
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-based core@shell catalysts for guaiacol hydro conversion: use of salicylic acid as a sacrificial modulator of the interaction between the metal phase and the silica support†
Diego A.
Aguirre-Abarca‡
 abc,
César
Pazo-Carballo
abc,
Dorothée
Laurenti
*d,
Alejandro
Karelovic
ce,
Ana B.
Dongil
f and
Néstor
Escalona
*abcg
abc,
César
Pazo-Carballo
abc,
Dorothée
Laurenti
*d,
Alejandro
Karelovic
ce,
Ana B.
Dongil
f and
Néstor
Escalona
*abcg
aDepartamento de Química Física, Facultad de Química y de Farmacia, Pontificia Universidad Católica de Chile, Avenida Vicuña Mackenna 4860, Macul, Santiago, Chile
bDepartamento de Ingeniería Química y Bioprocesos, Pontificia Universidad Católica de Chile, Avenida Vicuña Mackenna 4860, Macul, Santiago, Chile
cANID - Millennium Science Initiative Program – Millennium Nuclei on Catalytic Processes towards Sustainable Chemistry (CSC), Chile
dIRCELYON, UMR 5256 CNRS, Université Lyon 1, 2 Avenue A. Einstein, Villeurbanne Cedex 69626, France. E-mail: dorothee.laurenti@ircelyon.univ-lyon1.fr
eCarbon and Catalysis Laboratory (CarboCat), Department of Chemical Engineering, University of Concepción, Concepción, Chile
fInstituto de Catálisis y Petroleoquímica, CSIC, C/Marie Curie, 2 L10, Cantoblanco, Madrid 28049, Spain
gCentro de Investigación en Nanotecnología y Materiales CIEN-UC, Pontificia Universidad Católica de Chile, Avenida Vicuña Mackenna 4860, Macul, Santiago, Chile. E-mail: neescalona@uc.cl
First published on 18th July 2024
Abstract
In this work, four cobalt-based catalysts were obtained using two routes of preparation which differed in the expected degree of interaction with the silica matrix, using salicylic acid as a sacrificial modifier during the synthesis to modulate the interaction between the cobalt phase and the silica support due to its complexing properties. The catalysts were tested for the hydroconversion of guaiacol at 300 °C and 5 MPa of H2, with clear differences between each procedure. It is found that the synthesis route has a deep impact on the catalytic activity, with the catalysts prepared using a one-pot route being less active and having an increased selectivity to cyclohexane. In contrast, the ones prepared using a two-step process have increased yield towards phenol and cyclohexanol under the conditions used herein. Salicylic acid increases the catalytic activity in comparison to the untreated material for both families of materials, without changing the observed selectivity between samples with the same preparation.
Introduction
There is an increasing concern about the consequences of the rising levels of atmospheric CO2 caused by fossil oil consumption, with climate change caused by global warming being the most noticeable. An option to lessen our dependence on oil is to use the residues of the agroindustry as a substitute feedstock for some of the growing demands of the transportation and chemical industries. A rather mature approach for converting agroindustry residues using a fast pyrolysis process, which transforms those residues into a liquid called bio-oil. However, the bio-oil thus obtained requires further upgrading, due to its low chemical stability, low heating value, and corrosiveness, requiring a hydrodeoxygenation (HDO) process1–4 for its further use.There have been many studies with various catalytic systems for this upgrading process. Among them, alumina-supported CoMoS and NiMoS catalysts, used in the oil hydrodesulfurization process, have been extensively studied. While those catalysts are quite active, they tend to deactivate in the presence of nitrogen-containing compounds, high amounts of water, or due to sulfide loss from the active phase.5–8 Still, some formulations based on supported CoMoS and NiMoS phases over zirconia are stable under 100 ppm of H2S in the feed, with a high yield of benzenoid products9,10 with either pure guaiacol or a mixed feed of guaiacol and straight run gas oil (SRGO).11 Considering these limitations, catalysts like supported metals, metal phosphides, or carbides have been studied. For example, Ni2P over SiO2 allows for the obtention of phenol and benzene from the hydrotreating of guaiacol,12 and in more acidic support it leads to the obtention of catechol.13 Mo2C supported over Mxene nano sheets leads to the obtention of phenol and methyl anisole14 or phenolic compounds when supported over activated carbon.15 Systems based in either Ni or Ir supported on ZrO2 show high selectivity towards cyclohexanol coupled with gasification of guaiacol as methane and ethane.16 Rh-based catalysts supported on zirconia yield mainly cyclohexane,17,18 while a Ru-based catalyst produces cyclohexanol as the major product of the HDO of guaiacol.19 Finally, systems supported over activated carbon containing a mixture of Pd and Re allow benzene and cyclohexene to be obtained in high yields from the hydroconversion of guaiacol,20 and a ReOx catalyst has also exhibited yields of BTX up to 30% in the conversion of guaiacol over different supports and metal loadings.21
Core@shell materials based on non-noble metals are good options for converting guaiacol or other biomass-derived model compounds since the presence of the shell allows for increased stability and a certain extent of tunability of the catalysts’ activity. For example, using a MOF shell over Pd for the conversion of vanillin to produce creosol,22 or the use of MOF as the precursor of the shell with a ZnO/Co for the same reaction23 have been disclosed. Also, the conversion of vanillin to 2-methoxy-4-methyl phenol in ethanol using a Ni2P-based system with a yolk structure has been described.24 Finally, catalytic systems for the conversion of glucose to 5-hydroxymethylfurfural with a magnetic core25 and the conversion of octanoic acid to alkanes in the presence of a hollow@Ni/ZrO2 system have been reported.26
The usage of organic or inorganic modifiers as a tool for tuning the activity of catalysts has been explored in the formulation of hydrotreating catalysts, including sulfur-containing additives as well as polyhydroxy alcohols, used as promoting agents for the activation procedure, protectors towards loss of active phase27 and dispersion agents for the reactivation of CoMo sulfide-based catalysts.28 Furthermore, the usage of carboxylic acids as selectivity-tuning agents for aerobic ammoxidation29 as well as modifiers of supports like alumina30 also uses phosphonic acids for tuning of Pt and Pd-based catalysts for the conversion of benzyl alcohol and vanillin.31,32 However, the potential of such additives is still underexplored, especially their usage as sacrificial templates for tuning the active phase. Herein, two different families of catalysts based on Co were prepared using a one-pot or a two-step approach, with or without salicylic acid as a sacrificial modifier. Their catalytic performance was studied using the hydrodeoxygenation of guaiacol as a bio-oil model compound, and the results suggest that this approach could enable the obtention of catalysts with fine-tuned activity and selectivity.
Experimental section
Synthesis of the catalysts
The procedure was partially adapted from 33,33–36 using tetraethyl orthosilicate (TEOS, Sigma 98,0%) as the silica source; all procedures were carried out in the fume hood. The materials used for the synthesis were used as received from the chemical supplier, except salicylic acid which was purified by recrystallization in a mixture of ethanol and deionized water. Briefly, a solution of Co2+ in deionized water at 0.1 mole L−1 was precipitated with concentrated ammonia. The obtained solid was separated, washed thrice with deionized water, and dried. The green solid was then calcined and used to prepare the core@shell materials using the two-step route. The calcined material was suspended using ultrasound in a mixture of ethanol, water, and concentrated ammonia (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:3 by volume). The suspension was heated to 60 °C, and then the TEOS (Co:Si 1:1.2 mole:mole) dissolved in ethanol was added to the suspension with stirring and kept at 60 °C for 4 hours. The solids were centrifuged, washed with water and ethanol, and air-dried overnight. The materials thus prepared were then calcined in air at 250 °C (ramp 10 °C min−1) for 2 hours per gram of material; afterward, the material was heated to 550 °C under a 100 mL min−1 flow of pure hydrogen and kept at this temperature for 2 hours. Finally, the sample was passivated in a liquid nitrogen/isopropanol slurry with a flow of 60 mL min−1 of 5% O2 in N2 for 90 minutes for storage. The material was identified as Co@SiO2-2S. The modified catalyst was prepared by treating the calcined cobalt oxide with a small amount of aqueous Co2+ (1 mL per gram of cobalt oxide of a 1% by weight solution of Co(NO3)2·6H2O, Merck, 99%) for 1 hour. Then, salicylic acid (Co:salicylic acid 1:0.064 mole:mole) dissolved in 10 mL of ethanol was added to this mixture, and it was left to equilibrate overnight before creating a suspension via ultrasound and TEOS treatment. The catalyst was identified as Co@SiO2-SA-2S.
:20:3 by volume). The suspension was heated to 60 °C, and then the TEOS (Co:Si 1:1.2 mole:mole) dissolved in ethanol was added to the suspension with stirring and kept at 60 °C for 4 hours. The solids were centrifuged, washed with water and ethanol, and air-dried overnight. The materials thus prepared were then calcined in air at 250 °C (ramp 10 °C min−1) for 2 hours per gram of material; afterward, the material was heated to 550 °C under a 100 mL min−1 flow of pure hydrogen and kept at this temperature for 2 hours. Finally, the sample was passivated in a liquid nitrogen/isopropanol slurry with a flow of 60 mL min−1 of 5% O2 in N2 for 90 minutes for storage. The material was identified as Co@SiO2-2S. The modified catalyst was prepared by treating the calcined cobalt oxide with a small amount of aqueous Co2+ (1 mL per gram of cobalt oxide of a 1% by weight solution of Co(NO3)2·6H2O, Merck, 99%) for 1 hour. Then, salicylic acid (Co:salicylic acid 1:0.064 mole:mole) dissolved in 10 mL of ethanol was added to this mixture, and it was left to equilibrate overnight before creating a suspension via ultrasound and TEOS treatment. The catalyst was identified as Co@SiO2-SA-2S.
Two other catalysts, Co@SiO2-1S and Co@SiO2-SA-1S were synthesized using a one-pot route. The cobalt precursor was dissolved in a mixture of deionized water and ethanol (8:2 by volume) and then the concentrated ammonia (25%, Loba Chemie) was added to precipitate the hydroxide. The obtained solid was suspended in the mother liquor by ultrasound for 30 minutes. The suspension was heated to 60 °C and the TEOS (1:1.2 Co:Si mole to mole) dissolved in ethanol was added and left to react for 4 hours. The precursor thus obtained was calcined in air at 250 °C (ramp 10 °C min−1) for 2 hours per gram of material; afterward, the material was heated to 550 °C under a 100 mL min−1 flow of pure hydrogen and kept at said temperature reduced at 550 °C for 2 hours. Finally, the sample was passivated in a liquid nitrogen/isopropanol slurry with a flow of 60 mL min−1 of 5% O2 in N2 for 90 minutes for storage. The modification with salicylic acid was achieved by adding the desired amount of the acid (Co:Si 1:0.064 mole:mole) to the precipitated mixture and allowing it to stand overnight before the treatment with ultrasound.
Characterization of the catalysts
The nitrogen adsorption–desorption isotherms, TPR-H2, TPD-NH3, and chemisorption of CO were measured using Micromeritics 3Flex equipment. For the TPR-H2 and TPD-NH3, MS signals of relevant compounds were measured.N2-physisorption
Before analysis, the samples were degassed under vacuum at 90 °C for 30 minutes to remove water and held at 300 °C for 4 hours. The physisorption isotherms were measured at the temperature of the liquid nitrogen using N2 as the adsorbate. The SBET estimation was done in the range P/P0 from 0.05 to 0.25 and the pore volume was determined at P/P0 = 0.99.CO chemisorption analysis
The samples were evacuated at 110 °C for 30 minutes, reduced at 550 °C under flowing H2 (60.0 mL min−1) for 90 minutes, and then allowed to reach the analysis temperature (35 °C) before the analysis. Then, the CO was admitted in 50.0 mm Hg increments from 0 to 450 mm Hg to obtain a first isotherm. Afterwards, vacuum was applied, and a second isotherm was measured using the same dosing. The CO uptake was determined from the difference between the first and second isotherms, fitted between 100 to 450 mm Hg.TPR-H2 analysis
The sample was first treated with 5% H2 in an Ar mixture, with a flow of 100 mL min−1 at 50 °C for 30 minutes. After this treatment, the baseline was allowed to stabilize and the TPR-H2 analysis was carried out using a 5% H2 in Ar mixture, with a flow of 100 mL min−1 and a heating ramp of 10 °C min−1 up to 1050 °C. The effluent was analyzed using a TCD detector and a mass spectrometer.TPD-NH3 analysis
The samples were degassed under flowing He (50 mL min−1) with a heating ramp of 10 °C min−1 up to a final temperature of 200 °C for 30 minutes. Then, the samples were cooled to 100 °C under He and a flow of NH3 (40 mL min−1) was passed over the sample for 15 minutes. The weakly adsorbed species were removed by flowing He (100 mL min−1) until a stable baseline was obtained. Then the sample was heated at a rate of 10 °C min−1 up to 500 °C, and the TCD and MS signals were recorded.Transmission electronic microscopy (TEM)
The TEM images were acquired using a JEOL JEM-1200 EX II, by dispersing the sample in either ethanol or acetone and putting a drop on a graphite-covered copper grid for analysis.XPS spectra measurement
XPS spectra were collected with a SPECS GmbH with UHV system, energy analyzer PHOIBOS 150 9MCD under a non-monochromatic X-ray source of Mg (200W, 1253.6eV) for the Co@SiO2-2S and Co@SiO2-SA-2S. For the samples, Co@SiO2-1S and Co@SiO2-SA-1S, a KRATOS ultra DLD with an Al monochromatic source (225 W, 1486.6 eV) was used for the measurement. The spectral data for each sample were analyzed using CASA XPS software. The binding energy (BE) was referenced to the C 1s peak at 284.6 eV. The equipment error in the energy determinations is less than 0.01 eV. The resulting spectra were deconvoluted and fitted to a mix of Gaussian and Lorentzian curves.
FTIR-ATR spectra
The samples were dried at 80 °C overnight and kept in a nitrogen box until analyzed. The FTIR-ATR spectra were collected with an Agilent Cary 630 FTIR spectrometer equipped with an ATR accessory; the range of measure was from 650 to 4000 cm−1 with a resolution of 4 cm−1.XRD diffractometry
The powder X-ray diffractograms were acquired using a Bruker D8 Advance diffractometer with a LYNXEYE-XE-T detector from 2 theta from 4.0° to 80.0° and electron discrimination of 0.18–0.25 V with λ = 0.154 nm; the phase identification was achieved by comparison against the PDF-4+ 2021 database with the DIFFRAC EVA software.Elemental analysis
The samples were digested with a mixture of HF and HNO3, evaporated to dryness, and redissolved with a HNO3 H2SO4 mixture. The total cobalt content was measured using ICP-OES in a Jobin Yvon “Activa” apparatus, with a measuring wavelength λ = 228.62 nm.Catalytic activity tests
In a 100 mL stainless steel vessel (Reactor PARR 4646) 100 mg of catalyst was added, and a solution of 1.7 g of guaiacol (MERCK, 99%) in 32 g of n-dodecane (MERCK, 99%) with hexadecane (Aldrich, 99%) as an internal standard for quantification of products by GC. The reactor was closed, purged with N2 for 15 minutes, closed, and heated to 300 °C. At this temperature, hydrogen gas (INDURA; 99.999%) was charged to a pressure of 50 bar, and liquid samples were collected at definite intervals. The samples were analyzed by GC, using a Shimadzu 2030 GC system equipped with an autosampler, a capillary column (Perkin-Elmer Elite 1, 0.53 mm ID, 30 m length, and dimethylpolysiloxane as stationary phase), nitrogen as a carrier gas and FID detector. The volume analyzed for each sample was 1 µL.The conversion and yield of a product are determined using the following expressions:
 | (1) |
 | (2) |
The initial rate of reaction, r0GUA, and TOF were determined according to the following formulas:
 | (3) |
 | (4) |
Results and discussion
Characterization of materials
Table 1 summarizes the textural properties of the prepared materials, and Fig. S1 (ESI†) shows the nitrogen adsorption–desorption isotherms of the catalysts reported. The main feature for all the samples is the characteristic Type IV isotherm linked to the presence of mesoporous materials, and all of them exhibit the same type of hysteresis, an H3 loop, that is related to plate-like particles or to a network of macropores partially filled with nitrogen.37 For the Co@SiO2-2S catalyst, the SBET diminished in comparison with the parent oxide nanoparticle, as shown by the data in Table 1. From the method of obtention of the precursor for the two-step approach, the porosity could be attributed to the interstitial voids between nonporous oxide particles, in concordance with reports of similar systems involving cobalt oxide phases.38,39 Thus, the lowering of the surface is caused by the growth of the silica shell over those particles that reduce the interparticle space. Linked to this, the formed silica layer has a different pore structure, leading to the difference observed in the isotherms. However, for the Co@SiO2-SA-2S catalyst, there is a slight increment of the surface area in comparison with the parent oxide, as well as a slightly bigger pore volume in comparison with the untreated catalyst.Regarding the catalysts Co@SiO2-1S and Co@SiO2-SA-1S, there is an increased surface area compared to the catalysts prepared using the two-step route, and the one with the highest surface area is the salicylic acid sample, Co@SiO2-SA-1S. In this case, the presence of the hydrated cobalt hydroxide leads to the formation of a gel where water is occluded inside the bulk of the sample. This leads to an increment in the porosity after the thermal treatment due to the loss of water, with the subsequent increase in the pore volume. In contrast, the use of salicylic acid in the Co@SiO2-SA-1S material leads to a rise in the surface area linked with a diminished pore volume. Thus, the formation of a layer of adsorbed acid over the surface of the hydrated hydroxide leads to a local hydrophobic environment due to the presence of the aromatic ring moiety; this enhances the cross-linking between the silica oligomer,40–42 leading to a more compact silica matrix, with fewer pores.
The normalized TCD signal for the temperature-programmed reduction with H2 (TPR-H2) profiles is shown in Fig. 1, and the MS signal spectra are included in the ESI† (Fig. S2). For the Co@SiO2-SA-2S material, there is a low-temperature signal linked to the production of CO2 and H2O from the decomposition of remnants of the salicylic acid used.43 Afterward, there is a smaller signal related to the formation of Co3O4 from the CoO(OH) species, then a sharp signal related to the reduction of Co3O4 and CoO species to Co around 300 °C.44 Finally, the reduction of Co2+ species with strong interaction with the silica shell appears as a broad signal around 700 °C.45,46 In the Co@SiO2-2S catalyst, there are two signals, one centered around 230 °C and the other at around 690 °C. The signal at low temperature has been related to the reduction of CoO(OH) to Co3O446 according to eqn (1), with subsequent reduction to CoO and Co. The broad high-temperature signal is related to Co2+ as a cobalt silicate species.45,47
| 6CoO(OH) + H2 → 2Co3O4 + 4H2O | (5) |
 | ||
| Fig. 1 TPR-H2 profiles of the prepared catalysts; TCD signal normalized per gram of cobalt in the samples. | ||
For the materials prepared using the one-pot procedure, there is a higher proportion of strong-interacting silica–cobalt species, with a more intense signal around 700 °C for both materials. Thus, the one-pot approach increases the interactions between the silica matrix and the cobalt phase; adding salicylic acid reduces this interaction, reflected as a lower reduction temperature for the peak at around 700 °C, independently of the preparation method. Finally, the degree of reduction was determined for the Co@SiO2-2S and Co@SiO2-SA-2S samples; the calculations and data are given in Table S1 (ESI†); it is observed that the salicylic acid treatment enhanced the reducibility, since for the Co@SiO2-2S, the reducibility was around 10%, whereas, for the Co@SiO2-SA-2S sample, it raised to 60%.
To analyze the changes in the morphology, the obtained catalysts were observed using TEM, and the results are shown in Fig. 1. Fig. 2A and B show the materials prepared using a one-step route, where the morphology obtained is a mixture of quasi-spherical particles and needle-like particles. Herein the formation of strong interacting cobalt species, similar to cobalt silicate species is expected due to a higher interaction between the silica precursor oligomers and the Co2+ and Co(OH)2 species, a situation reported for similar reaction systems.48,49 For the salicylic acid catalyst, the needle-like structures are blurred, pointing to an impaired formation of the cobalt silicate by the presence of the salicylic acid. Fig. 2C and D show the images of the catalysts prepared using a two-step procedure, and the presence of a distinct silica shell covering the cobalt phase for the catalysts prepared using the two-step route is clear. The catalyst Co@SiO2-2S-SA shows a slight separation between the cobalt phase and the silica shell; this space is derived from the layer of salicylic acid over the oxide nanoparticles that inhibits the coverage of the cobalt core by the silica. This layer is destroyed during the thermal treatment leaving behind a void, raising the surface area and pore volume, as observed in the data from Table 1.
 | ||
| Fig. 2 TEM images of Co@SiO2-1S (A) and Co@SiO2-SA-1S (B) catalysts Co@SiO2-2S (C) and Co@SiO2-SA-2S (D) catalysts. | ||
Table 2 shows the data obtained for the cobalt and oxygen species from the XPS measurements, and fitted spectra are given in Fig. 3. Treatment with salicylic acid increases the percentage of Co0 species on the surface of the Co@SiO2-SA-2S material (Fig. 3A) compared to the Co@SiO2-2S sample (Fig. 3B). There is a significant shift of the BE for the Co2+ signal in the Co@SiO2-SA-2S catalyst, coupled with a lowering of its Oox/Co2+ ratio, indicative of a deficiency of oxygen species on the surface of the catalyst. The displacement towards higher BE has been associated with the formation of vacancies in bimetallic CoNi systems50 and enhanced activity. However, there is no measurable presence of surface Co0 species for the catalysts prepared using the one-step route, and only the Co2+ signal is observed (Fig. 3C and D). This indicates the presence of hardly reducible cobalt species, presumably cobalt silicates, which is supported by the TPR-H2 profiles, where the most pronounced signal was the peak related to such silicate species.
| Catalyst | Cobalt species BE, eV | Oxygen species O 1s BE, eV | ||
|---|---|---|---|---|
| Co0 2p3/2 | Co2+ 2p3/2 | CoOx | SiO2 | |
| Co@SiO2-2S | 778.7 (41) | 780.4 (59) | 529.7 (15) | 532.1 (85) |
| Co@SiO2-SA-2S | 779.2 (51) | 781.2 (49) | 529.8 (7) | 531.9 (93) |
| Co@SiO2-1S | N.D. | 781.3 | N.D. | 531.8 |
| Co@SiO2-SA-1S | N.D. | 781.3 | N.D. | 531.8/534.1 |
 | ||
| Fig. 3 XPS spectra of the Co 2p region of the Co@SiO2 catalysts. (A) Co@SiO2-SA-2S, (B) Co@SiO2-2S, (C) Co@SiO2-SA-1S, and (D) Co@SiO2-1S. | ||
Regarding the O 1s data, the materials obtained using the two-step route exhibit the presence of metallic oxide and silica oxygen species, as seen in Fig. S3 and S4 (ESI†). For the materials prepared using the one-step procedure, there is no presence of metallic oxide species; in the Co@SiO2-1S material (Fig. S5, ESI†), only the silica oxygen species related to silicate species in a non-bridging structure are detected.51,52 For the Co@SiO2-SA-1S sample (Fig. S6, ESI†), there is another signal at around 534 eV; related to oxygen atoms of the silica matrix,51–53 and the presence of this signal indicates that the salicylic acid treatment induces local structure modifications of the silica in the proximity of the cobalt phase. It might be possible to argue that this signal is related to carbonate or hydroxy species; however, considering the carbon content in the catalyst, below 0.03%, it is dubious that CO32− species are relevant. For the hydroxyl species, the absence of the Si–OH signal around 950 cm−154 in the catalysts’ samples, rules out the presence of this group; thus, the signal around 534 eV could be related to a Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, due to the increased electronegativity of the sp2 oxygen. Additional evidence of the modification of the interaction between the core and the silica matrix lies in the Si 2s spectra of the samples. In those spectra, there is a two-component signal for the catalyst Co@SiO2-SA-1S (Fig. S7, ESI†), and only one for the Co@SiO2-1S catalyst (Fig. S8, ESI†). This supports the perturbation of the formation of Si–O–Co species by using salicylic acid, observed in the TPR-H2 profiles as a shift of the peak to lower temperature for the Co@SiO2-SA-1S.
O bond, due to the increased electronegativity of the sp2 oxygen. Additional evidence of the modification of the interaction between the core and the silica matrix lies in the Si 2s spectra of the samples. In those spectra, there is a two-component signal for the catalyst Co@SiO2-SA-1S (Fig. S7, ESI†), and only one for the Co@SiO2-1S catalyst (Fig. S8, ESI†). This supports the perturbation of the formation of Si–O–Co species by using salicylic acid, observed in the TPR-H2 profiles as a shift of the peak to lower temperature for the Co@SiO2-SA-1S.
The XRD diffractograms of the prepared catalysts are displayed in Fig. 4A. The main features are the peaks at around 2 Theta = 36, 42, 44, and 76 degrees. The signal at 36° is linked to CoO for the catalysts prepared using the two-step route. In the case of the catalysts made using a one-pot procedure, there is an additional contribution at around 33° that could be related to cobalt silicate-like species. Thus, the precipitated cobalt hydroxide reacts with the silica oligomers to produce some cobalt silicate. At 42°, the reflection for the (200) plane of CoO is responsible for this peak, and at 44° the presence of Co is indicated by the reflection due to the (111) plane, while the peak at 77° is caused by the reflection of the (220) plane. Rietveld's refinement of the XRD patterns allows for better quantification and identification of the different phases present in the material, derived from the Rietveld analysis, where both the hcp and fcc phases of Co were detected. The samples treated with salicylic acid showed an increment of the hcp phase of around 60% compared to the untreated sample, which is an interesting feature, pointing to a potential effect of this modifier over the activity of the materials. The analyzed diffractograms are included in the ESI,† with each assigned phase indicated (Fig. S9, ESI†). The results from Rietveld's analysis are coherent with the TPR-H2 results, where the catalysts prepared using a two-step route show typical signals for the reduction of cobalt oxides, whereas, for the ones prepared using a one-step route, there is no signal of cobalt oxides in the TPR-H2 analysis.
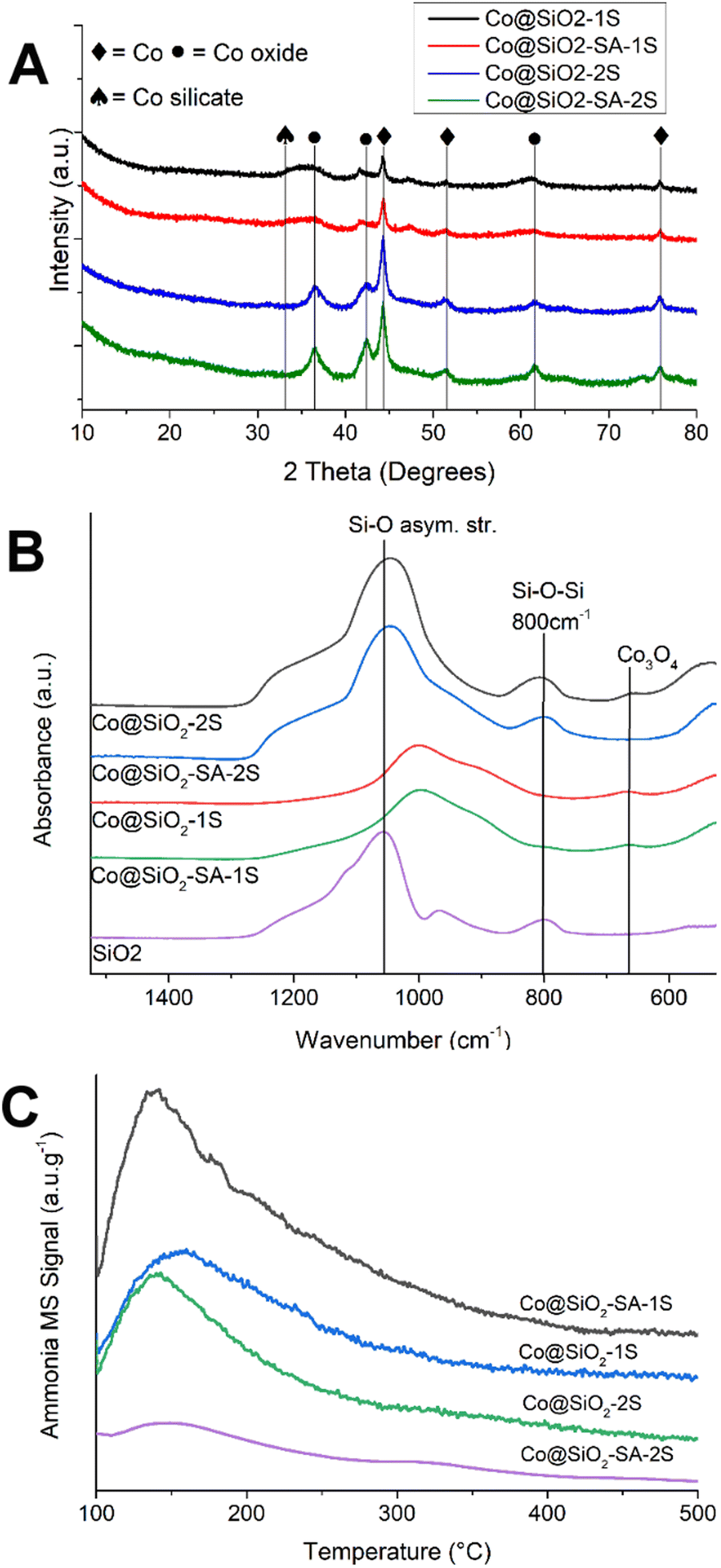 | ||
| Fig. 4 (A) XRD patterns of the passivated catalysts. (B) FTIR-ATR spectrum of the prepared passivated catalysts in the region 1525 to 525 cm−1 (C) TPD-NH3 profiles (signal MS = 17) of the reported catalysts; signals normalized per gram of catalyst. | ||
Further information is obtained from the FTIR spectra, shown in Fig. 4B from 1525 to 650 cm−1, with the spectrum of a silica sample prepared without salicylic acid. The most relevant features are the bands at around 800 cm−1 due to the symmetric stretch of the Si–O–Si bond and the signal around 1100 cm−1, associated with the Si–O asymmetric stretching mode. This latter signal is highly sensitive to modifications in the silica matrix and can give qualitative information about the degree of substitution of the silica, with lower wavenumbers indicating a higher substitution.55–57 The catalysts Co@SiO2-2S and Co@SiO2-SA-2S show a little lowering of this signal compared to the silica sample. In the salicylic acid treated material, there is no shift of the band, supporting the hypothesis of the shielding effect of the cobalt oxide against the silicate formation by the salicylic acid. In the case of the materials prepared using the one-pot procedure, Co@SiO2-1S, and Co@SiO2-SA-1S, there is a shift of the Si–O band of 90 cm−1, indicative of a high degree of substitution of the silica matrix with cobalt ions.55 Also, the signal at 800 cm−1 is weak or negligible, caused by short silica chains with fewer Si–O–Si bonds capable of absorption in this region. For all materials, there is a broad band at 650 cm−1, related to the presence of Co3O4 species.46
Fig. 4C shows the temperature-programmed desorption of NH3 (TPD-NH3) profiles for Co@SiO2 catalysts normalized per gram of catalyst; in all catalysts, there is a wide signal centered around 150 °C, which indicates the presence of weak acidic sites in the materials.58 In the case of the catalysts prepared using the one-pot route, the profiles show little difference between them, with a higher amount of ammonia (mass 17) desorbed from the Co@SiO2-SA-1S evidence of a higher quantity of acid sites in the materials. In the case of the materials prepared using a one-step approach, the formation of either Co–Si–OH species with stronger acidity, or an increase in the amount of silanol groups due to a branching of the silica matrix by the intercalation cobalt cations could result in their higher total acidity. The first proposal is not supported by the TPD-NH3 data, since the maximum for the peak is not shifted significantly (less than 3 °C) between samples. The second hypothesis is supported by the FTIR-ATR and XPS data, which show the strong interaction present in the one-pot route and, for the SA-1S catalyst the presence of different Si–O bonds. Considering the maximum temperature, the weak Bronsted-acid type is dominant in our materials; there is a negligible or not detectable quantity of Lewis-acid sites.59 This leads to a favored demethoxylation to produce phenol, followed by hydrogenation to cyclohexanol with a final dehydration step to yield cyclohexene.
The absence of Co0 species in the XPS data for the catalysts prepared using the one-pot method, coupled with its detection on the XRD pattern of the samples, suggests a limitation of the XPS technique for the analysis where the silica matrix is extended and covers the cobalt phase in a wide range. This behavior can be related to the working depth of the XPS technique, which is around 7 nm. Regarding the different proportion of Co species obtained from Rietveld's refinement, it has been reported that, for the hcp phase, the B5 type sites exhibit a diminished sterically impediment, leading to an incremented TOF for the ammonia decomposition reaction and the Fisher-Tropsch synthesis.60,61 Thus, a rise in the quantity of the hcp phase could increase the activity in the catalysts treated with salicylic acid. In addition, the particle size measured via TEM (Table S3, ESI†) indicates that the catalysts’ particle size is around 20 to 30 nm. In this range, the presence of both fcc and hcp phases of Co are expected to appear in variable proportions.62 Also, for the materials prepared using the two-step route, the reduction of CoO, under our reduction conditions is expected to form a mixture of the Co phases indicated above,63 which could have an increased proportion of the more active Co hcp phase in the salicylic acid-treated material.
Some chemical properties are shown in Table 3, including total acidity and chemisorbed CO. For the samples prepared using the two-step procedure the amount of CO chemisorbed is the same. Considering only Co0 as the adsorption site for CO, a difference in CO uptake between both samples was expected, from the XPS data. Thus, other species of cobalt might be involved, in addition to Co0, in the CO uptake of the catalysts.64,65 The reversible chemisorption of the CO over Co2+ based on a MgO/CoO solid solution has been described,65,66 which explains why materials with undetected surface Co0 can chemisorb this probe molecule. In conclusion, the CO uptake reflects the total active sites, being either Co0 or Co2+ species. For the samples obtained using a one-pot approach, there is a diminished chemisorption of CO on the salicylic acid-treated catalyst; this may be linked to a different coordination mode of CO on the fcc and hcp surfaces of the Co,67,68 thus adsorbing less CO in the hcp phase.
| Catalyst | Cobalt contenta | Co phases presentb | Total acidityc | CO uptaked | Initial ratee | TOF (s−1) |
|---|---|---|---|---|---|---|
| a Mass percentage. b From Rietveld analysis of XRD patterns. c mmol NH3 g−1 cat. d 10−5 moleCO g−1 cat. e 10−4 molGUA s−1 g−1 cat. f As a mixture of fcc and hcp phases; overlapping of cobalt oxide peaks does not allow for separate quantification of the phases. | ||||||
| Co@SiO2-2S | 69.0 | Co 36.3f | 0.510 | 1.3 | 6.4 | 49 |
| CoO 62.7 | ||||||
| Co@SiO2-SA-2S | 59.6 | Co 41.3 | 0.0532 | 1.0 | 9.6 | 96 |
| CoO 58.7 | ||||||
| Co@SiO2-1S | 50.4 | Co (FCC) 80.8 | 0.805 | 2.3 | 1.8 | 7.8 |
| Co (HCP) 19.2 | ||||||
| Co@SiO2-SA-1S | 47.2 | Co (FCC) 68.9 | 1.05 | 1.3 | 1.1 | 8.5 |
| Co (HCP) 31.1 | ||||||
Catalytic activity: conversion of guaiacol
Fig. 5 shows the conversion of guaiacol in the presence of the treated and untreated catalysts for all the samples. As can be observed, both catalysts prepared using the two-step route are highly active in the conversion of guaiacol, while the two prepared using the 1-step route are less active. The initial rates of reaction for all the catalysts as well as their TOF are summarized in Table 3. The main feature is the disparity between initial rates based on the synthetic route: for the materials prepared using a two-step approach, the catalytic activity is around 5 times higher than for the materials synthesized using the 1-step approach. This in turn reflects the fact that the formation of cobalt silicate species, which was enhanced in the 1-step route, is detrimental to the catalytic activity of the materials. | ||
| Fig. 5 Evaluation of the catalytic activity of the Co@SiO2 family of materials. Reaction conditions: 300 °C, 5 MPa H2, and 100 mL of a 0.3 mole per liter guaiacol solution, with a catalyst load of 100 mg in each run. | ||
The treatment with salicylic acid has a marked impact over the initial rate; the catalyst prepared using a two-step route has an increase of the initial rate of around 50%, while, for the material prepared using a 1-step route, there is a slight decrease in the initial rate of the treated catalyst compared with the untreated sample, but a raise of the TOF of almost 50%.
Regarding the selectivity of the catalysts, the primary product obtained is phenol for the conversion of guaiacol in all cases. Afterwards, hydrogenation leads to cyclohexanol/one, followed by dehydration and hydrogenation to yield cyclohexene and cyclohexane. Cyclohexanol remains the main secondary product in the case of the 2S catalysts, while for the 1S catalysts, this compound is converted quickly to cyclohexene and then to cyclohexane. The absence of catechol in the samples is striking, considering that the methyl–O bond is expected to be easier to break than the Ar–O bond, indicating that some phenomenon is inhibiting the detection of catechol during the analyses, presumably the grafting of said compound over the surface of the metal through chelation. A possible drawback of the core@shell structure would be a limited diffusion of the reactants towards the active sites; for the 2S family of materials, the Mears’ and Weisz-Pratter criteria were calculated (data and calculations are given in the ESI,† Table S2) and it was concluded that, for the materials analyzed, there was no diffusion-controlled rate, thus the conversion of guaiacol proceeded in the chemical-control regime. Also, calculations for carbon balance (results are given in the ESI†) indicate that more than 90% of the guaiacol was converted and detected during our catalytic activity tests below 30% of conversion, except for the Co@SiO2-SA-1S material, which has a carbon balance of around 80%; this could be caused by adsorption of the catechol formed during the reaction, limiting the number of products detected, thus lowering the carbon balance.
The effects of the treatment with salicylic acid are related to its chelating ability, which modulates the formation of cobalt silicate, enhances the reducibility of the active phase, and tunes the active sites. This is supported by the XPS data, where the shift of the Co2+ to higher BE for the Co@SiO2-SA-2S indicates the formation of vacancies in the cobalt's phase surface for the two-step synthesis catalyst. Those vacancies increase the activity of Co@SiO2-SA-2S compared to Co@SiO2-2S. For the Co@SiO2-AS-1S catalyst, the increased activity compared to the untreated Co@SiO2-1S is related to the diminished formation of inactive cobalt silicate, instead of modifications of the active surface. Also, the increased presence of Co0 species leads to a higher rate of conversion of phenol into cyclohexanol for the Co@SiO2-SA-2S catalyst compared to the Co@SiO2-2S system. The low acidity of the catalysts, linked with a low quantity of Lewis-acid sites, diminishes the ability of the material to completely deoxygenate the substrate, which favors the production of phenol through the demethoxylation of guaiacol. The produced phenol is hydrogenated to form cyclohexanol, which accumulates due to the low acidity of the materials that limit its conversion to cyclohexene and finally, to cyclohexane. In addition, the increased hcp phase content for the salicylic acid treated catalyst impacts the TOF value, due to the increased activity of the surface sites of the hcp Co phase, leading to an increased activity. Thus, the combination of oxygen vacancies on the surface of the catalyst synthesized, the increased presence of Co0 species, and lower acidity, allow for a higher selectivity towards cyclohexanol for the Co@SiO2-SA-2S sample. Also, the rise in the hcp Co phase leads to a higher activity in the guaiacol conversion for the catalysts modified with salicylic acid, coupled with an enhanced reducibility of the materials treated with salicylic acid, as observed in the TPR-H2 results.
The product distribution at a conversion of 30% for all catalysts is given in Fig. S10 (ESI†). At this conversion, the main product in all cases is phenol, with nearly the same yield, indicating that the first step of the conversion of guaiacol over the obtained catalysts is a demethoxylation reaction. For the catalysts with the highest acidity, namely Co@SiO2-1S, and Co@SiO2-SA-1S, there is also an increased conversion of cyclohexanol into cyclohexene and cyclohexane. Considering the reaction network depicted in Fig. 6, it is expected that the most acidic catalyst, Co@SiO2-SA-1S, will exhibit the highest combined yield of cyclohexene and cyclohexane. This proposal supports the role of the acid sites in the conversion of cyclohexanol into cyclohexene.
 | ||
| Fig. 6 Simplified proposed reaction network for the conversion of guaiacol over the Co@SiO2 catalysts researched in this work. | ||
Conclusion
To conclude, the impact of the synthesis procedure is reflected in three aspects of the obtained catalysts: morphology, acidity, and interaction with the silica matrix. In the case of the morphology of the catalysts, a well-defined core@shell structure is obtained for the materials synthesized using a two-step route while, for the materials prepared for a one-pot route, the morphology comprises a mixture of quasi-spherical particles immersed in an extended matrix of silica. The materials of the 1S family are the most acidic, due to the higher degree of interaction between the cobalt ions and the silica matrix favored by the interaction between the Co(OH)2 and the silicate oligomer species. This rise in acidity is reflected in the higher proportion of cyclohexene and cyclohexane, emphasizing the role played by the acid sites in the conversion of cyclohexanol to cyclohexane. The effect of the salicylic is to block the surface from reacting with silicate ions, reducing the formation of cobalt silicate species; then, the salicylic acid is decomposed, creating unsaturated coordination sites which raise the activity of the catalysts. Also, the usage of salicylic acid as a modifier increases the proportion of the more active hcp Co phase, leading to increased activity of the catalysts for the conversion of guaiacol under the conditions used.Overall, the combination of the synthesis route and the use of salicylic acid as a surface modifier can be regarded as a reasonable strategy to develop catalysts with high activity from inexpensive metals, although in a high load, for the hydrotreatment of biomass derivatives, showcased here for the conversion of guaiacol.
Author contributions
Investigation, methodology, visualization, and draft writing performed by Diego-Alejandro Aguirre-Abarca. XPS analysis measurements and analysis were performed by Ana B. Dongil, and TEM analyses were done by Alejandro Karelovic. Funding acquisition, supervision, and resources were undertaken by Nestor Escalona and Dorothée Laurenti. Rietveld analysis of the samples was carried out by César Pazo. Review and editing were performed by all authors, who also approved the final version of the manuscript.Data availability
The O 1s and Si 2s XPS spectra and Rietveld's analyses supporting this article have been included in the ESI† as Fig. S3 to S9.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding from ANID Chile for the following projects: Fondecyt No. 1220763, Fondequip No. EQM160070, and ANID-ACE210012. Additional funding was provided under the ANID Millennium Science Initiative Program – NCN2021_090. DAAA is grateful for the support received from Universidad de Costa Rica through the scholarship granted by the Office of International Affairs and External Cooperation.Notes and references
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- P. M. Mortensen, J. D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS.
- M. Høj, T. M. H. Dabros, A. D. Jensen, P. A. Jensen, P. M. Mortensen, J.-D. Grunwaldt, J. Gabrielsen and M. Z. Stummann, Prog. Energy Combust. Sci., 2018, 68, 268–309 CrossRef.
- X. Li, G. Chen, C. Liu, W. Ma, B. Yan and J. Zhang, Renewable Sustainable Energy Rev., 2017, 71, 296–308 CrossRef CAS.
- E. Laurent and B. Delmon, J. Catal., 1994, 146(1), 281–291 CrossRef CAS.
- M. Ferrari, R. Maggi, B. Delmon and P. Grange, J. Catal., 2001, 198, 47–55 CrossRef CAS.
- E. Laurent and B. Delmon, Ind. Eng. Chem. Res., 2005, 32, 2516–2524 CrossRef.
- E. Laurent and B. Delmon, Appl. Catal., A, 1994, 109, 97–115 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- V. N. Bui, G. Toussaint, D. Laurenti, C. Mirodatos and C. Geantet, Catal. Today, 2009, 143, 172–178 CrossRef CAS.
- X. Lan, E. J. M. Hensen and T. Weber, Appl. Catal., A, 2018, 550, 57–66 CrossRef CAS.
- S. Ted Oyama, T. Onkawa, A. Takagaki, R. Kikuchi, S. Hosokai, Y. Suzuki and K. K. Bando, Top. Catal., 2015, 58, 201–210 CrossRef CAS.
- E. Blanco, A. Rosenkranz, R. Espinoza-González, V. M. Fuenzalida, Z. Zhang, S. Suárez and N. Escalona, Catal. Commun., 2020, 133, 105833 CrossRef CAS.
- Z. Cai, F. Wang, X. Zhang, R. Ahishakiye, Y. Xie and Y. Shen, Mol. Catal., 2017, 441, 28–34 CrossRef CAS.
- M. Alda-Onggar, P. Mäki-Arvela, A. Aho, I. L. Simakova and D. Y. Murzin, React. Kinet., Mech. Catal., 2019, 126, 737–759 CrossRef CAS.
- Y. He, Y. Bie, J. Lehtonen, R. Liu and J. Cai, Fuel, 2019, 239, 1015–1027 CrossRef CAS.
- Y. He, R. Liu, D. Yellezuome, W. Peng and M. Tabatabaei, Renewable Energy, 2022, 184, 487–497 CrossRef CAS.
- X. Du, W. Liu, X. Zhang, F. Hu, A. J. Ragauskas, W. Mu, H. Ben and Y. Deng, Bioresour. Technol., 2014, 173, 6–10 CrossRef PubMed.
- S. T. Thompson and H. H. Lamb, Appl. Catal., A, 2018, 563, 105–117 CrossRef CAS.
- K. Leiva, R. Garcia, C. Sepulveda, D. Laurenti, C. Geantet, M. Vrinat, J. L. Garcia-Fierro and N. Escalona, Catal. Today, 2017, 296, 228–238 CrossRef CAS.
- V. R. Bakuru, D. Davis and S. B. Kalidindi, Dalton Trans., 2019, 48, 8573–8577 RSC.
- V. Ranaware, D. Verma, R. Insyani, A. Riaz, S. M. Kim and J. Kim, Green Chem., 2019, 21, 1021–1042 RSC.
- G. Li and H. Li, Energy Fuels, 2021, 35, 4158–4168 CrossRef CAS.
- I. Elsayed, M. Mashaly, F. Eltaweel, M. A. Jackson and E. B. Hassan, Fuel, 2018, 221, 407–416 CrossRef CAS.
- H. Chen, Y. Wu, S. Qi, Y. Chen and M. Yang, Appl. Catal., A, 2017, 529, 79–90 CrossRef CAS.
- L. Oliviero, F. Maugé, P. Afanasiev, C. Pedraza-Parra and C. Geantet, Catal. Today, 2021, 377, 3–16 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret and J. M. Basset, J. Am. Chem. Soc., 2001, 123, 2062–2063 CrossRef CAS PubMed.
- X. Jia, J. Ma, F. Xia, Y. Xu, J. Gao and J. Xu, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- T. Roy, M. Corral-Valero, T. Corre, O. Delpoux, G. Pirngruber and G. Lefèvre, Colloids Surf., A, 2022, 634, 127923 CrossRef CAS.
- P. D. Coan, M. B. Griffin, P. N. Ciesielski and J. W. Medlin, J. Catal., 2019, 372, 311–320 CrossRef CAS.
- P. Hao, D. K. Schwartz and J. W. Medlin, Appl. Catal., A, 2018, 561, 1–6 CrossRef CAS.
- X. Yu, J. Zhang, X. Wang, Q. Ma, X. Gao, H. Xia, X. Lai, S. Fan and T. S. Zhao, Appl. Catal., B, 2018, 232, 420–428 CrossRef CAS.
- M. A. Ermakova and D. Y. Ermakov, Appl. Catal., A, 2003, 245, 277–288 CrossRef CAS.
- M. Nasrollahzadeh, Z. Issaabadi, H. Ali Khonakdar and U. Wagenknecht, Sep. Purif. Technol., 2018, 203, 185–192 CrossRef CAS.
- M. Nasrollahzadeh, M. Sajjadi and H. A. Khonakdar, J. Mol. Struct., 2018, 1161, 453–463 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- R. Xu, J. Wang, Q. Li, G. Sun, E. Wang, S. Li, J. Gu and M. Ju, J. Solid State Chem., 2009, 182, 3177–3182 CrossRef CAS.
- H. Che, A. Liu, C. Liu, R. Jiang, Q. Fu, C. Wang, L. Wang, A. Liu, C. Liu, R. Jiang, Q. Fu, C. Wang, L. W. Facile, H. Che, A. Liu, C. Liu, R. Jiang, Q. Fu, C. Wang and L. Wang, Mater. Res. Innovations, 2014, 18, 380–385 CrossRef CAS.
- S. Yang and A. Navrotsky, Chem. Mater., 2004, 16, 3682–3687 CrossRef CAS.
- T. Okubo, T. Miyamoto, A. Otake, M. Suda and A. Tsuchida, Colloid Polym. Sci., 2004, 282, 1341–1346 CrossRef CAS.
- E. J. A. Pope and J. D. Mackenzie, J. Non-Cryst. Solids, 1986, 87, 185–198 CrossRef CAS.
- K. Rissanen, J. Valkonen, P. Kokkonen and M. Leskelä, Acta Chem. Scand., 1987, 21, 299–309 CrossRef.
- J. Ilsemann, A. Straß-Eifert, J. Friedland, L. Kiewidt, J. Thöming, M. Bäumer and R. Güttel, ChemCatChem, 2019, 11, 4884–4893 CrossRef CAS.
- G. Bagnasco, C. Cammarano, M. Turco, S. Esposito, A. Aronne and P. Pernice, Thermochim. Acta, 2008, 471, 51–54 CrossRef CAS.
- C. W. Tang, C. Bin Wang and S. H. Chien, Thermochim. Acta, 2008, 473, 68–73 CrossRef CAS.
- R. Xie, H. Wang, P. Gao, L. Xia, Z. Zhang, T. Zhao and Y. Sun, Appl. Catal., A, 2015, 492, 93–99 CrossRef CAS.
- W. Cheng, F. Rechberger, G. Ilari, H. Ma, W. I. Lin and M. Niederberger, Chem. Sci., 2015, 6, 6908–6915 RSC.
- M. Wolf, E. K. Gibson, E. J. Olivier, J. H. Neethling, C. R. A. Catlow, N. Fischer and M. Claeys, Catal. Today, 2020, 342, 71–78 CrossRef CAS.
- M. Liu, J. Zhang, L. Zheng, G. Fan, L. Yang and F. Li, ACS Sustainable Chem. Eng., 2020, 8, 6075–6089 CrossRef CAS.
- K. N. Dalby, H. W. Nesbitt, V. P. Zakaznova-Herzog and P. L. King, Geochim. Cosmochim. Acta, 2007, 71, 4297–4313 CrossRef CAS.
- A. Mekki, D. Holland, C. F. McConville and M. Salim, J. Non-Cryst. Solids, 1996, 208, 267–276 CrossRef CAS.
- A. Mekki and M. Salim, J. Electron Spectrosc. Relat. Phenom., 1999, 101, 227–232 CrossRef.
- R. Ellerbrock, M. Stein and J. Schaller, Sci. Rep., 2022, 12, 11708 CrossRef CAS PubMed.
- P. Innocenzi, J. Non-Cryst. Solids, 2003, 316, 309–319 CrossRef CAS.
- J. W. De Haan, H. M. van den Bogaert, J. J. Ponjeé and L. J. M. van de Ven, J. Colloid Interface Sci., 1986, 110, 591–600 CrossRef CAS.
- G. Ortega-Zarzosa, C. Araujo-Andrade, M. E. Compeán-Jasso and J. R. Martinez, J. Sol-Gel Sci. Technol., 2002, 24, 23–29 CrossRef CAS.
- I. T. Ghampson, G. Pecchi, J. L. G. Fierro, A. Videla and N. Escalona, Appl. Catal., B, 2017, 208, 60–74 CrossRef CAS.
- P. Yan, M. Meng-Jung Li, E. Kennedy, A. Adesina, G. Zhao, A. Setiaman and M. Stockenhuber, Catal. Sci. Technol., 2020, 10, 810–825 RSC.
- R. Agrawal, P. Phatak and L. Spanu, Catal. Today, 2018, 312, 174–180 CrossRef CAS.
- M. Zybert, A. Tarka, W. Patkowski, H. Ronduda, B. Mierzwa, L. Kępiński and W. Raróg-Pilecka, Catalysts, 2022, 12(10), 1285 CrossRef CAS.
- O. Kitakami, H. Sato, Y. Shimada, F. Sato and M. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 13849–13854 CrossRef CAS.
- L. J. Garces, B. Hincapie, R. Zerger and S. L. Suib, J. Phys. Chem. C, 2015, 119, 5484–5490 CrossRef CAS.
- A. Zecchina, G. Spoto, S. Coluccia and E. Guglielminotti, J. Phys. Chem., 1984, 88, 2575–2581 CrossRef CAS.
- A. Zecchina, G. Spoto, E. Garrone and A. Bossi, J. Phys. Chem., 1984, 88, 2587–2591 CrossRef CAS.
- A. Zecchina, G. Spoto, E. Borello and E. Giamello, J. Phys. Chem., 1984, 88, 2582–2587 CrossRef CAS.
- W. Li, X. Nie, H. Yang, X. Wang, F. Polo-Garzon, Z. Wu, J. Zhu, J. Wang, Y. Liu, C. Shi, C. Song and X. Guo, Appl. Catal., B, 2022, 315, 121529 CrossRef CAS.
- J. X. Liu, H. Y. Su, D. P. Sun, B. Y. Zhang and W. X. Li, J. Am. Chem. Soc., 2013, 135, 16284–16287 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Nitrogen physisorption isotherms, XPS spectra of the O 1s region, Si 2s region, Rietveld's analyses results, isoconversion product distribution, calculations for Mears’, Weisz-Pratter criterion and carbon balance. See DOI: https://doi.org/10.1039/d4nj02689f |
| ‡ Current address: Universidad de Costa Rica, Sede del Atlántico, Ruta 10. Turrialba, Cartago, Costa Rica, 030501. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |