
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCalix[4]arene with a stiff upper rim bridge: spontaneous macrocyclization, structure, and dynamic behaviour†
Vladimir A.
Azov
 *a,
Jonas
Warneke
bc,
Ziyan
Warneke
b,
Matthias
Zeller
d and
Linette
Twigge
a
*a,
Jonas
Warneke
bc,
Ziyan
Warneke
b,
Matthias
Zeller
d and
Linette
Twigge
a
aDepartment of Chemistry, University of the Free State, P.O. Box 339, 9300 Bloemfontein, South Africa. E-mail: azovv@ufs.ac.za
bWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, 04103 Leipzig, Germany
cLeibniz-Institut für Oberflächenmodifizierung e. V. (IOM), Permoserstrasse 15, 04318 Leipzig, Germany
dDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
First published on 14th June 2024
Abstract
Macrocyclisation of the upper-rim distal disubstituted bromomethylene calix[4]arene derivative proceeds spontaneously on a silica gel due to the spatial proximity of two reaction centers, affording the formation of a bridged calixarene derivative. Due to the short span of the bridge, the product represents an extreme example of calix[4]arenes fixed in the pinched cone conformation with two aromatic rings steeply inclined into the center of the calixarene cavity. Dynamic NMR spectroscopy was used to investigate the temperature-dependent rocking motion of the bridgehead and thermochemical parameters of this motion were determined experimentally, as well as estimated using computational methods. Mass spectrometry was employed to study the upper rim macrocyclization reaction in the gas phase.
Introduction
Calixarenes are well known easily accessible macrocycles comprising several aromatic units interconnected by methylene bridges1,2 The number of aromatic units can vary, with the smallest calix[4]arene consisting of just four aromatic units. Due to the presence of the hydrophobic cavity inside the macrocycle and the possibility to attach ligands to the hydroxyl groups on the lower rim, calixarenes have been widely explored as host molecules, and proved to serve as successful building blocks in supramolecular chemistry in particular for the construction of molecular recognition systems of different types and purposes.3–8Calix[4]arenes are known to exist in four different conformations, cone, partial cone, 1,2-alternate, and 1,3-alternate,2 depending on the relative orientations of hydroxyl groups on the aromatic units. Interconversion between these four conformations occurs for the unprotected calix[4]arenes spontaneously at room temperature in solution, but can be hindered by the installation of protective groups on the hydroxyls of the lower rim. Since the backbone of calix[4]arene consist of four identical structural elements, it may be assumed that calix[4]arene should demonstrate a four-fold rotational symmetry. However, calix[4]arenes with four identically substituted HO-groups are desymmetrized due to the steric congestion of the R-groups and exist in two degenerate interconverting pinched cone conformations (Scheme 1), in which a pair of two opposite aromatic rings is almost parallel or even pinched into the inner cavity, whereas the two other aromatic rings are widely open and tilt outside.9–11
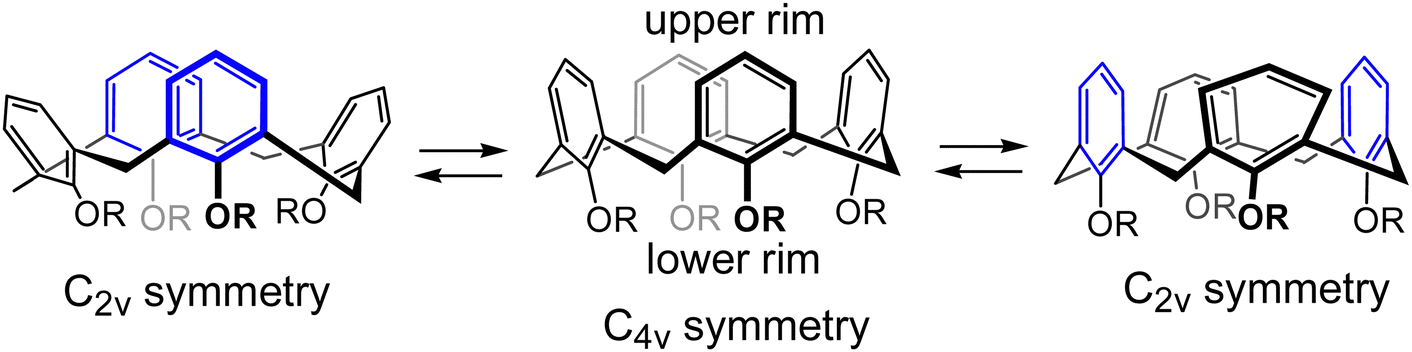 | ||
| Scheme 1 Equilibrium between two degenerate pinched cone conformations of a calix[4]arene with protected hydroxyl groups on the lower rim. Pairs of the quasi-parallel aromatic rings are shown in blue. | ||
This conformational behavior leads to the situation where the 1,3-distal positions (two opposite para-positions, or 5,17-positions according to the standard calix[4]arene numbering scheme) on the upper calix[4]arene rim may come into the close proximity with each other. This leads to surprisingly fast and efficient macrocyclization reactions with the formation of upper rim bridged calixarene derivatives. In particular, it was reported that 5,17-bis-hydroxymethyl calix[4]arene undergos facile macrocyclization by the formation of –CH2OCH2– bridges between two hydroxymethyl groups. First instances of this macrocyclization reaction were reported almost three decades ago,12–15 reporting formation of the products, differing from each other by the nature of the R-groups on the lower rim. Two such derivatives were characterized, but their molecular geometries and dynamic behavior have never been investigated in detail. In a report published in 2008 it was demonstrated that such a macrocyclization on the upper rim may indeed be a very efficient reaction.16 It was shown that the upper rim short ether bridge formation is much preferred to both the formation of derivatives with longer bridges and the oligomerization of several calix[4]arene monomer units into larger oligomeric calixarene macrocycles. Such high macrocyclization reaction rates were explained by the very high effective molarity (EM) for this intramolecular reaction17,18 due to the rigidity of the calixarene backbone and the limited number of rotatable bonds involved in the formation of the macrocycle, which drastically decreases the entropic component of the reaction. Several other instances of enhanced reactivity of the functional groups attached to the distal upper rim positions of calix[4]arene have been reported, such as an exceptionally fast and efficient intramolecular Cannizzaro reaction of the 5,17-bis-formyl calix[4]arene19 and intramolecular ester formation of the 5-carboxy-17-hydroxymethyl calix[4]arene derivative.20 The same effect was also used for chemical locking und unlocking of the cone conformation of 5,17-diaminocalix[4]arene.21
Results and discussion
During the synthesis of the dibromomethyl calix[4]arene derivative 1 by the bromination of 5,17-bis-hydroxymethyl calix[4]arene 2, we encountered a facile formation of the cyclization product 3 during the purification phase. Bromination, performed using PBr3 in dichloromethane, was followed by the basic workup of the reaction mixture. The expected bromination product 1 formed almost quantitatively, as evidenced by the TLC of the crude reaction mixture. Still, compound 1 partially decomposed during the purification by column chromatography on a neutral silica gel stationary phase, affording the new nonpolar substance 3. Its identity could be established using a combination of NMR and MS spectra (including HR-MS, see the ESI†), supported by the characterization data in one of the previous reports,16 demonstrating that the formation of macrocyclic ether 3 had occurred (Scheme 2). Significant amounts (yield up to 35%) of the cyclized product could be separated in the case of slowly performed gravity chromatography.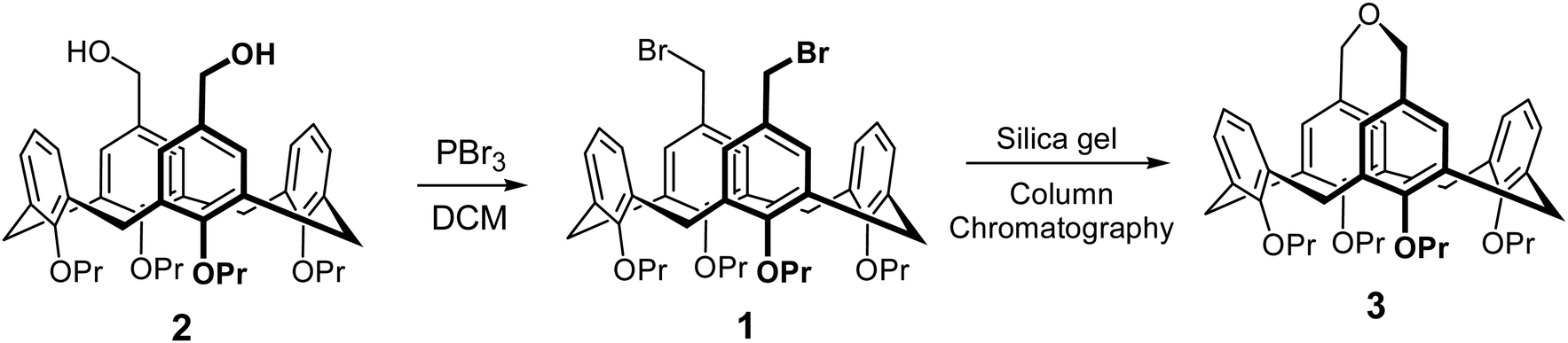 | ||
| Scheme 2 Bromination of 2, which was followed by the cyclization of the product 1 with formation of 3. | ||
Such ether formation is not a reaction expected upon chromatography of a benzyl bromide derivative, in particular taking into account that it required the presence of water to complete, as well as it was not expected to happen under neutral conditions. We can only hypothesize about the possible mechanism of this cyclization reaction on SiO2. It seems obvious that, first, one bromomethyl group should be hydrolyzed, followed by the cyclization of the hydroxymethyl-bromomethyl intermediate. Two factors might contributed to the facile decomposition/macrocyclization of the bis-bromomethyl derivative 1. First, the presence of Si–OH groups on the surface of silica gel give it a Brønsted acid nature, rendering it a catalytic activity even without impregnation by any additional reagents. It has been shown that SiO2 can promote acid-catalyzed reactions in its non-modified state,22,23 and it can be used as a catalyst in a variety of synthetic transfromations.24 Second, imposed proximity of two reaction centers may facilitate the initial step, heterolitic dissociation of the C–Br bond with the formation of the stabilized benzylic carbocation via the mechanism of the neighboring group participation by the cyclic bromonium ion bridging the two upper rim methylene groups. Though commonly known halonium ions (e.g., the intermediates of the halogenation reactions) are tricyclic, formation of larger 5-membered halonium ions upon the neighbouring group participation has been reported before,25 and the geometry for the formation of halonium cation is also favorable on this calix[4]arene platform. Afterwards, the benzylic cation is hydrated by water absorbed on silica and then cyclize by the substitution of the bromide on the neighboring methylene group.
The final proof of the molecular structure came from the X-ray structure determination of the crystals, which were grown by slow evaporation of a chloroform/ethanol solution. The structure unveiled the presence of two asymmetric calix[4]arene molecules with partially disordered propyl chains, as well as a disordered ethanol molecule (Fig. 1). It demonstrated an asymmetric disposition of the upper rim –CH2–O–CH2– bridge, which can be tilted to either of the sides of the calixarene backbone (Fig. 1b), as evidenced by the disorder of the bridge in the crystal structure. This implies that the side-tilted positions of the bridge represent the energetic minima, whereas the central, symmetric disposition is energetically disfavored. Another striking feature is the strong inclination of the two aromatic rings, interconnected by the ether bridge, into the calix[4]arene cavity. The interplanar angles between the planes of the bridged aromatic rings amount to −39.14° for one and −39.41° for the second asymmetric calix[4]arene molecule, respectively. The interplanar angles between the planes of two aromatic rings tilted outwards were found to be 72.85° for one and 73.38° for the other molecule, respectively. The calix[4]arene backbones of both asymmetric molecules adopt the conformations that are geometrically very close to each other, differing only by the disposition of the propyl substituents on the lower rim.
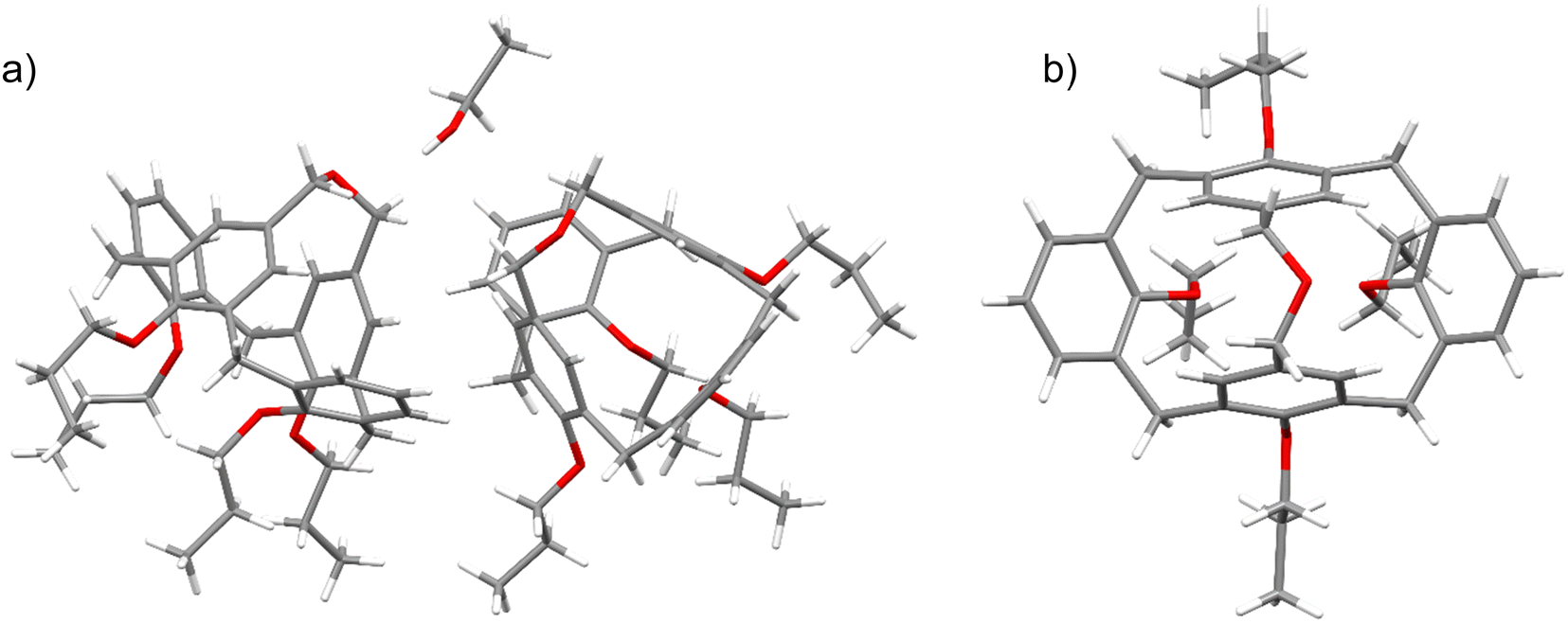 | ||
| Fig. 1 (a) The two crystallographically independent molecules of 3 and an ethanol molecule in the asymmetric unit. Only major orientations of the –CH2–O–CH2– bridges, propyl chains and ethyl alcohol molecule are shown. (b) Top view into the cavity of one of the molecules of 3 depicting the –CH2–O–CH2– bridge pointing to the right side. | ||
The molecular geometry of bridged calix[4]arene 3 looks very differently from the commonly assumed vase form of the cone conformation of calix[4]arene, and it appears to be one the most pinched calix[4]arene derivatives reported up to date. Though calix[4]arene derivatives sometimes adopt the strongly pinched geometry in the solid state with two aromatic rings inclining into the calixarene cavity,26–28 the most prominent examples come from the calix[4]arenes with the upper rim bridges.29 CCDC search returned three X-ray structures of calix[4]arene derivatives in the cone conformation with particularly short two-atom upper rim bridges,30–32 for which the angles between the planes of the pinched benzene rings are even more extreme, ranging from −49.50° to −53.03°.
One of the interesting properties of 3 was the broadening of the NMR signals of benzylic CH2 groups, which represents a classical example of a dynamical exchange effect in NMR.33,34 It takes place between two equally populated sites (conformations), and is caused by the side-to-side rocking movement of the upper rim bridge. The exchange is fast at elevated temperatures, but slows down upon cooling of a solution of compound 3 (Scheme 3), fixing the calix[4]arene backbone in either of two energetically degenerate conformations.
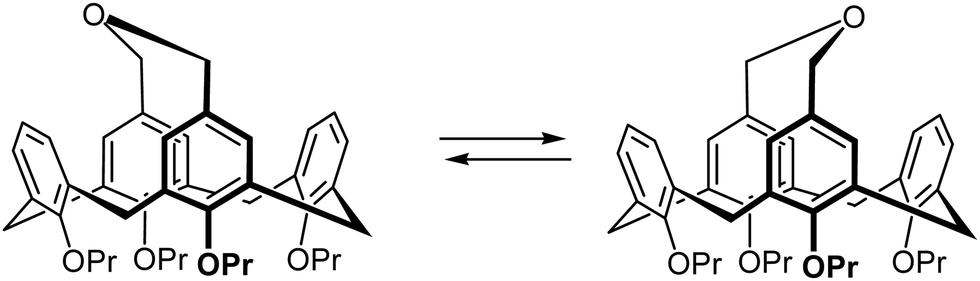 | ||
| Scheme 3 Switching between two degenerate conformations of 3 due to the rocking motion of the upper rim bridge. | ||
To estimate the parameters of the rocking movement of the bridge, a VT-NMR study in the temperature range of 213–328 K in CDCl3 has been performed. VT-NMR demonstrated splitting of several broadened NMR signals into a more complex NMR pattern upon cooling of the solution (Fig. 2). This splitting was very prominent for the aromatic protons in the ortho-position to the upper rim ether bridge, which appeared as a singlet at 5.80 ppm at 328 K, and as two sets of finely split doublets at 5.60 and 5.99 ppm at 263 K and below (see Fig. S2c, ESI† respectively). Dynamic behavior was also seen for the benzylic protons of the –CH2–O–CH2– bridges, which appeared as a very broad singlet at 3.97 ppm at 328 K, and as two sets of doublets at 3.54 and 4.61 ppm at 263 K.
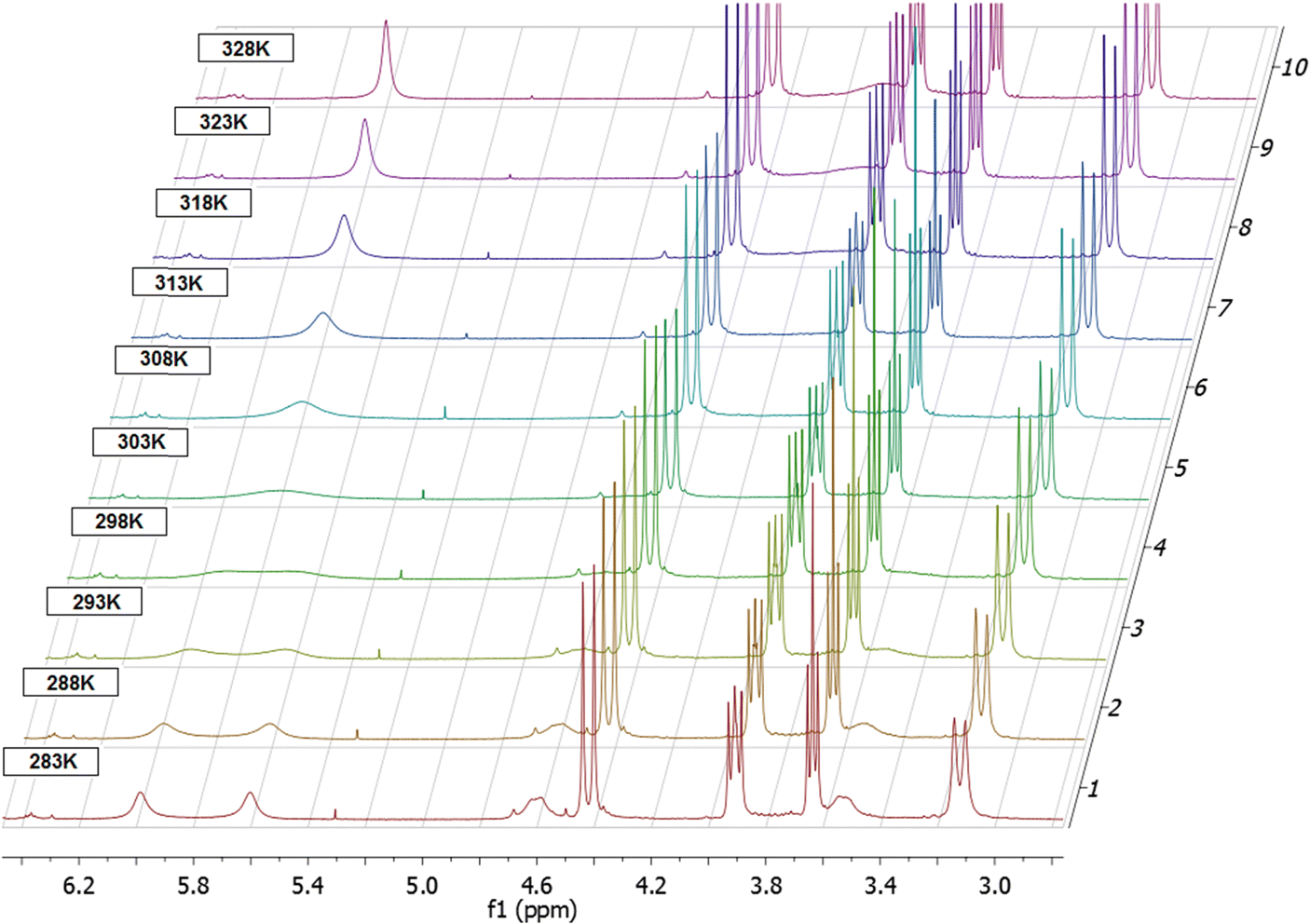 | ||
| Fig. 2 1H VT-NMR scan of 3 in the 283–328 K range in CDCl3. Note the behavior of signals at 5.6–6 ppm range. | ||
The temperature-dependent splitting of the signal at 5.80 ppm (protons in the ortho-position to the ether bridge) at 328 K into the pair of doublets at 5.60 and 5.99 ppm at 263 K and below was particularly sensitive and allowed for precise determination of the coalescence temperature at 299 K. It gave a Gibbs activation energy ΔG‡ of 58.97 kJ mol−1 for the rocking process by using the equation for the determination of the activation energy at the NMR signal coalescence temperature (see ESI† for details).
The same set of NMR signals was used for the fitting of the NMR spectra at different temperatures in order to determine the rate constants for the exchange process between two degenerate conformations of the calix[4]arene (see ESI† for the details). Using the rate constants, the activation parameters of the process could be determined using the Eyring plot method as described before,35–39 giving an activation enthalpy and entropy of ΔH‡ = 66.16 kJ mol−1 and ΔS‡ = 22.04 J K−1 mol−1, respectively (Fig. 3). Using these enthalpy and entropy values at 299 K, the free energy of activation ΔG‡ is calculated to be 59.57 kJ mol−1, almost matching the value determined from the coalescence temperature.
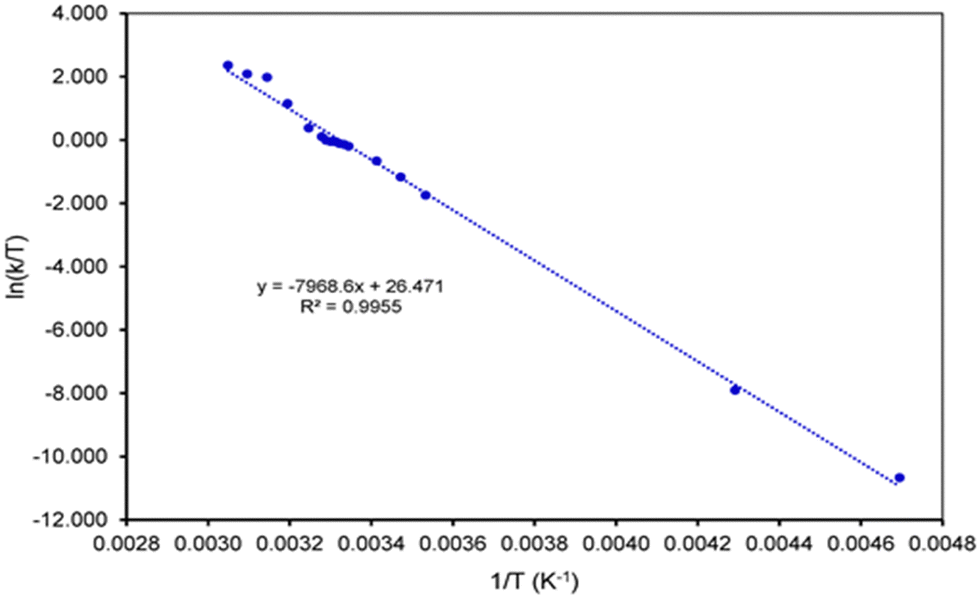 | ||
| Fig. 3 Eyring plot used to determine the activation parameters for the rocking movements of the upper rim bridge of 3. | ||
The energetics of the flipping of the ether bridge is dominated by the enthalpic component, whereas the contribution of the entropic term is relatively small. This implies that the distortion of the bridge valence angles during the flipping process is a rather high energy process.
To gain a deeper insight into the process of the conformational interconversion, we performed a theoretical investigation at the B3LYP/6-31g(d,p) level of theory (using a D3-BJ dispersion correction40,41 and the polarizable continuum model42 (PCM) for chloroform) using the Gaussian 1643 software (see the ESI† for details). In our model, we replaced the Pr-groups on the lower rim with Me-groups to reduce the number of conformers which are possible due to the flexibility of the propyl chains. First, we optimized the geometry of the initial low energy conformation, obtaining a geometry close to that of the molecules in the crystal state (Fig. 4). In particular, the angles between the planes of the two pinched aromatic units at −37.55° showed an almost perfect match with the angle observed in the crystal structure, while the angle between the rings tilted outwards was found to be 64.71° (this angle is much more flexible because it is determined only by the movement of the torsional angles of the calix[4]arene bowl and steric interactions of substituents on the lower rim). Subsequently, a transition state (TS) was found, and its geometry was optimized, and its authenticity was verified by frequency calculations. It turned out that the TS did not have the expected C2v symmetry, but was desymmetrized to C2, presumably to reduce the strain in the valence angles of the ether bridge. The energy of the transition state for the conformational interconversion process was found to be 55.7 kJ mol−1, which is in reasonably good agreement with the experimental data.
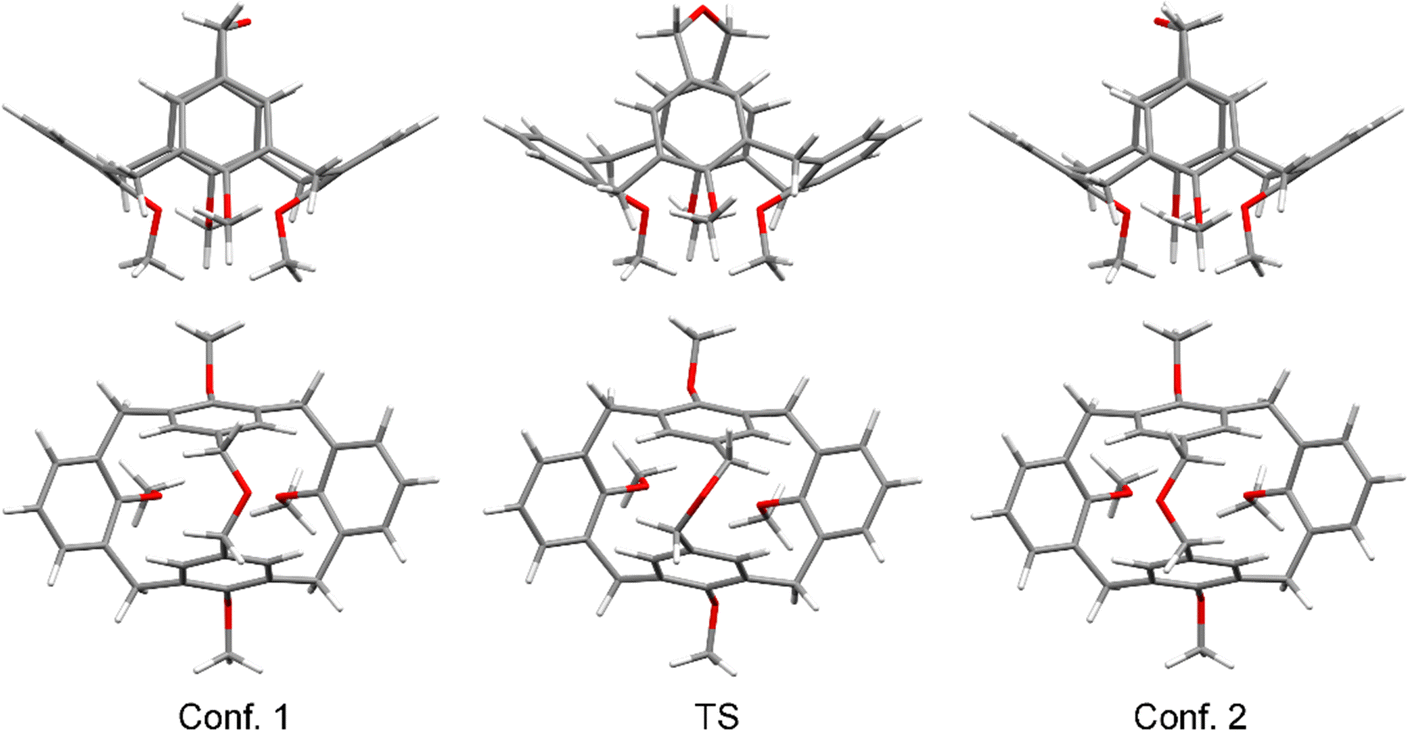 | ||
| Fig. 4 Representations of two degenerate low energy conformations of 3 and the transition state for the process of their conformational interconversion, top and side views. Pr groups on the lower rim were replaced with Me groups for the calculations. | ||
The ease of intramolecular macrocyclization of the upper rim distal substituted calix[4]arene derivatives could be evidenced also in the gas phase. We could not detect compound 2 in its protonated form [2 + H]+ in ESI(+) mode, though the [2 + Na]+ and [2 + K]+ aggregates were abundant in the spectra (see the ESI† for details). Presence of the signal at m/z 635, which corresponds to [3 + H]+ implied that 2 immediately loses a water molecule upon protonation in the gas phase, presumably forming the cyclized compound 3 in its protonated form. Mechanistically, this reaction can be explained by the protonation of one of the HO-groups followed by fast intramolecular cyclization upon nucleophilic attack of the distal methylene group, accompanied by simultaneous loss of a water molecule. (Scheme 4). The [2 + Na]+ and [2 + K]+ aggregates are stable in the gas phase, presumably because alkali metal cations prefer to be bound between the oxygen atoms of the calix[4]arene lower rim. Fragmentation of ions with m/z 635 isolated after ESI(+) of both compounds 2 and 3 showed identical fragmentation patterns, giving a strong support to the hypothesis that protonated compound 2 should undergo facile intramolecular cyclization in the gas phase upon loss of a water molecule.
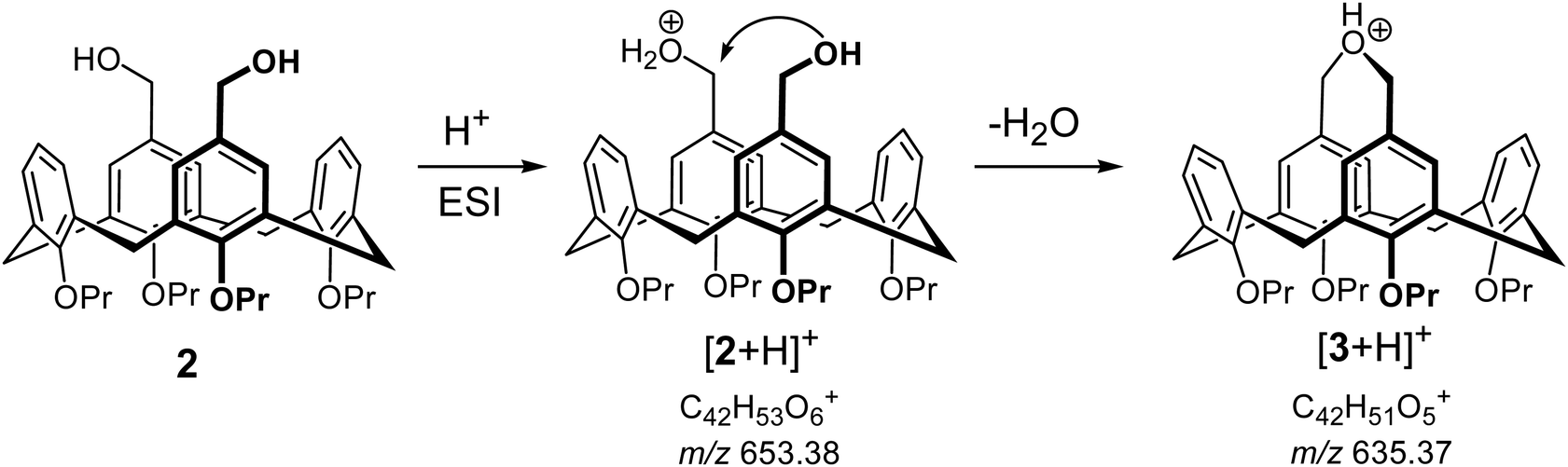 | ||
| Scheme 4 Macrocyclization of 2 in the gas phase after H+ adduct formation in ESI(+). | ||
Conclusions
To summarize, a calix[4]arene 3 derivative was discovered by serendipity upon chromatographic purification of an upper rim bis-bromomethyl calix[4]arene derivative 1. The crystal structure of calix[4]arene 3 evidenced that this compound is an extreme example of a pinched cone conformation of calix[4]arene, with two opposite rings demonstrating a very strong incline into the calixarene cavity. The rigidity of the short bridge does not allow for its free movement; it rather prefers to tilt to either side of the calix bowl, giving rise to two degenerate conformations. Conformational interconversion was investigated using VT-NMR and computational methods, allowing to determine the energetics of the process. An MS fragmentation study gave evidence that such upper-rim macrocyclization may be facilitated also in the gas phase. Our study represents another case of the enhanced reactivity between the functional groups at the distal upper rim positions of a calix[4]arene derivative, complementing the previously reported examples.16,19–21Experimental section
General
NMR characterization was performed on Bruker Avance DPX-200 and Bruker Avance DRX600 spectrometers. VT-NMR experiments were performed on a Bruker Avance DRX600 spectrometer. The sample was equilibrated for 10 min at each temperature before each measurement was performed. Chemical shifts (δ) are reported in parts per million (ppm) relative to TMS, the residual solvent signals were used as reference: CDCl3 (7.26 ppm for 1H, 77.0 ppm for 13C). 1H NMR coupling constants (J) are reported in Hertz (Hz) and multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), m (multiplet). EI-MS spectrum (EI, 70 EV) was measured on a Finnigan MAT 8200 spectrometer. High resolution MS (HRMS) measurements were performed on a LTQ Orbitrap XL (Thermo Fisher Scientific, Bremen) spectrometer (ESI+ mode). The sample was dissolved in acetonitrile at concentrations of approx. 10−5 to 10−6 mol L−1 and injected via a syringe pump at flow rates of 5 μL min−1. Fragmentation experiments (ESI+ mode) were performed on a Esquire 3000Plus (Bruker Daltonics, Bremen) ion trap spectrometer.5,17-Dibromomethyl-25,26,27,28-tetra(1-propoxy)-calix[4]arene 1 was prepared as previously reported by the bromination of corresponding dihydroxy derivative 2, which was prepared as described before.44,45 A typical procedure was done as following.45 Phosporus tribromide (86 mg, 30 μl, 0.32 μmol) was added dropwise to a solution of 5,17-dihydroxymethyl-25,26,27,28-tetra-(1-propoxy)calix[4]arene 2 (200 mg, 0.306 μmol) in dichloromethane (30 ml), and the reaction mixture was stirred for further 10–60 min (TLC control). Then the mixture was washed with NaHCO3 solution (30 ml), water, brine, dried over Na2SO4 and evaporated to dryness. Dibromide 1 is obtained in 85–95% yield and was usually sufficiently clean to be used in the next steps without further purification. Further chromatographic separation of the product leads to the partial decomposition of 1 with the formation of the bridged calix[4]arene 3. Compound 3 was isolated upon flash chromatography of the crude carried out using 230–440 mesh (particle size 36–70 μm) silica gel in CH2Cl2/petroleum ether (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), coming as the fraction eluted before the fraction of dibromide 1. Depending on the eluent flow rate through the chromatography column and the thickness of the silica layer, the yield could reach up to 35%.
:1), coming as the fraction eluted before the fraction of dibromide 1. Depending on the eluent flow rate through the chromatography column and the thickness of the silica layer, the yield could reach up to 35%.
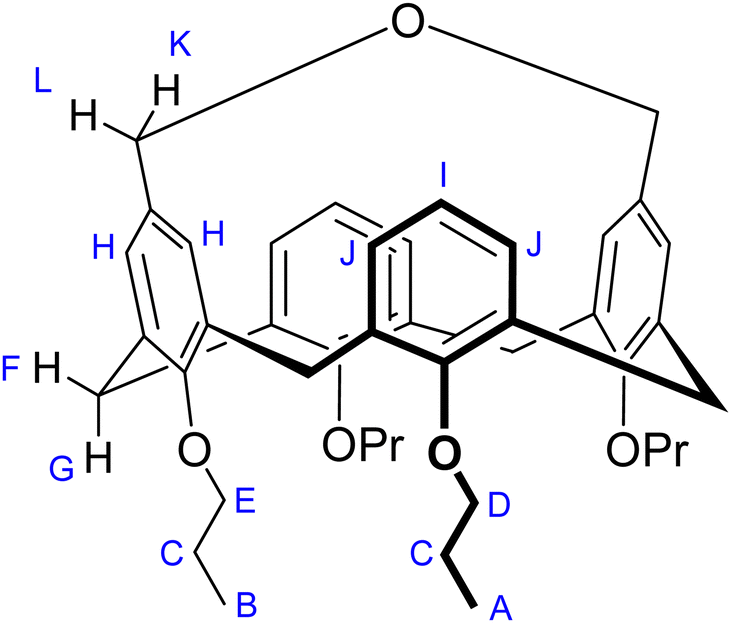
1H-NMR(CDCl3, 600 MHz, 328 K): δ 7.15 (d, J = 7.4 Hz, 4H, HJ), 6.98 (t, J = 7.4 Hz, 2H, HI), 5.80 (broad s, 4H, HH), 4.45 (d, J = 14.0 Hz, 4H, HG), 4.07 (v broad s, 4H, HKL), 3.94 (t, J = 8.0 Hz, 4H, HF), 3.67 (t, J = 6.5 Hz, 4H, HE), 3.12 (d, J = 14.0 Hz, 4H, HD), 1.79–1.89 (m, 8H, HC), 1.12 (t, J =7.4 Hz, 6H, HB), 0.86 (t, J = 7.5 Hz, 6H, HA) ppm. 1H-NMR (CDCl3, 600 MHz, 293 K): δ 7.15 (d, J = 7.4 Hz, 4H, HJ), 6.98 (t, J = 7.4 Hz, 2H, HI), 5.96 (broad s, 2H, HH), 5.63 (broad s, 2H, HH), 4.61 (v broad s, 2H, HL), 4.43 (d, J = 14.0 Hz, 4H, HG), 3.92 (t, J = 8.0 Hz, 4H, HF), 3.64 (t, J = 6.5 Hz, 4H, HE), 3.52 (v broad s, 2H, HK), 3.12 (d, J = 14.0 Hz, 4H, HD), 1.78–1.86 (m, 8H, HC), 1.11 (t, J =7.4 Hz, 6H, HB), 0.83 (t, J = 7.5 Hz, 6H, HA) ppm. 1H-NMR (CDCl3, 600 MHz, 263 K): δ 7.17 (d, J = 7.5 Hz, 4H, HJ, two sets), 6.96–7.02 (m, 2H, HI), 5.99 (d, J = 1.5 Hz, 2H, HH), 5.60 (d, J = 1.5 Hz, 2H, HH), 4.61 (d, J = 13.3 Hz, 2H, HL), 4.40 (d, J = 14.1 Hz, 2H, HG), 4.39 (d, J = 14.0 Hz, 2H, HG), 3.85–3.90 (m, 4H, HF), 3.61 (t, J = 6.3 Hz, 4H, HE), 3.54 (d, J = 13.3 Hz, 2H, HK), 3.14 (d, J = 13.6 Hz, 2H, HD), 3.11 (d, J = 13.9 Hz, 2H, HD), 1.77–1.85 (m, 8H, HC), 1.10 (t, J =7.4 Hz, 3H, HB), 0.79 (t, J = 7.5 Hz, 6H, HA), 0.78 (t, J = 7.5 Hz, 3H, HA) ppm. 13C-NMR (CDCl3, 50 MHz, 293 K): δ 158.7, 154.6, 137.6, 133.2, 129.1, 129.0, 127.3, 121.6, 76.2 (O–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2–CH2–CH3), 75.9 (O–H2–CH2–CH3), 31.0 (ArH2Ar), 23.5 (O–CH2–H2–CH3), 23.0 (O–CH2–H2–CH3), 10.9 (O–CH2CH2H3), 9.8 (O–CH2CH2H3) ppm.
H2–CH2–CH3), 75.9 (O–H2–CH2–CH3), 31.0 (ArH2Ar), 23.5 (O–CH2–H2–CH3), 23.0 (O–CH2–H2–CH3), 10.9 (O–CH2CH2H3), 9.8 (O–CH2CH2H3) ppm.
M.p. 161–164 °C. Rf = 0,33 (SiO2, CH2Cl2/PE, 7:1). IR (NaCl plate): ν 2961, 2919, 2874, 1465, 1383, 1278, 1215, 1124, 1080, 1036, 1007, 1008, 965, 765 cm−1. MS (EI, 70 eV) m/z (I%): 634 (100%) [M]+, 591 (40%) [M–C3H7]+, 549 (30%) [M–C3H7–C3H6]+, 507 (10%) [M–C3H7–2C3H6]+. HR-MS (ESI+) m/z calcd for [C42H50O5 + H]+: 635.372 [M + H]+, found: 635.372.
Crystal structure
Crystals were grown by slow evaporation of chloroform/ethanol solution. Disorder of several of the propyl side chains and of the ether oxygen atoms is observed, and a site partially occupied by ethanol is present. The moieties of the disordered propyl side chains were restrained to have geometries similar to that of a well-defined propyl group. The oxygen atoms adjacent to the propyl groups were included in the disorder refinement.One of the propyl groups was refined to be disordered over three sites. Uij components of ADPs for disordered atoms closer to each other than 2.0 Å were restrained to be similar (SIMU restraint of Shelxl). Subject to these conditions the occupancy ratios refined to values of 0.873(3) to 0.127(3) (O1), 0.811(4) to 0.189(4) (O6) and 0.802(4) to 0.198(4) (O8). For the three fold disordered site occupancies refined to 0.607(2), 0.298(2) 0.095(2) (O3). The ether oxygen atoms of the bridging units are disordered in both crystallographically independent molecules. Occupancy ratios refined to 0.514(3) to 0.486(3) (O5) and 0.707(3) to 0.293(3) (O10).
A partially occupied site with ethanol molecules is present. The molecule is present in two orientations, hydrogen bonded to either O5B (the minor occupied ether atom of molecule one), or to O10B (the minor occupied ether atom of molecule two). The two ethanol molecules were restrained to have similar geometries, Uij components of ADPs for disordered atoms closer to each other than 2.0 Å were restrained to be similar (SIMU restraint of Shelxl). Subject to these restraints the occupancy rates refined to 0.086(4) and 0.069(4). CCDC Deposition Number 2347668.†
Author contributions
AA: conceptualization, synthesis/isolation of the compounds 2 and 3, growth of crystals, analysis of the data, writing of the paper. JW: VT-NMR measurements, writing of the paper, ZW: MS characterization and fragmentation experiments; MZ: crystallography; LT: VT-NMR fittingData availability
Crystal structure was deposited to Cambridge Crystallographic Data Centre, CCDC Deposition Number 2347668. Processed NMR and MS spectra are available in the ESI.† Molecular coordinates for the computed key molecular structures are available in the ESI.† Raw spectral data and molecular coordinates for all computed structures are available from the authors on request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to Mr Christian Herbst, who contributed to the preparation of compounds 1, 2 and 3 during his work on the undergraduate research project. JW is grateful to the Volkswagen foundation for a Freigeist Fellowship. JW and ZW are grateful for Technical support from the MS Core Facility MS UL at Leipzig University. XRD data had been collected on an instrument at Youngstown State University which was funded by NSF Grants DMR 1337296, CHE 0087210, Ohio Board of Regents Grant CAP-491, and by Youngstown State University.References
- C. D. Gutsche, Calixarenes, Royal Society of Chemistry, Cambridge, 2008 Search PubMed.
- V. Böhmer, Macrocycles with (Almost) Unlimited Possibilities, Angew. Chem., Int. Ed. Engl., 1995, 34, 713–745, DOI:10.1002/anie.199507131.
- A. Ikeda and S. Shinkai, Novel Cavity Design Using Calix[n]arene Skeletons: Toward Molecular Recognition and Metal Binding, Chem. Rev., 1997, 97, 1713–1734, DOI:10.1021/cr960385x.
- S. E. Matthews and P. D. Beer, Calixarene-Based Anion Receptors, Supramol. Chem., 2005, 17, 411–435, DOI:10.1080/10610270500127089.
- J. Harrowfield, Calixarenes and Cations, Chem. Commun., 2013, 49, 1578–1580, 10.1039/c3cc38667h.
- A. Dondoni and A. Marra, Calixarene and Calixresorcarene Glycosides: Their Synthesis and Biological Applications, Chem. Rev., 2010, 110, 4949–4977, DOI:10.1021/cr100027b.
- H. J. Kim, M. H. Lee, L. Mutihac, J. Vicens and J. S. Kim, Host–Guest Sensing by Calixarenes on the Surfaces, Chem. Soc. Rev., 2012, 41, 1173–1190, 10.1039/C1CS15169J.
- D. Ajami, L. Liu and J. Rebek, Jr., Soft Templates in Encapsulation Complexes, Chem. Soc. Rev., 2014, 44, 490–499, 10.1039/C4CS00065J.
- M. Conner, S. L. Regen and V. Janout, Pinched-Cone Conformers of Calix[4]arenes, J. Am. Chem. Soc., 1991, 113, 9670–9671, DOI:10.1021/ja00025a041.
- A. Soi, W. Bauer, H. Mauser, C. Moll, F. Hampel and A. Hirsch, Investigations on the Dynamic Properties of 25,26,27,28-Tetraalkoxycalix[4]arenes: Para-Substituent- and Solvent-Dependent Properties of Paco Conformers and Determination of Thermodynamic Parameters of the Pinched Cone/Pinched Cone Conversion, J. Chem. Soc. Perkin Trans, 1998, 2, 1471–1478, 10.1039/a708606g.
- J. Scheerder, R. H. Vreekamp, J. F. J. Engbersen, W. Verboom, J. P. M. Van Duynhoven and D. N. Reinhoudt, The Pinched Cone Conformation of Calix[4]arenes: Noncovalent Rigidification of the Calix[4]arene Skeleton, J. Org. Chem., 1996, 61, 3476–3481, DOI:10.1021/jo9600262.
- A. Arduini, S. Fanni, G. Manfredi, A. Pochini, R. Ungaro, A. R. Sicun and F. Ugozzoli, Direct Regioselective Formylation of Tetraalkoxycalix[4]arenes Fixed in the Cone Conformation and Synthesis of New Cavitands, J. Org. Chem., 1995, 60, 1448–1453, DOI:10.1021/jo00110a054.
- A. Arduini, S. Fanni, A. Pochini, A. R. Sicuri and R. Ungaro, Highly Distorted Cone Calix[4]arenes through Intramolecular McMurry Coupling Reaction, Tetrahedron, 1995, 51, 7951–7958, DOI:10.1016/0040-4020(95)00411-Z.
- A. Dondoni, A. Marra, M. C. Scherrmann, A. Casnati, F. Sansone and R. Ungaro, Synthesis and Properties of O-Glycosyl Calix[4]arenes (Calixsugars), Chem. – Eur. J., 1997, 3, 1774–1782, DOI:10.1002/chem.19970031108.
- H. D. Banks, A. Dondoni, M. Kleban and A. Marra, Computational and Experimental Studies of Di- and Tetrasubstituted Calix[4]arenes, Chirality, 2002, 14, 173–179, DOI:10.1002/chir.10059.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, On the ‘Livingness’ of a Dynamic Library of Cyclophane Formaldehyde Acetals Incorporating Calix[4]arene Subunits, J. Phys. Org. Chem., 2008, 21, 688–693, DOI:10.1002/poc.1357.
- S. Di Stefano and L. Mandolini, The Canonical Behavior of the Entropic Component of Thermodynamic Effective Molarity. An Attempt at Unifying Covalent and Noncovalent Cyclizations, Phys. Chem. Chem. Phys., 2019, 21, 955–987, 10.1039/C8CP06344C.
- L. Mandolini, Advances in Physical Organic Chemistry, Academic Press, 1986, vol. 22, pp. 1–111 Search PubMed.
- M. Galli, J. A. Berrocal, S. Di Stefano, R. Cacciapaglia, L. Mandolini, L. Baldini, A. Casnati and F. Ugozzoli, Highly Efficient Intramolecular Cannizzaro Reaction between 1,3-Distal Formyl Groups at the Upper Rim of a Cone-Calix[4]arene, Org. Biomol. Chem., 2012, 10, 5109–5112, 10.1039/C2OB25458A.
- J. A. Berrocal, M. B. Baker, L. Baldini, A. Casnati and S. Di Stefano, Inherently Chiral Cone-Calix[4]Arenes via a Subsequent Upper Rim Ring-Closing/Opening Methodology, Org. Biomol. Chem., 2018, 16, 7255–7264, 10.1039/C8OB01813H.
- D. Del Giudice, E. Spatola, R. Cacciapaglia, A. Casnati, L. Baldini, G. Ercolani and S. Di Stefano, Time Programmable Locking/Unlocking of the Calix[4]arene Scaffold by Means of Chemical Fuels, Chem. – Eur. J., 2020, 26, 14954–14962, DOI:10.1002/chem.202002574.
- P. J. Kropp, G. W. Breton, S. L. Craig, S. D. Crawford, W. F. Durland, J. E. Jones and J. S. Raleigh, Surface-Mediated Reactions. 6. Effects of Silica Gel and Alumina on Acid-Catalyzed Reactions, J. Org. Chem., 1995, 60, 4146–4152, DOI:10.1021/jo00118a035.
- G. Szalóki and L. Sanguinet, Silica-Mediated Synthesis of Indolinooxazolidine-Based Molecular Switches, J. Org. Chem., 2015, 80, 3949–3956, DOI:10.1021/acs.joc.5b00282.
- V. A. Basiuk, Organic Reactions on the Surface of Silicon Dioxide: Synthetic Applications, Russ. Chem. Rev., 1995, 64, 1003–1019, DOI:10.1070/RC1995v064n11ABEH000190.
- P. E. Peterson, Cyclic Halonium Ions with Five-Membered Rings, Acc. Chem. Res., 1971, 4, 407–413, DOI:10.1021/ar50048a003.
- M. Larsen, F. C. Krebs, N. Harrit and M. Jørgensen, Synthesis and Conformational Studies of a Series of 5,17-bis-Aryl-25,26,27,28-tetrapropoxycalix[4]arenes: The Influence of π–π Interactions on the Molecular Structure, J. Chem. Soc. Perkin Trans, 1999, 2, 1749–1758, 10.1039/a901881f.
- P. F. Hudrlik, S. T. Hailu, A. M. Hudrlik and R. J. Butcher, Reactions of Calix[4]arenes with 1,3-Dibromopropane and 1,5-Dibromopentane. Identification of Products Using 1D and 2D NMR Techniques, and X-Ray Crystallography, J. Mol. Struct., 2013, 1054–1055, 271–281, DOI:10.1016/j.molstruc.2013.09.040.
- F. Elaieb, D. Sémeril, D. Matt and A. Hedhli, Crystal Structure of 5,17-bis-Cyano-25,26,27,28-tetrapropyloxy-calix[4]arene, C42H46N2O4, Z. Kristallogr. – New Cryst. Struct., 2016, 231, 943–945, DOI:10.1515/ncrs-2016-0015.
- D. A. Tan and M. Mocerino, Calix[4]arenes and Resorcinarenes Bridged at the Wider Rim, in Calixarenes and Beyond, Springer International Publishing, Cham, 2016, pp. 235–253 DOI:10.1007/978-3-319-31867-7_10.
- L. Baldini, F. Sansone, G. Faimani, C. Massera, A. Casnati and R. Ungaro, Self-Assembled Chiral Dimeric Capsules from Difunctionalized N,C-Linked Peptidocalix[4]arenes: Scope and Limitations, Eur. J. Org. Chem., 2008, 869–886, DOI:10.1002/ejoc.200700943.
- M. Zajícová, V. Eigner, J. Budka and P. Lhoták, Intramolecular Bridging of Calix[4]arene Dialdoximes, Tetrahedron Lett., 2015, 56, 5529–5532, DOI:10.1016/j.tetlet.2015.08.032.
- M. Řezanková, J. Budka, J. Mikšátko, V. Eigner, I. Císařová, P. Cuřínová and P. Lhoták, Anion Receptors Based on Intramolecularly Bridged Calix[4]arenes Bearing Ureido Functions, Tetrahedron, 2017, 73, 742–749, DOI:10.1016/j.tet.2016.12.054.
- A. D. Bain and Prog Nucl, Chemical Exchange in NMR, Magn. Reson. Spectrosc., 2003, 43, 63–103, DOI:10.1016/j.pnmrs.2003.08.001.
- J. Sandström, Dynamic NMR Spectroscopy, Academic Press, London, 1982 Search PubMed.
- K. Damodaran, S. D. Nielsen, S. J. Geib, W. Zhang, Y. Lu and D. P. Curran, Aryl−Csp3 Bond Rotation Barriers of 2-Aryl Perhydropyrrolo[3,4-c]pyrrole-1,3-diones, J. Org. Chem., 2009, 74, 5481–5485, DOI:10.1021/jo901123c.
- D. B. Guthrie, K. Damodaran, D. P. Curran, P. Wilson and A. J. Clark, Bond Rotation Dynamics of N-Cycloalkenyl-N-benzyl α-Haloacetamide Derivatives, J. Org. Chem., 2009, 74, 4262–4266, DOI:10.1021/jo900491w.
- S. Szymański, H. Dodziuk, M. Pietrzak, J. Jaźwiński, T. B. Demissie and H. Hopf, Dynamics of [n.3]Paracyclophanes (n = 2 - 4) as Studied by NMR. Obtaining Separate Arrhenius Parameters for Two Dynamic Processes in [4.3]Paracyclophane, J. Phys. Org. Chem., 2013, 26, 596–600, DOI:10.1002/poc.3137.
- K. Damodaran, X. Li, X. Pan and D. P. Curran, Dynamic Behavior of N-Heterocyclic Carbene Boranes: Boron–Carbene Bonds in B,B-Disubstituted N,N-Dimethylimidazol-2-ylidene Boranes Have Substantial Rotation Barriers, J. Org. Chem., 2015, 80, 4465–4469, DOI:10.1021/acs.joc.5b00324.
- P. Roncucci, L. Pirondini, G. Paderni, C. Massera, E. Dalcanale, V. A. Azov and F. Diederich, Conformational Behavior of Pyrazine-Bridged and Mixed-Bridged Cavitands: A General Model for Solvent Effects on Thermal “Vase–Kite” Switching, Chem. – Eur. J., 2006, 12, 4775–4784, DOI:10.1002/chem.200600085.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104, DOI:10.1063/1.3382344.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32, 1456–1465, DOI:10.1002/jcc.21759.
- B. Mennucci, Polarizable Continuum Model, WIREs Comput. Mol. Sci., 2012, 2, 386–404, DOI:10.1002/wcms.1086.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. MontgomeryJr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, 2016, Gaussian 16, Revision C.01, Gaussian, Inc., Wallin Search PubMed.
- K. Araki and H. Hayashida, Guest Inclusion Properties of a Novel Cage Molecule Composed of Two Calix[4]arenes, Tetrahedron Lett., 2000, 41, 1209–1213, DOI:10.1016/S0040-4039(99)02232-7.
- M. H. Düker, F. Kutter, T. Dülcks and V. A. Azov, Calix[4]arenes with 1,2- and 1,3-Upper Rim Tetrathiafulvalene Bridges, Supramol. Chem., 2014, 26, 552–560, DOI:10.1080/10610278.2013.872245.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347668. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj01698j |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |