
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of a model phyllobilin bearing an optical marker†
Anh Thu
Nguyen Tran
 a,
Pengzhi
Wang
a,
Shaofei
Zhang
a,
Milena
Jovanovic
a,
Bianka
Siewert
b,
Simone
Moser
b and
Jonathan S.
Lindsey
*a
a,
Pengzhi
Wang
a,
Shaofei
Zhang
a,
Milena
Jovanovic
a,
Bianka
Siewert
b,
Simone
Moser
b and
Jonathan S.
Lindsey
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu; Tel: 919-515-6406
bDepartment of Pharmacognosy, Institute of Pharmacy, University of Innsbruck, Center for Chemistry and Biomedicine, Innsbruck 6020, Austria
First published on 6th June 2024
Abstract
Phyllobilins, derived from the breakdown of chlorophyll in plants, are acyclic tetrapyrroles that bear the signature ring E of the parent macrocycle. Few synthetic studies have concerned members of this emergent class of natural products, which now number >70. A prior synthesis of southern rim structures relied on Knoevenagel and Nazarov reactions to construct a dipyrromethane bearing the annulated ring E. Here, the synthesis has been extended to linear tetrapyrroles containing ring E as well as an appended dicyanovinyl unit. The latter serves as an optical marker with clearly observable absorption near 400 nm, rendering all compounds yellow, orange or red, thereby facilitating isolation and reaction monitoring. Knoevenagel condensation was carried out with a DC half (gem-dimethyl-substituted dihydrodipyrrin-1-carboxaldehyde bearing a 9-carboethoxy group) and an AB half (dipyrromethane equipped with a β-ketoester at the 8-position and the dicyanovinyl group at the 1-position). The resulting propenone is blocked by the ester and dicyanovinyl groups from reactions at the respective pyrrolic termini. Subsequent Yb(OTf)3-catalyzed Nazarov reaction accompanied by unexpected prototropic rearrangement afforded the isolated phyllobilin (8) wherein conjugation shifted from the DC dihydrodipyrrin moiety to the CB southern rim, thereby resembling the conjugation of a native phyllolumibilin. The expected direct products of Nazarov cyclization, the cis and trans products (pre-8-cis, pre-8-trans) due to the configuration of substituents in ring E, were not observed. Calculations by density functional theory showed the stability order as 8 > pre-8-trans > pre-8-cis. Access to phyllobilin analogues may open opportunities for fundamental studies pertaining to the native constituents, which are germane to plant sciences, nutrition, and ecology.
Introduction
Chlorophylls a and b are the chief light-harvesting pigments in plants. The annual production of such chlorophylls is estimated to be 1015 g.1 The breakdown products of chlorophylls until recently were unknown – chlorophyll was regarded as simply vanishing in the global ecosphere. In the past 35 years, the chlorophyll breakdown process has begun to be characterized and found to entail oxidative scission of the macrocycle and companion transformations to yield a class of products known as phyllobilins. The companion reactions include oxidation and/or reduction of the π-system as well as modification of the peripheral substituents. More than 70 phyllobilins have been identified, which generally are acyclic tetrapyrroles that retain the telltale annulated ring of the parent chlorophyll a (Chart 1).2–4 The phyllobilins have been grouped into categories on the basis of optical properties and corresponding molecular structures. Two general classifications originate on the basis of the structure of ring A: type I comprises an α-formylpyrrole unit whereas type II comprises an α-pyrrolinone unit (containing an α-oxo rather than α-formyl substituent). Within each type are subgroups of phyllolumibilins (and side-chain modified derivatives), phylloleucobilins, phylloxanthobilins, and phylloroseobilins. Examples of type I phyllobilins are shown in Chart 1. It warrants mention that the numbering system for phyllobilins is different from that of the parent chlorophylls.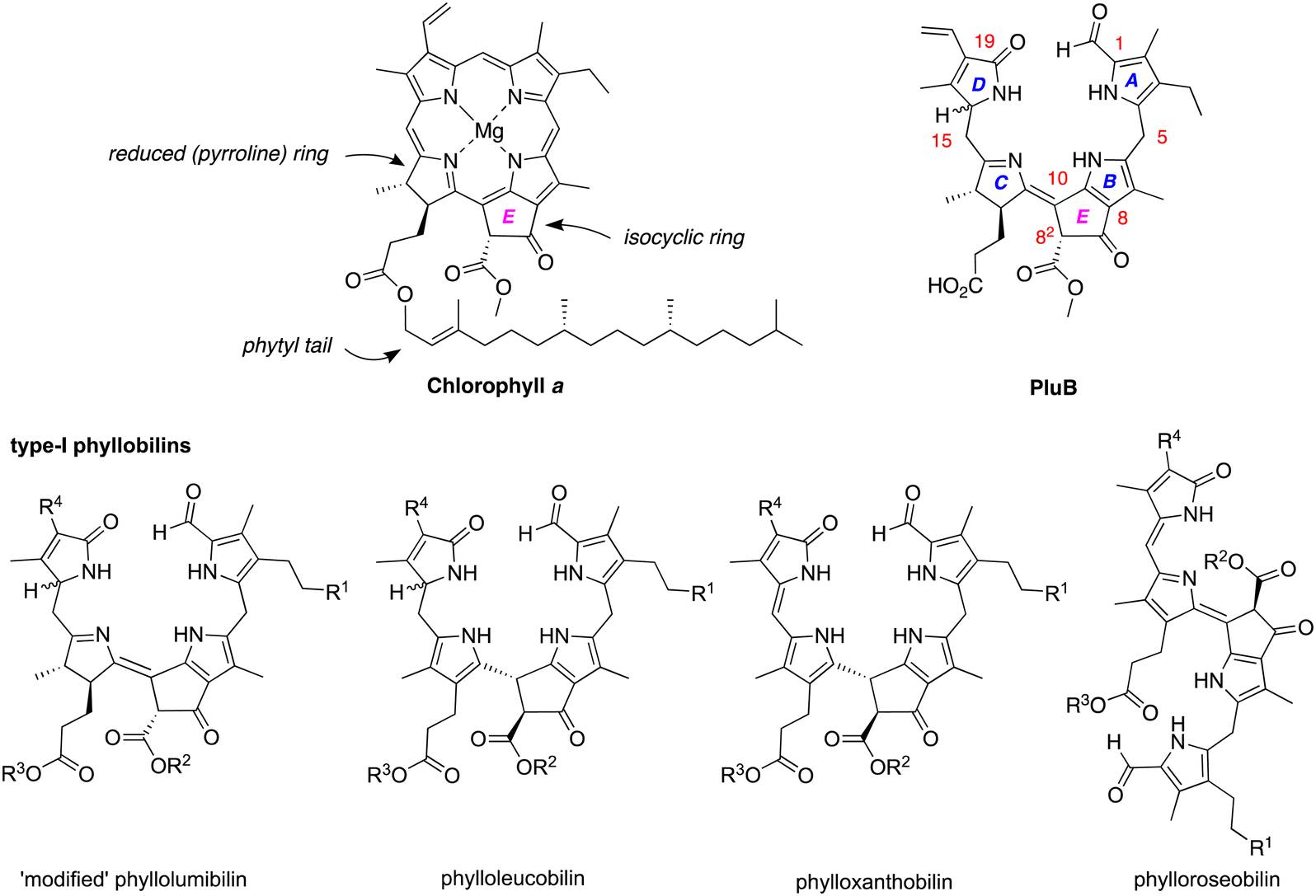 | ||
| Chart 1 Structures of chlorophyll a and representative phyllobilins. The general nomenclature and numbering system for phyllobilins, illustrated for PluB, is shown at upper right. | ||
For some time, phyllobilins were suspected to be mere catabolites of little interest. Recent studies, however, have revealed antioxidant,5,6 anti-inflammatory,7,8 and anti-proliferative9 activities among the diverse phyllobilin family. Phenotypic screening for anti-migratory action in cancer cells has revealed actin as a protein target for phyllobilins.10 Phyllobilins are thus an interesting class of natural products from phytochemical, biochemical, and photophysical viewpoints.8,11 The evolutionary origin of the chlorophyll degradation pathway is proposed to have occurred commensurate with the emergence of land plants.12 Despite their global abundance, ecological relevance, and potential human benefits in a plant-based diet, none of the known phyllobilins has yet been synthesized.
Both classes of natural products – chlorophylls and phyllobilins – have been relatively neglected as targets of chemical synthesis.13 Studies of phyllobilins have remained dependent on isolation from natural sources, which typically has entailed laborious fractionation procedures. As part of a program to develop a total synthesis of native (bacterio)chlorophylls14–19 and natural products derived therefrom such as phyllobilins, we have carried out a number of model studies to better understand the constraints associated with new synthetic routes. A model bacteriochlorophyll has been accessed via Knoevenagel condensation of two dihydrodipyrrins (I, II-DHDP) to form an enone (III-BC), which upon one-flask reaction undergoes Nazarov cyclization, electrophilic aromatic substitution, and elimination of methanol to yield the tetrapyrrole macrocycle bearing the annulated ring (isocyclic ring E) (Scheme 1, top panel).14 Application of one dihydrodipyrrin (I) and one dipyrromethane (II-DPM) afforded enone III-C and the model chlorophyll (Scheme 1, middle panel).15 A more recent study employed a pyrrole-carboxaldehyde (IV) and pyrrole-β-ketoester (V) to form the enone (VI), which upon Nazarov cyclization afforded the annulated dipyrromethane (VII) (Scheme 1, bottom panel).20 The products resemble the southern rim of phylloleucobilins and phylloxanthobilins (Chart 1). The Knoevenagel condensation of IV and V gave reasonable yields (43–92%). The Nazarov cyclization of compound VI in the presence of 2,6-di-tert-butylpyridine (2,6-DTBP), In(OTf)3, and LiClO4 at 40–50 °C afforded the southern rim structure VII in 51–80% yield.
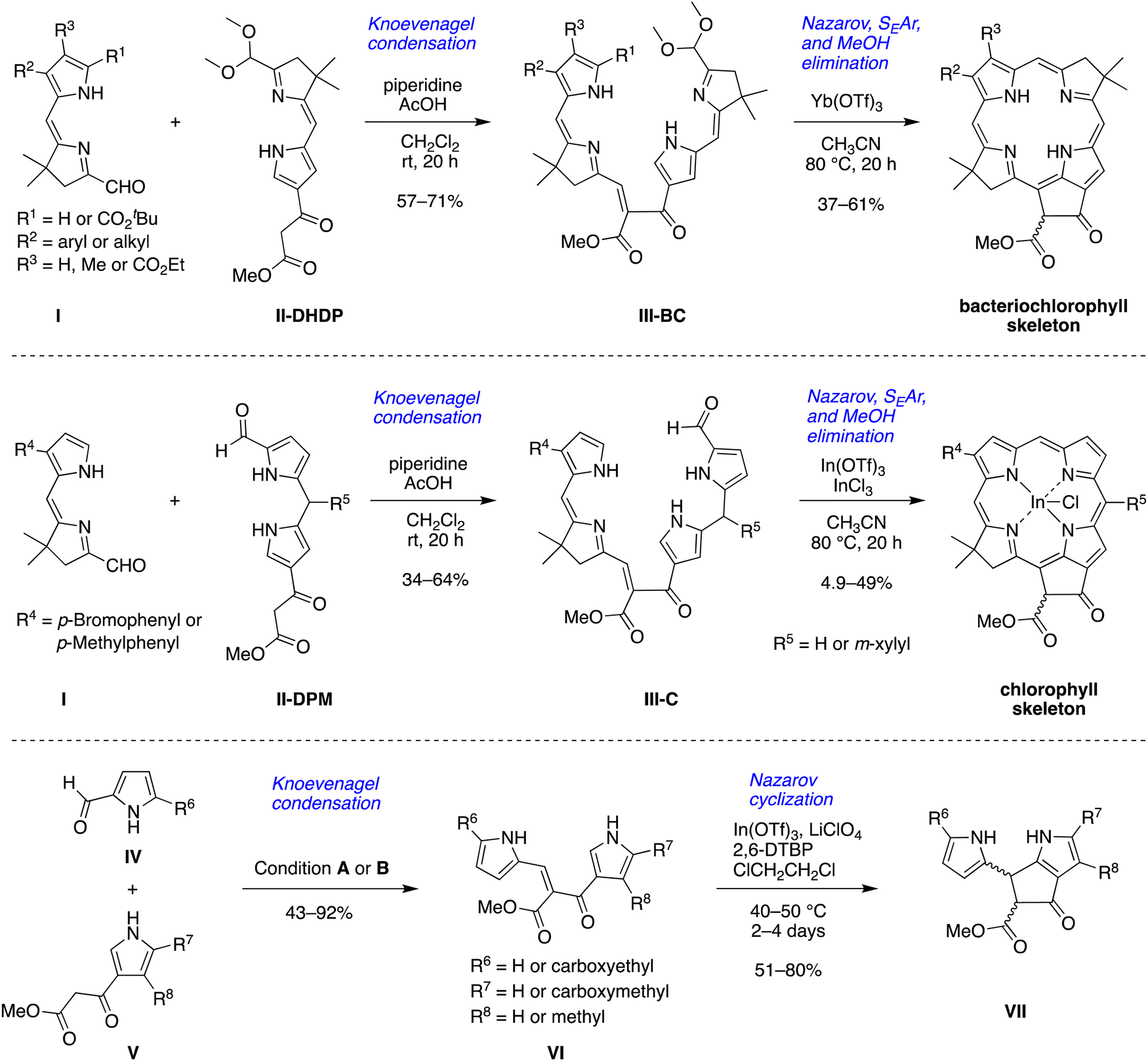 | ||
| Scheme 1 Routes to the skeleton of bacteriochlorophylls, chlorophylls and southern rim structures. Condition A: piperidine, AcOH, MS 4 Å, CH2Cl2, 40 °C, 20 h; condition B: piperidine, MeOH, 50 °C, 24 or 48 h. | ||
This paper reports an extension of the strategies shown in Scheme 1 to prepare a model phyllobilin. The model phyllobilin contains a gem-dimethyl group, rather than a trans-dialkyl group, in the pyrroline ring. The tetrapyrrolic intermediate formed upon Nazarov cyclization underwent unexpected in situ prototropic rearrangement to obtain a southern rim architecture similar to that of native type-I phyllolumibilins. The long-term objective of the work described herein is to gain access to diverse phyllobilins for fundamental chemical, ecological, and nutritional studies.
Results and discussion
Synthesis of a dipyrromethane bearing a 2,2-dicyanovinyl group
To study the formation of phyllobilin-like compounds, targets were sought that contain four pyrrolic rings yet also are equipped with a marker for visible absorption spectroscopic monitoring. For the latter objective, the 2,2-dicyanovinyl moiety attached to a pyrrole was attractive. The 2,2-dicyanovinyl unit was first employed nearly a century ago in the chemistry of pyrroles,21 and was later used by Rapoport in the synthesis of deoxophylloerythroetioporphyrin.22 Woodward also reported the use of the 2,2-dicyanovinyl unit in the synthesis toward chlorophyll as a masked formyl group of the ring B pyrrole.23 Subsequent use has been sparing,24–30 with applications in the synthesis of porphocyanines31 and dipyrromethanes.32–37 The sparing use may stem from the emergence of diverse protecting groups for pyrroles38 and the challenges of deprotection of the dicyanovinyl group. Previous reports relied solely on absorption spectroscopy for characterization of pyrroles bearing a 2-(2,2-dicyanovinyl) substituent.22,23 The 2,2-dicyanovinyl moiety is inert under acidic and other conditions,38 which is attractive for use in synthesis as described in the following.Pyrroles 1,39,402,25 and 325 are known compounds. Pyrrole 3 was previously prepared from 2-ethoxycarbonyl-3,4,5-trimethylpyrrole.25 Here, a new route to pyrrole 3 was developed from inexpensive tert-butyl acetoacetate (Scheme 2). Treatment of tert-butyl acetoacetate with sodium nitrite solution in acetic acid followed by addition of 3-methyl-2,4-pentanedione and zinc dust39 gave pyrrole 1 in 50% yield. Treatment of 1 with benzoyl chloride in DMF at reflux gave 2-formyl-3,4,5-trimethylpyrrole, which upon reaction with malononitrile in ethanol at 70 °C afforded 2-(2,2-dicyanovinyl)pyrrole 2 in 43% yield over the two steps.25 Pyrrole 2 was charactered by single-crystal X-ray diffraction (see the ESI†). Chlorination of 2 with sulfuryl chloride in acetic acid at 55 °C25 yielded pyrrole 3 in 73% yield upon simple filtration.
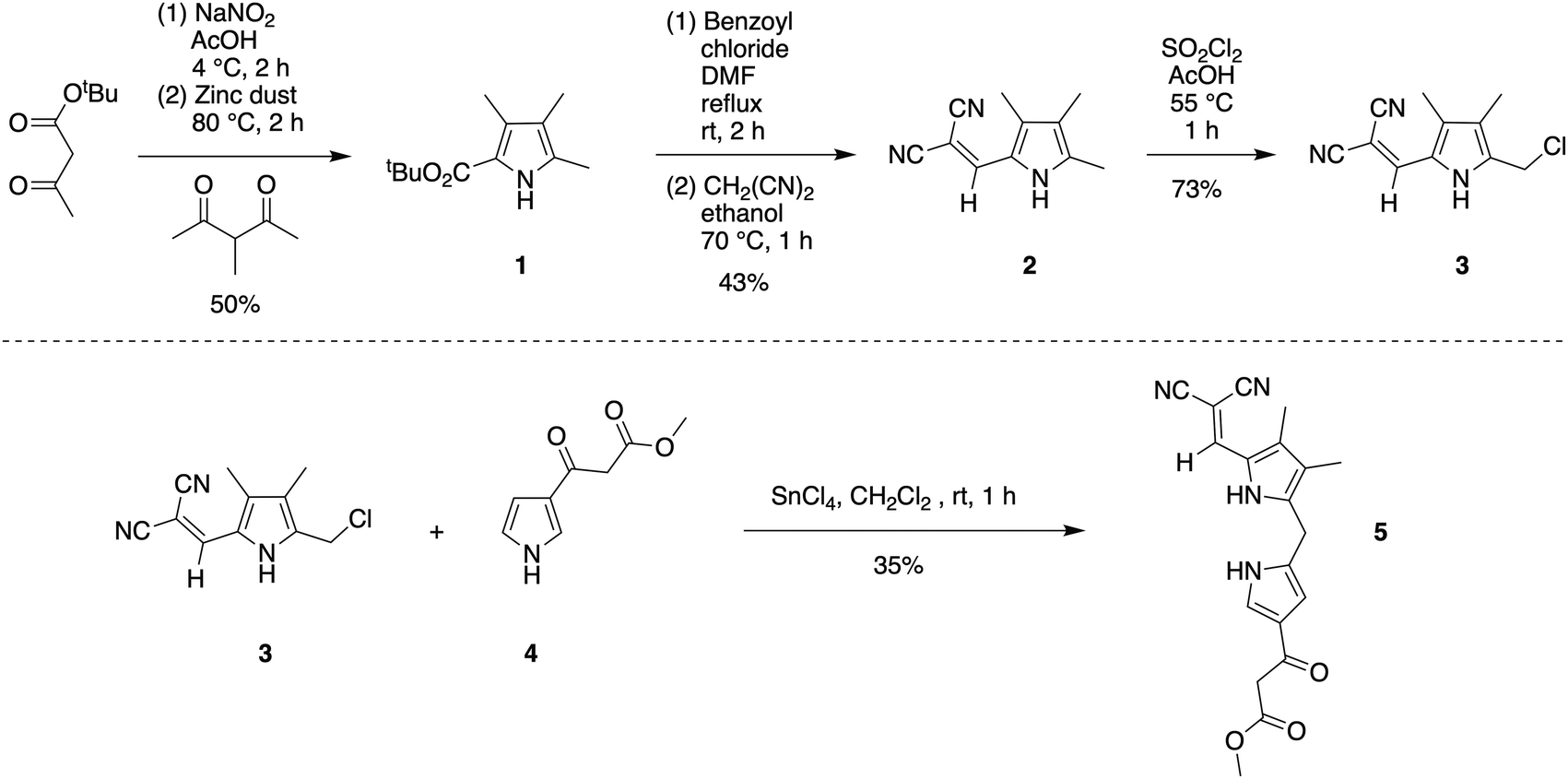 | ||
| Scheme 2 Synthesis of 2,2-dicyanovinyldipyrromethane 5. | ||
The condensation41 of 5-chloromethyl-2-(2,2-dicyanovinyl)pyrrole 3 and known β-ketoester-substituted pyrrole 417 in CH2Cl2 containing SnCl4 afforded dipyrromethane 5 in 35% yield. Other reported acidic conditions, such as refluxing HCl/ethanol,23 led to hydrolysis of the ester and cleavage of the carboxylic acid, thereby converting the β-ketoester to an acetyl group (not shown). The absorption spectra of the 2,2-dicyanovinyl-substituted compounds 2, 3, and 5 are shown in Fig. 1. The 2,2-dicyanovinyl chromophore is responsible for the strong absorption peak at 403 nm, 389 nm and 402 nm of 2, 3, and 5, respectively.
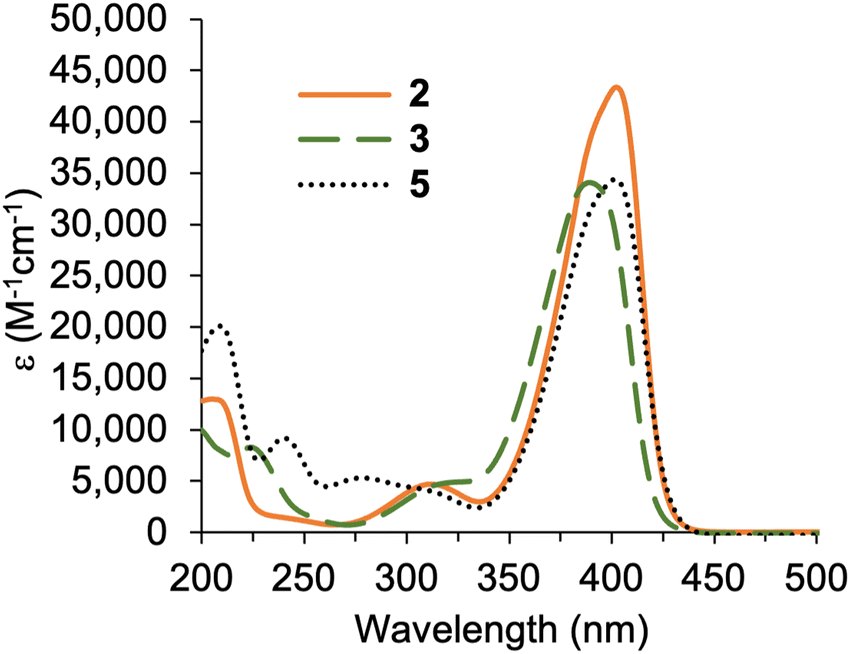 | ||
| Fig. 1 Absorption spectra in acetonitrile at room temperature of 2, 3, and 5. | ||
Formation of a model phyllobilin
Knoevenagel condensation42,43 of dipyrromethane 5 and known dihydrodipyrrin–carboxaldehyde 644 in the presence of AcOH/piperidine gave the desired α,β-unsaturated ketone 7 in 31% yield (Scheme 3). The stereochemical configuration of the alkene in 7 could not be determined unambiguously. The E isomer is often formed preferentially in Knoevenagel reactions of heteroaromatic β-keto esters; regardless, interconversion of the E and Z isomers occurs during the course of the Nazarov cyclization.16,17,45 Subsequent Nazarov cyclization45–47 catalyzed by Yb(OTf)3 afforded phyllobilin 8 in 46% yield. The yield was consistent with the tetrapyrrole intermediates that typically gave bacteriochlorins (via Nazarov, SEAr, and methanol elimination) in ∼50% yield.14–16 However, the previous conditions for the double-ring closure employed ten equivalents of the Lewis acid (Yb(OTf)3),14 whereas here the cyclization of enone 7 only required one equivalent of Yb(OTf)3. The Nazarov reaction of enone 7 with ten equivalents of Yb(OTf)3 afforded phyllobilin 8 in only 11% yield (not shown). The model phyllobilin 8 was characterized by NMR spectroscopy, ESI-MS, and absorption spectroscopy.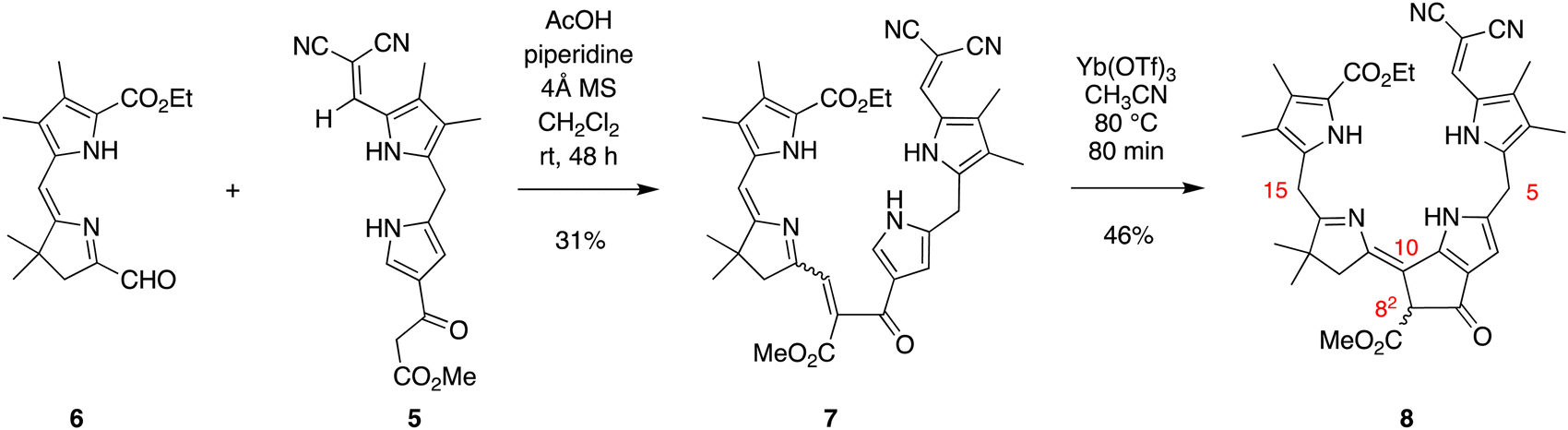 | ||
| Scheme 3 Synthetic route to a model phyllobilin. | ||
The absorption spectra of enone 7 and model phyllobilin 8 are shown in Fig. 2. The distinct peaks allowed the reaction to be monitored by absorption spectroscopy. Thus, the time course of the transformation of 7 to 8 (one equivalent of Yb(OTf)3) was monitored by removal of 50 μL samples from the reaction mixture ([7] = 0.2 mM initially), dilution to 1 mL, and analysis by absorption spectroscopy. The spectral evolution is shown in Fig. 2. The key features are as follows: (1) within 5 min upon treatment with Yb(OTf)3, the peak at 468 nm declined along with those at ∼403 and 279 nm. The disappearance of the peak at 468 nm, which is characteristic of the enone π-system, reflects the ring closure of the Nazarov reaction, which affords a saturated center at position 82. (2) A broad peak at ∼350 nm gradually arose after 5 min and also reached a maximum after 80 min. (3) The absorption spectrum observed at 80 min remained constant for a subsequent hour (not shown). All of the time-evolution traces are provided in the ESI.†
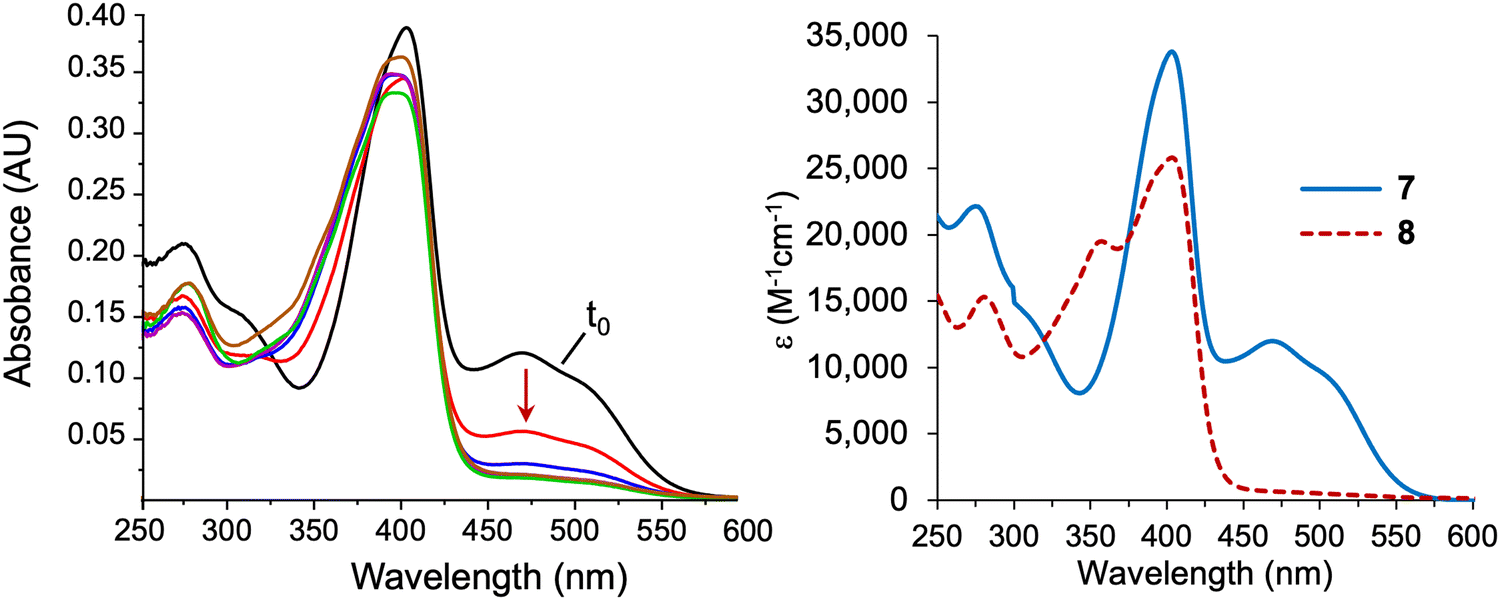 | ||
| Fig. 2 Absorption spectra in acetonitrile at room temperature. Left: Time-course spectral evolution of the conversion of 7 to 8 upon treatment under the Nazarov cyclization (0 min, black; 5 min, red; 10 min, blue; 20 min, magenta; 40 min, green; 80 min, brown). Right: Purified 7 and 8. | ||
Characterization
Enone 7 and model phyllobilin 8 were examined by 1H NMR spectroscopy including COSY and NOESY analysis. Expected features in the 1H NMR spectra of 7 and 8 are as follows (Fig. 3): (i) Each spectrum revealed signals corresponding to 38 protons. Among the signals are seven singlets due to the seven methyl groups. The four methyl groups on the two Northern pyrroles of both appear as four singlets in the region δ = 2.0–2.3 ppm. The two methyl groups of the gem-dimethyl moiety in phyllobilin 8 (δ = 1.25 and 1.27 ppm) are diastereotopic owing to the stereocenter in the isocyclic ring (position 82) while the two methyl groups are equivalent in enone 7 (δ = 1.08 ppm). (ii) The methylene unit at position 12 in model phyllobilin 8 resonates as AB peaks (δ = 2.42–2.44 and 2.53–2.55 ppm) due to the stereocenter at position 82, while that of enone 7 appears as a singlet (δ = 2.48 ppm). (iii) The meso methylene unit at position 5′ in enone 7 appears as a singlet at δ = 3.99 ppm, whereas that at position 5 of model phyllobilin 8 appears as a doublet at δ = 4.25 ppm (with a nOe observed with the proton at position 7).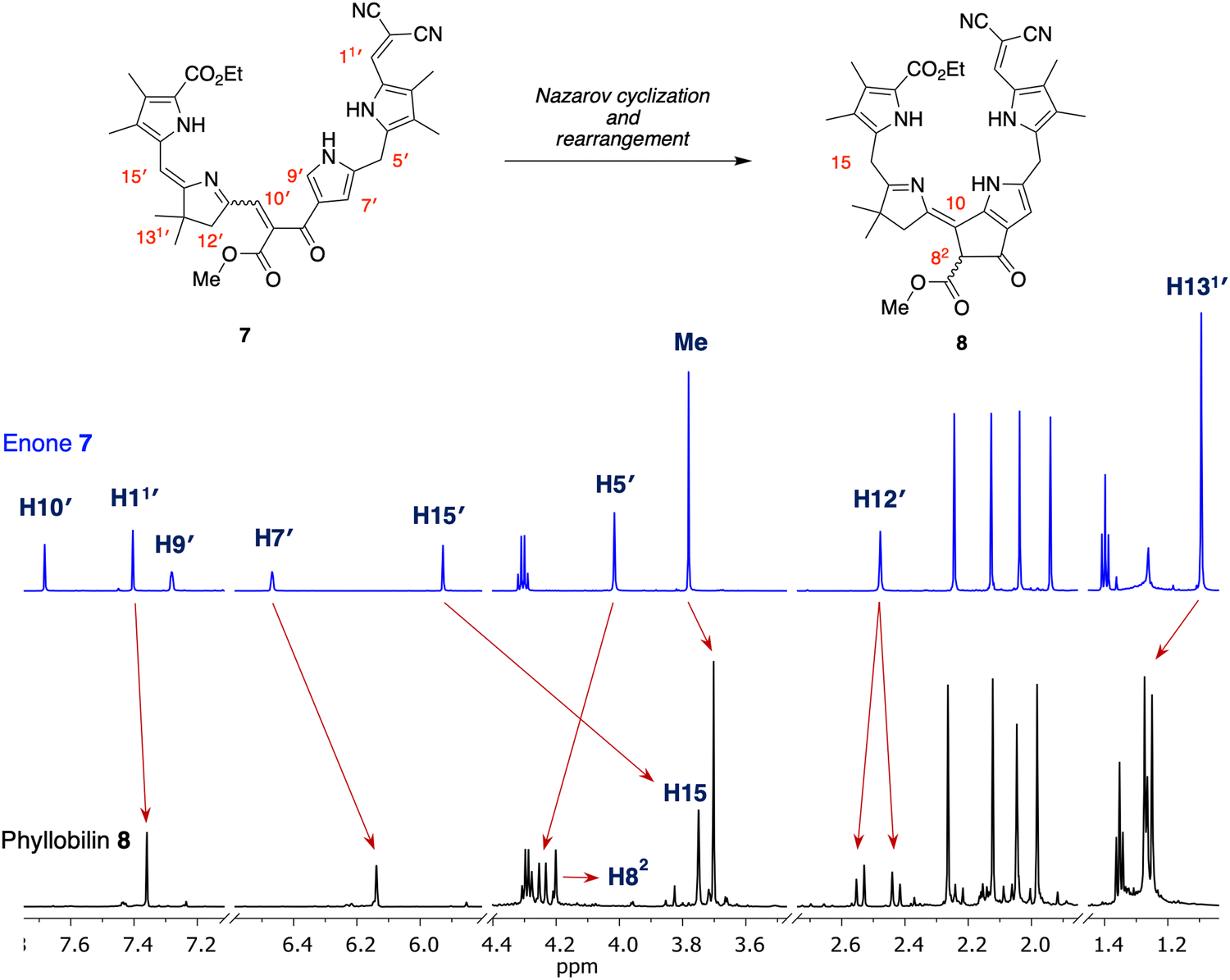 | ||
| Fig. 3 Correspondence of 1H NMR spectra (in CD2Cl2 at room temperature, 700 MHz) of reactant 7 (top) and isolated product 8 (bottom). | ||
On the other hand, key unexpected features are as follows: (i) The resonance due to the meso proton at position 15′ in enone 7 (δ = 5.89 ppm) is not seen in model phyllobilin 8, which instead shows an upfield resonance (δ = 3.70 ppm) due to two protons. (ii) The resonance due to the methine at position 9′ in enone 7 disappears as expected in model phyllobilin 8. On the other hand, the signal due to the proton at position 10′ in enone 7 is unexpectedly missing in model phyllobilin 8. (iii) The proton at position 82 of 8 is observed at δ = 4.20 ppm, consistent with the β-ketoester motif of the phyllobilin, but resonates as a singlet. (iv) A COSY spectrum of 8 shows no interaction between other protons and the proton at position 82, indicating the absence of a proton at the adjacent meso-carbon (position 10). (v) A NOESY spectrum of 8 shows interaction between the gem-dimethyl protons and the two protons at position 15. The spectral data are consistent with saturation at position 15 and unsaturation at position 10, which can only occur upon prototropic rearrangement (i.e., double-bond migration) following Nazarov cyclization.
Density functional theoretical (DFT) calculations48 were carried out to gain insight concerning the driving force for the prototropic rearrangement. Three compounds were considered: the isolated product 8 and the two expected products upon Nazarov cyclization without rearrangement, pre-8-trans, and pre-8-cis. The geometry optimization was performed at the PBE0/def2-TZVP level of theory49–51 using Orca 5.0.452 in vacuum at 0 K. Note that each compound 8, pre-8-trans, and pre-8-cis has an enantiomer (not shown), which were not the subject of calculation. The optimized structures of pre-8-cis, pre-8-trans and 8 are shown in Fig. 4. Each compound exhibited a somewhat helical architecture, like that of a lock washer, which is not unexpected given the constrained geometry of the southern rim and the presence of two appended pyrrole units (indeed, helical chirality is known for bilins.53). The results showed the order of stability to be 8 > pre-8-trans > pre-8-cis. The difference in energy between 8 and pre-8-trans is 2.016 kcal mol−1, while that of pre-8-trans and pre-8-cis is 5.449 kcal mol−1. Hence, the final product 8 is predicted to be energetically favored, which is consistent with experimental observation.
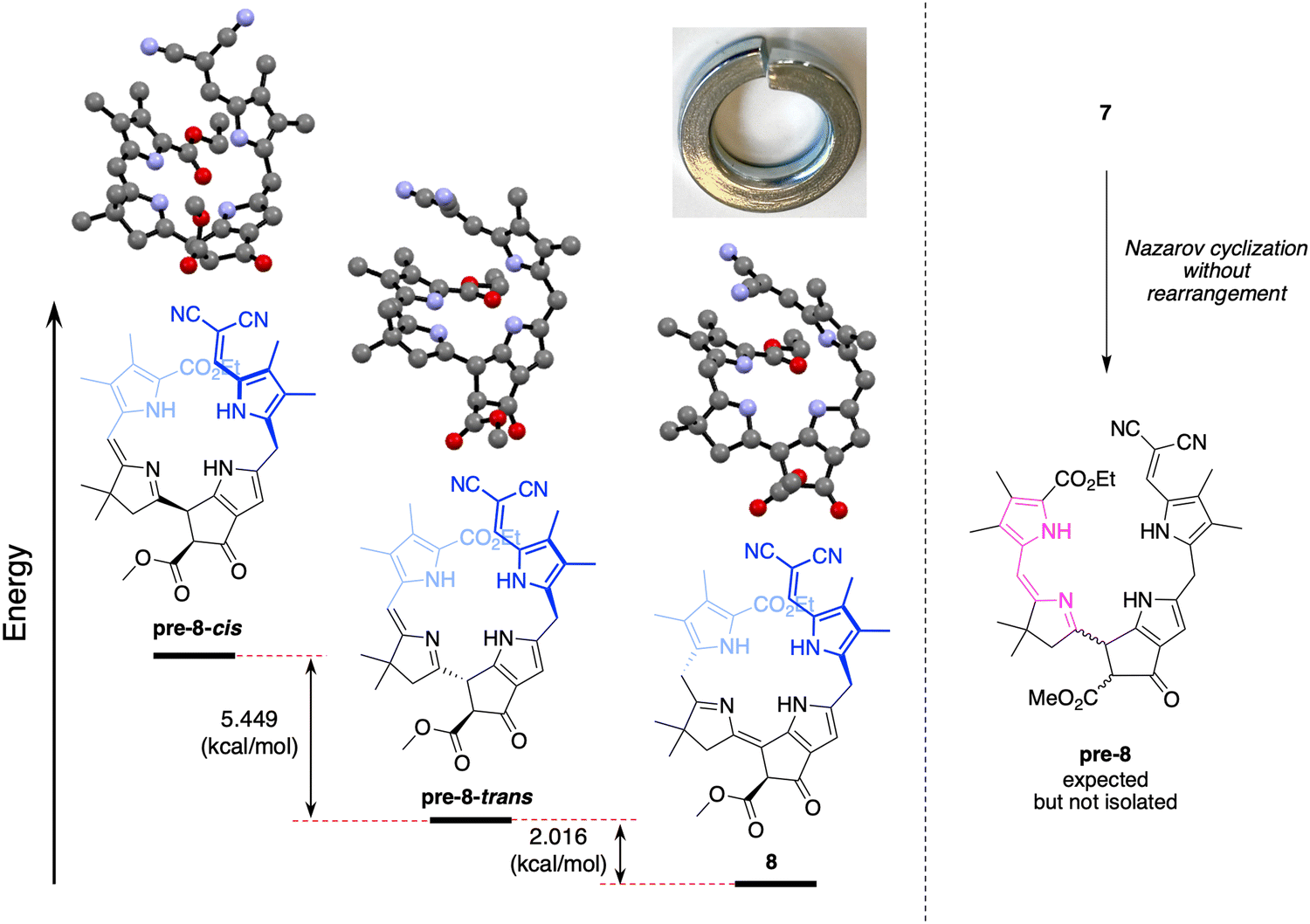 | ||
| Fig. 4 Optimized structures of compound pre-8-cis, pre-8-trans and 8 (hydrogen atoms omitted for clarity); a picture of a lock washer; and scheme showing expected formation of pre-8 upon Nazarov cyclization of 7. | ||
The prototropic rearrangement in model phyllobilin 8 in effect shifts the double bond from the dihydrodipyrrin constituent (originating in 6) to the lower rim. In other words, the dihydrodipyrrin unit has shifted from encompassing rings DC to rings CB. The mechanism is not known. While a simple mechanism would entail abstraction of the proton at position 10 (H10), forming an enamino structure, a more interesting mechanism may involve the more acidic proton of the β-ketoester. A corresponding formal mechanism is outlined in Scheme 4 as one proposal. In step (i), keto–enol tautomerism of the β-ketoester of pre-8 affords I-1. In step (ii), abstraction of H10, which is allylic, benzylic, and located at the imine α-position, may trigger the prototropic rearrangement to give I-2. In step (iii), the enol reverts to the β-ketoester. The two keto–enol tautomeric processes (steps (i) and (iii)) are expected to be reversible, whereas the conversion of I-1 to I-2 in step (ii) is likely to be the source of exergonicity. While the keto–enol tautomerism of the β-ketoester of chlorophylls and related macrocycles has been examined over several decades,54–58 more in-depth calculations would be required to provide insight concerning possible mechanisms.
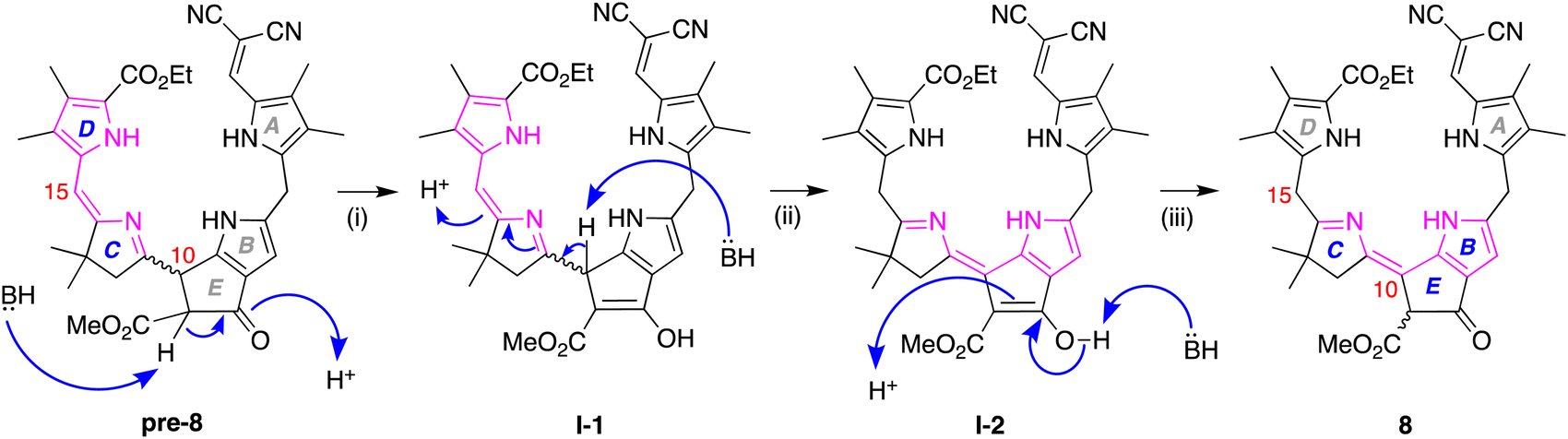 | ||
| Scheme 4 Proposed rearrangement pathway leading to model phyllobilin 8. | ||
The normalized absorption spectrum of a native phyllolumibilin (PluB)4 and model phyllobilin 8 are shown in Fig. 5. The native and synthetic compounds share a similar absorption peak at ∼355 nm. The absorption band near 355 nm is a feature in common with the B/C chromophore of fluorescent chlorophyll catabolites, such as PluB4. The typical absorption (312 nm) for the α-formylpyrrole unit (ring A) in phyllolumibilins is absent in 8, replaced instead by the absorption at 403 nm corresponding to the α-(2,2-dicyanovinyl)pyrrole unit.
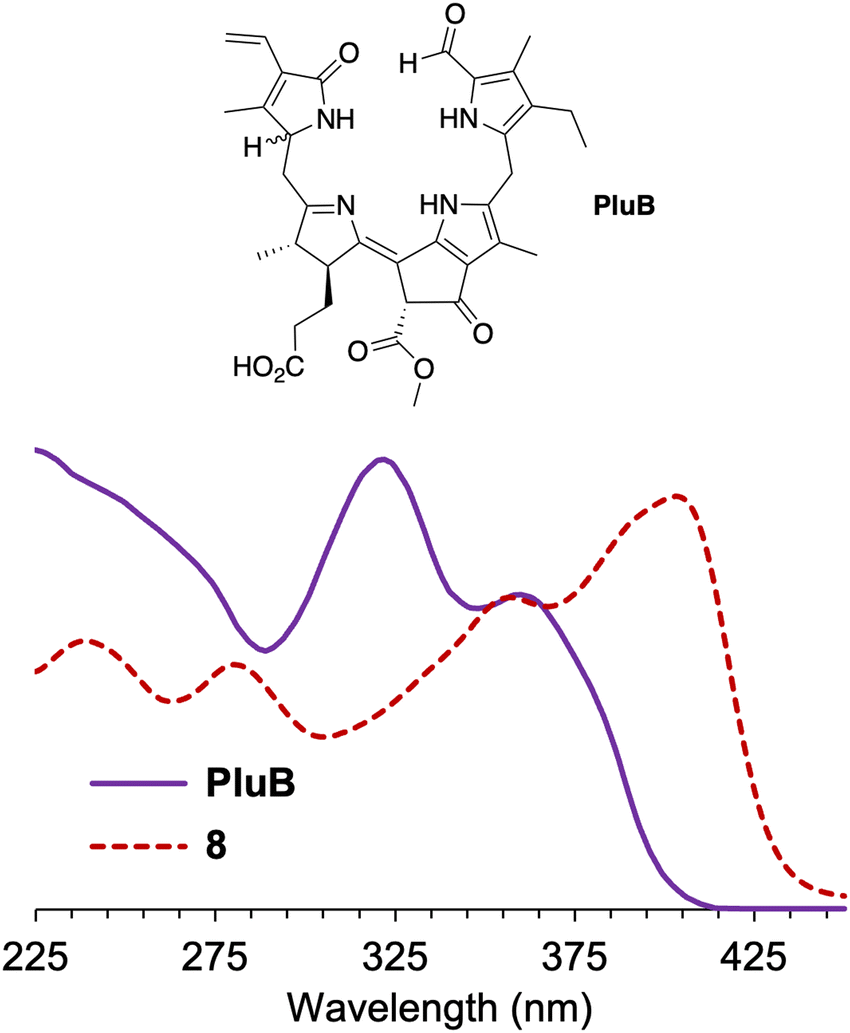 | ||
| Fig. 5 Absorption spectra at room temperature of phyllolumibilin PluB4 in aqueous MeOH (potassium phosphate buffer, 20 mM, pH 7) and 8 in acetonitrile (normalized at 355 nm). | ||
The structures of typical native phyllobilins59–62 are shown in Scheme 5. The formation of the native phyllobilins begins with chlorophyll a and proceeds via oxidation and reduction to give the observed products. For example, phyllolumibilins (PluBs), which contain a dihydrodipyrrin chromophore encompassing rings B and C, undergo double prototropic rearrangement upon treatment with acid to give the dipyrromethane wherein the pyrroline ring has become unsaturated (Scheme 5, left panel). In so doing, the trans-dialkyl stereochemistry of ring C is lost. Similarly, spontaneous rearrangement of dioxophyllolumibilins (DPluBs) converts the pyrroline to a pyrrole and saturates position 10 to give the dioxophylloleucobilins (DPleBs, Scheme 5, middle panel).62 For conversion of expected product pre-8 to the isolated model phyllobilin 8, the gem-dimethyl group at position 13 precludes the tautomerization involving the β-position of the pyrroline motif; instead, the tautomerization engages with the flanking meso site (position 15) (Scheme 4). Thus, due to the gem-dimethyl group at the 13-position in 8, isomerization typical in the natural system that converts a phyllolumibilin to a phylloleucobilin, or a dioxophyllolumibilin to a dioxophylloleucobilin, is not possible.
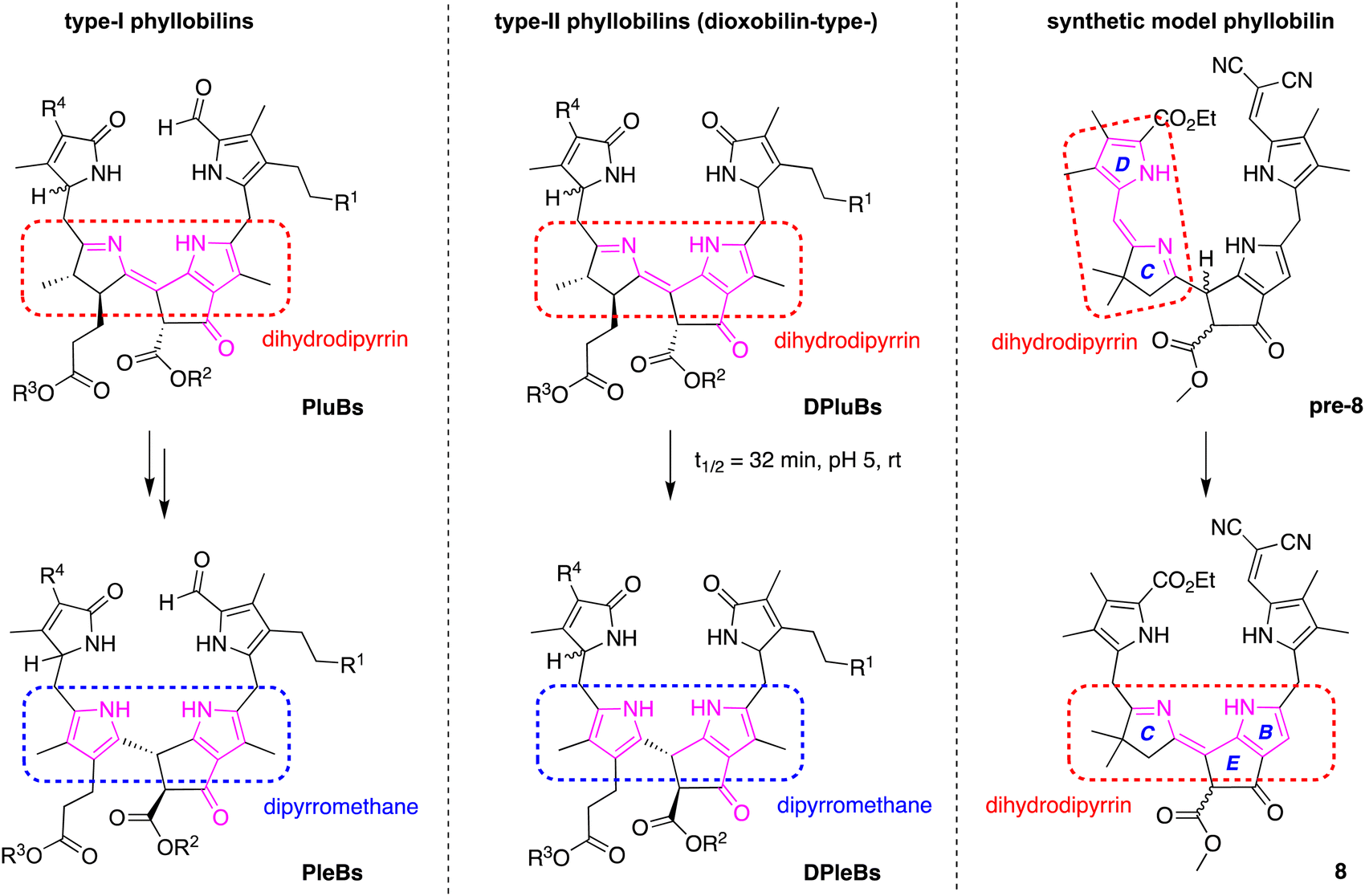 | ||
| Scheme 5 Natural phyllobilins and a synthetic phyllobilin. | ||
Conclusions
A route toward a synthetic phyllobilin has been developed via Knoevenagel condensation of a dihydrodipyrrin-carboxaldehyde (DC half) and a dipyrromethane-β-ketoester (AB half) followed by Nazarov cyclization. The route has been extended from that under investigation for the total synthesis of native (bacterio)chlorophylls. The A and D pyrroles (of the respective AB and DC halves) bear substituents at the α-positions to block reactions. The dicyanovinyl group blocks macrocyclization and provides a convenient chromophore (∼400 nm) that facilitates compound isolation and reaction monitoring. The synthetic phyllobilin contains the characteristic BCE ring architecture of native phyllobilins. The π-conjugation resembles a phyllolumibilin and forms by prototropic rearrangement following Nazarov cyclization. Further studies will be required to gauge the general occurrence of such rearrangements, which heretofore seem to have not been described. On the other hand, several distinctions from the native structures include (1) the presence of a gem-dimethyl group in pyrroline ring C, (2) a tetrahydrodipyrrin π-system encompassing rings C and D, and (3) methyl groups at four β-positions of the pyrroles. A present limitation is the low yield of the Nazarov cyclization. The synthetic work provides an entrée for further studies related to the chemistry of phyllobilins, which ultimately impinges on the fields of plant biology, nutrition, ecology, and agricultural sciences.Experimental section
General methods
1H NMR and 13C NMR spectra were collected at room temperature in CDCl3 unless noted otherwise. Absorption spectra were collected in 1-cm pathlength quartz cuvettes at room temperature. Electrospray ionization mass spectrometry (ESI-MS) data are reported for the molecular ion or protonated molecular ion. Molecular sieves (4 Å, powder) were heated (120 °C) overnight prior to use. Silica (40 μm average particle size) was used for column chromatography. THF used in all reactions was freshly distilled from Na/benzophenone ketyl unless noted otherwise. THF (ACS-grade) was used as received. Solutions that were concentrated were done so under reduced pressure using a rotary evaporator. All commercially available compounds were used as received. Non-commercial and known compounds 1,392,25 and 418 were prepared as described in the literature. Known compound 325 was prepared by a new route described herein.Computational methods
Density function calculations were performed using Orca 5.0.4.48 The geometry optimization calculations were done at the PBE0/def2-TZVP level of theory,49–51 followed by frequency calculations at the same level of theory to confirm that all structures are true minima. The energies of 8, pre-8-trans, and pre-8-cis were −2097.720, −2097.717, and −2097.708 Hartree.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1).
000 M−1 cm−1).
000 M−1 cm−1).
:1)] to afford a red solid (29.9 mg, 31%): mp 160–165 °C; 1H NMR (600 MHz, CDCl3) δ 1.08 (s, 6H), 1.40–1.42 (t, J = Hz, 3H), 1.93 (s, 3H), 2.04 (s, 3H), 2.11 (s, 3H), 2.25 (s, 3H), 2.25 (s, 3H), 2.48 (s, 2H), 3.79 (s, 3H), 3.99 (s, 2H), 4.31–4.35 (q, J = Hz, 2H), 5.89 (s, 1H), 6.46 (s, 1H), 7.25 (s, 1H), 7.32 (s, 1H), 7.72 (s, 1H), 9.16 (s, 1H), 9.36 (s, 1H), 10.91 (s, 1H); 13C{1H} NMR (100 MHz) δ 8.8, 9.0, 9.9, 10.5, 14.6, 25.5, 29.0, 29.8, 42.2, 49.8, 53.0, 60.0, 64.5, 106.8, 107.7, 115.8, 117.3, 120.4, 120.6, 121.2, 124.3, 126.7, 126.8, 126.9, 128.8, 131.2, 136.1, 136.9, 138.7, 139.9, 141.3, 161.8, 163.2, 165.5, 170.3, 188.0; ESI-MS m/z: [M + H]+ calcd for C36H39 N6O5 635.29674; found 635.2962. UV-Vis λmax (MeCN) (ε) 243 (26000), 278 (22000), 403 (34000) and 469 (nm) (12000 M−1 cm−1).
:1)] afforded an orange solid (10.3 mg, 46%): mp 135–140 °C dec.; 1H NMR (700 MHz, CD2Cl2) δ 1.25 (s, 3H), 1.27 (s, 3H), 1.34–1.36 (t, J = 7 Hz, 3H), 1.98 (s, 3H), 2.05 (s, 3H), 2.12 (s, 3H), 2.26 (s, 3H), 2.42–2.44 (d, J = 16.8 Hz, 1H), 2.53–2.55 (d, J = 17.5 Hz, 1H), 3.70 (s, 3H), 3.75 (s, 2H), 4.20 (s, 1H), 4.23–4.25 (d, J = 15.4 Hz, 2H), 4.28–4.31 (q, J = 7 Hz, 2H), 6.14 (s, 1H), 7.36 (s, 1H), 9.87 (s, 1H), 10.16 (s, 1H), 10.05 (s, 1H), 11.37 (s, 1H); 13C{1H} NMR (150 MHz, CD2Cl2) δ 9.0, 9.2, 10.1, 10.9, 14.7, 24.9, 25.8, 25.9, 26.3, 30.3, 42.6, 50.9, 53.0, 61.2, 62.0, 64.4, 102.1, 114.5, 116.8, 117.6, 117.9, 120.8, 124.8, 126.7, 128.8, 129.3, 137.3, 138.9, 141.8, 141.9, 147.2, 154.6, 164.9, 169.3, 186.4, 189.4; ESI-MS m/z: [M + H]+ calcd for C36H38N6O5 635.2976; found 635.2957. UV-Vis λmax (MeCN) (ε) 237 (14000), 280 (15000), 357 (20000) and 403 (nm) (26000 M−1 cm−1).
000 M−1 cm−1).
000 M−1 cm−1).
000 M−1 cm−1).
:1)] to afford a red solid (29.9 mg, 31%): mp 160–165 °C; 1H NMR (600 MHz, CDCl3) δ 1.08 (s, 6H), 1.40–1.42 (t, J = Hz, 3H), 1.93 (s, 3H), 2.04 (s, 3H), 2.11 (s, 3H), 2.25 (s, 3H), 2.25 (s, 3H), 2.48 (s, 2H), 3.79 (s, 3H), 3.99 (s, 2H), 4.31–4.35 (q, J = Hz, 2H), 5.89 (s, 1H), 6.46 (s, 1H), 7.25 (s, 1H), 7.32 (s, 1H), 7.72 (s, 1H), 9.16 (s, 1H), 9.36 (s, 1H), 10.91 (s, 1H); 13C{1H} NMR (100 MHz) δ 8.8, 9.0, 9.9, 10.5, 14.6, 25.5, 29.0, 29.8, 42.2, 49.8, 53.0, 60.0, 64.5, 106.8, 107.7, 115.8, 117.3, 120.4, 120.6, 121.2, 124.3, 126.7, 126.8, 126.9, 128.8, 131.2, 136.1, 136.9, 138.7, 139.9, 141.3, 161.8, 163.2, 165.5, 170.3, 188.0; ESI-MS m/z: [M + H]+ calcd for C36H39 N6O5 635.29674; found 635.2962. UV-Vis λmax (MeCN) (ε) 243 (26000), 278 (22000), 403 (34000) and 469 (nm) (12000 M−1 cm−1).
:1)] afforded an orange solid (10.3 mg, 46%): mp 135–140 °C dec.; 1H NMR (700 MHz, CD2Cl2) δ 1.25 (s, 3H), 1.27 (s, 3H), 1.34–1.36 (t, J = 7 Hz, 3H), 1.98 (s, 3H), 2.05 (s, 3H), 2.12 (s, 3H), 2.26 (s, 3H), 2.42–2.44 (d, J = 16.8 Hz, 1H), 2.53–2.55 (d, J = 17.5 Hz, 1H), 3.70 (s, 3H), 3.75 (s, 2H), 4.20 (s, 1H), 4.23–4.25 (d, J = 15.4 Hz, 2H), 4.28–4.31 (q, J = 7 Hz, 2H), 6.14 (s, 1H), 7.36 (s, 1H), 9.87 (s, 1H), 10.16 (s, 1H), 10.05 (s, 1H), 11.37 (s, 1H); 13C{1H} NMR (150 MHz, CD2Cl2) δ 9.0, 9.2, 10.1, 10.9, 14.7, 24.9, 25.8, 25.9, 26.3, 30.3, 42.6, 50.9, 53.0, 61.2, 62.0, 64.4, 102.1, 114.5, 116.8, 117.6, 117.9, 120.8, 124.8, 126.7, 128.8, 129.3, 137.3, 138.9, 141.8, 141.9, 147.2, 154.6, 164.9, 169.3, 186.4, 189.4; ESI-MS m/z: [M + H]+ calcd for C36H38N6O5 635.2976; found 635.2957. UV-Vis λmax (MeCN) (ε) 237 (14000), 280 (15000), 357 (20000) and 403 (nm) (26000 M−1 cm−1).
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the NSF (CHE-1760839, CHE-2054497). NMR spectroscopy and mass spectrometry measurements were carried out in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University. Single-crystal X-ray diffraction data were obtained at the University of Tennessee at Knoxville (by Dr Phattananawee Nalaoh). Computing resources were provided by North Carolina State University High Performance Computing Services Core Facility (RRID:SCR_022168). This work used Bridges-2 at Pittsburgh Supercomputing Center through allocation [CHE240037] from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, and #2138296.References
- G. A. F. Hendry, J. D. Houghton and S. B. Brown, New Phytol., 1987, 107, 255–302 CrossRef CAS PubMed.
- B. Kräutler, Chem. Soc. Rev., 2014, 43, 6227–6238 RSC.
- P. Wang, C. A. Karg, N. Frey, J. Frädrich, A. M. Vollmar and S. Moser, Arch. Pharm., 2021, 354, e2100061 CrossRef PubMed.
- C. A. Karg, M. Taniguchi, J. S. Lindsey and S. Moser, Planta Med., 2023, 89, 637–662 CrossRef CAS PubMed.
- T. Müller, M. Ulrich, K. Ongania and B. Kräutler, Angew. Chem., Int. Ed., 2007, 46, 8699–8702 CrossRef PubMed.
- C. A. Karg, L. Parráková, D. Fuchs, H. Schennach, B. Kräutler, S. Moser and J. M. Gostner, Antioxidants, 2022, 11, 2056 CrossRef CAS PubMed.
- M. Roca, Food Chem., 2012, 130, 134–138 CrossRef CAS.
- C. A. Karg, C. Doppler, C. Schilling, F. Jakobs, M. C. S. Dal Colle, N. Frey, D. Bernhard, A. M. Vollmar and S. Moser, Food Chem., 2021, 359, 129906 CrossRef CAS PubMed.
- C. A. Karg, P. Wang, F. Kluibenschedl, T. Müller, L. Allmendinger, A. M. Vollmar and S. Moser, Eur. J. Org. Chem., 2020, 4499–4509 CrossRef CAS.
- C. A. Karg, S. Wang, N. Al Danaf, R. P. Pemberton, D. Bernard, M. Kretschmer, S. Schneider, T. Zisis, A. M. Vollmar, D. C. Lamb, S. Zahler and S. Moser, Angew. Chem., Int. Ed., 2021, 60, 22578–22584 CrossRef CAS PubMed.
- C. A. Karg, P. Wang, A. M. Vollmar and S. Moser, Phytomedicine, 2019, 60, 152969 CrossRef CAS PubMed.
- I. Schumacher, D. Menghini, S. Ovinnikov, M. Hauenstein, N. Fankhauser, C. Zipfel, S. Hörtensteiner and S. Aubry, Plant J., 2022, 109, 1473–1488 CrossRef CAS PubMed.
- Y. Liu, S. Zhang and J. S. Lindsey, Nat. Prod. Rep., 2018, 35, 879–901 RSC.
- S. Zhang and J. S. Lindsey, J. Org. Chem., 2017, 82, 2489–2504 CrossRef CAS PubMed.
- P. Wang, F. Lu and J. S. Lindsey, J. Org. Chem., 2020, 85, 702–715 CrossRef CAS PubMed.
- K. Chau Nguyen, P. Wang, R. D. Sommer and J. S. Lindsey, J. Org. Chem., 2020, 85, 6605–6619 CrossRef PubMed.
- D. T. M. Chung, P. V. Tran, K. Chau Nguyen, P. Wang and J. S. Lindsey, New J. Chem., 2021, 45, 13302–13316 RSC.
- K. Chau Nguyen, A. T. Nguyen Tran, P. Wang, S. Zhang, Z. Wu, M. Taniguchi and J. S. Lindsey, Molecules, 2023, 28, 1323 CrossRef CAS PubMed.
- K. Chau Nguyen and J. S. Lindsey, J. Org. Chem., 2023, 88, 11205–11216 CrossRef PubMed.
- A. T. Nguyen Tran, Z. Wu, D. T. M. Chung, P. Nalaoh and J. S. Lindsey, New J. Chem., 2023, 47, 13626–13637 RSC.
- H. Fischer and M. Neber, Justus Liebigs Ann. Chem., 1932, 496, 1–26 CrossRef CAS.
- M. E. Flaugh and H. Rapoport, J. Am. Chem. Soc., 1968, 90, 6877–6879 CrossRef CAS.
- R. B. Woodward, W. A. Ayer, J. M. Beaton, F. Bickelhaupt, R. Bonnett, P. Buchschacher, G. L. Closs, H. Dutler, J. Hannah, F. P. Hauck, S. Itǒ, A. Langemann, E. Le Goff, W. Leimgruber, W. Lwowski, J. Sauer, Z. Valenta and H. Volz, Tetrahedron, 1990, 46, 7599–7659 CrossRef CAS.
- Y. Joh and K. Makino, Jikeikai Med. J., 1971, 18, 121–131 CAS.
- J. B. Paine III, R. B. Woodward and D. Dolphin, J. Org. Chem., 1976, 41, 2826–2835 CrossRef.
- T. P. Wijesekera, J. B. Paine III and D. Dolphin, J. Org. Chem., 1985, 50, 3832–3838 CrossRef CAS.
- B. Morgan and D. Dolphin, Angew. Chem., Int. Ed. Engl., 1985, 24, 1003–1004 CrossRef.
- B. Morgan, D. Dolphin, R. H. Jones, T. Jones and F. W. B. Einstein, J. Org. Chem., 1987, 52, 4628–4631 CrossRef CAS.
- B. Morgan and D. Dolphin, J. Org. Chem., 1987, 52, 5364–5374 CrossRef CAS.
- C. Haubmann, H. Hübner and P. Gmeiner, Bioorg. Med. Chem. Lett., 1999, 9, 1969–1972 CrossRef CAS PubMed.
- L. Y. Xie, R. W. Boyle and D. Dolphin, J. Am. Chem. Soc., 1996, 118(20), 4853–4859 CrossRef CAS.
- S. David, D. Dolphin, B. R. James, J. B. Paine III, T. P. Wijesekera, F. W. B. Einstein and T. Jones, Can. J. Chem., 1986, 64, 208–212 CrossRef CAS.
- T. P. Wijesekera, J. B. Paine III and D. Dolphin, J. Org. Chem., 1988, 53, 1345–1352 CrossRef CAS.
- D. Dolphin, S. J. Rettig, H. Tang, T. Wijesekera and L. Y. Xie, J. Am. Chem. Soc., 1993, 115, 9301–9302 CrossRef CAS.
- R. W. Boyle, V. Karunaratne, A. Jasat, E. K. Mar and D. Dolphin, Synlett, 1994, 939–940 CrossRef CAS.
- L. Y. Xie and D. Dolphin, J. Chem. Soc., Chem. Commun., 1994, 1475–1476 RSC.
- T. Okawara, R. Kawano, H. Morita, A. Finkelstein, R. Toyofuku, K. Matsumoto, K. Takehara, T. Nagamura, S. Iwasa and S. Kumar, Molecules, 2017, 22, 1816 CrossRef PubMed.
- B. Jolicoeur, E. E. Chapman, A. Thompson and W. D. Lubell, Tetrahedron, 2006, 62, 11531–11563 CrossRef CAS.
- E. Bullock, A. W. Johnson, E. Markham and K. B. Shaw, J. Chem. Soc., 1958, 1430–1440 RSC.
- H. Falk, O. Hofer and H. Lehner, Monatsh. Chem., 1973, 104, 925–932 CrossRef CAS.
- J. B. Paine III and D. Dolphin, Can. J. Chem., 1976, 54, 411–414 CrossRef.
- G. Jones, Org. React., 1967, 15, 204–599 CAS.
- B. List, Angew. Chem., Int. Ed., 2010, 49, 1730–1734 CrossRef CAS PubMed.
- S. Zhang, M. N. Reddy, O. Mass, H.-J. Kim, G. Hu and J. S. Lindsey, New J. Chem., 2017, 41, 11170–11189 RSC.
- T. Mietke, T. Cruchter, V. A. Larionov, T. Faber, K. Harms and E. Meggers, Adv. Synth. Catal., 2018, 360, 2093–2100 CrossRef CAS.
- Z. A. Krasnaya, I. V. Torgov and N. S. Prostakov, Herald Russ. Acad. Sci., 2006, 76, 370–375 CrossRef.
- T. Vaidya, R. Eisenberg and A. J. Frontier, ChemCatChem, 2011, 3, 1531–1548 CrossRef CAS.
- F. Neese, WIREs Comp. Mol. Sci., 2022, 12, e1606 CrossRef.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- S. E. Boiadjiev and D. A. Lightner, Tetrahedron: Asymmetry, 1999, 10, 607–655 CrossRef CAS.
- H. H. Strain and W. M. Manning, J. Biol. Chem., 1942, 146, 275 CrossRef CAS.
- A. S. Holt and E. E. Jacobs, Plant Physiol., 1955, 30, 553–559 CrossRef CAS PubMed.
- J. L. Burdett and M. T. Rogers, J. Am. Chem. Soc., 1964, 86, 2105–2109 CrossRef CAS.
- M. Nsangou, A. B. Fredj, N. Jaïdane, M. G. Kwato Njock and Z. B. Lankhdar, J. Mol. Struct.: THEOCHEM, 2004, 681, 213–224 CrossRef CAS.
- J. J. Ríos, M. Roca and A. Pérez-Gávez, J. Agric. Food Chem., 2014, 62, 10576–10584 CrossRef PubMed.
- B. Kräutler and S. Hörtensteiner, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Advances in Photosynthesis and Respiration, Springer: Dordrecht, The Netherlands, 2006, vol. 25, pp. 237–260 Search PubMed.
- M. Oberhuber, J. Berghold and B. Kräutler, Angew. Chem., Int. Ed., 2008, 47, 3057–3061 CrossRef CAS PubMed.
- B. Kräutler, in Tetrapyrroles: Birth, Life and Death, ed. M. J. Warren and A. G. Smith, Landes Bioscience: Austin, TX, 2009, pp. 274–285 Search PubMed.
- I. Süssenbacher, S. Hörtensteiner and B. Kräutler, Angew. Chem., Int. Ed., 2015, 54, 13777–13781 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption spectra and determination of the molar absorption coefficients for 2 and 5–8; 1H NMR and 13C{1H} NMR data for all new compounds; single-crystal X-ray diffraction data. CCDC 2341280 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj01533a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |