DOI:
10.1039/D4NJ01621A
(Paper)
New J. Chem., 2024,
48, 13276-13288
Copper(II) complexes derived from naphthalene-based halogenated Schiff bases: synthesis, structural analysis, DFT computational studies and in vitro biological activities†
Received
8th April 2024
, Accepted 3rd July 2024
First published on 5th July 2024
Abstract
Two halogenated Schiff bases namely, (E)-1-(((2,6-dichlorophenyl)imino)methyl)naphthalen-2-ol (L1) and (E)-1-(((4-bromo-2,6-dichlorophenyl)imino)methyl)naphthalen-2-ol (L2) were synthesized and reacted with copper(II) nitrate trihydrate in ethanol to give Cu(L1)2 (1) and Cu(L2)2 (2). All the ligands and complexes were successfully characterized using FT-IR, UV-Vis, 1H & 13C-NMR as well as mass spectra and the purity was ascertained by elemental analysis. Electron paramagnetic resonance (EPR) was used to confirm the paramagnetic nature of 1 and 2. Furthermore, the single X-ray crystal structures of 1 and 2 were determined, confirming the formation of mononuclear species in which the Cu(II) center was bonded to two bidentate Schiff bases (L1 or L2) adopting a slightly distorted square planar geometry. Density Function Theory studies revealed that, complex 2 with lowest energy band gap (ΔE) of 2.49 eV is the most reactive among the compounds. α-Amylase and α-glucosidase assays were used to evaluate the antidiabetics potential of the compounds. Complexes 1 and 2 displayed very promising antidiabetic activities with IC50 values of 148.126 mM and 107.786 mM for α-amylase assay while it was 171.559 mM for acarbose (reference drug). The antioxidant potential of the compounds was investigated using nitric oxide (NO), ferric reducing ability power (FRAP) and 2,2-diphenyl-1-picrylhydrazyl (DPPH) assays. The compounds showed moderate to good antioxidant activities with 1 and 2 having IC50 values of 100.044 mM and 247.463 mM for NO scavenging assay, which relatively surpass Vanillin with IC50 value of 466.626 mM. All the compounds showed poor to moderate results against Escherichia coli, Staphylococcus aureus, Bacillus substilis, and Pseudomonas aeruginosa. However, none of them were active against Klebsiella pneumoniae. Generally, 1 and 2 displayed better antidiabetes, antioxidant, and antibacterial potential than L1 and L2. Predicted physicochemical and pharmacokinetic properties of all the compounds showed minimal violation of Lipinski's Ro5.
1. Introduction
Diabetes mellitus (DM) is a deadly disease which arises due to unusual excessive glucose in the body due to insulin resistance or irregularities in the production of insulin, and, in some patients, the two factors might be the cause.1,2 There are three types of diabetes, namely, type 1 (insulin-dependent), type 2 (insulin-independent) and gestational diabetes.3 It has been projected that, by the year 2030, 7.7% of the total grown-up population (439 million individuals) would be experiencing diabetic problems if not checkmated.4 Inhibition of hydrolytic enzymes such as intestinal α-glucosidase as well as pancreatic α-amylase have been employed over the years as a diagnostic therapy for diabetes.5 These enzymes are responsible for the breaking down of carbohydrate to glucose and inhibiting these enzymes would decrease the use of dietary carbohydrates which in turn results in reduction of glucose in the body as well as suppression of postprandial glycemia, hence a remedy for diabetic illness.6 Commercially available drugs such as acarbose and miglitol have been effectively used in curing diabetes by retarding the body tendency to breakdown starches and some sugars to glucose. However, side effects such as farting, diarrhea, stomach pain, and meteorism had drastically reduce their usage as potential antidiabetic drugs.7,8 Interestingly, free radicals have been associated with the cause of diabetes as well as deadly diseases such as cancer, liver cirrhosis, atherosclerosis among others. It has been reported that, compounds with the tendency to scavenge free radicals have the great potential in ameliorating diabetes.9 Thus, a non-toxic antioxidant could play an important chemoprotective role in the treatment of diabetes.10 Hence, the need to discover novel antidiabetic and antioxidant drugs with little or no side effects.
Furthermore, research on the discovery of new antibiotic drugs is on the rise in the past decades. Infectious diseases were known to be the major contributor to high mortality and morbidity rate all over the world prior to 20th century.11 Fleming's discovery of Penicillin in 192912 was a breakthrough in modern medicine towards the fight against pathogens and between 1940 to 1965 several antibiotics were discovered which resulted in the revolutionization of bacterial infectious diseases treatment.13 Unfortunately, the overuse and misuse of antibiotics led to the emergence of antimicrobial resistance, a problem ravaging around the world and posing serious threats to mankind.14,15 Annually, 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths were recorded in the United States due to antimicrobial resistance.16 Of more concern is the unavailability or misrepresentation of this data in underdeveloped countries where misuse of antibiotics is on the high side. Therefore, the need for the development of novel antibiotics with distinct mechanism of actions which differs from those well-known antibiotics to which relevant pathogens resist.17
000 deaths were recorded in the United States due to antimicrobial resistance.16 Of more concern is the unavailability or misrepresentation of this data in underdeveloped countries where misuse of antibiotics is on the high side. Therefore, the need for the development of novel antibiotics with distinct mechanism of actions which differs from those well-known antibiotics to which relevant pathogens resist.17
The medicinal properties of Schiff bases and their metal complexes have been studied extensively.18–21 The imine bond in Schiff bases plays a vital role in their medicinal properties and these are greatly improved upon complexation with metal ions due to increase in the lipophilic character of the metal complexes when compared to the free ligands.15,22 Considering all transition metal ions, copper(II) ion plays a crucial role in all living organisms i.e. major structural component in enzymes such as laccase, ceruloplasmin, ascorbate oxidase, superoxide dismutase among others.23,24 Copper complexes have been used as antimicrobial,25,26 antioxidant,27 anticancer,28 anti-inflammatory,29 antituberculosis30 among others. In this research work, we designed halogenated Schiff bases and their copper(II) complexes. Our curiosity stems from the fact that substitution of hydrogen with chlorine atoms in drug molecules increases their medicinal potency by 10-fold and even beyond in some cases and this could be attributed to the increase in lipophilicity of the halogenated compounds.17,31 Herein, we report the in vitro antibacterial, antidiabetic and antioxidant studies of halogenated Schiff base metal complexes.
2. Experimental section
2.1 Materials
Solvents explored for this study were A.C.S. grade (purity ≥99.5%) and used without further purification. Reagents used are 2,6-dichloro-phenylamine (≥98%), (4-bromo-2,6-dichlorophenyl)amine (97%), 2-hydroxyl-1-naphthaldehyde (98%) and copper(II) nitrate trihydrate (99%). All chemicals were purchased from Sigma-Aldrich.
2.2 Instrumentation
Bruker AvanceIII 600 MHz spectrometer were used to analyze 1H and 13C NMR spectra of L1 & L2 at 25 °C. Deuterated chloroform was used as a solvent to obtain the 1H N.M.R. and 13C N.M.R. data and peaks at δ 7.26 and δ 77.00 ppm are ascribed to residual d-chloroform respectively. Vario elemental E.L. cube CHNS analyser was used for the elemental analysis and I.R. spectra were obtained on a PerkinElmer Universal A.T.R. spectrum 100 FT-IR spectrometer. Mass spectra of L1 and L2 as well as complexes 1 and 2 were recorded using Waters Synapt G2 coupled to a Waters UPLC., ESI probe, ESI Positive, Cone Voltage 15 V while Shimadzu UV-Vis-NIR spectrophotometer was used to process the UV-Visible spectra. The electron paramagnetic resonance was obtained using JEOL FA200 spectrometer. Bruker APEX II DUO CCD with MoKα radiation (λ = 0.71073) was used to obtain the single crystal X-ray data and structure refinement parameters of complexes 1 and 2 at 100 K are presented in Table 1.
Table 1 X-ray crystal data collection and structure refinement parameters for 1 and 2
| Compound |
1
|
2
|
| Empirical formula |
C34H20Cl4CuN2O2 |
C34H18Br2Cl4CuN2O2 |
| Formula weight |
693.86 |
851.66 |
| Temperature/K |
100(2) |
298(2) |
| Crystal system |
Orthorhombic |
Monoclinic |
| Space group |
Pbca
|
P21/c |
|
a/Å |
8.3287(6) |
18.9437(6) |
|
b/Å |
15.8649(11) |
9.0738(3) |
|
c/Å |
21.7887(16) |
22.7712(7) |
|
α/° |
90 |
90 |
|
β/° |
90 |
98.1150(10) |
|
γ/° |
90 |
90 |
| Volume/Å3 |
2879.0(4) |
3875.0(2) |
|
Z
|
4 |
4 |
|
ρ
calc g cm−3 |
1.601 |
1.460 |
|
μ/mm−1 |
1.167 |
2.931 |
|
F(000) |
1404 |
1676 |
| Crystal size/mm3 |
0.198 × 0.176 × 0.080 |
0.528 × 0.103 × 0.058 |
| 2Θ range for data collection/° |
1.869 to 27.517 |
1.807 to 27.491 |
| Index ranges |
−10 ≤ h ≤ 10, −20 ≤ k ≤ 19, −28 ≤ l ≤ 26 |
−24 ≤ h ≤ 21, −11 ≤ k ≤ 11, −29 ≤ l ≤ 27 |
| Reflections collected |
37728 |
50048 |
| Independent reflections |
3311 [Rint = 0.0501] |
8878 [Rint = 0.1034] |
| Data/restraints/parameters |
3311/0/196 |
8878/0/406 |
| Goodness-of-fit on F2 |
1.120 |
0.920 |
| Final R indexes [I >= 2σ (I)] |
R
1 = 0.0439, wR2 = 0.1071 |
R
1 = 0.0604, wR2 = 0.1515 |
| Final R indexes [all data] |
R
1 = 0.0512, wR2 = 0.1106 |
R
1 = 0.1515, wR2 = 0.1970 |
| Largest diff. peak/hole/e Å−3 |
0.495 and −0.726 |
0.778 and −0.895 |
2.3 Synthesis of ligands
The Schiff bases ligands L1 and L2 were synthesized using a conventional protocol we previously reported.32,33 Generally, appropriate mass of 2-hydroxyl-1-naphthaldehyde was dissolved in 20 ml of methanol in a round-bottom flask and stirred vigorously to obtain a homogeneous solution. To the resulting mixture, an equimolar mass of 2,6-dichloro-phenylamine (for L1) and (4-bromo-2,6-dichlorophenyl)amine (for L2) was added. Four drops of glacial acetic acid were added to catalyze the reaction and the mixture stirred for 4 h at room temperature. A yellow and an off-yellow precipitate was obtained as crude products for L1 and L2 respectively. They were rinsed with diethyl ether three times to remove unreacted anilines and the pure products stored in a desiccator for further use.
2.3.1 Synthesis of (E)-1-(((2,6-dichlorophenyl)imino)methyl)naphthalen-2-ol (L1).
The reaction of 2-hydroxyl-1-naphthaldehyde (0.40 g, 2.0 mmol) and 2,6-dichloro-phenylamine (0.38 g, 2.0 mmol) in 20 mL of ethanol furnished L1 as a yellow powder. Yield: 85%, m.p 112–113 °C. 1H-NMR (CDCl3, 600 MHz): 7.13 (t, 1H, JH,H = 12, Ar–H), 7.27 (d, 1H, JH,H = 12, Ar–H), 7.41 (t, 1H, JH,H = 12, Ar–H), 7.44 (d, 2H, JH,H = 12, Ar–H), 7.57 (t, 1H, JH,H = 12, Ar–H), 7.83 (d, 1H, JH,H = 6, Ar–H), 7.94 (d, 1H, JH,H = 6, Ar–H), 8.15 (d, 1H, JH,H = 6, Ar–H), 9.54 (s, 1H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 14.40 (s, 1H, Ar–OH). 13C-NMR (CDCl3, 600 MHz): 119.43, 120.00, 123.82, 126.24, 127.70, 127.85, 128.27, 128.81, 129.33, 133.08, 136.09, 144.07, 164.10 and 165.39. FT-IR ν (cm−1): 3693 (O–H), 2912 (C–H), 1623 (–CN), 1556 (CC), 1179 (C–O). UV-Vis (dichloromethane, λmax, nm): 325 nm and 377 nm. ESI-MS TOF m/z (%): [M]+ 316.02, [M + 2]+ 318.02, MS Calc. analysis: 315.02 (100.0%), 317.02 (63.90%). Anal. calc. for C17H11Cl2NO: C, 64.58, H, 3.51, N, 4.43. Found: C, 63.96, H, 3.64, N, 4.626.
N), 14.40 (s, 1H, Ar–OH). 13C-NMR (CDCl3, 600 MHz): 119.43, 120.00, 123.82, 126.24, 127.70, 127.85, 128.27, 128.81, 129.33, 133.08, 136.09, 144.07, 164.10 and 165.39. FT-IR ν (cm−1): 3693 (O–H), 2912 (C–H), 1623 (–CN), 1556 (CC), 1179 (C–O). UV-Vis (dichloromethane, λmax, nm): 325 nm and 377 nm. ESI-MS TOF m/z (%): [M]+ 316.02, [M + 2]+ 318.02, MS Calc. analysis: 315.02 (100.0%), 317.02 (63.90%). Anal. calc. for C17H11Cl2NO: C, 64.58, H, 3.51, N, 4.43. Found: C, 63.96, H, 3.64, N, 4.626.
2.3.2 Synthesis of (E)-1-(((2,6-dichlorophenyl)imino)methyl)naphthalen-2-ol.
The reaction of 2-hydroxyl-1-naphthaldehyde (0.40 g, 2.0 mmol) and (4-bromo-2,6-dichlorophenylamine)amine (0.56 g, 2.0 mmol) in 20 mL of methanol furnished L2 as a off-yellow powder. Yield: 88%, m.p 119–120 °C. 1H-NMR (CDCl3, 600 MHz): 7.27 (t, 1H, JH,H = 6, Ar–H), 7.42 (t, 1H, JH,H = 12, Ar–H), 7.57 (t, 1H, JH,H = 6, Ar–H), 7.59 (s, 2H, Ar–H), 7.83 (d, 1H, JH,H = 12, Ar–H), 7.95 (d, 1H, JH,H = 12, Ar–H), 8.10 (d, 1H, JH,H = 6, Ar–H), 9.51 (s, 1H, CN), 14.19 (s, 1H, Ar–OH). 13C-NMR (CDCl3, 600 MHz): 117.70, 119.36, 119.85, 123.94, 127.90, 128.39, 128.42, 129.37, 131.44, 132.99, 136.37, 143.48, 163.89 and 165.70. FT-IR ν (cm−1): 3709 (O–H), 3067 (C–H), 1624 (–CN), 1567 (CC), 1169 (C–O). UV-Vis (dichloromethane, λmax, nm): 327 nm and 378 nm. ESI-MS TOF m/z (%): [M]+ 395.93, [M + 2]+ 397.93, MS calc. analysis: 392.93 (100.0%), 394.93 (97.30%). Anal. calc. for C17H10BrCl2NO: C, 51.68, H, 2.55, N, 3.55. Found: C, 52.06, H, 3.09, N, 3.63.
2.4 Synthesis of copper complexes [Cu(L)2]
The respective Schiff base ligands, L1 and L2 (2 mmol) were dissolved in 10 mL of ethanol. Thereafter, 1 mmol of copper(II) nitrate trihydrate dissolve in 5 mL of ethanol in a vial was added dropwise and a sudden brown colour was observed. The resultant mixture was stirred for 24 h at room temperature. A rotary evaporator was used to remove solvent from the solution and the left-over solid was thoroughly washed with hexane and filtered to afford a brown fine crystalline solid which was dried and kept in the desiccator for further use.
2.4.1 Synthesis of [Cu(L1)2] (1).
The reaction of L1 (0.3 g, 1.0 mmol) and Cu(NO3)2·3H2O (0.09 g, 0.50 mmol) in ethanol furnished complex 1 as a brown powder. Yield: 73%, m.p: 195–196 °C. FT-IR ν (cm−1): 3065 (C–H), 1601 (–CN), 1583 (CC), 1092 (C–O), 557 (Cu–O) and 464 (Cu–N). UV-Vis (dichloromethane, λmax, nm): 322 nm and 393 nm. ESI-MS TOF m/z (%): [M]+ 693.96, MS calc. analysis: 690.96 (100.0%), 692.95 (63.90%). Anal. calc. for C34H20Cl4CuN2O2: C, 58.85, H, 2.91, N, 4.04. Found: C, 58.87, H, 3.32, N, 4.17.
2.4.2 Synthesis of [Cu(L2)2] (2).
The reaction of L2 (0.3 g, 1 mmol) and Cu(NO3)2·3H2O (0.11 g 0.5 mmol) in ethanol furnished complex 2 as a brown powder. Yield: 76%, m.p: 199–201 °C. FT-IR ν (cm−1): 3067 (C–H), 1599 (–CN), 1535 (CC), 1093 (C–O), 512 (Cu–O) and 467 (Cu–N). UV-Vis (dichloromethane, λmax, nm): 324 nm and 395 nm. ESI-MS TOF m/z (%): [M]+ 851.78, MS calc. analysis: 850.77 (100.0%), 848.78 (78.20%). Anal. calc. for C34H18Br2Cl4CuN2O2: C, 47.95, H, 2.13, N, 3.29. Found: C, 48.14, H, 2.69, N, 3.56.
2.5 DFT studies methodology
3D models of synthesized compounds were built using GaussView v6.034 graphical interface program. This program helps to create input files through model buildup, to set up modelled structures for Gaussian calculation, and to visualize structures before and after calculations. For the geometry optimization of the selected substrate, the M06-L density functional theory method with the 6-31G(d) basis set for the main group elements (C, H, N, O, Cl) and the def2-TZVp basis set for the Cu atom and the Br atom respectively were employed. All these were inclusive in the Gaussian 16 program package available on the Lengua cluster of the centre for high performance computing (CHPC), Cape Town, South Africa.
2.6
In vitro antidiabetes studies
2.6.1 α-Glucosidase enzyme inhibitory activity.
The inhibitory effect of the synthesized compounds on α-glucosidase enzyme was studied following the procedure reported by Olofisan et al.35 with a slight modification. The activity of the compounds was evaluated using p-nitrophenol glucopyranoside (pNPG) as the substrate. In summary, 0.4 mL of yeast α-glucosidase (1 U mL−1) was added to 0.5 mL solution of the tested compounds or acarbose, (0.25–2 mM) along with 500 μL of one hundred millimolar buffer solution (Mono/Dibasic sodium phosphate salt, pH 6.8), and the mixture was incubated at 37 °C for 15 min. After adding 0.2 ml of pNPG (0.005 M) to the resulting solution, the amount of yellow p-nitrophenol emitted from the substrate was then measured at 405 nm.
2.6.2 α-amylase enzyme inhibitory activity.
Exploring a modified method reported by Ibitoye et al.,36 the inhibitory effect of L1–L2 and 1–2 on α-amylase enzyme activity was investigated. In a 20000 μM buffer solution (Mono/Dibasic sodium phosphate salt, pH 6.8) with 5 U mL−1 pancreatic α-amylase, increasing doses (0.25–2 mM) of the tested compounds or acarbose were incubated for 10 min at 37 °C. The solution was equilibrated further for 20 min at 37 °C after adding 200 μL of 10 mg mL−1 starch solution. A 0.6 mL of DNSA before boiling at 100 °C in a water bath for 10 minutes. The optical density of the coloured mixture formed was read relatively to a control solution devoid of the inhibitors at 540 nm.
2.7
In vitro antioxidant activity experiment
2.7.1 Nitric oxide (NO) scavenging capacity.
The Kurian et al. method37 was employed to investigate nitric oxide radical scavenging potential of the tested compounds. A 0.50 mL aliquot of various concentrations (0.25–2 mM) of tested compounds or Vanillin was equilibrated at 25 °C for 2 h with 10 millimolar sodium nitroprusside dispersed in pH 7.4 PBS. Griess reagent (0.20 mL) was added to the solution, before further equilibration at 25 °C for 5 min. Then, using a blank solution devoid of the test samples, the NO scavenging abilities of L1–L2, 1–2 and the reference drug (Vanillin) were recorded at 546 nm.
2.7.2 2,2′-diphenyl-1-picrylhydrazyl (DPPH) mopping capacity.
This assay was done by determining the capacity of the compounds to release electrons to purple coloured DPPH free radical using Turkoglu et al.38 with slight modification. 400 μL aliquot of 0.25–2 mM concentrations of the tested compounds or Vanillin and DPPH solution (300 μM) in DMSO were maintained away from light for 30 min at ambient laboratory temperature. Then, absorbance was read at 516 nm against a control solution devoid of the samples or Vanillin.
2.7.3 Ferric (Fe3+) reducing power capacity.
This ferric reducing capacity of L1–L2 and 1–2 was evaluated using potassium ferricyanide K3Fe(CN)6 in accordance with Tan and Chan's method.39 300 μL of two hundred millimolar phosphate buffer was incubated with 500 μL solution of each tested compounds or Vanillin (0.25–2 mM) and 500 μL of 1% K3Fe(CN)6 solution at 50 °C for 25 min. Afterwards, 0.4 millimolar of 0.1 g mL−1 TCA solution was added before dilution with 0.3 mL distilled water. The optical density of the resultant mixture was read at 700 nm after adding 0.2 mL FeCl3 (0.1%). From a standard curve made with vanillin (y = 0.0243x − 0.0586; R2 = 0.9852), the compounds’ capacity to reduce ferric ion was approximated.
2.8
In vitro biological studies
2.8.1
In vitro antibacterial activity of the compounds.
The antibacterial activities of L1–L2, 1–2 and reference drugs (gentamicin and ciprofloxacin) were evaluated against Pseudomonas aeruginosa ATCC 27853, Escherichia coli ATCC 35218, Bacillus subtilis SL34 and Staphylococcus aureus ATCC 6571 based on the zone of inhibition by disc diffusion method. A loop full of each test strain was inoculated separately in 50 ml of nutrient broth and incubated for 24 h in a shaking incubator at 30 °C and 120 rpm. Overnight grown culture (50 μL) of each bacterium was spread on solidified nutrient agar (about 20 mL of molten nutrient agar media was added into a 120 mm diameter Petri dish). 3.125 mg mL−1, 6.250 mg mL−1, and 12.50 mg mL−1 of each compound were tested. Under the same conditions, the reference drugs were also tested at highest concentration (12.50 mg mL−1) to compare their activity with tested compounds. Non-antibiotic-impregnated discs dipped in different ppm of the compounds were placed on the solidified nutrient agar, and the plates were incubated at 37 °C for 24 h. The antibacterial activity potential of the compounds was determined by measuring the diameter (in mm) of the inhibition zone around the compounds-impregnate disc.40 DMSO was used as a negative control, and it displayed no antibacterial activity against any of the bacteria strains used for this study at different concentrations.
3. Results and discussion
3.1 Synthesis of the Schiff base ligands and their Cu(II) metal complexes
The synthetic pathway for ligands L1 and L2 as well as the one for Cu(II) complexes 1 and 2 are shown in Scheme 1. Synthesis of the Schiff base ligands was achieved by reacting equimolar amount of 2-hydroxyl-1-naphthaldehyde with appropriate primary amines, mainly 2,6-dichloro-phenylamine and (4-bromo-2,6-dichlorophenyl)amine under suitable reaction conditions. The metal complexes 1 and 2 were prepared by the reaction of Cu(NO3)2·3H2O with ligands L1 and L2 in the ratio 1:2 respectively. All the synthesized compounds are air stable. The synthesized compounds displayed good solubility in solvents such as chloroform, dichloromethane, dimethyl sulfoxide and toluene.
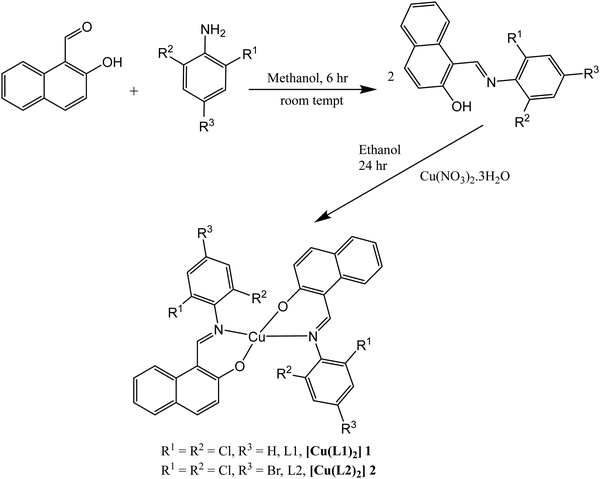 |
| | Scheme 1 Synthesis of Schiff bases L1 and L2 and their Cu(II) complexes 1 and 2. | |
3.2 Spectroscopy studies
3.2.1
1H and 13C NMR.
The proton NMR data for L1 and L2 were recorded in d-chloroform and 2D NMR was used to assign all the peaks. The azomethine or imine proton for L1 and L2 appeared at 9.54 ppm and 9.51 ppm respectively and these values are consistent with related compounds previously reported.32,41,42 The peak for the hydroxyl proton (–OH) attached to the naphthalene ring appeared downfield at 14.40 ppm and 14.19 ppm for L1 and L2 respectively (see Fig. S5 and S7, ESI†). Generally, the aromatic protons appeared as doublets and triplets within the range of 7.13–8.15 ppm, though a singlet was observed around 7.59 ppm due to the presence of a bromine atom on para position of the benzene ring of L2. No peak was observed in the upfield region (within 0–6 ppm) and this affirms that both structures have no aliphatic protons. The 13C-NMR spectra of L1 and L2 displayed peaks for the imine carbon (–CN) at 164.10 ppm and 163.89 ppm respectively (see Fig. S6 and S8, ESI†). Also, the peak for the carbon atom of the naphthalene ring directly bonded to the –OH group appeared at the far downfield region, 165.39 ppm and 165.70 ppm for L1 and L2 respectively. The aromatic carbon peaks for benzene and naphthalene rings appeared between 117.70 –144.07 ppm.
3.2.2 FT-IR and mass spectra.
We further characterized the synthesized compounds using FT-IR spectroscopy to identify their characteristic functional group. The IR spectra were recorded in the range of 4000–400 cm−1. The ν(O–H) vibration frequency of L1 and L2 appeared as a less intense band at 3693 cm−1 and 3709 cm−1 respectively. This weak band arises due to the participation of the –OH functional group in intramolecular hydrogen bonding with the nitrogen atom of the azomethine group (–CN) and in agreement with the ones previously reported.43,44 Upon complexation, this ν(O–H) vibration frequency disappeared in the spectra of complexes 1 and 2 and this affirms the deprotonation of the hydrogen atom of the –OH group on the naphthalene ring and the coordination of the naphtholate oxygen to the Cu(II) ion in the complexes. The vibrational frequency of the imine group (–CN) appeared as a strong peak at 1623 cm−1 for L1 and 1624 cm−1 for L2 (see Fig. S1 and S2, ESI†). However, it appeared at a lower wave number in the spectra of the complexes, 1601 cm−1 for 1 and 1599 cm−1 for 2 (see Fig. S3 and S4, ESI†). This shift could be attributed to the reduction in the electron density on nitrogen atom of the imine group due to the donation of its lone pair of electrons to the Cu(II) metal center.19 Similarly, the vibrational frequency of ν(C–O) in 1 and 2 is lower when compared to the one for L1 and L2 and this confirms the participation of oxygen in the complexation. The vibrational frequency for ν(Cu–O) and ν(Cu–N) appeared around 557–512 cm−1 and 464–467 cm−1 respectively.
The molecular ion peak [M]+ could be seen in the mass spectra of all the synthesized compounds. It appeared as 316.02, 395.93, 693.96 and 851.78 for L1, L2, 1 and 2 respectively (see Fig. S11–S14, ESI†). The mass spectra of L1 and L2 also displayed [M]+, [M + 2]+, and [M + 4]+ peaks with intensity of 9:6:1. These peaks could be attributed to the presence of two chlorine atoms in both compounds.45
3.2.3 Electronic absorption spectra.
The dichloromethane solution of the compounds was used to record their respective electronic absorption spectra (Fig. 1). The spectra of L1–L2 displayed two major absorption bands in the UV region. The band around 325–327 nm is assigned to π → π* transition due to the conjugated CC chromophore while the band around 376–378 nm is assigned to n → π* transition due to CN and the O–H on the naphthalene ring.18 These two bands were also observed in the spectra of 1 and 2. However, the first band was blue shifted to 322 nm – 324 nm and can be ascribed to π → π* intraligand charge transfer (ILCT) transition while the second band was red shifted to 392–395 nm, which can be assigned to ligand-to-metal charge transfer (LMCT).46 In addition to these two major bands, a weak and broad d–d transition band was observed around 520–530 nm in the electronic spectra of 1 and 2, at high concentration and this can be assigned to 2B1g → 2E1g.47
 |
| | Fig. 1 Electronic absorption spectra for ligands (L1 and L2) and metal complexes (1 and 2). | |
3.2.4 Electron paramagnetic resonance (EPR) spectra for complexes 1 and 2.
To further confirm the paramagnetic nature of complexes 1 and 2, X-band EPR spectra were recorded at 298 K as shown in Fig. 2. The EPR spectra of complexes 1 and 2 are perfectly isotropic with a single line and g-value of approximately 2.05 which is equivalent to the g-factor of free electron (g = 2.0023). This depicts that there is a wholly symmetric environment where the electrons in separate d-orbitals interact in all directions.26 The g values obtained in this study are similar to those reported in literature.48 The absence of half field signal in the spectra rule out any pronounced Cu⋯Cu interactions and deduced that both complexes devoid ms = ±2 transition.26
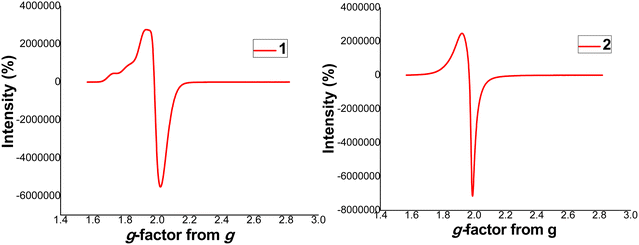 |
| | Fig. 2 EPR spectra for complexes 1 and 2. | |
3.3 Single X-ray crystal analysis
Single crystals of 1 and 2 used for this analysis were obtained by slow evaporation of toluene solution of both compounds at room temperature. The crystal structures of 1 and 2 are given in Fig. 3 while the selected bond distances and bond angles are given in Table 2. The asymmetric unit of 1 entails half of the molecule of which the whole molecule is generated by an inverse symmetry operation, while for 2, it was the whole molecule. Complexes 1 and 2 are mononuclear as well as neutral. The ligands bonded to the Cu2+ center via the azomethine nitrogen and napthanolate oxygen in a κ2N:O coordination mode to adopt a slightly distorted square planar geometry i.e. the O–Cu–N bond angles are 90.22° and 89.78° for 1 which deviates from ideal 90° for square planar geometry. The bond lengths and bond angles of 1 and 2 are comparable to similar Schiff base Cu(II) complexes previously reported.18,19Fig. 3 revealed that, N2O2 (coordinating atoms) from the two Schiff base ligands in both complexes are arrayed in a trans-configuration. The plane of the chelate rings with Cu(II) at the center are parallel to each other. In the packing diagram of the complexes, 1 has 16 asymmetric structures in a unit cell while 2 has 4 asymmetric structures in a unit cell.
 |
| | Fig. 3 Crystal structures of 1 and 2 with thermal ellipsoids drawn at 50% and 30% probability for 1 and 2 respectively. Symmetry code for equivalent atoms generated in 1 is 2 − x, 1 − y, 1 − z. | |
Table 2 Selected bond length (Å) and angles (°) for complex 1 and 2
| Parameters |
1
|
2
|
| Symmetry code for equivalent atoms generated in 1 (i) = 2 − x, 1 − y, 1 − z. |
| Bond lengths |
| Cu–O |
1.969(1) |
2.2023(5) |
| Cu–N |
1.906(1) |
2.2045(3) |
| Bond angles |
| O–Cu–O |
180.00(1)i |
172.2(2) |
| O–Cu–N |
90.22(11)i |
91.2(2) |
| 89.78i |
90.6(2) |
|
|
89.4(2) |
| N–Cu–N |
180.00(1)i |
172.2(2) |
| Torsion angles |
| O1–Cu1–N1–C11 |
21.0(2) |
13.0(5) |
3.4 Density functional theory (DFT) studies
Quantum chemical descriptor.
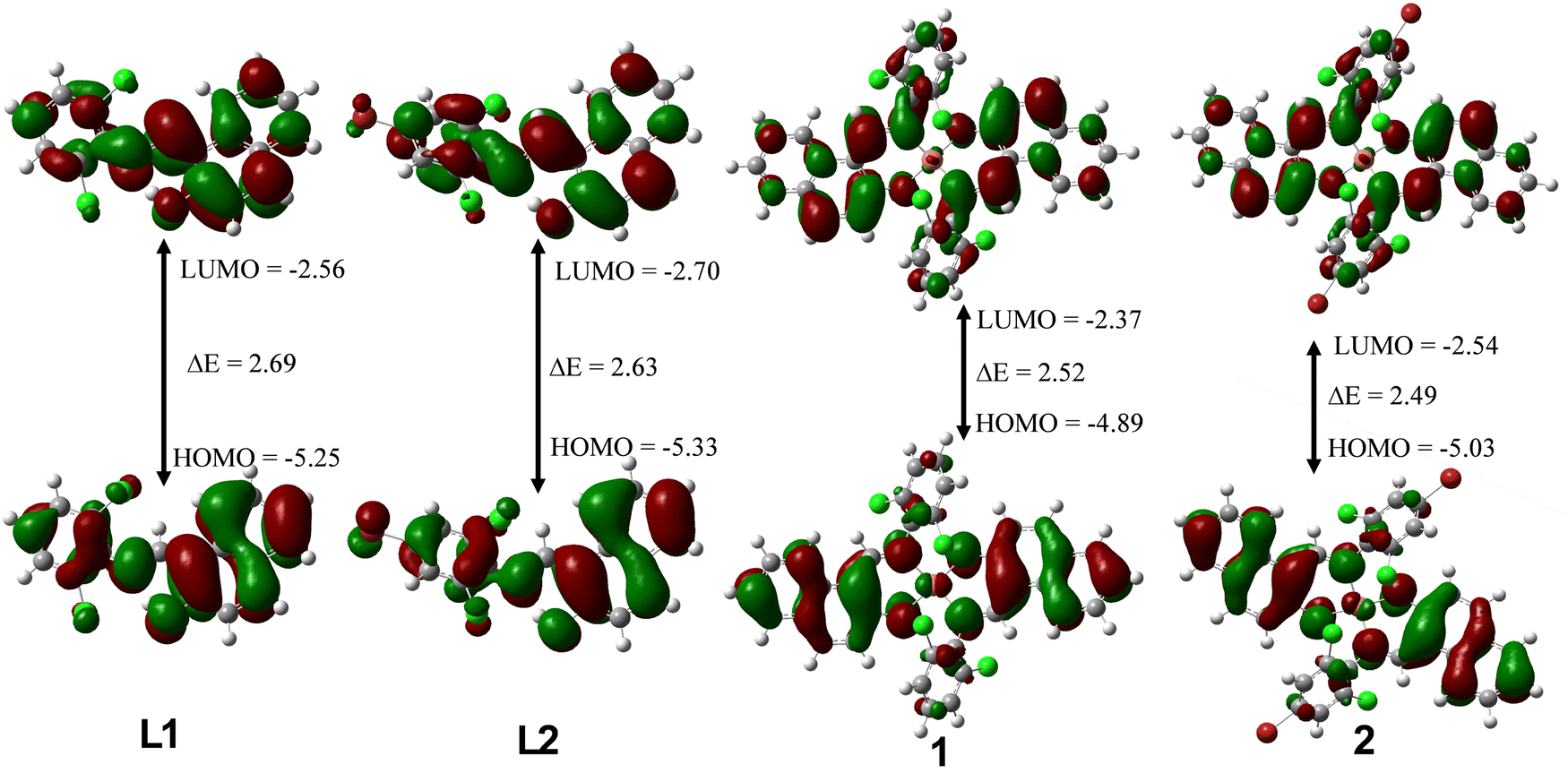
We summarized the results of the quantum chemical calculations in Table 3 and give the isodensity plots for molecular orbitals in Fig. 4. The optimized structures for all the compounds can be found in the ESI† (Fig. S15). Quantum chemical descriptors have been explored to understand the selectivity, stability, reactivity, and various physical properties of compounds.49 The energies of the highest occupied molecular orbital (EHOMO) as well as lowest unoccupied molecular orbital (ELUMO) are the basic parameters explored to calculate other parameters using Koopman's theorem.50,51EHOMO value gives insight into the tendency of a molecule to donate its most loosely bound electron. Thus, as EHOMO increases, the electron-donating capacity of compounds increases. However, higher ELUMO depicts better electron-accepting capacity. Complex 1 with EHOMO value of −4.89 eV could probably be the one with highest electron-donating ability while L2 with EHOMO value of −5.33 eV might be the least. The energy band gap (ΔE) is a vital metric for quantifying chemical stability and reactivity of molecules. Literature has it that, lower ΔE signifies higher chemical reactivity and stability Generally, complexes 1 and 2 have a lower energy band gap when compared to free ligand L1 and L2. Complex 2 has the lowest energy band gap (ΔE = 2.49 eV) and therefore the most reactive and stable among the four compounds, followed by 1, L2 and L1. The global hardness measures how electron cloud of molecules can resist polarization or deformation in response to slight chemical perturbation. It is directly proportional to energy band gap, thus the higher the ΔE the higher the global hardness. However, global softness is the inverse of ΔE, the lower the ΔE, the higher the global softness. For example, complex 2 with lowest ΔE of 2.49 eV has the highest global softness of 0.80 eV. The electrophilicity (ω) properties for L1, L2, 1 and 2 are 5.66 eV, 6.12 eV, 5.22 eV and 5.74 eV respectively. Thus, L2 displayed the strongest electrophilic prowess, while 1 exhibited the strongest nucleophilic ability. Electron affinity (E.A.) measures the electron attraction power of molecules, L2, with the EA value of 2.70 eV has the highest ability to attract electrons when compared to others.
Table 3 Quantum chemical descriptors of L1, L2, L3 and L4 at M06-L/def2-TZVP level of theory
| Parameters |
Description |
L1
|
L2
|
1
|
2
|
|
E
HOMO (eV) |
Highest occupied molecular orbital |
−5.25 |
−5.33 |
−4.89 |
−5.03 |
|
E
LUMO (eV) |
Lowest unoccupied molecular orbital |
−2.56 |
−2.70 |
−2.37 |
−2.54 |
| ΔE (eV) |
Energy band gap |
2.69 |
2.63 |
2.52 |
2.49 |
|
A (eV) |
Electron affinity |
2.56 |
2.70 |
2.37 |
2.54 |
|
I (eV) |
Ionization potential |
5.25 |
5.33 |
4.89 |
5.03 |
|
η (eV) |
Hardness |
1.34 |
1.32 |
1.26 |
1.25 |
|
S (eV−1) |
Softness |
0.74 |
0.76 |
0.79 |
0.80 |
|
μ
|
Chemical potential |
−3.90 |
−4.01 |
−3.63 |
−3.78 |
|
χ (eV) |
Electronegativity |
3.90 |
4.01 |
3.63 |
3.78 |
|
ω (eV) |
Electrophilicity index |
5.66 |
6.12 |
5.22 |
5.74 |
| ΔNmax |
Maximum no of electrons |
2.90 |
3.05 |
2.88 |
3.03 |
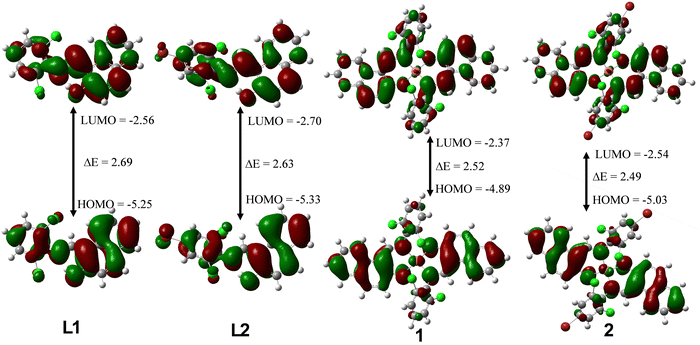 |
| | Fig. 4 LUMO and HOMO plots for L1, L2, 1 and 2 at the M06-L/def2-TZVP level of theory. | |
3.5 Antidiabetic studies
α-Glucosidase assay.
α-Glucosidase assay has been extensively explored in the development of therapeutics for diabetes treatment. Antidiabetic drugs could function by inhibiting α-glucosidase and α-amylase, these are hydrolyzing enzymes which are responsible for the breaking down of carbohydrate into glucose during digestion.52,53 This act drastically reduces the intake of glucose in the body as well as blunting the postprandial plasma glucose rise.8 The α-glucosidase prowess for the ligands (L1 and L2) and metal complexes (1 and 2) are evaluated using IC50 (the lower the IC50 the better the antidiabetic potential) as presented in Table 4. Our findings showed that, the antidiabetics potential of L1 and L2 was enhanced upon chelation with Cu2+i.e., the IC50 of L1 is 0.682 mM while the one for 1 is 0.073 mM. None of the synthesized compounds outshined the α-glucosidase inhibition ability of acarbose (the reference drug) but it is important to note that, the IC50 of 1 and 2 are almost equivalent to the one for acarbose i.e., IC50 of 1, 2 and acarbose are 0.073 mM, 0.064 mM and 0.054 mM respectively. Fig. 5 depicts that, the % α-glucosidase inhibition increases with concentration. Based on the calculated IC50 value, the order of their increasing α-glucosidase inhibition ability is L2 < L1 < 1 < 2 < acarbose.
Table 4 Antidiabetic potential of Schiff bases and their Cu(II) metal complexes
| Compounds |
α-Glucosidase IC50 (mM) |
α-Amylase IC50 (mM) |
|
L1
|
0.682 |
170.559 |
|
L2
|
5.359 |
330.491 |
|
1
|
0.073 |
148.126 |
|
2
|
0.064 |
107.786 |
|
Acarbose
|
0.054 |
171.559 |
 |
| | Fig. 5 % α-glucosidase inhibition vs. concentration. The mean activity ± standard deviation of each compound represented by bars at each concentration with different alphabets (a)–(d) are significantly different at p < 0.05. | |
α-Amylase assay.
Antidiabetic potential of all the compounds were further investigated by α-amylase assay. All the compounds displayed good to excellent α-amylase inhibition activity. Complexes 1 and 2 exhibited the highest suppressive effect of α-amylase, as indicated by their IC50 values of 148.126 mM and 107.786 mM, which is significantly lower than that of acarbose (IC50 = 171.559 mM) and the free ligands, Table 4. As displayed in Fig. 6, it is observed that as concentration increases, the % α-amylase inhibitory activity increases. The order of their increasing α-amylase inhibitory activity as evaluated from calculated IC50 is L2 < L1 < acarbose< 1 < 2.
 |
| | Fig. 6 % α-amylase inhibition vs. concentration. The mean activity ± standard deviation of each compound represented by bars at each concentration with different alphabets (a)–(c) are significantly different at p < 0.05. | |
3.6 Antioxidant studies
DPPH assay.
DPPH assay is one of the most used assays to determine the radical scavenging ability of natural and synthetic compounds due to its simplicity and require shorter time when compared to other methods.54 It is mainly based on the ability of tested compounds to donate hydrogen or electron radicals to stabilize the unpaired electron in the structure of DPPH.33 Compounds with the ability to scavenge free radicals have the potential to slow down or prevent oxidative damage in the body. Thus, their therapeutic role in preventing diseases such as cancer, asthma, diabetes, Parkinson and Alzheimer diseases, majorly caused by oxidative damage.55 Herein, we investigated the free radical scavenging ability of halogenated Schiff bases and their Cu2+ metal complexes at different concentrations.
The IC50 values (Table 5) of the synthesized compounds were calculated from the % free radicals scavenging values and the lower the IC50 values the higher the antioxidant activities. As seen from Table 5 and Fig. 7 the metal complexes 1 and 2 displayed better free radical scavenging ability than their respective ligands L1 and L2i.e., the IC50 for 1 is 0.187 while the one for L1 is 0.871. This could be ascribed to the presence of Cu2+ in 1 and 2 enhancing their ligand proton donating tendency. We found out that, 1 has the least IC50 value (IC50 = 0.187 mM) and the highest value of vanillin equivalent antioxidant capacity (VEAC) followed by 2, L1 and L2 respectively. This could be ascribed to the fact that complex 1 with highest EHOMO would probably have the highest electron-donating ability to stabilize the free radical in DPPH structure. The replacement of the hydrogen atom in 1 and L1 (at the para position of the phenyl group in both compounds) with bromine atom in 2 and L2 seems not to enhance free radical scavenging capability. It was also observed that, as the concentration of the tested compounds increases from 0.25 mM to 2 mM, the % free radical scavenging ability also increases.
Table 5 Antioxidant potential of Schiff bases and their Cu(II) metal complexes
| Compounds |
DPPH IC50 (mM) |
FRAP IC50 (mM) |
NO IC50 (mM) |
|
L1
|
0.871 |
>500 |
>500 |
|
L2
|
1.132 |
>500 |
>500 |
|
1
|
0.187 |
>500 |
100.044 |
|
2
|
0.445 |
>500 |
247.463 |
|
Vanillin
|
0.188 |
>500 |
466.627 |
 |
| | Fig. 7 % free radical scavenging ability vs. concentration. The mean activity ± standard deviation of each compound represented by bars at each concentration with different alphabets (a)–(d) are significantly different at p < 0.05. | |
Nitric oxide assay
Nitric oxide radical has one unpaired electron and its one of the reactive nitrogen species (RON). In the body, it has a beneficial role in physiological processes such as defense mechanism, immune regulation, neurotransmission and blood pressure regulation while in low concentrations. However, in high concentrations, it may lead to nitrosylation reaction which distorts protein structures resulting in great adverse effects on body systems.56 NO˙ is a reactive nitrogen species that contains one unpaired electron on the antibonding site of 2π*y orbital.57 The excess production of nitric oxide radical as well as other reactive nitrogen species is called nitrosative stress.58 This may lead to nitrosylation reactions, which alter protein structures and so have an adverse effect on body systems. Hence, the need of novel drugs which can mop nitric oxide radical in the body. Interestingly, 1 and 2 showed promising nitric oxide scavenging ability compared to L1 and L2 and even outshined the vanillin (reference drug) i.e., the IC50 value for vanillin is 466.627 mM while it was 100.044 mM and 247.463 mM for 1 and 2 respectively (Table 5). This revealed that, chelation of the free ligands with Cu2+ enhance the nitric oxide radical scavenging ability. Generally, it was observed that, as the concentration increases, the ability of the reported compounds to scavenge nitric oxide radical also increases (Fig. 8).
 |
| | Fig. 8 % nitric oxide scavenging ability vs. concentration. The mean activity ± standard deviation of each compound represented by bars at each concentration with different alphabets (a)–(e) are significantly different at p < 0.05. | |
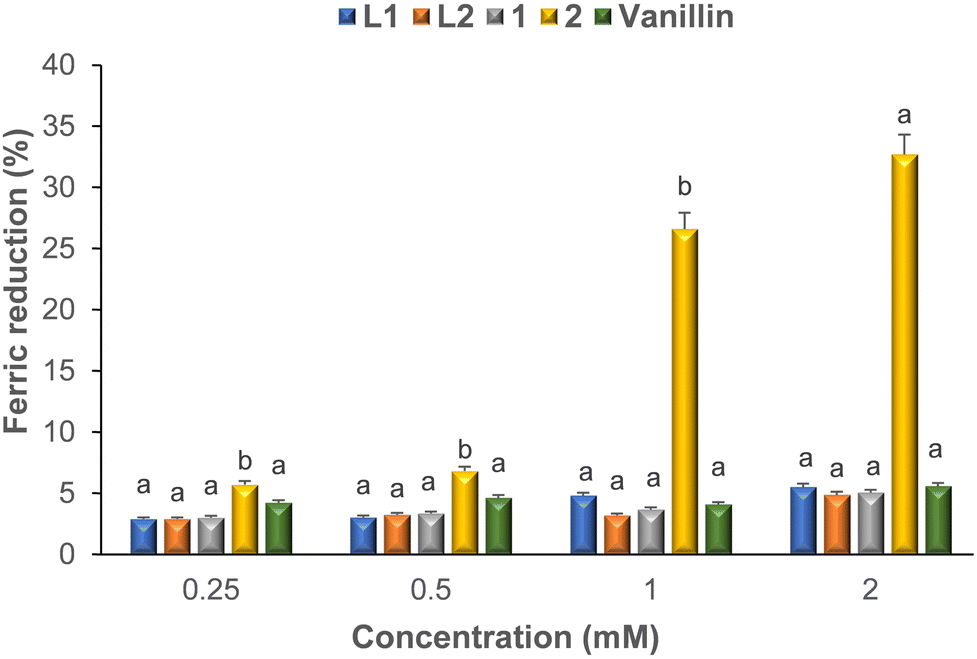
FRAP assay.
Ferric reducing antioxidant power (FRAP) assay has been proving to be one of the reliable methods to evaluate the antioxidant potential of compounds. Basically, FRAP assay quantifies the ability of compounds to reduce Fe3+ to Fe2+, thus revealing its electron donating prowess and free radical scavenging capabilities.59 As shown in Fig. 9, both the free ligands and their complexes displayed poor ferric reducing power activity when compared to the reference drug, vanillin. It could also be observed that, concentration did not have pronounced effect on the FRAP ability of the tested compounds while it does for the standard drug.
 |
| | Fig. 9 % ferric reducing ability power vs. concentration. The mean activity ± standard deviation of each compound represented by bars at each concentration with different alphabets (a), (b) are significantly different at p < 0.05. | |
3.7 Antibacterial studies
The free ligands and their complexes were screened against two Gram-positive bacteria, i.e., S. aureus and B. subtilis as well as three Gram-negative bacteria, i.e., E. coli, P. aeruginosa and K. pneumoniae. The zone of inhibition (ZI) was used to evaluate the antibacterial potency and results were compared against reference drugs i.e., the higher the zone of inhibition values the higher the antibacterial potential. At highest concentrations used for this study, all the compounds showed moderate antibacterial potential. However, they did not outshine the reference drug (gentamicin and ciprofloxacin) except complexes 1 and 2 which displayed better antibacterial activity than the standard against P. aeruginosa. Generally, complexes 1 and 2 showed better antibacterial activity than L1 and L2. For example, against E. coli at lowest concentration used for this study, the ZI for L1 and L2 are 6 mm and 0 mm while it was 10 mm and 5 mm for complexes 1 and 2 and this was observed at all concentrations (Table 6). This might be attributed to fact that; lipophilic character of metal complexes increases upon chelation of ligands.15 Hence, the ease in permeability of the complexes through lipid layers of cell membranes when compared to the ligands.22 It was observed that, none of the compounds displayed antibacterial activities against K. pneumonia strain at all concentrations. This could be that the complexes were being damaged or modified as they enter the cell wall of K. pneumoniae thus resulting in their inactivity.14 Except in a few cases, L1 and 1 exhibited better antibacterial activity than their counterparts L2 and 2i.e., against E. coli (at 3.125 mg ml−1) the ZI values for L1 and 1 are 6 mm and 10 mm while it was 0 mm and 5 mm for L2 and 2 respectively. We could therefore conclude that the replacement of a hydrogen atom (in L1 and 1) with bromine atom (in L2 and 2) did not enhance antibacterial activity. As seen in Fig. 10, the antibacterial potential increases as the concentration increases.
Table 6 Antibacterial activity of the ligands (L1 & L2) and their metal complexes (1 & 2)
| Zone of inhibition (mm) |
| Concentrations |
Compounds |
E. coli
|
S. aureus
|
B. substilis
|
P. aeruginosa
|
K. pneumoniae
|
| 3.125 mg ml−1 |
L1 |
6 |
0 |
0 |
0 |
0 |
| L2 |
0 |
0 |
0 |
0 |
0 |
| 1 |
10 |
10 |
12 |
0 |
0 |
| 2 |
5 |
0 |
0 |
7 |
0 |
|
|
| 6.250 mg ml−1 |
L1 |
9 |
0 |
8 |
9 |
0 |
| L2 |
8 |
6 |
7 |
0 |
0 |
| 1 |
12 |
11 |
16 |
10 |
0 |
| 2 |
9 |
9 |
0 |
11 |
0 |
|
|
| 12.50 mg ml−1 |
L1
|
11 |
0 |
12.5 |
12 |
0 |
|
L2
|
12 |
10 |
12 |
9.5 |
0 |
|
1
|
14 |
15 |
19.5 |
14 |
0 |
|
2
|
13 |
11 |
10 |
13 |
0 |
|
GEN
|
21 |
20 |
16 |
12 |
22 |
|
CIP
|
35 |
25 |
24 |
28 |
29 |
 |
| | Fig. 10 Zone of inhibition vs. concentration of Schiff bases ligands and their Cu(II) complexes. | |
3.8 Physicochemical and pharmacokinetics parameters of L1, L2, 1 and 2
The synthesized compounds exhibited promising medicinal properties, thus, the need to evaluate their drug-likeness properties. SwissADME, a common web-based tool was explored to approximate the physicochemical and pharmacokinetic parameters of the compounds (Table 7), which would later give insights into their drug-like nature.60 The results were compared with the acceptable threshold of Lipinski's Ro5 to predict the bioavailability of the synthesized compounds. Promising drug candidates are expected not to exceed acceptable threshold of Lipinski's Ro5 or better still have a minimal violation. Molecular weight (M.W.), aqueous solubility (LogS), lipophilicity (LogP), hydrogen bond acceptor (H.B.A.) and donor (HBD) ability, topological polar surface area (TPSA), rotatable bonds (RotB) as well as skin permeation (LogKp) were predicted for their physicochemical parameters. We also estimated the pharmacokinetic parameters such as P-glycoprotein (P-gp) substrate, blood–brain barrier (B.B.B.) permeant and gastrointestinal (G.I.) absorption.
Table 7 Predicted physicochemical and pharmacokinetic properties of compounds L1, L2, 1 and 2
|
|
L1
|
L2
|
1
|
2
|
Acceptable threshold (Ro5) |
| Physicochemical properties |
| Molecular weight (Da) |
316.18 |
395.08 |
693.89 |
851.68 |
<500 |
| LogP |
5.02 |
5.63 |
6.91 |
7.97 |
<5 |
| LogS (mol L−1) |
−6.14 |
−7.05 |
−11.47 |
−13.29 |
0 → −6 |
| TPSA (A2) |
32.59 |
32.59 |
43.18 |
43.18 |
≤140 |
| HBA |
2 |
2 |
2 |
2 |
≤10 |
| HBD |
1 |
1 |
0 |
0 |
≤5 |
| Rotatable bonds |
2 |
2 |
2 |
2 |
<10 |
|
|
| Pharmacokinetics properties |
| GI absorption |
High |
High |
Low |
Low |
|
| BBB permeant |
Yes |
Yes |
No |
No |
|
| P-gp substrate |
No |
No |
Yes |
Yes |
|
The Mw of L1 and L2 does not violate Lipinski's Ro5, which ascribes 500 g mol−1 as the M.W. of a promising drug molecule while the ones for 1 and 2 slightly exceeded the threshold.17 This means that L1 and L2 will find it easier to be transported to biomolecular targets when compared to 1 and 2. The LogS values for L1 and L2 deviate minimally from the acceptable standard of 0 → −6 mol L−1 while 1 and 2 violate Ro5. The LogP values for all the compounds have minimal violation for Lipinski's Ro5. L1 with the LogP value of 5.02 almost fell within the acceptable range of Lipinski's Ro5 and this indicates that it will be more lipophilic than other compounds. For all the compounds, the estimated values for TPSA are within the accepted values of (≤140) and this implies their tendency to be transported through a lipid bilayer that is densely packed, i.e., the gastrointestinal tract.61L1 and L2 with smaller TPSA values will tend to penetrate through the cells easily relative to 1 and 2. The TPSA values showed that all the compounds are bioavailable since they fell within the acceptable threshold of Lipinski's Ro5. HBDs, HBAs as well as RotBs are also vital parameters to measure the suitability of compounds as promising drug candidates. In Table 7, the estimated values for H.B.D.s, H.B.A.s, and RotBs fell within the acceptable threshold of Lipinski's Ro5. A potential drug candidate should be easily absorbed at the intestine and while developing an orally available drug, this stage is very critical. L1–L2 and 1–2 displayed high and low G.I. absorption respectively. Only L1–L2 have the tendency to penetrate through the brain–blood barrier (B.B.B.). The bioavailability of a wide range of drugs are drastically reduced in the intestine by P-glycoprotein. Generally, a good drug candidate should not bind to P-glycoprotein (not P-gp substrate). L1 and L2 are not P-gp substrates while 1 and 2 are P-gp substrates. Interestingly, excipients such as vitamin E-TPGS can be added to drug formulations to disrupt intestinal P-gp.62 Thus, the bioavailability of 1 and 2 can be improved by adopting this method.
4. Conclusion
Two halogenated Schiff bases (L1 and L2) and their Cu(II) complexes (1 and 2) were successfully synthesized and comprehensively characterized by spectroscopic techniques and structural analysis. Single X-ray crystal elucidation revealed that Cu(II) metal centre in complexes 1 and 2 are directly bonded to a pair of oxygen and nitrogen atoms to adopt a distorted square planar geometry. Aside L2, all the compounds exhibited excellent α-amylase enzyme inhibitory activity and even outshined acarbose. Complexes 1 and 2 displayed good α-glucosidase enzyme inhibitory activity while L1 and L2 exhibited moderate activity. However, none of the compounds showed better inhibited α-glucosidase than acarbose. All the compounds displayed moderate to good antioxidant potential, with 1 showing better DPPH and nitric oxide radical scavenging activity than other compounds and vanillin (reference drug). The ZI values showed that all the compounds showed moderate antibacterial activities against all the bacterial strains except K. pneumoniae where no activity was observed. It can be concluded that, the antibacterial potential of the reported compounds is dosage dependent.
Author contributions
Conceptualization, S. D. O. and RCL; methodology; S. D. O., and RCL.; software, S. D. O., and RCL validation, S. D. O., and R. C. L.; formal analysis; S. D. O., and RCL; resources’ RCL.; data curation, S. D. O., and RCL.; writing original draft preparation; S. D. O.; writing review and editing, S. D. O, and RCL., visualization, S. D. O and R. C. L., supervision, R. C. L., project administration, R. C. L., funding acquisition, S. D. O and R. C. L.
Data availability
CCDC 2338587 and CCDC 2338588 contain supplementary crystallographic data for compounds 1 and 2.†
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The authors acknowledged the funding from the National Research Foundation (NRF), South Africa (grant number: 129284) as well as Stellenbosch University. The first author is a recipient of the postdoctoral fellowship award from the NRF and based at Stellenbosch University. The authors are also grateful to the South African Centre for High-Performance Computing, CHPC [https://www.chpc.ac.za] for resources. We wish to also thank Mr Marvin Zitha for his assistance in obtaining the EPR spectra.
References
- A. T. Kharroubi and H. M. Darwish, World J. Diabetes, 2015, 6, 850 CrossRef PubMed
 .
.
- M. Naser, M. M. Nasr and L. H. Shehata, Int. J. Prog. Sci. Tech., 2021, 24, 586–596 Search PubMed .
- T. Sivakumar and B. Deepa, J. Univ. Shanghai Sci. Technol., 2023, 25, 303–314 Search PubMed .
- M. Karamanou, A. Protogerou, G. Tsoucalas, G. Androutsos and E. Poulakou-Rebelakou, World J. Diabetes, 2016, 7, 1 CrossRef .
- S. D. Oladipo, R. C. Luckay, K. A. Olofinsan, V. A. Obakachi, S. J. Zamisa, A. A. Adeleke, A. A. Badeji, S. A. Ogundare and B. P. George, Heliyon, 2024, 10, e23174 CrossRef CAS PubMed .
- E. J. Prpa, B. H. Bajka, P. R. Ellis, P. J. Butterworth, C. P. Corpe and W. L. Hall, Crit. Rev. Food Sci. Nutr., 2021, 61, 3783–3803 CrossRef CAS PubMed .
- C. Rosak and G. Mertes, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2012, 12, 357–367 CrossRef PubMed .
- H. O. Omoregie, T. L. Yusuf, S. D. Oladipo, K. A. Olofinsan, M. B. Kassim and S. Yousuf, Polyhedron, 2022, 217, 115738 CrossRef CAS .
- M. C. Sabu and R. Kuttan, J. Ethnopharmacol., 2002, 81, 155–160 CrossRef CAS .
- M. K. Logani and R. E. Davies, Lipids, 1980, 15, 485–495 CrossRef CAS .
- Z. Breijyeh and R. Karaman, Antibiotics, 2023, 12, 628 CrossRef CAS .
- J. W. Bennett and K. T. Chung, Adv. Microbiol., 2021, 49, 163–196 Search PubMed .
- M. I. Hutchings, A. W. Truman and B. Wilkinson, Curr. Opin. Microbiol., 2019, 51, 72–80 CrossRef CAS .
- S. D. Oladipo, B. Omondi and C. Mocktar, Polyhedron, 2019, 170, 712–722 CrossRef CAS .
- S. D. Oladipo, B. Omondi and C. Mocktar, Appl. Organomet. Chem., 2020, 34, e5610 CrossRef CAS .
- G. R. Aguilar, L. R. Swetschinski, N. D. Weaver, K. S. Ikuta, T. Mestrovic, A. P. Gray, E. Chung, E. E. Wool, C. Han, A. G. Hayoon and D. T. Araki, Lancet Reg. Health, 2023, 25, 100561 Search PubMed .
- S. D. Oladipo, F. A. Olotu, M. Soliman, C. Mocktar and B. Omondi, J. Mol. Struct., 2020, 219, 128553 CrossRef .
- T. L. Yusuf, D. C. Akintayo, S. D. Oladipo, A. A. Adeleke, K. Olofinsan, B. Vatsha and N. Mabuba, New J. Chem., 2022, 46, 12968–12980 RSC .
- T. L. Yusuf, S. D. Oladipo, S. Zamisa, H. M. Kumalo, I. A. Lawal, M. M. Lawal and N. Mabuba, ACS Omega, 2021, 6, 13704–13718 CrossRef CAS .
- A. A. Adeleke, M. S. Islam, O. Sanni, C. Mocktar, S. J. Zamisa and B. Omondi, J. Inorg. Biochem., 2021, 214, 111266 CrossRef CAS PubMed .
- A. A. Adeleke, M. S. Islam and B. Omondi, Arch. Pharm., 2023, 356, 2200308 CrossRef CAS .
- A. Gölcü, Transition Met. Chem., 2006, 31(3), 405–412 CrossRef .
- I. Bento, C. Peixoto, V. N. Zaitsev and P. F. Lindley, Acta Crystallogr.,
Sect. D: Biol. Crystallogr., 2007, 63, 240–248 CrossRef CAS .
- L. Esmaeili, M. G. Perez, M. Jafari, J. Paquin, P. Ispas-Szabo, V. Pop, M. Andruh, J. Byers and M. A. Mateescu, J. Inorg. Biochem., 2019, 192, 87–97 CrossRef CAS PubMed .
- S. D. Oladipo, C. Mocktar and B. Omondi, Arabian J. Chem., 2020, 13, 6379–6394 CrossRef CAS .
- W. A. Munzeiwa, S. D. Oladipo, C. U. Ibeji, C. Mocktar and B. Omondi, J. Inorg. Biochem., 2021, 225, 111600 CrossRef CAS .
- S. D. Oladipo and B. Omondi, Inorganics, 2022, 10, 79 CrossRef CAS .
- A. Climova, E. Pivovarova, M. Szczesio, K. Gobis, D. Ziembicka, A. Korga-Plewko, J. Kubik, M. Iwan, M. Antos-Bielska, M. Krzyżowska and A. Czylkowska, J. Inorg. Biochem., 2023, 240, 112108 CrossRef CAS .
- Q. U. Sandhu, M. Pervaiz, A. Majid, U. Younas, Z. Saeed, A. Ashraf, P. R. Khan, S. Ullah, F. Ali and S. Jelani, J. Coord. Chem., 2023, 76, 1094–1118 CrossRef CAS .
- A. Bielenica, A. Głogowska, E. Augustynowicz-Kopeć, J. Orzelska-Górka, D. Kurpios-Piec and M. Struga, Tuberculosis, 2023, 143, 102412 CrossRef CAS PubMed .
- D. Chiodi and Y. Ishihara, J. Med. Chem., 2023, 66, 5305–5331 CrossRef CAS .
- S. D. Oladipo, T. L. Yusuf, S. J. Zamisa, M. Shapi and T. J. Ajayi, J. Mol. Struct., 2021, 1241, 130620 CrossRef CAS .
- S. D. Oladipo, T. L. Yusuf, S. J. Zamisa, G. F. Tolufashe, K. A. Olofinsan, Z. Tywabi-Ngeva and N. Mabuba, Eur. J. Chem., 2021, 12, 204–215 CrossRef CAS .
-
R. Dennington, T. A. Keith and J. M. Millam, GaussView, version 6.0. 16, Semichem Inc, Shawnee Mission KS, 2016 Search PubMed .
- K. A. Olofinsan, O. L. Erukainure, N. Z. Msomi and M. S. Islam, Asian Pac. J. Trop. Biomed., 2022, 12, 300–3011 CrossRef CAS .
- O. B. Ibitoye, K. A. Olofinsan, K. Teralı, U. M. Ghali and T. O. Ajiboye, J. Food Biochem., 2018, 42, e12627 CrossRef .
- G. A. Kurian, S. Suryanarayanan, A. Raman and J. Padikkala, Chin. Med., 2010, 5, 1–7 CrossRef PubMed .
- A. Turkoglu, M. E. Duru, N. Mercan, I. Kivrak and K. Gezer, Food Chem., 2007, 10, 267–273 CrossRef .
- Y. P. Tan and E. W. C. Chan, Food Biosci., 2014, 6, 17–23 CrossRef CAS .
- D. K. Vizhi, N. Supraja, A. Devipriya, N. V. Tollamadugu and R. Babujanarthanam, Adv. Nano Res., 2016, 4, 129 CrossRef .
- S. D. Oladipo and B. Omondi, Transition Met. Chem., 2020, 45, 391–402 CrossRef CAS .
- S. A. Olagboye, T. L. Yusuf, S. D. Oladipo and S. J. Zamisa, Z. Kristallogr. – New Cryst. Struct., 2020, 235, 833–836 CrossRef CAS .
- I. Waziri, T. L. Yusuf, H. A. Zarma, S. O. Oselusi, L. C. Coetzee and A. S. Adeyinka, Inorg. Chim. Acta, 2023, 552, 121505 CrossRef CAS .
- A. A. Adeleke, S. D. Oladipo, R. C. Luckay, E. O. Akintemi, K. A. Olofinsan, O. I. Babatunde, S. T. Yussuf, S. A. Ogundare, O. M. Adeleke and K. I. Babalola, ChemistrySelect, 2024, 9, e202304967 CrossRef CAS .
-
L. Ryan and R. Norris,Cambridge International AS and A Level Chemistry Coursebook with CD-ROM, Cambridge University Press, 2014 Search PubMed .
- M. Hazra, T. Dolai, A. Pandey, S. K. Dey and A. Patra, Bioinorg. Chem. Appl., 2014, 2014, 1–13 CrossRef .
- A. A. Osowole, R. Kempe, R. Schobert and K. Effenberger, Synth. React. Inorg. Met.-Org. Chem., 2011, 41, 825–833 CrossRef CAS .
- D. J. Hodgson, Inorg. Chim. Acta, 1983, 75, 225–228 CrossRef CAS .
- S. D. Oladipo, S. J. Zamisa, A. A. Badeji, M. A. Ejalonibu, A. A. Adeleke, I. A. Lawal, A. Henni and M. M. Lawal, Sci. Rep., 2023, 13, 13414 CrossRef CAS .
- T. Tsuneda, J. W. Song, S. Suzuki and K. Hirao, J. Chem. Phys., 2010, 133, 174101 CrossRef PubMed .
- S. D. Oladipo, G. F. Tolufashe, C. Mocktar and B. Omondi, Inorg. Chim. Acta, 2021, 520, 120316 CrossRef CAS .
- U. Hossain, A. K. Das, S. Ghosh and P. C. Sil, Food Chem. Toxicol., 2020, 145, 111738 CrossRef CAS .
- F. Hasaninezhad, Z. Tavaf, F. Panahi, M. Nourisefat, A. Khalafi-Nezhad and R. Yousefi, Diabetes Metab. Syndr., 2020, 19, 1505–1515 CAS .
- I. Gulcin and S. H. Alwasel, Processes, 2023, 11, 2248 CrossRef CAS .
- A. M. Pisoschi, A. Pop, F. Iordache, L. Stanca, G. Predoi and A. L. Serban, Eur. J. Med. Chem., 2021, 209, 112891 CrossRef CAS PubMed .
- W. K. Alderton, C. E. Cooper and R. G. Knowles, Biochem. J., 2001, 357, 593–615 CrossRef CAS PubMed .
- L. Beneš, Z. Ďuračková and M. Ferenčik, Life Sci., 1999, 65, 1865–1874 CrossRef .
- I. Kostova and L. Saso, Cur. Med. Chem., 2013, 20, 4609–4632 CrossRef CAS .
- T. Ak and I. Gülçin, Chem. Biol. Interface, 2008, 174, 27–37 CrossRef CAS .
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef .
- S. Shityakov, W. Neuhaus, T. Dandekar and C. Förster, Int. J. Comput. Biol. Drug Des., 2013, 6, 146–156 CrossRef CAS PubMed .
- L. Yu, A. Bridgers, J. Polli, A. Vickers, S. Long, A. Roy, R. Winnike and M. Coffin, Pharm. Res., 1999, 16, 1812–1817 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a