
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular designs with PEG groups for water-solubilization of sparsely substituted porphyrins†
Phuong-Lien Doan
Cao
a,
Zhiyuan
Wu
a,
Phattananawee
Nalaoh
 b and
Jonathan S.
Lindsey
*a
b and
Jonathan S.
Lindsey
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
bDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
First published on 5th June 2024
Abstract
Molecular designs that achieve solubility of porphyrins in aqueous media are attractive for diverse applications. The presence of 4-sulfophenyl or 4-N-methylpyridinium groups at the four meso-positions is effective, but the use of fewer substituents is desirable for custom tailoring. Here, five target porphyrins (along with selected copper or zinc chelates) were prepared bearing PEG groups to understand how distinct designs affect aqueous solubility (where “PEG” refers to an oligoethylene glycol unit). One objective was to employ only one or two pegylated meso-aryl groups so that other meso-positions would be open for synthetic elaboration while retaining a compact structure. The key design features examined include (i) 2,6- versus 3,5-dipegylated aryl groups; (ii) one versus two 2,6-dipegylated aryl groups; (iii) a nonpolar versus ionizable terminus of the PEG moiety; and (iv) length of the PEG moiety. In each case, the PEG groups were attached on a porphyrin scaffold bearing one or two bis(2-propynyloxy)aryl groups. Assessment entailed octanol–aqueous solution partitioning (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values) and aggregation over the concentration range of 0.2–200 μM. A trans-A2 free base porphyrin bearing two 2,6-dialkoxyphenyl groups equipped with methyl-terminated PEG6 groups gave ∼3:1 partitioning in water versus octanol but high overall solubility (at least 12 mM) in water alone; the analogous porphyrin with carboxylic acid-terminated PEG6 groups gave >100:1 partitioning in phosphate-buffered saline (PBS, pH 7.4). A trans-AB porphyrin bearing a single 2,6-dialkoxyphenyl group equipped with carboxylic acid-terminated PEG6 groups gave 43:1 partitioning in PBS versus octanol but was prone to self-aggregation at 20–200 μM in PBS alone. The results point to new molecular designs of potential value in the life sciences.
P values) and aggregation over the concentration range of 0.2–200 μM. A trans-A2 free base porphyrin bearing two 2,6-dialkoxyphenyl groups equipped with methyl-terminated PEG6 groups gave ∼3:1 partitioning in water versus octanol but high overall solubility (at least 12 mM) in water alone; the analogous porphyrin with carboxylic acid-terminated PEG6 groups gave >100:1 partitioning in phosphate-buffered saline (PBS, pH 7.4). A trans-AB porphyrin bearing a single 2,6-dialkoxyphenyl group equipped with carboxylic acid-terminated PEG6 groups gave 43:1 partitioning in PBS versus octanol but was prone to self-aggregation at 20–200 μM in PBS alone. The results point to new molecular designs of potential value in the life sciences.
Introduction
The design and synthesis of water-soluble tetrapyrrole macrocycles have been of longstanding interest.1–4 The chief challenge is to attach groups at the perimeter of the hydrophobic, tetrapyrrole macrocycle so that the entire structure can be dissolved in aqueous solution. The tetrapyrrole macrocycle is disk-shaped and essentially planar, where the core free base porphine macrocycle, lacking any peripheral substituents, has formula C20N4H14. Early designs have relied on incorporation of polar groups at the meso-positions of porphyrins. Indeed, meso-tetrakis(4-sulfophenyl)porphyrin (TPS-por)5 and the meso-tetrakis(4-N-methylpyridinium)porphyrin (TPyr-por)6 are water soluble and remain in common use (Chart 1). These designs have attractive features, but the occupancy of all four meso-positions leaves diminished opportunities for convenient synthetic elaboration. The uroporphyrins, derived from biosynthesis of tetrapyrrole macrocycles, are water soluble but have been little used for synthetic elaboration or physicochemical studies.7 | ||
| Chart 1 Water-soluble porphyrins. | ||
We have pursued several design features over the years in an effort to create compact, water-soluble, bioconjugatable porphyrins. The features include (1) use of a trans-AB porphyrin architecture, with A and B substituents at the 5- and 15-positions of the macrocycle and no substituents at any other position, (2) emphasis on substituent types that cause polar groups to project above and below the face of the hydrophobic porphyrin, and ideally, (3) reliance on a “single-junction water-solubilization unit”.8 The work has been intermittent as objectives and synthetic methods have evolved, key examples of which are shown in Chart 2.9
 | ||
| Chart 2 Representative prior trans-AB porphyrins.9 | ||
• The first design contained a compact, swallowtail substituent, which is a symmetrically branched alkyl group; the preferred conformation contains the C–H unit in the plane of the macrocycle and the two alkyl groups project above and below the plane of the porphyrin.10 The porphyrins (e.g., Por-A) appeared reasonably soluble in aqueous solution, with the key drawback, however, of the rather lengthy synthesis of the swallowtail units.11
• The second design relied on a 2,4,6-trialkoxyaryl unit, which was attractive given the inexpensive phloroglucinaldehyde (2,4,6-trihydroxybenzaldehyde) and ease of synthesis. The earliest set of porphyrins (e.g., Por-B) exhibited broadened absorption spectra both in aqueous as well as organic media.12
• The third design extended the 2,4,6-trialkoxyaryl unit to include PEG groups (e.g., Por-C).8,9 The resulting porphyrin was water-soluble but in ensuing years uncertainty arose as to how much solubilization was provided by the carboxylic acid in the bioconjugation motif versus the three neutral PEG groups.
The use of PEG groups to impart aqueous solubility of hydrophobic compounds has become widespread across the molecular sciences.13,14 PEG groups have long been used in the pharmaceutical industry15–17 to increase aqueous solubility of hydrophobic drugs and to prolong the systemic circulation of rapidly excreted drugs. A striking feature of PEG groups is solubility in both aqueous and organic media.13 The origin of the solubility in water is not merely due to the preponderance of oxygen atoms, as the polymer with twice the number of oxygen atoms (repeating unit –CH2O–) is not soluble in water. The solubility likely stems from the preferred gauche conformation of the –OCH2CH2O– moiety and all of the consequences therefrom including local dipole moments, hydrogen-bonding patterns, and kinked or partially coiled conformations.18 The PEG groups rarely achieve the fully extended conformation as are shown (for clarity) in Chart 2. The synthetic installation of PEG groups has been aided by the advent of heterotelechelic, monodisperse PEG reagents,15,17,19,20 which are available from commercial sources.
In this paper, the synthesis and characterization of a dozen porphyrins are described. The motivation for the work has been to revisit and extend studies over the past two decades concerning the role of substitution patterns and substituent composition in engendering aqueous solubility. The issues addressed include the advantages and disadvantages of the following: (i) the pattern of 2,6- versus 3,5-dipegylated aryl groups; (ii) the terminus of the PEG moiety (methyl versus carboxylic acid); (iii) one versus two 2,6-dipegylated aryl groups; and (iv) a very short versus intermediate length PEG moiety. Zinc and copper chelates also were prepared given utility in photochemistry21,22 and radiotherapeutics,23 respectively. The attributes were assessed by measurements of aqueous–organic partitioning and concentration-dependent self-aggregation in aqueous solution.
Results
Synthesis
 | ||
| Scheme 1 Pegylation of a meta-aryl substituted trans-A2 porphyrin. | ||
A facially encumbered porphyrin was prepared by a MacDonald-type condensation27–31 (Scheme 2). Thus, condensation of 2,6-bis(prop-2-yn-1-yloxy)benzaldehyde (3)32 and dipyrromethane (4)33 was carried out upon catalysis by BF3·O(Et)2 followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and subsequent neutralization of the Lewis acid with triethylamine (TEA). In this manner, the trans-A2 porphyrin 5,15-bis(2,6-bis(2-propynyloxy)phenyl)porphyrin (5) was obtained in 21% yield. Porphyrin 5 was metalated with Zn(OAc)2·2H2O in refluxing N,N-dimethylformamide (DMF)34 to give the zinc chelate Zn5.
 | ||
| Scheme 2 Synthesis of a facially encumbered trans-A2 porphyrin scaffold. | ||
 | ||
| Scheme 3 Synthesis of trans-A2 pegylated porphyrins. | ||
Analogues of porphyrins 6, Zn6, and Cu6 were prepared wherein each PEG group is terminated with an ionizable group. The carboxylic acid was chosen given ionization as the carboxylate at physiological pH as well as commercial availability of the required PEG synthons. Thus, the click reaction of porphyrin Zn5 with azido-PEG6-CO2t-Bu (II) gave Zn7 in 81% yield (Scheme 3). Treatment of the latter with TFA caused removal of the tert-butyl protecting groups and the zinc chelate, affording the free base porphyrin bearing four carboxylic acids (8) in 58% yield. Finally, exposure to copper acetate gave the copper porphyrin Cu8 in 62% yield. Zinc porphyrin Zn8 was prepared in small scale for comparative studies by zincation of free base porphyrin 8.
 | ||
| Scheme 4 Synthesis of trans-AB pegylated porphyrins. | ||
Characterization
The porphyrins were typically characterized by absorption spectroscopy, 1H NMR and 13C{1H} NMR spectroscopy, and mass spectrometry. The absorption spectra of trans-A2 and trans-AB free base porphyrins closely resemble those of well-known A4 free base porphyrins, with a strong B (Soret) band and a progression of bands in the visible region typically in a phyllo35 pattern. The zinc and copper chelates exhibit absorption spectra, particularly in the visible region, that are distinct from the parent free base porphyrin and also distinct from each other. For 6, for example, the most intense visible band is found at 503 nm versus that of Cu6 at 531 nm and of Zn6 at 545 nm. The 1H NMR spectrum of each free base or zinc porphyrin exhibited the characteristic resonance of the two meso-protons at δ ∼ 10.12 ppm, and for the free base porphyrins, the NH resonances near δ −3.13 ppm. The β-pyrrole protons resonated as two doublets in the aromatic region (δ ∼ 9.29 and ∼ 8.98 ppm) for trans-A2 porphyrins and four doublets (δ ∼ 9.36, 9.26, 9.03 and 8.94 ppm) for trans-AB porphyrins. The PEG resonances are described below. The copper chelates were not characterized by NMR spectroscopy given the line broadening caused by relaxation from the copper center.36,37The 1H NMR spectrum of the zinc trans-A2 porphyrin with 3,5-dipegylated aryl groups (Zn2) is shown in Fig. 1 (top panel). The resonances from the PEG –OCH2CH2– units and the terminal methyl group appear in the range δ ∼ 3.7–3.0 ppm. The 1H NMR spectrum of the zinc trans-A2 porphyrin with 2,6-dipegylated aryl groups (Zn6) is shown in Fig. 1 (bottom panel). Here, the resonances from the PEG –OCH2CH2– units and the terminal methyl group encompass a much larger range, spanning δ 3.7–2.0 ppm, versus that of Zn2. The peaks in the upfield region of Zn6 are attributed to –OCH2CH2– units thrust over the faces of the macrocycle, thereby experiencing the aromatic ring current. The –OCH2– unit linking the meso-aryl and triazole moieties of Zn6 resonates downfield (δ 5.04 ppm) versus that of Zn2 (δ 4.45 ppm) which is attributed also to the ring current but at the outer edge of the macrocycle. Similarly, the triazolyl-H proton of Zn6 resonates downfield (δ 5.99 ppm) versus that of Zn2 (δ 5.12 ppm), again shifted by the porphyrin ring current. The minimum conclusions are that the 2,6-disubstitution pattern (Zn6) affords facial encumbrance by the attached PEG chains that is not present in the 3,5-disubstitution pattern (Zn2).
 | ||
| Fig. 1 1H NMR spectra in CDCl3 at room temperature of zinc porphyrins Zn2 (top) and Zn6 (bottom). Peak assignments: (a) the resonance of the triazolyl–H. (b) The resonance of the protons of the –OCH2– linking the meso-aryl and triazole moieties. All other peaks derive from the PEG chains. | ||
The structure of Zn5 was confirmed by single-crystal X-ray diffraction upon crystallization from THF at −20 °C (Fig. 2). The zinc ion is hexacoordinate with a THF molecule at each apical site. The meso-aryl dihedral plane is ca. 71° (70.98(4)°). The 2-propynyl groups project over the two faces of the macrocycle. Other analogues include the following substituents on the oxy moieties of 2,6-dialkoxyarylporphyrins: methyl,38–45 ethyl,46 butyl,47–49 octyl,50 dodecyl,51 3,3-dimethylbutyl,52 4-hydroxybutyl,53 4-oxa-3-oxoheptyl,54tert-butyldimethylsilyl,55 and pentafluorophenylmethyl.56 Strapped porphyrins with 2,6-dialkoxyaryl units include doubly strapped meso–meso-linked arrays57 and basket-handle thiolate Fe(III) porphyrins.58
 | ||
| Fig. 2 Crystal structure of Zn5 (crystallized from THF at −20 °C). Disorder is omitted for clarity. Labels: C = grey, N = blue, O = red, Zn = cyan, and H = white. | ||
Assessments in aqueous solution
A set of tests was carried out with selected porphyrins to gauge suitability of the designs for use in aqueous solution.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) P measurements.
The partitioning of an organic compound between octanol and aqueous solution is a standard proxy to gauge aqueous-membrane partitioning in biological systems.59 The partition coefficient (P) for a given porphyrin was examined by allowing a minute quantity of porphyrin to partition between deionized water (or PBS) and octanol at room temperature (∼10−4 M total porphyrin concentration).60 A sample was removed from each phase and examined by absorption spectroscopy in dimethylsulfoxide (DMSO) to determine the concentration. The results are shown in Table 1. Representative photographs are provided in Fig. 3. Two porphyrins can be regarded as benchmarks: TPS-por is found entirely in the aqueous layer (logP < −2) whereas the fully tert-butyl protected tetraester zinc porphyrin Zn7 is found entirely in the octanol layer (logP > 2).
P measurements.
The partitioning of an organic compound between octanol and aqueous solution is a standard proxy to gauge aqueous-membrane partitioning in biological systems.59 The partition coefficient (P) for a given porphyrin was examined by allowing a minute quantity of porphyrin to partition between deionized water (or PBS) and octanol at room temperature (∼10−4 M total porphyrin concentration).60 A sample was removed from each phase and examined by absorption spectroscopy in dimethylsulfoxide (DMSO) to determine the concentration. The results are shown in Table 1. Representative photographs are provided in Fig. 3. Two porphyrins can be regarded as benchmarks: TPS-por is found entirely in the aqueous layer (logP < −2) whereas the fully tert-butyl protected tetraester zinc porphyrin Zn7 is found entirely in the octanol layer (logP > 2).
| Compd | Type | Projection | PEG n | Terminus | LogP |
|---|---|---|---|---|---|
| a Deionized water as the aqueous phase. b PBS as the aqueous phase. c Not determined because of insolubility in aqueous solution. | |||||
| TPS-por | A4 | — | 0 | — | <−2 |
| Zn2 | A2 | 3,5- | 6 | –OMe | NDc |
| 6 | A2 | 2,6- | 6 | –OMe | −0.46 |
| Zn6 | A2 | 2,6- | 6 | –OMe | −0.40 |
| Cu6 | A2 | 2,6- | 6 | –OMe | −0.37 |
| Zn7 | A2 | 2,6- | 6 | –CO2tBu | >2 |
| 8 | A2 | 2,6- | 6 | –CO2H | <−2 |
| Zn8 | A2 | 2,6- | 6 | –CO2H | <−2 |
| Cu8 | A2 | 2,6- | 6 | –CO2H | <−2 |
| 11 | AB | 2,6- | 6 | –CO2H | −1.6 |
| Zn11 | AB | 2,6- | 6 | –CO2H | −1.1 |
| Cu11 | AB | 2,6- | 6 | –CO2H | NDc |
| 14 | AB | 2,6- | 2 | –CO2H | NDc |
 | ||
| Fig. 3 Photographs of results upon logP evaluations with free base porphyrins (TPS-por, 8), copper porphyrin Cu8, and zinc porphyrins (Zn7, Zn8, Zn11). | ||
Porphyrin Zn2, which bears neutral PEG groups at the 3,5-positions of the two aryl rings, exhibited an extensively broadened Soret band (see the ESI†) in water, which is suggestive of aggregation; hence, a logP value was unobtainable. All of the other porphyrins examined bear PEG groups at the 2,6-positions and gave sharp absorption bands at very dilute concentration (2 μM) in aqueous media. The three porphyrins 6, Zn6, and Cu6 each showed preferential partitioning in water versus octanol by a ratio of ∼2.5:1. No significant difference was observed as a function of the metalation state: zinc(II), copper(II), or free base. Similar phenomena were observed for the series of 8, Zn8, and Cu8; however, the aqueous partitioning decreased along the series 11 (logP = −1.6), Zn11 (−1.1), and Cu11 (insoluble in aqueous media). The lack of solubility of the copper chelate Cu11versus the zinc chelate Zn11 and free base 11 was surprising and may indicate beneficial solubilization interactions in the latter two cases that are not available in the copper chelate. Such interactions could include aqueous (or PEG carboxylate/carboxylic acid) oxygenic coordination to the apical zinc site of Zn11, or aqueous (or PEG carboxylate/carboxylic acid) hydrogen-bonding with the free base N–H moieties of 11.
Porphyrin 8, which bears four PEG groups terminated with carboxylic acid moieties (as opposed to methyl groups), was not detected in the organic layer. We denote the ratio as >100:1 and the logP value as <−2. For porphyrins with only two PEG groups, the PEG6 groups imparted preference for aqueous solution (11, logP −1.6) whereas the PEG2 groups (14) resulted in aggregation in PBS; hence, a logP value could not be determined.
 | ||
| Fig. 4 Absorption spectra of porphyrins in PBS at room temperature. | ||
During the course of experimentation, stock solutions of porphyrins 6 and 11 were prepared in aqueous solution (see ESI†). Porphyrin 6 in water (12 mM) gave an optically clear, bright red solution. Porphyrin 11 (1 mM) in PBS gave a transparent albeit muddy red solution. A muddy appearance is consistent with broadened absorption bands,61 and the aforementioned concentration-dependent studies indicated aggregation even at substantially lower concentrations.
Discussion
The development of water-soluble porphyrins has been an ongoing activity since the 1950s. Early studies required only a simple porphyrin devoid of substituents other than those that impart aqueous solubility, which could be satisfied by uroporphyrins and the synthetic porphyrins shown in Chart 1. In the decades since, designs have been sought that achieve aqueous solubilization while maintaining sites on the porphyrin macrocycle for other substituents such as auxochromes and bioconjugatable groups. In some cases, very compact designs are desired for biomedical applications.PEG groups are widely used in seemingly ad hoc fashion to impart aqueous solubility without studies to explore the effects of the number of PEGs, the length of PEGs, and the terminal groups of PEGs. Here, three porphyrin scaffolds bearing 2-propynyloxy groups (Zn1, Zn5, Zn9) have been derivatized with PEG groups via click chemistry. It may be little appreciated beyond the aficionado that PEG molecules can afford both organic and aqueous solubility, although a core text states that “PEG will partition in favor of water in a water–benzene system and in favor of methylene chloride in a water–methylene chloride system.”13
Assessments pertaining to aqueous solubilization entailed partitioning between octanol and aqueous solution and examination of the absorption spectra as a function of concentration. Methods for calculation of clogP values often give wildly disparate results with porphyrins;62 hence, measured values are essential. It warrants emphasis that a logP value is not an indication of solubility per se but rather a partitioning in lipophilic versus aqueous media. Aqueous–organic partitioning and solubility are related yet distinct phenomena. The former represents competition between solubilization in two distinct liquid phases, whereas the latter represents competition between homogeneous dispersion in a liquid phase versus the affinity for the aggregated solid state. Aqueous–organic partitioning is given by a dimensionless value whereas solubility has units such as g cm−3. The overall solubility in water could be low or high but give the same ratio for aqueous–organic partitioning. The key findings are as follows:
(i) 2,6-diaryl substitution (Zn6) is superior to 3,5-diaryl substitution (Zn2) for aqueous solubilization. An interpretation is provided in Fig. 5. The 2,6-dialkoxyaryl group has constrained motion causing the groups to project above and below the plane, whereas the 3,5-dialkoxyaryl group is not so constrained and can rotate toward planarity with the macrocycle.63 The latter motion opens the possibility of intermolecular hydrophobic interactions leading to self-aggregation and therefore limited solubility. We had surmised that facial encumbrance was invaluable for solubilization but had heretofore not done a direct test in aqueous solution with non-ionizable substituents.
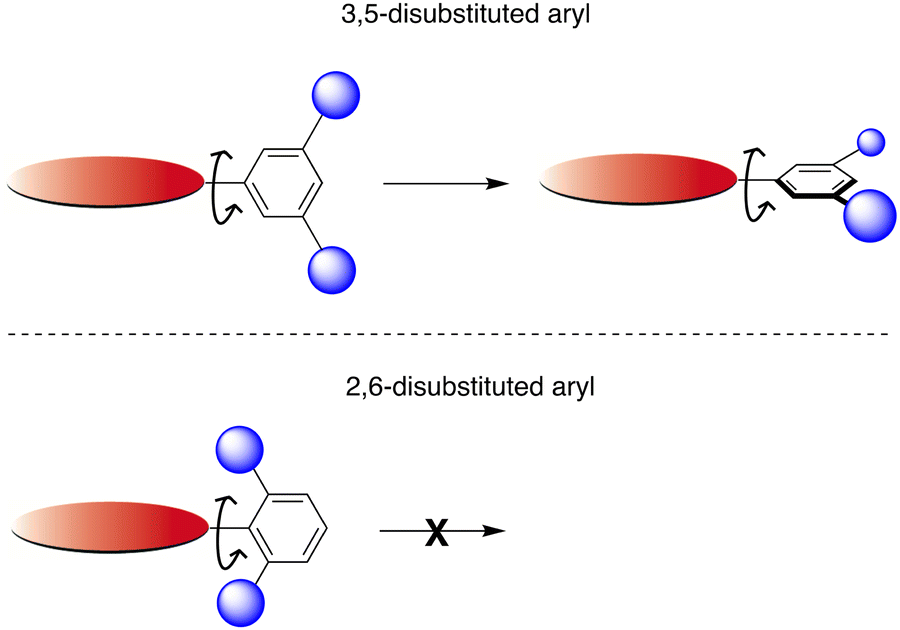 | ||
| Fig. 5 Wide conformational motion is available in the 3,5-disubstituted aryl porphyrin (top) versus limited motion for the 2,6-disubstituted aryl porphyrin (bottom). | ||
(ii) PEG groups terminated with carboxylic acid groups provide enhanced aqueous solubility versus simple methyl termination at physiological pH, where the former are predominantly in the carboxylate form. Both trans-A2 free base porphyrins 6 (methyl termini) and 8 (carboxylate termini) are aqueous soluble, as evidenced by the logP values (Table 1) and the retention of sharp absorption spectra over a 1000-fold concentration range (Fig. 4). The logP values show a key distinction, however the aqueous:octanol ratio is 100:35 for 6versus >100:1 for 8.
(iii) One rather than two water-solubilization motifs suffices to impart a degree of aqueous solubility, which enables use of a trans-AB porphyrin (e.g., 11, two PEG-carboxylates) rather trans-A2 porphyrins (e.g., 8, four PEG-carboxylates). Both 8 and 11 exhibit quite negative logP values (<−2, −1.6; i.e., high aqueous partitioning) but 11 does show signs of self-aggregation at concentrations in the range of 20–200 μM.
(iv) The porphyrin bearing two carboxylate-terminated PEG2 groups (14) rather than PEG6 groups (11) was insufficiently soluble to obtain a logP measurement. Despite the self-aggregation of 11 in the 20–200 μM range, a derivative thereof that contains a folic acid moiety tethered via a PEG5 group (clicked onto the 4-ethynylphenyl moiety) did not show signs of self-aggregation across the concentration range of 4.8–480 μM in PBS at room temperature.32 Such results indicate that incorporation of a polar motif with 11 substantially increases the aqueous solubility.
In summary, the studies reported herein point to molecular designs for achieving aqueous solubilization with porphyrins. The use of only one or two meso-substituents leaves other sites available for substitution. More extensive comparisons beyond the pairwise evaluations reported herein are required to explore the generality of the molecular design heuristics.
Experimental section
General methods
1H and 13C{1H} NMR spectra were recorded (Bruker Ascend™ 500 and Bruker Ascend™ 700 MHz instruments) in CD2Cl2 or CDCl3 at room temperature unless noted otherwise. Absorption spectra were collected in toluene or DMSO at room temperature. Matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) was recorded using a Bruker autoflex® max with the matrix α-cyano-4-hydroxycinnamic acid (α-CHCA). Electrospray ionization mass spectrometry (ESI-MS) data were recorded using a Thermo Fisher Scientific Exactive Plus MS, a benchtop full-scan orbitrap mass spectrometer using heated electrospray ionization. Data are reported for the molecular ion or cationized molecular ion. Single crystal X-ray diffraction (SCXRD) data were collected using a Bruker D8 Venture, running at 100 K with MoKα (λ = 0.71073 Å). All crystal data and graphics were refined and generated by OLEX2 and Mercury software, respectively. THF was freshly distilled from sodium/benzophenone ketyl and used immediately. Commercially available compounds were used as received. Silica gel (40 μm average particle size) was used for column chromatography. PBS buffer (1×) was employed at pH 7.4. Compounds Zn1,244,33Zn9,32 and Zn1032 were prepared as described in the literature.Determination of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P values
P values
Values were determined at room temperature following a reported method.60 The quantity of porphyrin employed was ∼0.2–0.4 mg in a total octanol–aqueous solution of 2 mL giving concentrations of 40–150 μM (i.e., ∼10−4 M). In each case, octanol refers to 1-octanol.
Examination of concentration-dependent self-aggregation
Values were determined by reciprocal change of concentration and cuvette pathlength in aqueous media at room temperature following a reported method.60Synthesis procedures and characterization data
:1) to (19:1) to (15:1)] to afford a red oil (5.4 mg, 15%): 1H NMR (500 MHz, CDCl3) δ 10.21 (s, 2H), 9.32 (d, J = 4.4 Hz, 4H), 9.08 (d, J = 4.4 Hz, 4H), 7.81 (s, 4H), 7.45 (s, 4H), 7.04 (s, 2H), 5.13 (s, 4H), 4.45 (s, 8H), 3.73 (s, 8H), 3.37–2.86 (m, 96H); 13C{1H} NMR (175 MHz, CDCl3) δ 156.3, 148.6, 148.5, 144.0, 142.4, 131.2, 130.6, 123.2, 118.0, 114.5, 104.9, 100.2, 70.4, 69.3–68.2, 61.0, 57.6, 53.2, 49.3; λabs (toluene) 416, 544, 580 nm; MALDI-MS obsd 2024.02, calcd 2024.90 [M+]; ESI-MS obsd 1035.4384, calcd 1035.4393 [(M + 2Na)2+], M = C96H136N16O28Zn.
:1) to (15:1) to (9:1)] to afford a red oil (29 mg, 71%): 1H NMR (500 MHz, CDCl3) δ 10.12 (s, 2H), 9.29 (d, J = 4.4 Hz, 4H), 8.97 (d, J = 4.4 Hz, 4H), 7.79 (t, J = 8.5 Hz, 2H), 7.25 (d, J = 8.5 Hz, 4H), 5.99 (s, 4H), 5.04 (s, 8H), 3.66 (t, J = 5.2 Hz, 8H), 3.47–3.35 (m, 32H), 3.26 (s, 12H), 3.20 (t, J = 5.0 Hz, 8H), 3.05–3.03 (m, 8H), 2.83 (t, J = 5.0 Hz, 8H), 2.58 (t, J = 5.0 Hz, 8H), 2.28 (t, J = 5.0 Hz, 8H), 2.15 (t, J = 4.0 Hz, 8H), 2.06 (t, J = 4.5 Hz, 8H); 13C{1H} NMR (175 MHz, CD2Cl2) δ 159.5, 150.4, 149.2, 143.5, 131.7, 131.6, 130.3, 122.9, 121.9, 111.6, 106.9, 105.1, 72.0–63.2, 58.7, 58.6, 50.9, 49.5; λabs (toluene) 414, 545, 581 nm; MALDI-MS obsd 2025.40, calcd 2024.90 [M+]; ESI-MS obsd 2047.8895, calcd 2047.8894 [(M + Na)+], M = C96H136N16O28Zn.
:2) was treated with Cu(OAc)2·H2O (54 mg, 0.27 mmol). The reaction mixture was stirred overnight at room temperature under an argon atmosphere. The mixture was concentrated under reduced pressure, diluted in CH2Cl2, and neutralized by the addition of saturated aqueous NaHCO3. The organic phase was washed with water and brine, and then dried (Na2SO4). The combined organic extract was concentrated to give a red oil (5.4 mg, 100%) λabs (toluene) 407, 531, 563 nm; MALDI-MS obsd 2046.98, calcd 2046.90 [(M + Na)+]; ESI-MS obsd 2046.8931, calcd 2046.8898 [(M + Na)+], M = C96H136CuN16O28.
:1) to (16:1)] to afford a red oil (40 mg, 81%): 1H NMR (700 MHz, CDCl3) δ 10.13 (s, 2H), 9.29 (d, J = 4.3 Hz, 4H), 8.98 (d, J = 4.3 Hz, 4H), 7.78 (t, J = 8.7 Hz, 2H), 7.27–7.17 (m, 4H), 5.99 (s, 4H), 5.05 (s, 8H), 3.69–3.64 (m, 16H), 3.53–3.49 (m, 16H), 3.44 (t, J = 4.9 Hz, 8H), 3.34 (t, J = 4.9 Hz, 8H), 3.18 (t, J = 4.9 Hz, 8H), 2.99 (t, J = 4.9 Hz, 8H), 2.81 (t, J = 4.9 Hz, 8H), 2.47–2.45 (m, 16H), 2.13 (t, J = 4.9 Hz, 8H), 2.04 (t, J = 4.9 Hz, 8H), 1.92 (t, J = 4.9 Hz, 8H), 1.43 (s, 36H); 13C{1H} NMR (175 MHz, CDCl3) δ 170.9, 159.3, 150.3, 149.0, 143.5, 131.8, 131.5, 130.3, 122.8, 121.7, 111.5, 106.8, 104.9, 80.5, 70.4–68.8, 68.1, 66.8, 63.3, 49.2, 36.2, 28.1; λabs (toluene) 414, 544, 582 nm; MALDI-MS obsd 2481.87, calcd 2481.17 [M+]; ESI-MS obsd 2504.1593, calcd 2504.1617 [(M + Na)+], M = C120H176N16O36Zn.
:2) was treated with Cu(OAc)2·H2O (54 mg, 0.27 mmol). The mixture was stirred overnight at room temperature, then concentrated under reduced pressure. The crude residue was dissolved in CH2Cl2 and neutralized by the addition of saturated aqueous NaHCO3. The organic layer was washed with water and brine, dried (Na2SO4), and concentrated to give a red oil (2.6 mg, 62%): λabs (toluene) 408, 530, 568 nm; ESI-MS obsd 2254.9144, calcd 2254.9152 [(M − H)−], M = C104H144CuN16O36.
:1) until pH = 11. The aqueous phase was washed once with CH2Cl2. The aqueous phase was acidified with aqueous 1 M HCl until pH = 2, then extracted with CH2Cl2 (2 × 15 mL). The combined organic extract was dried (Na2SO4) and concentrated to give a red solid (2.5 mg, 94%): 1H NMR (700 MHz, CDCl3) δ 10.28 (s, 2H), 9.41 (d, J = 4.4 Hz, 2H), 9.34 (d, J = 4.4 Hz, 2H), 9.05 (d, J = 4.4 Hz, 2H), 8.99 (d, J = 4.4 Hz, 2H), 8.23 (d, J = 7.8 Hz, 2H), 7.96 (d, J = 7.8 Hz, 2H), 7.80 (t, J = 8.7 Hz, 1H), 7.28 (d, J = 8.7 Hz, 2H), 5.91 (s, 2H), 5.11 (s, 4H), 3.66 (t, J = 5.2 Hz, 4H), 3.61 (t, J = 6.0 Hz, 4H), 3.50–3.37 (m, 12H), 3.36 (s, 1H), 3.32–3.27 (m, 4H), 3.21–3.14 (m, 4H), 3.05–2.99 (m, 4H), 2.87 (t, J = 5.1 Hz, 4H), 2.76 (t, J = 4.7 Hz, 4H), 2.48–2.44 (m, 8H), 2.29–2.24 (m, 4H), 2.15–2.7 (m, 4H), −3.11 (s, 2H); 13C{1H} NMR (175 MHz, CDCl3) δ 174.0, 159.1, 143.7, 142.0, 134.7, 131.9, 131.6, 131.1, 131.0, 130.8, 130.7, 122.8, 121.8, 119.8, 118.1, 111.4, 107.0, 105.0, 83.6, 70.4, 70.2, 70.1, 70.0, 70.0, 69.6, 69.4, 69.3, 69.3, 68.4, 66.5, 63.2, 49.5, 35.1; ESI-MS obsd 1375.5843, calcd 1375.5857 [(M + Na)+], M = C70H84N10O18.
:1) to (1:9) then pure ethyl acetate] to afford a red non-crystalline solid (24 mg, 73%): 1H NMR (500 MHz, CDCl3) δ 10.05 (s, 2H), 9.28 (d, J = 4.5 Hz, 2H), 9.13 (d, J = 4.4 Hz, 2H), 8.99 (d, J = 4.4 Hz, 2H), 8.78–8.65 (m, 2H), 8.14 (d, J = 7.5 Hz, 2H), 7.89 (d, J = 7.5 Hz, 2H), 7.60 (t, J = 8.5 Hz, 1H), 6.79–6.61 (m, 2H), 4.88 (s, 2H), 3.91 (s, 4H), 2.99 (s, 4H), 2.36–2.11 (m, 8H), 1.75–1.50 (m, 8H), 1.39–1.31 (m, 4H), 1.28 (s, 21H), 1.16 (s, 18H); 13C{1H} NMR (125 MHz, CDCl3) δ 170.3, 158.6, 150.3, 149.4, 149.1, 149.0, 143.4, 143.1, 134.5, 131.8, 131.7, 131.4, 130.2, 122.5, 121.6, 121.3, 119.0, 111.1, 107.3, 106.4, 105.3, 91.6, 80.3, 68.5, 68.4, 67.4, 65.6, 62.2, 48.6, 35.0, 27.8, 18.9, 11.5; ESI-MS obsd 1331.5653, calcd 1331.5662 [(M + H)+], M = C71H86N10O10SiZn.
:1 to 1:2)] to afford a dark red solid (20 mg, 93%): 1H NMR (500 MHz, CDCl3) δ 10.04 (s, 2H), 9.28 (d, J = 4.4 Hz, 2H), 9.11 (d, J = 4.4 Hz, 2H), 8.98 (d, J = 4.4 Hz, 2H), 8.70 (d, J = 4.4 Hz, 2H), 8.20–8.14 (m, 2H), 7.94–7.87 (m, 2H), 7.57 (t, J = 8.5 Hz, 1H), 6.65 (d, J = 8.6 Hz, 2H), 4.88 (s, 2H), 3.74 (s, 4H), 3.34 (s, 1H), 2.95 (t, J = 5.4 Hz, 4H), 2.37–2.24 (m, 8H), 1.81–1.67 (m, 8H), 1.45 (t, J = 6.5 Hz, 4H), 1.18 (s, 18H); 13C{1H} NMR (125 MHz, CDCl3) δ 170.4, 158.7, 150.4, 149.5, 149.3, 149.1, 144.1, 143.1, 134.7, 131.9, 131.8, 131.6, 130.4, 130.2, 121.5, 121.4, 121.2, 118.8, 111.2, 106.3, 105.5, 84.0, 80.4, 78.2, 68.8, 68.6, 67.5, 65.8, 62.1, 48.7, 35.2, 28.0; ESI-MS obsd 1175.4308, calcd 1175.4328 [(M + H)+], M = C62H66N10O10Zn.
:1) until pH = 11. The aqueous phase was washed once with CH2Cl2. The aqueous phase was acidified with aqueous 1 M HCl until pH = 2, then extracted with CH2Cl2 (2 × 15 mL). The combined organic extract was dried (Na2SO4) and concentrated to give a red solid (4.4 mg, 44%): 1H NMR (500 MHz, CD2Cl2) δ 10.29 (s, 2H), 9.41 (d, J = 4.5 Hz, 2H), 9.36 (d, J = 4.5 Hz, 2H), 9.05 (d, J = 4.4 Hz, 2H), 8.95 (d, J = 4.4 Hz, 2H), 8.24 (d, J = 7.9 Hz, 2H), 7.96 (d, J = 7.9 Hz, 2H), 7.78 (t, J = 8.7 Hz, 1H), 7.25 (d, J = 8.7 Hz, 2H), 6.16 (s, 2H), 5.06 (s, 4H), 3.72 (t, J = 5.0 Hz, 4H), 3.42 (s, 1H), 2.95 (t, J = 5.0 Hz, 4H), 2.82 (t, J = 6.1 Hz, 4H), 2.49–2.42 (m, 4H), 2.39–2.33 (m, 4H), 2.03 (t, J = 6.1 Hz, 4H); 13C{1H} NMR (125 MHz, CD2Cl2) δ 175.4, 159.0, 143.5, 142.0, 134.8, 132.0, 131.6, 130.8, 122.9, 121.6, 119.7, 118.2, 111.2, 106.8, 105.0, 83.5, 78.2, 69.3, 69.2, 68.4, 65.6, 63.0, 49.6, 34.4; ESI-MS obsd 1023.3746, calcd 1023.3760 [(M + Na)+], M = C54H52N10O10.
For logP measurements, the following porphyrins were prepared in small scale by zincation or cupration of the corresponding free base porphyrin. The products were characterized by absorption spectroscopy, fluorescence spectroscopy (for the zinc chelates), and mass spectrometry prior to examination in logP measurements.
:1). The reaction mixture was stirred at room temperature for 3 h. The mixture was diluted with CH2Cl2 (10 mL). The organic phase was washed with water (10 mL) and brine, dried (Na2SO4), and concentrated to dryness: λabs (DMSO) 427, 556, 571 nm; λem 600, 652 nm; MALDI-MS obsd 2257.65, calcd 2257.92 [(M + H)+], M = C104H144N16O36Zn.
:1). The reaction mixture was stirred at room temperature for 3 h. The mixture was diluted with CH2Cl2 (10 mL). The organic phase was washed with water (10 mL) and brine, dried (Na2SO4), and concentrated to dryness: λabs (DMSO) 419, 549, 584 nm; λem 591, 643 nm; MALDI-MS obsd 1415.46, calcd 1415.51 [(M + H)+], M = C70H82N10O18Zn.
:1). The reaction mixture was stirred at room temperature for 30 min. The mixture was diluted with CH2Cl2 (10 mL). The organic phase was washed with water (10 mL) and brine, dried (Na2SO4), and concentrated to dryness: λabs (DMSO) 407, 528, 559 nm; MALDI-MS obsd 1436.66, calcd 1436.51 [(M + Na)+], M = C70H82N10O18Cu.
Conflicts of interest
The authors declare competing financial interests.Acknowledgements
Funding was provided by the National Science Foundation to Oncurie, Inc. (NSF 2136700), and by NC State University. NMR spectroscopy and mass spectrometry measurements were carried out in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University.References
- P. Hambright, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, CA, 2000, vol. 3, pp. 129–210 Search PubMed.
- F. Dumoulin, M. Durmuş, V. Ahsen and T. Nyokong, Coord. Chem. Rev., 2010, 254, 2792–2847 CrossRef CAS.
- S. Pisarek, K. Maximova and D. Gryko, Tetrahedron, 2014, 70, 6685–6715 CrossRef CAS.
- M. Luciano and C. Brückner, Molecules, 2017, 22, 980 CrossRef PubMed.
- J. Winkelman, Cancer Res., 1962, 22, 589–596 CAS.
- P. Hambright and E. B. Fleischer, Inorg. Chem., 1970, 9, 1757–1761 CrossRef CAS.
- P. A. Carapellucci and D. Mauzerall, Ann. N. Y. Acad. Sci., 1975, 244, 214–238 CrossRef CAS PubMed.
- N. Matsumoto, M. Taniguchi and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2020, 24, 362–378 CrossRef CAS.
- A. K. Mandal, T. Sahin, M. Liu, J. S. Lindsey, D. F. Bocian and D. Holten, New J. Chem., 2016, 40, 9648–9656 RSC.
- P. Thamyongkit, M. Speckbacher, J. R. Diers, H. L. Kee, C. Kirmaier, D. Holten, D. F. Bocian and J. S. Lindsey, J. Org. Chem., 2004, 69, 3700–3710 CrossRef CAS PubMed.
- K. E. Borbas, P. Mroz, M. R. Hamblin and J. S. Lindsey, Bioconjugate Chem., 2006, 17, 638–653 CrossRef CAS PubMed.
- A. Z. Muresan and J. S. Lindsey, Tetrahedron, 2008, 64, 11440–11448 CrossRef CAS PubMed.
- J. M. Harris, Poly(ethylene glycol) Chemistry: Biotechnical and Biomedical Applications, Plenum Press, New York, NY, 1992, pp. 1–14 Search PubMed.
- D. Sato, Z. Wu, H. Fujita and J. S. Lindsey, Organics, 2021, 2, 161–273 CrossRef CAS.
- N. Larson and H. Ghandehari, Chem. Mater., 2012, 24, 840–853 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- R. Begum and H. Matsuura, J. Chem. Soc., Faraday Trans., 1997, 93, 3839–3848 RSC.
- S. Zalipsky and J. M. Harris, in Introduction to Chemistry and Biological Applications of Poly(ethylene glycol); In Poly(ethylene glycol) Chemistry and Biological Applications; ACS Symposium Series, American Chemical Society, Washington, DC, 1997, vol. 680, pp. 1–13.
- M. S. Thompson, T. P. Vadala, M. L. Vadala, Y. Lin and J. S. Riffle, Polymer, 2008, 49, 345–373 CrossRef CAS.
- M. Taniguchi, J. S. Lindsey, D. F. Bocian and D. Holten, J. Photochem. Photobiol., C, 2021, 46, 100401 CrossRef CAS.
- Y. Kuramochi and A. Satake, Catalysts, 2023, 13, 282 CrossRef CAS.
- M. Shokeen and T. J. Wadas, Med. Chem., 2011, 7, 413–429 CrossRef CAS PubMed.
- P.-L. D. Cao, Z. Wu, J. Rong and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2023, 27, 1049–1058 CrossRef CAS.
- F. Dumoulin and V. Ahsen, J. Porphyrins Phthalocyanines, 2011, 15, 481–504 CrossRef CAS.
- R. Liu, M. Liu, D. Hood, C.-Y. Chen, C. J. MacNevin, D. Holten and J. S. Lindsey, Molecules, 2018, 23, 130 CrossRef PubMed.
- G. P. Arsenault, E. Bullock and S. F. MacDonald, J. Am. Chem. Soc., 1960, 82, 4384–4389 CrossRef CAS.
- C. Brückner, J. J. Posakony, C. K. Johnson, R. W. Boyle, B. R. James and D. Dolphin, J. Porphyrins Phthalocyanines, 1998, 2, 455–465 CrossRef.
- B. J. Littler, Y. Ciringh and J. S. Lindsey, J. Org. Chem., 1999, 64, 2864–2872 CrossRef CAS PubMed.
- G. R. Geier III, B. J. Littler and J. S. Lindsey, J. Chem. Soc., Perkin Trans. 2, 2001, 701–711 RSC.
- C. K. Chang, Isr. J. Chem., 2016, 56, 130–143 CrossRef CAS.
- H. A. Houson, Z. Wu, P.-L. D. Cao, J. S. Lindsey and S. E. Lapi, Mol. Pharm., 2024, 21, 2441–2445 CrossRef CAS PubMed.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process Res. Dev., 2003, 7, 799–812 CrossRef CAS.
- A. D. Adler, F. R. Longo, F. Kampas and J. Kim, J. Inorg. Nucl. Chem., 1970, 32, 2443–2445 CrossRef CAS.
- J. E. Falk, Porphyrins and Metalloporphyrins, Elsevier Publishing Co., Amsterdam, 1964, p. 74 Search PubMed.
- G. M. Godziela and H. M. Goff, J. Am. Chem. Soc., 1986, 108, 2237–2243 CrossRef CAS PubMed.
- G. N. La Mar and F. A. Walker (Jensen), in The Porphyrins, ed. D. Dolphin, Academic Press, New York, 1979, vol. 4, pp. 61–157 Search PubMed.
- K. W. Gold, D. J. Hodgson, A. Gold, J. E. Savrin and G. E. Toney, J. Chem. Soc., Chem. Commun., 1985, 563–564 RSC.
- M. V. Peters, R. Goddard and S. Hecht, J. Org. Chem., 2006, 71, 7846–7849 CrossRef CAS PubMed.
- D. Conrad, J. DeCoskey, C. Yeisley, M. Zeller, A. D. Hunter and E. P. Zovinka, Acta Crystallogr., 2007, E63, m2824 Search PubMed.
- D. Conrad, J. DeCoskey, S. Mock, B. C. Noll, J. Petrovic and E. P. Zovinka, Acta Crystallogr., 2007, 63, m3058 CAS.
- S. Maji and S. Sarkar, Inorg. Chim. Acta, 2010, 363, 2778–2785 CrossRef CAS.
- S. Nowakowska, F. Mazzola, M. N. Alberti, F. Song, T. Voigt, J. Nowakowski, A. Wäckerlin, C. Wäckerlin, J. Wiss, W. B. Schweizer, M. Broszio, C. Polley, M. Leandersson, S. Fatayer, T. Ivas, M. Baljozovic, S. F. Mousavi, A. Ahsan, T. Nijs, O. Popova, J. Zhang, M. Muntwiler, C. Thilgen, M. Stöhr, I. A. Pasti, N. V. Skorodumova, F. Diederich, J. Wells and T. A. Jung, ACS Nano, 2018, 12, 768–778 CrossRef CAS PubMed.
- C. Li, K. Lang, H. Lu, Y. Hu, X. Cui, L. Wojtas and X. P. Zhang, CCDC 1894934: Experimental Crystal Structure Determination, 2019 Search PubMed.
- R. Inoue, M. Yokoyama, I. Maruyama and Y. Morisaki, Chem. – Eur. J., 2023, 29, e202301717 CrossRef CAS PubMed.
- S. Choi, S. H. Chae, J. H. Shin, Y. Kim, S.-J. Kim, D. H. Choi and S. J. Lee, Chem. Commun., 2013, 49, 3994–3996 RSC.
- C. Arunkumar, P. Bhyrappa and B. Varghese, Tetrahedron Lett., 2006, 47, 8033–8037 CrossRef CAS.
- S. J. Lee, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, Adv. Mater., 2008, 20, 3543–3549 CrossRef CAS.
- S. H. Chae, K. Y. Lee, S.-J. Kim, S. J. Lee and Y. Kim, Inorg. Chem. Commun., 2016, 69, 40–44 CrossRef CAS.
- A. Meindl, S. Plunkett, A. A. Ryan, K. J. Flanagan, S. Callaghan and M. O. Senge, Eur. J. Org. Chem., 2017, 3565–3583 CrossRef CAS.
- S. Choi, C. H. Kim, J.-O. Baeg, H.-J. Son, C. Pac and S. O. Kang, ACS Appl. Energy Mater., 2020, 3, 11581–11596 CrossRef CAS.
- M. P. Nikiforov, U. Zerweck, P. Milde, C. Loppacher, T.-H. Park, H. T. Uyeda, M. J. Therien, L. Eng and D. Bonnell, Nano Lett., 2008, 8, 110–113 CrossRef CAS PubMed.
- A. Zingg, B. Felber, V. Gramlich, L. Fu, J. P. Collman and F. Diederich, Helv. Chim. Acta, 2002, 85, 333–351 CrossRef CAS.
- P. J. Dandliker, F. Diederich, M. Gross, C. B. Knobler, A. Louati and E. M. Sanford, Angew. Chem., Int. Ed. Engl., 1994, 33, 1739–1742 CrossRef.
- A. Sen and K. S. Suslick, J. Am. Chem. Soc., 2000, 122, 11565–11566 CrossRef CAS.
- R. W. Wagner, J. S. Lindsey, I. Turowska-Tyrk and W. R. Scheidt, Tetrahedron, 1994, 50, 11097–11112 CrossRef CAS.
- T. Ikeda, J. M. Lintuluoto, N. Aratani, Z. S. Yoon, D. Kim and A. Osuka, Eur. J. Org. Chem., 2006, 3193–3204 CrossRef CAS.
- P. Li, K. Alenezi, S. K. Ibrahim, J. A. Wright, D. L. Hughes and C. J. Pickett, ChemSusChem, 2012, 5, 2361–2375 CrossRef CAS PubMed.
- A. Leo, C. Hansch and D. Elkins, Chem. Rev., 1971, 71, 525–616 CrossRef CAS.
- Q. Liu, M. Taniguchi, S. Goel and J. S. Lindsey, Dyes Pigm., 2024, 223, 111914 CrossRef CAS.
- S. F. Mason, in The Chemistry of Synthetic Dyes, ed. K. Venkataraman, Academic Press, NY, 1970, vol. III, pp. 169–221 Search PubMed.
- A. R. M. Soares, Y. Thanaiah, M. Taniguchi and J. S. Lindsey, New J. Chem., 2013, 37, 1087–1097 RSC.
- J. S. Lindsey, Chem. Rev., 2015, 115, 6534–6620 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C{1H} NMR, and MALDI-MS spectra for new compounds; absorption spectra and photographs of selected compounds in solution; single-crystal X-ray diffraction data. CCDC 2321457 (Zn5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj01178c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |