DOI:
10.1039/D4NJ01350F
(Paper)
New J. Chem., 2024,
48, 9890-9898
Synthesis, and optical and electrochemical properties of 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives with the same or different aryl substituents on the thiophene rings†
Received
22nd March 2024
, Accepted 29th April 2024
First published on 8th May 2024
Abstract
We designed and synthesized 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives, 1,1′,3,3′-PhtBu-4,4′-BBT (BBT-PhtBu4), 1,1′,3,3′-PhCN-4,4′-BBT (BBT-PhCN4), and 1,1′-PhtBu-3,3′-PhCN-4,4′-BBT (BBT-PhtBu2PhCN2), which have four electron-donating tert-butylphenyl groups, four electron-withdrawing cyanophenyl groups, and two tert-butylphenyl groups and two cyanophenyl groups, respectively, on each thiophene ring by the Stille coupling reaction using 1,1′-diaryl-3,3′-distannyl-4,4′-BBT or 1,1′,3,3′-tetrastannyl-4,4′-BBT. It was found that the photoabsorption and fluorescence maximum wavelengths (λabsmax and λflmax) of BBT-PhtBu4, BBT-PhCN4, and BBT-PhtBu2PhCN2 appear in a longerwavelength region, in comparison with those of 1,1′-diaryl-4,4′-BBT derivatives BBT-PhtBu2 with a tert-butylphenyl group and BBT-PhCN2 with a cyanophenyl group on each thiophene ring. Moreover, the cyclic voltammetry (CV) curves of the 4,4′-BBT derivatives indicated that the introduction of the electron-donating tert-butylphenyl group and/or the electron-withdrawing cyanophenyl group into the benzo[c]thiophene skeleton leads to the lowering of the oxidation potential. In addition, an electron-donating phenyl substituent is more effective than an electron-withdrawing phenyl substituent in the lowering of the oxidation potential. Density functional theory (DFT) calculations as well as the experimental results revealed that increasing the number of electron-donating and electron-withdrawing phenyl groups on the thiophene ring results in the increase of the HOMO energy level and the lowering of the LUMO energy level, respectively, that leads to a decrease in the HOMO–LUMO band gap, that is, a bathochromic shift of the photoabsorption band. Thus, this work provides not only facile synthetic methods for 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives with the same or different aryl substituents on the thiophene rings but also useful methods to precisely adjust their optical and electrochemical properties.
Introduction
Benzo[c]thiophene and its derivatives have attracted significant attention as promising fluorophores for highly efficient bioimaging and phototheranostics1 and key constituents of emitters, semiconductors and photosensitizers for organic optoelectronic devices such as organic field-effect transistors (OFETs),2 organic photovoltaics (OPVs),3 organic light-emitting diodes (OLEDs),4 and dye-sensitized solar cells (DSSCs),5 as well as scientific interest in synthetic organic chemistry, polymer chemistry, photochemistry, electrochemistry, and theoretical chemistry.6 For this reason, some benzo[c]thiophene derivatives with substituents on the thiophene ring and/or the benzene ring, including 1,3- and 5,6-disubstituted benzo[c]thiophenes, have been synthesized, and their bioimaging, phototheranostic, and optoelectronic device performances as well as their photophysical and electrochemical properties were investigated so far.1,7–9 Moreover, a few synthetic methods for 3,3′-disubstituted-1,1′-bibenzo[c]thiophenes as the 1,1′-dimer of benzo[c]thiophene have been developed by Cava,10 Mohanakrishnan,11 and Ono et al.5 (Fig. 1a), although there are no reports on the synthesis of unsubstituted 1,1′-bibenzo[c]thiophenes. In particular, the introduction of an aryl substituent with an electron-donating or electron-withdrawing group on the benzo[c]thiophene chromophore allows us to adjust their optical and electrochemical properties due to the perturbation to the HOMO and the LUMO as well as the expansion of the π-conjugated system. Indeed, as a prior work for the synthetic methods, Mohanakrishnan et al. reported that 1,3-diarylbenzo[c]thiophenes and 3,3′-diaryl-1,1′-bibenzo[c]thiophenes have been prepared from 3-arylphthalide and diphthalide, respectively, via the lactone ring opening with an aryl Grignard reagent (ArMgBr) followed by thionation and concurrent intramolecular cyclization using Lawesson's reagent, and they revealed their optical and electrochemical properties (Fig. 1b).9,11
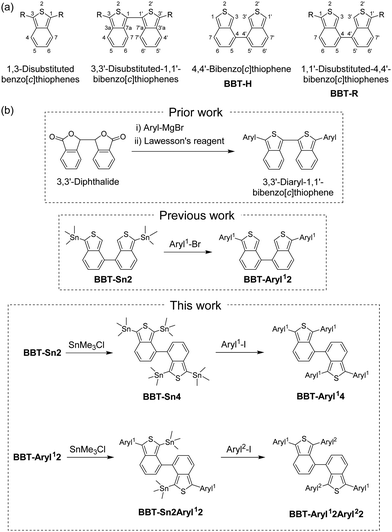 |
| | Fig. 1 (a) Chemical structures of benzo[c]thiophenes, 1,1′-bibenzo[c]thiophenes, and 4,4′-bibenzo[c]thiophenes. (b) Prior work on 3,3′-diaryl-1,1′-bibenzo[c]thiophenes by Mohanakrishnan et al. (ref. 11), previous work (ref. 12) on 1,1′-diaryl-4,4′-bibenzo[c]thiophenes, and this work on 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophenes. | |
On the other hand, in our previous work, we offered new synthetic methods for 1,1′-diaryl-4,4′-bibenzo[c]thiophene (abbreviated as 1,1′-diaryl-4,4′-BBT), 1,1′-di-tert-butylphenyl-4,4′-BBT (BBT-PhtBu2) and 1,1′-dicyanophenyl-4,4′-BBT (BBT-PhCN2), using a Stille coupling reaction of 1,1′-distannyl-4,4′-BBT (BBT-Sn2) with an aryl halide and Suzuki coupling reaction of 1,1′-dibromo-4,4′-BBT (BBT-Br2) with arylboronic acid. Furthermore, we revealed their photophysical properties in the solution and the solid state, electrochemical properties, and X-ray crystal structures (Fig. 1b).12 It was found that the bathochromic shift of the photoabsorption band from 4,4′-bibenzo[c]thiophene (BBT-H)13 to BBT-PhtBu2 and BBT-PhCN2 is mainly attributed to the destabilization of the HOMO energy level for BBT-PhtBu2 and both the destabilization of the HOMO energy level and the stabilization of the LUMO energy level for BBT-PhCN2 due to the introduction of electron-donating tert-butylphenyl groups and the electron-withdrawing cyanophenyl groups, respectively, into the thiophene rings, leading to a decrease in the HOMO–LUMO band gap.
In this work, to further gain insight into not only the efficient synthetic route to aryl-substituted 4,4′-bibenzo[c]thiophene derivatives but also the effect of aryl substituents on the molecular structure and the photophysical and electrochemical properties, we designed 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives, 1,1′,3,3′-PhtBu-4,4′-BBT (BBT-PhtBu4), 1,1′,3,3′-PhCN-4,4′-BBT (BBT-PhCN4), and 1,1′-PhtBu-3,3′-PhCN-4,4′-BBT (BBT-PhtBu2PhCN2), which have four electron-donating tert-butylphenyl groups, four electron-withdrawing cyanophenyl groups, and two tert-butylphenyl groups and two cyanophenyl groups, respectively, on each thiophene ring (Fig. 1b). Herein, we provide facile synthetic methods for 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives with the same or different aryl substituents on the thiophene rings using the Stille coupling reaction employing 1,1′-diaryl-3,3′-distannyl-4,4′-BBT or 1,1′,3,3′-tetrastannyl-4,4′-BBT with an aryl halide and reveal their photophysical properties in the solution and the solid state, electrochemical properties, and X-ray crystal structures, in comparison with those of 1,1′-diaryl-4,4′-BBT derivatives.
Results and discussion
Synthesis
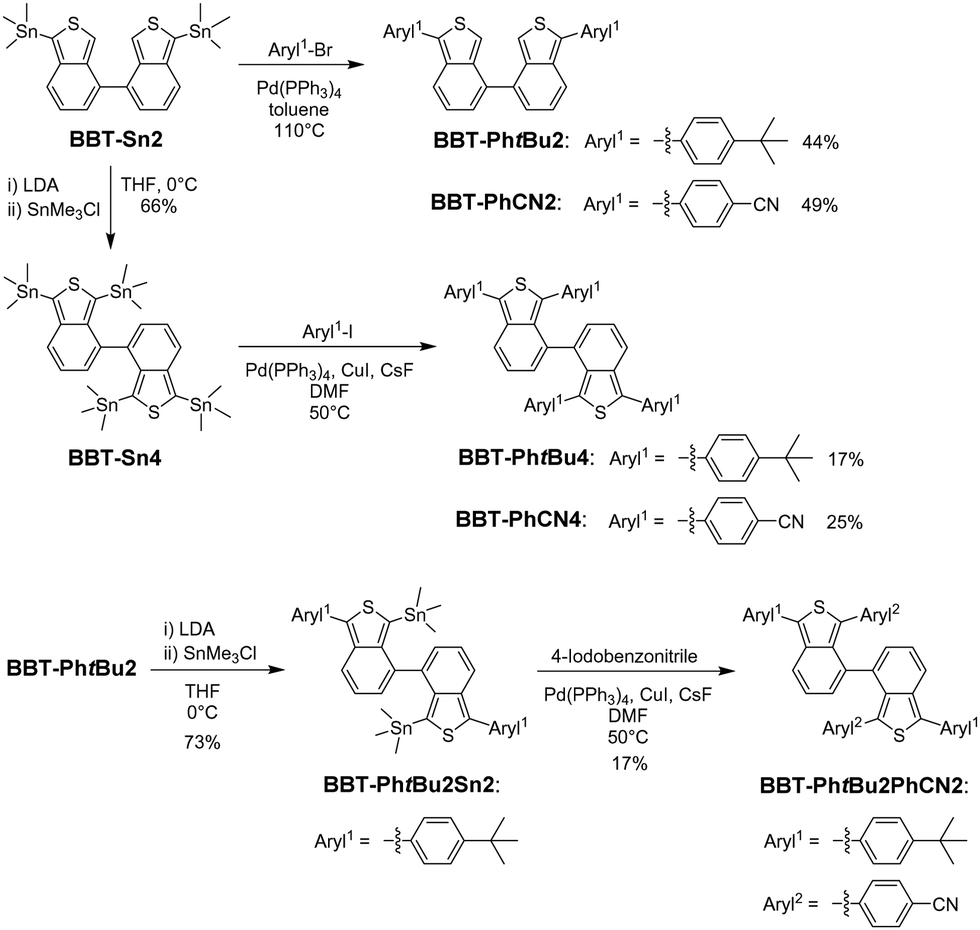
We found that BBT-Sn2 is a useful intermediate to produce 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives as well as 1,1′-diaryl-4,4′-BBT derivatives through the Stille coupling reaction with an aryl halide. In fact, our previous work demonstrated that BBT-PhtBu2 and BBT-PhCN2 are obtained in moderate yields (44% and 49%, respectively) using the Stille coupling reaction of 1,1′-distannyl-4,4′-BBT (BBT-Sn2) with the corresponding aryl halide (Scheme 1).12 Moreover, for the preparation of 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives with four same aryl substituents on the thiophene rings, 1,1′,3,3′-tetrastannyl-4,4′-BBT (BBT-Sn4) was derived from the reaction of BBT-Sn2 with lithium diisopropylamide (LDA), followed by treatment with trimethyltin chloride. Thus, we conducted the Stille coupling reaction of BBT-Sn4 with 1-tert-butyl-4-iodobenzene or 4-iodobenzonitrile using CuI and CsF in the presence of Pd(PPh3)4, because Baldwin et al. demonstrated that the combination of CuI and CsF can significantly promote the Stille reaction.14 As a result, BBT-PhtBu4 and BBT-PhCN4 were successfully prepared, although the yields (17% and 25%) of the two tetraaryl-4,4′-BBT derivatives are lower than those of the corresponding diaryl-4,4′-BBT derivatives BBT-PhtBu2 and BBT-PhCN2 using Stille coupling reactions. The relatively low yields of BBT-PhtBu4 and BBT-PhCN4 are due to the production of the corresponding 1,1′,3-triaryl-4,4′-BBT derivative as well as the 1,1′-diaryl-4,4′-BBT derivatives BBT-PhtBu2 or BBT-PhCN2 as by-products. On the other hand, for the preparation of 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives with two different aryl substituents on each thiophene ring, we prepared 1,1′-tert-butylphenyl-3,3′-distannyl-4,4′-BBT (BBT-Sn2PhtBu2) using the reaction of BBT-PhtBu2 with LDA, followed by treatment with trimethyltin chloride. The Stille coupling of BBT-PhtBu2Sn2 with 4-iodobenzonitrile gave BBT-PhtBu2PhCN2 with a 4-cyanophenyl group and a tert-butylphenyl group on each thiophene ring with a yield of 17%. However, we could not produce 1,1′-PhCN-3,3′-PhtBu-4,4′-BBT (BBT-PhCN2PhtBu2) in the current stage because the dehydration of BBT-PhCN2 by LDA, followed by di-stannylation did not proceed. Nevertheless, this result provides the stepwise synthetic method for the introduction of aryl substituents into the thiophene rings of the 4,4′-BBT skeleton, leading to the preparation of 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives with the same or different aryl substituents on the thiophene rings.
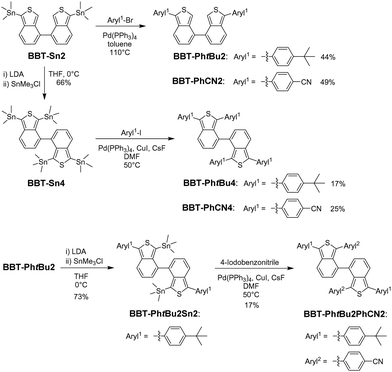 |
| | Scheme 1 Synthetic routes to 1,1′-diaryl-4,4′-bibenzo[c]thiophene derivatives BBT-PhtBu2 and BBT-PhCN2 (ref. 12) and 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives BBT-PhtBu4, BBT-PhtBu2PhCN2, and BBT-PhCN4. | |
X-ray crystal structures
In our previous work,12 it was found that the crystal structure of BBT-PhtBu2 has two crystallographically independent molecules in which the dihedral angles between the two benzo[c]thiophene units are 127.61 and 126.90°, respectively (Fig. 2a and c). For 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives in this work, single-crystal X-ray structural analysis of BBT-PhtBu4 was successfully made, while unfortunately we could not obtain single crystals of BBT-PhCN4 and BBT-PhtBu2PhCN2 with sufficient size to make the X-ray structural analysis possible. A single crystal of BBT-PhtBu4 was grown from a mixed solvent of ethanol/dichloromethane. The obtained yellow plate crystal contained the CH2Cl2 solvate in the crystal packing (see the CIF file, ESI†). The crystal structure of BBT-PhtBu4 has two crystallographically independent molecules, as with the crystal structure of BBT-PhtBu2, but in which the dihedral angles between the two benzo[c]thiophene units are 66.90 and 68.41°, respectively (Fig. 2b and d). This result shows that the two benzo[c]thiophene units in BBT-PhtBu2 and BBT-PhtBu4 have an anti-clinal (ac) and a syn-clinal (sc) conformation, respectively. Thus, 1,1′,3,3′-tetrasubstituted-4,4′-BBT derivatives seem to have a sc conformation because 1,1′,3,3′-tetrakis(tert-butyldimethylsilyl)-4,4′-BBT, which has been developed in our previous work, also forms a sc conformation in the crystal structure. On the other hand, the formation of one-dimensional continuous molecular chains by the intermolecular CH⋯S hydrogen bonding interactions12 between the thiophene rings of neighboring molecules was observed in the crystal structure of BBT-PhtBu2 (Fig. S16a, ESI†). Meanwhile, the short interatomic contacts between the carbon atom of the benzo[c]thiophene unit and the chlorine atom of CH2Cl2 were observed in the crystal structure of BBT-PhtBu4 (Fig. S16b, ESI†). However, there are no short π–π contacts of less than 3.60 Å between the benzo[c]thiophene skeletons of the neighboring molecules of BBT-PhtBu2 and BBT-PhtBu4, which indicates the absence of π–π interactions between the 4,4′-BBT fluorophores.
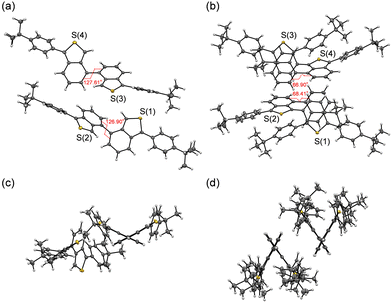 |
| | Fig. 2 Crystal structures from the (a) top view and (c) side view of BBT-PhtBu2, and the (b) top view and (d) side view of BBT-PhtBu4. The CH2Cl2 solvate in the crystal packing of BBT-PhtBu4 was omitted to view more clearly the conformation of BBT-PhtBu4. | |
Optical properties in the solution and the solid state
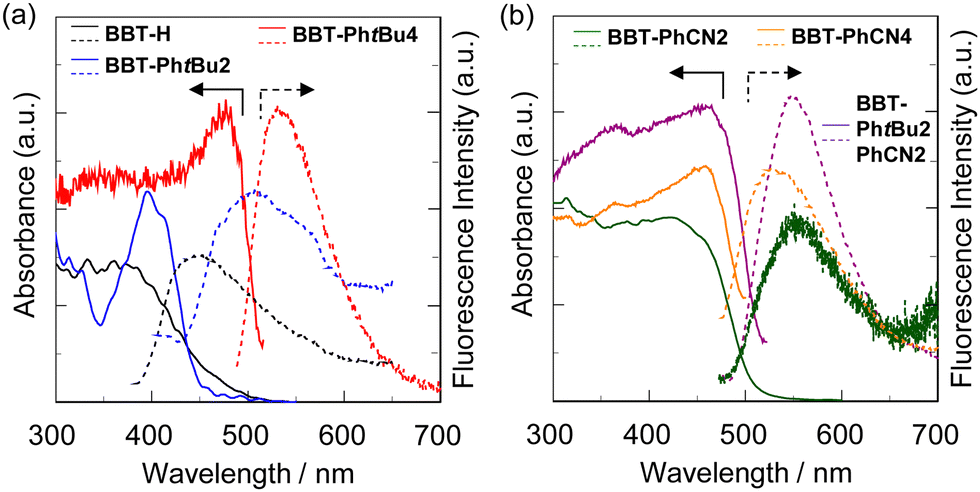
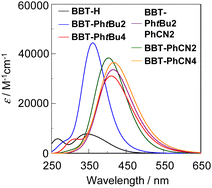
The photoabsorption and fluorescence spectra of BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, and BBT-PhCN2 in toluene, and BBT-PhCN4 in dichloromethane are shown in Fig. 3, and their photophysical data are summarized in Table 1. It was found that the 1,1′,3,3′-tetraaryl-4,4′-BBT as well as 1,1′-diaryl-4,4′-BBT show an intense photoabsorption maximum wavelength (λabsmax = 386–415 nm) with a relatively high molar extinction coefficient (εmax = 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600–27000 M−1 cm−1) in a longer wavelength region, in comparison with BBT-H (λabsmax = 359 nm, εmax = 7500 M−1 cm−1). This result is due to the perturbation to the HOMO and the LUMO by the introduction of an aryl substituent with an electron-donating or electron-withdrawing group on the benzo[c]thiophene chromophore, as discussed later in density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. Indeed, the λabsmax values of 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives BBT-PhtBu4 and BBT-PhCN4 appear in a longer wavelength region by 23 nm and 16 nm, respectively, in comparison with those of 1,1′-diaryl-4,4′-BBT derivatives BBT-PhtBu2 and BBT-PhCN2. Meanwhile, one can see that BBT-PhCN2 and BBT-PhCN4 with electron-withdrawing cyanophenyl groups exhibit λabsmax values in a longer wavelength region by 13 nm and 6 nm, respectively, in comparison with BBT-PhtBu2 and BBT-PhtBu4 with electron-donating tert-butylphenyl groups. Moreover, it was found that the λabsmax value of BBT-PhtBu2PhCN2 with both cyanophenyl groups and tert-butylphenyl groups is the same as that of BBT-PhCN4, but appear in a longer wavelength region by 16 nm, compared to that of BBT-PhtBu4. As with the case of the photoabsorption band, the corresponding fluorescence maximum wavelengths (λflmax) of the 4,4′-BBT derivatives appear in longer wavelength regions in the order of BBT-H (410 nm) < BBT-PhtBu2 (469 nm) < BBT-PhCN2 (472 nm) < BBT-PhtBu4 (519 nm) < BBT-PhCN4 (536 nm) ≈ PhtBu2PhCN2 (537 nm). Meanwhile, the fluorescence quantum yield (Φfl) decreased in the order of BBT-H (0.41) > BBT-PhtBu2 (0.39) > BBT-PhtBu4 (0.28) ≈ BBT-PhCN2 (0.27) > BBT-PhCN4 (0.11) ≈ PhtBu2PhCN2 (0.11).
600–27000 M−1 cm−1) in a longer wavelength region, in comparison with BBT-H (λabsmax = 359 nm, εmax = 7500 M−1 cm−1). This result is due to the perturbation to the HOMO and the LUMO by the introduction of an aryl substituent with an electron-donating or electron-withdrawing group on the benzo[c]thiophene chromophore, as discussed later in density functional theory (DFT) and time-dependent DFT (TDDFT) calculations. Indeed, the λabsmax values of 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives BBT-PhtBu4 and BBT-PhCN4 appear in a longer wavelength region by 23 nm and 16 nm, respectively, in comparison with those of 1,1′-diaryl-4,4′-BBT derivatives BBT-PhtBu2 and BBT-PhCN2. Meanwhile, one can see that BBT-PhCN2 and BBT-PhCN4 with electron-withdrawing cyanophenyl groups exhibit λabsmax values in a longer wavelength region by 13 nm and 6 nm, respectively, in comparison with BBT-PhtBu2 and BBT-PhtBu4 with electron-donating tert-butylphenyl groups. Moreover, it was found that the λabsmax value of BBT-PhtBu2PhCN2 with both cyanophenyl groups and tert-butylphenyl groups is the same as that of BBT-PhCN4, but appear in a longer wavelength region by 16 nm, compared to that of BBT-PhtBu4. As with the case of the photoabsorption band, the corresponding fluorescence maximum wavelengths (λflmax) of the 4,4′-BBT derivatives appear in longer wavelength regions in the order of BBT-H (410 nm) < BBT-PhtBu2 (469 nm) < BBT-PhCN2 (472 nm) < BBT-PhtBu4 (519 nm) < BBT-PhCN4 (536 nm) ≈ PhtBu2PhCN2 (537 nm). Meanwhile, the fluorescence quantum yield (Φfl) decreased in the order of BBT-H (0.41) > BBT-PhtBu2 (0.39) > BBT-PhtBu4 (0.28) ≈ BBT-PhCN2 (0.27) > BBT-PhCN4 (0.11) ≈ PhtBu2PhCN2 (0.11).
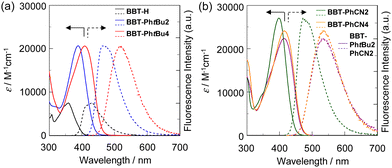 |
| | Fig. 3 (a) Photoabsorption (solid line) and fluorescence (dotted line) spectra (λex = λabsmax) of BBT-H, BBT-PhtBu2, and BBT-PhtBu4 (3.0 × 10−5 M) in toluene. (b) Photoabsorption (solid line) and fluorescence (dotted line) spectra (λex = λabsmax) of BBT-PhtBu2PhCN2 and BBT-PhCN2 (ca. 3.0 × 10−5 M) in toluene, and BBT-PhCN4 (3.0 × 10−5 M) in dichloromethane. | |
Table 1 Photophysical and electrochemical data and HOMO and LUMO energy levels of 4,4′-bibenzo[c]thiophene derivatives in solution
| Dye |
λ
absmax/nm (εmax/M−1 cm−1) |
λ
flmax/nm (Φfl)c |
τ
fl
/ns |
k
r
/s−1 |
k
nr
/s−1 |
k
nr
/k
r
|
E
oxonset
/V |
E
optg
/eV |
HOMOi/eV |
LUMOj/eV |
|
In toluene.
In dichloromethane.
Fluorescence quantum yields (Φfl) were determined by using a calibrated integrating sphere system (λex = 359 nm, 386 nm, 409 nm, 415 nm, 399 nm, and 415 nm for BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4, respectively).
Fluorescence lifetime.
Radiative rate constant (kr = Φfl/τfl).
Nonradiative rate constant (knr = (1 − Φfl)/τfl).
Onset (Eoxonset) versus Fc/Fc+ of the oxidation potential.
Optical energy gaps (Eoptg) were determined from the intersection (393 nm, 432 nm, 459 nm, 466 nm, 439 nm, and 468 nm for BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4, respectively) of the photoabsorption and fluorescence spectra in toluene.
Versus vacuum level.
Previous work (ref. 12).
|
|
BBT-H
|
359 (7500)a |
410 (0.41)a |
3.46a |
1.18 × 108a |
1.70 × 108a |
1.44a |
0.75a |
3.16a |
−5.55 |
−2.39 |
|
BBT-PhtBu2
|
386 (20 700)a |
469 (0.39)a |
2.33a |
1.68 × 108a |
2.62 × 108a |
1.56a |
0.42a |
2.87a |
−5.22 |
−2.35 |
|
BBT-PhtBu4
|
409 (20 600)a |
519 (0.28)a |
2.14a |
1.31 × 108a |
3.36 × 108a |
2.56a |
0.26a |
2.70a |
−5.06 |
−2.36 |
|
BBT-PhtBu2PhCN2
|
415 (22 700)a |
537 (0.11)a |
1.34a |
8.21 × 107a |
6.64 × 108a |
8.09a |
0.54a |
2.66a |
−5.34 |
−2.68 |
|
BBT-PhCN2
|
399 (27 000)a |
472 (0.27)a |
1.27a |
2.13 × 108a |
5.75 × 108a |
2.70a |
0.59a |
2.82a |
−5.39 |
−2.57 |
|
BBT-PhCN4
|
415 (24 100)b |
536 (0.11)b |
1.88b |
5.85 × 107b |
4.73 × 108b |
8.09b |
0.70b |
2.65b |
−5.50 |
−2.85 |
Thus, in order to understand the difference in Φfl values between the 4,4′-BBT derivatives in the solutions, we performed time-resolved fluorescence spectroscopy. It was found that the fluorescence lifetimes (τfl = 1.27–1.88 ns) of PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are shorter than those (2.14–3.46 ns) of BBT-H, BBT-PhtBu2, and BBT-PhtBu4. The radiative rate constant (kr = 2.13 × 108 s−1) of BBT-PhCN2 is somewhat larger than those (1.18–1.63 × 108 s−1) of BBT-H, BBT-PhtBu2, and BBT-PhtBu4, meanwhile kr values (5.85–8.21 × 107 s−1) for PhtBu2PhCN2 and BBT-PhCN4 are about one-half those of BBT-H and BBT-PhtBu2. On the other hand, the nonradiative rate constants (knr = 4.73–6.64 × 108 s−1) of PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are two to four times larger than those (1.70–3.36 × 108 s−1) of BBT-H, BBT-PhtBu2, and BBT-PhtBu4. Consequently, the ratios of the nonradiative constant to the radiative constant (knr/kr = 8.09, 2.70, and 8.09) of PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are larger than those (1.44, 1.56, and 2.56) of BBT-H, BBT-PhtBu2, and BBT-PhtBu4, indicating that the lower Φfl values of PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are mainly due to the larger knr value, compared to the Φfl values of BBT-H, BBT-PhtBu2, and BBT-PhtBu4. Therefore, this result provides a useful insight into the photophysical properties of 4,4′-BBT derivatives that increasing the number of phenyl substituents with electron-donating or electron-withdrawing groups on the thiophene ring leads to a bathochromic shift of λabsmax and λflmax and lowering of the Φfl value. In addition, the increase in knr values of PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 may be induced by rotation between electron-withdrawing cyanophenyl groups and the 4,4′-BBT fluorophore, leading to excited-state intramolecular charge transfer (ICT)-based fluorescence quenching.15
The solid-state photophysical properties of the as-recrystallized 4,4′-BBT derivatives have been investigated by solid-state UV-Vis diffuse reflection–photoabsorption and fluorescence spectral measurement (Fig. 4), and their photophysical data are summarized in Table 2. The photoabsorption spectrum of BBT-PhtBu2 in the solid state shows a photoabsorption maximum wavelength (λabs-solidmax) at 400 nm which is similar to that in the solution. On the other hand, the photoabsorption bands of BBT-H, BBT-PhtBu4, PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 in the solid state are broadened in a longer wavelength region. In particular, the λabs-solidmax values of BBT-PhtBu4, PhtBu2PhCN2, and BBT-PhCN4 showed significant bathochromic shifts by 68 nm, 50 nm, and 44 nm, respectively, compared to those in the solution. Furthermore, the corresponding solid-state fluorescence spectra revealed that BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, and BBT-PhCN2 except BBT-PhCN4 show a fluorescence maximum wavelength (λfl-solidmax) in a longer wavelength region by 45 nm, 41 nm, 12 nm, 10 nm, and 76 nm, respectively, compared to those in solution. The Φfl-solid values of BBT-H, BBT-PhtBu4, PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 in the solid state are <0.02, 0.04, 0.09, 0.04, <0.02, and <0.02, respectively, which are significantly lower than those in solution (Table 1). Thus, the precise evaluations of the τfl-solid values for the 4,4′-BBT derivatives were difficult due to their feeble solid-state fluorescence properties. Consequently, for these 4,4′-BBT derivatives, the bathochromic shifts of λabsmax and λflmax and the lowering of the Φfl value by changing from the solution to the solid state could be attributed to the formation of intermolecular π–π interactions between the 4,4′-BBT fluorophores, although it was not observed in the crystal structures of BBT-PhtBu2 and BBT-PhtBu4, continuous intermolecular CH⋯S hydrogen bonding interactions between the fluorophores for BBT-PhtBu2, and/or the short interatomic contacts between the 4,4′-BBT fluorophore and CH2Cl2 solvate for BBT-PhtBu4 in the solid state and consequent delocalization of excitons or excimers.16
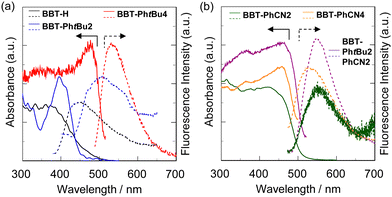 |
| | Fig. 4 Solid-state UV-vis diffuse reflection–absorption (solid line) and fluorescence (dotted line) spectra (λex = 360 nm for BBT-H and BBT-PhtBu2, 477 nm for BBT-PhtBu4, 467 nm for BBT-PhtBu2PhCN2, 422 nm for BBT-PhCN2, and 459 nm for BBT-PhCN4) of (a) the as-recrystallized BBT-H, BBT-PhtBu2, BBT-PhtBu4, (b) BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4. | |
Table 2 Photophysical data of 4,4′-bibenzo[c]thiophene derivatives in the solid state
| Dye |
λ
abs-solidmax/nm |
λ
fl-solidmax/nm (Φfl-solid)a |
|
Fluorescence quantum yields (Φfl-solid) were determined by using a calibrated integrating sphere system (λex = 360 nm for BBT-H and BBT-PhtBu2, 477 nm for BBT-PhtBu4, 467 nm for BBT-PhtBu2PhCN2, 422 nm for BBT-PhCN2, and 459 nm for BBT-PhCN4).
Previous work (ref. 12).
|
|
BBT-H
|
360 |
455 (<0.02) |
|
BBT-PhtBu2
|
400 |
510 (0.04) |
|
BBT-PhtBu4
|
477 |
531 (0.09) |
|
BBT-PhtBu2PhCN2
|
465 |
547 (0.04) |
|
BBT-PhCN2
|
422 |
548 (<0.02) |
|
BBT-PhCN4
|
459 |
527 (<0.02) |
Electrochemical properties
The electrochemical properties of BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 were investigated using cyclic voltammetry (CV) in acetonitrile, dichloromethane or DMF containing 0.1 M tetrabutylammonium perchlorate (Bu4NClO4). The potentials were internally referenced to ferrocene/ferrocenium (Fc/Fc+). The cyclic voltammograms of the 4,4′-BBT derivatives are shown in Fig. 5, and their electrochemical data and the estimated HOMO and LUMO energy levels are summarized in Table 1. The cyclic voltammograms of the 4,4′-BBT derivatives showed an irreversible oxidation wave at 0.88 V for BBT-H, 0.55 V for BBT-PhtBu2, 0.52 V for BBT-PhtBu4, 0.76 V for BBT-PhtBu2PhCN2, 0.73 V for BBT-PhCN2, and 0.83 V for BBT-PhCN4, while any obvious reduction wave did not appear within the potential window (−1.5 to 0 V versus Fc/Fc+). Thus, the oxidation waves of BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are cathodically shifted by 0.33 V, 0.36 V, 0.12 V, 0.15 V, and 0.05 V, respectively, compared to that of BBT-H. Indeed, this result indicates that the introduction of the electron-donating tert-butylphenyl group and/or the electron-withdrawing cyanophenyl group into the thiophene ring leads to the lowering of the oxidation potential. In addition, an electron-donating phenyl substituent is more effective than an electron-withdrawing phenyl substituent in the lowering of the oxidation potential.
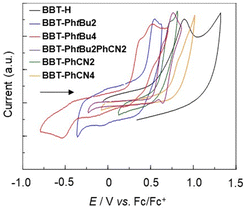 |
| | Fig. 5 Cyclic voltammograms of BBT-H and BBT-PhtBu2 in acetonitrile containing 0.1 M Bu4NClO4, BBT-PhtBu2 and BBT-PhtBu2PhCN2 in dichloromethane containing 0.1 M Bu4NClO4, and BBT-PhCN2 and BBT-PhCN4 in DMF containing 0.1 M Bu4NClO4 at a scan rate of 100 mV s−1. The arrow denotes the direction of the potential scan. | |
The HOMO energy levels (−[Eoxonset + 4.8] eV) versus vacuum level were estimated from the onset potentials (Eoxonset) of the oxidation waves, and the LUMO energy levels were estimated from the Eoxonset and the intersections (optical energy gap: Eoptg) of the photoabsorption and fluorescence spectra in solution (Table 1). The HOMO energy levels rise in the order of BBT-H (−5.55 eV) < BBT-PhCN4 (−5.50 eV) < BBT-PhCN2 (−5.39 eV) < BBT-PhtBu2PhCN2 (−5.34 eV) < BBT-PhtBu2 (−5.22 eV) < BBT-PhtBu4 (−5.06 eV), so that the HOMO energy levels of BBT-PhtBu2 and BBT-PhtBu4 are significantly higher than those of the other 4,4′-BBT derivatives. On the other hand, the LUMO energy levels lower in the order of BBT-PhtBu2 (−2.35 eV) ≈ BBT-PhtBu4 (−2.36 eV) > BBT-H (−2.39 eV) > BBT-PhCN2 (−2.57 eV) > BBT-PhtBu2PhCN2 (−2.68 eV) > BBT-PhCN4 (−2.85 eV). Thus, this result indicates that the LUMO energy levels of BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are much lower than those of BBT-H, BBT-PhtBu2, and BBT-PhtBu4, although the LUMO energy levels of BBT-H, BBT-PhtBu2, and BBT-PhtBu4 are similar to each other. The fact reveals that the bathochromic shift of the photoabsorption band from BBT-H to BBT-PhtBu2 and BBT-PhtBu4 is mainly attributed to the destabilization of the HOMO energy level through the introduction of electron-donating tert-butylphenyl groups into the benzo[c]thiophene skeleton, leading to a decrease in the HOMO–LUMO band gap. In contrast, the HOMO and LUMO energy levels of BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are somewhat higher and significantly lower, respectively, than those of BBT-H. Consequently, the bathochromic shift of the photoabsorption band from BBT-H to BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 is mainly attributed to the stabilization of the LUMO energy level through the introduction of the electron-withdrawing cyanophenyl group into the benzo[c]thiophene skeleton, resulting in a decrease in the HOMO–LUMO band gap. Therefore, it was found that increasing the number of electron-donating or electron-withdrawing phenyl groups on the thiophene ring results in the increase of the HOMO energy level or the lowering of the LUMO energy level, respectively, so this result provides a useful method to precisely adjust the HOMO and LUMO energy levels of 4,4′-BBT derivatives.
Theoretical calculations
The electronic structures and molecular orbitals of BBT-H, BBT-PhtBu2, BBT-PhtBu4, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 were derived from DFT calculations at the B3LYP/6-31G(d,p) level17 (Fig. 6). The DFT calculations demonstrated that the HOMO of BBT-H is delocalized on each benzo[c]thiophene unit, but the HOMOs of the other 4,4′-BBT derivatives are mainly delocalized on each benzo[c]thiophene unit, although the HOMOs of two benzo[c]thiophenes are practically independent because the HOMO−1 energy level is very close to the HOMO energy level (Fig. S18, ESI†). The LUMOs of BBT-H, BBT-PhtBu2, and BBT-PhtBu4 are mainly delocalized over the whole benzo[c]thiophene skeletons through 4,4′-positions. On the other hand, it is worth mentioning here that the LUMOs of BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 are delocalized over the whole benzo[c]thiophene skeletons containing the cyanophenyl groups through 4,4′-positions. It was found that the HOMO energy levels of BBT-PhtBu2 and BBT-PhtBu4 are higher than that of BBT-H, but their LUMO energy levels are similar to each other. The HOMO and LUMO energy levels of BBT-PhCN2 and BBT-PhCN4 are significantly lower than those of BBT-H, but the lowering of the LUMO energy levels is larger than that of the HOMO energy levels, although the CV showed that the HOMO energy levels of BBT-PhCN2 and BBT-PhCN4 are higher than that of BBT-H. Meanwhile, the HOMO energy level of BBT-PhtBu2PhCN2 is nearly equal to that of BBT-H, but its LUMO energy level is much lower than that of BBT-H. Consequently, the HOMO–LUMO band gaps decrease in the order of BBT-H (3.74 eV) > BBT-PhtBu2 (3.56 eV) > BBT-PhCN2 (3.32 eV) > BBT-PhtBu4 (3.19 eV) > BBT-PhtBu2PhCN2 (3.12 eV) ≈ BBT-PhCN4 (3.10 eV). Thus, the DFT calculation suggested that compared to BBT-H, the increase of the HOMO energy level for BBT-PhtBu2 and BBT-PhtBu4 and the lowering of the LUMO energy level for BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 result in a decrease in the HOMO–LUMO band gap from BBT-H to the 1,1′-diaryl-4,4′-BBT and 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives, as in the case of experimental results.
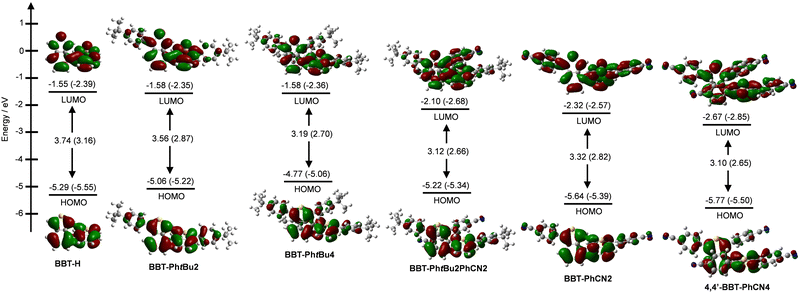 |
| | Fig. 6 Energy level diagram, HOMO and LUMO of BBT-H, BBT-PhtBu2, BBT-PhtBu2, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 derived from DFT calculations at the B3LYP/6-31G(d,p) level. For all the 4,4′-BBT derivatives the optimized geometries are a syn-clinal (sc) conformation. Numbers in parentheses are the experimental values (Table 1). | |
Furthermore, the TDDFT calculations were performed to elucidate the photophysical properties of the 4,4′-BBT derivatives (Fig. 7). The calculated λabs-calcdmax and εcalcd values of the 4,4′-BBT derivatives are 345 nm and 7800 M−1 cm−1 for BBT-H, 361 nm and 44300 M−1 cm−1 for BBT-PhtBu2, 410 nm and 31100 M−1 cm−1 for BBT-PhtBu4, 416 nm and 33600 M−1 cm−1 for BBT-PhtBu2PhCN2, 403 nm and 38300 M−1 cm−1 for BBT-PhCN2, and 417 nm and 36500 M−1 cm−1 for BBT-PhCN2. The S0 → S1 transitions are mainly attributed to the transitions from the HOMO to the LUMO (67% for BBT-H, 88% for BBT-PhtBu2, 93% for BBT-PhtBu4, 88% for BBT-PhtBu2PhCN2, 93% for BBT-PhCN2, and 90% for BBT-PhCN4). Indeed, the TD-DFT calculations are in good agreement with the experimental results of the bathochromic shift of the photoabsorption band from BBT-H to BBT-PhtBu2, BBT-PhCN2, BBT-PhtBu4, BBT-PhtBu2PhCN4, and BBT-PhCN4, although there are differences in the εcalcd values between the experimental and TD-DFT calculation results.
 |
| | Fig. 7 Photoabsorption spectra of BBT-H, BBT-PhtBu2, BBT-PhtBu2, BBT-PhtBu2PhCN2, BBT-PhCN2, and BBT-PhCN4 derived from TD-DFT calculations. | |
Experimental
General
Melting points were measured with an AS ONE ATM-02. IR spectra were recorded using a SHIMADZU IRTracer-100 by the ATR method. 1H NMR spectra were recorded using a Varian-400 FT NMR spectrometer. High-resolution mass spectral data by APCI was acquired using a Thermo Fisher Scientific LTQ Orbitrap XL. Recycling gel permeation chromatography (GPC) was performed using an RI-detector (SHIMADZU RID-20A), a UV-detector (SHIMADZU SPD-20A), and a pump (SHIMADZU LC-20A) with two columns (Shodex GPC FP-2002). Photoabsorption spectra of solutions were recorded with a Shimadzu UV-3600 plus spectrophotometer. Photoabsorption spectra of solids were recorded using a Shimadzu UV-3600 plus spectrophotometer with a calibrated integrating sphere system. Fluorescence spectra of solutions and solids were recorded with a HORIBA FluoroMax-4 spectrofluorometer. The fluorescence quantum yields in solution and in the solid state were determined using a HORIBA FluoroMax-4 spectrofluorometer with a calibrated integrating sphere system. Fluorescence decay measurements were performed with a HORIBA DeltaFlex modular fluorescence lifetime system, using a Nano-LED pulsed diode excitation source (370 nm). Cyclic voltammetry (CV) curves were recorded in an acetonitrile/Bu4NClO4 (0.1 M), dichloromethane/Bu4NClO4 (0.1 M), or DMF/Bu4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ (AgNO3 in acetonitrile/Bu4NClO4, dichloromethane/Bu4NClO4, or DMF/Bu4NClO4) as the reference electrode, a Pt plate as the working electrode and Pt wire as the counter electrode using an Electrochemical Measurement System HZ-7000 (HOKUTO DENKO).
Synthesis
1,1′,3,3′-Tetra(trimethylstannyl)-4,4′-bibenzo[c]thiophene (BBT-Sn4).
To a THF solution (0.43 ml) of BBT-Sn2 (0.10 g, 0.17 mmol) under a nitrogen atmosphere at 0 °C was added dropwise a 1.0 M hexane/THF solution of lithium diisopropylamide (1.0 ml, 1.0 mmol). After stirring for 3.5 h, a 1.0 M hexane solution of trimethyltin chloride (1.0 ml, 1.0 mmol) was added dropwise. The reaction mixture was further stirred for 19 h at room temperature. The reaction mixture was quenched with water, and then, the solution was extracted with ethyl acetate. The ethyl acetate extract was dried over anhydrous MgSO4, filtered and concentrated to give BBT-Sn4 (0.10 g, yield 66%) as a brown solid; m.p. 188–189 °C; FT-IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 2959, 2918, 2853, 1508, 1470 cm−1; 1H NMR (400 MHz, acetone-d6): δ = −0.21 (s, 18H), 0.54 (s, 18H), 7.36 (d, J = 5.4 Hz, 2H), 7.58 (dd, J = 6.4 and 8.7 Hz, 2H), 8.18 (d, J = 7.6 Hz, 2H) ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C28H42S2Sn4, 922.88886; found 922.89098.
= 2959, 2918, 2853, 1508, 1470 cm−1; 1H NMR (400 MHz, acetone-d6): δ = −0.21 (s, 18H), 0.54 (s, 18H), 7.36 (d, J = 5.4 Hz, 2H), 7.58 (dd, J = 6.4 and 8.7 Hz, 2H), 8.18 (d, J = 7.6 Hz, 2H) ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C28H42S2Sn4, 922.88886; found 922.89098.
1,1′,3,3′-Tetra(4-(tert-buthyl)phenyl)-4,4′-bibenzo[c]thiophene (BBT-PhtBu4).
A solution of BBT-Sn4 (0.16 g, 0.17 mmol), 1-tert-butyl-4-iodobenzene (0.30 ml, 1.7 mmol), CsF (0.15 g, 1.0 mmol), CuI (0.0065 g, 0.034 mmol), and Pd(PPh3)4 (0.020 g, 0.017 mmol) in DMF (35 ml) was stirred for 20 h at 50 °C under a nitrogen atmosphere. After vacuum filtration of the reaction mixture, the solution was extracted with dichloromethane. The dichloromethane extract was dried over anhydrous MgSO4, filtered and concentrated. Recycling GPC (CHCl3 as the eluent) for the residue was performed to give BBT-PhtBu4 (0.024 g, yield 17%) as a yellow solid; m.p. 230–231 °C; FT-IR (ATR): = 2959, 2901, 2866, 1520, 1458, 1362, 1267 cm−1; 1H NMR (400 MHz, acetone-d6): δ = 0.98 (s, 18H), 1.39 (s, 18H), 6.75 (d, J = 8.1 Hz, 4H), 6.80 (d, J = 8.7 Hz, 4H), 7.10 (dd, J = 6.5 and 8.9 Hz, 2H), 7.19 (d, J = 5.5 Hz, 2H), 7.49 (d, J = 8.9 Hz, 2H), 7.52 (d, J = 8.7 Hz, 4H), 7.56 (d, J = 8.7 Hz, 4H) ppm; it was difficult to obtain the 13C NMR spectrum due to the low solubility of BBT-PhtBu4 in any deuterated solvents; HRMS (APCI): m/z (%): [M + H+] calcd for C56H58S2, 795.40527; found 795.40717.
1,1′,3,3′-Tetra(4-cyanophenyl)-4,4′-bibenzo[c]thiophene (BBT-PhCN4).
A solution of BBT-Sn4 (0.18 g, 0.19 mmol), 4-iodobenzonitrile (0.44 g, 1.9 mmol), CsF (0.17 g, 1.1 mmol), CuI (0.0072 g, 0.038 mmol), and Pd(PPh3)4 (0.022 g, 0.019 mmol) in DMF (40 ml) was stirred for 20 h at 50 °C under a nitrogen atmosphere. After vacuum filtration of the reaction mixture, the solution was extracted with dichloromethane. The dichloromethane extract was dried over anhydrous MgSO4, filtered and concentrated. The residue was chromatographed on silica gel (dichloromethane:hexane = 3:1 as the eluent) to give BBT-PhCN4 (0.033 g, yield 25%) as a yellow solid; m.p. 300 °C or more; FT-IR (ATR): = 2922, 2222, 1599, 1510 cm−1; 1H NMR (400 MHz, CD2Cl2): δ = 6.88 (d, J = 7.8 Hz, 4H), 7.06 (d, J = 8.5 Hz, 4H), 7.27 (dd, J = 6.6 and 8.9 Hz, 2H), 7.40 (d, J = 5.8 Hz, 2H), 7.59 (d, J = 8.9 Hz, 2H), 7.65 (d, J = 8.5 Hz, 4H), 7.83 (d, J = 8.6 Hz, 4H) ppm; it was difficult to obtain the 13C NMR spectrum due to the low solubility of BBT-PhCN4 in any deuterated solvents; HRMS (APCI): m/z (%): [M+] calcd for C44H22N4S2, 670.12804; found 670.12933.
1,1′-Bis(4-(tert-butyl)phenyl)-[4,4′-bibenzo[c]thiophene]-3,3′-bis(trimethylstannyl) (BBT-PhtBu2Sn2).
To a THF solution (0.49 ml) of BBT-PhtBu2 (0.12 g, 0.22 mmol) under a nitrogen atmosphere at 0 °C was added dropwise a 1.0 M hexane/THF solution of lithium diisopropylamide (1.3 ml, 1.3 mmol). After stirring for 3.5 h, a 1.0 M hexane solution of trimethyltin chloride (1.3 ml, 1.3 mmol) was added dropwise. The reaction mixture was further stirred for 15 h at room temperature. The reaction mixture was quenched with water, and then, the solution was extracted with ethyl acetate. The ethyl acetate extract was dried over anhydrous MgSO4, filtered, and concentrated to give BBT-PhtBu2Sn2 (0.14 g, yield 73%) as a brown solid; m.p. 197–198 °C; FT-IR (ATR): = 2961, 2866, 1605, 1508, 1458, 1395, 1362 cm−1; 1H NMR (400 MHz, acetone-d6): δ = −0.10 (s, 18H), 1.41 (s, 18H), 7.07 (d, J = 6.4 Hz, 2H), 7.27 (dd, J = 6.4 and 8.8 Hz, 2H), 7.64 (d, J = 8.7 Hz, 4H), 7.68 (d, J = 8.6 Hz, 4H), 7.98 (d, J = 8.9 Hz, 2H) ppm; HRMS (APCI): m/z (%): [M + H+] calcd for C42H50S2Sn2, 859.14706; found 859.14829.
1,1′-Bis(4-(tert-butyl)phenyl)-[4,4′-bibenzo[c]thiophene]-3,3′-bis(4-cyanophenyl) (BBT-PhtBu2PhCN2).
A solution of BBT-PhtBu2Sn2 (0.081 g, 0.095 mmol), 4-iodobenzonitrile (0.11 g, 0.48 mmol), CsF (0.044 g, 0.29 mmol), CuI (0.0036 g, 0.019 mmol), and Pd(PPh3)4 (0.011 g, 0.0095 mmol) in DMF (35 ml) was stirred for 23 h at 50 °C under a nitrogen atmosphere. After vacuum filtration of the reaction mixture, the solution was extracted with dichloromethane. The dichloromethane extract was dried over anhydrous MgSO4, filtered and concentrated. The residue was chromatographed on silica gel (dichloromethane:hexane = 1:1 as the eluent) to give BBT-PhtBu2PhCN2 (0.012 g, yield 17%) as a yellow solid; m.p. 300 °C or more; FT-IR (ATR): = 2961, 2866, 2224, 1601, 1460, 1364 cm−1; 1H NMR (400 MHz, CD2Cl2): δ = 1.40 (s, 18H), 6.87 (d, J = 8.0 Hz, 4H), 7.06 (d, J = 8.5 Hz, 4H), 7.16 (dd, J = 6.6 and 8.9 Hz, 2H), 7.34 (d, J = 6.4 Hz, 2H), 7.45 (d, J = 8.3 Hz, 4H), 7.55–7.58 (m, 6H) ppm; it was difficult to obtain the 13C NMR spectrum due to the low solubility of BBT-PhtBu2PhCN2 in any deuterated solvents; HRMS (APCI): m/z (%): [M + H+] calcd for C50H40N2S2, 733.27057; found 733.27228.
X-ray crystallographic analysis
The reflection data of BBT-PhtBu4 were collected at 100 K on a Rigaku XtaLAB Synergy-R/DW diffractometer using monochromated Mo-Kα (λ = 0.71073 Å). The structure was solved by the SHELXT 2014/5 method and refined based on full-matrix least squares on F2 using SHELXL-2018/3. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were fixed geometrically and not refined. Crystallographic data have been deposited in the Cambridge Crystallographic Data Centre (CCDC 2337948†).
Crystal of BBT-PhtBu4.
A single crystal of BBT-PhtBu4 was recrystallized from a mixed solvent of ethanol/dichloromethane as a yellow plate crystal, and was air stable. Crystallographic data: C57H60Cl2S2, M = 880.07, triclinic, a = 14.4868(3), b = 15.5221(5), c = 22.5994(6) Å, α = 75.521(2)°, β = 80.754(2)°, γ = 84.376(2)°, V = 4847.6(2) Å3, Dcalcd = 1.206 g cm−3, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), Z = 4, 22621 reflections measured, 23124 unique (Rint = 0.0521), which were used in all calculations. The final R1(reflections) = 0.0623 (16714) [I > 2σ(I)], wR2(reflections) = 0.1634 (23124). GOF = 1.015.
(no. 2), Z = 4, 22621 reflections measured, 23124 unique (Rint = 0.0521), which were used in all calculations. The final R1(reflections) = 0.0623 (16714) [I > 2σ(I)], wR2(reflections) = 0.1634 (23124). GOF = 1.015.
Conclusions
We developed 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives, BBT-PhtBu4 with four electron-donating tert-butylphenyl groups, BBT-PhCN4 with four electron-withdrawing cyanophenyl groups, and BBT-PhtBu2PhCN2 with two tert-butylphenyl groups and two cyanophenyl groups on each thiophene ring using the Stille coupling reaction with 1,1′-diaryl-3,3′-distannyl-4,4′-BBT or 1,1′,3,3′-tetrastannyl-4,4′-BBT and revealed their photophysical and electrochemical properties and X-ray crystal structures, in comparison with those of 1,1′-diaryl-4,4′-BBT derivatives BBT-PhtBu2 with a tert-butylphenyl group and BBT-PhCN2 with a cyanophenyl group on each thiophene ring. It was found that increasing the number of phenyl substituents with electron-donating or electron-withdrawing groups on the thiophene ring led to not only the bathochromic shift of λabsmax and λflmax but also the lowering of the Φfl value. The DFT calculations, as well as the cyclic voltammetry and photophysical analyses, revealed that increasing the number of electron-donating and electron-withdrawing phenyl groups on the thiophene ring results in the increase of the HOMO energy level and the lowering of the LUMO energy level, respectively, which leads to a decrease in the HOMO–LUMO band gap, that is, a bathochromic shift of the photoabsorption band. This finding offers a useful method to precisely adjust the HOMO and LUMO energy levels of 4,4′-BBT derivatives. Therefore, this work provides not only facile synthetic methods for 1,1′,3,3′-tetraaryl-4,4′-bibenzo[c]thiophene derivatives with the same or different aryl substituents on the thiophene rings but also useful methods to precisely adjust their optical and electrochemical properties. Further studies on the development of donor–π–acceptor (D–π–A)-type 1,1′,3,3′-tetraaryl-4,4′-BBT derivatives exhibiting intermolecular charge transfer (ICT) characteristics are now in progress to gain insight into the effects of electron-donating and electron-withdrawing aryl substituents on the optical and electrochemical properties of 4,4′-BBT derivatives.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number 22H02123.
Notes and references
-
(a) Y. Tan, Y. Sun, W. Huang, D. Zhu, D. Yan, D. Wang and B. Z. Tang, Luminescence, 2024, 39, e4606 CrossRef CAS PubMed;
(b) K.-W. Lee, Y. Cao, W.-C. Wei, J.-H. Tan, Y. Wan, Z. Feng, Y. Zhang, Y. Liu, X. Zheng, C. Cao, H. Chen, P. Wang, S. Li, K.-T. Wong and C.-S. Lee, Adv. Mater., 2023, 35, 2211632 CrossRef CAS PubMed;
(c) D. Yan, M. Wang, Q. Wu, N. Niu, M. Li, R. Song, J. Rao, M. Kang, Z. Zhang, F. Zhou, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e202202614 CrossRef CAS PubMed;
(d) D. Yan, W. Xie, J. Zhang, L. Wang, D. Wang and B. Z. Tang, Angew. Chem., Int. Ed., 2021, 60, 26769–26776 CrossRef CAS PubMed.
-
(a) X. Chen, D. Zhang, Y. He, M. U. Ali, Y. Wu, C. Zhao, P. Wu, C. Yan, F. Wudl and H. Meng, Mater. Chem. Front., 2020, 4, 3578–3584 RSC;
(b) K. Yamamoto, Y. Ie, M. Nitani, N. Tohnai, F. Kakiuchi, K. Zhang, W. Pisula, K. Asadi, P. W. M. Blom and Y. Aso, J. Mater. Chem. C, 2018, 6, 7493–7500 RSC;
(c) C. P. Yu, R. Kimura, T. Kurosawa, E. Fukuzaki, T. Watanabe, H. Ishii, S. Kumagai, M. Yano, J. Takeya and T. Okamoto, Org. Lett., 2019, 21, 4448–4453 CrossRef CAS PubMed.
-
(a) Y. Qin, J. Y. Kim, C. D. Frisbie and M. A. Hillmyer, Macromolecules, 2008, 41, 5563–5570 CrossRef CAS;
(b) M. R. Raj and S. Anandan, RSC Adv., 2013, 3, 14595–14608 RSC;
(c) J. D. Douglas, G. Griffini, T. W. Holcombe, E. P. Young, O. P. Lee, M. S. Chen and J. M. J. Fréchet, Macromolecules, 2012, 45, 4069–4074 CrossRef CAS.
- Y.-C. Hu, Z.-L. Lin, T.-C. Huang, J.-W. Lee, W.-C. Wei, T.-Y. Ko, C.-Y. Lo, D.-G. Chen, P.-T. Chou, W.-Y. Hung and K.-T. Wong, Mater. Chem. Front., 2020, 4, 2029–2039 RSC.
-
(a) Q. Liu, F.-T. Kong, T. Okujima, H. Yamada, S.-Y. Dai, H. Uno, N. Ono, X.-Z. You and Z. Shen, Tetrahedron Lett., 2012, 53, 3264–3267 CrossRef CAS;
(b) Q. Liu, Q.-Y. Feng, H. Yamada, Z.-S. Wang, N. Ono, X.-Z. You and Z. Shen, Chem. – Asian J., 2012, 7, 1312–1319 CrossRef CAS PubMed.
-
(a) F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382–3384 CrossRef CAS;
(b) H. Meng and F. Wudl, Macromolecules, 2001, 34, 1810–1816 CrossRef CAS;
(c) O. D. Abrbanel, J. Rozon and G. R. Hutchison, J. Phys. Chem. Lett., 2022, 13, 2158–2164 CrossRef PubMed;
(d) G. Grover, J. D. Tovar and M. Kertesz, J. Phys. Chem. C, 2022, 126, 5302–5310 CrossRef CAS;
(e) G. Grover, G. M. Peters, J. D. Tovar and M. Kertesz, Phys. Chem. Chem. Phys., 2020, 22, 11431–11439 RSC.
-
(a) S. T. Meek, E. E. Nesterov and T. M. Swager, Org. Lett., 2008, 10, 2991–2993 CrossRef CAS PubMed;
(b) K. Kawabata and H. Goto, J. Mater. Chem., 2012, 22, 23514–23524 RSC;
(c) U. Mitschke and P. Bäuerle, J. Chem. Soc., Perkin Trans. 1, 2001, 740–753 RSC;
(d) T. V. Hughes and M. P. Cava, J. Org. Chem., 1999, 64, 313–315 CrossRef CAS PubMed.
-
(a) M. Hori, T. Kataoka, H. Shimizu, J. Hongo and M. Kido, J. Chem. Soc., Perkin Trans. 1, 1989, 1611–1618 RSC;
(b) R. H. L. Kiebooms, P. J. A. Adriaensens, D. J. M. Vanderzande and J. M. J. V. Gelan, J. Org. Chem., 1997, 62, 1473–1480 CrossRef CAS;
(c) J. W. Terpstra and A. M. van Leusen, J. Org. Chem., 1986, 51, 230–238 CrossRef CAS;
(d) R. M. El-Shishtawy, K. Fukunishi and S. Miki, Tetrahedron Lett., 1995, 36, 3177–3180 CrossRef CAS.
-
(a) A. K. Mohanakrishnan and P. Amaladass, Tetrahedron Lett., 2005, 46, 4225–4229 CrossRef CAS;
(b) A. K. Mohanakrishnan, N. S. Kumar and P. Amaladass, Tetrahedron Lett., 2008, 49, 4792–4795 CrossRef CAS;
(c) J. A. Clement, P. Gunasekaran and A. K. Mohanakrishnan, Tetrahedron, 2009, 65, 4113–4123 CrossRef;
(d) N. S. Kumar and A. K. Mohanakrishnan, Tetrahedron, 2010, 66, 5660–5670 CrossRef CAS;
(e) G. G. Rajeshwaran, M. Nandakumar, R. Sureshbabu and A. K. Mohanakrishnan, Org. Lett., 2011, 13, 1270–1273 CrossRef CAS PubMed.
-
(a) Y. Okuda, M. V. Lakshmikantham and M. P. Cava, J. Org. Chem., 1991, 56, 6024–6026 CrossRef CAS;
(b) E. Aqad, M. V. Lakshmikantham and M. P. Cava, Org. Lett., 2004, 6, 3039–3041 CrossRef CAS PubMed.
-
(a) A. K. Mohanakrishnan, P. Amaladass and J. A. Clement, Tetrahedron Lett., 2007, 48, 779–784 CrossRef CAS;
(b) P. Amaladass, J. A. Clement and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2008, 3798–3810 CrossRef CAS.
- T. Higashino, Y. Hara, K. Imato, S. Akiyama, M. Ishida and Y. Ooyama, New J. Chem., 2023, 47, 9555–9559 RSC.
-
(a) K. Obayashi, T. Higashino, K. Imato and Y. Ooyama, New J. Chem., 2021, 45, 13258–13261 RSC;
(b) K. Obayashi, K. Imato, S. Aoyama, T. Enoki, S. Akiyama, M. Ishida, S. Suga, K. Mitsudo and Y. Ooyama, RSC Adv., 2021, 11, 18870–18880 RSC;
(c) K. Obayashi, S. Miho, M. Yasui, K. Imato, S. Akiyama, M. Ishida and Y. Ooyama, New J. Chem., 2021, 45, 17085–17094 RSC.
- S. P. H. Mee, V. Lee and J. E. Baldwin, Angew. Chem., Int. Ed., 2004, 43, 1132–1136 CrossRef CAS PubMed.
-
(a) D. Liese and G. Haberhauer, Isr. J. Chem., 2018, 58, 813–826 CrossRef CAS;
(b) C. Yan, Z. Guo, W. Chi, W. Fu, S. A. A. Abedi, X. Liu, H. Tian and W.-H. Zhu, Nat. Commun., 2021, 12, 3869 CrossRef CAS PubMed.
-
(a) H. Langhals, T. Potrawa, H. Nöth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 28, 478–480 CrossRef;
(b) H.-C. Yeh, W.-C. Wu, Y.-S. Wen, D.-C. Dai, J.-K. Wang and C.-T. Chen, J. Org. Chem., 2004, 69, 6455–6462 CrossRef CAS PubMed;
(c) Y. Ooyama, T. Nakamura and K. Yoshida, New J. Chem., 2005, 29, 447–456 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01 and C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Seiji
Akiyama
b,
Mio
Ishida
b and
Yousuke
Ooyama
a,
Seiji
Akiyama
b,
Mio
Ishida
b and
Yousuke
Ooyama