in aHF and the binary superacidic system HF/SbF5, 1H, 19F, and 13C NMR spectra were measured at −60 °C and −70 °C, respectively, with acetone-d6 as an external standard. In Table 1, the observed NMR shifts of both starting materials, 1, and 3 are summarized. The measured NMR spectra and the complete NMR data of F2AcF, F3AcF, 1, and 3 are listed in the ESI† (Fig. S5–S14).| F2AcFa | 1 | F3AcFa |
3
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 13 |
|
|---|---|---|---|---|
| a aHF as solvent. b In HF/SbF5. | ||||
| δ 1H obs [ppm] | 5.72 (td) (CHF2) | 9.81 (s) (CF2OH2) | — | 9.82 (s) (CF2OH2) |
| 5.60 (t) (CHF2) | ||||
| δ 19F obs [ppm] | 17.74 (t) (COF) | −90.62 (s) (CF2OH2) | 11.07 (q) (COF) | −88.53 (s) (CF2OH2) |
| −132.65 (dd) (CHF2) | −141.33 (d) (CHF2) | −78.86 (d) (CF3) | −90.97 (s) (CF3) | |
| δ 13C obs [ppm] | 155.1 (dt) (COF) | 115.6 (tt) (CF2OH2) | 147.0 (dq) (COF) | 115.9 (qt) (CF3) |
| 104.7 (td) (CHF2) | 105.8 (tt) (CHF2) | 112.6 (qd) (CF3) | 113.3 (tq) (CF2OH2) | |
The 1H, 19F, and 13C NMR spectra of F2AcF dissolved in aHF display an equilibrium in the solution between the acyl fluoride and the corresponding α-fluoroalcohol that is formed after HF-addition. As depicted in Scheme 1, the acyl fluoride is the predominant species under these conditions as the equilibrium is strongly shifted to the side of F2AcF (see Fig. S5 and S6, ESI†).
 | ||
| Scheme 1 Equilibrium of F2AcF dissolved in aHF at −60 °C. | ||
By first dissolving equimolar amounts of SbF5 compared to F2AcF in HF and then adding the acyl fluoride, 1 is formed. The 1H NMR spectrum shows a triplet at 5.60 ppm, which is assigned to the CHF2 moiety, and a singlet at 9.81 ppm for the OH2+ group. The 19F NMR spectrum displays a singlet at −90.62 ppm and a doublet at −141.33 ppm for the CF2OH2 and CHF2 moieties, respectively. The NMR resonance of the CF2OH2 moiety is consistent with values reported in the literature.13,15 In the 13C NMR spectrum two triplets of triplets are observed at 115.6 ppm and 105.8 ppm for the CF2OH2 and CHF2 moieties, respectively. Further, in the 19F NMR spectrum, the resonance at −129.19 ppm is assigned to the anion [SbF6]−.16
The 1H, 19F, and 13C NMR spectra of F2AcF dissolved in HF/SbF5 also show an equilibrium between the oxonium species 1 and F2AcF in the solution (see Fig. S7–S9, ESI†). According to Scheme 2, the equilibrium is strongly shifted to the side of the protonated α-fluoroalcohol 1.
 | ||
| Scheme 2 Equilibrium of F2AcF dissolved in HF/SbF5 at −60 °C. | ||
Interestingly, protonated F2AcF is not observed under these conditions. This contrasts with the observed reactivity of dichloroacetyl fluoride (Cl2AcF) in the binary superacidic system HF/SbF5. Here, an equilibrium between the analog oxonium species [CCl2HCF2OH2][SbF6] and protonated Cl2AcF was observed in the superacidic solution at −60 °C.15 This is discussed in the theoretical section below.
In the case of F3AcF, similar results to those of F2AcF were found. The observed NMR resonances are in accordance with values previously reported by Christe.13 The measured NMR spectra and the complete NMR data of F3AcF dissolved in aHF and HF/SbF5 are listed in the ESI† (see Fig. S10–S14).
Vibrational Spectra of [CHF2CF2OH2][SbF6] (1) and [CHF2CF2OD2][SbF6] (2)
The low-temperature Raman (Ra) and Infrared (IR) spectra of [CHF2CF2OX2][SbF6] (1, 2) (X = H, D) and F2AcF are illustrated in Fig. 1. In Table 2, selected observed Raman and IR frequencies of 1 and 2 are listed together with quantum chemically calculated frequencies of the HF-complexed cation [CHF2CF2OH2]+·2HF, which is discussed later. The complete vibrational frequencies of 1 and 2 as well as difluoroacetyl fluoride are provided in the ESI† (Fig. S1 and Tables S1–S3). | ||
| Fig. 1 Low-temperature Raman and IR spectra of [CHF2CF2OX2][SbF6] (1, 2) (X = H, D) and vibrational spectra of CHF2COF. | ||
| [CHF2CF2OH2][SbF6] (1) | [CHF2CF2OD2][SbF6] (2) | [CHF2CF2OH2]+·2HF | Assignments | ||||
|---|---|---|---|---|---|---|---|
| Raman | IR | Raman | IR | calc.ab (IR/Raman)c | |||
| a Calculated at ωB97XD/aug-cc-pVTZ-level of theory. b Frequencies are scaled with a factor of 0.956. c IR intensity in [km mol−1] and Raman intensity in [Å4 u−1].Abbreviations for IR intensities: vs = very strong, s = strong, m = medium, w = weak, vs = very weak. | |||||||
| 3055(25) | 3055(50) | 3053 m | 2983(5/81) | v 1 | A | v(C–H) | |
| 1362(13) | 1364 s | 1359(19) | 1358 w | 1406(14/1) | v 5 | A | v(C–F) |
| 1251(11) | 1256 s | 1251(12) | 1256 m | 1235(236/2) | v 6 | A | v(C–F) |
| 1164(5) | 1151 s | 1144(6) | 1152(181/2) | v 9 | A | v(C–F) | |
| 1124(13) | 1125(15) | 1121 m | 1102(6/4) | v 10 | A | v(C–F) | |
| 836(34) | 835 s | 816(30) | 814 m | 1045(89/3) | v 12 | A | v(C–O) |
| 758(3) | 756 w | 755(67/5) | v 14 | A | v(C–C) | ||
For the [CHF2CF2OH2]+ cation with C1 symmetry 24 fundamental vibrational modes are expected, all of which are Raman and IR active. The ν(O–H) is superposed by condensed water in the IR spectra due to the measuring method. Further, the O–H stretching vibration shows low intensity in the Raman spectra due to the poor polarizability of the O–H group, which does not apply to the O–D group.17 The O–D stretching vibrations of the D-isotopomeric species 2 are observed at 2403 cm−1 and 2286 cm−1 in the Raman spectra and at 2403 cm−1 and 2330 cm−1 in the IR spectra. The O–D stretching vibrations are in good agreement with values reported in the literature.14,15 The C–F stretching vibrations of the CF2 moiety appear significantly blue-shifted by up to 110 cm−1 in comparison to the neutral compound at 1362 cm−1, 1251 cm−1 (1), 1359 cm−1, and 1251 cm−1 (2) (Ra) as well as 1364 cm−1, 1256 cm−1 (1), 1358 cm−1, and 1256 cm−1 (2) (IR).18 They are in good agreement with the values reported for [CCl2HCF2OH2][SbF6].15 Further, the formation of the oxonium species is indicated by the C–O stretching vibration at 836 cm−1 and 816 cm−1 (2) in the Raman spectra and at 835 cm−1 (1) and 814 cm−1 (2) in the IR spectra. The ν(C–O) is in accordance with values reported for protonated alcohols in the literature19,20 but significantly red-shifted by approximately 200 cm−1 compared to the νs(C–O) of the analog oxonium salt [CCl2HCF2OH2][SbF6].15 The ν(C–C) of 2 is observed at 758 cm−1 (Ra) and 756 cm−1 (IR) red-shifted by nearly 100 cm−1 compared to F2AcF.18
For the anion [SbF6]− with ideal octahedral symmetry more vibrations are observed than expected due to interionic interactions leading to a symmetry distortion.17
Crystal structure of [CHF2CF2OH2][SbF6] (1)
[CHF2CF2OH2][SbF6] (1) crystallizes in the triclinic space group P with two formula units per unit cell. The molecular unit of 1 is illustrated in Fig. 2. Crystal data and structure refinement are provided in the ESI† (Table S4). Selected geometric data are listed in Table 3.
 | ||
| Fig. 2 Molecular unit of [CHF2CF2OH2][SbF6] (1) (displacement ellipsoids with 50% probability). | ||
| Bond lengths [Å] | |||
| C1–C2 | 1.524(5) | C2–F3 | 1.341(4) |
| C1–F1 | 1.318(4) | C2–F4 | 1.346(4) |
| C1–F2 | 1.325(4) | C1–O1 | 1.424(4) |
| Bond angles [°] | |||
| C2–C1–O1 | 109.1(3) | F1–C1–O1 | 106.3(3) |
| F1–C1–C2 | 111.9(3) | F1–C1–F2 | 110.0(3) |
| F3–C2–C1 | 108.1(3) | F3–C2–F4 | 108.5(3) |
| Dihedral angles [°] | |||
| F3–C2–C1–F1 | 55.0(3) | F3–C2–C1–O1 | –62.4(3) |
| F3–C2–C1–F2 | 178.0(2) | F4–C2–C1–F1 | 171.8(2) |
| Interatomic contacts [Å] | |||
| O1(–H1)⋯F6i | 2.471(3) | O1⋯F9ii | 2.689(3) |
| O1(–H2)⋯F5 | 2.452(3) | F5⋯F3v | 2.888(3) |
| C2(–H3)⋯F7iii | 3.129(4) | F5⋯F9iv | 2.890(2) |
| C2(–H3)⋯F10iv | 3.166(4) | ||
The bond length C1–O1 (1.424(4) Å) is significantly elongated compared to difluoroacetyl fluoride (1.180(5) Å)18,21 and is in the range of a formal C–O single bond (1.432 Å).22 The C1–O1 bond length is consistent with the corresponding C–O bond length reported for [CCl2HCF2OH2][SbF6] (1.418(3) Å)15 but significantly shorter than values of oxonium species or protonated alcohols reported in the literature.19,23,24 The bond lengths C1–F1 (1.318(4) Å) and C1–F2 (1.325(4) Å) are slightly shortened compared to the starting material (1.342(7) Å)18,21 and are in accordance with the C–F bond lengths of [CCl2HCF2OH2][SbF6] (1.325(2) Å).15 Further, the bond C1–C2 (1.524(5) Å) remains unchanged compared to F2AcF (1.514(7) Å).18,21
The Sb–F bond lengths of [SbF6]− are in the range between 1.853(2) Å and 1.925(2) Å and correspond with values reported in the literature.25–27 Due to solid-state effects, the anion displays distorted octahedral symmetry. The bonds Sb1–F5 and Sb1–F6 are significantly longer than the other Sb–F bonds because they are involved in hydrogen bonding.
In the crystal structure of 1 the ions are arranged into chains along the a-axis by the strong hydrogen bonds O1(–H1)⋯F6i (2.471(3) Å) and O1(–H2)⋯F5 (2.452(3) Å) (see Fig. 3 and Fig. S2, ESI†).28 The ions also form chains along the c-axis by the weak hydrogen bond C2(–H3)⋯F7ii (3.129(4) Å) and the O⋯F interaction O1⋯F9iv (2.689(3) Å) (see Fig. 3 and Fig. S3, ESI†). Further, dimers of cation–anion pairs are formed by the intermolecular F⋯F contacts F5⋯F3v (2.888(3) Å) and F5⋯F9iv (2.890(2) Å) as well as the weak hydrogen bond C2(–H3)⋯F10iv (3.166(4) Å) (see Fig. S4, ESI†). All interatomic O⋯F and F⋯F contacts are below the sum of their van der Waals radii (2.99 Å, and 2.94 Å).29 Interatomic distances are listed in Table 3.
 | ||
| Fig. 3 Interionic contacts of 1 (displacement ellipsoids with 50% probability). Symmetry codes: i = −1 + x, y, z; ii = 1 − x, −y, 2 − z; iii = 1 − x, 1 − y, 1 − z; iv = x, 1 + y, z. | ||
Theoretical calculations
Quantum chemical calculations were performed at the ωB97XD/aug-cc-pVTZ-level of theory.30 F2AcF, F3AcF, and [CHF2CF2OH2]+·2HF were calculated in the gas phase for a better assignment of the vibrational modes and to compare the theoretical and experimentally determined geometric parameters. Solid-state effects and intermolecular interactions were simulated by adding additional HF molecules to the cation in the gas phase.31 In the ESI† (Fig. S15), [CHF2CF2OH2]+·2HF is illustrated with selected bond lengths and angles in comparison to the crystal structure. The calculated values of the HF-complexed cation are in good agreement with the experimentally obtained data.As observed in the experimental section, the NMR spectra of F2AcF and F3AcF dissolved in HF/SbF5 both show equilibria between the oxonium species 1 and 3, respectively, and their corresponding haloacetyl fluorides. In both cases, the protonated haloacetyl fluorides were not observed. This is in contrast with recent studies of our group on the reactivity of dichloroacetyl fluoride in the binary superacidic system HF/SbF5.15 Here, the measured NMR spectra of Cl2AcF dissolved in HF/SbF5 at −60 °C displayed an equilibrium between [CCl2HCF2OH2][SbF6] and protonated Cl2AcF.15 The addition of HF to the carbonyl bond of Cl2AcF was also only observed after the protonation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, as the carbonyl carbon displayed an increased electrophilicity due to the protonation.15 To get a more detailed insight into the versatile reactivity of the haloacetyl fluorides in HF/SbF5, quantum chemical calculations were performed at the MP2/aug-cc-pVTZ-level of theory. Therefore, Cl2AcF, F2AcF, F3AcF, as well as [CCl2HC(OH)F]+·HF, [CHF2C(OH)F]+·HF, and [CF3C(OH)F]+·HF were optimized and molecular electrostatic potentials (MEPs) were calculated alongside natural population analysis charges (NPAs) (see Tables S6, S7 and S9–S13, ESI†). The calculated MEPs are shown in Fig. 4 and the calculated NPA charges are listed in Table S13 (ESI†).
O bond, as the carbonyl carbon displayed an increased electrophilicity due to the protonation.15 To get a more detailed insight into the versatile reactivity of the haloacetyl fluorides in HF/SbF5, quantum chemical calculations were performed at the MP2/aug-cc-pVTZ-level of theory. Therefore, Cl2AcF, F2AcF, F3AcF, as well as [CCl2HC(OH)F]+·HF, [CHF2C(OH)F]+·HF, and [CF3C(OH)F]+·HF were optimized and molecular electrostatic potentials (MEPs) were calculated alongside natural population analysis charges (NPAs) (see Tables S6, S7 and S9–S13, ESI†). The calculated MEPs are shown in Fig. 4 and the calculated NPA charges are listed in Table S13 (ESI†).
 | ||
| Fig. 4 Molecular 0.0004 bohr−3 3D isosurfaces with mapped electrostatic potential as a color scale from −91.9 kJ mol−1 (red) to 113.9 kJ mol−1 (blue) (top), and 249.4 kJ mol−1 (red) to 679.8 kJ mol−1 (blue) (bottom). The electrostatic potential isosurfaces have been calculated for Cl2AcF, F2AcF, F3AcF (top), [CCl2HC(OH)F]+·HF, [CHF2C(OH)F]+·HF, and [CF3C(OH)F]+·HF (bottom). Calculated at the MP2/aug-cc-pVTZ-level of theory. | ||
As illustrated, the MEPs of the haloacetyl fluorides as well as of the protonated species show electron-deficient moieties, so-called π-holes, in the region of the carbonyl carbon.14,15,32–35 These indicate that electrophilic regions of the molecules are located at the carbon atoms. To quantify the influence of the electron-withdrawing substituents on the electrophilicity of Cl2AcF, F2AcF, and F3AcF as well as the protonated species, the associated MEP values at the π-holes of the calculated molecules are highlighted in Fig. 4. Accordingly, the most positive MEP values that were found on the MEP surfaces of the haloacyl fluorides are 73.2 kJ mol−1 (Cl2AcF), 113.9 kJ mol−1 (F2AcF), and 140.8 kJ mol−1 (F3AcF). Thus, the π-holes become significantly larger the more electron-withdrawing the adjacent substituent is. Further, the electrophilicity of the carbonyl carbon is therefore significantly increased the more electron-withdrawing the adjacent substituent is.
In the case of the protonated species, the observed MEP values at the π-holes are significantly higher than for the acyl fluorides.14,15,35 Thus, the electrophilicity of the haloacetyl fluorides is significantly increased due to the protonation.14,15,35 Interestingly, as depicted in Fig. 4, the MEP value for the π-hole of [CHF2C(OH)F]+·HF is significantly higher than for [CF3C(OH)F]+·HF. We assume that this is due to the stabilizing effects of CF3 moieties due to hyperconjugation and was observed before for CF3-substituted carbocations.36 In Table S13 (ESI†), the NPA charges of the calculated protonated species are listed. Accordingly, as observed before for protonated acyl fluorides, the NPA charges of the fluorine atoms that are bound to the carbonyl groups are strongly increased compared to the neutral compounds. This indicates that the positive charges located at the carbon atoms are stabilized by electron-donation of the directly bound fluorine atoms (“+R-effect”).14,15,32–35 To further examine these trends and to quantify the influence of the electron-withdrawing substituents on the back-donation of the fluorine lone-pair electrons, NBO analyses were performed at the MP2/aug-cc-pVTZ-level of theory for Cl2AcF, F2AcF, F3AcF, and all protonated species. Thus, for all calculated molecules strong interactions between the π*(CO) and the in-plane fluorine lone-pairs were found.14,15,35 The stabilization energies of these interactions, according to the second-order perturbation theory analyses, are summarized in Table 4.
O) interactions from second-order perturbation theory analyses of Cl2AcF, F2AcF, F3AcF, [CCl2HC(OH)F]+·HF, [CHF2C(OH)F]+·HF, and [CF3C(OH)F]+·HF
As expected, the stabilization energies are significantly higher in the case of the protonated acyl fluorides compared to the neutral compounds due to the increased back-donation of fluorine lone-pair electrons.14,15,35 Furthermore, for both the acyl fluorides and the protonated species, the stabilization energies of the n(F) → π*(CO) interactions increase the more electron-withdrawing the adjacent substituents become. Only in the case of F2AcF and F3AcF, approximately the same stabilization energies were found, which is probably again due to the hyperconjugation of the CF3 moiety.36 Thus, these trends also suggest that the π-holes and therefore the electrophilicities of the CO moieties increase as the neighboring substituents become more electron-withdrawing.
We therefore consequently assume that the reactivity of Cl2AcF, F2AcF, and F3AcF dissolved in HF/SbF5 is directly correlated to the electrophilicity of their carbonyl carbons. As illustrated in Fig. 4, the substitution of the CCl2H moiety for the CHF2 and CF3 moieties leads to a significantly increased electrophilicity of the neutral compounds as well as the protonated species. This results in the favoring of the HF-addition to the carbonyl bond and consequently the shift of the observed equilibria in HF/SbF5 to the side of the protonated oxonium species. Since the π-hole in the case of protonated Cl2AcF is significantly lower than in the cases of protonated F2AcF or F3AcF, the addition of HF to the CO bond is less favored and proceeds much slower. This enables the observation of the protonated intermediate in the solution and even the isolation of the species as a solid.15 In the cases of protonated F2AcF and F3AcF, the significantly increased electrophilicity of the species leads to a more favored and faster HF-addition to the carbonyl bonds. Therefore, the protonated haloacyl fluorides were not observed in the solutions even at low temperatures. Accordingly, the substitution of the CCl2H moiety for the CHF2 moiety represents the limit that allows the isolation of electron-deficient protonated haloacetyl fluorides from the binary superacidic system HF/SbF5.
In addition to the calculations relating to the HF-addition, quantum chemical calculations were performed to evaluate the stability of the oxonium species 1. As observed in the crystallographic section, the solid-state phase of 1 is strongly influenced by O(–H)⋯F and C(–H)⋯F hydrogen bonds. Interestingly, the optimization of the free cation [CHF2CF2OH2]+ in the gas phase as an energetic minimum was only possible with a strongly extended C–O bond length compared to the crystal structure of 1. Only the addition of HF molecules to the cation in the gas phase to simulate the strong hydrogen bonds enabled the optimization with an appropriate C–O bond length. This indicates that the intermolecular contacts have a significant influence on the bond lengths of the cation of 1 as well as on the stabilization of the compound in the solid-state phase. To further investigate this, a Hirshfeld surface analysis37,38 of the intermolecular contacts was performed for the cation of 1. In Fig. 5, the mapped Hirshfeld surface with dnorm of the [CHF2CF2OH2]+ cation in 1 is shown, whereas the strong hydrogen bonds O1(–H1)⋯F6i (2.471(3) Å) and O1(–H2)⋯F5 (2.452(3) Å) as well as the weak hydrogen bond C2(–H3)⋯F7ii (3.129(4) Å) are highlighted. The 2D fingerprint plots of the intermolecular contacts are depicted in the ESI† (see Fig. S18).
 | ||
| Fig. 5 Interatomic contacts and Hirshfeld surface of the [CHF2CF2OH2]+ cation in 1 (mapped with dnorm).37,38 Color coding of the Hirshfeld surface: white (distance d equals VDW), blue (d exceeds VDW distance), and red (d is smaller than VDW distance). For the hydrogen bond C2(–H3)⋯F7ii, only the contacting fluorine atom is shown for a better visualization. Symmetry codes: i = −1 + x, y, z; ii = 1 − x, 1 − y, 1 − z. | ||
As illustrated, the O(–H)⋯F and C(–H)⋯F hydrogen bonds are the predominant intermolecular contacts in the coordination sphere of the cation in 1, with their distances being below the sum of their van der Waals radii (2.99 Å, 3.17 Å).29 This is confirmed by the 2D fingerprint plots of the intermolecular contacts (see Fig. S18, ESI†). Thus, although the F⋯F contacts occur more often in the coordination sphere of the cation in 1 in terms of area, they have a significantly smaller influence on the structure and stability of the cation, due to their distances being equal or exceeding the van der Waals radii (2.94 Å).29 The H⋯F contacts occur less than the F⋯F contacts in terms of area, but are stronger and thus more relevant for the crystal structure of 1.
Conclusions
The reactivity of difluoroacetyl fluoride and trifluoroacetyl fluoride was investigated in aHF and the binary superacidic system HF/SbF5. As indicated by low-temperature NMR spectroscopy, the haloacetyl fluorides form equilibria with their corresponding α-fluoroalcohols in aHF. While the acyl fluorides are the predominant species in aHF at low temperatures, the addition of the strong Lewis acid SbF5 enables the shift of the equilibria to the side of the α-fluoroalcohols and the stabilization of these compounds as oxonium salts. Protonated 1,1,2,2-tetrafluoroethanol was isolated as a solid and characterized by low-temperature vibrational spectroscopy as well as single-crystal X-ray diffraction. Protonated perfluoroethanol is only stable in solution. Quantum chemical calculations reveal that the substitution of the CCl2H moiety for the CHF2 and CF3 moieties leads to a significant increase in the electrophilicity of the protonated acyl fluorides and represents the limit that allows the isolation of electron-deficient protonated acyl fluorides from HF/SbF5. The O(–H)⋯F and C(–H)⋯F hydrogen bonds have a significant influence on the structure and stability of protonated tetrafluoroethanol, as demonstrated by Hirshfeld surface analysis of the intermolecular contacts.Data availability
The data supporting this article have been included as part of the ESI.† For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details. Crystallographic data for [CHF2CF2OH2][SbF6] has been deposited at the CCDC under CCDC 2312629† and can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Department of Chemistry of the Ludwig Maximilian University, the Deutsche Forschungsgemeinschaft (DFG), and F-Select GmbH for the financial support of this work. Special thanks go to Prof. Dr Konstantin Karaghiosoff for the help with this work after Prof. Dr Andreas J. Kornath passed away.References
- T. W. Bentley and P. V. R. Schleyer, Adv. Phys. Org. Chem., 1977, 14, 1 CrossRef CAS.
- W. J. Middleton and R. V. Lindsey, J. Am. Chem. Soc., 1964, 86, 4948 CrossRef CAS.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490 CrossRef CAS PubMed.
- T. J. Barbarich, S. T. Handy, S. M. Miller, O. P. Anderson, P. A. Grieco and S. H. Strauss, Organometallics, 1996, 15, 3776 CrossRef CAS.
- J. J. Rockwell, G. M. Kloster, W. J. DuBay, P. A. Grieco, D. F. Shriver and S. H. Strauss, Inorg. Chim. Acta, 1997, 263, 195 CrossRef CAS.
- U. P. Preiss, G. Steinfeld, H. Scherer, A. M. T. Erle, B. Benkmil, A. Kraft and I. Krossing, Z. Anorg. Allg. Chem., 2013, 639, 714 CrossRef CAS.
- W. A. Sheppard and C. M. Sharts, Organic Fluorine Chemistry, W. A. Benjamin, Inc., New York, 1969 Search PubMed.
- M. T. Nguyen, M. H. Matus, V. T. Ngan, R. Haiges, K. O. Christe and D. A. Dixon, J. Phys. Chem. A, 2008, 112, 1298 CrossRef CAS PubMed.
- K. Seppelt, Angew. Chem., Int. Ed. Engl., 1977, 16, 322 CrossRef.
- K. O. Christe, J. Hegge, B. Hoge and R. Haiges, Angew. Chem., Int. Ed., 2007, 46, 6155 CrossRef CAS.
- S. Andreades and D. C. England, J. Am. Chem. Soc., 1961, 83, 4670 CrossRef CAS.
- A. F. Baxter, J. Schaab, K. O. Christe and R. Haiges, Angew. Chem., Int. Ed., 2018, 57, 8174 CrossRef CAS.
- A. F. Baxter, J. Schaab, J. Hegge, T. Saal, M. Vasiliu, D. A. Dixon, R. Haiges and K. O. Christe, Chem. – Eur. J., 2018, 24, 16737 CrossRef CAS.
- S. Steiner, C. Jessen and A. J. Kornath, Z. Anorg. Allg. Chem., 2022, 648, e202200060 CrossRef CAS.
- S. Steiner, A. Nitzer, C. Jessen and A. J. Kornath, Z. Anorg. Allg. Chem., 2024, 650, e202400013 CrossRef CAS.
- P. A. W. Dean and R. J. Gillespie, J. Am. Chem. Soc., 1969, 91(26), 7260 CrossRef CAS.
- J. Weidlein, U. Müller and K. Dehnicke, Schwingungsspektroskopie. Eine Einführung, Thieme, Stuttgart, 1988 Search PubMed.
- J. R. Durig, G. A. Guirgis and T. A. Mohamed, J. Mol. Struct., 1998, 444, 165 CrossRef CAS.
- R. Minkwitz and S. Reinemann, Z. Anorg. Allg. Chem., 1999, 625, 121 CrossRef CAS.
- R. Minkwitz and V. Gerhard, Z. Anorg. Allg. Chem., 1991, 603, 95 CrossRef CAS.
- B. P. van Eijck, J. Mol. Struct., 1977, 37, 1 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- R. Minkwitz and S. Schneider, Z. Anorg. Allg. Chem., 1998, 624, 1989 CrossRef CAS.
- A. Karmakar, L. M. D. R. S. Martins, M. F. C. G. da Silva, S. Hazra and A. J. L. Pombeiro, Catal. Lett., 2015, 145, 2066 CrossRef CAS.
- R. Minkwitz, N. Hartfeld and C. Hirsch, Z. Anorg. Allg. Chem., 1999, 625, 1479 CrossRef CAS.
- R. Minkwitz and S. Schneider, Angew. Chem., Int. Ed., 1999, 38, 210 CrossRef CAS.
- R. Minkwitz, C. Hirsch and T. Berends, Eur. J. Inorg. Chem., 1999,(12), 2249 CrossRef CAS.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, 1997 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Klene, J. E. Know, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. O. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzweski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian16, Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- T. Soltner, N. Goetz and A. J. Kornath, Eur. J. Inorg. Chem., 2011, 5429 Search PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- G. Frenking, W. Koch and H. Schwarz, J. Comput. Chem., 1986, 7(4), 406 CrossRef CAS.
- G. A. Olah, G. Liang and Y. K. Mo, J. Org. Chem., 1974, 39(16), 2394 CrossRef CAS.
- M. Bayer, C. Kremser, C. Jessen, A. Nitzer and A. J. Kornath, Chem. – Eur. J., 2022, 28(15), e202104422 CrossRef CAS PubMed.
- A. J. Fernandes, A. Panossian, B. Michelet, A. Martin-Mingot, F. R. Leroux and S. Thibaudeau, Beilstein J. Org. Chem., 2021, 17, 343 CrossRef CAS PubMed.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer 17.5, Revision: f4e298a, University of Western Australia, 2017 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: For full details on vibrational spectroscopy, NMR spectroscopy, X-ray diffraction refinement, and computational details. CCDC 2312629. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj02884h |
| ‡ Prof. Dr Andreas J. Kornath passed away in March 2024. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |