
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Differential insights into structural dynamics and photophysical behavior of two phosphorus-containing diesters
Elena
Perju
 *,
Diana
Serbezeanu
,
Mihaela
Homocianu
,
Mihaela
Avadanei
and
Tăchiţă
Vlad-Bubulac
*,
Diana
Serbezeanu
,
Mihaela
Homocianu
,
Mihaela
Avadanei
and
Tăchiţă
Vlad-Bubulac
“Petru Poni” Institute of Macromolecular Chemistry, Grigore Ghica Voda Alley 41A, 700487, Iasi, Romania. E-mail: elena.perju@icmpp.ro
First published on 4th June 2024
Abstract
Understanding the phase transitions and structural dynamics of monomers is essential for designing advanced materials. In this study, the phase transitions of two phosphorus-containing monomers with methoxycarbonyl functional end groups were investigated. Polarized light optical microscopy, differential scanning calorimetry, and UV-vis absorption and fluorescence spectroscopy were used to examine microstructural changes and molecular conformational rearrangements during temperature variations. Additionally, attenuated total reflectance Fourier-transform infrared spectroscopy was used to explore these changes with high sensitivity. The specific sensitive absorption bands offered significant information regarding the phase transition, even within a narrow transition temperature range, demonstrating the efficiency of such a method in the investigation of polymorphic and liquid crystalline materials. Interestingly, although both molecules absorb light at similar wavelengths regardless of the solvent, their emission properties are different, suggesting that the presence of an extra oxygen atom linked to the central phosphorus atom influences the movement of electrons in the excited state.
Introduction
Fourier transform infrared (FTIR) spectroscopy is a powerful tool frequently used to investigate conformational changes, polymorphisms, and phase transitions in crystals and liquid crystal compounds.1–3 In the supramolecular organization of organic compounds, the vibrations of the functional groups are closely related to the molecular geometry, molecular packing, and intermolecular or intramolecular interactions.4–6 Liquid crystals have become a significant area of focus in supramolecular chemistry, presenting intensive research in the fields of organic electronics, optoelectronics, semiconductors, photovoltaic materials, and medicine.7–9Throughout the years, interest in the structure–property relationships of liquid crystals has been constantly observed. Minor changes to the structural segments that form the molecular structure generate drastic changes in the physical properties of liquid crystal materials.10 Owing to their outstanding fluidity, translational, orientational, and conformational order within the mesophase, liquid crystal materials can be easily manipulated by light, temperature, and electric fields.11–14 This tunability allows their use in various applications such as liquid crystal displays (LCDs), where an electric field reorients the liquid crystals, altering light transmission and creating the image. The most intensely studied liquid crystals are calamitic ones, which generally contain a rigid core, flexible chains, and linkage units such as ester, ether, azo, and azomethine.15–20 The ester linkage unit is widely used in liquid crystals due to its stability and easy synthesis pathways. On the other hand, the reversible direction of the polar ester linking group influences the polarizability of the molecule and gives rise to significant changes in thermotropic behavior.21 A considerable change in the polarizability can be caused by the introduction of heteroatoms in the molecular structure.
Phosphorus, a key element in biological processes and industrial applications, offers a rich array of reactivity and functionality, facilitating the design of monomers with diverse properties and applications.22–26 Of particular interest are phosphorus-containing diesters, a subclass of organic compounds notable for their diverse chemical reactivities and significant roles in biological and synthetic processes.27–29 Therefore, phosphorus-containing monomers with methoxycarbonyl end functional groups represent a compelling area of research at the intersection of organic chemistry, polymer science, and materials engineering. These monomers are distinguished by their unique molecular architecture, which combines the chemical versatility of phosphorus with the tunability afforded by methoxycarbonyl end groups.30 The incorporation of phosphorus within the diester structure introduces enhanced thermal stability and flame-retardant capabilities, which are attributed to the ability of phosphorus to undergo coordination reactions and form intumescent char layers under fire conditions. Phosphorus-containing LCs, especially those based on phosphonates or phosphates, are relatively underexplored in liquid crystal research. The incorporation of phosphorus atoms can introduce unique properties, leading to novel mesophases and enhanced functionalities. Despite their potential, there is a significant gap in the synthesis and characterization of organophosphorus-based LCs. This research addresses this issue by synthesizing new P-containing LC molecules and characterizing their phase behavior. Additionally, ATR-FTIR spectroscopy has been used to study the phase transitions in these materials, a technique that has not been previously applied to organophosphorus-based LCs. This dual approach advances liquid crystal science and broadens its application potential in advanced technologies. Through careful molecular design and manipulation, phosphorus-containing diesters can be tailored to display liquid crystal properties, offering opportunities for innovative applications in fields such as display technologies, optical materials, and biomedical devices. Moreover, liquid crystalline dimers bridged with a phosphonic group may serve as promising vertical alignment agents and develop functionalized liquid crystalline materials with a twist-bend nematic phase.31 Ongoing research in this field aims to expand the scope of phosphorus-containing monomers with methoxycarbonyl end functional groups, explore new synthetic methodologies, elucidate structure–property relationships, and advance their practical utility in emerging technologies.
In this context, the phase transitions and structural changes of two phosphorus-containing monomers, bearing methoxycarbonyl functional end groups, namely bis(4-(methoxycarbonyl) phenyl)phenylphosphonate and bis(4-(methoxycarbonyl)phenyl)phenylphosphate, denoted as 3a and 3b, under thermal treatment were studied. Moreover, the investigation of the sensitive absorption bands during the phase transition provides valuable information regarding the sequential order change in the molecular structure. This research focused on using ATR-FTIR spectroscopy as an important and complementary tool to investigate and improve the understanding of the phase transitions of these phosphorus-containing monomers, and how structural changes at the molecular level influence their optical properties.
Experimental section
A. Materials
Phenyl phosphonic dichloride, phenyl dichlorophosphate, p-hydroxybenzoic acid, triethylamine, acetic acid, sulfuric acid (H2SO4 98%), tetrahydrofuran, ethanol, diethyl ether, methylene chloride, cyclohexane, methanol, 2-propanol, acetonitrile, and 1,4-dioxane were commercially available and used as received without further purification (Sigma-Aldrich). CuCl (Sigma-Aldrich) was purified by washing successively with acetic acid, ethanol, and diethyl ether, followed by drying under vacuum at 60 °C before use. Methyl 4-hydroxybenzoate was prepared using p-hydroxybenzoic acid, methanol, and sulfuric acid. The reaction yield: 68%; m.p.: 125–128 °C. 1H NMR (DMSO-d6, ppm): 10.33 (s, 1H, –OH); 8.03–8.01 (d, 2H); 6.87–6.81 (d, 2H); 3.79 (s, 3H, –CH3).B. Methods
The melting points of the monomers and intermediates were measured using Melt-Temp II (Laboratory Devices).Infrared spectra were recorded on an FTIR Bruker Vertex 70 spectrophotometer in the transmission mode at wavenumbers ranging from 400 cm−1 to 4000 cm−1. The samples were then mixed with KBr and pressed into pellets.
Proton nuclear magnetic resonance spectra were recorded using a Bruker Advance DRX 400 MHz spectrometer, equipped with a 5 mm, direct detection, multinuclear probe, operating at 400.1, 161.9, and 100.6 MHz for 1H, 31P and 13C nuclei, respectively. 1H and 13C chemical shifts were reported in δ units (ppm) relative to the residual peak of the solvent (ref. 1H: DMSO-d6: 2.51 ppm, 13C: DMSO-d6: 39.47 ppm). 31P chemical shifts are electronically referred to as 85% H3PO4 (0 ppm).
Polarized light optical microscopy (PLM) was performed using a Zeiss Axio Imager M2 microscope (Carl Zeiss AG). A 10×, 20× or 40× objective lens was used for all measurements. The samples were prepared by casting a small amount of the compound on a glass plate, which was subjected to two heating–cooling cycles. For 3a, the temperature regime was as follows: from room temperature at a heating rate of 10 °C min−1 up to 110 °C and with 1 °C min−1 up to 150 °C, then cooled to 25 °C at a cooling rate of 1 °C min−1; for t 3b, a heating/cooling rate of 10 °C min−1 was used; xC – cooling scan and xH – heating scan (x – number of the thermic treatment cycle).
Differential scanning calorimetry (DSC) was carried out using a NETZSCH DSC 214 instrument for 3a, and a PerkinElmer Pyris Diamond instrument for 3b, using nitrogen as a carrier gas at a flow rate of 10 mL min−1. For 3a, several heating/cooling cycles were recorded using a temperature regime as follows: first heated from room temperature at a heating rate of 10 °C min−1 up to 110 °C and with 0.1 °C min−1 up to 150 °C, then cooled to 25 °C at a cooling rate of 1 °C min−1. 3b was heated up to 110 °C from −20 °C and cooled to an equal negative temperature at a heating/cooling rate of 10 °C min−1. The phase transition temperatures of the diester monomers were taken as the maxima of the endothermic and exothermic peaks.
FTIR spectroscopy in attenuated total internal reflectance (ATR-FTIR) was used to study the phase transitions of the phosphorus-containing monomers in the temperature range of 24–150 °C using a Vertex 70 spectrometer equipped with a Golden Gate® ATR accessory (diamond crystal, Specac Ltd) and a temperature controller. The scans were performed in the 4000–600 cm−1 spectral range at a heating/cooling rate of 1 °C min−1, with 128 scans at a resolution of 2 cm−1. A mixed Gaussian–Lorentzian was used to resolve the peaks of interest.
An Analytic Jena 210+ spectrophotometer was used to measure the absorption spectra, while the fluorescence spectra were obtained using a PerkinElmer LS55 fluorescence spectrophotometer. The emission spectra were measured between 250 and 500 nm, with an excitation wavelength of 235 nm. The experiments were carried out at ambient temperature using quartz cuvettes with a 10 mm optical path length. Spectroscopic grade solvents, such as cyclohexane (CYHEX), methanol (MeOH), 2-propanol (IPA), acetonitrile (ACN), and 1,4-dioxane (DIOX), were used without requiring additional purification.
C. Synthesis and characterization of bis(4-(methoxycarbonyl)phenyl)phenylphosphonate (3a) and bis(4-(methoxycarbonyl)phenyl)phenylphosphate (3b)
6.08 g of methyl 4-hydroxybenzoate (1), 33 mL THF (tetrahydrofuran), 5.56 mL TEA (triethylamine), and 0.10 g of CuCl were placed in a 100 mL three-necked flask equipped with a magnetic stirrer and nitrogen inlet and outlet. The mixture was then cooled to 0 °C in an ice bath. To this mixture, 3 mL of phenyl phosphoric dichloride (2a) or phenyl dichlorophosphate (2b) was added dropwise during a 30 min period, and the solution was allowed to react for 2 h at 0 °C and 48 h at room temperature. Finally, the mixture was filtered and concentrated under reduced pressure, and the crude product was purified by silica gel chromatography. The product was eluted with methylene chloride to give a clear colorless liquid, which was concentrated in order to obtain the desired compounds 3a and 3b.30![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H stretch of aromatic rings); 2948, 2847 (C–H stretch of aliphatic chains); 1726 (CO stretch of ester groups); 1603, 1501 (aromatic CC bands); 1131 (P–Caryl); 1278, 1018 (ester C–O–C); 962/951 (P–O–Caryl); 1195 (PO). 1H NMR (δ, ppm): 3.83 (6H, s, CH3), 7.39 (4H, d, 8.0 Hz, H-2), 7.61–7.66 (2H, m, H-9), 7.75 (1H, t, 8.0 Hz, H-10), 7.99 (4H, d, 8.0 Hz, H-3), 7.97–8.02 (2H, m, H-8, overlap with H-3). 13C NMR (δ, ppm): 52.1 (CH3), 120.6 (d, 3J(P, C) = 4.7 Hz, C-2), 125.1 (d, 1J(P, C) = 191 Hz, C-7), 126.8 (C-4), 129.2 (d, 3J(P, C) = 20.0 Hz, C-9), 131.4 (C-3), 132.1 (d, 2J(P, C) = 10.0 Hz, C-8), 133.1 (d, 4J(P, C) = 3.0 Hz, C-10), 153.3 (d, 2J(P, C) = 7.0 Hz, C-1), 165.3 (COO). 31P NMR (δ, ppm): 12.1 (t, 13 Hz).
C–H stretch of aromatic rings); 2955, 2853 (C–H stretch of aliphatic chains); 1726 (CO stretch of ester groups); 1603, 1504 (aromatic CC bands); 1279, 1013 (ester C–O–C); 962/951 (P–O–Caryl); 1197 (PO). 1H NMR (δ, ppm): 3.86 (6H, s, CH3), 7.31–7.34 (3H, m, H-8 and H-10), 7.47 (4H, d, 8.0 Hz, H-2), 7.46–7.50 (2H, m, H-9), 8.06 (4H, d, 8.0 Hz, H-3). 13C NMR (δ, ppm): 52.2 (CH3), 119.9 (d, 3J(P, C) = 4.8 Hz, C-8), 120.2 (d, 3J(P, C) = 5.3 Hz, C-2), 126.2 (C-10), 127.3 (C-4), 130.3 (C-9), 131.6 (C-3), 149.5 (d, 2J(P, C) = 7.5 Hz, C-7), 153.1 (d, 2J(P, C) = 6.8 Hz, C-1), 165.2 (COO). 31P NMR (δ, ppm): 18.5 (s).
C–H stretch of aromatic rings); 2948, 2847 (C–H stretch of aliphatic chains); 1726 (CO stretch of ester groups); 1603, 1501 (aromatic CC bands); 1131 (P–Caryl); 1278, 1018 (ester C–O–C); 962/951 (P–O–Caryl); 1195 (PO). 1H NMR (δ, ppm): 3.83 (6H, s, CH3), 7.39 (4H, d, 8.0 Hz, H-2), 7.61–7.66 (2H, m, H-9), 7.75 (1H, t, 8.0 Hz, H-10), 7.99 (4H, d, 8.0 Hz, H-3), 7.97–8.02 (2H, m, H-8, overlap with H-3). 13C NMR (δ, ppm): 52.1 (CH3), 120.6 (d, 3J(P, C) = 4.7 Hz, C-2), 125.1 (d, 1J(P, C) = 191 Hz, C-7), 126.8 (C-4), 129.2 (d, 3J(P, C) = 20.0 Hz, C-9), 131.4 (C-3), 132.1 (d, 2J(P, C) = 10.0 Hz, C-8), 133.1 (d, 4J(P, C) = 3.0 Hz, C-10), 153.3 (d, 2J(P, C) = 7.0 Hz, C-1), 165.3 (COO). 31P NMR (δ, ppm): 12.1 (t, 13 Hz).
C–H stretch of aromatic rings); 2955, 2853 (C–H stretch of aliphatic chains); 1726 (CO stretch of ester groups); 1603, 1504 (aromatic CC bands); 1279, 1013 (ester C–O–C); 962/951 (P–O–Caryl); 1197 (PO). 1H NMR (δ, ppm): 3.86 (6H, s, CH3), 7.31–7.34 (3H, m, H-8 and H-10), 7.47 (4H, d, 8.0 Hz, H-2), 7.46–7.50 (2H, m, H-9), 8.06 (4H, d, 8.0 Hz, H-3). 13C NMR (δ, ppm): 52.2 (CH3), 119.9 (d, 3J(P, C) = 4.8 Hz, C-8), 120.2 (d, 3J(P, C) = 5.3 Hz, C-2), 126.2 (C-10), 127.3 (C-4), 130.3 (C-9), 131.6 (C-3), 149.5 (d, 2J(P, C) = 7.5 Hz, C-7), 153.1 (d, 2J(P, C) = 6.8 Hz, C-1), 165.2 (COO). 31P NMR (δ, ppm): 18.5 (s).
Results and discussion
Two phosphorus-containing monomers 3a and 3b were prepared via an O-acylation reaction of methyl 4-hydroxybenzoate with phenyl phosphonic dichloride or phenyl dichlorophosphate, respectively, in the presence of CuCl, as shown in Scheme 1. In our previous study, the crystal structures of 3a and 3b were evidenced as belonging to a monoclinic system and a triclinic system, respectively.30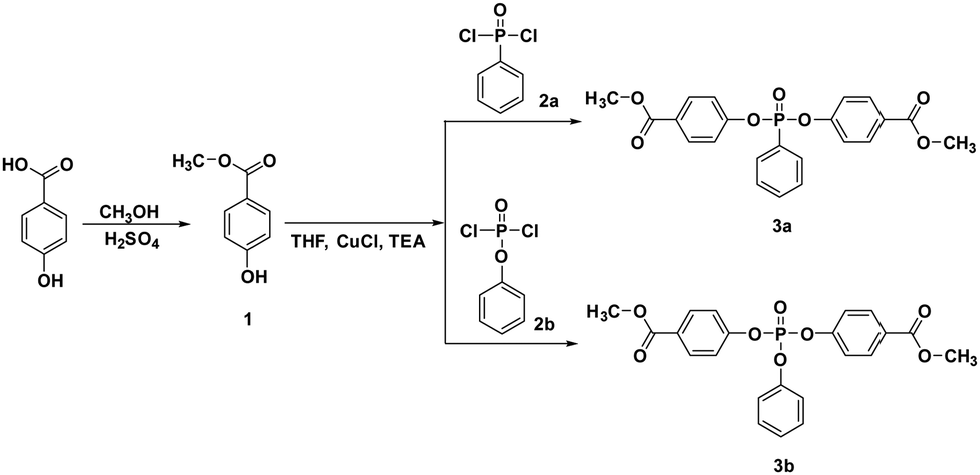 | ||
| Scheme 1 Synthesis of bis(4-(methoxycarbonyl)phenyl) phenylphosphonate (3a) and bis(4-(methoxycarbonyl)phenyl) phenylphosphate (3b). | ||
The phase transition behavior of phosphorus-containing monomers were thoroughly examined by PLM in several heating and cooling scans. The PLM observations correlated with DSC measurements. The transition temperatures recorded using both methods with similar heating/cooling rates are presented in Table 1.
| Code | PLM | DSC | ||
|---|---|---|---|---|
| H | C | H | C | |
| H – heating scan; C – cooling scan; Cr – crystalline; LC – liquid crystalline; I – isotropic.a Frozen isotropic state, which gradually crystallized overnight at 5 °C.b Frozen isotropic state. | ||||
| 3a | Cr 134 LC 138 I | I 65 Cr | Cr 135 LC 140 I | I 70 Cr1 68 Cr2 67 Cr3 65 Cr4 |
| 3b | Cr 95 I | —a | Cr 93 I | —b |
Under PLM, 3a presented small birefringent areas at room temperature, which became more pronounced in the whole mass during the first heating cycle due to different crystallization arrangements (Fig. 1a). The transition to a liquid crystalline state appeared as a typical nematic marble texture at around 133 °C (Fig. 1b), followed by the isotropic phase at 138 °C. At a cooling rate of 1 °C min−1, the crystallization of 3a appeared in the isotropic state at 56 °C (Fig. 1c). During the second heating–cooling cycle, a similar behavior was observed with the appearance of a liquid crystalline phase at 134 °C, which demonstrates the reversible thermotropic behavior of 3a (Fig. 1d). DSC measurements of 3a showed two endothermic peaks at 135 °C due to the crystalline–liquid crystalline phase transition, and at 140 °C, corresponding to the liquid crystalline–isotropic phase transition (Fig. 2). Upon cooling, only a strong exothermic peak appeared at 70 °C, followed by multiple small exothermic peaks associated with different crystalline–crystalline transitions. As expected, the enthalpy of the nematic–isotropic transition compared to the crystalline–nematic and crystalline–isotropic transition is lower, in line with the lower energy requirements of the transition between two states with closer ordering degrees.32 Due to the change of the heating rate, a drop in the curve and a change of the baseline appeared at 110 °C in the DSC thermogram.
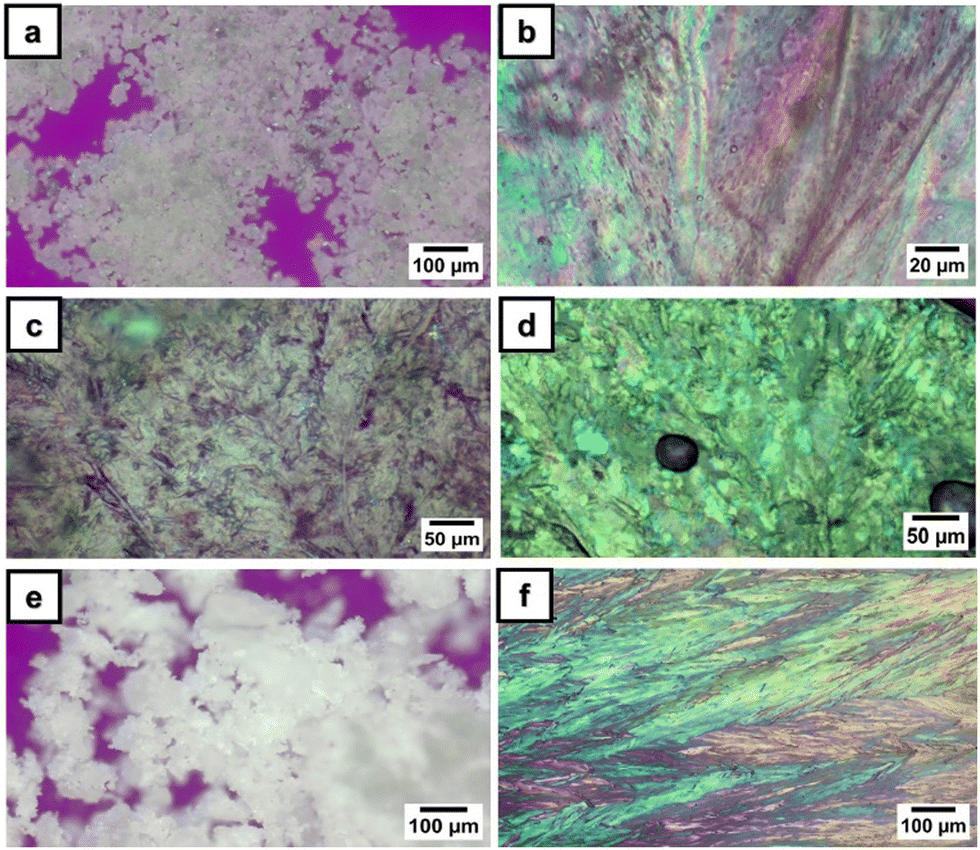 | ||
| Fig. 1 Polarized optical light microscopy photographs of phosphorus-containing monomers: (a) 1H, 114 °C, 10×; (b) 1H, 134 °C, 40×; (c) 1C, 25 °C, 20×; (d) 2H, 136 °C, 20×; (e) 1H, 25 °C, 10×; and (f) 1C, 25 °C, 10×. | ||
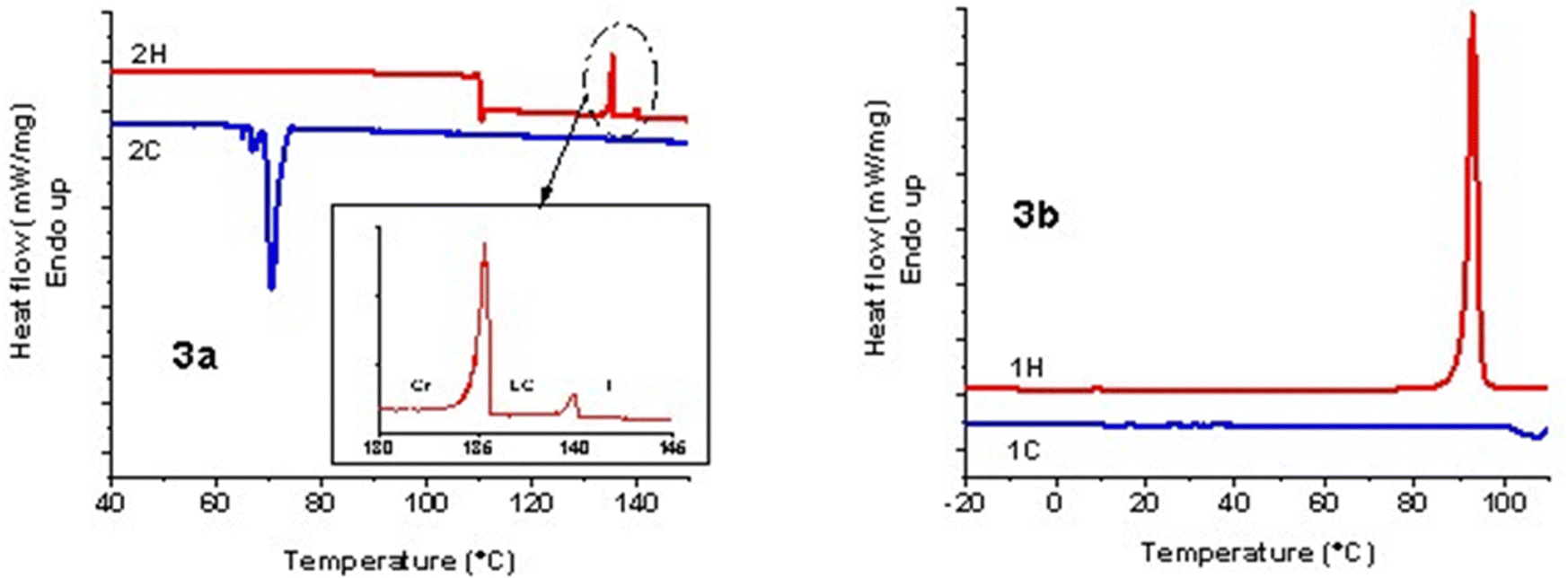 | ||
| Fig. 2 DSC charts of phosphorus-containing diesters. | ||
Instead, 3b showed only a melting process at 94 °C (Fig. 1e), while under cooling, the isotropic state persisted at room temperature even for several days. This behavior was confirmed by the DSC curves, which showed one endothermic peak at 93 °C, corresponding to the melting temperature, and no exothermic peak in the DSC cooling scan (Fig. 2). Even if crystallization was not observed in the DSC traces, PLM revealed a crystallization process that occurred when the sample was maintained at 5 °C overnight (Fig. 1f). The second heating–cooling run confirmed the crystalline–isotropic transition and the frozen isotropic state of 3b.
ATR-FTIR spectroscopy is a powerful tool for investigating the microstructural changes and molecular dynamics of phosphorus-containing monomers with highly sensitive methoxycarbonyl end functional groups. By monitoring the intensity distribution and positions of specific absorption bands during temperature changes, ATR-FTIR spectroscopy provides unique insights into the molecular conformational rearrangements associated with phase transitions.4–6 The distinctive signatures of phase transitions revealed by ATR-FTIR spectroscopy offer valuable information regarding the structural transformations and thermodynamic properties of the monomers. Thus, the structural changes during phase transitions of the studied compounds at the molecular level were investigated by ATR-FTIR spectroscopy in order to obtain support information on the modification around the chemical groups and the order–disorder transitions. Fig. 3 shows the thermal variation of some selected regions of the FTIR spectra of 3a and 3b, with appropriate designations of the phases and the relevant absorption bands. As a general feature, the spectrum intensities of both compounds 3a and 3b increased as the temperature increased to 60 °C. This behavior can be seen as a type of reorganization and stabilization of the ATR crystal to accommodate the temperature change. One could then consider that the real thermal transformation begins after 50 °C. The introduction of an oxygen atom between phosphorus and phenyl led to differences in the FTIR spectra. The breaking of electron delocalization by the oxygen atom increases the flexibility of the phosphonate unit, allowing free rotation around the O–P–O bonds. This high degree of freedom has been expanded throughout the molecular structure and affect the vibration frequencies of the most fundamental modes.
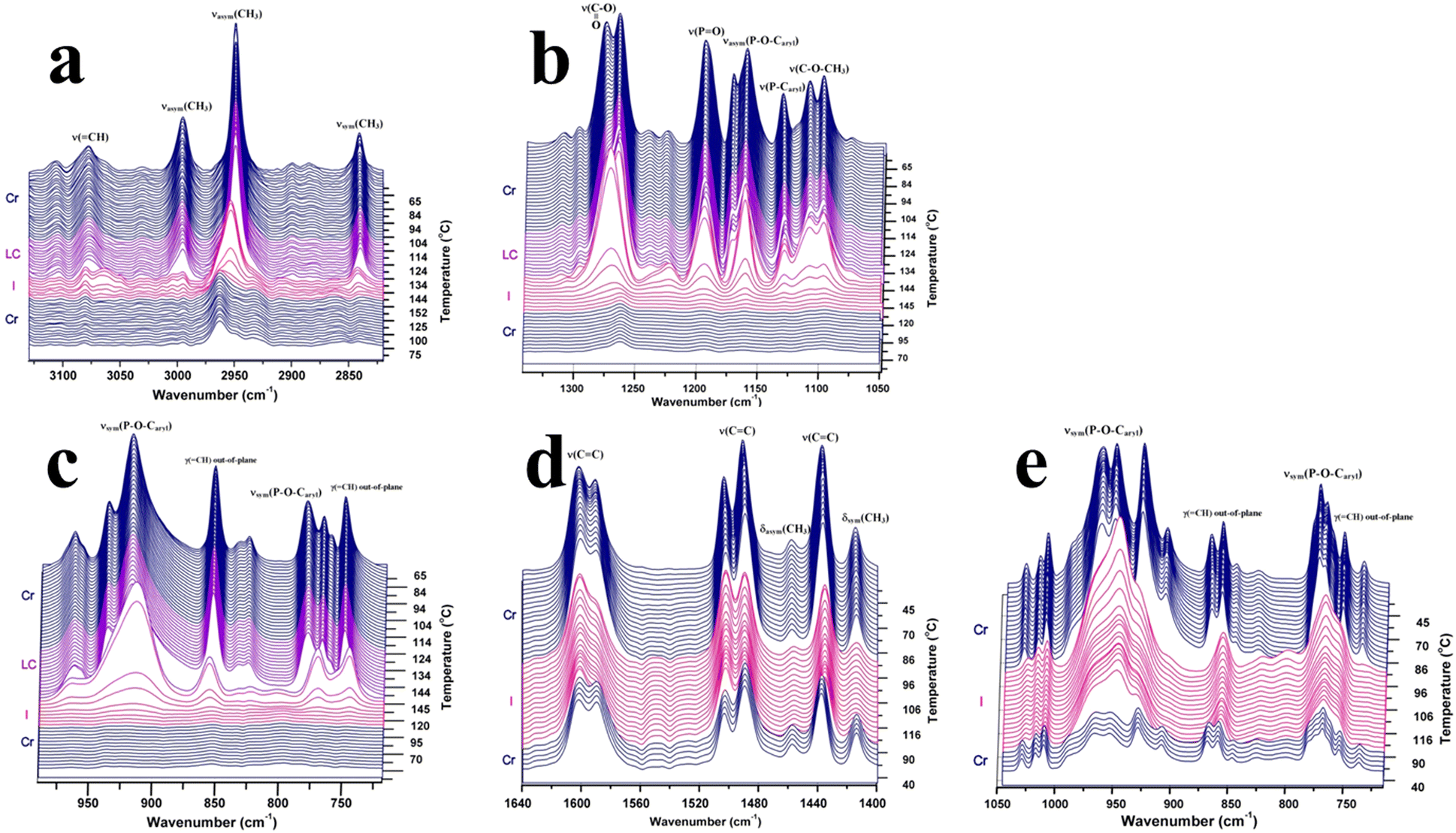 | ||
| Fig. 3 Relevant fragments of the FTIR spectra showing the spectral evolution from the crystalline state to the isotropic liquid state of various chemical groups in 3a: (a) CH stretching region, connected to the methoxy extremities; (b) and (c) significant bands of the central phosphorus group; and in 3b: (d) aromatic stretching vibrations and (e) symmetric stretching vibration of the phosphorus group. | ||
The vibrations representative of the central unit are ν(PO) (1195 cm−1 in 3a) and νasym(P–O–Caryl) (1172/1160 cm−1), which are slightly displaced towards lower energies for 3b. In addition, ν(P–Caryl) in 3a was observed at 1131 cm−1, while the corresponding νsym(P–O–Caryl) of the phenoxy unit in 3b was detected as an intense doublet at 962/951 cm−1. Apparently decoupled from the motion of the molecular skeleton, the stretching vibrations of the CH3 terminal groups were displaced to higher frequencies in bis(4-(methoxycarbonyl)phenyl)phenyl phosphate (3b) than in bis(4-(methoxycarbonyl) phenyl)phenylphosphonate (3a). This is due to the less-ordered arrangement of the bis(4-(methoxycarbonyl)phenyl)phenyl phosphate molecules in the crystalline state. The methyl ester moiety, which is common to both compounds, shows a doublet structure for ν(O–CH3) (1111/1098 cm−1) and ν(COO) (1277/1265 cm−1). Some of the singlet absorption bands in the spectrum of 3a were replaced by doublets in the spectrum of 3b, as observed by the aromatic ν(CC) vibrations, γr(O–CH3) (1015 vs. 1017/1010 cm−1) or νsym(P–O–Caryl) (853 vs. 868/858 cm−1).
Inspection of the FTIR spectra revealed bands whose parameters are sensitive to the crystalline-to-liquid crystal state transition. Spectral changes were observed for some bands in the heating run, while other groups showed a lack of change. In the case of 3a, the sharp aspect of the absorption bands in the crystalline state was preserved in the mesophase, indicating a similar molecular environment and conformational order. The absorption modes followed during the temperature variation for both compounds provided different views of the phase transitions, as experienced by specific parts of the molecule. Thus, the chemical group dynamics can be separated into those belonging to the molecular core and those belonging to the flexible methyl ester moiety.
Although the whole spectrum of 3a changes according to the thermal variations induced by the temperature increase, a few bands were sensitive enough to mark the crystalline–liquid crystal transition. The most dynamic bands were found in those given by ν(P–O–Caryl), ν(COO) and ν(O–CH3), indicating variations in the P–O–Caryl and P–Caryl angles and some conformational changes in the methyl ester segment. Surprisingly, two vibrational modes connected to the aromatic ester moiety, i.e. ν(COO) and ν(CO), were found to be highly coupled with liquid crystalline ordering. As seen in Fig. 3, with increasing temperature, ν(COO) of 3a gradually loses its high-frequency component. Then, an abrupt frequency shift of 6 cm−1 in the band maximum occurred at the melting point, and the band achieved a symmetric contour. The continuous decrease of the high-frequency peak with temperature and the growth in the cooling run may suggest its involvement in a highly ordered structure. The thermally induced torsional motions partly destroy this ordering and affect the planarity of this group. The 3b spectrum shows the ν(COO) vibration as a single and sharp band, which slowly decreases in intensity during heating and with no change in frequency. The νasym(P–O–Caryl) vibration (1172/1160 cm−1) in 3a abruptly transforms from a doublet at room temperature into a single band at the liquid crystal–isotropic phase transition. The persistence of the peak at 1171 cm−1 in both crystalline and liquid crystal states, although constantly diminishing during heating, shows the existence of the same molecular environment and almost the same P–O–CC dihedral angle. Except for the constant variations in the contour of the ν(COO) and ν(P–O–Caryl) bands, the spectral signature of the liquid crystal phase is not different from that of the crystalline state, as the band shape persists until the isotropic state.
The variation in the integrated intensity and frequency of the absorption bands as a function of temperature reveals the dynamics of various parts of the molecule. The plots presented in Fig. 4 show the thermotropic response of the main vibration modes and allow the detection of the beginning of the mesophase and the final transition to the isotropic phase. For the phosphate group, the spectral variation in νasym(P–O–Caryl) and ν(PO) defines two regions: the first one ends at the melting temperature (144 °C), showing a continuous decrease in integrated intensity. The second region consists of an abrupt decrease of intensity as a result of compound melting (144 °C to 152 °C). Only a small hump is detected at the onset of the mesophase, especially for νasym(P–O–Caryl), and the progression of the LC ordering brings no additional change.
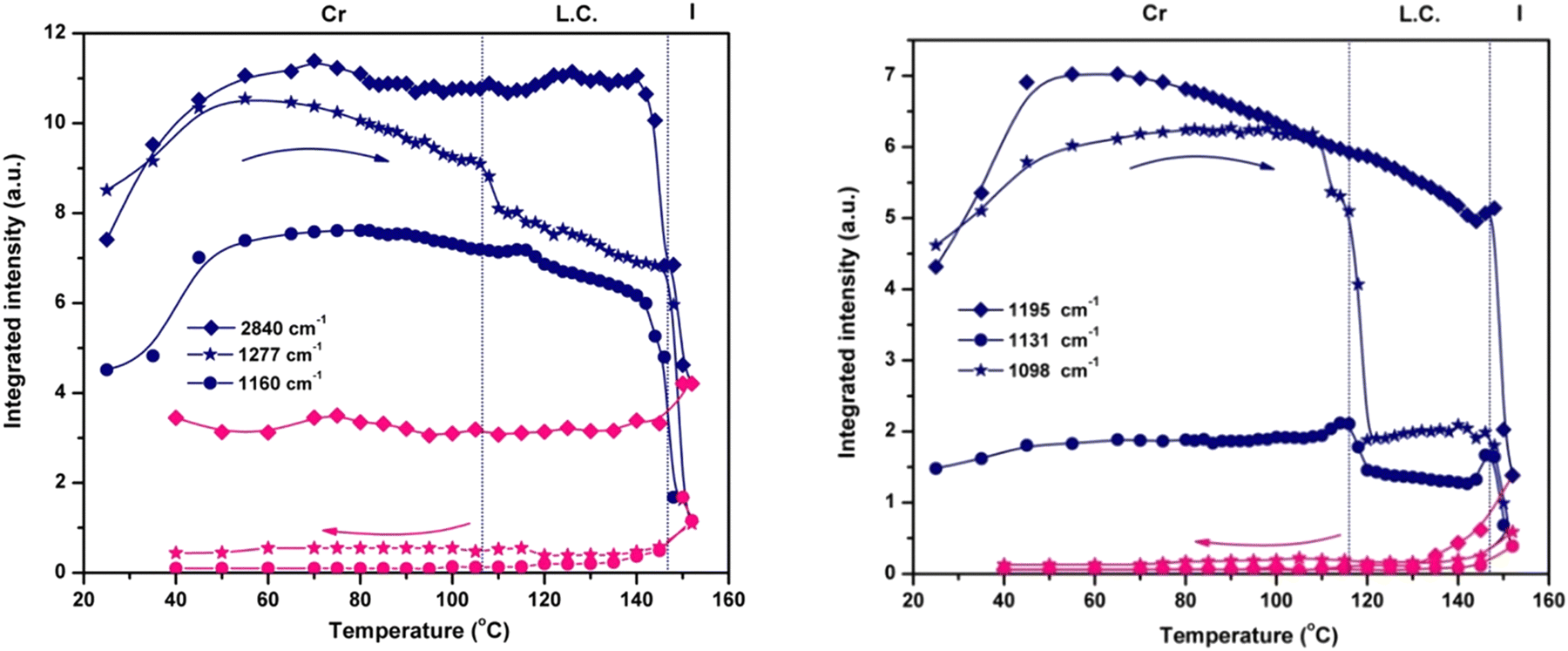 | ||
| Fig. 4 The dynamics as a function of temperature of the integrated intensity of the signals in 3a for the flexible methoxy moiety – νsym(CH3) (2840 cm−1) and ν(O–CH3) (1098 cm−1); aromatic ester group – ν(COO) (1277 cm−1) and the central unit – ν(PO) (1195 cm−1), νasym(P–O–Caryl) (1160 cm−1) and ν(P–Caryl) (1131 cm−1). | ||
Instead, the thermal profiles of the ν(COO), ν(O–CH3) and ν(P–Caryl) bands exhibit a clearly defined LC regime but span their temperature domains for every vibration mode monitored. The transition into the liquid crystal phase started at 110 °C for the methoxy and phenyl groups, and at 106 °C for the ester group. These features indicate the presence of different degrees of flexibility for every chemical group, in close connection with the electronic and structural effects. Although electronically coupled with the central phosphonate group, the phenyl ring has a certain degree of freedom to rotate around the P–CC bond. Its coupling with the LC phase organization is evident from the evolution of the integrated intensity of the ν(P–Caryl) vibration (Fig. 4), which indicates a variation in the P–CC angle induced by torsional motion. The temperature range over which the pendant phenyl ring adjusted itself to attain conformation in the LC phase was ≈6 °C. For the O–CH3 linkage, the crystalline–liquid crystal transition occurred within a ≈12 °C interval. At the COO group level, the phase transition was even faster, in a ≈4 °C range. The motion of the terminal methyl groups is decoupled from the rest of the molecule and is not sensitive to LC ordering because the integrated intensity of the symmetric and asymmetric stretching vibrations shows a constant evolution with temperature (Fig. 4) until they finally drop at the melting point.
The frequency representation as a function of temperature in Fig. 5 displays another perspective on how liquid crystal ordering influences the chemical group dynamics in 3a. The position of P–C stretching (1131 cm−1) is gradually displaced to lower frequencies, with the same small hump around the Cr → LC temperature and showing a 3.2 cm−1 decrease until complete melting. A similar change in frequency is shown by the ν(P–O–C) band (1160 cm−1), where the transition into the mesophase is very smooth.
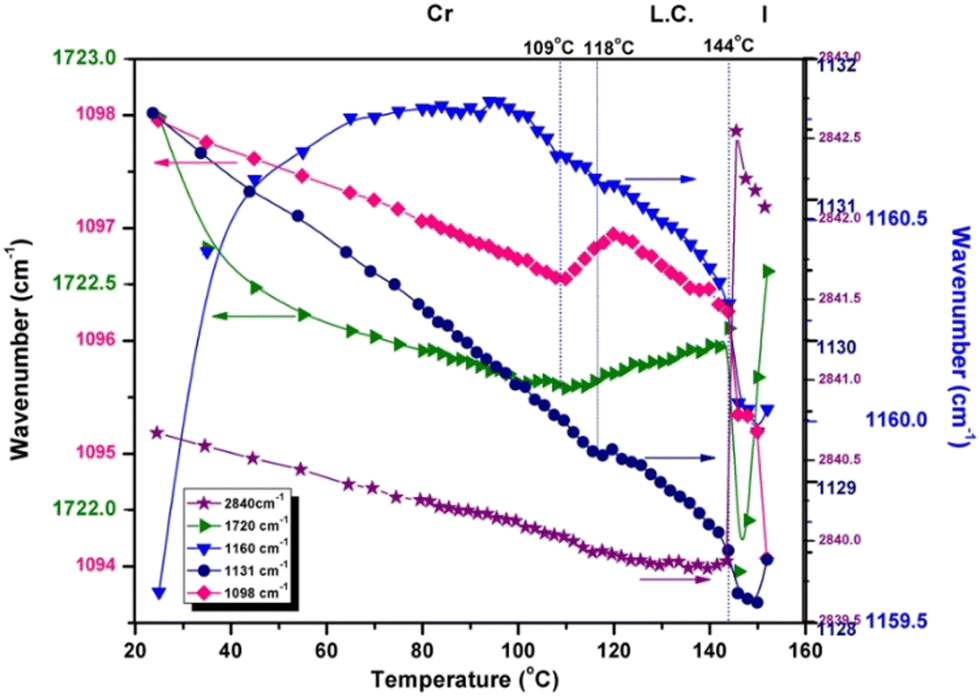 | ||
| Fig. 5 Frequency shift in various signals during the heating run: νsym(CH3) (2840 cm−1), ν(COO) (1720 cm−1), ν(P–O–C) (1160 cm−1), ν(P–C) (1131 cm−1) and ν(O–CH3) (1095 cm−1), indicating the variation in intermolecular interaction strength with increasing temperature for 3a. | ||
The motion of the molecule core, monitored through νasym(P–O–Caryl) and ν(PO) vibrations, whose monotonous evolution with temperature ultimately shows the rigidity of the phosphonate moiety and its strongly coupled vibrations. There is no clear indication of some conformational changes in the liquid crystal phase.
Understanding the relationship between the structure of a material, its optical properties, and its phase transitions is critical for the design of advanced devices. Phosphorus-containing diesters (3a and 3b) possess specific molecular structures that influence their optical properties. 3a contains a phosphonate group (PO) as the central atom, while 3b contains a phosphate group (PO4−); this small difference in the chemical structure has significant effects on the overall molecular behavior. Phosphonate groups typically exhibit stronger electron-withdrawing properties than phosphate groups due to the presence of a double bond between the phosphorus and oxygen atoms. This difference in electron density distribution can affect the molecular interactions and packing arrangements within the material. Additionally, the nature of the substituents attached to the central phosphorus atom may also play a role. In 3a, the methoxycarbonyl groups may contribute to the increased molecular asymmetry and flexibility, which are favorable for liquid crystal formation. Moreover, the overall molecular shape and size of 3a may be more conducive to liquid crystal formation than those of 3b. Subtle differences in molecular geometry can have profound effects on the ability of molecules to self-organize into liquid crystalline phases. Although both groups contain oxygen atoms and are potentially polar, the phosphonate group in 3a is generally considered to be more hydrophobic and less polar than the phosphate group in 3b. By monitoring the changes in these characteristics, we can gain insight into the structural modifications that occur at the molecular level during the phase transitions. These materials are crucial for various applications like LCDs,33 OLEDs,34 imaging,35 and sensors.36,37 To design and optimize such devices, it is crucial to comprehend the relationships between their optical properties, phase transitions, and structural changes. Fig. 6 shows the UV-Vis absorption and fluorescence spectra of 3a and 3b in the selected solvents. All spectra show an absorption band maximum of around 235 nm, which is expected to correspond to π–π* transitions within the aromatic backbone of the molecule and n–π* transitions of the free electron pairs.
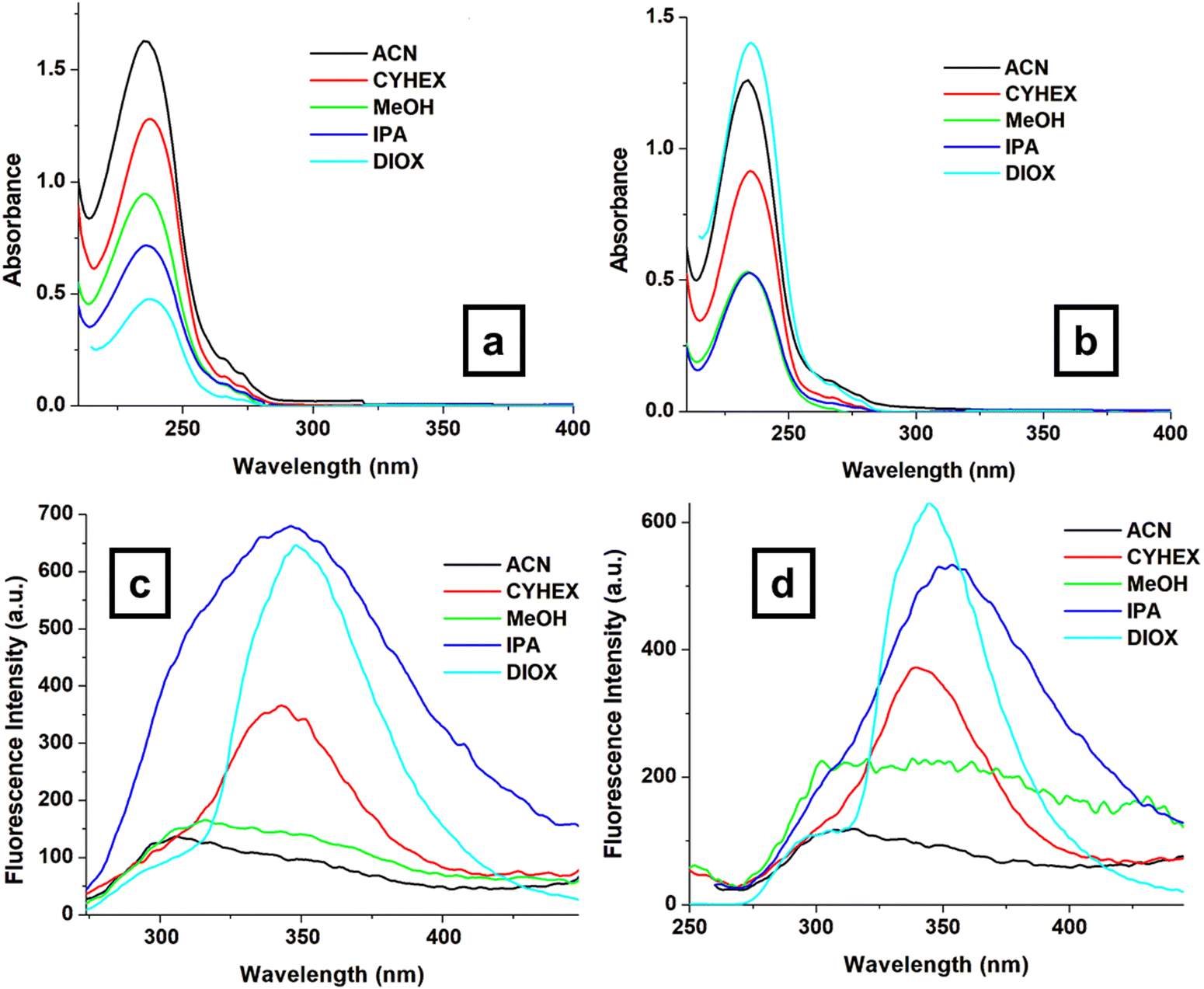 | ||
| Fig. 6 UV-Vis absorption spectra of compounds 3a (a) and 3b (b) as well as the emission spectra of compounds 3a (c) and 3b (d) in selected solvents. | ||
Both 3a and 3b molecules have similar absorption maxima (λmax) between 234–238 nm; this domain remains unaffected by the solvents used. This suggests that the solvent has a minimal influence on the ground-state electronic transitions. However, there are differences in their emission maxima and Stokes shifts (SS, nm), as shown in Fig. 6c, d and Table 2, indicating variations in their excited state properties. The emission properties of both molecules vary depending on the solvent, with emission maxima ranging from 340–354 nm and Stokes shift values ranging from 102 nm to 120 nm (Table 2). Stokes shift values provide information on the relaxation processes that occur upon excitation, which may be influenced by molecular conformation, solvent interactions, and electronic transitions. Larger Stokes shift values indicate lower excited-state energy loss, potentially due to less interaction with the solvent environment. The differences in the SS values can be due to solvation effects or possibly different excited-state processes. The main difference between 3a (phosphonate) and 3b (phosphate) is in the central P–O bond. The extra oxygen atom in 3b (phosphate) can influence the conjugation within the molecule, potentially affecting the excited-state behavior.
| Samples | CYHEX | MeOH | IPA | ACN | DIOX | |
|---|---|---|---|---|---|---|
| SS – Stokes shift (nm); CYHEX – cyclohexane; MeOH – methanol; IPA – 2-propanol; ACN – acetonitrile; and DIOX – 1,4-dioxane.a Very broad emission band. | ||||||
| 3a | λ max (nm) | 235 | 236 | 236 | 235 | 238 |
| λ em (nm) | 343 | 340a | 346 | 340a | 348 | |
| SS (nm) | 108 | 104 | 110 | 105 | 110 | |
| 3b | λ max (nm) | 235 | 234 | 234 | 234 | 235 |
| λ em (nm) | 340 | 340a | 354 | 340a | 345 | |
| SS (nm) | 105 | 102 | 120 | 102 | 110 | |
Conclusions
In this study, we investigated the phase transitions of two monomers using a comprehensive approach, employing PLM and DSC. These techniques provide valuable insights into the microstructural changes occurring within the monomers as they undergo temperature variations, elucidating the molecular conformational rearrangements associated with phase transitions. Only the bis(4-(methoxycarbonyl)phenyl)phenyl phosphonate monomer exhibits a liquid crystalline behavior, while the other monomer exhibits a crystalline state. Furthermore, ATR-FTIR spectroscopy was employed to investigate the microstructural changes and molecular dynamics of the monomers with high sensitivity. By monitoring the intensity distribution and positions of specific absorption bands during temperature changes, we elucidated the distinctive signatures of phase transitions in these materials. The most dynamic bands were found in those given by ν(P–O–Caryl), ν(COO) and ν(O–CH3), variations in the P–O–Caryl and P–Caryl angles, and some conformational changes in the methyl ester segment. Specific ordering was evidenced by monitoring the vibrational modes connected to the aromatic ester moiety. The motion of the molecular core showed an insignificant response to the conformational changes in the liquid crystal phase due to its rigidity. Furthermore, although both types of monomers shared similar absorption maxima around 234–238 nm regardless of the solvent used, their emission properties differed significantly. This suggests that the surrounding solvent molecules have minimal influence on the ground-state electronic structure of the monomers but significantly impact their behavior in the excited state. The main difference between these derivatives lies in the central P–O bond, which can influence excited-state behavior. The insights gained from this study are invaluable for the rational design and optimization of these monomers for application in advanced materials, including flame-retardant polymers, liquid crystal displays, and biomedical devices.Author contributions
D. S., T. V.-B. and E. P. coordinated and supervised the work. D. S. conducted the synthesis and characterization of the compounds. M. A., M. H. and E. P. performed the investigation and analysis of the data. The manuscript was written through contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by a grant from the Ministry of Research, Innovation 839 and Digitization, CCCDI – UEFISCDI, project number PN-III-P2-2.1-PED-2021-2700, within PNCDI 840 III.References
- S. G. Kazarian and K. L. A. Chan, Applications of ATR-FTIR spectroscopic imaging to biomedical samples, Biochim. Biophys. Acta, 2006, 1758(7), 858–867, DOI:10.1016/j.bbamem.2006.02.011.
- Z. Li and B. Zhang, Investigation of Glycine Polymorphic Transformation by In Situ ATR-FTIR and FT-Raman Spectroscopy, Crystals, 2022, 12(8), 1141, DOI:10.3390/cryst12081141.
- A. Hatta, H. Nohara and W. Suëtaka, Application of Infrared ATR Spectroscopy to Liquid Crystals. II. Surface-induced Orientation of Nematic MBBA and Reorientation in Electric Fields, Bull. Chem. Soc. Jpn., 1987, 51(4), 967–973, DOI:10.1246/bcsj.51.967.
- V. Cozan, M. Avadanei, E. Perju and D. Timpu, FTIR investigations of phase transitions in an asymmetric azomethine liquid crystal, Phase Transitions, 2009, 82(8), 607–619, DOI:10.1080/01411590903133396.
- M. Avadanei, E. Perju, V. Cozan and M. Bruma, Phase transitions of a monotropic azomethine liquid crystal investigated by ATR-FTIR spectroscopy, Phase Transitions., 2014, 87(3), 243–254, DOI:10.1080/01411594.2013.807430.
- V. Cozan, M. Avadanei, S. Shova and M.-F. Zaltariov, Crystal smectic E revisited for (E)-N-(biphenyl-4-ylmethylene)-4-butylaniline – mesomorphism, crystal structure and FTIR study, Liq. Cryst., 2019, 46(3), 492–501, DOI:10.1080/02678292.2018.1512665.
- J. I. Sohn, W.-K. Hong, S. S. Choi, H. J. Coles, M. E. Welland, S. N. Cha and J. M. Kim, Emerging Applications of Liquid Crystals Based on Nanotechnology, Materials, 2014, 7(3), 2044–2061, DOI:10.3390/ma7032044.
- H.-W. Chen, J.-H. Lee, B.-Y. Lin, S. Chen and S.-T. Wu, Liquid crystal display and organic light-emitting diode display: present status and future perspectives, Light Sci. Appl., 2018, 7, 17168, DOI:10.1038/lsa.2017.168.
- Z. Zhang, X. Yang, Y. Zhao, F. Ye and L. Shang, Liquid Crystal Materials for Biomedical Applications, Adv. Mater., 2023, 35(36), 2300220, DOI:10.1002/adma.202300220.
- G. W. Gray, Molecular Structure and Properties of Liquid Crystals, Academic Press, New York, 1962 Search PubMed.
- R. Rosenhauer, T. Fischer, J. Stumpe, R. Giménez, M. Piñol, J. L. Serrano, A. Viñuales and D. Broer, Light-Induced Orientation of Liquid Crystalline Terpolymers Containing Azobenzene and Dye Moieties, Macromolecules, 2005, 38(6), 2213–2222, DOI:10.1021/ma048259f.
- Y. Yang, C. Liu, Y. Ding, B. Ding, J. Xu, A. Liu, J. Yu, L. Grater, H. Zhu, S. S. Hadke, V. K. Sangwan, A. S. R. Bati, X. Hu, J. Li, S. Min Park, M. C. Hersam, B. Chen, M. K. Nazeeruddin, M. G. Kanatzidis and E. H. Sargent, A thermotropic liquid crystal enables efficient and stable perovskite solar modules, Nat. Energy, 2014, 9, 316–323, DOI:10.1038/s41560-023-01444-z.
- M. T. Máthé, B. Farkas, L. Péter, Á. Buka, A. Jákli and P. Salamon, Electric field-induced interfacial instability in a ferroelectric nematic liquid crystal, Sci. Rep., 2023, 13, 6981, DOI:10.1038/s41598-023-34067-1.
- J. H. Lee, J.-Y. Lee, J.-S. Yu and J.-H. Kim, Electrical Energy Harvesting from the Flexible Liquid Crystal Cells, J. Phys. Chem. C, 2021, 125(41), 22429–22434, DOI:10.1021/acs.jpcc.1c05372.
- Y. Arakawa, Y. Ishida and H. Tsuji, Ether- and Thioether-Linked Naphthalene-Based Liquid-Crystal Dimers: Influence of Chalcogen Linkage and Mesogenic-Arm Symmetry on the Incidence and Stability of the Twist-Bend Nematic Phase, Chem. – Eur. J., 2020, 26(17), 3767–3775, DOI:10.1002/chem.201905208.
- M. Alaasar, M. Prehm, M. Brautzsch and C. Tschierske, 4-Methylresorcinol based bent-core liquid crystals with azobenzene wings – a new class of compounds with dark conglomerate phases, J. Mater. Chem. C, 2014, 2, 5487, 10.1039/C4TC00533C.
- Y. Arakawa, K. Komatsu, J. Feng, C. Zhu and H. Tsuji, Distinct twist-bend nematic phase behaviors associated with the ester-linkage direction of thioether-linked liquid crystal dimers, Mater. Adv., 2021, 2, 261–272, 10.1039/D0MA00746C.
- V. Jevtovic, H. A. Ahmed, M. T. Khan, S. A. Al-Zahrani, N. Masood and Y. A. Jeilani, Preparation of Laterally Chloro-Substituted Schiff Base Ester Liquid Crystals: Mesomorphic and Optical Properties, Crystals, 2023, 13(5), 835, DOI:10.3390/cryst13050835.
- H. Mali, V. S. Sharma, A. S. Sharma, P. S. Shrivastav, V. A. Rana and H. R. Prajapati, Azo-linked room temperature columnar liquid crystals with bisphenol A core: Structure property relationship and photophysical properties, J. Mol. Struct., 2024, 1303, 137505, DOI:10.1016/j.molstruc.2024.137505.
- E. Perju and L. Marin, Mesomorphic Behavior of Symmetric Azomethine Dimers Containing Different Chromophore Groups, Molecules, 2021, 26, 2183, DOI:10.3390/molecules26082183.
- K. D. Katariya, K. J. Nakum, H. Soni, S. Nada and M. Hagar, Imine based Four-Ring Chalcone-Ester liquid Crystals: Synthesis, Characterization, mesomorphic behaviour and DFT approach, J. Mol. Liq., 2023, 380, 121719, DOI:10.1016/j.molliq.2023.121719.
- P. Strasser and I. Teasdale, Main-Chain Phosphorus-Containing Polymers for Therapeutic Applications, Molecules, 2020, 25(7), 1716, DOI:10.3390/molecules25071716.
- H. Liu, K. Hu, D. Yan, R. Chen, Y. Zou, H. Liu and S. Wang, Recent Advances on Black Phosphorus for Energy Storage, Catalysis, and Sensor Applications, Adv. Mater., 2018, 30(32), 1800295, DOI:10.1002/adma.201800295.
- M. Batmunkh, M. Bat-Erdene and J. G. Shapter, Phosphorene and Phosphorene-Based Materials – Prospects for Future Applications, Adv. Mater., 2016, 28(39), 8586–8617, DOI:10.1002/adma.201602254.
- Y. Yi, Z. Sun, J. Li, P. K. Chu and X.-F. Yu, Optical and Optoelectronic Properties of Black Phosphorus and Recent Photonic and Optoelectronic Applications, Small Methods, 2019, 3(10), 1900165, DOI:10.1002/smtd.201900165.
- H. Tian, J. Wang, G. Lai, Y. Dou, J. Gao, Z. Duan, X. Feng, Q. Wu, X. He, L. Yao, L. Zeng, Y. Liu, X. Yang, J. Zhao, S. Zhuang, J. Shi, G. Qu, X.-F. Yu, P. K. Chu and G. Jiang, Renaissance of elemental phosphorus materials: properties, synthesis, and applications in sustainable energy and environment, Chem. Soc. Rev., 2023, 52(16), 5388–5484, 10.1039/D2CS01018F.
- B. C. Saunders and G. J. Stacey, Esters containing phosphorus; diisopropyl fluorophosphonate, J. Chem. Soc., 1948, 695–699, 10.1039/jr9480000695.
- M. Voráčová, M. Zore, J. Yli-Kauhaluoma and P. Kiuru, Harvesting phosphorus-containing moieties for their antibacterial effects, Bioorg. Med. Chem., 2023, 96, 117512, DOI:10.1016/j.bmc.2023.117512.
- Y. Iwasaki, C. Wachiralarpphaithoon and K. Akiyoshi, Novel Thermoresponsive Polymers Having Biodegradable Phosphoester Backbones, Macromolecules, 2007, 40(23), 8136–8138, DOI:10.1021/ma0715573.
- D. Serbezeanu, I.-D. Carja, A. Nicolescu, M. Aflori, T. Vlad-Bubulac, M. Butnaru, R.-F. Damian, S. Dunca and S. Shova, Synthesis, crystal structure and biological activity of new phosphoester-p-substituted-methylparabens, J. Mol. Struct., 2019, 1196, 637–646, DOI:10.1016/j.molstruc.2019.07.001.
- D. Wang, J. Liu, W. Zhao, Y. Zeng, J. Huang, J. Fang and D. Chen, Facile Synthesis of liquid crystal dimers bridged with a phosphonic group, Chem. – Eur. J., 2022, 28, e202202146, DOI:10.1002/chem.202202146.
- J. Thoen, G. Cordoyiannis and C. Glorieux, Investigations of phase transitions in liquid crystals by means of adiabatic scanning calorimetry, Liq. Cryst., 2009, 36, 669–684, DOI:10.1080/02678290902755564.
- X. Yu, V. V. Vashchenko, M. F. Prodanov and A. K. Srivastava, Monomolecular vertical alignment layer with room temperature processibility for flexible liquid crystal displays, J. Mol. Liq., 2022, 367, 120535, DOI:10.1016/j.molliq.2022.120535.
- J. Köhling, V. Kozel, V. Jovanov, R. Pajkert, S. N. Tverdomed, O. Gridenco and V. Wagner, Synthesis and Characterization of Oxazaborinin Phosphonate for Blue OLED Emitter Applications, ChemPhysChem, 2019, 20(5), 665–671, DOI:10.1002/cphc.201801087.
- H. Ehtesabi and S. O. Kalji, Metal Phosphate and Phosphonate Application for Imaging and Diagnosis, Metal Phosphates and Phosphonates: Fundamental to Advanced Emerging Applications, Springer International Publishing, Cham, 2023, pp. 373–391 Search PubMed.
- A. Oshchepkov, M. Oshchepkov, G. Pavlova, A. Ryabova, S. Kamagurov, S. Tkachenko and E. A. Kataev, Naphthalimide-functionalized bisphosphonates for fluorescence detection of calcification in soft tissues, Sens. Actuators, B, 2020, 314, 128047, DOI:10.1016/j.snb.2020.128047.
- D. Chakraborty, S. Bej, S. Sahoo, S. Chongdar, A. Ghosh, P. Banerjee and A. Bhaumik, Novel nanoporous Ti-phosphonate metal–organic framework for selective sensing of 2,4,6-trinitrophenol and a promising electrode in an energy storage device, ACS Sustainable Chem. Eng., 2021, 9(42), 14224–14237, DOI:10.1021/acssuschemeng.1c04877.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |