
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational studies of ring cleavage and deoxygenation reactions of a σ3λ3-oxaphosphirane†
Antonio
García Alcaraz
 a,
Arturo
Espinosa Ferao
*a and
Rainer
Streubel
*b
a,
Arturo
Espinosa Ferao
*a and
Rainer
Streubel
*b
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Murcia, Campus de Espinardo, 30071, Spain. E-mail: artuesp@um.es
bIntitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität Bonn, Gerhard-Domagk-Str. 1, 53121 Bonn, Germany. E-mail: r.streubel@uni-bonn.de
First published on 24th September 2024
Abstract
The thermal stability of a σ3λ3-oxaphosphirane is computationally explored with regard to their ring-opening reactions. The possibility of deoxygenation by trivalent phosphorus reagents, leading to phosphaalkenes, is found to be disfavoured by direct attack of P to O but, instead, the initial C-attack is kinetically preferred leading to a 1,2,4-oxadiphosphetane via the corresponding betaine. Epimerization of this four-membered ring, from the initial erythro to the most stable threo diastereomeric pair, enables [2+2] cycloreversion to the most stable final E-phosphaalkene as the final deoxygenation product.
Introduction
The structure and high reactivity of three-membered rings have fascinated both experimental and theoretical chemists over the years.1 These qualities have led to the synthesis and characterisation of numerous small-sized organic2 and inorganic3 heterocycles in recent decades. The high reactivity and instability of these species arises mainly from the high ring strain energy (RSE), which is key, for example, in ring-opening polymerisations of oxiranes and other small-sized rings,4 and in ring-opening reactions.5 Oxiranes (epoxides)6 are of paramount importance in molecular organic and polymer chemistry, but only one example of the heavier three-membered ring analogues oxasiliranes I7 (Fig. 1) was reported so far. Similarly, oxaziridines8 are versatile organic reagents, but their heavier analogues, σ3λ3-oxaphosphiranes II are almost unexplored even though, owing to their extraordinary structural and electronic flexibility, organophosphorus compounds are highly versatile reagents with a fundamental role in today's synthetic chemistry.9 Part of their structural versatility may be considered to derive from their diagonal analogy in the periodic table with carbon, which led to the coining of the expression ‘Phosphorus: the carbon copy’.10 On the contrary, P-ligated versions of oxaphosphiranes III11 are firmly established since the early discover by Mathey using epoxidation of phosphaalkene P-complexes,12 the phosphinidene complex transfer reaction to carbonyls13 and the most recent and convenient protocol using Li/Cl-phosphinidenoid complexes.14 Also, σ5λ5-oxaphosphiranes IV derivatives have been known for a long time as unstable intermediates in the reaction of alkyl phosphites or phosphanes with carbonyl compounds, earlier proposed by Perkow,15 Ramirez,16 Mironov17 and, theoretically, later by Espinosa Ferao.18 Only in one case, reported by Barrans,19 a 31P-NMR signal at −25 ppm was attributed to a type IV intermediate, but without providing further analytical data. Even a σ4λ5-oxaphosphirane P-imide V was prepared from iminophosphanes and fluorinated ketones.13a Recently, the first example of an unligated σ3λ3-oxaphosphirane II was prepared by decomplexation of a κP-pentacarbonylmolybdenum(0) complex III using bis(diphenylphosphino)ethane (DPPE), and only adduct formation with borane and various reactions under oxidative conditions were explored.20 Even more recently, the decomplexation was most conveniently achieved with N-methyl imidazole and a wider set of reactions thoroughly studied showing an interesting reactivity feature of the P fragment behaving as a formal singlet phosphinidene, albeit being still part of the ring.21 | ||
| Fig. 1 Heavier group 14 and 15 three-membered rings, including those with higher coordination at phosphorus; external lines denote organic substituents. | ||
Several preliminary theoretical studies on oxaphosphiranes have already highlighted the ease of P-complexation22 and their high ring strain energy (RSE).23 Indeed, strained small heterocycles are prone to undergoing ring-opening reactions by the action of external initiators, as occurs at the heart of oxirane chemistry6 and as has also been shown recently with very simple model oxaphosphiranes.23b The latter have revealed their potential to also undergo closed-shell isomerisation reactions by breaking one of their endocyclic bonds, with preferential cleavage of the endocyclic C–O bond over the P–C bond.24 In addition, the recent report on the PZ3 reagent-promoted deoxidation of oxiranes to alkenes25 makes oxaphosphiranes an interesting potential source of phosphaalkenes.
Herein, theoretical results on endocyclic bond cleavage processes and reactions of an unligated 3,5-difluorophenyl-substituted oxaphosphirane towards P(III) reagents are reported.
Results and discussion
A simplified model of a potentially accessible derivative bearing a tert-butyl group as a P-substituent and the experimentally used 3,5-difluorophenyl group as a C-substituent21 (1) was chosen to explore the thermal stability of the ring by looking at the cleavage processes of the three endocyclic bonds (Scheme 1). The cleavage of the C–O bond exergonically produced the open-chain σ3λ5-isomer phosphaalkene P-oxide 2 in a kinetically favoured process (Fig. 2). Phosphaalkene P-oxides (alkylidene(oxo)phosphoranes) are phospha-analogues of nitrones26 (imine N-oxides), that are useful intermediates in chemical synthesis and formally derived from C–O cleavage in the much more strained oxaziridine rings (computed RSE = 26.4 kcal mol−1 for the parent ring).27 This isomer 2 is stable towards dissociation (after intersystem crossing, ISC) into the triplet carbene 3t and tert-butyl phosphinidene oxide 4. In contrast, cleavage of the P–C bond gave rise to the zwitterionic species 5 in an endergonic process with a moderately high activation barrier (34.5 kcal mol−1). Although not experimentally observed so far in free oxaphosphiranes, the related C–O and P–C bond cleavage processes constitute the initial step in many reactions displayed by their κ-P-complexes.28 The less favoured ring opening process was found to be the P–O bond cleavage leading to the endergonic dissociation into aldehyde 6 and triplet state phosphinidene 7t (Fig. 2), with high intersystem crossing energy. The first step is very much related to the observed singlet phosphinidene-like reactivity of the oxaphosphirane P fragment.21 These results show that oxaphosphirane 1 is stable towards the cleavage of its P–C and P–O bonds, but the most labile C–O bond only displays significant kinetic protection (ΔG‡ = 29.4 kcal mol−1).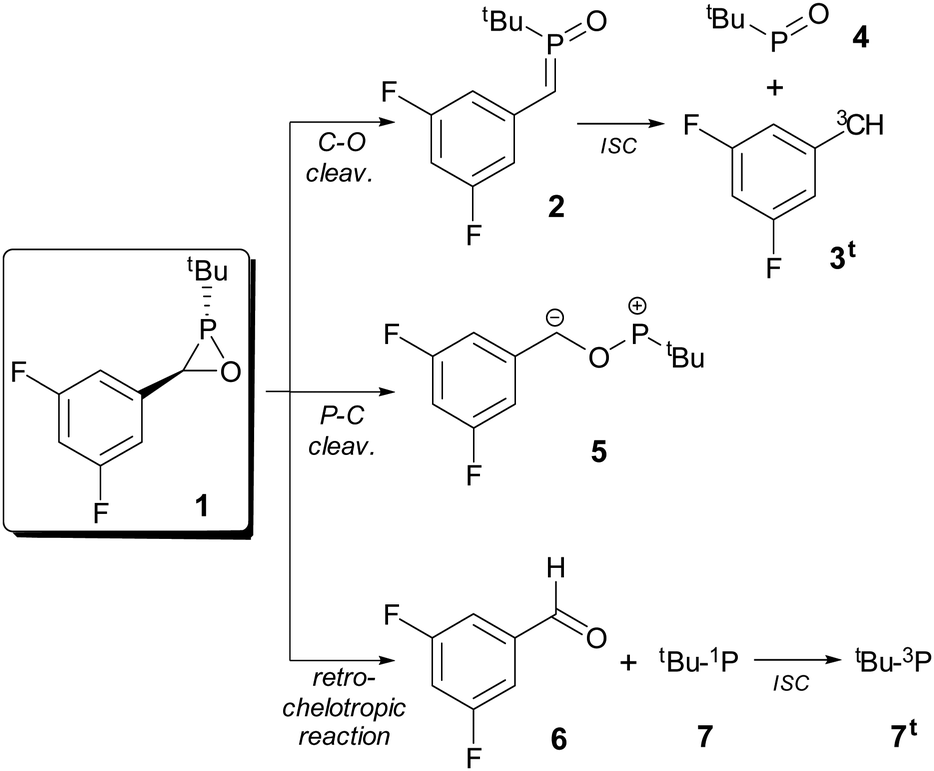 | ||
| Scheme 1 Studied ring-opening reactions from oxaphosphirane 1. | ||
 | ||
| Fig. 2 Computed (CPCM(tol)/PWPB95-D3/def2-QZVPP) relative Gibbs free energy profile for endocyclic bond cleavage processes in 1. | ||
Next, the reaction of 1 with different trivalent phosphorus compounds (PMe3, P(OMe)3 and P(NMe2)3) 8a–c was explored. First, the abstraction of the O atom from oxaphosphirane 1 by the P(III) reagent was studied, leading to the formation of phosphane oxides 9 and phosphaalkene 10E, owing to the assumed trans stereochemistry of the starting oxaphosphirane (Scheme 2). According to the very recent published work on the deoxygenation of oxiranes,25 it seems reasonable to posit that this reaction occurs by direct nucleophilic attack of the phosphorus centre of the PZ3 reagent to the O atom of oxaphosphirane 1. The values reported in the aforementioned work on ![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) hermodynamic
hermodynamic ![[O with combining low line]](https://www.rsc.org/images/entities/i_char_004f_0332.gif) xygen atom transfer
xygen atom transfer ![[P with combining low line]](https://www.rsc.org/images/entities/i_char_0050_0332.gif) otentials (TOPH, the superscript ‘H’ referring to the use of enthalpies for energy evaluation) allow for predictions. Here, three PZ3 reagents were tested, PMe38a (TOPH = −100.1 kcal mol−1), P(OMe)38b (TOPH = −113.9 kcal mol−1), and P(NMe2)38c (TOPH = −111.1 kcal mol−1), and they should be able to deoxygenate oxaphosphiranes, which are the oxidized species of the redox couple with the corresponding phosphaalkene (TOPH = −63.7 kcal mol−1 reported for 2,2,2-trimethyloxaphosphirane).27,29 Indeed, at the calculation level used in this work (CPCM(tol)/PWPB95-D3/def2-QZVPP), a TOPH of −73.1 kcal mol−1 has been estimated for the 10E/1 redox pair and −99.6, −109.0 and −110.1 kcal mol−1 for the 8/9 redox pairs for PMe3, P(OMe)3 and P(NMe2)3, respectively, versus the H2O/H2O2 pair used as a reference. Consistent with these TOPH oxophilicity parameters, and the difference ΔTOPH = −26.5, −35.9 and −37.1 kcal mol−1 for PMe3, P(OMe)3 and P(NMe2)3, respectively, markedly exergonic values have been obtained for the deoxygenation of 1 (ΔG = −31.8, −38.7 and −40.8 kcal mol−1, respectively), with very energetic transition states, the kinetically most favourable case being that of PMe3 (Scheme 2). These results demonstrate that the use of the phosphorus compounds PMe3, P(OMe)3 and P(NMe2)3 to form phosphaalkenes from oxaphosphiranes could be experimentally feasible, but only under very harsh conditions.
otentials (TOPH, the superscript ‘H’ referring to the use of enthalpies for energy evaluation) allow for predictions. Here, three PZ3 reagents were tested, PMe38a (TOPH = −100.1 kcal mol−1), P(OMe)38b (TOPH = −113.9 kcal mol−1), and P(NMe2)38c (TOPH = −111.1 kcal mol−1), and they should be able to deoxygenate oxaphosphiranes, which are the oxidized species of the redox couple with the corresponding phosphaalkene (TOPH = −63.7 kcal mol−1 reported for 2,2,2-trimethyloxaphosphirane).27,29 Indeed, at the calculation level used in this work (CPCM(tol)/PWPB95-D3/def2-QZVPP), a TOPH of −73.1 kcal mol−1 has been estimated for the 10E/1 redox pair and −99.6, −109.0 and −110.1 kcal mol−1 for the 8/9 redox pairs for PMe3, P(OMe)3 and P(NMe2)3, respectively, versus the H2O/H2O2 pair used as a reference. Consistent with these TOPH oxophilicity parameters, and the difference ΔTOPH = −26.5, −35.9 and −37.1 kcal mol−1 for PMe3, P(OMe)3 and P(NMe2)3, respectively, markedly exergonic values have been obtained for the deoxygenation of 1 (ΔG = −31.8, −38.7 and −40.8 kcal mol−1, respectively), with very energetic transition states, the kinetically most favourable case being that of PMe3 (Scheme 2). These results demonstrate that the use of the phosphorus compounds PMe3, P(OMe)3 and P(NMe2)3 to form phosphaalkenes from oxaphosphiranes could be experimentally feasible, but only under very harsh conditions.
 | ||
| Scheme 2 Computed (CPCM(tol)/PWPB95-D3/def2-QZVPP) pathways in the reaction of 1 with phosphanes 8. Relative Gibbs free energies (kcal mol−1) in square brackets and colour coded according to the phosphane used: blue (8a), red (8b) and green (8c). | ||
Approach of the P(III) reagent 8 to oxaphosphirane 1 takes places by interacting the basic P centre of 8 with one of the two lobes of the LUMO, mostly located at a phosphorus p-type atomic orbital, of 1,21 and leading to an encounter complex 1·8.
Taking PMe3 as a case in point, its interaction with 1 can be predicted using DFT-rooted parameters30 related with Pearson theory of hard and soft acids and bases (HSAB).31 The (local) quadratic difference in softness (ΔS)2 is much lower for the interaction with the ring P atom (0.14) than with the O (1.62) or C (1.96) ring atoms. Also, the initial interaction energy ΔEint, resulting from two-step sequential chemical potential-equalization and reshuffling of the charge distribution,32 points to a most favourable initial interaction with P over O and C (−87.1, −42.5 and −40.9 kcal mol−1, respectively). Indeed, the attack of 8a to the O atom of 1 to promote deoxygenation was found to required formation of the encounter complex 1·8a(O) with rather exothermic P⋯P interaction (ΔEZPE = −23.6 kcal mol−1, ΔGrel = 8.4 kcal mol−1). However, another similar most stable encounter complex 1·8a(C) was found at the C-side of the ring (ΔEZPE = −25.7 kcal mol−1, ΔGrel = 7.1 kcal mol−1). Both initial species 1·8a display a P⋯P pattern corresponding to a van der Waals complex according to a recent definition,33 on the basis of the small positive value of Laplacian of the electron density at the bond critical points (BCPs) and the relative position of the donor (PD) and acceptor (PA) valence-shell charge concentration (VSCC) bands at both sides of the BCP (Fig. 3).33b
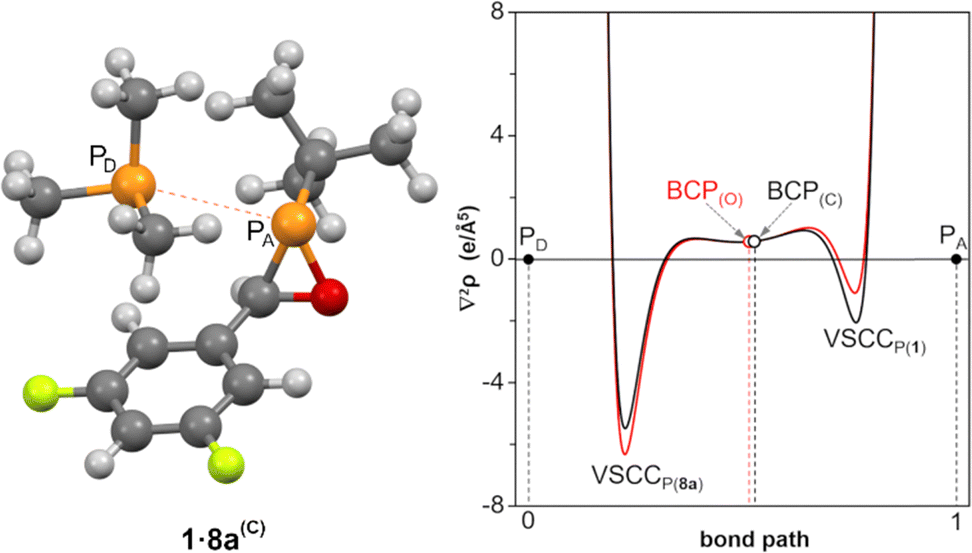 | ||
| Fig. 3 Computed (B3LYP/def2-TZVPP//CPCM(tol)/PBEh-3c) structure of van der Waals complex 1·8a(C) and variation of the Laplacian of the electron density along the P⋯P bond path for 1·8a(C) (black) and 1·8a(O) (red). | ||
The alternative reaction of the phosphorus attack of the PZ3 reagent 8 at the ring C atom of the oxaphosphirane 1 starts from the van der Waals complex 1·8(C) and gives rise to the zwitterionic species (betaines) 11ery (Scheme 2). Compared to O-abstraction, this process is thermodynamically less favourable, although the activation barriers are noticeably smaller, making them the kinetic products of reactions with PZ3. These betaines can cyclise to the corresponding 1,2,4-oxadiphosphetanes 12ery through a process with low activation barrier, which in turn, may undergo a [2+2] cycloreversion producing the elimination of the phosphane oxide O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PZ3 (9) and leading exergonically to a phosphaalkene, the product of the oxaphosphirane 1 deoxygenation, but now with the opposite Z-configuration (10Z). In the case of oxiranes, also the attack of PZ3 to the O atom (deoxygenation) is less favoured than to the ring C atom but, in the latter case, carbene transfer with the elimination of the carbonyl component (PZ3 → R2C = PZ3) usually occurs due to the low stability of the betaine C–C bond.25 The other possible [2+2] cycloreversion in erythro 1,2,4-oxadiphosphetanes 12ery leads to phosphorane 13 and phosphinidene oxide 4. A variety of [2+2] cycloreversion reactions in related 1,2-oxaphosphetanes with the P centre in different oxidation states and coordination numbers have recently been reported,34 the σ5λ5-P centre being the most favoured for PO bond formation. Recombination of 13 and 4 through the opposite molecular face in one of the reagents would give rise to the diastereomeric 1,2,4-oxadiphosphetane 12thr, hence opening the path to phosphaalkene 10E by loss of phosphane oxide 9 (Scheme 2). All herein studied [2+2] cycloreversion reactions of 1,2,4-oxadiphosphetanes 12 affording phosphorane 13 and phosphinidene oxide 4 were found to be barrierless processes.
PZ3 (9) and leading exergonically to a phosphaalkene, the product of the oxaphosphirane 1 deoxygenation, but now with the opposite Z-configuration (10Z). In the case of oxiranes, also the attack of PZ3 to the O atom (deoxygenation) is less favoured than to the ring C atom but, in the latter case, carbene transfer with the elimination of the carbonyl component (PZ3 → R2C = PZ3) usually occurs due to the low stability of the betaine C–C bond.25 The other possible [2+2] cycloreversion in erythro 1,2,4-oxadiphosphetanes 12ery leads to phosphorane 13 and phosphinidene oxide 4. A variety of [2+2] cycloreversion reactions in related 1,2-oxaphosphetanes with the P centre in different oxidation states and coordination numbers have recently been reported,34 the σ5λ5-P centre being the most favoured for PO bond formation. Recombination of 13 and 4 through the opposite molecular face in one of the reagents would give rise to the diastereomeric 1,2,4-oxadiphosphetane 12thr, hence opening the path to phosphaalkene 10E by loss of phosphane oxide 9 (Scheme 2). All herein studied [2+2] cycloreversion reactions of 1,2,4-oxadiphosphetanes 12 affording phosphorane 13 and phosphinidene oxide 4 were found to be barrierless processes.
Using the reaction of 1 with PMe3 (8a) as a case in point, the latter behaves as a nucleophile as demonstrated by the (Mulliken) electric charge transferred from the phosphane to the oxaphosphirane in the TS for both the O- (ΔqM = 0.31e) and C-attack (ΔqM = 0.25e). The initial betaine formation (1 + 8a → 11aery) is the rate-determining step, all subsequent steps having lower activation barriers (Fig. 4) which would lead to the formation of the most stable E-phosphaalkene (10E). The Z-isomer 10Z would be formed somewhat faster, following a slightly lower energy profile, but the activation energy for the reverse reaction (10Z + 9a → 12aery) is almost identical to that of the initial step (Fig. 4). Hence, in the required thermal regime all the subsequent steps could reach equilibrium and thus favour the overall preferential formation of the more stable phosphaalkene 10E. As expected, cleavage of both diastereomeric 1,2,4-oxadiphosphetanes (12a) to the corresponding phosphaalkene (10) occurs through less stable intermediately formed isomers with the ring O atom in the equatorial position around the trigonal bipyramidal σ5λ5-P atom (12a−eq). Worth mentioning is that the regioselectivity of the [2+2] cycloaddition of 13a and 4 can be explained in terms of the lower quadratic difference in local softness (2.99) compared to the other (not shown) interaction that would lead to regioisomer 1,2,3-oxadiphosphetane (6.47).
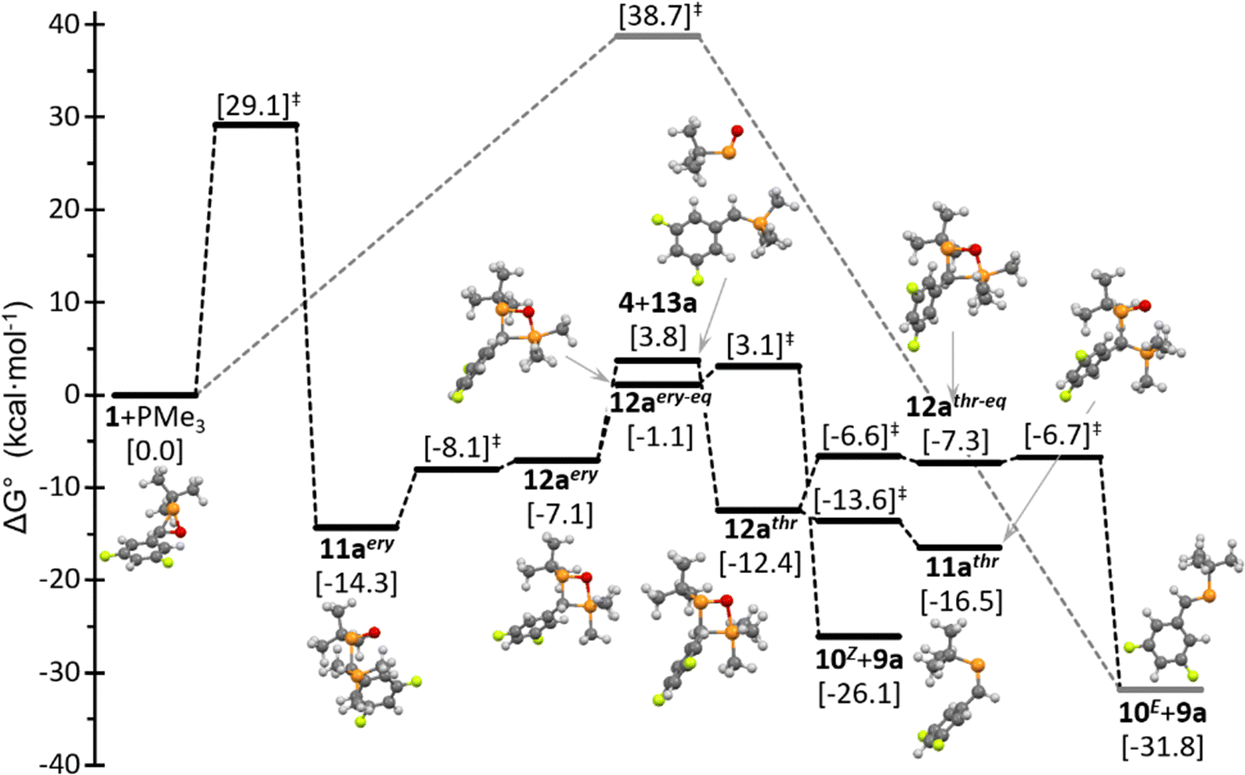 | ||
| Fig. 4 Computed (CPCM(tol)/PWPB95-D3/def2-QZVPP) relative free Gibbs energy profile for the reaction of 1 with PMe3. | ||
In the case of the reaction of P(OMe)3 (8b) with 1 (Fig. 5), although the potential energy surface is qualitatively similar, excepting the higher barrier for the initial rate-determining step, formation of the Z-phosphaalkene from the initially produced 1,2,4-oxadiphosphetanes (12bery) is kinetically disfavoured compared to the alternative [2+2] cycloreversion affording 13b and 4, which in turn opens the lower energy pathway finally leading to the most stable E-phosphaalkene 10E (Fig. 5). No O-equatorial isomers were found for these 1,2,4-oxadiphosphetanes 12b, but three different rotamers for the P-methoxy substituents were located for each diastereomer.
 | ||
| Fig. 5 Computed (CPCM(tol)/PWPB95-D3/def2-QZVPP) relative free Gibbs energy profile for the reaction of 1 with P(OMe)3. | ||
Finally, when the phosphorus reagent is P(NMe2)3, the activation energies for both possible pathways are in between those of the reactions with PMe3 and P(OMe)3 and following the same preference for the attack to the ring C atom over the O atom attack (Fig. 6). In this case, the initially formed erythro 1,2,4-oxadiphosphetane 12cery is rather destabilized and is converted into the most stable threo isomer 12cthr through two consecutive barrierless [2+2] cycloreversion and [2+2] cycloaddition steps, hence enabling the preferential formation of the most stable E-phosphaalkene 10E (Fig. 6).
 | ||
| Fig. 6 Computed (CPCM(tol)/PWPB95-D3/def2-QZVPP) relative free Gibbs energy profile for the reaction of 1 with P(NMe2)3. | ||
Moreover, the reductive potential reported for the unsubstituted σ3λ3-oxaphosphirane itself (TOPH = −90.6 kcal mol−1),25 to be converted into the corresponding P-oxide, must be sufficient to deoxygenate the oxaphosphirane ring (vide supra). Indeed, at the working level of theory, the TOPH for the 1/14 pair is −93.8 kcal mol−1 and the disproportionation of two molecules of 1 to give 10E and the corresponding oxaphosphirane P-oxide 14 was computed to be exergonic by ΔG = −23.8 kcal mol−1.
Computational methods
DFT calculations were performed with the ORCA electronic structure program package (version 4.2.1).35 All geometry optimizations were run in redundant internal coordinates with tight convergence criteria, in the gas phase and using Grimme's dispersion-corrected composite PBEh-3c level.36 For the mechanistic studies (i.e., excluding TOPH evaluations that were computed in the gas phase), solvent (toluene) effects were taken into consideration with the CPCM solvation method,37 as implemented in ORCA. Harmonic frequency calculations at the optimization level were used to obtain thermal correction contributions and verified the nature of ground or transition states (TS) having all positive frequencies or only one imaginary frequency, respectively. TS structures were further confirmed by following the intrinsic reaction path in both directions of the negative eigenvector. Final energies were obtained by means of single-point energy calculations with the double-hybrid-meta-GGA functional PWPB95,38 the Grimme's semiempirical D3 correction39 and the def2-QZVPP basis set,40 using the RI-JK approximation.41 The topological analysis was performed with the electron density at B3LYP/def2-TZVPP and using the Multiwfn program.42Conclusions
The stability of the σ3λ3-oxaphosphirane ring system towards thermal ring cleavage was theoretically explored for trans-3-(3,5-difluorophenyl)-2-tert-butyl-oxaphosphirane. Only the C–O bond cleavage is slightly exergonic (ΔG = −9.6 kcal mol−1) and proceeds over a moderate barrier (ΔG‡ = 29.4 kcal mol−1).Reaction of this derivative with P(III)-reagents leading to a deoxygenation (P → O attack) is always kinetically disfavoured (by 7.3–9.6 kcal mol−1). The preferred P → C attack is the rate-limiting step of the stepwise, alternative deoxygenation involving betaine formation, and a ring-closure to give an erythro-1,2,4-oxadiphosphetane, while [2+2]-cycloreversion affords the less stable Z-configured phosphaalkene. The stable E-phosphaalkene derivative can be easily accessed through the barrierless alternative oxadiphosphetane [2+2]-cycloreversion which opens the path to the [2+2]-cycloaddition to yield the threo diastereomer. In the thermal conditions required for overcoming the first rate-limiting step, the equilibrium conditions should favour the formation of the most stable E-phosphaalkene as the major final product. Further mechanistic investigations on the thermodynamically favoured oxaphosphirane disproportionation leading to a phosphaalkene and an oxaphosphirane P-oxide are currently ongoing.
Author contributions
A. E. F. and R. S. are responsible for the conceptualization. A. G. A. and A. E. F. performed all calculations, data analysis and wrote the ESI.† All authors contributed to writing the manuscript from the first draft.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the computational resources used at the computation centre at Servicio de Cálculo Científico (SCC – University of Murcia).Notes and references
- (a) D. Cremer and E. Kraka, J. Am. Chem. Soc., 1985, 107, 3800–3810 CrossRef; (b) J. A. Boatz and M. S. Gordon, J. Phys. Chem., 1989, 93, 3025–3029 CrossRef CAS; (c) A. Skancke, D. Van Vechten, J. F. Liebman and P. N. Skancke, J. Mol. Struct., 1996, 376, 461–468 CrossRef CAS; (d) M. Alcamí, O. Mó and M. Yáñez, J. Comput. Chem., 1998, 19, 1072–1086 CrossRef; (e) T. P. M. Goumans, A. W. Ehlers, K. Lammertsma and E.-U. Würthwein, Eur. J. Org. Chem., 2003, 2941–2946 CrossRef CAS.
- A. Hassner, The Chemistry of Heterocyclic Compounds, Small Ring Heterocycles, Wiley, 2009 Search PubMed.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- (a) P. Kubisa, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 457–468 CrossRef CAS; (b) I. C. Stewart, C. C. Lee, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 17616–17617 CrossRef CAS PubMed.
- C.-Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed.
- (a) A. Wurtz, Comptes Rendus, 1859, 48, 101–105 Search PubMed; (b) L. G. Wade Jr. and J. W. Simek, Organic Chemistry, Pearson Education Ltd, Essex, England, 9th edn, 2017 Search PubMed.
- D. Gau, R. Rodriguez, T. Kato, N. Saffon-Merceron, F. P. Cossío and A. Baceiredo, Chem. – Eur. J., 2010, 16, 8255–8258 CrossRef CAS PubMed.
- K. S. Williamson, D. J. Michaelis and T. P. Yoon, Chem. Rev., 2014, 114, 8016–8036 CrossRef CAS.
- (a) Organophosphorus Reagents in Organic Synthesis, ed. J. I. G. Cadogan, Academic Press, London, New York, 1979 Search PubMed; (b) R. Engel, Handbook of Organophosphorus Chemistry, CRC Press, 1992 Search PubMed; (c) P. Savignac and B. Iorga, Modern Phosphonate Chemistry, CRC Press, 2003 CrossRef; (d) P. J. Murphy, Organophosphorus Reagents: A Practical Approach in Chemistry, Practical Approach in Chemistry, 2004 CrossRef.
- K. B. Dillon, F. Mathey and J. F. Nixon, Phosphorus: The Carbon Copy. From Organophosphorus to Phospha-organic Chemistry, Wiley, 1998 Search PubMed.
- N. Volk, P. Malik, A. García Alcaraz, A. Espinosa Ferao and R. Streubel, Coord. Chem. Rev., 2021, 437, 213818 CrossRef CAS.
- S. Bauer, A. Marinetti, L. Ricard and F. Mathey, Angew. Chem., Int. Ed., 1990, 29, 1166–1167 CrossRef.
- (a) G.-V. Röschenthaler, K. Sauerbrey and R. Schmutzler, Chem. Ber., 1978, 111, 3105–3111 CrossRef; (b) E. Niecke, D. Gudat, W. W. Schoeller and P. Rademacher, J. Chem. Soc., Chem. Commun., 1985, 1050–1051 RSC; (c) F. U. Seifert and G.-V. Röschenthaler, Z. Naturforsch., B: Chem. Sci., 1993, 48, 1089–1093 CrossRef CAS; (d) R. Streubel, A. Kusenberg, J. Jeske and P. G. Jones, Angew. Chem., Int. Ed. Engl., 1995, 33, 2427–2428 CrossRef; (e) R. Streubel, N. Volk, G. Schnakenburg, A. García Alcaraz and A. Espinosa Ferao, Eur. J. Inorg. Chem., 2021, 252–257 CrossRef.
- A. Schmer, P. Junker, A. Espinosa Ferao and R. Streubel, Acc. Chem. Res., 2021, 54, 1754–1765 CrossRef PubMed.
- (a) W. Perkow, K. Ullerich and F. Meyer, Naturwissenschaften, 1952, 39, 353 CrossRef; (b) W. Perkow, Chem. Ber., 1954, 87, 755–758 CrossRef.
- F. Ramirez, P. C. Smith, J. F. Pilot and A. S. Gulati, J. Org. Chem., 1968, 33, 3787–3794 CrossRef.
- (a) R. M. Aminova, V. F. Mironov, M. E. Filatov and L. I. Savostina, Zh. Obshch. Khim., 1996, 66, 1042 CAS; (b) R. M. Aminova, V. F. Mironov and M. E. Filatov, Zh. Obshch. Khim., 1999, 69, 58 Search PubMed; (c) R. M. Aminova, G. A. Shamov, V. N. Mushkin, V. F. Mironov, A. I. Konovalov and A. V. Aganov, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 147, 265 CrossRef.
- (a) A. Espinosa Ferao, J. Phys. Chem. A, 2017, 121, 6517–6522 CrossRef CAS; (b) A. Espinosa Ferao, Inorg. Chem., 2018, 57(14), 8058–8064 CrossRef CAS.
- M. T. Boisdon and J. Barrans, J. Chem. Soc., Chem. Commun., 1988, 615–617 RSC.
- J. Faßbender, N. Volk, A. García Alcaraz, S. Balasubramaniam, A. Espinosa Ferao and R. Streubel, Chem. Commun., 2023, 59(10), 1285–1288 RSC.
- N. Volk, A. García Alcaraz, S. Balasubramaniam, Y. Heumann, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Chem. Sci. Search PubMed , submitted (ID SC-EDG-09-2024-006185).
- A. Espinosa, E. de las Heras and R. Streubel, Inorg. Chem., 2014, 53, 6132–6140 CrossRef CAS PubMed.
- (a) O. Krahe, F. Neese and R. Streubel, Chem. – Eur. J., 2009, 15, 2594–2601 CrossRef CAS; (b) A. Espinosa Ferao, A. Rey Planells and R. Streubel, Eur. J. Inorg. Chem., 2021, 348–353 CrossRef CAS.
- A. Espinosa Ferao and R. Streubel, Eur. J. Inorg. Chem., 2017, 2707–2712 CrossRef CAS.
- A. Espinosa Ferao, ChemPlusChem, 2024, 89, e202300474 CrossRef CAS.
- (a) J. Hamer and A. Macaluso, Chem. Rev., 1964, 64(4), 473–495 CrossRef CAS; (b) G. R. Delpierre and M. Lamchen, Chem. Soc. Rev., 1965, 19(4), 329–348 RSC.
- A. Rey Planells and A. Espinosa Ferao, Chem. – Eur. J., 2023, 29, e202302243 CrossRef PubMed.
- (a) R. Streubel, C. Murcia-García, G. Schnakenburg and A. Espinosa Ferao, Organometallics, 2015, 34, 2676–2682 CrossRef CAS; (b) M. Klein, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Dalton Trans., 2016, 45, 2085–2094 RSC; (c) C. Murcia García, A. Espinosa Ferao, G. Schnakenburg and R. Streubel, Dalton Trans., 2016, 45, 2378–2385 RSC; (d) P. Malik, A. Espinosa Ferao, G. Schnakenburg and R. Streubel, Angew. Chem., Int. Ed., 2016, 55, 12693–12697 CrossRef CAS PubMed; (e) J. Faßbender, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Dalton Trans., 2018, 47, 9347–9354 RSC.
- It should be noted that the above displayed values for PMe3 and P(NMe2)3 do not correspond to those previously reported (TOPH = −97.0 and −43.2 kcal mol−1, respectively) and, therefore, were recalculated in the present work.
- (a) R. G. Parr and W. Yang, in Density-functional theory of atoms and molecules, Oxford University Press, New York, 1989 Search PubMed; (b) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef; (c) J. L. Gázquez, in Chemical Hardness, ed. K. D. Sen, Struct. Bonding, 1993, 80, pp. 27–43 Search PubMed.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef; (b) T. L. Ho, Chem. Rev., 1975, 75, 1–20 CrossRef.
- (a) P. K. Chattaraj, H. Lee and R. G. Parr, J. Am. Chem. Soc., 1991, 113, 1855–1856 CrossRef; (b) R. G. Parr and J. L. Gázquez, J. Phys. Chem., 1993, 97, 3939–3940 CrossRef.
- (a) M. W. Stanford, J. I. Schweizer, M. Menche, G. S. Nichol, M. C. Holthausen and M. J. Cowley, Angew. Chem., Int. Ed., 2019, 58, 1329 CrossRef PubMed; (b) A. Espinosa Ferao, A. García Alcaraz, S. Zaragoza Noguera and R. Streubel, Inorg. Chem., 2020, 59, 12829 Search PubMed; (c) V. Nesterov, R. Baierl, F. Hanusch, A. Espinosa Ferao and S. Inoue, J. Am. Chem. Soc., 2019, 141, 14576 Search PubMed.
- A. W. Kyri, F. Gleim, A. García Alcaraz, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Chem. Commun., 2018, 54, 7123–7126 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 Search PubMed.
- S. Grimme, J. G. Brandenburg, C. Bannwarth and A. Hansen, J. Chem. Phys., 2015, 143, 54107 CrossRef PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef.
- (a) L. Goerigk and S. Grimme, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef PubMed; (b) L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, J. Comput. Chem., 2008, 29, 167–175 CrossRef CAS PubMed.
- Multiwfn, v. 3.8 CrossRef CAS; T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS Website: https://sobereva.com/multiwfn/.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates and energies for all computed species. See DOI: https://doi.org/10.1039/d4nj02773f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |