
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional modification of graphene nanoparticles: covalent grafting of peptides and π bonding for drug loading and delivery†
Kaiyue
Hu
a,
Luigi
Brambilla
 a,
Paola
Moretti
a,
Chiara
Bertarelli
ab,
Chiara
Castiglioni
a,
Giuseppe
Pappalardo
c and
Giuseppina
Sabatino
*c
a,
Paola
Moretti
a,
Chiara
Bertarelli
ab,
Chiara
Castiglioni
a,
Giuseppe
Pappalardo
c and
Giuseppina
Sabatino
*c
aPolitecnico di Milano, Dip. di Chimica, Materiali e Ing. Chimica Giulio Natta, 20133 – Milano, Italy
bIstituto Italiano di Tecnologia, CNST@PoliMi, 20133 – Milano, Italy
cInstitute of Crystallography, National Research Council Italy, 95126 Catania, Italy. E-mail: giuseppina.sabatino@cnr.it
First published on 18th September 2024
Abstract
Graphene nanoparticles (GNPs) can serve as a versatile platform for the development of drug-delivery systems by means of suitable functionalization strategies. Using a green physical method, we prepared GNPs comprising N < 10 stacked graphene layers with an average lateral size of about 102 nm. These GNPs are naturally endowed with carboxyl groups decorating their edges, facilitating further functionalization. Thus, a cell permeable peptide (CPP) poly-arginine-11 (R11) was grafted onto the GNPs to obtain peptide-functionalized GNPs (R11@GNP). Moreover, the preparation of a non-covalent complex with 1-pyrene carboxylic acid (PyCA) demonstrated that the GNP surface can be loaded with small molecules for drug delivery purposes. The structure of R11@GNP and PyCA@GNP supramolecular systems and the presence of covalent and non-covalent bonds, respectively, was investigated. A thorough analysis of the functionalized GNPs through UV-vis, FTIR and Raman spectroscopy techniques as well as dynamic light scattering and Z-potential measurements well characterized their structures at the molecular level. Fluorescence spectroscopy allowed collecting further evidence of the formation of stable π–π complexes between GNPs and PyCA and provided a first test of the thermally induced release of absorbed molecules.
1. Introduction
Graphene nanoparticles (GNPs) can be used as a versatile platform for the development of effective and selective drug delivery systems.1–9 GNPs combine several different functions, enabled by their peculiar structural characteristics listed below:(i) GNPs feature a large surface, formed by an extended regular network of condensed aromatic rings. This surface provides docking sites for small molecules, which can adsorb on the graphene layers through π–π interaction.10,11 This non-covalent bonding can be exploited for drug loading/release.
(ii) Native COOH groups on the edges of the graphene layers are ideal anchoring sites for covalent functionalization with polymers or molecules, which can impart specific chemical–physical properties, e.g. increased bio-compatibility and/or targeting features.8,12–17
(iii) GNPs can form stable dispersions in water owing to the presence of polar groups (such as COOH), while their stability in specific environments can be modulated by functionalization strategies.
(iv) Owing to their nanometer size, GNPs can penetrate tissues and cells.
It is possible to obtain a more suitable GNP size by fine-tuning parameters that affect GNP production. For instance, physical methods based on graphite grinding and exfoliation allow the proper selection of the milling time as well as ultrasonication intensity and duration.
Despite the extensive literature on the potential therapeutic applications of GNPs, the exploration of the effectiveness of these materials for biomedical applications remains a challenge. While the availability of different chemical or physical methods for preparing GNPs represents a great opportunity, the resulting multitude of different GNPs slows down research progress. In several cases, a single GNP batch is intrinsically highly heterogeneous, showing a wide particle size distribution and individual particles with different chemical/structural characteristics. These features often make it difficult to compare the results of in vitro assays performed in different laboratories. Even more difficult is to correlate the properties of a given GNP sample (e.g. cytotoxicity) with the structural or chemical characteristics of the nanoparticles.
To guarantee the significance and the reliability of the tests on GNPs, it is mandatory to work with sufficiently homogeneous samples, consisting of particles with a narrow size distribution, and showing a limited variability of chemical and structural features. Furthermore, a detailed picture of the GNP structure at the molecular level and at each step of their preparation would be advantageous to develop reliable structure/property relationships.
In this context, we propose the use of a physical procedure developed previously,18 involving ball-milling and liquid-phase exfoliation of pure graphite, which harvests two different stable water dispersions of GNPs – hereafter referred to as B60 and T60. The method is environment-friendly, since only graphite and water are used to prepare GNPs. A careful analysis of B60 and T60 GNPs by transmission electron microscopy (TEM), dynamic light scattering, UV-vis absorption spectroscopy, FTIR spectroscopy, and multi-wavelength Raman spectroscopy confirmed that the preparation method is reproducible and that good control over GNP size and defects has been achieved.18
These results form the basis for the subsequent modification of T60 and B60 presented herein. This study deals with the successful functionalization of B60 and T60 nanoparticles, focusing on their chemical and/or structural modifications by means of thorough spectroscopic characterization.
We used two different strategies for the functional modification of GNPs: (i) R11@GNP conjugates were obtained through covalent grafting of poly-arginine peptide-11 (R11), which imparts specific cell targeting properties and (ii) PyCA@GNP conjugates were prepared by exploiting non-covalent π–π interactions of 1-pyrene-carboxylic acid (PyCA) with the graphene surface. PyCA@GNP mimics absorption on the GNPs surface of a drug featuring π electrons,19,20 like the anti-cancer drug doxorubicin.5,6 While GNP conjugates (i) and (ii) represent two specific study cases, we believe that the functional modification of graphene-based nanoparticles obtained by our physical approach and the spectroscopic characterization illustrated herein could be extended in the future for the development of different GNP conjugates for drug delivery.
Compared to commercially available GNP batches, mostly involving reduced graphene-oxide GNPs, we think that the use of drug delivery platforms based on our laboratory-made GNPs could guarantee a superior structural control and reproducibility. This feature is fundamental for the exploration and understanding of the relationship between GNP structures and their performances, in the perspective of the optimization of drug carriers.
2. Materials and methods
Materials
N,N-Dimethylformamide (DMF) for peptide synthesis, CH3CN for HPLC and methanol were purchased from Sigma-Aldrich. Rink amide AM resin, Fmoc-Arg(Pbf)-OH and the activators N,N′-diisopropylcarbodiimide (DIC) and oxyma pure were purchased from Iris Biotech. Trifluoroacetic acid (TFA), triisopropyl silane (TIS), diisopropyl ether (iPr2O), and 1-pyrenecarboxylic acid were purchased from Sigma Aldrich.Synthesis of GNPs
GNPs were synthesized by the method reported previously.18 In brief, as shown in Scheme S1 (ESI†), 500 mg of a graphite rod (Sigma-Aldrich 496561-240.5G, 99.995% carbon) was ground using a Retch Mixer Mill MM 400 for 200 minutes. Then 80 mg of the obtained powder was added to 24 mL of sterile water (molecular biology reagent grade water, Sigma-Aldrich: W4502-1L) in a vial for sonication for 5 min. The sonication was performed at room temperature using an Ultrasonic Processor GEX750 with a flat head tip, 450 W, at 20 kHz. A fraction of small-sized GNPs were selected via the first centrifugation procedure (5000 rpm for 10 min). In the second centrifugation cycle (5000 rpm for 60 min), GNPs were separated into two fractions: the supernatant named T60 and the sediment named B60. T60 and B60 were used separately for subsequent functionalization.Synthesis of R11@GNP
R11 was synthesized by a microwave-assisted solid-phase peptide synthesis MW-SPPS method, a convenient chemical process in terms of solvent and time demand, which allows us to obtain a good yield and purity of the peptides. MW-SPPS of the peptide was performed using an fully automated peptide synthesizer, Liberty Blue 2.0 (CEM Corporation, Matthews, NC, USA), following the fluorenylmethoxycarbonyl (Fmoc)/tert-butyl (tBu) strategy on 0.1 mmol of Rink amide AM resin (185 mg, 0.54 mmol g−1, 100–200 mesh). After resin swelling in DMF, Fmoc-Arg(Pbf)-OH was introduced through the following protocol: (1) Fmoc-deprotection in 10% piperidine in DMF (4 mL); (2) washing with DMF (4 × 4 mL); and (3) double couplings with Fmoc-Arg(Pbf)-OH (5 eq., 0.2 M in DMF, 2.5 mL), oxyma pure (5 eq., 1 M in DMF, 0.5 mL) and DIC (10 eq., 1 M in DMF, 1 mL) prepared in separated bottles. Both deprotection and coupling reactions were performed in a Teflon vessel with microwave energy and nitrogen bubbling; monitoring of the temperature was performed using an internal optical fiber. The resin was exposed to the microwave-assisted cycle described in Table 1.| Step | Temperature (°C) | Power (W) | Time (s) |
|---|---|---|---|
| Deprotection | 75 | 175 | 15 |
| 90 | 37 | 50 | |
| 1st coupling | 75 | 175 | 15 |
| 90 | 55 | 230 | |
| 2nd coupling | 75 | 175 | 15 |
| 90 | 55 | 230 |
The final cleavage of the peptide from the resin with concomitant side-chain deprotection was achieved by treatment of resin-bound peptide with a TFA/TIS/H2O solution (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5, 1 mL mixture/100 mg of resin). The cleavage was carried out for approximately 7 h at room temperature. The product was precipitated by addition of ice-cold diisopropyl ether (iPr2O) (40 mL). The crude peptide was washed with ice-cold iPr2O (3 × 30 mL), dried under vacuum, dissolved in H2O (10 mL) and lyophilized (using a Labconco FreeZone lyophilizer). The resulting crude peptide R11 was purified by preparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a SHIMADZU LC-20A chromatography system equipped with an SPD-M20A photodiode array detector with detection at 215 nm. A Kinetex XB-C18 (250 × 21.2 mm, 10 nm) column was used with solvent A (H2O + 0.1% TFA) and solvent B (CH3CN + 0.1% TFA). The peptide was eluted at a flow rate of 10 mL min−1 by the following method: 5% B in 3 min, gradient 5–50% B in 10 min. Fractions containing the desired product were obtained and lyophilized. The purity of the peptide was checked by analytical RP-HPLC using an Onyx Monolithic C18 column (100 × 4.6 mm) and the following method: 5% B in 3 min, gradient 5–50% B in 10 min, flow rate 1.5 mL min−1, Rt 15.7 min (Fig. S1, ESI†). Sample identity was confirmed by ESI-MS (Q Exactive Orbitrap Mass Spectrometer, Thermo Fisher Scientific) (m/z): [M+5H]5+ 348.033, [M+6H]6+ 290.195 (found), [M+5H]5+ 348.032, [M+6H]6+ 290.194 (calcd), as shown in Fig. S2 (ESI†).
:5:5, 1 mL mixture/100 mg of resin). The cleavage was carried out for approximately 7 h at room temperature. The product was precipitated by addition of ice-cold diisopropyl ether (iPr2O) (40 mL). The crude peptide was washed with ice-cold iPr2O (3 × 30 mL), dried under vacuum, dissolved in H2O (10 mL) and lyophilized (using a Labconco FreeZone lyophilizer). The resulting crude peptide R11 was purified by preparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a SHIMADZU LC-20A chromatography system equipped with an SPD-M20A photodiode array detector with detection at 215 nm. A Kinetex XB-C18 (250 × 21.2 mm, 10 nm) column was used with solvent A (H2O + 0.1% TFA) and solvent B (CH3CN + 0.1% TFA). The peptide was eluted at a flow rate of 10 mL min−1 by the following method: 5% B in 3 min, gradient 5–50% B in 10 min. Fractions containing the desired product were obtained and lyophilized. The purity of the peptide was checked by analytical RP-HPLC using an Onyx Monolithic C18 column (100 × 4.6 mm) and the following method: 5% B in 3 min, gradient 5–50% B in 10 min, flow rate 1.5 mL min−1, Rt 15.7 min (Fig. S1, ESI†). Sample identity was confirmed by ESI-MS (Q Exactive Orbitrap Mass Spectrometer, Thermo Fisher Scientific) (m/z): [M+5H]5+ 348.033, [M+6H]6+ 290.195 (found), [M+5H]5+ 348.032, [M+6H]6+ 290.194 (calcd), as shown in Fig. S2 (ESI†).
R11@GNP was covalently grafted onto the carboxylic group of GNPs using an EDC/NHS coupling reaction. EDC (0.13 mmol, 20 mg) and NHS (0.43 mmol, 50 mg) were added into the GNP dispersion (5 mg, in 1 mL of H2O) and sonicated for 40 min to generate semi-stable NHS ester linkage. Then, the R11 (0.3 μmol, 5 mg), dissolved in H2O, was added. After pH adjustment to 8 with 1 mM NaOH, the mixture was stirred in the darkness at room temperature overnight. Unbound R11 was removed from the GNP by centrifugation at 13000 rpm for 30 minutes. R11@GNP samples were purified with ultrapure water by centrifugation/decantation (several times, 13000 rpm, 15 min).
Synthesis of PyCA@GNP
PyCA@GNP conjugates were prepared following the procedure described in ref 21. GNPs (20 mg) and 1-pyrenecarboxylic acid (0.013 mmol, 3.3 mg) were added in methanol (10 mL) and sonicated for 45 min in an ultrasonic bath. Then 40 mL of sterile water was added to the dispersion, and the sonication was continued for 1 day.We obtained stable water dispersions (PyCA@B60_0W), which can be left on a laboratory bench top for weeks without evidence of precipitation (Scheme 1a). In order to remove the excess of PyCA and methanol and to test the stability of PyCA@B60 conjugates, we performed several washing cycles. The red arrows shown in Scheme 1.b describe the washing cycle steps.
 | ||
| Scheme 1 (a) Pictures of the PyCA@B60_MW conjugates after different washing cycles (M = 0–5; M = 0: no wash, M = 1–5: conjugates subjected to washing from 1 to 5 times). (b) Preparation steps of PyCA@B60_MW. | ||
PyCA@B60_0W (sample before washing) was centrifuged at 13000 rpm for 15 min, and then the supernatant was removed, and the same amount of fresh water was added into the remaining precipitate. A black stable dispersion was obtained after the first washing step, and it is named PyCA@B60_1W (see Scheme 1b). The washing procedure was repeated M times (M = 4–5 washing cycles) to obtain an increasingly clean supernatant, which should contain isolated PyCA molecules, methanol and very thin GNPs, possibly conjugated with PyCA. The concentration of all these species is expected to decrease at each subsequent cycle.
Characterization of R11@GNP and PyCA@GNP
Water dispersions of R11@GNP and PyCA@GNP samples were analyzed by UV-Visible absorption spectroscopy (Jasco V-570 spectrophotometer, Japan) in quartz cuvettes (volume of 1 mL, optical path of 1 cm).The chemical structure of the functionalized GNPs was investigated by infrared spectroscopy. The spectra were recorded in the specular reflection (SR) mode using a Thermo Nicolet 6700 FT-IR spectrometer, equipped with a ThermoElectron-Nicolet Continuum FT-IR microscope (15X Infinity Corrected Cassegrain objective, 512 scan, and 4 cm−1 resolution). The SR measurements were performed on samples prepared as flat and thick layers (hundreds of microns thickness) of pressed powders. To convert the reflectance spectrum into an absorption spectrum, we applied the Kramer–Koenig (KK) transformation22 using the OMNIC 8.0 software [see 269-032217 – Ver. 8.0 – OMNIC User Guide.pdf]. Band deconvolution of the FTIR spectra has been performed using the Fityk 9 software.23
Raman spectroscopy was carried out using a Jobin Yvon Labram HR800 Raman spectrometer, coupled to an Olympus BX41 microscope, with a 50 X objective and a He–Ne laser at 633 nm for excitation, and the structural integrity of GNPs was confirmed after functionalization. The powdered conjugates were deposited on an aluminum foil fixed on a glass slide. In each experiment, the power of the laser was set to prevent degradation or photo-induced changes of the samples during the acquisition of the spectra (120 s with a spectral resolution of 2 cm−1).
Fluorescence spectroscopy (Jasco FP-6600 Spectrofluorometer, Japan), with excitation at 290 nm was applied to the study of π–π interactions in PyCA@GNP. The average particle sizes of B60, R11@B60, and PyCA@B60_5W samples and their distribution were estimated by DLS using a Zetasizer Nano ZS Analyzer (Malvern Instruments Ltd, Worcestershire, UK). The samples were diluted with distilled water to adjust the GNP content to 0.067 wt% and directly placed in the cell. All measurements were carried out at 25 °C. Three measurements consisting of 3 scans of 5 s each were acquired in the backscattering (θ = 173°) mode. The same GNP dispersions were placed in Malvern disposable folded capillary cells (DTS1070) and the zeta potential was measured using the same instrument and software as those used for DLS.
3. R11@GNP: covalent functionalization of GNPs
Among the highly cationic peptides that can improve cellular uptake, we selected the cell penetrating peptide (CPP) R11 to prepare a GNP conjugate to be used as a potential delivery system for bladder cancer cell lines.24 Since the coupling of the arginine residue is considered difficult,25 the peptide R11 was synthesized using a double coupling strategy for the entire peptide sequence. R11 was covalently grafted to the GNPs (T60 and B60) via crosslinking amidation between the native carboxyl groups on the edges of the graphene layers and the N-terminal amino group of the peptide by an EDC/NHS coupling reaction, as shown in Scheme 2. | ||
| Scheme 2 Covalent functionalization of GNPs with the poly-arginine-11 peptide via an EDC/NHS coupling reaction between the native COOH groups on the edges of the GNPs and the N-terminal group of the peptide. | ||
3.1. Experimental evidence of covalent functionalization
R11@T60 and R11@B60 were subjected to several analyses, aiming to verify the following:(i) GNP integrity after functionalization. In particular, we verified that the grafting of R11 did not determine any significant modification of the structural characteristics of the graphene layers.
(ii) The achievement of covalent functionalization.
(iii) The possibly different behaviour of T60 and B60 in terms of functionalization yield.
Point (iii) is relevant because the two GNP samples present some different structural characteristics. T60 and B60 differ in size, showing an average lateral size 〈L〉 = 70 nm and 〈L〉 = 120 nm, respectively; their thickness is measured as the average number of stacked layers, and is 〈N〉 = 4 for T60 and 〈N〉 = 6 for B60.18 In addition, the edges of B60 and T60 are decorated by different amounts of oxygen-containing groups, which form spontaneously during the preparation process and provide anchoring sites for a subsequent functionalization.
 | (1) |
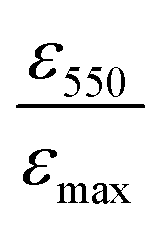 is the ratio between the absorbance value at λ = 550 nm and the absorbance value at the maximum. The 〈N〉 value of R11@T60, reported in Table 2, is close to that of T60 before functionalization. In the case of R11@B60, the shape of the spectrum shows more pronounced changes (Fig. 1(b)). Compared to T60, B60 GNPs are characterized by a larger amount of COOH groups, which promote some GNP aggregation in water dispersion, as proven by DLS.18 Because of the higher number of COOH, B60 should favour a higher degree of functionalization with R11, which in turn can affect the distribution of aggregates in solutions, thus modifying the profile of its UV-vis absorption spectrum.
is the ratio between the absorbance value at λ = 550 nm and the absorbance value at the maximum. The 〈N〉 value of R11@T60, reported in Table 2, is close to that of T60 before functionalization. In the case of R11@B60, the shape of the spectrum shows more pronounced changes (Fig. 1(b)). Compared to T60, B60 GNPs are characterized by a larger amount of COOH groups, which promote some GNP aggregation in water dispersion, as proven by DLS.18 Because of the higher number of COOH, B60 should favour a higher degree of functionalization with R11, which in turn can affect the distribution of aggregates in solutions, thus modifying the profile of its UV-vis absorption spectrum.
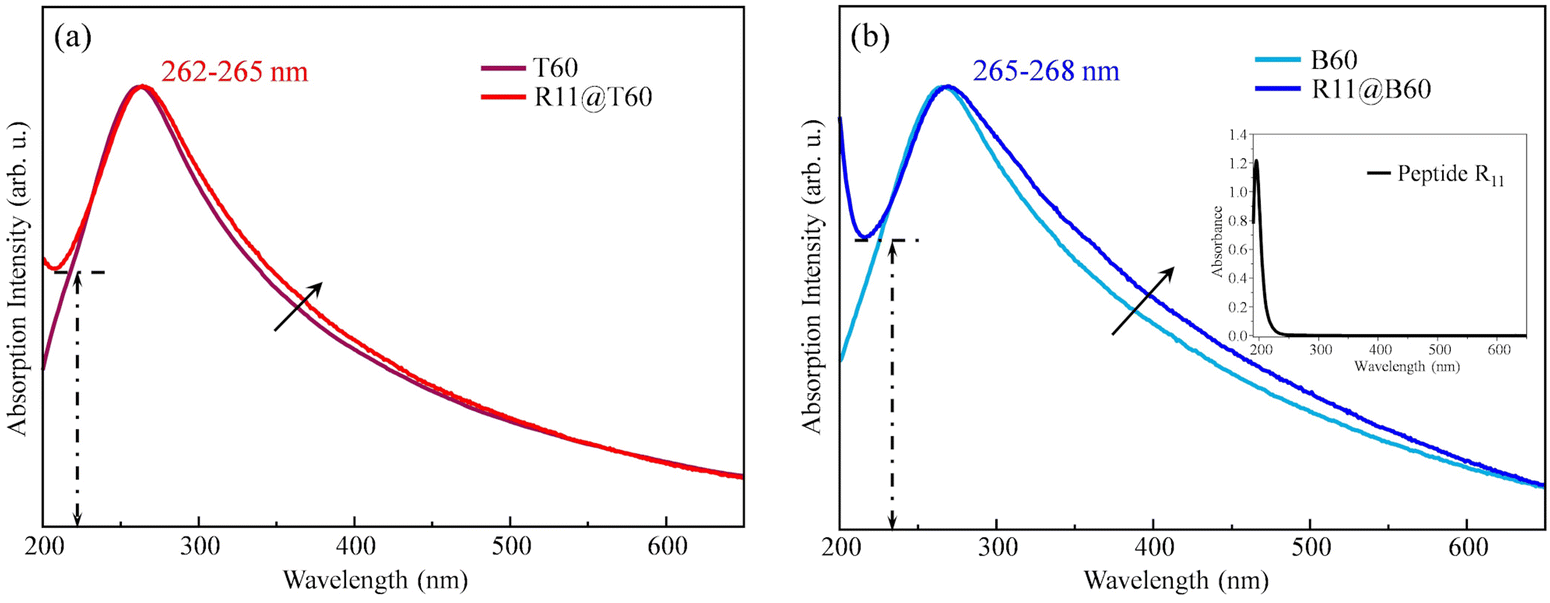 | ||
| Fig. 1 UV-vis absorption spectra of R11@T60 (a) and R11@B60 (b) compared with the spectra of pristine T60 and B60 water dispersions. | ||
| Sample | T60 | R11@T60 | B60 | R11@B60 |
|---|---|---|---|---|
| 〈N〉 | 3.6 | 4.9 | 5.7 | 7.2 |
Interestingly, the UV-vis spectra of the two R11@GNP samples show a steep rise of the absorbance while approaching λ = 200 nm, not observed in the case of bare GNPs. This feature is ascribed to the electronic transition characteristic of R11, peaking at λ < 200 nm, as proven by the UV-vis spectra of R11 in water solution, as reported in Fig. S3 (ESI†).
This evidence suggests successful covalent functionalization, more pronounced for R11@B60 with respect to T60.
As illustrated in Fig. 1, a quantitative comparison is provided by the ratio 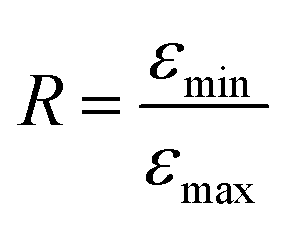 , where εmin is the absorbance value at the local minimum close to λ = 200 nm and εmax is the absorbance value at the GNP peak (λ = 265 nm). The relationship R (R11@B60) = 0.82 > R (R11@T60) = 0.69 confirms a greater functionalization of B60.
, where εmin is the absorbance value at the local minimum close to λ = 200 nm and εmax is the absorbance value at the GNP peak (λ = 265 nm). The relationship R (R11@B60) = 0.82 > R (R11@T60) = 0.69 confirms a greater functionalization of B60.
 | ||
| Fig. 2 Raman spectra of R11@GNP conjugates and GNPs before functionalization, excitation wavelength (λexc) = 633 nm. | ||
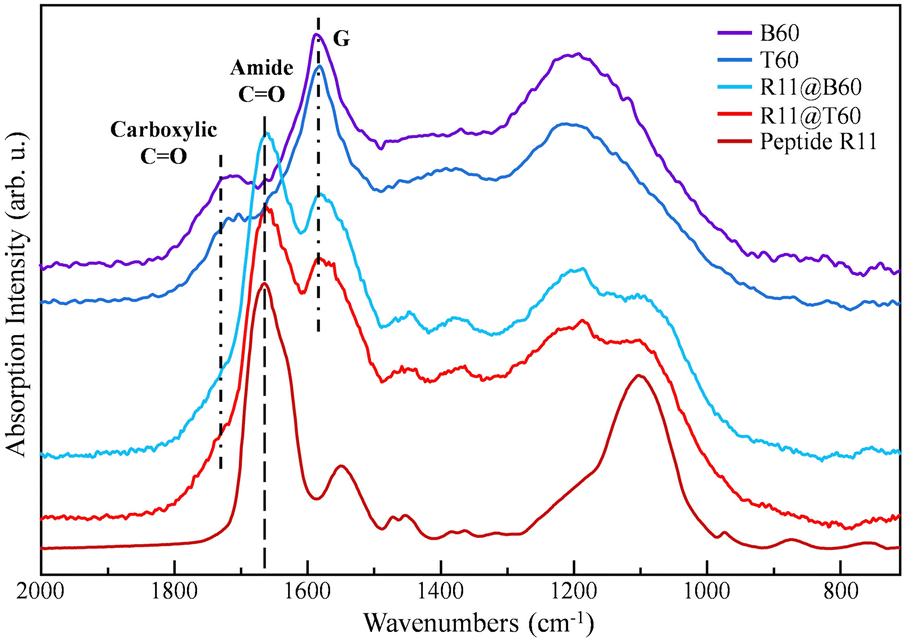 | ||
| Fig. 3 FTIR spectra of T60, B60, R11, R11@T60 and R11@B60. The spectra are vertically stacked to ease comparison. | ||
Fig. 3 allows recognizing the distinctive features of R11 in the FTIR spectra of the conjugates. More precisely, we identify the following:
(i) A strong amide I band at 1666 cm−1 and
(ii) Two structured bands (at about 1450 cm−1 and 1375 cm−1) in the region of 1500–1300 cm−1, where the FTIR spectra of pure GNPs do not show peaks.
The amide I band occurs at a lower wavenumber with respect to the broad and structured band peaking at 1720 cm−1, assigned to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds belonging to the COOH groups and to other functional groups – e.g. aldehydes – which decorate the edges of bare GNPs. The direct comparison among the spectra of T60 and B60 and the corresponding spectra of R11@GNP shows, as expected, a weakening of the 1720 cm−1 band after functionalization because of the grafting via the condensation reaction with COOH groups. Moreover, after conjugation with R11, the shape of the strong absorption band of GNPs in the 1250–1000 cm−1 region changes. This feature can be explained as a result of the convolution of the GNP absorption with the structured band of R11, peaking at about 1099 cm−1. The above-mentioned findings indicate that for the two R11@GNP samples, grafting is effective.
O bonds belonging to the COOH groups and to other functional groups – e.g. aldehydes – which decorate the edges of bare GNPs. The direct comparison among the spectra of T60 and B60 and the corresponding spectra of R11@GNP shows, as expected, a weakening of the 1720 cm−1 band after functionalization because of the grafting via the condensation reaction with COOH groups. Moreover, after conjugation with R11, the shape of the strong absorption band of GNPs in the 1250–1000 cm−1 region changes. This feature can be explained as a result of the convolution of the GNP absorption with the structured band of R11, peaking at about 1099 cm−1. The above-mentioned findings indicate that for the two R11@GNP samples, grafting is effective.
Further evidence is provided by the difference spectrum obtained by subtracting the spectrum of B60 from the spectrum of R11@B60, as shown in Fig. 4. The difference spectrum clearly shows all the main absorption peaks of R11 and shows a profile very similar to the spectrum of R11, despite the different set-up adopted for recording the IR spectra (specular reflection and KK transformation for B60 and R11@B60 and conventional transmission mode for R11).
 | ||
| Fig. 4 Comparison of the FTIR spectra of B60, R11@B60, their difference spectrum (subtraction of the spectrum of B60 from that of conjugate R11@B60) and R11. | ||
Unfortunately, at this stage of the analysis, it is hard to obtain direct evidence of the formation of the covalent bonds with GNPs. However, the change in the shape and intensity of the CO stretching band of GNPs (maximum at 1720 cm−1) after functionalization allows us to state that some COOH groups on the GNP edges are involved in the reaction with R11.
With the aim to quantitatively estimate the degree of GNP functionalization, we subjected the FTIR spectra to a band deconvolution procedure, which allow us to separate R11 and GNP components and to obtain parameters (e.g. band peak, band area, and bandwidth), which provide a measure of their relative importance. The results of the curve fitting for B60 and R11@B60 are illustrated in Fig. 5, while the fitting results for T60 and R11@T60 samples are shown in Fig. S5 (ESI†).
 | ||
| Fig. 5 Deconvolution of the FTIR spectra of B60, R11 and R11@B60. | ||
In Fig. 5, different colors are used to highlight the band components belonging to R11 (red line) and to GNPs (blue and yellow/orange lines). The individual components of the fitting are identified by a number, which corresponds to the band labelling of Tables 3 and 4, which list the characteristic band parameters. The fitting was carried out in the region of 700–2000 cm−1, in a supervised way, using the Fityk 9 software.23 We chose the minimum number of components which allows a good fitting: 7 bands are needed for the fitting of the spectra of T60 and B60, 11 components for R11, and 10 components for R11@T60 and R11@B60. Lorentzian or Voigt band shapes were adopted (Table S1, ESI†).
| # Band | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| T60 | Peak position in cm−1 | 1734 | 1701 | 1584 | 1455 | 1380 | 1201 | 1058 |
| Height | 0.13 | 0.20 | 1.00 | 0.25 | 0.43 | 0.74 | 0.27 | |
| Area | 0.05 | 0.13 | 1.00 | 0.26 | 0.54 | 0.89 | 0.41 | |
| FWHM (cm−1) | 41.99 | 74.30 | 112 | 118.88 | 140.10 | 198.89 | 171.30 |
| # Band | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| B60 | Peak position in cm−1 | 1734 | 1701 | 1583 | 1453 | 1371 | 1196 | 1056 |
| Height | 0.23 | 0.19 | 1.00 | 0.48 | 0.25 | 0.97 | 0.22 | |
| Area | 0.17 | 0.15 | 1.00 | 1.06 | 0.34 | 1.51 | 0.51 | |
| FWHM (cm−1) | 65.11 | 72.86 | 92 | 201.37 | 126.46 | 211.14 | 214.10 |
| # Band | 1 | 8 | 9 | 3 | 10 | 4/12 | 5/13 | 6 | 16 | 7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| R11@T60 | Peak position in cm−1 | 1734 | 1666 | 1630 | 1584 | 1534 | 1450 | 1379 | 1192 | 1098 | 1064 |
| Height | 0.25 | 1.07 | 0.23 | 1.00 | 0.38 | 0.33 | 0.34 | 0.86 | 0.10 | 0.21 | |
| Area | 0.12 | 0.37 | 0.06 | 1.00 | 0.32 | 0.22 | 0.30 | 1.43 | 0.04 | 0.16 | |
| FWHM (cm−1) | 55.67 | 57.20 | 30.29 | 112 | 95.64 | 76.29 | 98.41 | 273.65 | 40.02 | 82.61 |
| # Band | 1 | 8 | 9 | 3 | 10 | 4/12 | 13 | 6 | 16 | 7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| R11@B60 | Peak position in cm−1 | 1734 | 1666 | 1630 | 1583 | 1536 | 1451 | 1374 | 1201 | 1095 | 1050 |
| Height | 0.28 | 1.26 | 0.30 | 1.00 | 0.51 | 0.27 | 0.56 | 0.79 | 0.29 | 0.42 | |
| Area | 0.20 | 0.53 | 0.14 | 1.00 | 0.51 | 0.18 | 0.88 | 1.01 | 0.24 | 0.49 | |
| FWHM (cm−1) | 65.94 | 57.48 | 41.69 | 92 | 92.09 | 61.08 | 145.50 | 173.46 | 78.01 | 106.46 |
| # Band | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide R11 | Peak position in cm−1 | 1670 | 1631 | 1546 | 1471 | 1450 | 1370 | 1315 | 1207 | 1099 | 972 | 872 |
| Height | 1.00 | 0.61 | 0.32 | 0.05 | 0.10 | 0.05 | 0.04 | 0.19 | 0.72 | 0.05 | 0.03 | |
| Area | 1.00 | 0.74 | 0.56 | 0.02 | 0.08 | 0.07 | 0.05 | 0.39 | 1.53 | 0.03 | 0.02 | |
| FWHM (cm−1) | 48.59 | 40.3 | 58.75 | 10.77 | 27.03 | 44.76 | 48.36 | 94.96 | 102.72 | 23.39 | 24.29 |
As in the previous work on T60 and B60,18 in the spectra of GNPs and in the spectra of the conjugates, we identified the G band (1583 cm−1, band #3), and a broad band (band #6), centered at about 1200 cm−1, both characteristic of the graphene layers. Because the graphene layers are not affected by the bonding with R11, in the fitting of the spectra of the conjugates, the wavenumber and FWHM of the G band were kept fixed to the values obtained by fitting the T60 and B60 spectra.
The structured CO stretching band, associated with the oxygenated groups on the edges of the graphene layers, appeared in the region of 1700–1730 cm−1. In Fig. 5 and Fig. S5 (ESI†) (top panels), two components – labelled as #1, #2 – can be taken as markers of the native CO groups of GNPs belonging to the aldehyde and carboxyl groups, showing characteristic CO stretching bands at higher and lower frequencies respectively. The presence of structural disorder, which is typical of T60 and B60 GNPs, is evidenced by the occurrence of broad bands #4, #5 and #7. These three bands are also present in the spectra of R11@GNPs.
In the following analysis of the parameters obtained from the deconvolution of R11@GNP spectra (Tables 3 and 4), we will mainly focus on the CO stretching region in the different samples. The data from spectra deconvolution allow deriving some other useful parameters (Table 5). The amount of CO groups on GNPs was estimated by the total intensity (sum of the areas of the two CO stretching components at 1734 and 1701 cm−1) normalized to the area of the G band, ACO(1,2)/AG. This parameter indicates that B60 (ACO(1,2)/AG = 0.31) show a larger amount of native carboxylic groups with respect to T60 (ACO(1,2)/AG = 0.18). In Table 5, the parameter ACO(8,9)/AG of R11@GNPs describes the additional contribution to the total CO stretching intensity (bands #8, #9), ascribed to the CO groups of R11, and indicates that R11@B60 bears an excess of peptide functionalities of about 50% with respect to R11@T60.
O stretching band intensity and the IR G-band intensity of GNP and R11@GNP samples: region of amide CO groups (bands #8 and #9) and region of oxygen-containing functional groups of bare GNPs (bands #1 and #2). The intensity is measured as integrated area
| Samples |
A
CO(8,9)/AG |
A
CO(1,2)/AG |
|---|---|---|
| T60 | — | 0.18 |
| R11@T60 | 0.43 | 0.12 (Δ% = −33%) |
| B60 | — | 0.31 |
| R11@B60 | 0.67 | 0.20 (Δ% = −35%) |
A similar information could be obtained by considering the integral over the whole CO stretching region in R11@GNP (sum of the areas of bands #1, #2, #7, #8, i.e. ACO,total). In R11@T60 and R11@B60, ACO,total/AG is about two times and three times larger than that of the corresponding GNPs, respectively.
Considering the change in the intensity of bands #1 and #2 in the R11@GNP samples, we found that not all the oxygen-containing groups in the nanoparticles were exploited to link R11 (Fig. 5). Two bands (#1 and #2) fit the broad CO stretching feature of B60 and T60, while after grafting, one only component survives (band #1 at 1734 cm−1). This suggests that the COOH groups – responsible for component #2 – are the preferential sites for grafting R11 by condensation. A slight increase in the band area of component #1 going from the GNPs to the corresponding conjugates cannot be easily rationalized, but could be ascribed to the uncertainty in the fitting procedure. The relevant feature is the decrease in the value of the “global” parameter ACO(1,2)/AG, after grafting R11 (Table 5), because of the disappearance of band #2 in the R11@GNP spectra.
In conclusion, the IR pattern of R11@GNP samples proves that the functionalization has been successful and that COOH groups along the edges of GNPs are the suitable sites for grafting R11. This is further confirmed by the fact that the use of B60 allows raising the yield of the functionalization reaction by 50%, thanks to the larger amount of native COOH groups.
Table 6 allows the comparison of the relevant DLS parameters of R11@B60 with those of the aqueous dispersion of B60, while the histograms of the particle size distribution are compared in Fig. S6 (ESI†). The increase (of about 30%) in the average hydrodynamic radius, in conjunction with a wider size distribution (PDI), can be mainly ascribed to the occurrence of particle aggregation in R11@B60 samples, which is further proven by the presence of a non-negligible amount of very big aggregates, showing diameters of the order of a few microns. Furthermore, the zeta potential value of R11@B60 is low and suggests that the particles are prone to form aggregates in water. A remarkable proof of the successful grafting of B60 by condensation on the native COOH groups of B60 is the change in the sign of zeta potential. This result clearly indicates that the acid COOH groups, responsible for the large negative zeta potential of the B60 sample, have been removed after the formation of the covalent link with R11.
| Samples | 〈d〉 (nm) | PDI | ζ (mV) |
|---|---|---|---|
| B60 | 179.6 | 0.151 | −47.2 |
| R11@B60 | 243 | 0.256 | 11.3 |
| PyCA@B60_5W | 221.4 | 0.263 | −45.2 |
| PyCA@B60_5W (40C-55 min) | 176.2 | 0.156 | −29.1 |
4. PyCA@GNP: loading of molecules via π–π interactions
PyCA composed of condensed aromatic rings like several anti-cancer drugs is a simple model for the study of possible mechanisms of drug loading on GNPs and drug release. It is reported in the literature that PyCA can form stable complexes with graphene layers.21 In a polar medium (e.g. H2O) containing GNPs, the non-polar pyrene unit can stick on top of the graphene surface via π–π interactions (Scheme 3), or it can penetrate within the graphene layers to reduce hydrophobic interactions with water, while the polar –COOH groups PyCA should enhance the stability of nanoparticles dispersed in aqueous solutions. | ||
| Scheme 3 Structure of PyCA and sketch of the PyCA@GNP conjugate. The condensed aromatic rings are involved in the π–π interactions on the basal plane of a GNP. | ||
PyCA is a strongly fluorescent molecule, but its fluorescence emission is quenched by intermolecular π–π interaction as reported for several fluorescent dyes and organic molecules containing condensed aromatic rings.27,28 Fluorescence quenching can be exploited for monitoring the uptake and release of drugs from GNPs.
We can provide an estimate of the potential drug loading capability (DLC) of B60 GNPs by means of a simple structural model of PyCA@B60, which is illustrated in detail in the ESI.† In the hypothesis that PyCA molecules form a dense, ordered monolayer adsorbed on the exposed surfaces of the nanoparticles, and considering that B60 nanoparticles are formed, on average, by 6 staked graphene layers, we obtained an ideal DLC = 0.12 (DLC was calculated as the weight ratio between the amount of absorbed PyCA and the amount of GNP carriers). This remarkable loading capability is attributed to the large surface-to-volume ratio of B60 GNPs. Interestingly, the ideal DLC value is close to the relative amount of PyCA and GNPs we used for the preparation of PyCA@B60 (16%).
4.1. Experimental evidence of π–π interaction functionalization
PyCA@B60 conjugates were characterized by complementary spectroscopies, aiming to assess the following:(i) The successful conjugation of PyCA to B60 GNPs and
(ii) The reversibility of PyCA functionalization by means of thermal release.
 | ||
| Fig. 6 UV-vis absorption spectra of PyCA@B60_0W (no wash) and PyCA@B60_MW conjugates (M = 1–5: samples subjected to M washing cycles); spectra are stacked for clarity. Inset: Spectra of pure PyCA (in water) and a water dispersion of B60 GNPs. | ||
For a more effective comparison, we show in Fig. 7 the subtraction spectra obtained as the difference between the UV-vis spectrum of each PyCA@B60_MW and that of a water dispersion of B60 GNPs. All the subtraction spectra not only clearly display the absorption bands characteristic of PyCA in the solution but also reveal broad background and absorption features in the longer wavelength region of 360–450 nm, similar to those observed in the UV-vis spectrum of solid PyCA.
 | ||
| Fig. 7 Difference spectra obtained by subtracting the spectrum of B60 dispersion from that of PyCA@B60_MW (M = 0, 1, 2, 4, and 5). The spectra are normalized on the PyCA bands and are stacked for clarity. Spectra of a PyCA water/methanol solution and solid PyCA are shown for the sake of comparison. | ||
These observations suggest that the interaction of PyCA molecules with GNPs does not appreciably perturb the absorption features of the pyrene moiety. However, the broad absorption of PyCA@B60 at long wavelengths could be ascribed to PyCA molecules intercalated between graphene layers, which are experiencing a solid-state-like environment.
To confirm that the PyCA features observed in the PyCA@B60_MW spectra (Fig. 6) indicate a successful conjugation, we prepared a physical mixture of PyCA/GNPS in water (see ESI† for the preparation method, and Scheme S2) and compared the UV-vis spectra of PyCA@B60_0W with the one of the mixture (Fig. S9 and S10, ESI†).
An estimate of the relative concentration of PyCA and GNPs can be obtained from the UV-vis spectra (see Fig. 6) as the ratio (R) of the area of the strongest PyCA absorption band, floating on the GNP absorption feature in the region of 210–260 nm, with the whole integral of absorption in the same spectral range. Fig. S11 (ESI†) illustrates the procedure adopted, focusing on the spectrum of PyCA@B60_5W. Even if the peak heights of UV-vis spectra – or ratios between peak heights – are more commonly used for the sake of quantification, we preferred the use of integrals, which provide more stable results, particularly in the presence of weak absorption features.
The values of R reported in Table 7 provide the quantitative evidence that the relative contribution of PyCA molecules and of GNPs to the absorption spectrum of PyCA@B60_MW is quite stable, even after several washing steps. This proves that conjugates have a stable link between PyCA and GNPs. Moreover, the negligible effect of washing on the spectroscopic response of PyCA@B60_MW samples suggests that our protocol for the preparation of the conjugates allows reaching a very high reaction yield, and that the degree of covering of GNP surfaces is close to the ideal one illustrated in the ESI† (Scheme S3).
| PyCA@B60_MW | M = 1 | M = 2 | M = 3 | M = 4 | M = 5 |
|---|---|---|---|---|---|
| Relative intensity of PyCA R = APyCA/APyCA+B60 | 0.07 | 0.07 | 0.06 | 0.08 | 0.07 |
An accurate interpretation of the experimental evidence requires a preliminary characterization of PyCA in the solid state and solution with water and water/methanol, in order to discriminate fluorescence from aggregated or isolated PyCA molecules. The fluorescence spectrum of PyCA molecules in the solid state is very weak and shifts in the region at a wavelength longer than 500 nm, far from the characteristic spectral range where the fluorescence of isolated PyCA molecules and PyCA@B60 can be detected. In the ESI,† we discuss the fluorescence spectra of PyCA recorded in water and water/methanol mixtures (Fig. S12, ESI†): a distinctive fluorescence pattern, ascribed to the isolated PyCA molecules in solution, since PyCA aggregates precipitate or do not contribute to the fluorescence emission.
The fluorescence spectra of PyCA@B60_0W and a water dispersion of a PyCA/B60 physical mixture (prepared with the same concentration of GNPs and PyCA of PyCA@B60_0W sample) are shown in Fig. 8. PyCA@B60_0W shows a strong fluorescence reduction, which indicates the occurrence of quenching due to the π–π interaction between PyCA and B60, while the physical mixture has a comparable intensity to that of a PyCA water solution at the same concentration. In all the spectra, it is reasonable to ascribe the fluorescence signal to the presence of isolated PyCA molecules in water – it should be noticed that the PyCA@B60_0W sample was not subjected to any washing cycle, thus we expect that some amounts of free PyCA molecules are present. As alternative explanation, we can suggest that the PyCA–GNP interaction does not completely quench the pathway toward fluorescence emission. Irrespective of the two possible interpretations, the spectra in Fig. 8 provide additional evidence for the successful functionalization of B60 with PyCA, via π–π interactions.
 | ||
| Fig. 8 Fluorescence spectra of the water dispersions of PyCA@B60_0W (red line) and PyCA/GNPS (light blue line), and PyCA in the water solution (dark blue line). The PyCA concentration is the same for the three samples and the spectra are displayed after normalization to that of PyCA in water, showing the highest fluorescence signal. Inset: The same three spectra are shown in “full scale”, after normalization of each spectrum on the strongest peak. | ||
In Fig. 9, we compare the fluorescence spectrum of PyCA@B60_MW (M = 0 to 5). It is reasonable that, after 5 washing cycles, a negligible amount of free PyCA is present in the solution: in the hypothesis that the π–π interaction perfectly quenches the fluorescence signal, the spectrum could be explained as due to a small amount of PyCA linked to the edges of the GNP through hydrogen bonding with carboxylic acid groups. This type of interaction allows the pyrene moiety to experience an environment like in the solution, thus preventing, or limiting, the quenching effect.
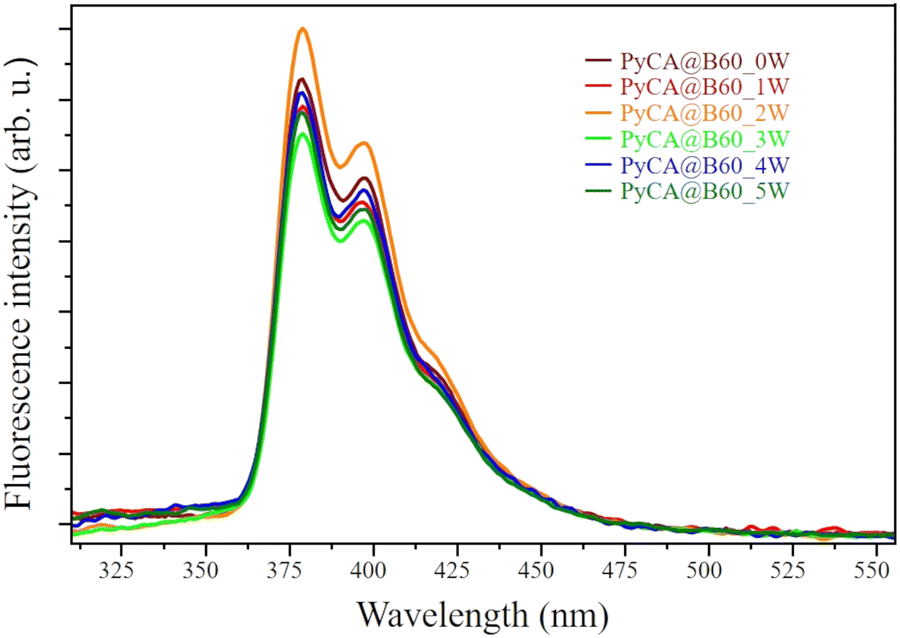 | ||
| Fig. 9 Fluorescence spectra of the PyCA@B60_MW conjugates (M = 0–5; 0: no wash, M = 1–5: after M washing cycles). The spectra are displayed after normalization to that of PyCA@B60_2W, showing the highest fluorescence signal. | ||
 | ||
| Fig. 10 Raman spectra of B60 GNPs and PyCA@B60_5W samples, excitation wavelength (λexc) = 633 nm. | ||
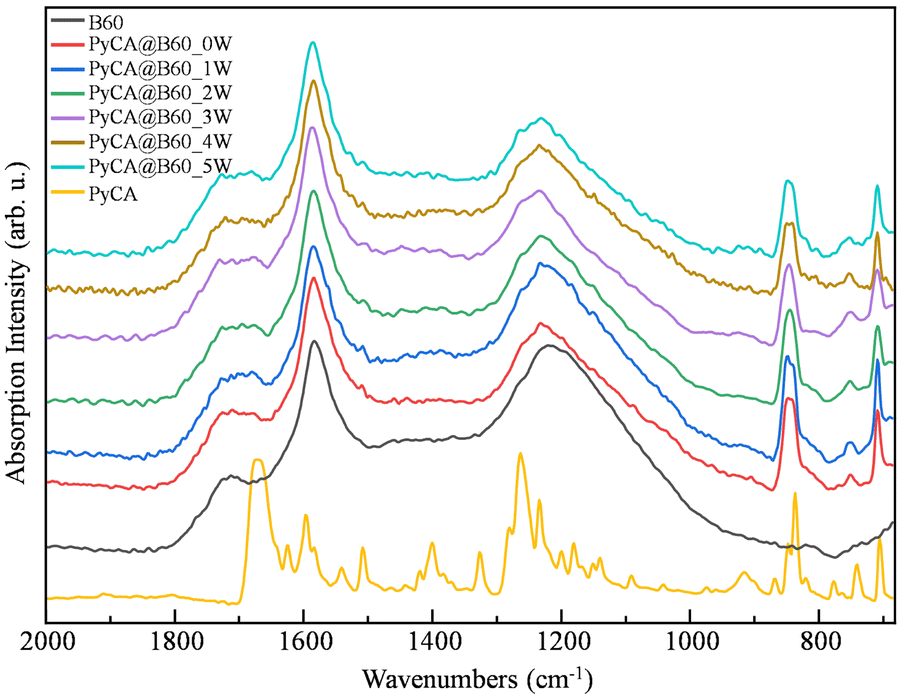 | ||
| Fig. 11 FTIR spectra of B60 GNPs, PyCA@B60_MW (with M = 0–5) and a solid (powder) PyCA sample. The spectra are stacked for clarity. | ||
PyCa shows a strong CO stretching band at a rather low wavenumber (∼1665 cm−1), which indicates the presence of strong intermolecular hydrogen bonds between carboxylic groups.
The CO stretching band of B60 (∼1720 cm−1) due to the native oxygen-containing groups of GNPs becomes broader in the spectra of all the PyCA@B60_MW samples, thus revealing the presence of new components. Moreover, the increase in the integrated absorption of the whole CO stretching region (area between 1650 cm−1 and 1800 cm−1) in the spectra of PyCA@B60_MW confirms that additional CO groups from PyCA contribute to the spectra of the conjugates.
The three peaks at 846, 757 and 709 cm−1 in the spectra of PyCA@B60_MW (Fig. 11) can be ascribed to the characteristic IR transitions of PyCA, and prove that PyCA is loaded on GNPs. The intensity of the strong band at 846 cm−1 (assigned to the CH out-of-plane bending mode), allows estimating the degree of PyCA uptake. As illustrated in Table 8, at each washing step, the relative amount of PyCA in the conjugates can be evaluated considering the peak height ratio, H = ICHopla/IG (or band area ratio, R = ACHopla/AG) of the 846 cm−1 band of PyCA and of the G-band (1586 cm−1) characteristic of GNPs. After three washing cycles, both H and R reach a stable value (H = 0.50, R = 0.27), showing that in subsequent washing steps, there is no further loss of PyCA molecules.
| PyCA@B60_MW | M = 1 | M = 2 | M = 3 | M = 4 | M = 5 |
|---|---|---|---|---|---|
|
H = ICHopla/IG |
0.71 | 0.63 | 0.51 | 0.50 | 0.50 |
|
R = ACHopla/AG |
0.41 | 0.33 | 0.27 | 0.27 | 0.27 |
The comparison between the IR spectrum of PyCA and that of B60 (see Fig. 11) suggests that, in the PyCA@B60 spectra, the characteristic bands of GNPs (bands 3–7, Fig. 5) could hide some components, arising from IR transitions of PyCA. A close look to the spectrum of PyCA@B60 shows that the shape of the G band (band 3) changes and there is an asymmetric broadening on the higher frequency side of the strong band peaking at ∼1200 cm−1 (band 6 in Fig. 5). This is not surprising, since vibrational normal modes localized on the pyrene moiety – consisting of four condensed aromatic rings – show analogies with vibrations of small graphene-like domains.
To better highlight the PyCA contributions in the IR spectrum of the conjugates, we carried out the subtraction between the spectrum of PyCA@B60_5W and that of B60 (Fig. 12). The difference spectrum reported in Fig. 12 is a good representative of PyCA@B60 conjugates. Indeed, Fig. S14 (ESI†) displays the whole set of spectra we obtained by subtracting the spectrum of B60 from that of PyCA@B60_MW (M = 0–5), showing that after three washing steps, the difference spectrum does not change.
 | ||
| Fig. 12 From the top to bottom: FTIR spectrum of PyCA@B60_5W, FTIR spectrum of B60, and the result from their subtraction (PyCA@B60_5W – B60); FTIR spectrum of PyCA in CHCl3 solution (bands of CHCl3 are removed for clarity) and as microcrystalline powder. | ||
In Fig. 12, we identify eight components in the difference spectrum (green line): four components lying in the spectral range 1800–1100 cm−1 are labelled following the same numbering of the four B60 bands close in frequency (bands 1′, 2′, 3′ and 6′), while the four lower frequency components (bands 8–11) do not find any correspondence to GNP vibrational features. Fig. 12 also reports the spectrum of PyCA in a CHCl3 solution as a representative of the isolated PyCA molecule: it shows that in a disordered environment, where the formation of directional and strong hydrogen bonds are unlikely, the CO stretching band (1712 cm−1) up-shifts to 14 cm−1 with respect to the corresponding band in the PyCA crystal (1668 cm−1) and is located between bands 1′ and 2′ of PyCA@B60. Interestingly, the spectrum of the solution displays only one strong CC stretching band at 1600 cm−1, showing rather good correspondence in frequency with the 3′ line.
In conclusion, based on the detection of three strong CH features below 900 cm−1 in the IR spectrum of PyCA@B60, we can state that PyCA is loaded on GNPs. In addition, the spectral features highlighted by means of spectra subtraction, and in particular, the CO stretching bands in the 1750–1650 cm−1 region suggest that PyCA molecules are loaded on the GNP surface mostly as single molecules, exploiting an effective π–π interaction with the graphene layer.
The deconvolution of the FTIR spectrum of the PyCA@B60_5W sample was carried out and it is illustrated in Fig. 13. Different colors are used to distinguish the components coming from GNPs (blue and yellow/orange lines, same as in Fig. 5) and from PyCA (red lines). The individual components resulting from the fitting are identified by a number which corresponds to the band labelling adopted in Table 9 and in Fig. 12.
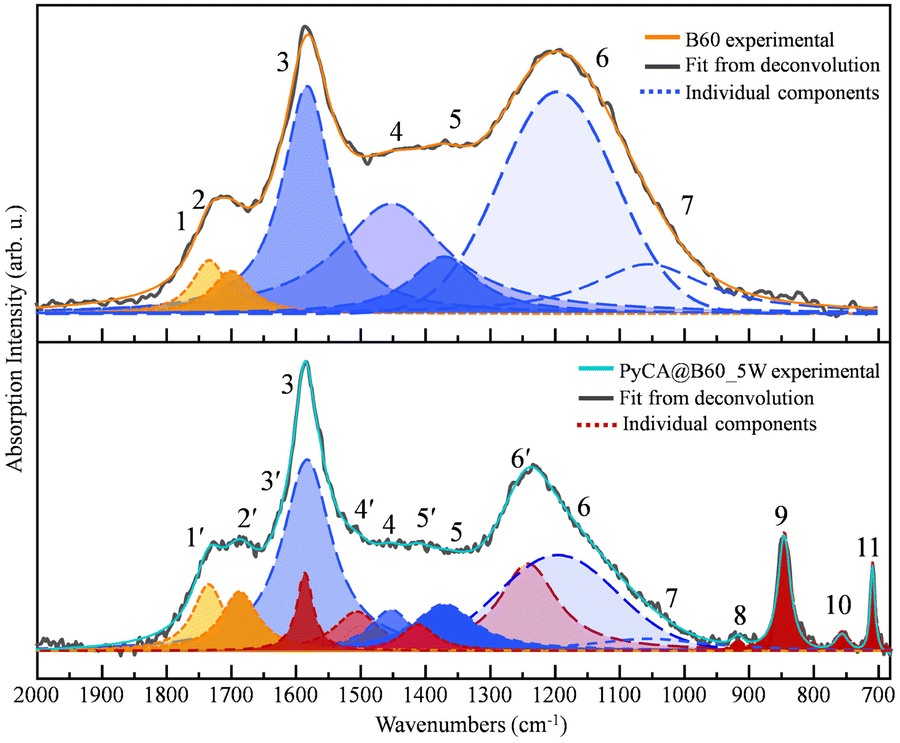 | ||
| Fig. 13 Deconvolution of the FTIR spectra of B60 and PyCA@B60_5W. | ||
| # Band | 1 | 2 | 3 | 4 | 5 | 6 | 7 | |
|---|---|---|---|---|---|---|---|---|
| B60 | Peak position in cm−1 | 1734 | 1701 | 1583 | 1453 | 1371 | 1196 | 1056 |
| Height | 0.23 | 0.19 | 1.00 | 0.48 | 0.25 | 0.97 | 0.22 | |
| Area | 0.17 | 0.15 | 1.00 | 1.06 | 0.34 | 1.51 | 0.51 | |
| FWHM (cm−1) | 65.11 | 72.86 | 92 | 201.37 | 126.46 | 211.14 | 214.10 |
| # Band | 1′ | 2′ | 3′ | 3 | 4′ | 4 | 5′ | 5 | 6′ | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PyCA@B60_5W | Peak position in cm−1 | 1735 | 1687 | 1586 | 1583 | 1506 | 1453 | 1412 | 1371 | 1242 | 1196 | 1056 | 917 | 846 | 757 | 709 |
| Height | 0.35 | 0.31 | 0.41 | 1.00 | 0.21 | 0.22 | 0.14 | 0.24 | 0.46 | 0.50 | 0.06 | 0.06 | 0.61 | 0.10 | 0.47 | |
| Area | 0.22 | 0.23 | 0.13 | 1.00 | 0.19 | 0.18 | 0.09 | 0.31 | 0.54 | 0.82 | 0.15 | 0.01 | 0.16 | 0.03 | 0.04 | |
| FWHM (cm−1) | 58.55 | 68.87 | 30.04 | 92.00 | 83.95 | 78.66 | 62.28 | 115.80 | 108.58 | 204.68 | 214.00 | 21.28 | 23.40 | 26.46 | 8.82 |
As already illustrated in Section 3 (see Fig. 5), curve fitting was performed in the region of 680–2000 cm−1 by adopting the minimum number (7) of components required for a good fitting of the B60 spectrum. In the case of PyCA@B60_5W, eight additional components are needed. The details of the fitting procedure carried out in a supervised way are reported in the ESI.†
A very important result from the fitting is that the components of B60, introduced in the fitting of the PyCA@B60 spectrum, allow a very nice fit, provided that some new components (4′, 5′, and 6′) are included to describe the contribution of PyCA. This is another indication that GNPs are little perturbed by the conjugation with PyCA.
Table 9 reports the relevant parameters of the individual components, featuring Lorentzian or Voigt band shapes (Table S2, ESI†). Table 10 shows that the total absorption intensity of the CO groups of PyCA@B60 increases after functionalization, since each PyCA carries an additional COOH group.
O stretching band intensity and the IR G-band intensity of the GNP and PyCA@B60_5W sample: contribution of the CO groups after the PyCA upload (bands 1′ and 2′) and contribution of the native oxygen-containing functional groups of GNPs (bands 1 and 2). The intensity is measured as the integrated area
| Samples |
A
CO(1/1′,2/2′)/AG |
|---|---|
| B60 | 0.32 |
| PyCA@B60_5W | 0.45 |
4.2 Thermal release
Since the π–π interaction is weak compared with that of a covalent bond, it can be easily dismantled by heating the samples. This phenomenon could be exploited for the release of drugs loaded on GNPs via a π–π bond. The proof-of-concept is obtained through the study of the thermal release of PyCA, by heating a PyCA@B60 sample. We performed three different experiments by heating 3 mL of PyCA@B60_5W water dispersion by means of a thermostatic water bath at different constant temperatures (T = 30 °C, 40 °C and 50 °C), and we monitored the fluorescence spectra at increasing times. The fluorescence intensity shows a remarkable increase after heating for about 1 hour (Fig. 14), suggesting that some PyCA molecules are detached from GNPs and contribute, as isolated molecules in solutions, to the growth of the fluorescence. Importantly, the observed phenomenon provides an additional proof that in PyCA@B60 samples, the PyCA molecules are attached on the GNP surface, forming π–π conjugates affected by fluorescence quenching.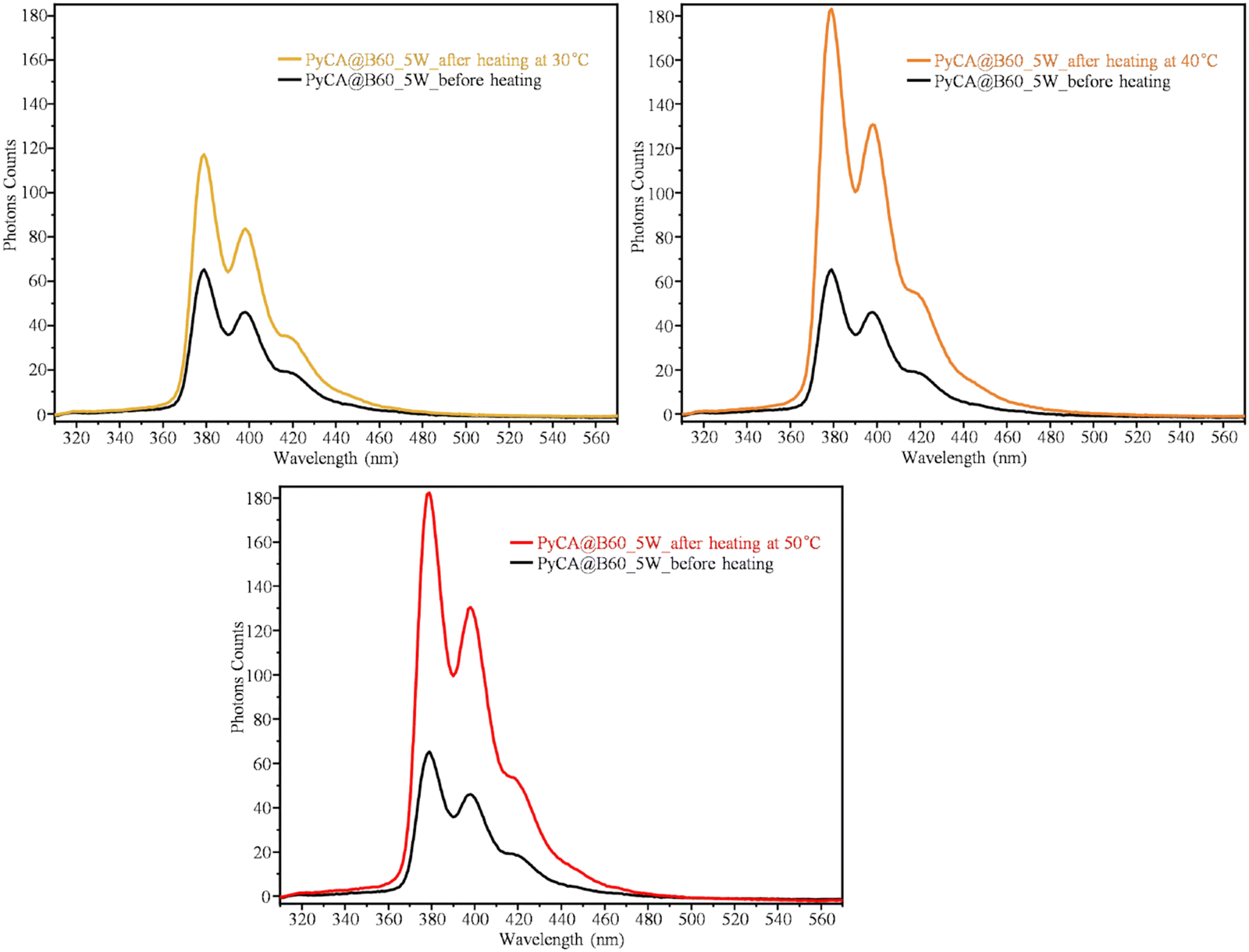 | ||
| Fig. 14 Fluorescence spectra of PyCA@B60_5W conjugates before heating and after local heating at T = 30 °C for 25 min, at T = 40 °C for 55 min, and at T = 50 °C for 55 min. | ||
The temperature and time dependence of the fluorescence intensity (measured as the height of the main emission peak, at 375 nm) is illustrated by the plots shown in Fig. 15. The PyCA release increases with time reaching a plateau value after heating for 50 minutes at 40 °C or 50 °C. The plateau value of the fluorescence emission is about three times higher than the initial one. After one night, during which the sample reaches the room temperature, the fluorescence intensity remains stable. As expected, at a lower temperature (T = 30 °C), the release is less effective and the maximum fluorescence signal recorded is about two times the initial one. Interestingly, also the UV-vis absorption spectrum shows an increase in the intensity of the PyCA bands during the thermal release (see Fig. S15, ESI†), suggesting that the interaction between PyCA and GNP lowers the dipole strength of the PyCA electronic transitions.
 | ||
| Fig. 15 Fluorescence intensity trend with increasing heating time of PyCA@B60_5W at three different temperatures. Spectra are recorded while keeping the sample at a constant temperature. The dotted segment corresponds to a time range during which the thermostat water bath was removed, reaching room temperature. | ||
As a further check, in order to confirm that the thermal release of PyCA is the mechanism responsible for the growth of the fluorescence signal during heating PyCA@B60_5W, we monitored the fluorescence spectrum of a solution of pure PyCA subjected to heating (Fig. S16, ESI†): in this case, the spectra at different times showed an almost constant fluorescence signal, proving that heating does not affect the fluorescence of isolated PyCA molecules.
The main achievement from the thermal release experiments is the evidence that a moderate heating can break the bonds between B60 nanoparticles and PyCA, as expected for non-covalent π–π interactions. A similar mechanism should work also for drugs loaded on B60 through π–π bonds. However, at this stage of the research, the suitable temperature range for a controlled drug release is unpredictable without specific experiments on drug@GNPs conjugates. In addition, the development of drug carriers will require considering the effect of the environment, e.g. biological fluids and tissues.
5. Conclusions
The physical methods used for preparing GNPs are simpler and more environmentally friendly than the chemical methods, and can enable a superior control of the chemical composition. However, the physical methods often result in samples with inhomogeneous structural features. Typically, ball-milling of graphite provides GNP samples with a wide distribution of particle sizes and shapes, possibly presenting different chemical–physical features and different degrees of cell toxicities. To overcome this issue, by means of sorting strategies followed by a thorough structural characterization, we developed a protocol18 that allowed us obtaining GNPs, namely T60 and B60 GNPs, featuring well-defined and reproducible structural characteristics.We demonstrated the covalent grafting on T60 and B60 of the cell-penetrating peptide poly-arginine-11 (R11) by the standard EDC/NHS activation of the carboxyl groups of GNPs and the subsequent formation of an amide bond with the N-terminal amino group of R11. This condensation reaction was optimized to achieve a sizeable amount of peptides attached on the GNP surface. The joint use of UV-vis, Raman and IR spectroscopies provided evidence of the successful synthesis of R11@GNPs and showed that the functionalization yield depends on the amount of COOH groups already present on bare GNPs.
Moreover, we studied the π–π interactions between GNPs and PyCA, a small PAH molecule characterized by the presence of few condensed aromatic rings. By using PyCA as the model compound, we mimed the interaction between the graphene platform and the condensed ring moiety of an anticancer-drug. Besides UV-vis, Raman and IR spectroscopies, the study of the fluorescence response of PyCA@GNP conjugates allowed us to demonstrate the upload of PyCA, which is revealed by the quenching of fluorescence because of π–π interactions. By monitoring over time the fluorescence signal of PyCA@GNP samples heated at different temperatures, we demonstrated that it is possible to trigger the drug release with a thermal input at a rather low temperature. By modelling the structure of a PyCA molecular mono-layer adsorbed on the GNP surfaces in PyCA@B60, we predicted an ideal DLC = 0.12, which is compatible with the experimental findings.
The goal of this research was the combination of facile, inexpensive and environmentally friendly preparation of GNPs with the set-up of several complementary characterization techniques. The study demonstrated that our method enables the cost-effective production of GNPs suitable for the subsequent functional modification with peptides and for the reversible loading of small molecules. Our laboratory-made drug delivery platforms boast a structure more precisely defined with respect to the widely used commercially available GNPs synthesized via the reduction of graphene oxide. In particular, T60 and B60 feature anchoring COOH sites along the periphery of the graphene layers, which guarantee a covalent functionalization with targeting units (e.g. with peptide) without affecting the flat perfect graphene surfaces. Such surfaces should allow effective loading by means of a fully reversible π–π bond with several drugs, as demonstrated with the test molecule PyCA.
Moreover, in this study we demonstrated that a thorough investigation of the functionalized graphene at the molecular level is possible by well-established and easily available spectroscopic techniques (i.e. UV-vis, FTIR and Raman).
We believe that this study could be of great help for the growing number of academic and industrial researchers working on the development of effective GNP platforms for cell targeting and drug upload. The results obtained pave the way for further studies, exploiting the methods of preparation and characterization to produce new GNP-based platforms covalently grafted with different peptides or other molecules able to improve the biocompatibility and/or targeting properties. Drug loading/release will be investigated in the future focusing on molecules relevant in therapies for the treatment of specific diseases.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Hu Kaiyue acknowledges the financial support from the China Scholarship Council (CSC, No. 202006560017).References
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano-Graphene Oxide for Cellular Imaging and Drug Delivery, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Graphene and graphene oxide: biofunctionalization and applications in biotechnology, Trends Biotechnol., 2011, 29, 205–212 CrossRef CAS PubMed.
- C. Chung, Y. K. Kim, D. Shin, S. R. Ryoo, B. H. Hong and D. H. Min, Biomedical applications of graphene and graphene oxide, Acc. Chem. Res., 2013, 46, 2211–2224 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, Graphene-based nanomaterials for drug delivery and tissue engineering, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- X. Y. Yang, X. Y. Zhang, Z. F. Liu, Y. F. Ma, Y. Huang and Y. Chen, High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide, J. Phys. Chem. C, 2008, 112, 17554–17558 CrossRef CAS.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, PEGylated nanographene oxide for delivery of water-insoluble cancer drugs, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- S. K. Singh, M. K. Singh, P. P. Kulkarni, V. K. Sonkar, J. J. Gracio and D. Dash, Amine-modified graphene: thrombo-protective safer alternative to graphene oxide for biomedical applications, ACS Nano, 2012, 6, 2731–2740 CrossRef CAS PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Stimuli-responsive nanocarriers for drug delivery, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- A. Gabizon, H. Shmeeda and Y. Barenholz, Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies, Clin. Pharmacokinet., 2003, 42, 419–436 CrossRef CAS PubMed.
- Q. Zhang, W. W. Li, T. Kong, R. G. Su, N. Li, Q. Song, M. L. Tang, L. W. Liu and G. S. Cheng, Tailoring the interlayer interaction between doxorubicin-loaded graphene oxide nanosheets by controlling the drug content, Carbon, 2013, 51, 164–172 CrossRef CAS.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Noncovalent Functionalization of Graphene and Graphene Oxide for Energy Materials, Biosensing, Catalytic, and Biomedical Applications, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- A. Raza, T. Rasheed, F. Nabeel, U. Hayat, M. Bilal and H. M. N. Iqbal, Endogenous and Exogenous Stimuli-Responsive Drug Delivery Systems for Programmed Site-Specific Release, Molecules, 2019, 24, 1117 CrossRef CAS PubMed.
- D. Liu, F. Yang, F. Xiong and N. Gu, The Smart Drug Delivery System and Its Clinical Potential, Theranostics, 2016, 6, 1306–1323 CrossRef CAS PubMed.
- A. Raza, U. Hayat, T. Rasheed, M. Bilal and H. M. N. Iqbal, “Smart” materials-based near-infrared light-responsive drug delivery systems for cancer treatment: A review, J. Mater. Res. Technol., 2019, 8, 1497–1509 CrossRef CAS.
- E. Khakpour, S. Salehi, S. M. Naghib, S. Ghorbanzadeh and W. Zhang, Graphene-based nanomaterials for stimuli-sensitive controlled delivery of therapeutic molecules, Front. Bioeng. Biotechnol., 2023, 11, 1129768 CrossRef PubMed.
- B. P. Timko, T. Dvir and D. S. Kohane, Remotely triggerable drug delivery systems, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- J. Song, J. Qu, M. T. Swihart and P. N. Prasad, Near-IR responsive nanostructures for nanobiophotonics: emerging impacts on nanomedicine, Nanomedicine, 2016, 12, 771–788 CrossRef CAS PubMed.
- K. Hu, L. Brambilla, P. Sartori, C. Moscheni, C. Perrotta, L. Zema, C. Bertarelli and C. Castiglioni, Development of Tailored Graphene Nanoparticles: Preparation, Sorting and Structure Assessment by Complementary Techniques, Molecules, 2023, 28, 565 CrossRef CAS PubMed.
- M. Cao, A. Fu, Z. Wang, J. Liu, N. Kong, X. Zong, H. Liu and J. J. Gooding, Electrochemical and Theoretical Study of π−π Stacking Interactions between Graphitic Surfaces and Pyrene Derivatives, J. Phys. Chem. C, 2014, 118, 2650–2659 CrossRef CAS.
- S. Setianto, C. Panatarani, D. Singh and I. M. Joni, Semi-empirical infrared spectra simulation of pyrene-like molecules insight for simple analysis of functionalization graphene quantum dots, Sci. Rep., 2023, 13, 2282 CrossRef CAS PubMed.
- X. An, T. Simmons, R. Shah, C. Wolfe, K. M. Lewis, M. Washington, S. K. Nayak, S. Talapatra and S. Kar, Stable aqueous dispersions of noncovalently functionalized graphene from graphite and their multifunctional high-performance applications, Nano Lett., 2010, 10, 4295–4301 CrossRef CAS PubMed.
- Z. M. Khoshhesab, Reflectance IR Spectroscopy, Infrared Spectrosc.: Mater. Sci., Eng. Technol., 2012, 11, 233–244 Search PubMed.
- M. Wojdyr, Fityk: a general-purpose peak fitting program, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- C. Ding, K. Wu, W. Wang, Z. Guan, L. Wang, X. Wang, R. Wang, L. Liu and J. Fan, Synthesis of a cell penetrating peptide modified superparamagnetic iron oxide and MRI detection of bladder cancer, Oncotarget, 2017, 8, 4718–4729 CrossRef PubMed.
- M. H. Cezari and L. Juliano, Studies on lactam formation during coupling procedures of N alpha-N omega-protected arginine derivatives, Pept. Res., 1996, 9, 88–91 CAS.
- C. Backes, K. R. Paton, D. Hanlon, S. Yuan, M. I. Katsnelson, J. Houston, R. J. Smith, D. McCloskey, J. F. Donegan and J. N. Coleman, Spectroscopic metrics allow in situ measurement of mean size and thickness of liquid-exfoliated few-layer graphene nanosheets, Nanoscale, 2016, 8, 4311–4323 RSC.
- Y. Liu, C. Y. Liu and Y. Liu, Investigation on fluorescence quenching of dyes by graphite oxide and graphene, Appl. Surf. Sci., 2011, 257, 5513–5518 CrossRef CAS.
- H. S. S. R. Matte, K. S. Subrahmanyam, K. V. Rao, S. J. George and C. N. R. Rao, Quenching of fluorescence of aromatic molecules by graphene due to electron transfer, Chem. Phys. Lett., 2011, 506, 260–264 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj02525c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |