
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of high-entropy oxides derived from metal–organic frameworks and their catalytic performance for total toluene oxidation†
Abid
Hussain
ab,
Yuhua
Zheng
*a,
Qianyu
Wang
ac and
Yanbin
Cui
 *ab
*ab
aState Key Laboratory of Mesoscience and Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: ybcui@ipe.ac.cn; Tel: +861062621607
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Beijing 102249, China
First published on 12th September 2024
Abstract
In this work, a series of high-entropy oxides (HEOs) was successfully prepared by the calcination of identical metal–organic framework (MOF) precursors. Thermal treatment above 400 °C led to the complete transformation of MOFs into HEOs. The performance of the obtained HEO catalysts in the removal of volatile organic compounds (VOCs) was studied with the catalytic oxidation of toluene as the probe reaction. The optimized Ce-HEO-400 catalyst showed impressive activity and stability over toluene catalytic oxidation, which resulted from the vast quantity of surface oxygen vacancies and the relatively variable metal valence. The T50 and T90 values of Ce-HEO-400 were 180 °C and 255 °C, respectively. Moreover, it also showed excellent stability for 120 h with a 98.5% toluene conversion rate at 260 °C. Combined with the characterizations of XRD, SEM, TEM, BET, H2-TPR, and XPS, it was found that the high dispersion of the active high-entropy structure and the synergistic effects of different metals are the key factors for improving the catalytic performance of the Ce-HEO-400 catalyst. This work not only presents a facile method for synthesizing HEO materials at low temperatures but also provides valuable technical support for the application of HEO catalysts in VOC removal. The findings of this study open up new possibilities for the development of efficient catalysts for the removal of VOCs.
1. Introduction
Volatile organic compounds (VOCs) emitted from industrial processes and automobile exhaust emissions are recognized as significant factors that cause air pollution due to their toxic nature and involvement in photochemical smog formation.1,2 Consequently, the removal of VOCs before emission into the atmosphere is an important and urgent task. Currently, many technologies are available for removal of VOCs, such as adsorption,3 photocatalysis,4 and catalytic oxidation,5 among which catalytic oxidation stands out for its beneficial characteristics, including low cost, high efficiency, high selectivity, and environmental friendliness.5–7Multicomponent solid solution materials known as high-entropy oxides (HEOs) include five or more elements in almost equiatomic proportions in order to maximize configurational entropy and stabilize their structures.8 High-entropy, lattice distortion effects, synergistic effects, remarkable chemical and thermal stability, a wide variety of cation compositions, and a range of redox behaviors are merely some of the attractive features of HEOs.9 HEOs have found applications in a broad array of fields, including catalysis, energy storage conversion, and environmental remediation. In the context of catalysis, the high-entropy effect can lead to several beneficial properties, including enhanced thermal stability, resistance to sintering, and unique surface characteristics that can improve catalytic performance. The random distribution of multiple metal elements in the lattice also introduces a variety of active sites, which can enhance the catalyst's activity, selectivity, and durability. HEO can act as efficient catalysts for various reactions, such as the oxygen evolution reaction (OER), CO oxidation, and decomposition of VOCs.10 Tianshan et al. synthesized (CuCoMnNiFeOx) HEOs from quinary layered double hydroxides by a co-precipitation method for the catalytic degradation of toluene. The temperature for 90% toluene conversion (T90) was recorded at 255 °C. However, upon undergoing a durability test over 10 cycles, a decline in performance was noted, and the T90 was shifted to 265 °C.11 Zhou et al. synthesized Ce-HEO (Ce/NiMgCuCoZnOx) for toluene oxidation and incorporated 5% Au as a catalyst. The catalyst exhibited complete combustion of toluene at 260 °C and maintained excellent stability for 60 h, achieving a 98% toluene conversion rate.12 Talluri et al. successfully synthesized nanoparticles of a high-entropy spinel metal oxide, denoted as (CoCrFeMnNi)3O4, which exhibits promising electrocatalytic properties for methanol oxidation. These nanoparticles are applied onto nickel foam to fabricate a working electrode. In their study, the catalyst demonstrated a specific current density of approximately 335 mA cm−2 for methanol oxidation. Moreover, the observed mass-activity of the catalyst was around 110 mA mg−1, with an onset potential of approximately 0.45 V.13
Different methods have been developed for preparing HEOs, such as nebulized spray pyrolysis (NSP),14 solid phase sintering,15 flame spray pyrolysis (FSP),16 and co-precipitation.13 Li et al. used a facile co-precipitation method to successfully synthesize a cobalt-based HEO (Co-HEO) with a spinel-type structure for N2O catalytic decomposition. The Co-HEO catalyst demonstrated superior catalytic performance and greater thermal stability in comparison to the single and binary Co-based oxides. Specifically, the temperature for 90% N2O decomposition (T90) was 356 °C for Co-HEO, whereas it was 406 °C for Co3O4.17 Chen et al. prepared a 0.3 wt% (Pt/NiMgCuZnCoOx) catalyst by co-precipitation for CO oxidation.18 The (Pt/NiMgCuZnCoOx) began showing activity at 60 °C, and complete conversion of CO occurred at 155 °C. Sarkar et al. conducted a study on HEOs for reversible energy storage by the NSP method. Their research revealed that the stabilization effect resulting from entropy contributes significantly to the retention of storage capacity in HEOs, thereby substantially enhancing cycling stability.14 Sarkar et al. also employed the FSP method to synthesize transition metal-based HEO (Co, Cu, Mg, Ni, Zn).19 The FSP methods are recognized for their capacity to effectively produce uniform nanocrystallite oxides. The FSP and NSP are rapid processes with short residence times, which facilitate quick synthesis and potential stabilization of metastable phases. In contrast, co-precipitation, a slower process, allows for sufficient time for diffusion and offers advantages in producing homogeneous multicomponent systems.14 Therefore, we used co-precipitation to prepare the HEO catalyst in our work.
A common issue faced by most of these techniques is that HEO is synthesized at high temperatures.20 (MnFeCoNiCuZn)3O4−x HEOs were synthesized by Dong et al. at 1227 °C using a rapid Joule heating method.21 The resultant HEOs have a large oxygen vacancy, leading to efficient electrocatalysts for water oxidation (i.e., OER). Similarly, (Al0.2CoCrFeMnNi)0.58O4−δ HEOs were synthesized by Xiang et al. at an optimal temperature of 650 °C by the method of solution combustion synthesis (SCS). Additional Al0.2 was doped into (CoCrFeMnNi)O4, which exhibited a reversible specific capacity of 554 mA h g−1 after 500 cycles at 200 mA g−1, and a rate capability of 634 mA h g−1 even at 3 A g−1, demonstrating good cycling performance.22 The high temperatures method for the synthesis of HEO limited the control of the HEO's shape and size, and the HEO had a large size and low surface area. The ineffective catalytic activities of HEOs produced at high temperatures were caused by their low intrinsic activities and a lack of exposed active sites. Gu et al. synthesized HEOs using a low-temperature plasma method. This method endowed the synthesized HEOs with high surface area, abundant oxygen vacancies, and nanosheet structure, which are favorable for the enhancement of electrocatalytic activity.23 It is well known that catalysts with large surface areas can produce more exposed active sites and improved mass diffusion efficiency, which improves catalytic performances.24 The achievement of high configurational entropy and avoidance of particle coarsening are key issues for the synthesis of HEOs. One effective approach for improving catalytic activity is defect engineering, for example, the occurrence of oxygen vacancies.25 Nevertheless, synthesizing HEOs with rich oxygen vacancies at low temperatures is difficult.26
Transition metal oxides derived from metal–organic frameworks (MOFs) prepared at high temperatures typically exhibit inherently high electrochemical activities.27 MOF-derived transition metal oxides have the freedom to diffuse through nanostructure pores, which in turn helps to enhance catalytic activities.28 A small amount of lattice disorder may be caused by the various ionic sizes of the components in polymetallic MOFs, allowing for unique features.29 HEOs designed and synthesized using MOF as a precursor represent an emerging class of catalysts that have gained attention due to their unique properties and potential applications. Zhao et al. showed that HEO (FeCuNiCoMnOx) formed from MOF increases the concentration of oxygen vacancies and lattice defects. These factors improve the electrocatalytic activity toward the oxygen evolution reaction in an alkaline system and act as catalysts for effective electrochemical water splitting.24
In this work, we report a class of HEOs synthesized from high-entropy MOF (HE-MOF). The HE-MOF was synthesized by a co-precipitation method from a mixed solution of five metal species (Mn2+, Ce3+, Co2+, Ni2+, and Cu2+) and an organic ligand, 1,4-benzendicarboxylic acid (1,4-BDC), at room temperature. This HE-MOF was then subjected to calcination at temperatures ranging from 300 °C to 700 °C. During calcination, the organic ligands within the MOF framework decomposed and were removed, resulting in the transformation of the HE-MOF into the Ce-HEO catalyst. Due to the significantly increased entropy effect and high oxygen vacancy, the as-prepared Ce-HEO-400 exhibited the lowest temperature for toluene oxidation, whose T50 and T90 were 180 °C and 255 °C, respectively.
2. Experimental section
2.1. Materials and chemicals
Manganese(II) nitrate tetrahydrate (Mn(NO3)2·4H2O, ≥98.0%), cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O, ≥99.5%), cobalt nitrate(II) tetrahydrate (Co(NO3)2·4H2O, ≥98.5%), nickel nitrate(II) tetrahydrate (Ni(NO3)2·4H2O, ≥99.0%), copper(II) nitrate trihydrate (Cu(NO3)2·3H2O, ≥99.0%), iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, ≥98.5%), and lanthanum(III) nitrate hexahydrate (La(NO3)3·6H2O, ≥99.5%) were bought from Aladdin Chemical Reagents. 1,4-Benzendicarboxylic acid (1,4-BDC, >99.0%) and triethylamine (TEA, >99.0%) were purchased from Tokyo Chemical Industry. Ethanol (CH3CH2OH, 99.5%) and N,N-dimethylformamide (DMF, 99.5%) were purchased from Aladdin Reagents. All chemicals were of analytical grade and were used directly without further purification.2.2. Synthesis of catalysts
Mn(NO3)2·4H2O (1 mmol, 0.251 g), Ce(NO3)2·6H2O (1 mmol, 0.403 g), Co(NO3)2·6H2O (1 mmol, 0.290 g), Ni(NO3)2·6H2O (1 mmol, 0.291 g), and Cu(NO3)2·3H2O (1 mmol, 0.241 g) were dissolved in a solution containing DMF (215 mL), ethanol (15 mL) and water (15 mL) under magnetic stirring. 1,4-BDC (6 mmol, 0.996 g) was dissolved in the above-mentioned solution, and then 5 mL of TEA was gradually added to the solution. After that, the mixture was agitated for 30 min to produce a consistent colloidal suspension. The colloidal solution was then continuously ultrasonicated for 1 h at 40 kHz. The final step was centrifugation to collect the product, followed by washing with ethanol and water and drying at 60 °C. The resulting samples were labeled as HE-MOF. The powders were calcined in a muffle furnace at 300 °C, 400 °C, 500 °C and 700 °C for 2 h in air with a heating rate of 4 °C min−1 (Fig. S1, ESI†). The obtained samples (CeCuNiCoMnOx) were denoted as Ce-HEO-T. For comparison, (FeCuNiCoMnOx) (denoted as Fe-HEO-T) and (LaCuNiCoMnOx) (denoted as La-HEO-T) (T refers to the calcination temperature) were also prepared by the same method, replacing Ce with Fe and La, respectively.2.3. Characterization
Using a Cu Kα radiation source, X-ray diffraction (XRD) characterization was carried out via a Rigaku Smart Lab (Japan). The catalysts were subjected to a 2θ scan at a rate of 10° min−1, between 10° and 90°, using 40 kV tube voltages and a 40 mA tube current. The N2 desorption/adsorption isotherms were obtained using an ASAP 2020HD88 device (Micromeritics Inc.). Before each measurement, the samples were outgassed at 200 °C for 12 h under vacuum. The Brunauer–Emmett–Teller (BET) method was used to calculate the surface area. The Barrett–Joyner–Halenda (BJH) method was employed to analyze the pore size distribution based on the desorption branch of the N2 adsorption isotherm. The functional groups of the samples were determined by Fourier transform infrared (FT-IR) spectroscopy (TENSOR 27, Bruker) within the range of 600–4000 cm−1. Analyses of the surface element distribution of the samples were conducted via scanning electron microscopy (SEM) using a Hitachi SU8020 microscope that was equipped with EDS equipment and operated at 5 kV. Before being scanned, a small film of Au was applied to the samples. An FEI Tecnai G2 F20 transmission electron microscope operating at 200 kV was used to acquire images for transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). For TEM observation, a drop of the suspension was placed on a carbon grid after each catalyst was disseminated into ethanol using ultrasonic treatment for five minutes. Using Thermo Scientific K-Alpha XPS analyzer equipped with an Al Kα X-ray radiation (1486.6 eV), X-ray photoelectron spectra (XPS) were used to measure the surface composition and binding energy (B.E.) of the catalysts. The binding energy values were calculated with reference to the C 1s peak of contaminant carbon at 284.6 eV. Using the peak area, the ratio of elements with various element valence states was calculated. The catalysts were characterized using H2-temperature programmed reduction (H2-TPR) in an AutoChem II2920 that has a thermal conductivity detector (TCD). Fresh samples were processed in N2 (60 mL min−1) at 200 °C for 30 min before being cooled to room temperature for each measurement. Subsequently, the catalyst was heated at a rate of 10 °C min−1 in 10 vol% H2/Ar at a flow rate of 30 mL min−1, from 50 °C to 800 °C.2.4. Catalytic performance tests
The toluene oxidation catalytic tests were conducted in a fixed-bed glass reactor (i.d. 10 mm) at atmospheric pressure. The glass reactor tube was filled with 100 mg of catalyst. The feed gas containing 1000 ppm toluene was produced by air bubbles in a conical flask containing pure toluene, and the total flow rate of the reaction gas was 100 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. A gas chromatograph (GC-7890A) fitted with a methane converter and FID was used to examine the gases online. The following equation was used to determine the toluene removal efficiency (X):
000 h−1. A gas chromatograph (GC-7890A) fitted with a methane converter and FID was used to examine the gases online. The following equation was used to determine the toluene removal efficiency (X):Cin and Cout are the concentrations of toluene in the inlet and outlet gases, respectively.
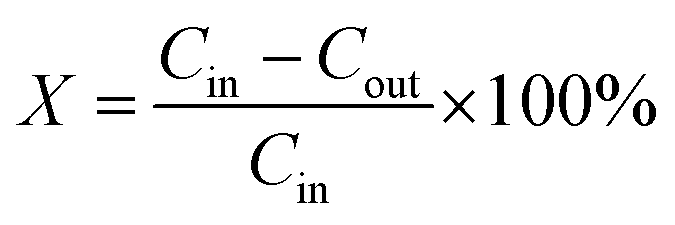
3. Results and discussion
3.1 Composition and structure of catalysts
The successful synthesis of HE-MOF was verified through FT-IR spectroscopy (Fig. 1). In the spectrum of commercial 1,4-BDC (depicted in Fig. 1b), the band located at 1690 cm−1 and the broad bands spanning from 2545 cm−1 to 3105 cm−1 are ascribed to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ν(OH) of the non-ionic carboxyl (–COO−) groups, respectively.24 Notably, the distinctive bands of pristine 1,4-BDC at 1690 cm−1 and 2545–3105 cm−1 vanish, with new bands emerging at 1502 cm−1, 1578 cm−1, and 1375 cm−1 in the resultant HE-MOF. The characteristic band of HE-MOF at 1502 cm−1 is assigned to the stretching vibrations of the para-aromatic C–H group, whereas the bands at 1578 cm−1 and 1375 cm−1 are respectively attributed to the asymmetric and symmetric vibrations of the coordinated carboxyl (–COO−) group.30,31 These two distinct modes confirm that the –COO− of 1,4-BDC is coordinated with metal ions in a bi-dentate manner.32,33
O) and ν(OH) of the non-ionic carboxyl (–COO−) groups, respectively.24 Notably, the distinctive bands of pristine 1,4-BDC at 1690 cm−1 and 2545–3105 cm−1 vanish, with new bands emerging at 1502 cm−1, 1578 cm−1, and 1375 cm−1 in the resultant HE-MOF. The characteristic band of HE-MOF at 1502 cm−1 is assigned to the stretching vibrations of the para-aromatic C–H group, whereas the bands at 1578 cm−1 and 1375 cm−1 are respectively attributed to the asymmetric and symmetric vibrations of the coordinated carboxyl (–COO−) group.30,31 These two distinct modes confirm that the –COO− of 1,4-BDC is coordinated with metal ions in a bi-dentate manner.32,33
 | ||
| Fig. 1 FT-IR spectra of (a) HE-MOF and (b) 1, 4-BDC. | ||
The TGA test for the MOF is shown in Fig. S2, ESI.† The TGA data show that the MOF precursor undergoes significant weight loss between 50 °C and 390 °C, indicating the gradual decomposition of organic components. The complete conversion to HEOs occurred at around 400 °C. The TGA results demonstrate that the synthesized MOF precursor is thermally stable up to 390 °C, and complete conversion to HEOs occurs at 400 °C. This temperature is critical because it matches the calcination temperature used to synthesize Ce-HEO-400. The stability observed in the TGA results implies that the HEOs synthesized at 400 °C are thermally robust, which contributes to their stability under catalytic reaction conditions, particularly during high-temperature VOC oxidation reactions The crystalline structures of the calcined Ce-HEO-T were characterized by XRD (Fig. 2). For all the samples, the diffraction peaks at 28.5°, 33.1°, 47.6° and 56.4° correspond to the (111), (200), (220), and (311) planes of CeO2 with a cubic fluorite structure (JCPDS no. 81-0792). The diffraction peaks at 18.4°, 30.8°, 36.8°, 37.5°, 44.8°, 57.5°, and 63.4° correspond to the (111), (220), (311), (222), (400), (511), and (440) planes of the spinel (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) structure. The Co, Cu, Mn, and Ce-based products exhibited a single-phase, the Fdm m spinel structure, which is in line with the (CoCuMnNi)O2 stoichiometry.24 The diffraction peaks of Ce-HEO-T catalysts obtained at different temperatures narrowed as the temperature increased, indicating the increased crystallinity of the samples.34 The narrowing of the diffraction peaks with increased calcination temperature suggests enhanced crystallinity. Higher crystallinity generally correlates with the improved stability and durability of the catalyst under reaction conditions. The combination of CeO2 with the spinel structure in Ce-HEO-T is particularly beneficial. CeO2 is known for its oxygen storage capacity and redox properties, while the spinel structure contributes additional oxygen vacancies and mobility, enhancing the overall catalytic performance.
m) structure. The Co, Cu, Mn, and Ce-based products exhibited a single-phase, the Fdm m spinel structure, which is in line with the (CoCuMnNi)O2 stoichiometry.24 The diffraction peaks of Ce-HEO-T catalysts obtained at different temperatures narrowed as the temperature increased, indicating the increased crystallinity of the samples.34 The narrowing of the diffraction peaks with increased calcination temperature suggests enhanced crystallinity. Higher crystallinity generally correlates with the improved stability and durability of the catalyst under reaction conditions. The combination of CeO2 with the spinel structure in Ce-HEO-T is particularly beneficial. CeO2 is known for its oxygen storage capacity and redox properties, while the spinel structure contributes additional oxygen vacancies and mobility, enhancing the overall catalytic performance.
 | ||
| Fig. 2 XRD patterns of Ce-HEO-700, Ce-HEO-500, Ce-HEO-400, and Ce-HEO-300. | ||
The morphologies of Ce-HEO-T were investigated using SEM and TEM. Ce-HEO-300 and Ce-HEO-400 (Fig. 3a and b) had particles with flake morphologies. With the increase in the calcination temperature from 300 to 700 °C, the flake structure of the catalyst became progressively compact and changed into a granular structure. Ce-HEO-500 in Fig. 3c shows the coexistence of flake and granular particles formed by the decomposition of the flake particles during high-temperature treatment. As the temperature further increased from 500 to 700 °C, the particles exhibited granular morphologies, as shown in Fig. 3d for Ce-HEO-700. The particle size gradually increased with the increase in calcination temperature (Table S1, ESI†). The corresponding EDS images (Fig. S3a–d, ESI†) demonstrated that all elements were uniformly dispersed in Ce-HEO-T. This homogeneous dispersion of elements is crucial for the synthesis of highly efficient catalysts. The transition from flake to granular morphology as the calcination temperature increases is significant for catalytic performance. The flake structure at lower temperatures may provide a greater surface area, which is beneficial for catalytic reactions. However, the transition to a more compact granular structure at higher temperatures could enhance the stability of the catalyst during high-temperature operations, which is crucial for long-term performance. The TEM image of Ce-HEO-400 (Fig. 3e) shows the flake particle structure and some dotted particles. As shown in the HRTEM images (Fig. 3f–h), the lattice fringes with inter-planar distances of 0.32, 0.28, and 0.20 nm were ascribed to the (111), (220), and (200) planes of CeO2, meaning that CeO2 has a predominantly hexahedral shape.35 The lattice fringes observed in the HRTEM images indicate well-defined crystalline structures of CeO2, which are crucial for providing active sites for the oxidation of VOCs. The presence of these well-structured crystalline planes suggests that the catalyst can effectively adsorb and activate reactant molecules, leading to higher catalytic efficiency. The combination of flake-like structures (which provide high surface area) and well-defined crystalline planes (which offer active sites) in Ce-HEO-400 could explain its superior catalytic performance compared to other samples.
 | ||
| Fig. 3 SEM images of (a) Ce-HEO-300, (b) Ce-HEO-400, (c) Ce-HEO-500, and (d) Ce-HEO-700. (e) TEM and (f)–(h) HRTEM images of Ce-HEO-400. | ||
The pore size distribution and N2 adsorption–desorption isotherms of the Ce-HEO samples are shown in Fig. 4. Similar mesoporous structures were shown by the type IV isotherms and unique H3 type hysteresis loops in all samples in relative pressure ranges of 0.63–1.0, 0.59–1.0, 0.72–1.0, and 0.85–1.0.36 Additionally, the average pore sizes of Ce-HEO-300, Ce-HEO-400, Ce-HEO-500, and Ce-HEO-700 were 12.62, 13.06, 22.47, and 32.05 nm, and the resultant specific BET surface areas were 136.18, 152.96, 66.33 and 15.83 m2 g−1 (Table S1, ESI†), respectively. The increase in calcination temperature caused an increase in the average pore size. Among these catalysts, Ce-HEO-400 exhibited the highest surface area, contributing to its superior catalytic activity.37
 | ||
| Fig. 4 (a) N2 adsorption–desorption isotherms and (b) the pore size distribution of Ce-HEO. | ||
3.2. Surface analysis and reduction properties
XPS was conducted to explore the chemical compositions and valence states of different elements in Ce-HEO-T. The XPS spectra were deconvoluted by the Gaussian fitting method;32,38–40 the high-resolution Mn 2p, Ce 3d XPS spectra of Ce-HEO-T are presented in Fig. 5. Table 1 shows the observed Ce-HEO-T surface element ratios. The eight peaks in the Ce 3d spectra (Fig. 5a) can be fitted. Two of the peaks, U′ and V′, correspond to Ce3+ species, while the remaining six, denoted by the symbols U, V, U″, V″, U‴, and V‴, were identified as Ce4+.12 The relative content of Ce3+ can be determined via Ce3+/(Ce3+ + Ce4+), where Ce3+ is equal to (U′ + V′) and Ce4+ is determined by (U + V + U″ + V″ + U‴ + V‴). Based on the fitting results, the relative Ce3+ content of Ce-HEO-300, Ce-HEO-400, Ce-HEO-500 and Ce-HEO-700 were 5.97%, 16.38%, 14.37% and 11.21%, respectively. The generation of oxygen vacancies and crystal defects is typically the result of nonstoichiometric Ce3+ species, which enhances the redox capabilities of the catalysts.41 Therefore, it was speculated that the Ce-HEO-400 with the highest Ce3+ content would have better performance for toluene catalytic oxidation (see Section 3.3.1).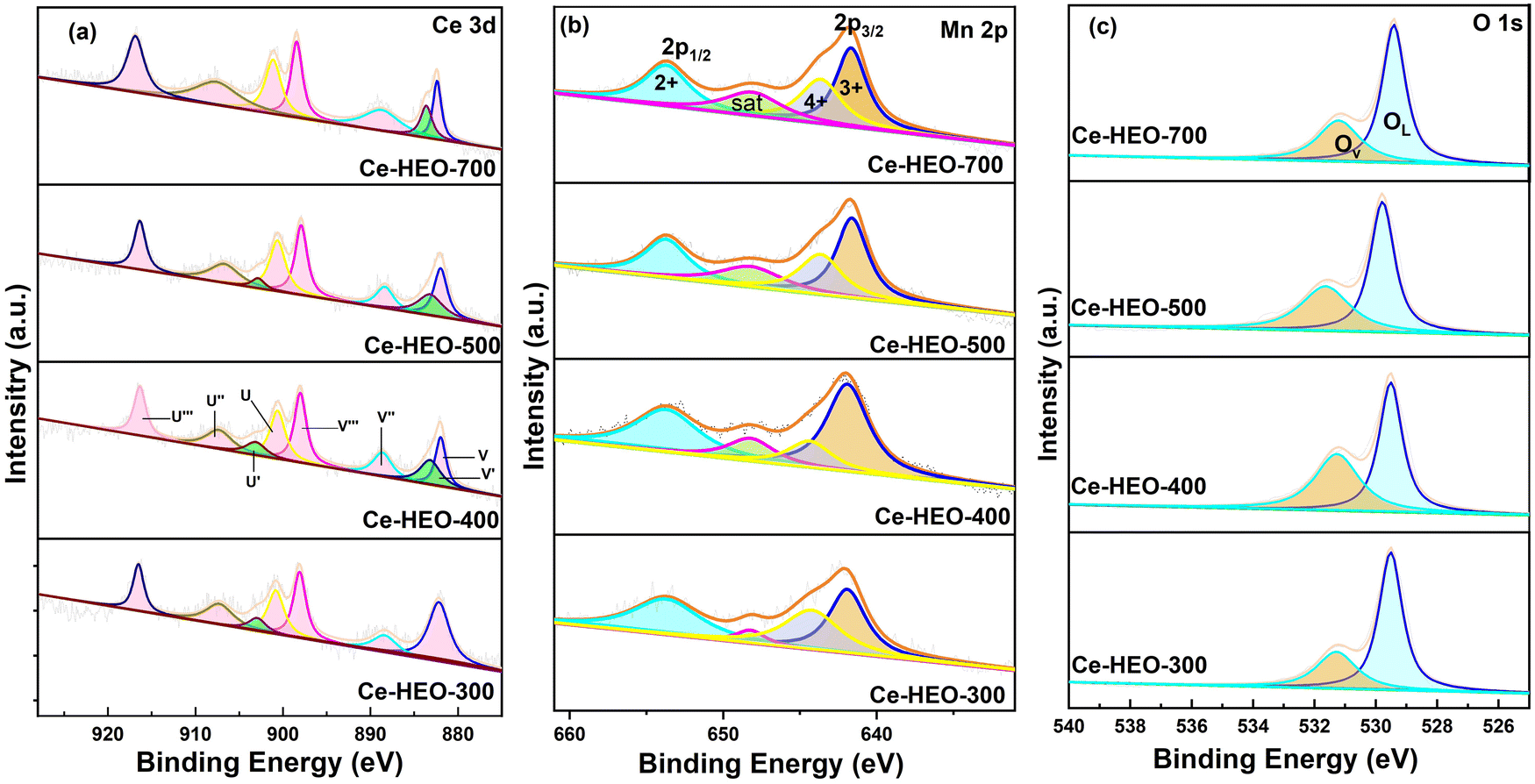 | ||
| Fig. 5 (a) Ce 3d, (b) Mn 2p, and (c) O 1s XPS spectra of Ce-HEO-T. | ||
| Surface element ratio | |||
|---|---|---|---|
| Catalysts |
|
|
|
| Ce-HEO-300 | 5.97 | 31.12 | 68.31 |
| Ce-HEO-400 | 16.38 | 45.13 | 84.82 |
| Ce-HEO-500 | 14.37 | 40.87 | 72.36 |
| Ce-HEO-700 | 11.21 | 34.64 | 70.01 |

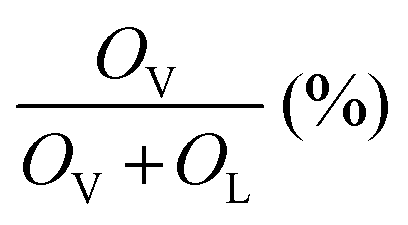

The Mn 2p core level XPS spectra (Fig. 5b) for Ce-HEO-T displayed four principal peaks at binding energies of 653.7 eV, 641.6 eV, 643.2 eV, and 648.2 eV. The first three peaks are attributed to the spin–orbit coupling signatures of Mn2+, Mn3+ and Mn4+ species, respectively and the peak with binding energy 648.2 eV is a satellite peak. The relative content of Mn3+ and Mn2+ can be determined using (Mn2+ + Mn3+)/(Mn2+ + Mn3+ + Mn4+). Based on the fitting results, the relative Mn3+ and Mn2+ contents of Ce-HEO-300, Ce-HEO-400, Ce-HEO-500 and Ce-HEO-700 were 68.31%, 84.82%, 72.36% and 70.01%, respectively. The presence of Mn2+ and Mn3+ weakened the binding capacity of the catalyst to oxygen, increased the oxygen mobility, and produced more oxygen vacancies, leading to more active oxygen species on the catalyst's surface. This correlation is associated with excellent catalytic activity and stability.42
The O 1s XPS spectra (Fig. 5c) of the Ce-HEO-T catalysts were fitted to two peaks at 529.5 and 531.2 eV, which are attributed to the surface lattice oxygen (OL) and oxygen vacancy (OV) (e.g. O2−, O22−or O−), respectively.43 The concentration of the oxygen vacancies can be determined by the ratio of (OV/(OV + OL)). The percentage of oxygen vacancy for Ce-HEO-400 (45.13%) was higher than those of the other samples, due to its high lattice oxygen mobility. Consequently, Ce-HEO-400 may easily produce additional oxygen vacancies, increasing the amount of surface-adsorbed oxygen species that are favorable for the oxidation of toluene.44
Fig. 6 presents the H2-TPR profiles of Ce-HEO-T catalysts. All samples exhibited three reduction peaks. The first peak is indicative of the reduction of Ce4+ to Ce3+, a process that is highly significant for understanding the redox properties of the catalyst. For Ce-HEO-400, due to its low calcination temperature, weak crystallinity, and the highest content of unstable Ce4+, the intensity of this peak is the highest. As the calcination temperature increases, the crystallinity is enhanced, making Ce4+ more stable and less prone to reduction. Therefore, the peak intensity gradually decreases. This demonstrates that Ce-HEO-400 is more conducive to electron mobility and is favorable for exerting a low-temperature catalytic effect during the toluene oxidation process.45 The reduction peaks observed at 154.4 °C could be associated with the reduction of Cu species.46 The reduction peak observed around 279.4 °C likely indicates the reduction of Co species.47 Meanwhile, for Ce-HEO-500, the reduction peaks at 160.9 °C and 305 °C, which are respectively attributed to Cu and Co species, shifted to higher temperatures. Similarly, the reduction peaks at 159.6 °C and 294.6 °C of Ce-HEO-700 also shifted to higher temperatures, suggesting a decrease in the catalyst's reduction ability. Hence, Ce-HEO-400 demonstrated commendable redox performance.46,48,49
 | ||
| Fig. 6 H2-TPR profiles of Ce-HEO-400, Ce-HEO-500, and Ce-HEO-700 catalysts. | ||
3.3 Catalytic performance
3.3.1 Catalytic activity
Fig. 7a indicates that the catalytic performances of the Ce-HEO-T catalysts for toluene oxidation (inlet concentration 1000 ppm) are as follows: Ce-HEO-400 > Ce-HEO-500 > Ce-HEO-700 > Ce-HEO-300. Among all the catalysts, Ce-HEO-400 showed the lowest temperature for toluene oxidation, with T50 and T90 of 180 °C and 255 °C, respectively (Table S2, ESI†). This result is also related to the specific surface area of the samples, and the BET surface area of Ce-HEO-400 is the highest among the catalysts (Table S1, ESI†). Moreover, Ce-HEO-400 has rich oxygen vacancies, which are conducive to improving the activation capacity of oxygen. The identical process employed to prepare Ce-HEO-400 was also used to synthesize Fe-HEO-400 and La-HEO-400. The Fe-HEO-400 and La-HEO-400 catalysts (Fig. 7b) showed lower catalytic activity as compared to Ce-HEO-400, whose T50 and T90 are 245 °C, 250 °C and 280 °C, 300 °C, respectively. | ||
| Fig. 7 Toluene conversions over (a) Ce-HEO catalysts obtained at different calcination temperatures, and (b) Ce-HEO-400, Fe-HEO-400, and La-HEO-400 catalysts. | ||
At approximately 280 °C, the Ce-HEO-400 catalyst totally converted toluene to CO2 and H2O, while the toluene oxidation reaction on other Ce-HEO-T catalysts had barely started. This good low-temperature reduction performance of Ce-HEO-400 shows that Ce3+ is also the active center of the Ce-HEO-400 catalyst. Table S3, ESI† compares the catalytic activity of toluene for Ce-HEO and other HEO catalysts that have been reported in the literature. The Ce-HEO-400 showed comparatively good catalytic activity to the reported HEO catalysts. Moreover, the preparation process of our catalyst is simple and the synthesis temperature is low, which can greatly reduce the cost of the catalyst. This is extremely important for practical application.
According to the Mars van Krevelen mechanism,50 toluene molecules adsorbed on the catalyst surface undergo oxidation and degradation by the lattice oxygen components within the metal oxide. The catalyst surface defects can constantly adsorb gas oxygen molecules as the reaction progresses, supplying more lattice oxygen to be consumed. Ce-HEO-400 exhibits a higher concentration of oxygen defect sites, enabling it to adsorb more oxygen species. This, in turn, accelerates the consumption and replenishment of the lattice oxygen, thereby enhancing the catalytic activity. Ce-HEO-400 also demonstrates high lattice oxygen mobility, significantly influencing the catalyst's activity. The mobility of the oxygen adsorbed on the surface impacts the activity of the catalyst.51 Metal oxide catalysts are constantly engaged in the adsorption and activation of gas-phase oxygen on their surface oxygen vacancies, generating surface-adsorbed oxygen. This process facilitates the rapid involvement of lattice oxygen in the reaction and regeneration.
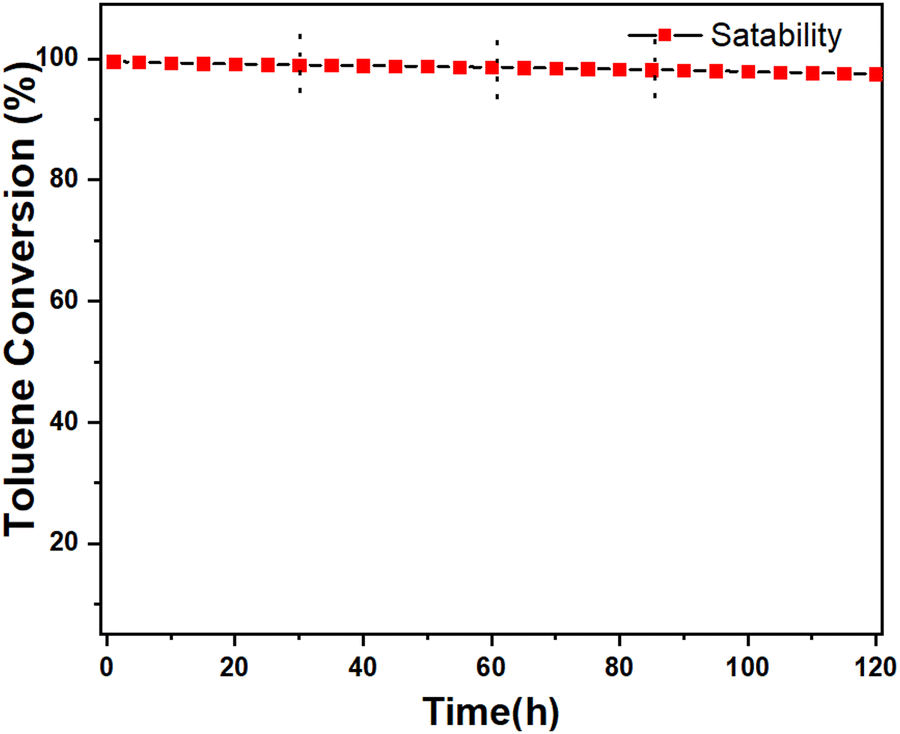 | ||
| Fig. 8 Catalytic stability of Ce-HEO-400 for toluene oxidation at 260 °C. | ||
4. Conclusion
A series of Ce-HEO-T catalysts were successfully synthesized using HE-MOFs as precursors. Among the Ce-HEO-T catalysts, Ce-HEO-400 demonstrated exceptional performance, with a lower temperature requirement for the complete combustion of toluene (255 °C) and high stability for 120 h, maintaining a toluene conversion rate above 98.5%. Additionally, Ce-HEO-400 exhibited an abundance of oxygen vacancies, which facilitated oxygen mobility and contributed to the outstanding performance. Furthermore, the presence of Mn2+ and Mn3+ weakened the binding capacity of the catalyst to oxygen, increasing its oxygen mobility and producing more oxygen vacancies, leading to more active oxygen species on the catalyst's surface, similar to the effect of Ce3+.Author contributions
Abid Hussain: investigation, methodology, data curation writing – original draft; Yuhua Zheng: formal analysis, writing-reviewing & editing, Qianyu Wang: writing – review & editing; Yanbin Cui: conceptualization, writing – review & editing, supervision.Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is financially supported by the financial support from National Natural Science Foundation of China (No. 22076189).References
- W. Li, W. Chu, M. Zhuang and J. Hua, Catal. Today, 2004, 93, 205 CrossRef.
- M. A. Bari and W. B. Kindzierski, Sci. Total Environ., 2018, 631, 627 CrossRef PubMed.
- Q. Zhao, Q. Du, Y. Yang, Z. Zhao, J. Cheng, F. Bi, X. Shi, J. Xu and X. Zhang, Chem. Eng. J., 2022, 433, 134510 CrossRef CAS.
- Y. Yang, S. Zhao, L. Cui, F. Bi, Y. Zhang, N. Liu, Y. Wang, F. Liu, C. He and X. Zhang, Green Energy Environ., 2022, 7, 654 Search PubMed.
- K. Zeng, Y. Wang, C. Huang, H. Liu, X. Liu, Z. Wang, J. Yu and C. Zhang, Ind. Eng. Chem. Res., 2021, 60, 6111–6120 CrossRef CAS.
- C. Zhang, H. Cao, C. Wang, M. He, W. Zhan and Y. Guo, J. Hazard. Mater., 2021, 402, 123473 CrossRef CAS PubMed.
- K. Zhang, L. Dai, Y. Liu, J. Deng, L. Jing, K. Zhang, Z. Hou, X. Zhang, J. Wang and Y. Feng, Appl. Catal., B, 2020, 279, 119372 CrossRef CAS.
- P. Sarker, T. Harrington, C. Toher, C. Oses, M. Samiee, J.-P. Maria, D. W. Brenner, K. S. Vecchio and S. Curtarolo, Nat. Commun., 2018, 9, 4980 CrossRef.
- A. Sarkar, Q. Wang, A. Schiele, M. R. Chellali, S. S. Bhattacharya, D. Wang, T. Brezesinski, H. Hahn, L. Velasco and B. Breitung, Adv. Mater., 2019, 31, 1806236 CrossRef.
- S. L. Fereja, Z. Zhang, Z. Fang, J. Guo, X. Zhang, K. Liu, Z. Li and W. Chen, ACS Appl. Mater. Interfaces, 2022, 14, 38727 CrossRef CAS PubMed.
- T. Xue, Y. Wang, L. Yang, Z. Li, Y. Gao and Q. Wang, Catalysts, 2023, 13, 119 CrossRef CAS.
- J. Zhou, Y. Zheng, G. Zhang, X. Zeng, G. Xu and Y. Cui, Environ. Technol., 2023, 23, 3016–3028 Search PubMed.
- B. Talluri, K. Yoo and J. Kim, J. Environ. Chem. Eng., 2022, 10, 106932 CrossRef CAS.
- A. Sarkar, L. Velasco, D. Wang, Q. Wang, G. Talasila, L. De Biasi, C. Kübel, T. Brezesinski, S. S. Bhattacharya and H. Hahn, Nat. Commun., 2018, 9, 3400 CrossRef.
- M. Biesuz, L. Spiridigliozzi, G. Dell’agli, M. Bortolotti and V. M. Sglavo, J. Mater. Sci., 2018, 53, 8074 CrossRef CAS.
- A. H. Phakatkar, M. T. Saray, M. G. Rasul, L. V. Sorokina, T. G. Ritter, T. Shokuhfar and R. Shahbazian-Yassar, Langmuir, 2021, 37, 9059 CrossRef CAS PubMed.
- B. Li, X. Duan, T. Zhao, B. Niu, G. Li, Z. Zhao, Z. Yang, D. Liu, F. Zhang and J. Cheng, Environ. Sci. Technol., 2024, 58, 2153 CrossRef CAS PubMed.
- H. Chen, J. Fu, P. Zhang, H. Peng, C. W. Abney, K. Jie, X. Liu, M. Chi and S. Dai, J. Mater. Chem. A, 2018, 6, 11129 RSC.
- A. Sarkar, R. Djenadic, N. J. Usharani, K. P. Sanghvi, V. S. Chakravadhanula, A. S. Gandhi, H. Hahn and S. S. Bhattacharya, J. Eur. Ceram. Soc., 2017, 37, 747 Search PubMed.
- Y. Ma, Y. Ma, S. L. Dreyer, Q. Wang, K. Wang, D. Goonetilleke, A. Omar, D. Mikhailova, H. Hahn and B. Breitung, Adv. Mater., 2021, 33, 2101342 Search PubMed.
- Q. Dong, M. Hong, J. Gao, T. Li, M. Cui, S. Li, H. Qiao, A. H. Brozena, Y. Yao and X. Wang, Small, 2022, 18, 2104761 CrossRef CAS PubMed.
- H. Z. Xiang, H. X. Xie, Y. X. Chen, H. Zhang, A. Mao and C. H. Zheng, J. Mater. Sci., 2021, 56, 8127 Search PubMed.
- K. Gu, D. Wang, C. Xie, T. Wang, G. Huang, Y. Liu, Y. Zou, L. Tao and S. Wang, Angew. Chem., 2021, 133, 20415 Search PubMed.
- D. Wang, Z. Liu, S. Du, Y. Zhang, H. Li, Z. Xiao, W. Chen, R. Chen, Y. Wang and Y. Zou, J. Mater. Chem. A, 2019, 7, 24211 RSC.
- H. Chu, D. Zhang, P. Feng, Y. Gu, P. Chen, K. Pan, H. Xie and M. Yang, Nanoscale, 2021, 13, 19518 RSC.
- K. Zeng, W. Li, Y. Zhou, Z. Sun, C. Lu, J. Yan, J. H. Choi and R. Yang, Open Chem. Eng. J., 2021, 421, 127831 CAS.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 Search PubMed.
- X. Tan, Y. Wu, X. Lin, A. Zeb, X. Xu, Y. Luo and J. Liu, Inorg. Chem. Front., 2020, 7, 4939 RSC.
- A. K. Cheetham, T. D. Bennett, F.-X. Coudert and A. L. Goodwin, Dalton Trans., 2016, 45, 4113 RSC.
- S. Maiti, A. Pramanik, U. Manju and S. Mahanty, ACS Appl. Mater. Interfaces, 2015, 7, 16357 CrossRef CAS PubMed.
- H. Hu, X. Lou, C. Li, X. Hu, T. Li, Q. Chen, M. Shen and B. Hu, New J. Chem., 2016, 40, 9746 RSC.
- F. L. Li, P. Wang, X. Huang, D. J. Young, H. F. Wang, P. Braunstein and J. P. Lang, Angew. Chem., 2019, 131, 7125 CrossRef.
- Q. Liu, L. Yu, Y. Wang, Y. Ji, J. Horvat, M. L. Cheng, X. Jia and G. Wang, Inorg. Chem., 2013, 52, 2817 CrossRef CAS PubMed.
- Q. Gan, X. Cheng, J. Chen, D. Wang, B. Wang, J. Tian, T. T. Isimjan and X. Yang, Electrochim. Acta, 2019, 301, 47 CrossRef CAS.
- S. Liu, X. Liao, Q. Zhang, Y. Zhang, H. Wang and Y. Zhao, Nanomaterials, 2022, 12, 762 CrossRef CAS PubMed.
- Z. Zhang, X. Yan, F. Gao, P. Thai, H. Wang, D. Chen, L. Zhou, D. Gong, Q. Li and L. Morawska, Environ. Pollut., 2018, 238, 452 Search PubMed.
- Y. Xia, M. Lin, D. Ren, Y. Li, F. Hu and W. Chen, J. Porous Mater., 2017, 24, 621 CrossRef CAS.
- X. Zhao, J. Jiang, Z. Xue, C. Yan and T. Mu, Chem. Commun., 2017, 53, 9418 RSC.
- K. Yuan, T. Song, D. Wang, Y. Zou, J. Li, X. Zhang, Z. Tang and W. Hu, Nanoscale, 2018, 10, 1591 Search PubMed.
- L. Zhao, Z. Zhang, Y. Li, X. Leng, T. Zhang, F. Yuan, X. Niu and Y. Zhu, Appl. Catal., B, 2019, 245, 502 Search PubMed.
- L. Yang, Z. Cai, L. Hao, Z. Xing, Y. Dai, X. Xu, S. Pan, Y. Duan and J. Zou, ACS Appl. Mater. Interfaces, 2017, 9, 22518 CrossRef CAS PubMed.
- S. Liu, J. Xing, H. Zhang, D. Ding, F. Zhang, B. Zhao, S. K. Sahu and S. Wang, Atmos. Environ., 2019, 218, 117020 CrossRef CAS.
- W. Hu, J. Lan, Y. Guo, X.-M. Cao and P. Hu, ACS Catal., 2016, 6, 5508 CrossRef CAS.
- S. Mo, Q. Zhang, J. Li, Y. Sun, Q. Ren, S. Zou, Q. Zhang, J. Lu, M. Fu and D. Mo, Appl. Catal., B, 2020, 264, 118464 CrossRef CAS.
- C. Tang, H. Zhang and L. Dong, Catal. Sci. Technol., 2016, 6, 1248 Search PubMed.
- Y. Shu, J. Bao, S. Yang, X. Duan and P. Zhang, AIChE J., 2021, 67, 17046 CrossRef.
- X. Zhang, J. Zhao, Z. Song, H. Zhao, W. Liu, Z. A. Ma, M. Zhao and H. Du, ChemistrySelect, 2019, 4, 8902 CrossRef CAS.
- Y. Wang, H. Ji, W. Liu, T. Xue, C. Liu, Y. Zhang, L. Liu, Q. Wang, F. Qi and B. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 20522 Search PubMed.
- X. Liu, Y. Du, C. Zou, L. Liu, B. Yang and X. Wu, ChemistrySelect, 2019, 4, 9488 CrossRef CAS.
- S. Behar, N.-A. Gómez-Mendoza, M.-Á. Gómez-García, D. Świerczyński, F. Quignard and N. Tanchoux, Appl. Catal., A, 2015, 504, 203 CrossRef CAS.
- L. Liotta, M. Ousmane, G. Di Carlo, G. Pantaleo, G. Deganello, G. Marcì, L. Retailleau and A. Giroir-Fendler, Appl. Catal., A, 2008, 347, 81 CrossRef CAS.
- J. Chen, X. Chen, W. Xu, Z. Xu, J. Chen, H. Jia and J. Chen, Chem. Eng. J., 2017, 330, 281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02650k |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |