
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancement of the electrochemical properties of vanadium dioxide via nitrogen-doped reduced graphene oxide for high-performance supercapacitor applications†
Samba
Sarr
 a,
Daba T.
Bakhoum
a,
Ndeye F.
Sylla
a,
Ndeye M.
Ndiaye
b,
Delvina J.
Tarimo
a,
Vusani M.
Maphiri
a,
Balla D.
Ngom
b and
Ncholu
Manyala
*a
a,
Daba T.
Bakhoum
a,
Ndeye F.
Sylla
a,
Ndeye M.
Ndiaye
b,
Delvina J.
Tarimo
a,
Vusani M.
Maphiri
a,
Balla D.
Ngom
b and
Ncholu
Manyala
*a
aDepartment of Physics, Institute of Applied Materials, SARChI Chair in Carbon Technology and Materials, University of Pretoria, Pretoria 0028, South Africa. E-mail: ncholu.manyala@up.ac.za
bLaboratoire de Photonique Quantique, d’Energie et de Nano-Fabrication, Faculté des Sciences et Techniques, Université Cheikh Anta Diop de Dakar (UCAD), B.P. 5005, Dakar-Fann Dakar, Senegal
First published on 3rd July 2024
Abstract
A one-step solvothermal approach was used to integrate nitrogen-doped reduced graphene oxide into vanadium dioxide (VO2) to prepare a V@XN-G (where X = 24, 48 and 96 and is the mass of urea as the nitrogen (N) source) nanocomposite material. After optimization, V@48N-G showed better performance with a specific capacitance of 197.4 F g−1 at 0.5 A g−1 in a wide working potential window of 0.0–0.8 V vs. silver/silver chloride and was adopted as a positive electrode. Thus, cocoa waste-based activated carbon (ACC) was investigated and considered a negative electrode in assembling an asymmetric full-cell V@48N-G//ACC. The device exhibited good specific energy and power of 28.8 W h kg−1 and 425.1 W kg−1, respectively, at a voltage window and specific current of 1.7 V and 0.5 A g−1, respectively. Its cycling stability resulted in a Coulombic efficiency (CE) and capacitance retention (CR) of 99.8% and 73%, respectively, over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 galvanostatic charge/discharge cycles at 10 A g−1. Therefore, the V@48N-G//ACC device shows excellent electrochemical performance and is suitable for energy storage application technology.
000 galvanostatic charge/discharge cycles at 10 A g−1. Therefore, the V@48N-G//ACC device shows excellent electrochemical performance and is suitable for energy storage application technology.
1. Introduction
The depletion of fossil fuels has motivated and accelerated intensive research to produce, convert and store energy efficiently, adequately and sustainably to secure socio-economic development.1,2 Renewable energy sources such as wind and sun drive the need for superior and sustainable energy technologies.3 For the efficient utilization of these energy sources, appropriate energy storage systems such as flywheels, batteries and supercapacitors are required to store excess generated energy and release the energy when low or no energy is being generated. Supercapacitor (SC) energy storage systems have attracted significant attention owing to their low maintenance, great specific power, rapid charge propagation dynamics and very long cycling life performance (up to 100000 cycles).4–7. In addition to its impressive specific power of 103 W kg−1, which is thousands of times greater than batteries (50–200 W kg−1), supercapacitors also have a specific energy of around of ∼5–10 W h kg−1. This specific energy is significantly more than that of electrostatic capacitors (<1 W h kg−1), but still lower than that of batteries.5,8,9 Thus, prodigious research efforts are being developed to improve the specific energy of supercapacitors to match that of batteries. This can be possible by optimising the performance of electrode materials since they play a key role in the overall device performance. Therefore, the study of materials with distinct properties has gained great interest in supercapacitor applications.
Transition metal oxides (TMOs) with multiple oxidation states produce higher specific capacitances (Csp) than conducting polymers and carbonaceous materials.10 TMOs, such as ruthenium dioxide (RuO2), cobalt oxide (Co3O4), manganese dioxide (MnO2), nickel oxide (NiO), and vanadium oxide (VOx), are typical electrode materials for supercapacitor applications and thus have been used as Faradaic/pseudocapacitor electrode materials.11–14 Particularly, vanadium dioxide (VO2) has attracted enormous interest owing to its several oxidation states (2+, 3+, 4+ and 5+), low cost, low toxicity, optoelectronic properties and unique electronic structure. VO2 has been widely used as an electrode material in energy storage applications.15,16 However, owing to its low electrical conductivity, slow ion diffusion and poor cycling stability, its practical application is limited.10,17,18
An effective technique to overcome this drawback of VO2 electrodes as other TMOs is through incorporating carbon materials (in other words, electrical double layer capacitors (EDLCs) nanomaterials) like carbon aerogels, activated carbon, carbon nanotubes and reduced graphene oxide to make a composite material. This can improve some TMOs properties including the electrical conductivity, cycling stability, and ion diffusion.19,20 For example, Liang et al.21 reported a coaxial-structured material based on CNTs enveloped by a few layers of VO2(B) sheath (VO2(B)/CNTs) by the controlled hydrolysis of vanadium triisopropoxide in situ. The composite VO2(B)/CNTs demonstrated a superior specific capacitance and better rate capability than the single component, showing 250 F g−1 as the specific capacitance in 1 M sodium sulphate neutral electrolyte at a specific current of 0.5 A g−1 and a CR of 71% at 10 A g−1.21 Xiao and co-workers22 reported VO2(B) nanobelts regularly embedded into the nanosheets of graphene by the hydrothermal method, in which GO was reduced to reduced graphene oxide (rGO). The VO2(B)/rGO composite electrode exhibited a mass capacitance value of 290.4 F g−1 linked to 209.2 F g−1 for the pure VO2(B) at 0.2 A g−1 in 1 M Na2SO4.22 Dhanda and colleagues23 synthesized VO2/g-C3N4 composites employing the hydrothermal method. The optimised 3VO2/g-C3N4 sample showed a specific capacitance value of 1416 F g−1 compared to 583.3 F g−1 for VO2(D) at a sweep rate of 2 mV s−1 in 1 M potassium hydroxide electrolyte.23 These studies concluded that the composite material performs better than pure VO2. These good electrochemical performances are ascribed to the synergistic outcome, which improves the electron transfer originated from the carbon-based material and charge storage capacities/capacitances due to VO2.24 On the other hand, carbon materials have the disadvantage of having a low specific capacitance. Therefore, in order to optimize the performances of composite materials, it is of interest to improve the EDLC materials performances and thus build a composite material. In particular, single-layer rGOs attract each other owing to the van der Waals force, resulting in the restacking of the material (rGO). This restacking reduces the surface area and then less capacitance is stored throughout the development of the double layer.25 Thus, the most universally appropriate strategy to enhance the specific capacitance for EDLC materials is the introduction of a spacer material or heteroatom doping.
Heteroatom doping such as sulphur (S), boron (B), oxygen (O) and nitrogen (N) can improve the capacitance properties of rGO by preventing the restacking by increasing the interlayer spacing. Moreover, doping with heteroatom increases the electronic conductivity of rGO.26 Consequently, the combined benefits amongst the pseudocapacitor (PC) material and the EDLC material doped with heteroatoms will provide excellent specific capacitance, interesting cycling properties, and high rate capability for the hybrid composite, showing great supercapacitive performances.27
Herein, we have established a low cost-effective, environment-friendly approach to enhance the electrochemical properties of vanadium dioxide (VO2) by the incorporation of N-doped reduced GO using a simple solvothermal process. The prepared material was characterized using different techniques such as X-ray diffraction (XRD) spectroscopy, Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy, energy dispersive X-ray (EDX) spectroscopy, field emission scanning electron microscopy (FESEM) and N2-adsorption/desorption isotherms together with the equivalent pore size distribution. The optimized composite (V@48N-G) was investigated in both three and two-electrodes configuration. In the two-electrode configuration, the assembled device combined V@48N-G and cocoa waste-based activated carbon (ACC) as the positive electrode and negative electrode, respectively, with a high specific energy of 28.8 W h kg−1. Thus, the prepared material is a suitable candidate for energy storage applications.
2. Experimental
2.1. Material synthesis
The materials utilized in this work were prepared similarly as those in our previous works.28,29 Vanadium dioxide nitrogen-doped reduced graphene oxide nanocomposites were produced using the solvothermal technique, where VO2 and reduced graphene oxide were prepared, as detailed in the ref 28 and 30. In summary, vanadium pentoxide (V2O5) was mixed with oxalic acid dihydrate (C2H2O4·2H2O) at a mass ratio of 1.2:3.6 g, respectively, under continuous stirring at a constant heat of 80 °C for 3 h till a uniform dark blue (vanadyl oxalate (VOC2O4)) solution was formed. Thereafter, nitrogen-doped reduced graphene oxide was prepared by dispersing 16 mg of reduced graphene oxide and different X amounts of urea in a volume of 10 mL deionized water (DW), then sonicated for 150 min. The obtained solution was labeled XN-G, where X = 24, 48 and 96 mg of urea as the nitrogen (N) source and G represent the reduced graphene oxide. Note that reduced graphene oxide was optimized in our previous work,29 exhibiting that the best amount of reduced graphene oxide added was 16 mg. 6 mL of VOC2O4 solution was mixed with 60 mL of isopropanol, then subsequently transferred into the XN-G mixture. The obtained solution was then homogenized by stirring for 20 min at a constant heat of 80 °C, shifted into a Teflon-lined autoclave and retained in an oven for 6 h at 200 °C. The autoclave was then allowed to naturally cool down to ambient conditions before collecting and thoroughly cleaning the products with DW and ethanol via centrifugal agitation and decantation of the liquid. After drying overnight in an electric oven at 60 °C, the obtained samples were named as V@24N-G, V@48N-G, and V@96N-G, where V represents vanadium dioxide. Scheme 1 shows different V@XN-G composite preparation stages.
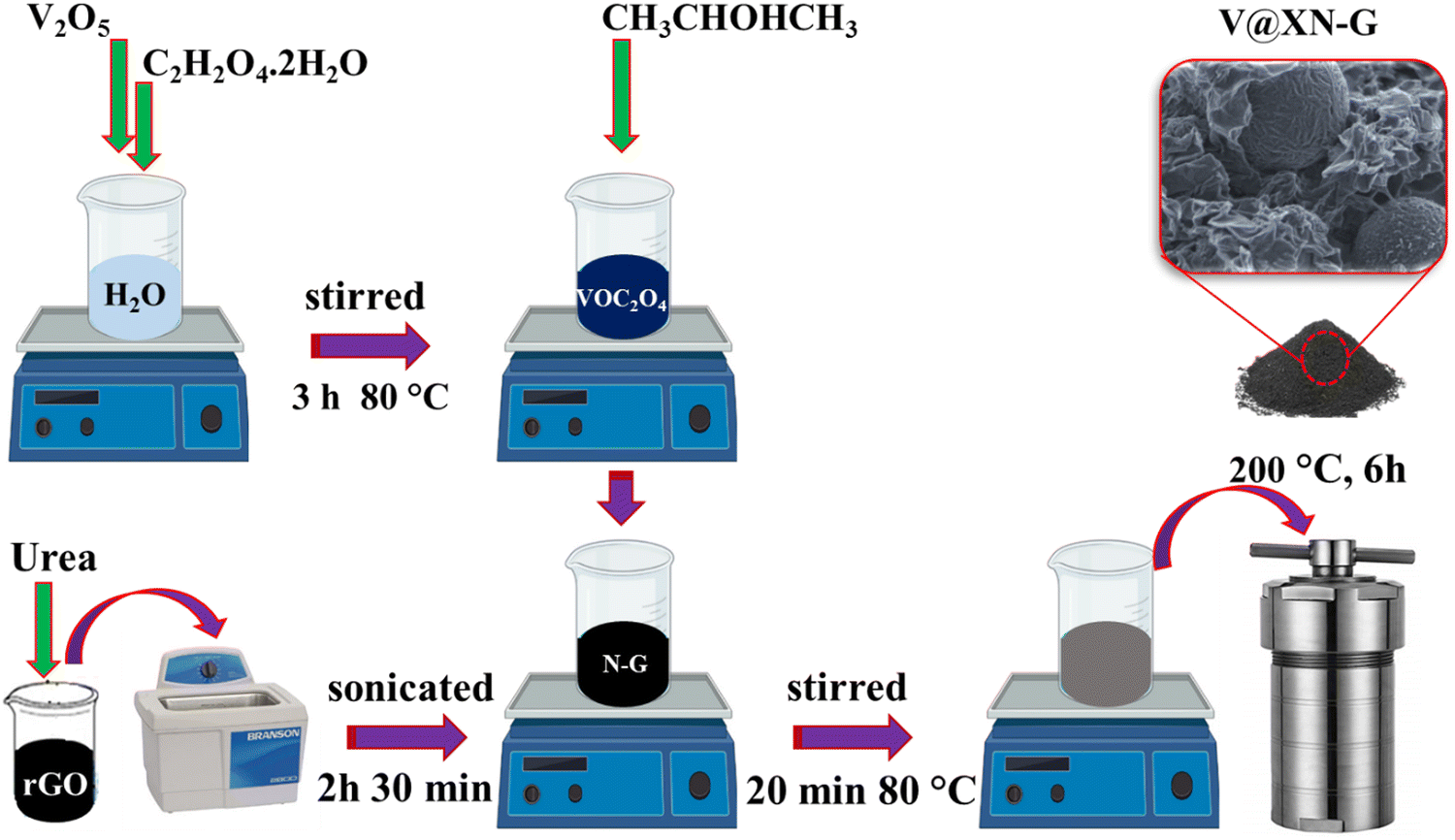 | ||
| Scheme 1 Illustration of the V@XN-G composite synthesis process. | ||
2.2. Material characterization
The structure of VO2 and V@XN-G composite materials was obtained by the aid of a Bruker BV 2D PHASER Benchtop powder X-ray diffraction instrument (PANalytical BV, Amsterdam, Netherland) operated with a Cu Kα1 (λ = 0.154056 nm) radiation source. The unit cell of the obtained VO2 structure was visualized using Vesta software and the corresponding crystallographic information file (CIF). A WITec alpha 300 RAS+ confocal micro-Raman microscope (Ulm, Germany) at a laser wavelength and power of 532 nm and 3.41 mW, respectively, and a spectral acquisition time of 60 s together with a Bruker Alpha-platinum-Attenuated Total Reflectance (Billerica, MA, USA) was used to perform Fourier transform infrared (FTIR) spectroscopy at 32 scans to analyse the diverse vibrations of the prepared materials. Quantachrome (NOVAtouch NT 2 LX-1, 220 V, USA) equipment was employed to analyze the specific surface area (SSA) alongside the pore size distribution (PSD) of the samples using the Brunauer–Emmett–Teller (BET) and density functional theory (DFT) techniques in the range of 0.05–0.95 relative pressure (P/P0) at 77 K. The samples micrographs followed by the energy dispersive X-ray (EDX) spectra were investigated using a Zeiss Ultra Plus 55 field emission scanning electron microscope (FE-SEM; Akishima-shi, Japan) coupled with an Oxford EDX operated at 2.0 and 20 kV accelerating potential, respectively. X-ray photoelectron spectroscopy (XPS) measurements were conducted with a VG Escalab 220i-XL instrument equipped with a monochromatic Al–K excited energy source (0.6 eV) and a hemispherical electron energy analyzer with an angle of 180°.2.3. Electrochemical characterization
In this study, the electrodes used were fabricated by mixing the as-prepared samples as active materials, acetylene carbon black (ACB) as the conductive additive and polyvinylidene difluoride (–(C2H2F2)n–, PVDF) as a binder in a mass ratio (in percentage) of 80:10:10, respectively, in an agate mortar with the dropwise addition of N-methyl pyrrolidone (NMP) to make a homogenous slurry. The resulting slurry was then pasted onto rectangular (1 × 2 cm2) and circular (1.8 cm2) clean nickel foam adopted as a current collector to fabricate electrodes for three and two-electrode measurements. The electrodes were then dried at 60 °C in an electric oven for 12 h. Afterwards, the supercapacitor performances of the fabricated electrodes were obtained using a Bio-Logic VMP-300 potentiostat (Knoxville, TN, USA) accompanied by the EC-Lab V11.33 software (Edmonton, AB, Canada) by means of the three and two-electrode configurations. The three-electrode test was done utilizing a glassy carbon as the counter electrode, silver/silver chloride (Ag/AgCl) as the reference electrode and the prepared samples representing the working electrode, while the two-electrode set-up was used to assemble an asymmetric device, in which a microfiber filter paper was used as a separator to sandwich between V@XN-G and ACC as the positive and negative electrodes, respectively, in a Swagelok cell. All the electrochemical tests, i.e., cyclic voltammetry (CV), galvanostatic charge/discharge (GCD) and electrochemical impedance spectroscopy (EIS), were performed in the range from 100 kHz to 10 mHz at room temperature using 0.5 M K2SO4 neutral electrolyte.
3. Results and discussion
3.1. Structural, morphological, and textural analysis
Fig. 1(a) depicts the powder X-ray diffraction (XRD) patterns of VO2 and V@XN-G composite samples. The patterns were indexed to the inorganic crystal structure database (ICSD) of VO2 (A) (card #1503)28 and to the peaks for the (002) and (100) planes of rGO material. The XRD pattern shows the co-existence of VO2 and rGO in all the three composite materials, confirming the good incorporation of rGO into the VO2 material.31 | ||
| Fig. 1 (a) XRD, (b) Raman, (c) FTIR patterns of VO2 and V@XN-G nanocomposites, and (d) the unit cell of VO2. | ||
The V@XN-G composites were further examined with Raman spectroscopy, as shown in Fig. 1(b), confirming the existence of VO2 through its different vibrational modes, as previously revealed in our work.28 Thus, V–O–V bending and stretching and V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching modes are attributed to the Raman shift range from 100 to 400 cm−1 and 400 to 800 cm−1, and ∼997 cm−1, respectively.28,32 Additionally, the Raman spectra show two strong characteristic bands attributed to the defects (D) and graphitic (G) bands caused by the defects (disordered) of the edges of graphite and graphene materials and the tangential vibration of the sp2 carbon atoms as a result of the highly ordered hexagonal structure within the graphite, respectively. These typical bands of carbon nanostructure demonstrate the existence of graphene in all the prepared nanocomposite materials.33,34 Consequently, the Raman results confirm the presence of carbon peaks revealed by the XRD analysis.
O stretching modes are attributed to the Raman shift range from 100 to 400 cm−1 and 400 to 800 cm−1, and ∼997 cm−1, respectively.28,32 Additionally, the Raman spectra show two strong characteristic bands attributed to the defects (D) and graphitic (G) bands caused by the defects (disordered) of the edges of graphite and graphene materials and the tangential vibration of the sp2 carbon atoms as a result of the highly ordered hexagonal structure within the graphite, respectively. These typical bands of carbon nanostructure demonstrate the existence of graphene in all the prepared nanocomposite materials.33,34 Consequently, the Raman results confirm the presence of carbon peaks revealed by the XRD analysis.
The intensity ratio (ID/IG) of the D and G-bands for the V@XN-G composite was investigated to estimate the degree of graphitization of the carbon materials.35 The calculated values of the ID/IG ratio together with the D and G-bands peak position are presented in Table 1. Note that the ID/IG ratio increases as the nitrogen content incorporated into the rGO increases. This can be explained through the interruption of the symmetry of the lattice caused by the introduction of the heteroatom into the graphitic structure. Therefore, the incorporation of N into the carbon lattice produces distortions, which transform the graphitic site (sp2) into the sp3 domain.35–37
| Samples | D-band position (cm−1) | G-band position (cm−1) | Intensity ratio (ID/IG) | BET SSA (m2 g−1) | Total pore volume (cm3 g−1) | Average pore size (nm) |
|---|---|---|---|---|---|---|
| VO2 | — | — | — | 24.23 | 0.085 | 13.05 |
| V@24N-G | 1353 | 1579 | 0.98 | 20.43 | 0.055 | 11.47 |
| V@48N-G | 1357 | 1606 | 0.99 | 9.44 | 0.028 | 12.61 |
| V@96N-G | 1357 | 1583 | 1.01 | 7.37 | 0.024 | 12.62 |
Fourier transform infrared spectroscopy (FTIR) measurements of pristine VO2 and V@XN-G were studied to investigate the chemical bonds between oxygen, vanadium, nitrogen and carbon atoms in the synthesized nanostructures. As exposed in Fig. 1(c), the peaks at about 983 and 478 cm−1 are assigned to the stretching vibration modes of VO and V–O–V, respectively.38,39 The peak at about 1606.7 cm−1 indicates the in-plane bending vibration mode of O–H while the peak at 3553.6 cm−1 is assigned to the O–H stretching vibration mode.40 These vibration bands identified for the pristine VO2 exist in the composite V@XN-G as well. However, a decrease in the intensity with a small shifting towards the lower wavenumbers is noted for these peaks such as O–H (1606.7–1565 cm−1), VO (983–961 cm−1), and V–O–V (478–431 cm−1). This is due to the interaction between VO2 and the XN-G. Furthermore, some additional peaks appear in the composites. Thus, the peak positioned at about 1407 cm−1 is ascribed to the CO vibration mode, while that at about 1066 cm−1 is associated with C–O stretching.41,42 Two absorption peaks at about 1540 cm−1 and 1066 cm−1 are observed as expected, which correspond to the typical absorption bands of CN and C–N, respectively, indicating the presence of nitrogen in the composites.43 An absorption band appears at about 1571 cm−1, partially overlapped with CN, corresponding to the aromatic skeletal CC vibrations.41 These results support the successful composite preparation via the solvothermal approach.
The unit cell of the VO2 structure is displayed in Fig. 1(d). In summary, the VO2 monoclinic crystal structure is visualized, in which the vanadium atoms occupy an octahedral coordination site. It can be seen that the vanadium atom lies at the centre surrounded by six oxygen atoms.44 The vibration modes are shown as the inset in the Raman spectrum (Fig. 1b).
The textural properties of VO2 and V@XN-G were analyzed through nitrogen sorption isotherms and PSD curves, as shown in Fig. 2. The isothermal analysis reveals type IV features associated with a H3 hysteresis loop for all the composites (V@24N-G, V@48N-G, and V@96N-G), suggesting the mesoporous structure of the samples, as shown in Fig. 2a.45,46 The pore size distribution curves of the samples (Fig. 2b) were recorded in the 2–16 nm pore diameter range, confirming the presence of a mesoporous structure, as also observed in the isotherms (Fig. 2a). The textural properties of the synthesized materials are displayed on Table 1. The pristine material (VO2) gives a higher specific surface area (SSA) and total pore volume than the composites (V@XN-G). It can be elicited that the introduction of XN-G into the VO2 sample decreases the SSA and total pore volume. This decrease is proportional to the amount of nitrogen added into the rGO matrix. Therefore, the high amount of nitrogen reduces the SSA by blocking some pores of the material.47 However, it is noted that the composite materials perform better as the electrode for supercapacitor applications.
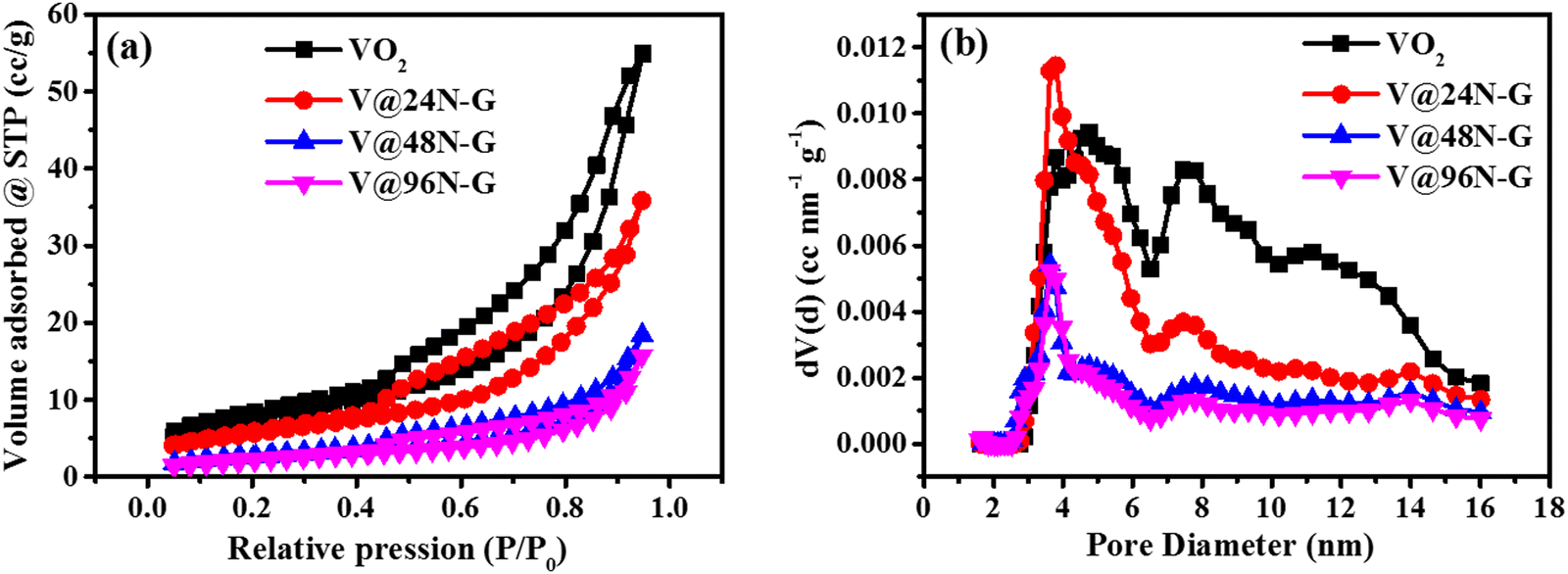 | ||
| Fig. 2 (a) N2-adsorption/desorption isotherm curves and (b) PSD curves of VO2 and V@XN-G nanocomposites. | ||
The morphology analysis of the composites (V@24N-G, V@48N-G and V@96N-G) materials is presented in Fig. 3 obtained by a field emission scanning electron microscope (FESEM). The FESEM micrograph of pristine VO2 revealed microspheres-like morphology assembled with nanograins on their surface (Fig. S1, ESI†).28Fig. 3(a)–(f) exhibit a similar morphology at low and high magnification for all the composites, showing a network of the VO2 morphology and an interconnected nanosheets-like structure of N-G.48,49 The SEM images demonstrate an embellishment of N-G nanosheets on the surface of the microspheres. In particular, Fig. 3(c) and (d) show a good incorporation of VO2 in the N-G sheets, demonstrating a great interaction between VO2 microspheres and N-G. This can provide better electrochemical performance due to the good synergistic effect.
 | ||
| Fig. 3 FESEM images of (a) and (b) V@24N-G, (c) and (d) V@48N-G, and (e) and (f) V@96N-G at low and high magnifications. | ||
Energy dispersive X-ray (EDX) spectroscopy was also investigated to carry out the elemental composition of the as-synthesized composites materials. Fig. 4 displays the EDX spectra of V@24N-G, V@48N-G and V@96N-G composite materials.
 | ||
| Fig. 4 EDX spectrum of (a) V@24N-G, (b) V@48N-G and (c) V@96N-G composite materials. | ||
It was observed that the synthesized composites contain the expected elements C, V, N and O. The insets in Fig. 4(a)–(c) display a description of the weight percent composition of each element.
The presence of sliver (Ag) and gold (Au) elements in the EDX spectra is ascribed to the Ag tape and Au conductive coat utilized in the EDX preparation. The elemental distribution mapping images confirms the good distribution of the elements (C, V, N and O) in the composites, as shown in Fig. S2 (ESI†), which confirms the fruitful integration of nitrogen into the rGO structure.
X-ray photoelectron spectroscopy (XPS) was conducted to further analyze the surface chemical property and the elemental composition of the synthesized pristine VO2 and composite materials V@XN-G. Fig. 5(a) displays the survey scan of the VO2 and V@XN-G composite samples. Herein, the XPS analysis of the V@48N-G sample that showed better features from previously discussed characterizations is detailed. The XPS survey spectrum reflects the elemental composition of the materials, showing four characteristic peaks of V 2p at 516.5 eV, O 1s at 529.3 eV, C 1s at 284.5 eV and N 1s at 400.7 eV, as illustrated in Fig. 5(a). This is in line with the EDS elemental composition results. The appearance of the additional VLMN and OKLL peaks (electrons auger) confirms the presence of V and O elements in the sample.50,51 The spectra were deconvoluted for in-depth analysis. The deconvoluted V 2p spectra has three V 2p3/2 peaks corresponding to the oxidation state of vanadium V3+ at 515.1 eV, V4+ at 516.5 eV and V5+ at 516.9 eV; similarly, the V 2p1/2 peak displays V3+ at 522.7 eV, V4+ at 524.0 eV and V5+ at 525.1 eV (Fig. 5b). The intense peaks at binding energy of 516.5 and 524.0 eV predict that V4+ is predominant in the material compared to V3+ and V5+, as shown in Fig. 5(b). The presence of V3+ can be due to the carbothermal reduction while V5+ comes from the V2O5 precursor.51,52 The O 1s spectra, deconvoluted in Fig. 5(b), shows the occurrence of V–O (529.1 eV), CO (529.8 eV) and C–O/OC–O (531.5 eV) interactions.53,54 The deconvoluted C 1s spectra (Fig. 5c) elucidate the presence of distinct types of carbon bonds. The carbon peak at 284.5 eV can be accredited to sp2 (CC) or sp3 (C–C) hybridized carbons.50,55 The peak at about 285.2 eV is attributed to C–N, while the peak at 287.2 eV is assigned to CO or C–O.52,56,57 The result indicates the formation of the N-doped carbon skeleton, conforming the FTIR result. The N 1s high-resolution XPS spectrum (see Fig. 5d) can be curve-fitted into three distinct peaks at approximately 399.5, 400.8 and 402.3 eV, associated with pyridinic-N, pyrrolic-N and graphitic-N, respectively.20,58 The high-resolution XPS results of the other samples (VO2, V@24N-G and V@96N-G) are shown in Fig. S3 and S4 (ESI†). Table 2 displays the description of the atomic percent composition of each element for different samples. The N-doping provides more active sites for ion adsorption and improves the electron transferability of the carbon material; thus, electrolyte ions are more conducive to diffuse when the charging/discharging behavior occurs to increase the energy storage capabilities.57
 | ||
| Fig. 5 XPS analysis: (a) survey scan of all the samples, (b) V 2p and O 1s, (c) C 1s and (d) high-resolution N 1s spectrum of V@48N-G. | ||
| Samples | Elements | |||
|---|---|---|---|---|
| V 2p (at% conc.) | O 1s (at% conc.) | C 1s (at% conc.) | N 1s (at% conc.) | |
| VO2 | 37.16 | 49.81 | 13.03 | — |
| V@24N-G | 40.68 | 31.31 | 27.25 | 0.77 |
| V@48N-G | 43.93 | 34.54 | 20.52 | 1.01 |
| V@96N-G | 39.62 | 32.39 | 26.81 | 1.18 |
Thus, the presence of pyridinic-N and pyrrolic-N favours charge storage owing to the enhanced charge accumulation at the electrode/electrolyte interface and the electrode surface wettability. Additionally, graphitic-N and pyridinic-N enhance electron transport, thus improving the electrical conductivity of the electrode materials.55,59 The various nitrogen functional groups may improve the electrochemical performance of the material.
As shown in Table 2, the wide scan XPS spectrum displays the main elements V, O, C and N of the different prepared materials. The observed carbon in VO2 may be due to the contamination from the precursors used.60 Similar to the EDX results, the amount of nitrogen in the composites follows the same trend. The moderate value of N might improve the electrochemical results.
3.2. Electrochemical performance
 | ||
| Fig. 6 Comparison of (a) CV at 30 mV s−1, (b) GCD at 0.5 A g−1, (c) Cspversus specific current, and (d) Nyquist plot (with an inset at high frequency) of VO2, N-G and V@XN-G. | ||
In the low-frequency region, they display a diffusion length short and close to the imaginary (Im(Z)) axis linked to VO2 (see Fig. 6d), implying a better capacitive characteristic.59
In summary, the V@48N-G composite showed a CV curve displaying a slight larger integrated area, a longer discharge time associated with the superior specific capacitance, and a lower Rs (2.5 Ω) and Rct (1 Ω) compared to the other composite materials. Therefore, this comparison proves that V@48N-G shows inferior electrolyte ions diffusion resistance and Rct and excellent capacitive performance, which are in accordance with the CV and GCD.61 Thus, the V@48N-G composite electrode outperforms the other composites. This can be attributed to the medium amount of nitrogen added, leading to the good incorporation between VO2 and N-G materials, as detected in the SEM analysis (Fig. 3c and d). Therefore, a good synergistic effect between VO2 and N-G can be noted for this particular doping. The better electrochemical results attained with the composite electrodes can be attributed to the synergistic effect due to the presence of effective channels, allowing the passage of charges and ions and also to the great electrical conductivity, good mechanical flexibility and high electrochemical stability arising from the rGO material and the pseudocapacitive behavior from VO2 and nitrogen-doping.64
Moreover, the addition of nitrogen atoms can significantly transform the chemical and electronic properties of the rGO lattice structure, leading to more active sites and improving the wettability through its pseudocapacitive behavior. The good surface wettability at the electrode/K2SO4 aqueous electrolyte interface reduces the diffusion resistance between the electrolyte ions and electrode material.57,65
Given that for the prepared composite materials, V@48N-G showed superior electrochemical performance, its detailed measurements were performed firstly in a half-cell (three-electrode) configuration by varying the scan rate, and the specific current for CV and GCD, respectively, in a working potential window from 0.0 to 0.8 V vs. Ag/AgCl, by assembling a full-cell device.
Fig. 7 displays further the three-electrode analysis of the V@48N-G composite. Fig. 7a shows the CV profiles at various scan rates values ranging from 5 to 100 mV s−1. They revealed a quasi-rectangular profile, approving the EDL capacitive behavior. From a low (5 mV s−1) to high (100 mV s−1) scan rate, the CV curves maintain a quasi-rectangular shape without any significant distortion, demonstrating the high rate capability of this electrode material.66 Obviously, the CV curves showed an increase in the current response and integrated area proportional to the scan rate. Fig. 7b relates the analogous GCD at various current densities, showing a linear variation of the charge and discharge path at all the current densities, designating a near-isosceles triangular profile confirming the EDL capacitor behavior seen in Fig. 7a. Fig. 7c displays the electrode's cyclic properties versus the cycle number. The electrode showed an excellent Coulombic efficiency of 99.9% and maintained 74.5% of its initial specific capacitance up to 5000 charge and discharge cycles at 10 A g−1. The cycling stability performance of the electrode material (V@48N-G) rapidly decreased after 1500 cycles. This decrease in the stability as a function of cycle number could be related to the formation of the vanadium species such as HV2O73−, V2O77−, V4O72−, or VO43− into the electrolyte owing to the over oxidation, which leads to the dissolution of V5+ in the oxide phase.28,67 The dissolution of the vanadium species into the electrolyte during cycling negatively affects the cycling performance, as reported by Shu et al.67 This might explain the immediate decrease in the capacitance retention after 1500 cycles, as observed in Fig. 7c. Thus, the cycling performance is affected by the amount of the vanadium species in the electrolyte, which might be significant after 1500 cycles.
 | ||
| Fig. 7 (a) CV and (b) GCD profiles at different scan rates and specific currents, and (c) cycling stability test of V@48N-G up to 5000 GCD cycles. | ||
The asymmetric supercapacitor was made using V@48N-G and ACC as the positive and negative electrode, respectively, in the neutral electrolyte mentioned above. Due to the difference in the specific capacitance of V@48N-G and ACC, a charge balance was carried out (Q+ = Q−) by calculating the mass of each electrode in the device using eqn (S5) and (S6) (ESI†). The optimal mass ratio between V@48N-G and ACC electrodes was ∼0.84 in the asymmetric cell. This leads to approximately 2.2 and 1.8 mg as the masses in the negative and positive, respectively, which exhibits in whole 4.0 mg (approximately 2.3 mg cm−2).
Fig. 8a relates the characteristic of the singular CV curves of ACC and V@48N-G composite electrodes tested in the three-electrode configuration, each in its operational potential windows range from −0.8 to 0.0 V and 0.0 to 0.8 V, respectively, using the same references cited above at 30 mV s−1. Fig. 8b represents the CV profile of the device at diverse sweep rates from 5 to 100 mV s−1 within a potential difference of 1.7 volts, revealing an EDLC behavior. From low (5 mV s−1) to high (100 mV s−1) sweep rate, the CV shapes of the device maintained a quasi-rectangular profile, implying its good capacitive behavior, which reveals good ion diffusion and great rate capability.68,69Fig. 8c shows the galvanostatic CD of the V@48N-G//ACC device at diverse current densities from 0.5 to 10 A g−1. All galvanostatic CD profiles exhibit semi-triangular shapes, suggesting good double-layer capacitive charge storage in the assembled single-ended supercapacitor, demonstrating the EDLC behavior characteristics of the single-ended device, as confirmed by the CV2.
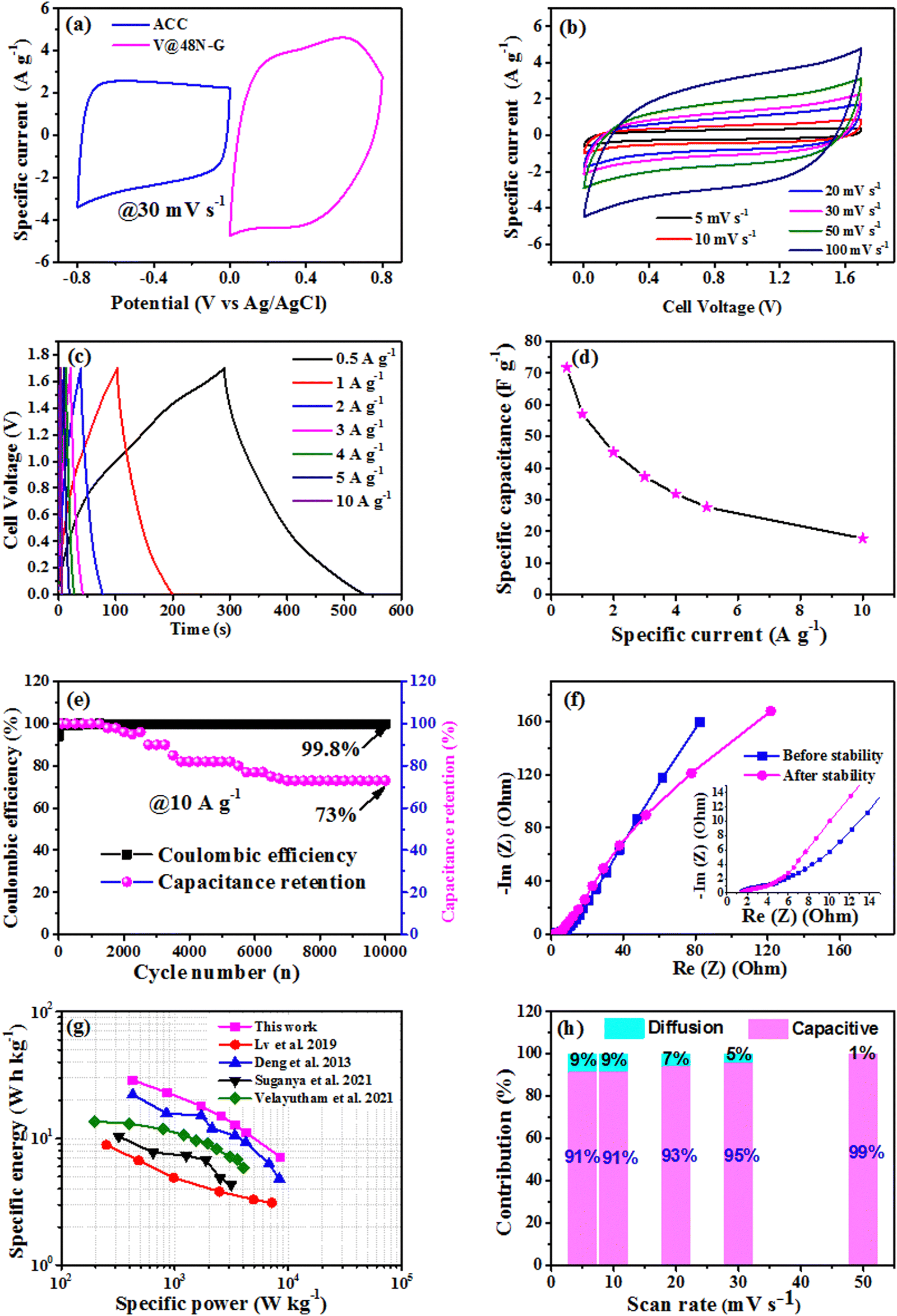 | ||
| Fig. 8 (a) CV of ACC and V@48N-G examined in the three-electrode set-up in their stable potential window. V@48N-G//ACC device: (b) CV curves, (c) GCD curves, (d) specific capacitance versus specific current, (e) Coulombic efficiency and capacitance retention as a function of the cycle number, (f) Nyquist plot studied before and after the stability test, (g) Ragone plots showing a comparison between this work and recently reported results in the literature, and (h) capacitive and diffusion-controlled contributions vs. scan rate. | ||
To deeply probe the contributions of capacitive and diffusion-controlled behaviors within the assembled asymmetric V@48N-G//ACC device, the CV curves displayed in Fig. 8b were explored to obtain the values of the current response (i) corresponding to the scan rate (ν) using various scan rates, according to the power law (i = aνb), where the values a and b are adjustable. Thus, there are two well-characterized values of b, where the b-value of 1 indicates that the current is controlled by the surface-controlled behavior, while the b-value of 0.5 indicates that the current is controlled by diffusion-controlled behavior. The value of b was determined by plotting the logarithm of the current (i) versus the logarithm of the scan rate (ν), as displayed in Fig. S6b (ESI†), whose slope represents the b-value at a certain cell voltage. The two values of b were determined at cell voltages of about 0.4 and 1.5 (Fig. S6a, ESI†), revealing b-values of 0.98 and 0.96, respectively. These values are approximately equal and very close to 1, implying the dominance of the capacitive behavior for the asymmetric device.70,71 Moreover, Trasatti's analysis was performed to estimate the diffusion-controlled and capacitive contributions within the prepared V@48N-G//ACC device using eqn (S7) and (S8) (ESI†). Based on previous works,71,72 the total capacitance (CT) was calculated by the aid of Fig. S7a (ESI†) displaying 1/C versus v0.5 using eqn (S9) (ESI†), whereas the EDL capacitance (CEDL) is determined from Fig. S7b (ESI†) exhibiting C versus v−0.5 using eqn (S10) (ESI†). The diffusion-controlled (Cdif) was estimated from the difference between CT and CEDL. Consequently, the capacitive and diffusion-controlled contributions were calculated at various scan rates and plotted (Fig. 8h). The results revealed that the capacitive contribution is higher than the diffusion-controlled contribution, as shown in Fig. 8h. Thus, Trasatti's analysis confirms the dominance of the capacitive behavior, as suggested by the power law, i.e., the V@48N-G//ACC is an EDLC-dominated device.
Fig. 8d displays the full-cell specific capacitance (Cd) against specific current, calculated from CD (Fig. 8c) using eqn (S2) (ESI†). The fabricated full-cell presented a high specific capacitance value of 71.8 F g−1 at 0.5 A g−1. At this specific current, it displayed a good specific energy of 28.8 W h kg−1, which corresponds to a power density of 425.1 W kg−1, calculated using eqn (S3) and (S4) (ESI†). Overall, the specific energy and power recorded with the V@48N-G//ACC full-cell are greater compared to those described for most reports on vanadium dioxide and other vanadium oxide/carbon-derived composite materials. For instance, Lv et al.24 manufactured free-standing thin vanadium oxide nanobelts and reduced graphene oxide (VO2/rGO) composite film. The VO2/rGO composite film and gel organic electrolyte (PVA/LiCl) were used to fabricate a symmetric supercapacitor. The solid-state supercapacitor exhibited an energy density of 8.96 W h kg−1 at a power density value of 250 W kg−1 24. Deng et al.73 prepared a composite material of graphene and VO2 (RG/VO2) via a hydrothermal approach by the aid of the precursors ammonium vanadate, graphite oxide (GO) and formic acid. The assembled asymmetric supercapacitor (RG/VO2//RG) used RG and RG/VO2 as the negative and positive electrodes, respectively, delivered an energy density of 22.8 W h kg−1 corresponding to a power density of 425 W kg−1 using 0.5 M K2SO4 neutral solution and a current density of 0.5 A g−1.73 Velayutham and co-workers74 reported the electrodeposition method to attain the binder-free vanadium pentoxide nanostructure on flexible CFC through the chronoamperometry method. The fabricated V2O5@CFC-30//O, N, S@AC device displayed energy and power densities of 17.7 W h kg−1 and 2728 W kg−1 at 1 A g−1 in 1 M Na2SO4 neutral medium, respectively.74 Suganya et al.75 reported N-doped graphene-anchored Zn3V2O8 composite synthesized by the hydrothermal process (Zn3V2O8@graphene). The assembled asymmetric supercapacitor combining the synthesized composite (positive) and activated carbon (AC) (negative) (Zn3V2O8@graphene//AC) recorded a specific energy of 10.4 W h kg−1 at a specific power of 312.5 W kg−1 in 0.5 M K2SO4 at 1 A g−1.75 This comparative study is summarized in Fig. 8g, which displays the so-called Ragone plot exhibiting the specific energy against the specific power.
The better performance arising from the V@48N-G//ACC device is attributed to the nitrogen-doping, where the conductivity of the sample increased owing to the extra lone pair of electrons in nitrogen atoms. This may offer a negative charge density for the sp2 hybrid carbon skeleton of the delocalized π system, thus facilitating electron transport.76,77 Also, it enhances the wettability of the electrode materials’ surface, which can enable the electrode/electrolyte ion contact.78
Furthermore, the electronegativity value of nitrogen atom (3.0) is superior to that of carbon atom (2.5), which leads to positive charge density near the adjacent carbon atoms.79 This increases the material's polarization, a characteristic representing the EDLC.80
To evaluate the stability performance, the device (V@48N-G//ACC) was constrained to 10000 GCD cycles at 10 A g−1. The cyclic property is a key test for the fabricated full-cell to be considered for practical applications in supercapacitors. Therefore, the plot of both Coulombic efficiency and capacitance retention as a function of cycle number is represented in Fig. 7e. It recorded an important cyclic performance with an excellent Coulombic efficiency of 99.8% and retained 73% of the initial specific capacitance over 10000 GCD cycles. The decrease in the capacitance retention with the number of charge/discharge cycles can be attributed to the destruction of the nanocomposite materials in the electrode and the mass loss of the electrode after 10000 charge/discharge cycles.81Fig. 7f shows the Nyquist plots of the asymmetric device before and after stability. At high frequency (inset in Fig. 7f), the device shows equivalent series resistances (Rs) approximately identical and equal to 1.42 Ω before stability and 1.47 Ω after stability. Still, in this region of high frequencies, the inset in Fig. 7f reveals the absence of a semicircle, indicating a very low charge transfer resistance, which results in good ionic conductivity. This favours the rapid formation of the EDL in these electrodes.82 At low frequency, a short diffusion path length and closeness to the Im(Z) axis is observed, indicating the capacitive behavior of the asymmetric cell.82 The low Rs attained for this device allowed to obtain an important maximum power device Pmax of 127.2 kW kg−1 estimated from eqn (S11) (ESI†).
The diffusion length faintly increased with a minor deviation from the Im(Z) axis. This is ascribed to a reduction in the ionic conductivity due to a drop of ions in the electrolyte solution after a long galvanostatic charge/discharge cycle. The degradation of the electrode surface (oxidation) after the stability test can similarly lower the ionic conductivity and lead to an increase in the diffusion length.83
4. Conclusion
In this study, vanadium dioxide nitrogen-doped rGO composite material (V@XN-G) has been effectively prepared utilizing a one-step solvothermal process via the variation of the amount of urea (N source) introduced into the rGO matrix. Thus, V@24N-G, V@48N-G and V@96N-G composite samples were obtained. The three-electrode measurements in 0.5 M K2SO4 electrolyte illustrated that the optimized sample corresponds to V@48N-G. This sample exhibited a Csp value of 197.4 F g−1 at 0.5 A g−1 within a large voltage window range of 0.0–0.8 V vs. Ag/AgCl. The V@48N-G electrode material recorded an excellent Coulombic efficiency of ∼99.9% with a capacitance retention of 74.5% over 5000 GCD cycles at 10 A g−1. An asymmetric full-cell device assembled using V@48N-G positive electrode and ACC negative electrode (V@48N-G//ACC) provided a good specific energy of 28.8 W h kg−1, which corresponds to a specific power of 425.1 W kg−1, recorded at 0.5 A g−1 at a large potential difference of 1.7 V. Due to the low equivalent series resistance, the device delivered a great maximum power Pmax of 127.2 kW kg−1. Moreover, the device showed remarkable cyclic properties with an excellent Coulombic efficiency of 99.8% and a capacitance retention of 73% after 10000 GCD cycles. These results elucidate that the prepared material has impressive potential for energy storage applications.
Data availability
Data can be available upon request from the authors.Conflicts of interest
The authors declare that they have no known competing financial interest or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was sponsored by the South African Research Chairs Initiative (SARChI) of the National Research Foundation (NRF) (Grant UID. 61056). Any opinion, finding and conclusion expressed in this work are those of the author(s) and NRF accepts no responsibility in this respect. S. Sarr acknowledges funding from the University of Pretoria and the National Research Foundation-The World Academy of Science (NRF-TWAS) for his doctoral studies. The authors acknowledge Prof Mohamed Chaker laboratory, the National Institute of the Scientific Research (INRS) – Energy Materials Telecommunication Centre (EMT), Canada, Quebec for XPS measurements.References
- H. Wang, H. Chen, X. Hou, H. Ye, Z. Guo, Z. Chen, Y. Jin, Y. Du and P. Ren, Diamond Relat. Mater., 2023, 136, 109888 CrossRef CAS.
- W. Zhang, R. Guo, L. Dang, J. Sun, Z. Liu and Z. Lei, J. Power Sources, 2021, 507, 230303 CrossRef CAS.
- S. R. Sinsel, R. L. Riemke and V. H. Hoffmann, Renewable Energy, 2020, 145, 2271–2285 CrossRef.
- M. K. Sahoo, P. Mane, B. Chakraborty and J. N. Behera, Inorg. Chem., 2024, 63, 6383–6395 CrossRef CAS.
- A. S. Lemine, M. M. Zagho, T. M. Altahtamouni and N. Bensalah, Int. J. Energy Res., 2018, 42, 4284–4300 CrossRef CAS.
- F. Niu, X. Han, H. Sun, Q. Li, X. He, Z. Liu, J. Sun and Z. Lei, ACS Sustainable Chem. Eng., 2021, 9, 4146–4156 CrossRef CAS.
- H. Sun, F. Niu, P. Yuan, X. He, J. Sun, Z. Liu, Q. Li and Z. Lei, Small Struct., 2021, 2, 1–9 Search PubMed.
- M. J. Madito, K. S. Matshoba, F. U. Ochai-Ejeh, N. Mongwaketsi, C. B. Mtshali, M. Fabiane and N. Manyala, Surf. Coat. Technol., 2020, 383, 125230 CrossRef CAS.
- C. Leng, Z. Zhao, Y. Song, L. Sun, Z. Fan, Y. Yang, X. Liu, X. Wang and J. Qiu, Nano-Micro Lett., 2021, 13, 1–11 CrossRef CAS.
- A. T. A. Ahmed, H. S. Chavan, Y. Jo, S. Cho, J. Kim, S. M. Pawar, J. L. Gunjakar, A. I. Inamdar, H. Kim and H. Im, J. Alloys Compd., 2017, 724, 744–751 CrossRef CAS.
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC.
- Q. Jiang, N. Kurra, M. Alhabeb, Y. Gogotsi and H. N. Alshareef, Adv. Energy Mater., 2018, 8, 1–10 Search PubMed.
- W. Guo, C. Yu, S. Li, Z. Wang, J. Yu, H. Huang and J. Qiu, Nano Energy, 2019, 57, 459–472 CrossRef CAS.
- S. Chen, H. Yu, L. Chen, H. Jiang and C. Li, Chem. Eng. J., 2021, 423, 130208 CrossRef CAS.
- R. Basu, S. Ghosh, S. Bera, A. Das and S. Dhara, Sci. Rep., 2019, 9, 4621 CrossRef PubMed.
- Z. Khan, P. Singh, S. A. Ansari, S. R. Manippady, A. Jaiswal and M. Saxena, Small, 2021, 17, 1–33 CrossRef.
- M. Guo, J. Balamurugan, T. D. Thanh, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2016, 4, 17560–17571 RSC.
- W. D. Wang, P. P. Zhang, S. Q. Gao, B. Q. Wang, X. C. Wang, M. Li, F. Liu and J. P. Cheng, J. Colloid Interface Sci., 2020, 579, 71–81 CrossRef CAS PubMed.
- B. D. Ngom, N. M. Ndiaye, N. F. Sylla, B. K. Mutuma, N. Manyala and M. Chaker, Philos. Trans. R. Soc., A, 2021, 379, 20200347 CrossRef CAS.
- R. Guo, J. Li, Y. Jia, F. Xin, J. Sun, L. Dang, Z. Liu and Z. Lei, J. Mater. Chem. A, 2019, 7, 4165–4174 RSC.
- L. Liang, H. Liu and W. Yang, J. Alloys Compd., 2013, 559, 167–173 CrossRef CAS.
- X. Xiao, S. Li, H. Wei, D. Sun, Y. Wu, G. Jin, F. Wang and Y. Zou, J. Mater. Sci.: Mater. Electron., 2015, 26, 4226–4233 CrossRef CAS.
- M. Dhanda, S. P. Nehra and S. Lata, Synth. Met., 2022, 286, 117046 CrossRef CAS.
- W. Lv, C. Yang, G. Meng, R. Zhao, A. Han, R. Wang and J. Liu, Sci. Rep., 2019, 9, 1–8 CrossRef.
- P. Bharathidasan, S. Devaraj and S. R. Sivakkumar, J. Solid State Electrochem., 2020, 24, 1095–1103 CrossRef CAS.
- Z. Liu, D. Li, Z. Li, Z. Liu and Z. Zhang, Appl. Surf. Sci., 2017, 422, 339–347 CrossRef CAS.
- S. R. Mangisetti, M. Kamaraj and S. Ramaprabhu, Electrochim. Acta, 2019, 305, 264–277 CrossRef CAS.
- S. Sarr, N. F. Sylla, D. T. Bakhoum, K. O. Oyedotun, B. D. Ngom and N. Manyala, J. Energy Storage, 2021, 43, 103294 CrossRef.
- S. Sarr, N. F. Sylla, D. T. Bakhoum, N. M. Ndiaye, D. J. Tarimo, V. M. Maphiri, B. D. Ngom and N. Manyala, J. Energy Storage, 2022, 55, 105666 CrossRef.
- D. J. Tarimo, K. O. Oyedotun, A. A. Mirghni and N. Manyala, Int. J. Hydrogen Energy, 2020, 45, 13189–13201 CrossRef CAS.
- A. A. Boochakravarthy, M. Dhanasekar and S. Venkataprasad Bhat, AIP Adv., 2020, 10, 85127 CrossRef CAS.
- N. M. Ndiaye, N. F. Sylla, B. D. Ngom, F. Barzegar, D. Momodu and N. Manyala, Electrochim. Acta, 2019, 316, 19–32 CrossRef CAS.
- S. Li, Y. Cen, Q. Xiang, M. K. Aslam, B. Hu, W. Li, Y. Tang, Q. Yu, Y. Liu and C. Chen, J. Mater. Chem. A, 2019, 7, 1658 RSC.
- N. F. Sylla, N. M. Ndiaye, B. D. Ngom, B. K. Mutuma, D. Momodu, M. Chaker and N. Manyala, J. Colloid Interface Sci., 2020, 569, 332–345 CrossRef CAS PubMed.
- C. S. Ramirez-Barria, D. M. Fernandes, C. Freire, E. Villaro-Abalos, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Nanomaterials, 2019, 9, 1761 CrossRef CAS PubMed.
- C. M. Chen, Q. Zhang, X. C. Zhao, B. Zhang, Q. Q. Kong, M. G. Yang, Q. H. Yang, M. Z. Wang, Y. G. Yang, R. Schlögl and D. S. Su, J. Mater. Chem., 2012, 22, 14076–14084 RSC.
- C. M. Masemola, N. Moloto, Z. N. Tetana, S. S. Gqoba, P. K. Mubiayi and E. C. Linganiso, Mater. Chem. Phys., 2022, 287, 126229 CrossRef CAS.
- X. Liu, J. Zheng, X. Jing, Y. Cheng and C. Meng, Appl. Sci., 2020, 10, 2742 CrossRef CAS.
- A. B. Habtemariam, A. Simo and M. Maaza, Appl. Phys. A: Mater. Sci. Process., 2020, 126, 1–9 CrossRef.
- X. Li, S. Zhang, L. Yang, X. Li, J. Chen and C. Huang, New J. Chem., 2017, 41, 15260–15267 RSC.
- L. Li, Y. Dou, L. Wang, M. Luo and J. Liang, RSC Adv., 2014, 4, 25658–25665 RSC.
- G. Nie, L. Zhang, J. Lei, L. Yang, Z. Zhang, X. Lu and C. Wang, J. Mater. Chem. A, 2014, 2, 2910–2914 RSC.
- J. Qin, J. Shen, X. Xu, Y. Yuan, G. He and H. Chen, Microchim. Acta, 2018, 185, 459 CrossRef PubMed.
- S. Guan, A. Rougier, M. R. Suchomel, N. Penin, K. Bodiang and M. Gaudon, Dalton Trans., 2019, 48, 9260–9265 RSC.
- H. C. Youn, S. M. Bak, M. S. Kim, C. Jaye, D. A. Fischer, C. W. Lee, X. Q. Yang, K. C. Roh and K. B. Kim, ChemSusChem, 2015, 8, 1875–1884 CrossRef CAS PubMed.
- S. I. El-Hout, S. G. Mohamed, A. Gaber, S. Y. Attia, A. Shawky and S. M. El-Sheikh, J. Energy Storage, 2021, 34, 102001 CrossRef.
- M. Mirzaeian, Q. Abbas, D. Gibson and M. Mazur, Energy, 2019, 173, 809–819 CrossRef CAS.
- J. Jayachandiran, J. Yesuraj, M. Arivanandhan, A. Raja, S. A. Suthanthiraraj, R. Jayavel and D. Nedumaran, J. Inorg. Organomet. Polym. Mater., 2018, 28, 2046–2055 CrossRef CAS.
- A. Bordbar-Khiabani, S. Ebrahimi and B. Yarmand, Appl. Surf. Sci., 2019, 486, 153–165 CrossRef CAS.
- D. T. Bakhoum, S. Sarr, V. M. Maphiri, N. F. Sylla, N. M. Ndiaye, M. Diop, B. D. Ngom, M. Chaker and N. Manyala, J. Energy Storage, 2024, 80, 110353 CrossRef.
- M. K. Sahoo, N. Bishoyi, K. Swain and J. N. Behera, Sustainable Energy Fuels, 2022, 6, 4779–4786 RSC.
- S. Li, Y. Cen, Q. Xiang, M. K. Aslam, B. Hu, W. Li, Y. Tang, Q. Yu, Y. Liu and C. Chen, J. Mater. Chem. A, 2019, 7, 1658–1668 RSC.
- D. Mohanadas, N. H. N. Azman and Y. Sulaiman, J. Energy Storage, 2022, 48, 103954 CrossRef.
- T. A. N. Bui, T. G. Nguyen, W. Darmanto and R. A. Doong, Electrochim. Acta, 2020, 361, 137018 CrossRef CAS.
- V. N. Kitenge, D. J. Tarimo, G. Rutavi, V. M. Maphiri, S. Sarr, M. Diop, M. Chaker and N. Manyala, J. Energy Storage, 2024, 81, 110453 CrossRef.
- D. T. Bakhoum, N. F. Sylla, S. Sarr, V. M. Maphiri, N. M. Ndiaye, D. J. Tarimo, A. Seck, B. D. Ngom, M. Chaker and N. Manyala, J. Energy Storage, 2023, 68, 107695 CrossRef.
- J. Zhao, W. Zhang, X. Guo, Z. Zhang, H. Xie, Y. Zheng, S. Wang, Q. Xu, Q. Fu and T. Zhang, Mater. Chem. Phys., 2022, 287, 126311 CrossRef CAS.
- S. Lu, L. Sui, M. Wu, S. Zhu, X. Yong and B. Yang, Adv. Sci., 2019, 6, 1801192 CrossRef.
- N. F. Sylla, S. Sarr, N. M. Ndiaye, B. K. Mutuma, A. Seck, B. D. Ngom, M. Chaker and N. Manyala, Nanomater., 2021, 11, 1056 CrossRef CAS PubMed.
- N. M. Ndiaye, T. M. Masikhwa, B. D. Ngom, M. J. Madito, K. O. Oyedotun, J. K. Dangbegnon and N. Manyala, Mater. Chem. Phys., 2018, 214, 192–200 CrossRef CAS.
- D. Gandla, X. Wu, F. Zhang, C. Wu and D. Q. Tan, ACS Omega, 2021, 6, 7615–7625 CrossRef CAS PubMed.
- G. A. Yakaboylu, C. Jiang, T. Yumak, J. W. Zondlo, J. Wang and E. M. Sabolsky, Renewable Energy, 2021, 163, 276–287 CrossRef CAS.
- L. Wang, S. Li, F. Huang, X. Yu, M. Liu and H. Zhang, J. Power Sources, 2019, 439, 227103 CrossRef CAS.
- N. M. Ndiaye, B. D. Ngom, N. F. Sylla, T. M. Masikhwa, M. J. Madito, D. Momodu, T. Ntsoane and N. Manyala, J. Colloid Interface Sci., 2018, 532, 395–406 CrossRef CAS.
- N. H. A. Rosli, K. S. Lau, T. Winie, S. X. Chin and C. H. Chia, Diamond Relat. Mater., 2021, 120, 108696 CrossRef CAS.
- S. Chen, X. Zhou, X. Ma, L. Li, P. Sun and M. Zhang, RSC Adv., 2019, 9, 30957–30963 RSC.
- D. Shu, H. Cheng, C. Lv, M. A. Asi, L. Long, C. He, X. Zou and Z. Kang, Int. J. Hydrogen Energy, 2014, 39, 16139–16150 CrossRef CAS.
- J. Du, L. Liu, Z. Hu, Y. Yu, Y. Zhang, S. Hou and A. Chen, ACS Sustainable Chem. Eng., 2018, 6, 4008–4015 CrossRef CAS.
- P. Man, Q. Zhang, J. Sun, J. Guo, X. Wang, Z. Zhou, B. He, Q. Li, L. Xie, J. Zhao, C. Li, Q. Li and Y. Yao, Carbon, 2018, 139, 21–28 CrossRef CAS.
- J. Liu, J. Wang, C. Xu, H. Jiang, C. Li, L. Zhang, J. Lin and Z. X. Shen, Adv. Sci., 2018, 5, 1700322 CrossRef.
- V. M. Maphiri, D. T. Bakhoum, S. Sarr, N. F. Sylla, G. Rutavi and N. Manyala, Nanomaterials, 2022, 12, 2211 CrossRef CAS.
- Y. Xu, C. Huang, A. Hu, Z. Fan, C. Chen, Y. Yang, Q. Tang, C. Jiang and X. Chen, Appl. Surf. Sci., 2021, 563, 150303 CrossRef CAS.
- L. Deng, G. Zhang, L. Kang, Z. Lei, C. Liu and Z. H. Liu, Electrochim. Acta, 2013, 112, 448–457 CrossRef CAS.
- R. Velayutham, R. Manikandan, C. J. Raj, A. M. Kale, C. Kaya, K. Palanisamy and B. C. Kim, J. Alloys Compd., 2021, 863, 158332 CrossRef CAS.
- B. Suganya, S. Maruthamuthu, J. Chandrasekaran, B. Saravanakumar, E. Vijayakumar, R. Marnadu, H. E. Ali and T. D. Nguyen, Surf. Interfaces, 2021, 24, 101129 CrossRef CAS.
- X. Sheng, N. Daems, B. Geboes, M. Kurttepeli, S. Bals, T. Breugelmans, A. Hubin, I. F. J. Vankelecom and P. P. Pescarmona, Appl. Catal., B, 2015, 176–177, 212–224 CrossRef CAS.
- Y. Gao, Q. Wang, G. Ji, A. Li and J. Niu, RSC Adv., 2021, 11, 5361–5383 RSC.
- X. Zhang, J. Zhao, X. He, Q. Li, C. Ao, T. Xia, W. Zhang, C. Lu and Y. Deng, Carbon, 2018, 127, 236–244 CrossRef CAS.
- T. Jiang, Y. Wang, K. Wang, Y. Liang, D. Wu, P. Tsiakaras and S. Song, Appl. Catal., B, 2016, 189, 1–11 CrossRef CAS.
- M. Duraivel, S. Nagappan, B. Balamuralitharan, S. Selvam, S. N. Karthick, K. Prabakar, C.-S. Ha and H.-J. Kim, New J. Chem., 2018, 42, 11093 RSC.
- H. Gul, A. U. H. A. Shah, U. Krewer and S. Bilal, Nanomaterials, 2020, 10, 118 CrossRef CAS.
- Z. J. Han, C. Huang, S. S. Meysami, D. Piche, D. H. Seo, S. Pineda, A. T. Murdock, P. S. Bruce, P. S. Grant and N. Grobert, Carbon, 2018, 126, 305–312 CrossRef CAS.
- E. Karaca, K. Pekmez and N. Ö. Pekmez, Electrochim. Acta, 2018, 273, 379–391 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj01029a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |