
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxazoline amino acid bioconjugates: one-pot synthesis and analysis of supramolecular interactions†
Marija
Bakija
 ,
Berislav
Perić
and
Srećko I.
Kirin
*
,
Berislav
Perić
and
Srećko I.
Kirin
*
Ruđer Bošković Institute, Bijenička cesta 54, HR-10000 Zagreb, Croatia. E-mail: Marija.Bakija@irb.hr; Srecko.Kirin@irb.hr
First published on 5th April 2024
Abstract
This publication describes oxazoline–amino acid bioconjugates 1 capable of supramolecular interactions. The bioconjugates contain three main building blocks: an oxazoline ring, a central aromatic unit and an amino acid substituent. Benzene, naphthalene or anthracene with several substitution motifs was used as the central aromatic unit. Two synthetic pathways to β-amino alcohol precursors 3–5 are presented; one-pot synthesis with various coupling reagents is compared to linearly sequenced synthesis using protecting groups. In the final step, a cyclization of precursors 3–5 to oxazolines 1 is described. Single crystal X-ray diffraction of seven oxazoline bioconjugates is reported (1p, 1m6, 1n2, 1n4, 1n5, 1a and 1t4), with an emphasis on supramolecular interactions in the solid state. The capability of bioconjugates 1 to participate in supramolecular interactions in solution was screened by NMR and CD spectroscopy, varying concentrations, temperatures and solvents. The results obtained by crystallography and spectroscopy were further corroborated by computational results for most interesting bioconjugate 1t1. Computational analysis was performed using a CREST/CENSO protocol. In particular, the free energy of formation, ΔG, as well as mean absolute error (MAE) values and correlations of experimental and GIAO calculated NMR parameters have been compared for 1t1 DFT models of monomers and dimers. These results revealed that for 1t1, the supramolecular dimer ensembles are more stable than monomer ensembles.
Introduction
Oxazolines have already been established as a versatile structural motif; their synthetic pathways, characterization methods and applications have been comprehensively reviewed as early as the 1970s.1 2-Oxazolines are particularly good building blocks characterized by predictable structures and modalities as well as straightforward synthetic procedures. 2-Oxazolines can easily be prepared from β-amino alcohols, enabling steric modifications via introduction of various substituents on positions of 1- and 4- on the planar and rigid oxazoline ring. It therefore does not come as a surprise that to this date 2-oxazolines have found broad application in numerous catalytic reactions,2,3 especially BOX-type oxazolines4 (also bearing the term “privileged”),5 as hybrid donor-type ligands,6 and as monomers in the synthesis of numerous polymers.7,8Compounds reported in the literature predominantly contain multiple oxazoline rings, while mono-oxazolines are less explored.9,10 In addition, published research concerning minimal oxazoline-containing compounds capable of undergoing supramolecular interactions is relatively scarce, pertaining mostly to various coordination compounds11 relying on complexation to facilitate supramolecular interactions between ligands,12,13 with applications in catalysis utilizing hydrogen bonding (SupraBox)14 and π–π stacking15 to increase the selectivity. Moreover, examples of non-polymers, oxazoline-containing supramolecular assemblies with no coordinative bonds are even fewer,16 and of these, only a handful are conjugated to biomolecules.17,18
Bioconjugates contain a stable covalent link between two molecules, at least one of which is a biomolecule, most often a peptide, a nucleic acid, a carbohydrate, a vitamin or a lipid.19 Bioconjugates often comprise a combination of properties of all of their structural units, giving rise to an array of molecules which are designed to overcome multiple difficulties in complex target systems. Amino acids and peptides are particularly interesting for bioconjugation as their starting materials are readily available and offer a facile method for incorporating chirality into the structure. In addition, these bioconjugates show a capacity for non-covalent interactions which have been utilized to form non-metal-containing supramolecular systems. Hydrogen bonding motifs of the amino acid strands in 1,2′-disubstituted ferrocene peptides have previously been described (Chart 1a),20 and ever since, various supramolecular structures bearing one or multiple of these motifs have been studied. Prominent examples include metal containing compounds;21–24 however, all-organic compounds containing aforementioned H-bonding motifs are known as well. In particular, such organic structures include dimeric structures composed of molecules containing a single (Chart 1b)25–27 or multiple amino acid substituents (Chart 1c).28–31
 | ||
| Chart 1 (a) Hydrogen bonding motifs in disubstituted ferrocene peptides; (b) and (c) “Herrick” hydrogen bonding in disubstituted (b) or trisubstituted amino acid bioconjugates (c); top view and side view for both (a) and (b/c) are boxed; the upper ring is indicated in bold. (d) Bioconjugates 1 discussed herein. Ox = oxazoline ring, Ar = central aromatic unit, Aa = amino acid(s). | ||
Oxazoline bioconjugates 1 presented in this study have the capacity to undergo supramolecular interactions (Chart 1d). The three main building blocks of bioconjugates 1, namely an oxazoline ring (blue), a central aromatic unit (gray) and an amino acid substituent (red), can form a variety of supramolecular interactions: the nitrogen atom of the oxazoline ring can act as a hydrogen bonding acceptor; the central aromatic ring can be involved in aromatic stacking, and the amino acid substituent contains both hydrogen bonding acceptors and donors. Synthetic approaches via the one-pot method and via protecting groups are compared. The amino alcohol substituents of precursors 3–5 are cyclized to yield a new set of 20 oxazoline–amino acid bioconjugates 1. The synthesized bioconjugates 1 are then screened for their ability to participate in supramolecular interactions in the solid state using SC-XRD diffraction and in solution using NMR and CD spectroscopies. Selected derivative 1t1 was subjected to a detailed computational analysis, from which free energies of formation, ΔG, and NMR parameters were derived to study potential supramolecular interactions.
Results and discussion
Synthesis of intermediates 3
A general approach to oxazoline amino acid bioconjugates 1 involves intermediates 3, which can be prepared from aromatic dicarboxylic acids P as precursors (Scheme 1). Amino acid derivatives 2 and amino alcohol derivatives 4 have been obtained as side products of the one-pot synthesis. In addition to disubstituted aromatic carboxylic acids, trimesic acid was used as a precursor to obtain trisubstituted derivatives of 1–4 (Scheme 1, R3 ≠ H). Two approaches to intermediates 3 were considered, namely a one-pot synthesis and a linear synthesis utilizing protecting groups. In the one-pot procedure, the diacid P is reacted simultaneously with two different amines (Scheme S1, ESI†). Generally, in this reaction, the expected ratio of the main products is the statistical distribution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, with the higher yield for the mixed product 3, containing both amines. However, this ratio is rarely obtained experimentally, due to different reactivity and solubility of the amines used.32–34
:2:1, with the higher yield for the mixed product 3, containing both amines. However, this ratio is rarely obtained experimentally, due to different reactivity and solubility of the amines used.32–34
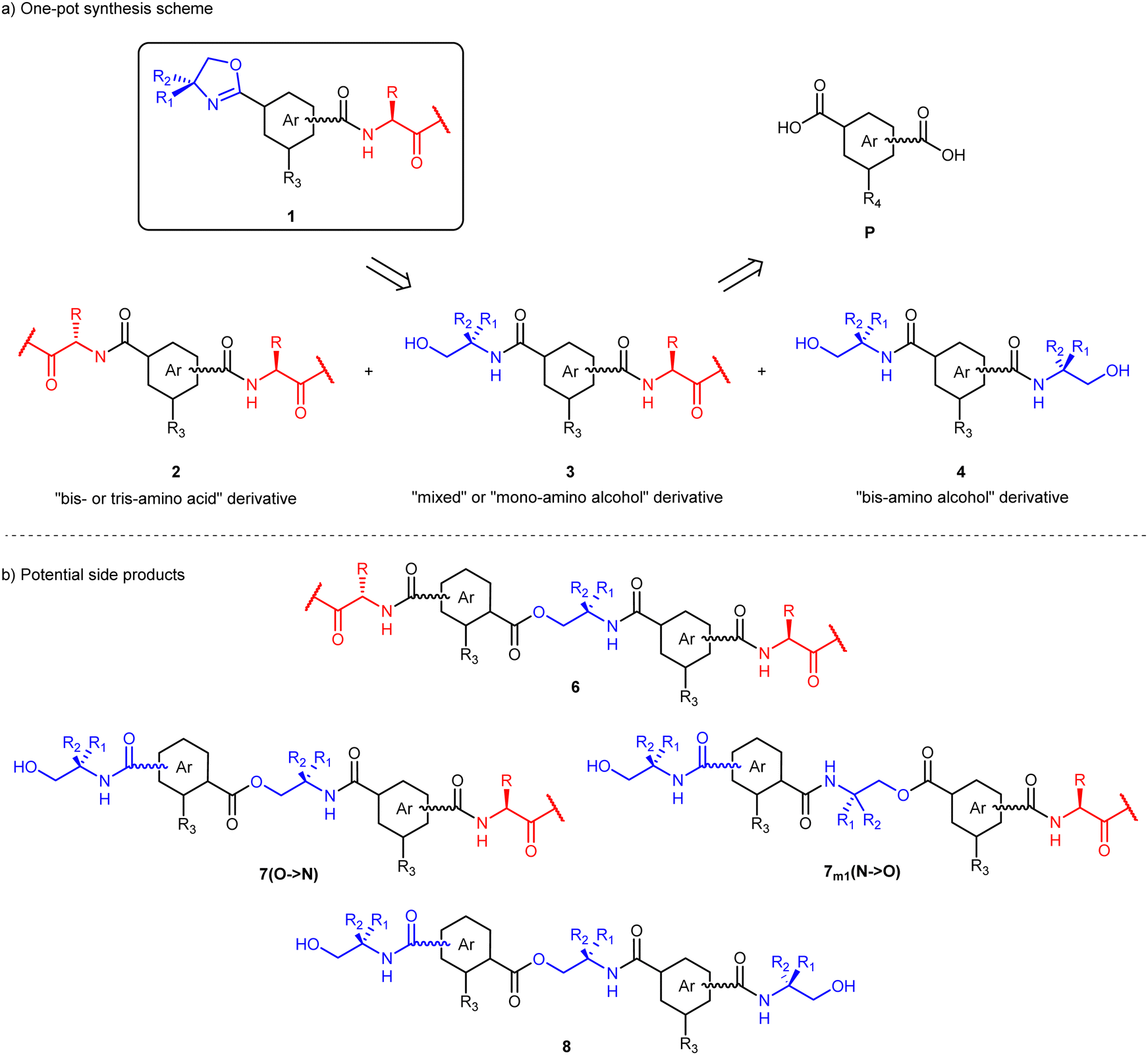 | ||
| Scheme 1 (a) Disconnection scheme with general structures of oxazoline amino acids 1, intermediates 2, 3 and 4 and precursors P. (b) Potential side products of the one-pot reaction, obtained by esterification. R3 = H, amino acid or amino alcohol. R4 = H or –COOH. | ||
The syntheses of intermediates 3 are divided into several sub-sections below. All obtained intermediates have been characterized by 1H and 13C NMR spectroscopies and ESI mass spectrometry. Additionally, two bis-amino acids (2m1 and 2n5), one bis-amino alcohol (4m5) and one mixed derivative (3t3) were characterized by single crystal X-ray diffraction (see Tables S6, S8, S9 and Fig. S13–S18, ESI†).
Synthesis of 3m1
For the initial optimization of the one-pot procedure, isophthalic acid was chosen as the diacid precursor, alanine methyl ester as the amino acid and 2-amino-2-methyl-propan-1-ol (AMP) as the amino alcohol. In the first attempt to synthesize the basic compound 3m1, the reaction was carried out with the commonly used benzotriazole reagents TBTU/HOBt (Scheme S1 and reaction 1, Table 1, ESI†).35 However, analysis of the isolated products revealed only the bis-alanine compound 2m1, while compounds 3m1 and 4m1 were not detected. Instead, ester-amide byproducts 6m1, 7m1 and 8m1 formed in trace amounts (Scheme 1b). Obviously, in the course of the reaction, the free alcohol group(s) in 3m1 and 4m1 can be further derivatized by the TBTU/HOBt protocol. Similarly, if DCC was used as a coupling reagent (reaction 2, Table 1), the desired derivative 3m1 was not obtained.In order to obtain 3m1 as well as to suppress the ester bond formation, other coupling reagents were explored (reactions 3–5, Table 1). In particular, reactions with pyAOP and COMU were performed in DMF, while DCM was used as a solvent for the reaction with HATU. Considering the similarity of yields of 3m1 obtained with all three successful coupling reagents (η ∼ 15%), HATU was used in the following reactions in order to avoid product isolation from DMF.
Since the bis-amino alcohol product 4m1 was not isolated in reactions 1–5, a direct attempt with isophthalic acid and two equivalents of AMP was performed, using the same procedure with HATU (reaction 6, Table 1). In this reaction, 4m1 was isolated with 27% yield. We note that 4m1 was highly insoluble in all solvents except for the particularly polar ones (MeOH, DMSO) and could be filtered off from the reaction solution. No such precipitation was observed in any of the reactions 1–5.
The linear approach to obtain the intermediate 3m1 was explored by using protecting groups (Scheme S2, ESI†). The overall yield was 9%, after five steps including two isolations by column chromatography, which is lower than the yield obtained by the one-pot procedure (15%). Considering the invested amount of time and materials in comparison to the one-pot synthesis, the linear synthesis sequence was not pursued further for simple disubstituted derivatives.
One-pot synthesis of other bis-derivatives 3
First, isomers of phthalic acid as the central aromatic unit have been considered (Table 2 and Chart S1, ESI†). Variations in the type and the number of amino acids and amino alcohols as well as the type and substitution motif of the central aromatic unit have been used.| Reaction | Ar | Amino acid | Amino alcohol | Yield (comp.)/% |
|---|---|---|---|---|
| a Isolated yield of product 2p was 7%. b Product contained significant amounts of 6p. c Isolated yield of product 4m5 was 3%. d Product contained significant amounts of 4m5. | ||||
| 7a | pC6H4 | H-Ala-OMe | 2-Amino-2-methyl propanol (AMP) | <13b (3p) |
| 8 | mC6H4 | H-Phe-OMe | 2-Amino-1-ethanol (ETA) | 22 (3m2) |
| 9 | mC6H4 | H-Ala-OMe | Valinol (Val#) | 16 (3m3) |
| 10 | mC6H4 | H-Ala-OMe | Phenylalaninol (Phe#) | 26 (3m4) |
| 11c | mC6H4 | H-Ala-OMe | Phenylglycinol (Phg#) | 13 (3m5) |
| 12 | mC6H4 | H-Gly–Val–Phe-OMe | Phenylglycinol (Phg#) | <52d (3m6) |
In reaction 7, p-substituted terephthalic acid was used as the central aromatic unit (Table 2). In this reaction, despite the use of HATU, an ester-amide byproduct 6p was present (Scheme 1b and Chart S5, ESI†). Moreover, it was not possible to separate the desired mixed product 3p from 6p by column chromatography. However, 6p was separated after the cyclization of 3p to the corresponding oxazoline (see the Oxazoline Synthesis section).
In reactions 8–12, isophthalic acid was used as the central aromatic unit, while alanine methyl ester, phenylalanine methyl ester or the -Gly–Val–Phe-OMe tripeptide was used as the amino acid component and AMP, 2-amino-1-ethanol, valinol, phenylalaninol and phenylglycinol were used as the amino alcohols (Table 2 and Chart S2, ESI†). When the syntheses of compounds 3m1 and 3p are compared to those of compounds 3m2–3m6 it can be seen that switching either the alanine amino acid or/and the AMP amino alcohol to derivatives with a bulkier, more hydrophobic group allowed for easier chromatographic isolation of target compounds. For 3m6, the corresponding bis-phenylglycinol product 4m5 was not separated by chromatography due to too similar chromatographic properties; comparable with 3p, the separation was performed after cyclization to the corresponding oxazolines (see below). The peptide amino acid sequence for the 3m6 derivative was chosen according to previously reported compounds containing the same sequence.23,36
Furthermore, the central aromatic unit was expanded from benzene to naphthalene or anthracene (reactions 13–18, Table 3 and Chart S3, ESI†). In particular, four naphthalene derivatives (with 1,4-; 1,5-; 2,6-; and 2,7- substitution patterns, respectively) and one anthracene derivative (with 9,10-substitution) were prepared. Bis-amino alcohols 4n3 and 4n4 that precipitated in reactions 11 and 12, respectively, have been filtered off from the reaction solution. The highest yields have been obtained for intermediates 3n1 and 3n4, 36% and 33%, respectively. The focus was on the synthesis of intermediates 3; therefore, no further attempts to synthesize other derivatives 4 were made. During the activation of the anthracene carboxylic acid groups, precipitation occurred even before the addition of amines (reaction 18, Table 3). To avoid loss of reagent before the reaction, in a second attempt, amines were added as soon as 9,10-anthracene dicarboxylic acid and HATU had visibly dissolved.
| Reaction | Ar | Yield (comp.)/% | |
|---|---|---|---|
| a H-Gly-OMe was used. b Isolated yield of product 4n3 was 17%. c Isolated yield of product 4n4 was 38%. | |||
| 13 | 1,4-Nph | 8 (2n1) | 36 (3n1) |
| 14a | 1,4-Nph | 17 (2n2) | 18 (3n2) |
| 15b | 1,5-Nph | 31 (2n3) | 15 (3n3) |
| 16c | 2,6-Nph | 5 (2n4) | 33(3n4) |
| 17 | 2,7-Nph | 18 (2n5) | 23 (3n5) |
| 18 | 9,10-Ant | 19 (2a) | 16 (3a) |
Synthesis of tris-derivatives 3t
A series of 1,3,5-substituted benzene derivatives was also prepared (Table 4 and Chart S4, ESI†). In anticipation of purification complexity of one-pot products with 1,3,5-substituted benzene, the linear approach was explored first (Scheme S3, ESI†). Derivatives 3t3 and 3t4 with amino acids L-phenylalanine and D-phenylalanine, respectively, and phenylglycinol as the amino alcohol were synthesized using protecting groups in a similar fashion as the linear synthesis of 3m1 (see above). However, there were significant difficulties with the isolation of carboxylic acid group-containing intermediates. It proved to be more efficient to skip the isolation of 14t3 and 16t3, and similarly to 3p and 3m6, to isolate the corresponding oxazolines after the cyclization (see below).Because of the difficult isolation of intermediates in the linear synthesis procedure, we synthesized trisubstituted derivatives 3t1, 3t2 and 3t5 using the two-step, one-pot method (Scheme 2 and Table 4). In contrast to the syntheses of disubstituted derivatives, in the trisubstituted case, the amino acid was added in the first step, while the amino alcohol was added in the second step, in order to avoid the formation of ester-amide byproducts. The ratio of added reagents in the two-step procedure proved to be crucial in facilitating formation of the preferred target compound. In reactions 19 and 20 (Table 4), using a 1:1 ratio of amino acid and amino alcohol, the target compounds 3t1 and 3t2 were successfully synthesized in higher yields than the other one-pot products, respectively. On the other hand, using a 1:2 ratio facilitated the formation of compound 4t5 with higher yield (reaction 21, Table 4).
 | ||
| Scheme 2 One-pot synthesis of t derivatives (i) amino acid, HATU, DIPEA, DCM, 1 day; (ii) amino alcohol, HATU, DIPEA, 1 day. | ||
Synthesis of derivative 3b
Preparation of the C2-symmetric derivative 3b, in which two simple 3m-like derivatives are linked by 1,4-diaminobutane, has also been performed (Scheme S4, ESI†). Considering the target product's complexity and all the possible byproducts and their polarities, the one-pot procedure was not attempted. Precursor 11m5 was easily synthesized by following the linear synthesis sequence as for compound 11m1 (see Scheme S2, ESI†). The methyl ester group was then cleaved to yield compound 12m5 and the reaction mixture was used in the next step without purification. Compound 17 was prepared from Boc-Val-OH and 1,4-diaminobutane in high isolated yield. In the next step, cleavage of the Boc-protecting group was performed and the obtained product was used in the next step without isolation. Coupling 12m5 and 18 with HATU successfully afforded derivative 3b.Oxazoline synthesis
A total of 20 oxazolines, divided into five groups, have been synthesized utilizing a method previously described by Gang Xu et al.,34 using DAST in DCM at −78 °C (Scheme 3 and Table S2, ESI†). It was found that the yield could be increased by adding excess DAST to the mixture (up to two equivalents). This is reflected in the results for the two attempts of synthesizing 1a, where with 1 equivalent of DAST the reaction yielded 36% of the product, while with 2 equivalents, it yielded 87%. The isolated yield of 1n5 is lower than expected due to difficulties in the separation of oxazoline 1n5 and starting material 3n5. As mentioned above, several intermediates were subjected to cyclization without purification (1p, 3m6, 14t3 and 16t3), resulting in lower isolated yields of the corresponding oxazolines. All synthesized oxazolines have been characterized by IR, 1H and 13C NMR spectroscopy and ESI-MS and ESI-HRMS spectrometry. | ||
| Scheme 3 Oxazoline cyclization reaction. (i) DAST, DCM, K2CO3, −78 °C (dry ice in acetone). R3, R4 = H, amino acid or oxazoline. *Not chromatographically purified in the previous step(s). | ||
Structures of oxazolines in the solid state
Single crystals of seven oxazoline bioconjugates have been obtained by diffusion from their respective DCM solutions layered with hexane, namely, two oxazolines with a disubstituted central benzene ring (1p and 1m6), three naphthalene-based oxazolines (1n2, 1n4 and 1n5), one anthracene oxazoline (1a) and one trisubstituted derivative (1t4). X-ray diffraction gave insights into the molecular structure and supramolecular interactions in the solid state; the ORTEP diagrams37 are shown in Fig. 1, experimental data for the X-ray diffraction studies are listed in Tables S6 and S7 (ESI†) while the packing diagrams are shown in Schemes S12 (ESI†). | ||
| Fig. 1 ORTEP-III drawings37 with 30% ellipsoid probability level for SCXRD determined structures (complete atom numbering schemes are shown in Fig. S11, ESI†). | ||
The crystal packing of the reported single crystal structures gives insight into intermolecular interactions in the solid state. The main intermolecular interaction in the reported structures is hydrogen bonding; in the discussion below, the Bernstein notation for hydrogen bonding patterns is used.38 Each individual oxazoline bioconjugate participates in hydrogen bonding forming infinite supramolecular polymeric chains,39 with oxazoline 1t4 as a partial exception (see below). Of all amide hydrogen atoms in these structures, only the glycine amide hydrogen atom in oxazoline tripeptide 1m6 does not participate in hydrogen bonding. In several cases, the oxazoline moiety is involved in hydrogen bonding as well. However, only the oxazoline nitrogen atoms participate in the hydrogen bonding, while the oxazoline oxygen atoms are almost never part of hydrogen bonding.40
Additionally, there are no structures in which ester atoms participate in hydrogen bonding, and the hydrogen bonding does not occur between separate supramolecular polymeric chains. Characteristic hydrogen bonded secondary structures present in oxazolines 1 are shown in Fig. S9 and S16–S19 (ESI†) and listed in Tables S5 and S9 (ESI†). In particular, four unitary graph set motifs (N1) appear in the reported structures; chain patterns C(4) and C(9), a finite pattern D and a ring pattern R22(16), while there is also an additional binary graph set motif R22(12), in the structure of oxazoline 1m6. Only one unitary N1 motif of hydrogen bonding occurs in all structures, apart from the larger oxazolines 1m6 and 1t4, which have more than one.
Within the presented structural motifs (Fig. S9, ESI†), in C(4) and R22(12) only amino acids participate in hydrogen bonding, while in C(9) and R22(16), both amino acids and oxazolines are involved. In addition, motif D describes hydrogen bonding of the oxazoline with the methanol solvent in oxazoline 1m6.
Within a polymeric chain, central aromatic units are offset, either in a perpendicular (1p, 1n2, 1t4) or in a parallel (1m6, 1n4, 1n5, 1a) manner. Therefore, aromatic stacking is not found within the hydrogen bonded chains. However, aromatic stacking41 is present in two cases, 1n2 and 1t4. In oxazoline 1n2, two molecules of neighboring polymeric hydrogen bonded chains interact by aromatic stacking with head-to-tail orientations of oxazoline and amino acid substituents. The distance between centers of the two naphthalene rings in the stack of 1n2 is 3.7019(6) Å, while the distance between two naphthalene ring planes is 3.5021(5) Å. Moreover, an aromatic stacking interaction is also present in 1t4. This stacking involves the central aromatic ring of one molecule with the (4S)-phenyl oxazoline substituent from a neighboring molecule. The distance between centers of the two benzene rings is 3.720(2) Å, the shortest contact is 3.130(3) (HAr-ox–CAr-t), and the angle between planes of the two benzene rings is 15.7(2)°.
The molecular structure of the oxazoline bioconjugates is dominated by six dihedral angles, α, φ, θ, ϕ, ψ, and χ. The definition of these angles is highlighted in Fig. S10 (ESI†), the corresponding data are collected in Table S6 (ESI†). Structures of similar compounds reported in the literature and the corresponding data are also listed in Table S10 (ESI†). In almost all structures of oxazoline bioconjugates 1 reported herein, the oxazoline double bond and the directly attached amide bond are nearly co-planar to the central aromatic unit, with a range of angles from 5° to 22° (Table S6, dihedral angles φ and θ, ESI†). Conversely, in the anthracene derivative 1a, the dihedral angles are 93° and 80°, respectively, due to the steric hindrance from neighboring anthracene aromatic hydrogen atoms.
Another finding present in all obtained structures is that the amino acid residues directly attached to the central aromatic unit are bent on the structural backbone (χ ∼ 90°) rather than stretched. Even in tripeptide 1m6, the χ(Gly–Val) angle is 87°, while χ(Val–Phe) and χ(Phe–ester) angles are 126° and 153°, respectively. This indicates that the tripeptide chain is bent the strongest at the glycine backbone, and gradually transitions into a stretched conformation towards the C-terminal end of the tripeptide.
Supramolecular interactions in solution
1H NMR spectra of all oxazolines 1 and derivatives 2 were measured; their amide hydrogen signals are collected in Table S3 (ESI†). Six oxazolines were selected to be screened for supramolecular interactions using NMR and CD spectroscopy in solution, based on their structural variations, namely 1m1, 1p, 1m5, 1m6, 1n4, 1t1 and 1t5. Their 1H NMR spectra were recorded at concentrations of 6 mM and 60 mM in CDCl3 and 6 mM d6-DMSO and their CD spectra at 0.06 mM and 0.6 mM in CH2Cl2 (Fig. S2–S8, ESI†). The CD results showed that there are no significant intermolecular interactions in solution at concentrations below c ∼ 0.6 mM, whereas the signal values were too large for the sensitivity of the detector (250–320 nm) at concentrations higher than c ∼ 0.6 mM. Furthermore, respective hydrogen bond acidity values, ANMR,42–44 were derived from 1H NMR data (Table S4, ESI†); the obtained results confirm that there is no significant hydrogen bonding in dilute solutions.However, in 1H NMR spectra at 60 mM, N–H chemical shifts of compounds 1m6 and 1t1 showed concentration dependence in CDCl3, shifting downfield by >0.40 ppm and 0.36 ppm, respectively (Table S4, ESI†). For this reason, oxazolines 1m6 and 1t1 were chosen for temperature-dependent NMR analysis at c = 60 mM. Spectra were collected every 20 °C in a range from −40 °C to 40 °C (Fig. 2). Individual amide peaks show a difference in the corresponding chemical shift of approximately ∼1 ppm at temperatures −40 °C and 40 °C, with the exception of 1m6(Val), where the difference is ∼0.7 ppm. Calculated temperature-dependent coefficients show that all analyzed amide hydrogen atoms are involved in hydrogen bonding interactions that are strongly affected by temperature, with Δδ/ΔT values considerably lower than −2.4 ppb K−1(Fig. 2).45
 | ||
| Fig. 2 Temperature dependency of amide hydrogen atom 1H NMR peak shifts in CHCl3 and corresponding amide proton 1H NMR temperature coefficients (ppb K−1, in the upper right corner). | ||
Interestingly, several aliphatic peaks shift with the decrease in temperature as well. The 1H NMR peak of the 1m6(Val) α-hydrogen atom shifted downfield with the decrease in temperature from 40 °C to −40 °C by 0.34 ppm. More substantial changes can be seen in the temperature-dependent 1H NMR spectra of oxazoline 1t1. The two distinct aromatic peaks gradually shift upfield and switch positions with decreasing temperature, indicating weak aromatic stacking interactions. A considerable change to the oxazoline methyl peaks can be seen as well, where the two methyl peaks are not chemically equivalent (Fig. 3, Hb). This lack of equivalency becomes larger with the decreasing temperature, from Δδ = 0.02 ppm to Δδ = 0.11 ppm. It is important to note that this finding has not been detected in any other 1H NMR spectrum of oxazolines derived from the AMP precursor. Additionally, the difference in the chemical shift of each methyl peak of compound 1t1 is even smaller at c = 6 mM, with the corresponding value being Δδ < 0.01 ppm. The oxazoline methylene group multiplicity also changes from a singlet to a strongly coupled AB spin system with decreasing temperature (Fig. 3, Ha). These findings indicate that the oxazoline ring faces are not equivalent. Moreover, there is only one set of alanine peaks even at −40 °C, which is in accordance with a conserved pseudo-C2 symmetry in the supramolecular structure in the solution.
 | ||
| Fig. 3 1H NMR spectra of 1t1 oxazoline in CHCl3 at 40 °C and −40 °C, c = 60 mM. | ||
Calculations
Oxazoline bioconjugate 1t1, that showed the most interesting supramolecular interactions in solution, was selected for a detailed computational study. Structural ensembles (SE) of 1t1 conformers, both monomeric and supramolecularly assembled, were obtained by applying a CREST/CENSO protocol to chosen starting geometries, as suggested by Grimme for non-rigid molecules (see Experimental and ESI† for details).46,47 Ensemble analysis reveals that among the obtained conformers, 29 monomer and 27 dimer 1t1 conformers are statistically populated within their respective ensembles (i.e. ΔG < 2 kcal mol−1). Cartesian coordinates of individual final 1t1 conformers from each obtained monomer and dimer ensemble are collected in two separate files (calc_mono_1t1.xyz, calc_dimer_1t1.xyz), readable with the free-of-charge program Mercury from CCDC.48Within the ensemble of dimer conformers, the dominating supramolecular interactions were found to be hydrogen bonding involving all four amide protons and π–π stacking of the central aromatic units. The hydrogen bonding pattern of the amides found in the dimer ensemble can be divided into five distinct groups: (a) hydrogen bonding with oxazoline nitrogen atom, (b) semi-Herrick conformation, (c) Herrick conformation, (d) hydrogen bonding with the methoxy oxygen and (e) van Staveren conformation (Fig. S20, ESI†). With respect to found hydrogen bonding patterns and relative positions of substituents on the stacked benzene rings, 2 major types of dimers I and II can be discerned within the ensemble with Boltzmann weight ≥10% (Fig. 4). Data for the types of dimers contributing to the remaining Boltzmann weight of 25% (all the considered dimer conformers make up for 97% of Boltzmann weight) are collected in Fig. S21 and Table S10 (ESI†). The statistically most populated type of dimers was found to be of type I, bearing two amide-oxazoline hydrogen bonds (a) and two semi-Herrick hydrogen bonds (b). Interestingly, conformers containing van Staveren hydrogen bonding were found to only make up about 8%. A more detailed description of structural characteristics of dimer ensemble conformers may be found in Fig. S21 and Table S10 (ESI†).
 | ||
| Fig. 4 DFT structures of 1t1 dimers I and II and corresponding hydrogen bonding with Boltzmann weight values greater than 10%. Types of hydrogen bonding: (a) HB with oxazoline, (b) semi-Herrick, and (c) Herrick. The arrowhead indicates the direction of the amide proton donation (each arrowhead on an arrow represents one amide proton). BW = Boltzmann weight, Ox = oxazoline ring, Aa = amino acid, HBA-hydrogen bond acceptor, HBD-hydrogen bond donor. | ||
Averaged free energies of the monomer and dimer ensembles were calculated by averaging in accordance with the obtained Boltzmann distribution across respective ensembles for several temperatures (233, 253, 273, 293, 298 and 313 K). The corresponding data are collected as shown in Tables S13 and S14 (ESI†). The free energies of formation, ΔG, calculated from these data show that this model predicts the dimeric structures to be more stable than the monomers at all six temperatures by ≥4.30 kcal mol−1 (Table S15, ESI†). Moreover, the free energies of formation gradually decrease in value (i.e. increase in absolute value) with the decreasing temperature, suggesting that the dimeric structures are increasingly more stable than monomers at lower temperatures.
For conformers with a Boltzmann population >2% in each structural ensemble, the 1H–1H J couplings and 1H and 13C shieldings were calculated using the GIAO approach.49 Complete details concerning the procedure of obtaining the final Boltzmann-weighted NMR parameters for each temperature are given in the Experimental part and in the ESI† (Tables S16, S17 and Fig. S24–S27). At low temperatures (233, 253 and 273 K), the calculated shieldings for protons which were experimentally found to be chemically inequivalent were left unaveraged. Fig. S24–S27 (ESI†) show the correlations of the calculated, Boltzmann averaged shieldings (σci) and experimental shifts (δoi), for monomer and dimer ensembles, respectively. For each correlation, the MAE (mean absolute error) value defined by50
 | (1) |
| T/K | MAE (monomer) | MAE (dimer) |
|---|---|---|
| 233 | 0.13881 | 0.0818 |
| 253 | 0.13778 | 0.08488 |
| 273 | 0.13402 | 0.08862 |
| 293 | 0.13506 | 0.09311 |
| 298 | 0.13488 | 0.0932 |
| 313 | 0.13024 | 0.10213 |
From data in Table 5, an opposite trend can be seen in MAE values of monomer and dimer correlations with increased temperature, i.e. this model predicts increasingly better compatibility of the dimer model at increasingly lower temperatures, while the monomer model predicts increasingly lower compatibility with the increasingly lower temperatures. Moreover, MAE values for monomeric correlations are generally greater in value than their dimeric counterparts at all temperatures. This is in agreement with the calculated free energies of formation, which was found to favor the dimer over monomer structures at all chosen temperatures.
Summary
Two synthetic pathways are presented for obtaining derivatives 2–5 and the amino alcohol substituent of the synthesized derivatives is cyclized to afford new oxazoline–amino acid bioconjugates 1. Generally, the one-pot syntheses proved to be more advantageous for all derivatives 3, apart from derivative 3b, for which the linear synthesis was chosen due to the number of constituent building blocks. Different substituents and substitution patterns on the central aromatic unit were used, in order to study their influence on the stacking capabilities of 1 in solution and in the solid state. Single crystal structures showed that all oxazolines participate in hydrogen bonding in the solid state, forming infinite supramolecular polymeric chains.In contrast, 1H NMR spectra in CHCl3 showed that there are no significant intermolecular interactions in solution at c = 6 mM regardless of the variation of structural elements in disubstituted derivatives with only one amino acid moiety. For larger oxazolines 1m6 and 1t1, however, NMR spectra at different temperatures showed that these compounds participate in highly temperature-dependent supramolecular interactions at c = 60 mM. The results for tripeptide 1m6 suggest that in disubstituted derivatives, more than one amino acid residue is necessary to stabilize supramolecular assemblies. In the case of tris-derivatives substituted with two amino acid moieties, small-non bulky substituents facilitate supramolecular interactions in solution.
In comparison to tris-amino acid compounds,51,52 which are known to form stacking interactions, we show herein that one amino acid chain can be switched with a small oxazoline building block while preserving the stacking potential. The performed computational studies also support these results, i.e. the relative free energy of interaction at all six studied temperatures shows that the dimer ensemble is expected to be more stable than the monomer ensemble (≥4.30 kcal mol−1). Additionally, the MAE values extracted from calculated and measured NMR parameters for the monomer and dimer ensembles show an opposite trend, suggesting better compatibility of the dimer model at lower temperatures and monomer compatibility at higher temperatures. However, the overall MAE values also suggest that the dimer model is generally more compatible than the monomer model, further supporting the experimentally observed results that 1t1 molecules participate in supramolecular interactions in solution.
Experimental
General remarks
Reactions were carried out in ordinary glassware and chemicals were used as purchased from commercial suppliers without further purification. All amino acids and amino acid-derived amino alcohols used have L-configuration, unless stated otherwise. All amino acid substituents are denoted with their standard abbreviations, while amino alcohol substituents are, if applicable, denoted with their parent amino acid abbreviation and the hash symbol (e.g. Phg# stands for phenylglycinol). Coupling reagent abbreviations refer to the following compounds: 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate (TBTU), 1-hydroxybenzotriazole (HOBt), 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU), N,N′-dicyclohexylcarbodiimide (DCC), (7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (pyAOP), and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate (COMU). The synthesis of compounds 10 and 12 was carried out in a microwave reactor (CEM Discover). Reactions were monitored by TLC on silica gel 60 F254 plates and detected with a UV lamp (254 nm); crude products were purified using classic column or flash chromatography. ESI mass spectra were recorded on an HPLC-MS system (Agilent Technologies 1200) coupled using a 6410 Triple-Quadrupole mass spectrometer, operating in a positive ESI mode. High-resolution mass spectra were obtained on a MALDI TOF-TOF instrument using a CHCA matrix. CD spectra were recorded using a spectropolarimeter in 1 cm and 0.1 cm quartz Suprasil cells. Stock solutions of the isolated compounds were prepared for CD measurements of ligands. The measured ellipticity θ [deg] (in CD) is converted to the concentration-independent Δε [M−1 cm−1] through the relation Δε = θ/(l × 32982 × c), where l [cm] is the path length and c [mM] is the concentration. NMR spectra were obtained using a Bruker Avance AV300 or AV600 spectrometer, operating at 300 or 600 MHz for 1H and 75 or 150 MHz for 13C; if not indicated further, the spectra were recorded at room temperature. Chemical shifts, δ (ppm), indicate a downfield shift from the internal standard, tetramethylsilane, TMS. Coupling constants, J, are given in Hz. Individual peaks are marked as singlet (s), doublet (d), triplet (t), quartet (q), quintet (quin.) or multiplet (m). IR spectra were recorded in solid state, using an ATR Agilent Cary 630 FT-IR spectrometer or KBr pellets with a Bruker Alpha FT-IR spectrometer, in the 4000–600 cm−1 (ATR) or 4000–350 cm−1 (KBr pellets) region.
General peptide coupling procedure in DMF
Isophthalic acid (1 equivalent), L-Ala-OMe·HCl (1 equivalent), AMP (1 equivalent) and DIPEA (4 equivalent) were dissolved in DMF (15 mL) and cooled to 0 °C in an ice bath. Peptide coupling reagent (pyAOP or COMU; 2 equivalents) was added slowly by continuously adding very small portions and stirring was continued overnight. The reaction mixture was then diluted with ethyl acetate (100 mL) and washed with NaHCO3 (sat. aq., 3 × 100 mL), citric acid (10% aq., 3 × 100 mL) and NaCl (sat. aq. 100 mL). The organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure to yield the crude product.General peptide coupling procedure in DCM
Dicarboxylic acid (1 equivalent) was dissolved in DCM (100 mL). Peptide coupling reagent (TBTU/HOBt, HATU or DCC; 1 equivalent) and DIPEA (2 equivalents) were added and stirring was continued for 60 min at room temperature (TBTU/HOBt, HATU). An amino acid hydrochloride (0.5 equivalent) and amino alcohol (0.5 equivalent) in DCM (10 mL) were added dropwise and stirring was continued for 24 hours. The procedure of adding the coupling reagent, DIPEA and amines were then repeated in the same manner as stated above and stirring was continued for another 24 hours. The reaction mixture was washed with NaHCO3 (sat. aq., 3 × 100 mL), citric acid (10% aq., 3 × 100 mL) and NaCl (sat. aq., 100 mL). The organic phase was dried over NaSO4, filtered and evaporated under reduced pressure to yield the crude product.Notes
The coupling procedure for trimesic acid was carried out in a similar manner, but with adjusted amounts of reagents used (indicated below in the description of individual reaction), and in each step, only amino acid or amino alcohol was added. Detailed spectroscopic data of precursors from reactions 1–21 can be found in the ESI.†:1 starting ratio, gradually changed to 1:9. Colorless oils 2m1 (8.0 mg, 1%) and 6m1 (55.5 mg, 4%) were isolated. Fractions containing mixtures of 7m1 and 8m1 were combined, evaporated and separated by another column chromatography in 4% MeOH in the DCM solvent system. Colorless oils 7m1 (94.4 mg, 4%) and 8m1 (22.6 mg, 2%) were isolated.
:8. Colorless oil 2m1 (130.4 mg, 20%) and white solid compound 3m1 (97.0 mg, 15%) were isolated.
:75. White powder 2p (434.2 mg, 17%), a slightly yellow powder mixture of 3p and 7p (184.2 mg in total, 116.1 mg and 59.0 mg, i.e. 9% and 5%, respectively, as calculated from NMR).
:6, the ratio gradually changed to 3:7. White powders 2n1 (189.6 mg, 8%) and 3n1 (333.0 mg, 36%) were isolated.
:8, the ratio gradually changed to pure ethyl acetate. White powders 2n2 (117.6 mg, 17%) and 3n2 (134.2 mg, 18%).
:1, the ratio gradually changed to 2:8. Yellow 2n5 (136.2 mg, 18%) and white 3n5 (31.9 mg, 23%) powders were isolated.
Synthesis of derivative 3b
:1 solution and stirred for 2 h at room temperature. The solvent was then evaporated under pressure, and the remaining TFA was neutralized with DIPEA (approx. 1.5 mL). The obtained mixture was used in the next step without any further manipulation.
Traces of tetramethylurea (TMU) or tri(pyrrolidin-1-yl)phosphine oxide (TPYRPO) were present in some precursors after column chromatography, δ (TMU)/ppm: 2.80 and δ (TPYRPO)/ppm: 3.16, 1.81 in 1H NMR (CDCl3). If there was a significant amount of TMU or TPYRPO present in the samples of precursors, corresponding precursor yields were calculated from their NMR spectra.
General oxazoline synthesis procedure
Diethylaminosulfur trifluoride (1.2 equivalents in 1 mL of dry DCM) was added dropwise to a cooled solution (−78 °C) of precursor (1 equivalent) in dry DCM (14 mL). Additionally, 1 mL of DMF is added to assist dissolution if necessary. After stirring for 1 h at −78 °C, anhydrous K2CO3 (1.5 equivalents) was added in one portion and the mixture was allowed to warm to room temperature. The reaction was quenched with saturated aqueous NaHCO3 (20 mL). The biphasic mixture was extracted with EtOAc (3 × 40 mL). The combined organic extracts were washed with water (100 mL) and NaCl (sat. aq., 100 mL), dried over anhydrous Na2SO4 and concentrated.34:3. Yield: 173.3 mg (0.6 mmol, 57%), colorless oil. Mr (C16H20N2O4) = 304.14. ESI-MS (m/z): 305.2 (M + H+). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.29 (s, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 6.77 (d, J = 6.6 Hz, 1H), 4.82 (quin., J = 7.2 Hz, 1H), 4.14 (s, 2H), 3.79 (s, 3H), 1.53 (d, J = 7.2 Hz, 3H), 1.40 (s, 6H). 13C NMR (151 MHz, CDCl3) δ/ppm: 173.64, 166.17, 161.46, 134.25, 131.47, 130.61, 128.94, 128.57, 126.24, 79.42, 67.94, 52.71, 48.70, 28.54, 28.52, 18.67. ESI-HRMS (m/z): expected 305.15, 327.13 (C16H20N2O4 + H+, C16H20N2O4 + Na+), observed 305.1492, 327.1307. IR (KBr): 3344 (w), 2970 (w), 1745 (m), 1650 (s), 1166 (m), 1540 (m), 1213 (m), 1064 (w), 973 (w), 712 (m).
:7. Yield: 62.1 mg (0.2 mmol, 57%), yellow powder. Mr (C16H20N2O4) = 304.14. ESI-MS (m/z): 305.1 (M + H+). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.01 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 8.5 Hz, 2H), 6.76 (d, J = 6.5 Hz, 1H), 4.81 (quin., J = 7.2 Hz, 1H), 4.13 (s, 2H), 3.80 (s, 3H), 1.54 (d, J = 7.1 Hz, 3H), 1.40 (s, 6H). 13C NMR (75 MHz, CDCl3) δ/ppm: 173.75, 166.19, 161.53–161.03 (m), 136.22, 131.30, 128.61, 127.16, 79.42, 68.00, 52.78, 48.71, 28.52, 18.77. ESI-HRMS (m/z): expected 305.15, 327.13, 631.27 (C16H20N2O4 + H+, C16H20N2O4 + Na+, 2(C16H20N2O4) + Na+), observed 305.1492, 327.1307, 631.2725. IR (KBr): 3273 (s), 3195 (m), 1654 (s), 1858 (m), 1534 (s), 1455 (m), 1368 (m), 1296 (m), 1038 (m), 759 (m), 608 (w), 533 (w), 442 (w).
:8. Yield: 197.8 mg (0.6 mmol, 68%), white solid. Mr (C20H20N2O4) = 352.14. ESI-MS (m/z): 353.10 (M + H+, 100%), 705.25 (2M + H+, 13%), 727.20 (2M + Na+, 9%). 1H NMR (600 MHz, CDCl3) δ/ppm: 8.27 (t, J = 1.8 Hz, 1H), 8.09 (dt, J = 7.8, 1.4 Hz, 1H), 7.89 (dt, J = 7.7, 1.5 Hz, 1H), 7.49 (t, J = 7.8 Hz, 1H), 7.34–7.21 (m, 3H), 7.18–7.10 (m, 2H), 6.63 (d, J = 7.6 Hz, 1H), 5.16–5.03 (m, 1H), 4.54–4.32 (m, 2H), 4.09 (t, J = 9.5 Hz, 2H), 3.76 (s, 3H), 3.36–3.16 (m, 2H). 13C NMR (151 MHz, CD3OD) δ/ppm: 173.52, 169.26, 166.18, 138.48, 135.82, 132.20, 131.55, 130.22, 129.88, 129.53, 128.25, 127.88, 69.32, 55.96, 55.28, 52.79, 38.14. MALDI-HRMS (m/z): expected 353.15 (C20H20N2O4 + H+), observed 353.1704. IR (ATR): 3273 (w), 2954 (m), 1759 (w), 1737 (m), 1646 (m), 1541 (m), 1439 (m), 1275 (m), 1256 (m), 1183 (m), 950 (m), 702 (m).
:3. Yield: 74.3 mg (0.2 mmol, 47%), white solid. Mr (C17H22N2O4) = 318.16. ESI-MS (m/z): 319.15 (M + H+, 100%). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.32 (s, 1H), 8.04 (ddt, J = 31.8, 7.7, 1.5 Hz, 2H), 7.50 (t, J = 7.8 Hz, 1H), 6.80 (d, J = 7.3 Hz, 1H), 4.82 (quin., J = 7.2 Hz, 1H), 4.44 (td, J = 7.3, 2.1 Hz, 1H), 4.23–4.00 (m, 2H), 3.79 (s, 3H), 1.87 (quin, J = 6.6 Hz, 1H), 1.54 (d, J = 7.2 Hz, 3H), 1.04 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H). 13C NMR (151 MHz, CD3OD) δ/ppm: 174.73, 169.20, 165.23, 135.76, 132.30, 131.65, 131.64, 129.92, 129.12, 128.42, 73.29, 71.55, 52.81, 50.18, 33.82, 18.84, 18.19, 17.21. MALDI-HRMS (m/z): expected 637.32 (2(C16H20N2O4) + H+), observed 637.3234. IR (ATR): 3312 (w), 2961 (w), 1741 (m), 1644 (m), 1536 (m), 1448 (m), 1213 (m), 1169 (m), 967 (m), 788 (m), 704 (m).
:7 → EtOAc: hexane = 1:1. Yield: 226,2 mg (0.6 mmol, 67%), white solid. Mr (C21H22N2O4) = 366.16. ESI-MS (m/z): 367.15 (M + H+, 100%), 733.25 (2M + H+, 11%). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.31 (t, J = 1.8 Hz, 1H), 8.04 (ddt, J = 26.5, 7.9, 1.5 Hz, 2H), 7.51 (t, J = 7.8 Hz, 1H), 7.40–7.19 (m, 5H), 6.80 (d, J = 7.3 Hz, 1H), 4.82 (quin., J = 7.2 Hz, 1H), 4.70–4.53 (m, 1H), 4.27 (dt, 2H), 3.79 (s, 3H), 3.32–2.65 (m, 2H), 1.54 (d, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ/ppm: 173.66, 166.20, 163.44, 137.89, 134.31, 131.50, 130.71, 129.38, 128.99, 128.82, 128.74, 128.22, 126.75, 126.42, 72.23, 68.04, 52.71, 48.72, 41.88, 18.64. MALDI-HRMS (m/z): expected 367.1658, 389.1469 (C21H22N2O4 + H+, C21H22N2O4 + Na+), observed 367.1649, 389.1469. IR (ATR): 3317 (w), 2960 (w), 1735 (m), 1649 (m), 1528 (m), 1265 (m), 1210 (m), 1165 (m), 972 (m), 734 (m), 700 (s).
:1 → pure EtOAc. Yield: 66.6 mg (0.2 mmol, 36%), colorless oil. Mr (C20H20N2O4) = 352.14. ESI-MS (m/z): 353.10 (M + H+, 100%), 705.25 (2M + H+, 17%), 727.20 (2M + Na+, 9%). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.43 (t, J = 1.8 Hz, 1H), 8.10 (ddt, J = 38.6, 7.8, 1.5 Hz, 2H), 7.54 (t, J = 7.8 Hz, 1H), 7.42–7.18 (m, 5H), 6.85 (d, J = 7.4 Hz, 1H), 5.41 (dd, J = 10.2, 8.2 Hz, 1H), 4.93–4.76 (m, 2H), 4.32 (t, J = 8.3 Hz, 1H), 3.78 (s, 3H), 1.53 (d, J = 7.2 Hz, 3H), 1.26 (t, 2H). 13C NMR (151 MHz, CDCl3) δ/ppm: 173.63, 166.12, 164.14, 142.10, 134.32, 131.66, 130.96, 129.04, 128.98, 127.97, 127.93, 126.90, 126.86, 126.57, 75.20, 70.35, 52.69, 48.74, 48.70, 18.58. MALDI-HRMS (m/z): expected 353.1501, 375.1315 (C20H20N2O4 + H+, C20H20N2O4 + Na+), observed 353.1493, 375.1316. IR (ATR): 3291 (w), 2954 (w), 1737 (m), 1638 (s), 1536 (m), 1265 (m), 1211 (m), 1169 (m), 699 (s).
:1. Yield: 74.3 mg (0.2 mmol, 42%), white powder. Mr (C20H22N2O4) = 354.16. ESI-MS (m/z): 355.2 (M + H+), 709.4 (2M + H+). 1H NMR (600 MHz, CDCl3) δ/ppm: 9.08 (d, J = 8.2 Hz, 1H), 8.33 (d, J = 7.9 Hz, 1H), 8.01 (d, J = 7.4 Hz, 1H), 7.66–7.56 (m, 3H), 6.53 (d, J = 7.3 Hz, 1H), 4.92 (quin., J = 7.2 Hz, 1H), 4.17 (s, 2H), 3.83 (s, 3H), 1.61–1.57 (m, 3H), 1.49 (s, 6H). 13C NMR (151 MHz, CDCl3) δ/ppm: 173.47, 168.78, 161.45, 136.97, 131.59, 130.49, 127.82, 127.69, 127.66, 127.40, 126.80, 125.70, 123.91, 78.58, 68.78, 52.78, 48.72, 28.67, 18.59. ESI-HRMS (m/z): expected 355.17, 377.15, 393.12 (C20H22N2O4 + H+, C20H22N2O4 + Na+, C20H22N2O4 + K+), observed 355.1643, 377.1463, 393.1202.
:1. Yield: 119.9 mg (0.4 mmol, 35%), white powder. Mr (C19H20N2O4) = 340.14. ESI-MS (m/z): 341.1 (M + H+). 1H NMR (300 MHz, CDCl3) δ/ppm: 9.12–9.01 (m, 1H), 8.41–8.28 (m, 1H), 8.01 (d, J = 7.4 Hz, 1H), 7.69–7.55 (m, 3H), 6.55 (t, J = 5.0 Hz, 1H), 4.34 (d, J = 5.4 Hz, 2H), 4.17 (s, 2H), 3.83 (s, 3H), 1.49 (s, 6H). 13C NMR (151 MHz, CD3OD) δ/ppm: 172.34, 171.68, 164.53, 138.67, 132.37, 131.64, 128.63, 128.59, 128.34, 127.12, 127.00, 125.08, 80.22, 69.24, 52.74, 42.28, 28.53. ESI-HRMS (m/z): expected 341.15, 363.13, 379.11 (C19H20N2O4 + H+, C19H20N2O4 + Na+, C19H20N2O4 + K+), observed 341.1488, 363.1305, 379.1045. IR (KBr): 3447 (w), 3300 (m), 2970 (w), 1736 (s), 1643 (m), 1528 (m), 1324 (w), 1276 (m), 1246 (m), 1113 (w), 1008 (w), 1004 (w), 865 (w), 778 (w).
:65. Yield: 147.1 mg (0.4 mmol, 42%), white powder. Mr (C20H22N2O4) = 354.16. ESI-MS (m/z): 355.1 (M + H+). 1H NMR (300 MHz, CDCl3) δ/ppm: 9.19 (d, J = 8.6 Hz, 1H), 8.47 (d, J = 8.6 Hz, 1H), 8.11–8.04 (m, 1H), 7.72–7.65 (m, 1H), 7.61–7.52 (m, 2H), 6.56 (d, J = 7.3 Hz, 1H), 4.91 (quin., J = 7.2 Hz, 1H), 4.16 (s, 2H), 3.82 (s, 3H), 1.59 (d, 3H), 1.48 (s, 6H). 13C NMR (75 MHz, CDCl3) δ/ppm: 134.25, 131.55, 130.56, 129.41, 129.36, 128.99, 126.06, 126.05, 125.54, 125.44, 78.51, 68.66, 52.75, 48.71, 28.69, 18.61. ESI-HRMS (m/z): expected 355.17, 377.15, 731.31 (C20H22N2O4 + H+, C20H22N2O4 + Na+, 2(C20H22N2O4) + Na+), observed 355.1642, 377.1461, 731.3026. IR (KBr): 3421 (w), 3341 (w), 2969 (w), 1745 (m), 1642 (m), 1528 (w), 1215 (w), 1197 (w), 1160 (w), 1040 (w), 797 (w).
:4, ratio gradually changed to 1:1. Yield: 76.3 mg (0.2 mmol, 43%), white powder. Mr (C20H22N2O4) = 354.16. ESI-MS (m/z): 355.2 (M + H+), 709.3 (2M + H+). Crystals suitable for single-crystal X-ray diffraction were obtained from solution in NMR tube after several months. 1H NMR (600 MHz, CDCl3) δ/ppm: 8.47 (s, 1H), 8.32 (s, 1H), 8.09 (d, J = 8.6 Hz, 1H), 7.95 (t, J = 8.4 Hz, 2H), 7.89 (d, J = 8.4 Hz, 1H), 6.89 (d, J = 6.8 Hz, 1H), 4.87 (quin., J = 7.1 Hz, 1H), 4.18 (s, 2H), 3.82 (s, 3H), 1.57 (d, J = 7.1 Hz, 3H), 1.43 (s, 6H). 13C NMR (151 MHz, CDCl3) δ/ppm: 173.82, 166.73, 161.96, 134.30, 133.94, 132.68, 129.56, 129.17, 128.45, 127.47, 127.34, 125.99, 124.40, 79.45, 67.98, 52.78, 48.77, 28.59, 18.82. ESI-HRMS (m/z): expected 355.17, 377.15, 731.31 (C20H22N2O4 + H+, C20H22N2O4 + Na+, 2(C20H22N2O4) + Na+), observed 355.1642, 377.1464, 731.3038. IR (KBr): 3474 (w), 3358 (w), 2973 (w), 1752 (m), 1639 (s), 1523 (m), 1207 (m), 1188 (m), 1161 (m), 1062 (m), 979 (w), 912 (w), 823 (w), 758 (w), 717 (w), 483 (w).
:1. Yield: 48.8 mg (0.1 mmol, 14%), white powder. Mr (C20H22N2O4) = 354.16. ESI-MS (m/z): 355.2 (M + H+). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.50 (d, J = 1.3 Hz, 1H), 8.37 (s, 1H), 8.17–8.09 (m, 1H), 7.94–7.86 (m, 3H), 6.88 (d, J = 7.3 Hz, 1H), 4.86 (quin., J = 7.2 Hz, 1H), 4.18 (s, 2H), 3.82 (s, 3H), 1.57 (d, J = 7.1 Hz, 3H), 1.43 (s, 6H). 13C NMR (75 MHz, CDCl3) δ/ppm: 173.80, 166.69, 161.90, 136.12, 132.11, 131.99, 129.62, 128.56, 128.51, 128.00, 127.00, 126.59, 125.26, 79.43, 67.96, 52.77, 48.78, 28.59, 18.82. ESI-HRMS (m/z): expected 355.17, 377.15, 731.31 (C20H22N2O4 + H+, C20H22N2O4 + Na+, 2(C20H22N2O4) + Na+), observed 355.1645, 377.1464, 731.3037. IR (KBr): 3444 (w), 3260 (w), 3045 (w), 2978 (w), 1740 (s), 1658 (s), 1637 (m), 1544 (m), 1316 (m), 1228 (m), 1168 (m), 1076 (m), 959 (w), 867 (w), 710 (w).
:8. Yield: 28.3 mg (0.1 mmol, 13%), yellow solid. 1H NMR (300 MHz, CD3CN) δ/ppm: 8.46 (d, J = 1.7 Hz, 2H), 8.38 (s, 1H), 7.93 (s, 1H), 7.62 (s, 2H), 4.50 (s, 1H), 4.27–4.02 (m, 6H), 3.71 (s, 6H), 1.11–0.87 (m, 6H). 13C NMR (151 MHz, CD3OD) δ/ppm: 171.77, 171.73, 169.02, 168.92, 168.75, 164.31, 137.39, 136.19, 135.98, 132.61, 131.02, 130.41, 130.37, 130.06, 129.85, 73.58, 71.86, 63.36, 63.13, 59.13, 52.75, 52.72, 42.47, 42.46, 33.89, 30.33, 20.09, 19.31, 18.86, 18.34. MALDI-HRMS (m/z): expected 420.19 (C20H25N3O7 + H+), observed 420.1796. IR (ATR): 3315 (w), 2961 (w), 2930 (w), 1741 (m), 1655 (m), 1534 (m), 1219 (m), 1088 (m), 734 (s), 704 (m).
:ethyl acetate = 1:1. Yield: 168.6 mg (0.3 mmol, 38%), colorless oil. Mr (C37H35N3O7) = 633.25. ESI-MS (m/z): 634.1 (M + H+), 1267.3 (2M + H+, 11%). 1H NMR (300 MHz, CDCl3) δ/ppm: 8.47 (d, J = 1.7 Hz, 2H), 8.27 (s, 1H) 7.47–7.03 (m, 15H), 6.74 (d, J = 7.7 Hz, 2H), 5.42 (d, J = 1.8 Hz, 1H), 5.09 (dt, J = 7.8, 6.0 Hz, 2H), 4.85 (dd, J = 10.2, 8.5 Hz, 1H), 4.33 (t, J = 8.4 Hz, 1H), 3.75 (s, 6H), 3.25 (qd, J = 13.9, 6.0 Hz, 4H). 13C NMR (75 MHz, (CD3)2CO) δ/ppm: 172.85, 166.24, 163.74, 143.74, 138.47, 135.97, 130.46, 130.25, 130.15, 129.59, 129.46, 129.34, 128.43, 127.76, 127.62, 76.00, 71.01, 55.52, 52.51, 38.02. MALDI-HRMS (m/z): expected 634.26 (C37H35N3O7 + H+), observed 634.2578. IR (ATR): 3312 (w), 3032 (w), 2954 (w), 1737 (m), 1655 (s), 1524 (m), 1215 (m), 1100 (m), 980 (m), 734 (m), 699 (s).
:ethyl acetate = 1:1. Yield: 363.0 mg (0.6 mmol, 57%), white solid. Mr (C37H35N3O7) = 633.25. ESI-MS (m/z): 634.2 (M + H+), 1267.4 (2M + H+, 13%). 1H NMR (300 MHz, CD3CN) δ/ppm: 8.42 (d, J = 1.7 Hz, 2H), 8.21 (t, J = 1.8 Hz, 1H), 7.50 (d, J = 8.0 Hz, 2H), 7.45–7.11 (m, 15H), 5.43 (dd, J = 10.1, 8.1 Hz, 1H), 4.92-4.85 (m, 3H), 4.27 (t, J = 8.3 Hz, 1H), 3.69 (s, 6), 3.39–3.00 (m, 4H). 13C NMR (151 MHz, CD3OD) δ/ppm: 173.44, 168.40, 165.47, 143.22, 138.36, 136.38, 131.10, 130.75, 130.22, 129.90, 129.57, 129.31, 128.94, 127.92, 127.86, 76.73, 71.06, 56.03, 52.84, 38.20. IR (KBr): 3419 (m), 3030 (w), 2953 (w), 2493 (w), 1742 (s), 1660 (s), 1600 (m), 1455 (s), 1435 (s), 1410 (m), 1357 (m), 1234 (m), 1204 (m), 1180 (m), 1100 (m), 983 (m), 758 (m), 701 (s).
:1. Yield: 97.9 mg (0.2 mmol, 34%), white solid. Mr (C35H31N3O5) = 573.23. ESI-MS (m/z): 574.1 (M + H+), 1147.4 (2M + H+, 13%). 1H NMR (300 MHz, CD3CN) δ/ppm: 8.63 (d, J = 1.6 Hz, 1H), 8.45 (d, J = 1.6 Hz, 2H), 7.54 (d, J = 8.1 Hz, 1H), 7.45–7.07 (m, 15H), 5.44 (dd, J = 10.1, 8.3 Hz, 2H), 4.89 (dd, J = 10.2, 8.5 Hz, 3H), 4.28 (t, J = 8.4 Hz, 2H), 3.69 (s, 3H), 3.38–2.95 (m, 2H). 13C NMR (151 MHz, CD3OD) δ/ppm: 173.40, 168.34, 165.43, 143.23, 138.42, 136.70, 131.93, 131.43, 130.23, 129.90, 129.59, 129.56, 128.93, 127.90, 127.84, 76.79, 71.06, 56.02, 52.84, 38.16. MALDI-HRMS (m/z): expected 574.23 (C35H31N3O5 + H+), observed 574.2406. IR (ATR): 3312 (w), 3066 (w), 3032 (w), 2954 (w), 2904 (w), 1737 (m), 1655 (m), 1541 (m), 1269 (m), 1215 (m), 980 (m), 734 (m), 697 (s).
:ethyl acetate = 6:4. Yield: 52.8 mg (0.2 mmol, 34%), white solid. Mr (C33H27N3O3) = 513.21. ESI-MS (m/z): 514.20 (M + H+, 100%), 1027.4 (2M + H+, 19%). 1H NMR (300 MHz, CDCl3) δ/ppm. 8.84 (s, 3H), 7.59–6.88 (m, 15H), 5.43 (dd, J = 10.2, 8.3 Hz, 3H), 4.83 (dd, J = 10.2, 8.5 Hz, 3H), 4.31 (t, J = 8.4 Hz, 3H). 13C NMR (151 MHz, CD3OD) δ/ppm: 165.33, 143.21, 132.16, 129.89, 129.86, 128.90, 127.81, 76.82, 71.07. MALDI-HRMS (m/z): expected 514.21 (C33H27N3O3 + H+), observed 514.2167. IR (ATR): 2963 (w), 2919 (m), 2851 (m), 1649 (m), 1269 (m), 1236 (m), 965 (m), 915 (m), 699 (s).
X-ray single crystal diffraction
X-ray intensity data for 2m1–1n5 were collected at room temperature (293 K) with an Xcalibur or XtaLAB diffractometer using monochromatic Cu-Kα radiation (λ = 1.54184 Å). The data were processed using the CrysalisPro program54 (unit cell determination and data reduction). The structures were solved with the program SXELXT55 and refined according to the least-squares procedure (F2 on all data) by the program SHELXL-2018.56 Basic experimental data are given in Tables S7 and S8 of the ESI.† Due to the faster scans used in the measurements of 2m1 and 1n4, a smaller number of reflections were obtained; however, the quality of the final structural parameters for these compounds remained very high (R values, min. and max. electron density, Goodness of fit, etc.). The absolute configurations of investigated compounds were known from the synthetic procedures, so the Friedel opposite reflections were not measured. All non-hydrogen atoms are refined in the anisotropic model of atomic displacement parameters (ADP). Terminal phenyl groups in 3t3 were treated as rigid rings with the ideal geometry (AFIX 66) and one of them was refined over two disorderly occupied positions (orientations), with refined occupancies of 0.55(2) and 0.45(2), respectively. Rigid body restraints for ADP parameters (RIGU, ISOR) were also used for the carbon atoms of terminal phenyl rings in the structure of 3t3. Hydrogen atoms bonded to carbon atoms were treated in the riding rigid body models, i.e. their positions were calculated from the positions of carbon atoms. Torsion angles of methyl and hydroxyl groups in all compounds (including the solvent methanol molecule in the structure of 1m6) were determined by the best fit to the difference in electron density (HFIX 137 or HFIX 147, respectively). Also, the rigid body restrains (RIGU) for ADP parameters for atoms of the solvent methanol molecule. Exceptions from the rigid body treatment were the hydrogen atoms bonded to the nitrogen atoms of the amide groups, due to their participation in the hydrogen bonds. These atoms were refined free, including their isotropic ADP parameters. Additional N–H distance restraints were used for 1m6, 1t3 and 1t4 structures. The CCDC 2335938–2335948† contains the supplementary crystallographic data for this paper.DFT calculations
Calculation of NMR parameters and structural ensembles (SE) of low energy conformers of 1t1 and supramolecular dimers of 1t1 in CHCl3 solution was calculated according to the literature suggested CREST/CENSO workflow in several steps:57,58Each starting structural model was used as an input in two independent CREST conformational search jobs,57 with the aim to find other possible, more stable dimer aggregates of 1t1 molecules. These jobs used the NCI-iMTD workflow, i.e. iterative meta-dynamic simulations (MTD) with additional ellipsoid wall potential.57 CREST jobs with vS1 or vS2 starting models ended with 95 or 93 conformers (i.e. the structural models of 1t1-dimer aggregates) within the energy threshold of 6 kcal mol−1, respectively.
The final Boltzmann-weighted NMR parameters for each temperature was calculated using the relation:
 | (2) |
 | (3) |
Calculated Boltzmann-weighted NMR parameters are given in Tables S16 and S17 (ESI†) for monomer and dimer ensembles, respectively. Fig. S24–S27 (ESI†) show the correlations of the calculated, Boltzmann averaged shieldings (σci) and experimental shifts (δoi), for monomer and dimer ensembles, respectively. For each correlation, the MAE (mean absolute error) values defined by eqn (1) (see results and discussion)50 were determined. Amide 1H calculated shifts using eqn (2) are also listed in Tables S16 and S17 (ESI†), as well as its absolute error (AE = |δci − δoi|) from the experimental value.
All single-point, geometry optimizations and GIAO calculations used by CREST (version 2.12) and CENSO (version 1.2.0) software were performed by the ORCA software (version 5.0.1).67 All calculations of averaged NMR parameters and linear regression calculations were performed using Excel software68 and they are available in two Excel tables in ESI† (NMR_mono_1t1.xlsx and NMR_dimer_1t1.xlsx).
Author contributions
M. B. prepared and characterized the compounds, performed the spectroscopic studies and wrote the manuscript draft. B. P. performed the crystallographic and DFT studies. S. I. K. conceived the study, supervised the experiments and finalized the manuscript. All authors have approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CAT Pharma (KK.01.1.1.04.0013), a project co-financed by the Croatian Government and the European Union through the European Regional Development Fund – the Competitiveness and Cohesion Operational Program; and Cage Cat (IP-2022-10-8456), a project financed by the Croatian Science Foundation. M. B. acknowledges the Croatian Science Foundation for a doctoral scholarship (DOK-2021–02-7366). We thank Dr Zoran Kokan, Dr Natalija Pantalon Juraj and Dr Saša Opačak for their helpful discussions.References
- J. A. Frump, Chem. Rev., 1971, 71, 483–505 CrossRef CAS.
- G. C. Hargaden and P. J. Guiry, Chem. Rev., 2009, 109, 2505–2550 CrossRef CAS PubMed.
- R. Connon, B. Roche, B. V. Rokade and P. J. Guiry, Chem. Rev., 2021, 121, 6373–6521 CrossRef CAS PubMed.
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726–728 CrossRef CAS.
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- T. Lorson, M. M. Lübtow, E. Wegener, M. S. Haider, S. Borova, D. Nahm, R. Jordan, M. Sokolski-Papkov, A. V. Kabanov and R. Luxenhofer, Biomaterials, 2018, 178, 204–280 CrossRef CAS PubMed.
- H. Wang, Z. Bai, T. Jiao, Z. Deng, H. Tong, G. He, Q. Peng and G. Chen, J. Am. Chem. Soc., 2018, 140, 3542–3546 CrossRef CAS PubMed.
- J. Holz, M. Ayerbe García, W. Frey, F. Krupp and R. Peters, Dalton Trans., 2018, 47, 3880–3905 RSC.
- M. W. Chojnacka, J. A. Adjei, A. J. Lough, G. G. Sacripante and R. A. Gossage, Z. Naturforsch., B: J. Chem. Sci., 2020, 75, 371–377 CrossRef CAS.
- C.-L. Liu, L.-P. Zhou, D. Tripathy and Q.-F. Sun, Chem. Commun., 2017, 53, 2459–2462 RSC.
- Y.-Q. Huang and W.-Y. Sun, CrystEngComm, 2018, 20, 6109–6121 RSC.
- M. Durini, E. Russotto, L. Pignataro, O. Reiser and U. Piarulli, Eur. J. Org. Chem., 2012, 5451–5461 CrossRef CAS.
- S. Dakovic, L. Liscic-Tumir, S. I. Kirin, V. Vinkovic, Z. Raza, A. Suste and V. Sunjic, J. Mol. Catal. A: Chem., 1997, 118, 27–31 CrossRef CAS.
- Y. Liu, Y. Wang, Y. Wang, J. Lu, V. Piñón and M. Weck, J. Am. Chem. Soc., 2011, 133, 14260–14263 CrossRef CAS PubMed.
- D. Obrecht, C. Abrecht, M. Altorfer, U. Bohdal, A. Grieder, M. Kleber, P. Pfyffer and K. Müller, Helv. Chim. Acta, 1996, 79, 1315–1337 CrossRef CAS.
- S. Dahiya and R. Dahiya, Eur. J. Med. Chem., 2021, 218, 113406 CrossRef CAS PubMed.
- K. Arora, P. M. Sherilraj and S. L. Mudavath, Comprehensive Analytical Chemistry, Elsevier, 2023, vol. 103, pp. 1–28 Search PubMed.
- S. I. Kirin, H.-B. Kraatz and N. Metzler-Nolte, Chem. Soc. Rev., 2006, 35, 348–354 RSC.
- N. Pantalon Juraj, T. Tandarić, V. Tadić, B. Perić, D. Moreth, U. Schatzschneider, A. Brozovic, R. Vianello and S. I. Kirin, Dalton Trans., 2022, 51, 17008–17021 RSC.
- M. Pernar, Z. Kokan, J. Kralj, Z. Glasovac, L.-M. Tumir, I. Piantanida, D. Eljuga, I. Turel, A. Brozovic and S. I. Kirin, Bioorg. Chem., 2019, 87, 432–446 CrossRef CAS PubMed.
- Z. Kokan and S. I. Kirin, RSC Adv., 2012, 2, 5729–5737 RSC.
- Z. Kokan, B. Kovačević, Z. Štefanić, P. Tzvetkova and S. I. Kirin, Chem. Commun., 2018, 54, 2094–2097 RSC.
- C. Saavedra, R. Hernández, A. Boto and E. Álvarez, J. Org. Chem., 2009, 74, 4655–4665 CrossRef CAS PubMed.
- S. E. Snyder, B.-S. Huang, Y.-T. Chen, H.-S. Lin and J. R. Carey, Org. Lett., 2012, 14, 3442–3445 CrossRef CAS PubMed.
- Z. Kokan, B. Perić, M. Vazdar, Ž. Marinić, D. Vikić-Topić, E. Meštrović and S. I. Kirin, Chem. Commun., 2017, 53, 1945–1948 RSC.
- X. Caumes, A. Baldi, G. Gontard, P. Brocorens, R. Lazzaroni, N. Vanthuyne, C. Troufflard, M. Raynal and L. Bouteiller, Chem. Commun., 2016, 52, 13369–13372 RSC.
- M. Raynal, Y. Li, C. Troufflard, C. Przybylski, G. Gontard, T. Maistriaux, J. Idé, R. Lazzaroni, L. Bouteiller and P. Brocorens, Phys. Chem. Chem. Phys., 2021, 23, 5207–5221 RSC.
- F. Perlitius, A. Walczak, M. Čonková, G. Markiewicz, J. Harrowfield and A. R. Stefankiewicz, J. Mol. Liq., 2022, 367, 120511 CrossRef CAS.
- B. Gong, C. Zheng and Y. Yan, J. Chem. Crystallogr., 1999, 29, 649–652 CrossRef CAS.
- Z. Kokan and S. I. Kirin, Eur. J. Org. Chem., 2013, 8154–8161 CrossRef CAS.
- S. I. Kirin, D. Wissenbach and N. Metzler-Nolte, New J. Chem., 2005, 29, 1168–1173 RSC.
- G. Xu, Q. Luo, S. Eibauer, A. F. Rausch, S. Stempfhuber, M. Zabel, H. Yersin and O. Reiser, Dalton Trans., 2011, 40, 8800–8806 RSC.
- S. Opačak, B. Perić, T. Gojšić, A. Čikoš, D. Vikić-Topić and S. I. Kirin, New J. Chem., 2022, 46, 13275–13285 RSC.
- S. Beheshti, S. Martić and H. Kraatz, Chem. – Eur. J., 2012, 18, 9099–9105 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- J. Bernstein, R. E. Davis, L. Shimoni and N. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- M. W. Powner, J. D. Sutherland and J. W. Szostak, J. Am. Chem. Soc., 2010, 132, 16677–16688 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- B. Ishimoto, K. Tonan and S. Ikawa, Spectrochim. Acta, Part A, 2000, 56, 201–209 CrossRef PubMed.
- L. Barišić, M. Čakić, K. A. Mahmoud, Y. Liu, H. Kraatz, H. Pritzkow, S. I. Kirin, N. Metzler-Nolte and V. Rapić, Chem. – Eur. J., 2006, 12, 4965–4980 CrossRef PubMed.
- M. H. Abraham and R. J. Abraham, New J. Chem., 2017, 41, 6064–6066 RSC.
- E. S. Stevens, N. Sugawara, G. M. Bonora and C. Toniolo, J. Am. Chem. Soc., 1980, 102, 7048–7050 CrossRef CAS.
- M. Bursch, J. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- S. Grimme, C. Bannwarth, S. Dohm, A. Hansen, J. Pisarek, P. Pracht, J. Seibert and F. Neese, Angew. Chem., Int. Ed., 2017, 56, 14763–14769 CrossRef CAS PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- P. H. Willoughby, M. J. Jansma and T. R. Hoye, Nat. Protoc., 2014, 9, 643–660 CrossRef CAS PubMed.
- P. P. Bose, M. G. B. Drew, A. K. Das and A. Banerjee, Chem. Commun., 2006, 3196–3198 RSC.
- A. Desmarchelier, M. Raynal, P. Brocorens, N. Vanthuyne and L. Bouteiller, Chem. Commun., 2015, 51, 7397–7400 RSC.
- H.-J. Kim, D. Moon, M. S. Lah and J.-I. Hong, Angew. Chem., Int. Ed., 2002, 41, 3174–3177 CrossRef CAS PubMed.
- CrysAlisPro (Version 1.171.39.46), Rigaku Oxford Diffraction Ltd, Yarnton, Oxfordshire, England., 2018.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, F. Bohle, A. Hansen, P. Pracht, S. Spicher and M. Stahn, J. Phys. Chem. A, 2021, 125, 4039–4054 CrossRef CAS PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, WIREs Comput. Mol. Sci., 2021, 11, e1493 CrossRef CAS.
- S. Ehlert, M. Stahn, S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 4250–4261 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, S. Ehlert and J.-M. Mewes, J. Chem. Phys., 2021, 154, 064103 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Spicher and S. Grimme, J. Chem. Theory Comput., 2021, 17, 1701–1714 CrossRef CAS PubMed.
- F. Jensen, Theor. Chem. Acc., 2010, 126, 371–382 Search PubMed.
- F. Jensen, J. Chem. Theory Comput., 2015, 11, 132–138 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- Microsoft Corporation, 2018. Microsoft Excel, Available at: https://office.microsoft.com/excel.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, schemes, comprehensive spectroscopic characterisation and computational details. CCDC 2335938–2335948. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj00995a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |