
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Yttria-stabilized zirconia (8YSZ) synthesis in a supercritical CO2-assisted process: a parametric study for achieving cubic phase stability
Loan
Avédikian
 a,
Julien
Vulliet
b,
Thomas
David
c and
Audrey
Hertz
*a
a,
Julien
Vulliet
b,
Thomas
David
c and
Audrey
Hertz
*a
aCEA, DES, ISEC, DMRC, Univ. Montpellier, Bagnols-sur-Cèze, F-30207, France. E-mail: audrey.hertz@cea.fr
bCEA, DAM, Le Ripault, Monts, F-37260, France
cCEA, LITEN, Univ. Grenoble Alpes, Grenoble, F-38054, France
First published on 22nd May 2024
Abstract
Yttria-stabilized zirconia (YSZ) with a yttria content beyond 8%mol is especially interesting for its high ionic conductivity in the high-temperature range (700–1000 °C). Thereby, cubic YSZ properties make it a useful ceramic material for electrodes or electrolytes in various applications, such as solid oxide cell (SOC) technologies for energy conversion or oxygen sensors. This work aims to propose an alternative microstructure, hopefully, to improve material features. Adapting a sol–gel-type process that involves the use of supercritical CO2 (scCO2) to stabilize doped-zirconia nanoparticles in their cubic structure allows the crystallization of powders at temperatures as low as 250 °C. The resulting powders are very fine, with large specific surface areas around 50–150 m2 g−1, low agglomeration, and small-sized crystallites around 8–10 nm. After studying the effect of the operating conditions on the stabilization of the cubic crystalline phase, a well-adapted powder was selected and characterized in detail with respect to its microstructure, and its thermal behavior was investigated to eventually prepare an electrode/electrolyte.
Introduction
Zirconia has aroused significant interest for its thermal resistance, chemical inertness and low thermal conductivity.1 These properties make zirconia an attractive ceramic material for high-temperature-resistant applications, alongside alumina and silica.2 The doping of zirconia with aliovalent cation oxides, such as CeO2, CaO, La2O3, Sc2O3 or Y2O3, makes it even more interesting. Indeed, the creation of atomic vacancies gives the material its ionic conductivity, allowing the transport of O2− oxide ions.3–5 Moreover, Zr4+ cation substitution also stabilizes the high-temperature crystalline phase (cubic or tetragonal) of zirconia at room temperature. The Kröger–Vink notation for the crystal defect creation induced by the substituting yttria Y2O3 is given below.6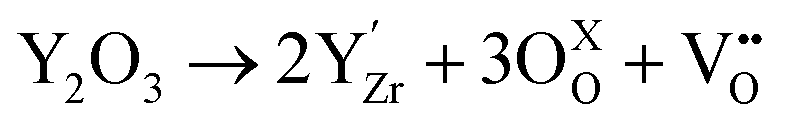 | (1) |
Significant efforts have been made in the past decades for the synthesis of YSZ nanoparticles in order to develop low-cost and robust synthesis methods and to improve their durability in high-temperature applications.12–17 Particular attention is generally paid to the powder microstructure as the morphology is closely linked to the properties of the material (ionic conductivity, sintering behavior, durability, and mechanical strength).14 Indeed, the ionic conductivity can be impacted by chemical parameters, such as the Y3+ concentration and oxygen partial pressure, or by structural parameters, such as the layer thickness, density, or particle sizes.18–20 The sinterability of the obtained powders is also a key parameter for material processing in order to achieve a highly dense material for an electrolyte or a porous support. Moreover, reducing the particle size induces the lowering of sintering temperatures as the surface energy increases owing to the increasing surface area.21,22 This provides a considerable advantage for large-scale industrialization in terms of reducing the manufacturing cost and time.
Besides energy-intensive processes such as solid-state synthesis or mechanosynthesis, YSZ powders are usually synthesized by co-precipitation and sol–gel routes.10,14,17,23 Even if these two techniques may provide relatively small particles, some drawbacks persist, like the poor phase control in the former and the agglomeration of particles in the latter.24,25 Moreover, these synthesis methods require costly and energy-consuming steps, such as solvent removal and a high-temperature treatment for powder crystallization. In this work, the supercritical CO2 (scCO2)-assisted synthesis was chosen as an alternative route due to its specific properties. Thanks to its high diffusivity, low surface tension and modular solvent power, scCO2 enables the synthesis of nano-structured powders with high specific areas.24,26–29 The use of scCO2 as a solvent could thus allow for avoiding the energy-intensive size-reduction step, implementing a bottom-up strategy for SOC manufacturing.30 Moreover, a supercritical treatment also allows the lowering of the crystallization temperature, which helps to avoid high-temperature treatments that are usually required for sol–gel and precipitation processes.
This work relies on the use of a supercritical route that was developed in previous works.31 The original route was optimized to elaborate tetragonal zirconia polycrystal (Y-TZP) with 4%mol of Y2O3. Using pentane as a co-solvent for the reactants solubilization, 50–250 nm wide particles constituted with 15–30 nm crystallites were obtained. The specific surface area of the powders was up to 100 m2 g−1. Nevertheless, it was observed that the increase in Y2O3 content up to 8 mol% in the crystal resulted in the destabilization of the cubic structure, leading to the formation of tetragonal and monoclinic zirconia after thermal treatment at 1150 °C.32 This unstable cubic structure was related to an inhomogeneous distribution of the Y3+ cations into the ZrO2 matrix. The purpose of the present work is to adapt this supercritical route to the synthesis of stabilized cubic YSZ. To do so, the feasibility of the synthesis at 250 °C was first demonstrated. The effect of different synthesis parameters, such as the reagent solubilization and the operating conditions (P & T), were investigated. Finally, a powder was selected and a study of its temperature behavior was carried out.
Experimental
Chemicals
YSZ syntheses were performed with zirconium(IV) acetate hydroxide (CH3CO2)xZr(OH)y, x + y ∼ 4 (Sigma-Aldrich) and yttrium(III) acetate tetrahydrate (CH3CO2)3Y·4H2O (Alfa-Aesar, 99.9%). The molar mass of zirconium acetate hydroxide was estimated to be 213 ± 3 g mol−1. The pre-solubilization was performed in pentane (Sigma) as a co-solvent and nitric acid (Supelco, 65%v/v) was added to the solution.After ageing the reactant solution for 20 h, the mixture is inserted in the reactor, which is then sealed. Some CO2 (between 40 and 50 bar) is added, the stirring is set up at 650 rpm and the heating is started (the pressure increases with the rising temperature). The pressure and temperature are maintained for 1 h. At the end of the stage, the heating and the stirring are stopped. The reactor is depressurized, and the pentane co-solvent and unreacted precursors are exhausted with CO2, enabling the recovery of solvent-free, dried powders.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–98000, 99+% hydrolyzed). The powders were dry-mixed using a mortar with a few ethanol droplets. The mixture was then compressed (25.5 kPa) into 10 mm diameter pellets and sintered in a muffle furnace under air. The sintering was performed at 1400 °C for 4 h, with a 10 °C min−1 heating rate.
000–98000, 99+% hydrolyzed). The powders were dry-mixed using a mortar with a few ethanol droplets. The mixture was then compressed (25.5 kPa) into 10 mm diameter pellets and sintered in a muffle furnace under air. The sintering was performed at 1400 °C for 4 h, with a 10 °C min−1 heating rate.
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and COD 1528644 served as a reference. The reference for the undesirable monoclinic phase was the P21/c (COD 2108450). Finally, the Scherrer equation (eqn (2)) was applied to the diffractograms in order to estimate the crystallite sizes.33
m space group and COD 1528644 served as a reference. The reference for the undesirable monoclinic phase was the P21/c (COD 2108450). Finally, the Scherrer equation (eqn (2)) was applied to the diffractograms in order to estimate the crystallite sizes.33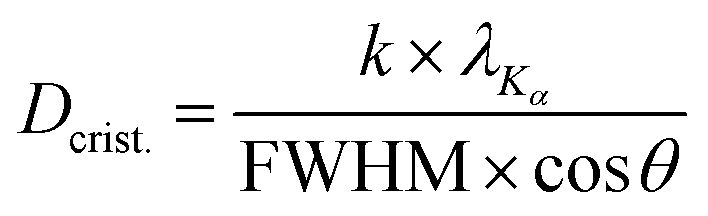 | (2) |
The morphology and microstructural aspect were studied by nitrogen porosimetry with an ASAP 2020 (micromeritics), and SEM imaging was done on a Zeiss Merlin either after resin embedding and polishing or simply observing the raw powder. The specific surface areas were calculated using the BET theory, while the pore size distributions were given according to the BJH theory (desorption branch). Moreover, the particle size distribution analysis was carried out with a Mastersizer 3000 (Malvern Panalytical) by dispersing the powder in water using ultrasound.
In addition, some thermogravimetric analyses (TGA) coupled with differential scanning calorimetry (DSC) were carried out to quantify the volatile/organic fraction of the samples. The samples were heated to 1000 °C with a 10 °C min−1 heating rate.
Results and discussion
Feasibility of crystallization, without calcination
During the maturation of the reactant solution, a gel appears after the addition of nitric acid. The gel formation is attributed to hydrolysis/condensation reactions, leading to the formation of a polymer network of metallic cation (Zr4+, Y3+) complexes.The XRD analysis of the blank experiment (furnace treatment without supercritical CO2) shows that the obtained powder is completely amorphous (Fig. 1), confirming that a 250 °C thermal treatment is not sufficient for crystallization.
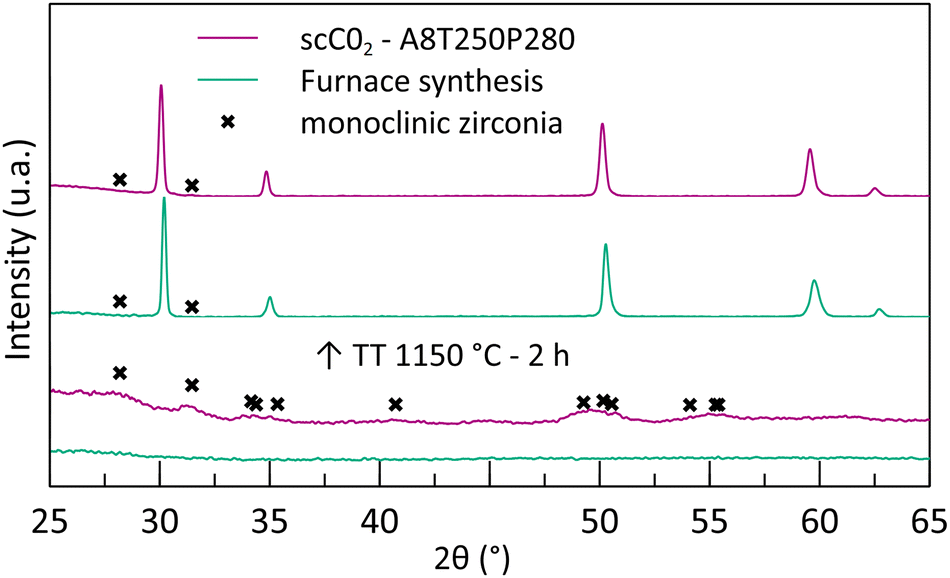 | ||
| Fig. 1 Diffraction patterns of the furnace-synthesized powder and the powder synthesized in supercritical CO2 before and after thermal treatment at 1150 °C. | ||
Thermogravimetric (TGA) experiments were conducted on the furnace powders (Fig. 2). The profile shows a total mass loss of 65% at 500 °C. The first slope, at around 150 °C, is attributed to the water loss (around 10%mass). The powders face a consequent mass loss of about 55%mass (equiv. to 60% from the dry powders). Given the fact that no crystal formation was observed, this mass drop may be the consequence of the ZrO2 and Y2O3 oxide formations (from intermediary species). The theoretical mass drop of the full oxidation of the unreacted hydroxyacetate/acetate Y & Zr precursors is 47%mass, which is not close to the 60%mass observed (eqn (3)). Assuming that all the organometallic Y & Zr compounds were converted into nitrate compounds (reaction with nitric acid), the theoretical mass loss associated with the oxide formation is about 63%mass (eqn (4)). In comparison, the mass loss starting from the hydroxide compounds is only 22%. The complex compounds must have almost all been converted into nitrate salts with a few hydroxide ligands.
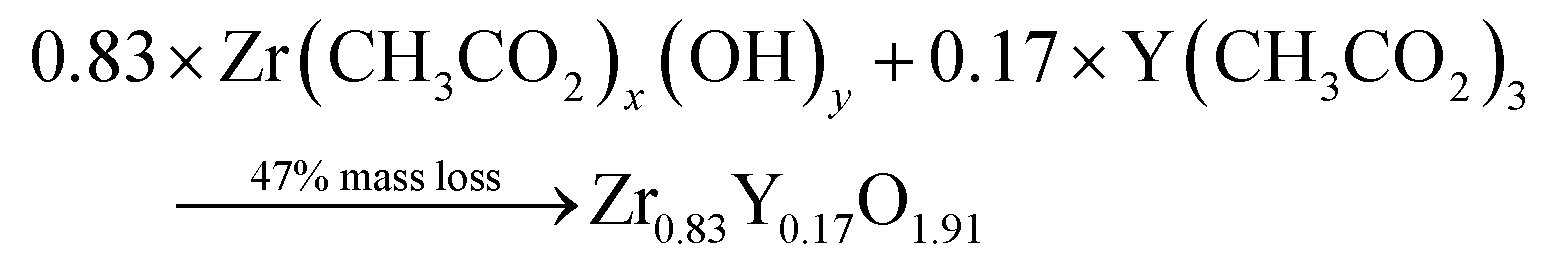 | (3) |
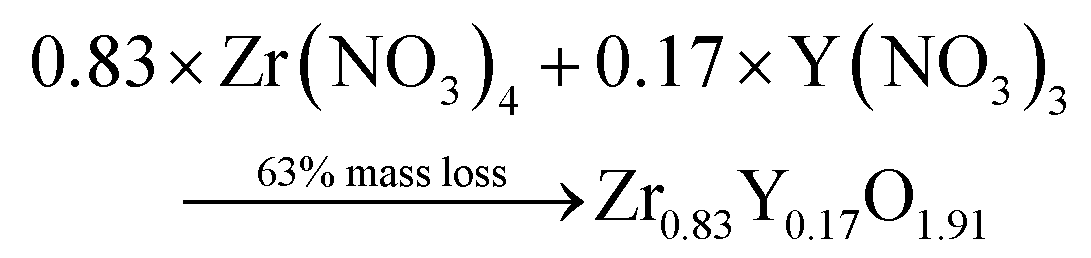 | (4) |
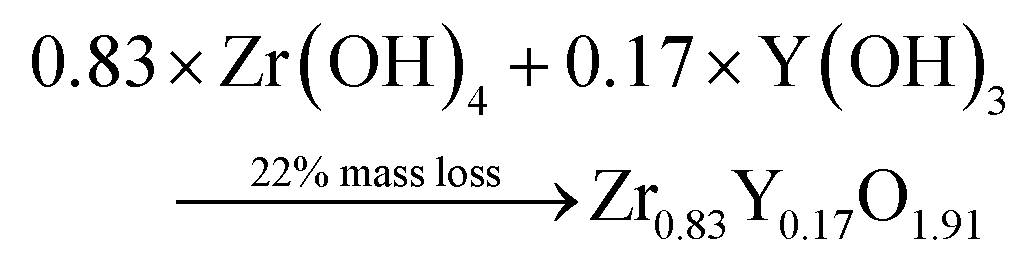 | (5) |
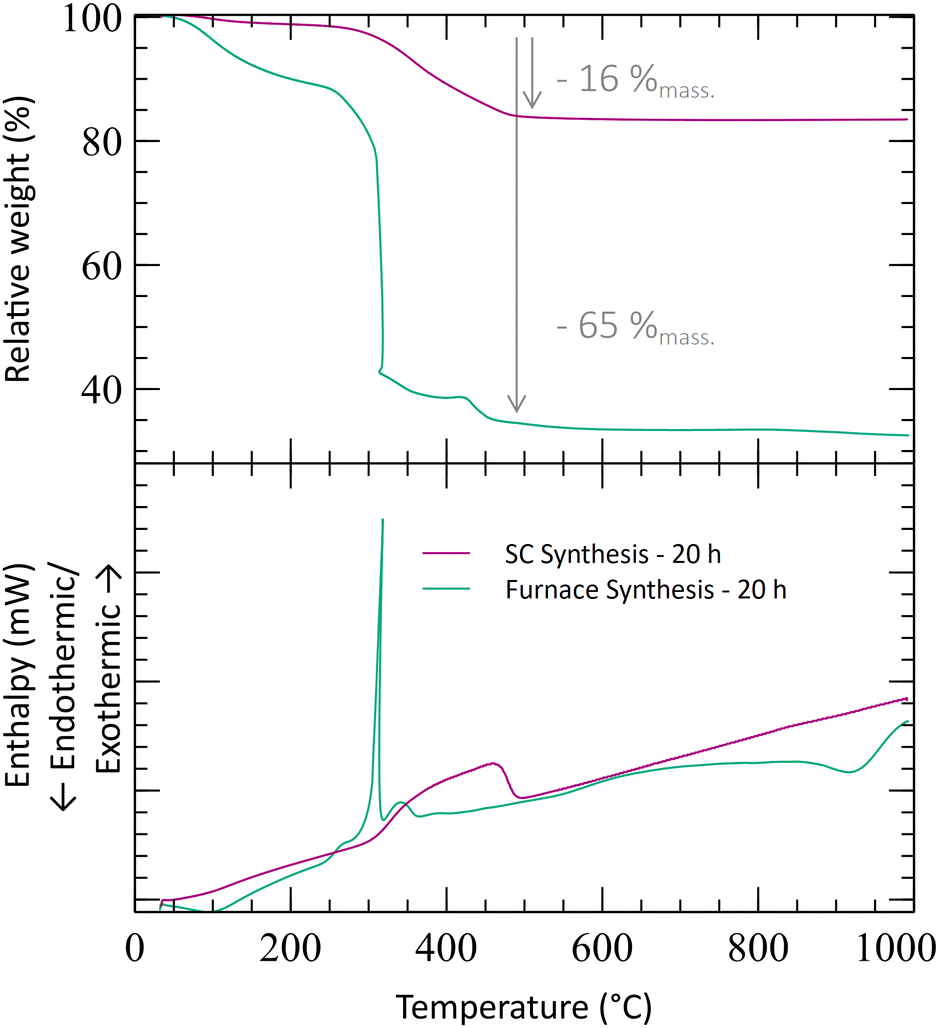 | ||
| Fig. 2 TGA analysis of the powders obtained by calcining Zr4+ and Y3+ solutions without supercritical treatment and treated in air at 250 °C for 1 h compared to the one obtained with the scCO2 treatment at 250 °C for 1 h. | ||
To illustrate the effect of the supercritical treatment, a synthesis was performed by applying supercritical treatment to a reactant mixture with the same acid amount (8 mL). The temperature was kept at 250 °C as well, and the CO2 pressure during this stage was maintained at 280 bar. The recovered powder shows a small TGA loss of mass at a low temperature (3%mass at 300 °C), followed by a slope at around 400 °C (Fig. 2). In comparison with the furnace treatment, the mass loss is much smaller, representing only 16%mass in total at 500 °C. This significant difference emphasizes that a better chemical conversion is achieved during the scCO2 synthesis in only 1 h and that the powders are already oxidized before the analysis. The mass loss associated with the supercritical powder can be attributed to the removal of organic compounds remaining from the precursors’ ligands and/or unreacted reactants. Moreover, the XRD analysis shows the appearance of a crystal structure corresponding to the formation of nano-crystallites of monoclinic zirconia (Fig. 1). Despite obtaining the undesirable phase of zirconia, the XRD analysis emphasizes that the crystallization can occur at low temperatures with scCO2, whereas calcination under air seems to only evaporate the solvent.
According to Adschiri and collaborators, in the case of supercritical water, the nanoparticle formation is induced by a decrease in the reactant solubility in the supercritical state, leading to a fast nucleation of oxide clusters.36 Also, the ability of the increasing pressure to lower the crystallization temperature and to generate nanoparticles is already observed for hydrothermal/solvothermal syntheses.37 The pressure effect is interpreted as a way to fasten the mass transfer, allowing fast reaction rates. Thus, a potential explanation for the monoclinic phase formation could be the nucleation of zirconia ZrO2 alone during the supercritical treatment. The poor yttrium content in the crystal then does not result in the stabilization of the cubic phase.
For both the furnace and the scCO2 powders, thermal treatment at 400 °C for 4 h allows the formation of cubic/tetragonal crystal structures with large peaks that prevent the identification of monoclinic peaks. Further thermal treatment at 1150 °C results in the sharpening of the diffraction peaks because of the crystallite growth. Thus, it permits revealing if any diffraction peaks overlap, especially when monitoring the possible presence of monoclinic zirconia. In both cases, the thermal treatment enables obtaining tetragonal/cubic yttria-doped zirconia with traces of the monoclinic phase.
SEM images show that furnace-synthesized powders do not have well-defined particles but a cloudy amorphous solid phase with large aggregates (Fig. 3). On the other hand, scCO2-synthesized powders exhibit small and well-defined particles with a likely homogeneous size distribution. These particles consist of primary grains aggregated between each other according to the crystallite size determined by the XRD analysis.
 | ||
| Fig. 3 SEM images of powders synthesized (a) by calcination and (b) in supercritical CO2 at the reactor outlet. | ||
scCO2 synthesis – chemical and process parameters influence
In the previous part, it was shown that the supercritical media promotes a high reactivity at temperatures as low as 250 °C, with some crystal structure formation. However, many parameters are to be optimized in order to obtain fully stabilized zirconia.The crystal structure analysis of the obtained powders appears to be in contradiction with what was expected. Indeed, as the acid content increases, the crystallinity of the as-synthesized powders decreases (Fig. 4). The A5-powder made from the less acidic solution (5 mL) exhibits diffraction peaks indicating a tetragonal/cubic phase, whereas the diffractograms of other powders show the presence of monoclinic phases, with very low intensity peaks. This analysis reveals that only a few Zr compounds are oxidized and crystallized into small monoclinic crystallites at high acid contents. Despite a potential reactivity between Y and Zr cations, no YSZ crystal formation is observed. The nitric acid does dissolve the organometallic complexes, but it prevents the reaction from taking place at high contents. Then, it was shown that the monoclinic phase remained even after the calcination of the powders at the highest temperature tested (1150 °C, 2 h) (Fig. 5 and 6). At this stage of the study, it seems that the lower the acid content, the better the crystallization. This behavior is attributed to a degradation of the gel by the high nitric acid content, leading to a weak interaction between the intermediate compounds Y and Zr.
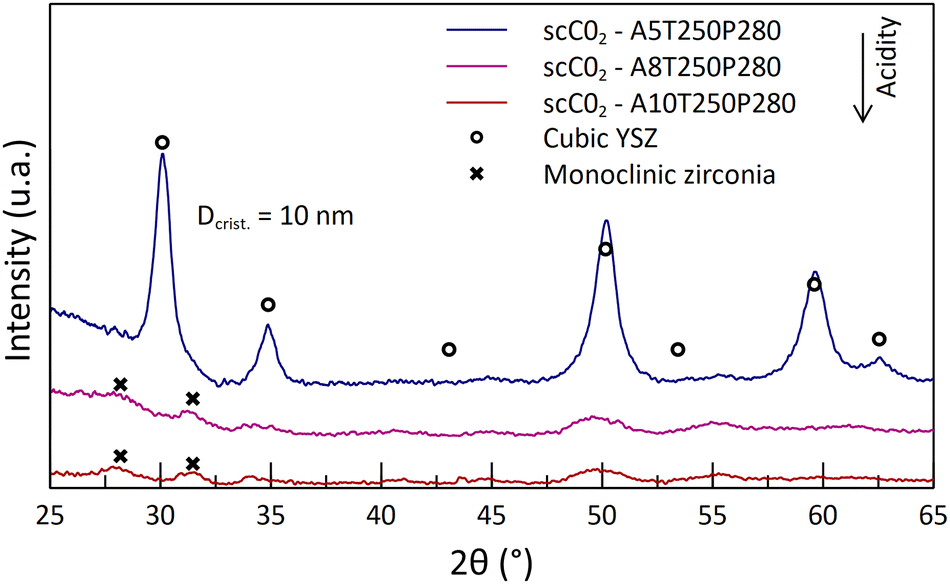 | ||
| Fig. 4 Diffractograms of the as-synthesized powders at 250 °C and 280 bar, with different nitric acid contents in the starting solution. | ||
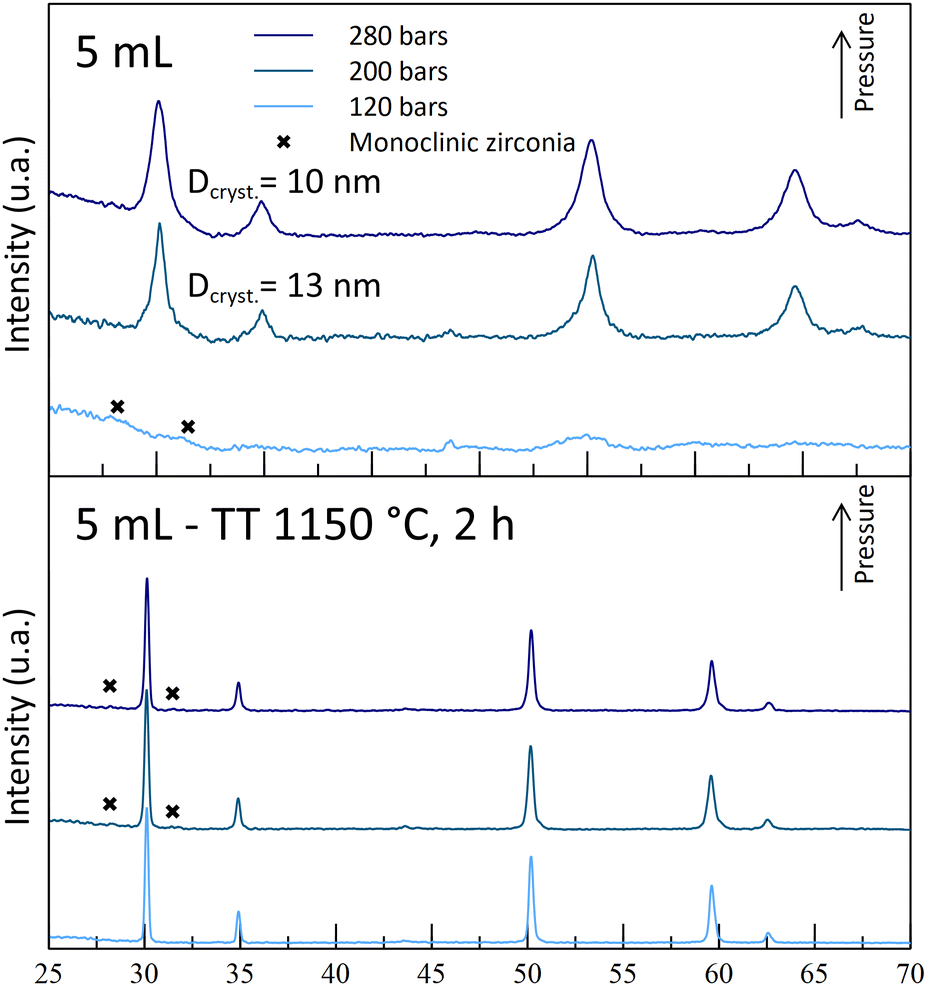 | ||
| Fig. 5 Diffractograms of powders synthesized with 5 mL nitric acid at 250 °C and at different operating pressures, before and after thermal treatments (400 °C for 4 h + 1150 °C for 2 h). | ||
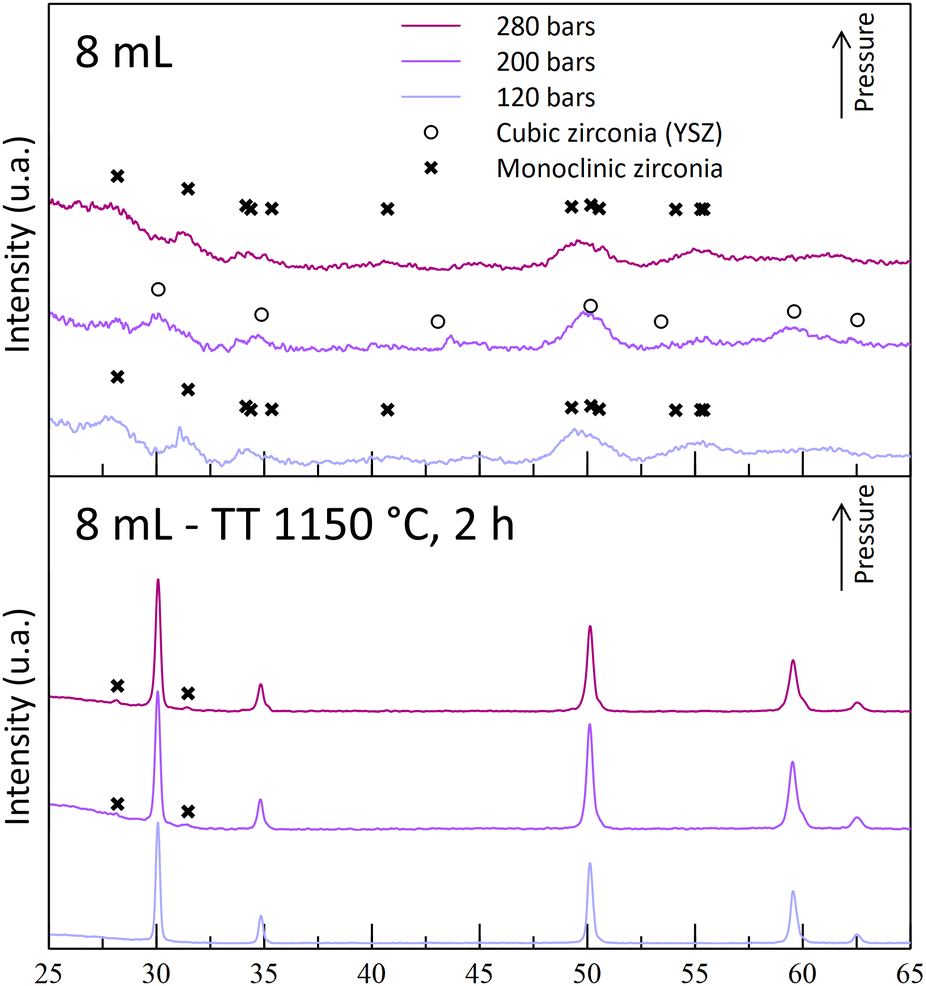 | ||
| Fig. 6 Diffractograms of powders synthesized with 8 mL nitric acid at 250 °C and at different operating pressures, before and after thermal treatments (400 °C for 4 h + 1150 °C for 2 h). | ||
This positive impact of the pressure on the crystallization was expected, as in supercritical CO2, the solubility of organometallic compounds increases with the increasing pressure.38 In addition to the fast diffusivity promoted by the supercritical media, the solubility enhancement is likely to promote the reaction rate of crystal oxide formation.
Moreover, besides the solubility increase, the diffusivity decreases with the increasing pressure.39 In these conditions, the nucleation of crystal clusters may be favored over the crystal growth. Thus, the grain size is expected to be smaller at high synthesis pressure. Indeed, a small crystallite size reduction is observed when raising the pressure from 200 bars to 280 bars (Table 1). However, no specific surface area evolution is observed. This unchanged surface may be due in part to measurement uncertainty, which has not been discussed.
| Sample reference | Acid quantity (mL) | Temperature (°C) | Pressure (bar) | SS BET (m2 g−1) | As synthesized | TT 1150 °C – 2 h | ||
|---|---|---|---|---|---|---|---|---|
| Crystal structure | “C” Crystallite size (nm) | Crystal structure | “C” Crystallite size (nm) | |||||
| C = cubic; T = tetragonal; M = monoclinic (lower case “m” in case of residual signal). SS BET = specific surface area evaluated with BET equation. | ||||||||
| A5T250P120 | 5 | 250 | 120 | 133 | m | C/T | 54 | |
| A5T250P200 | 200 | 81 | C/T | 13 | C/T + m | 42 | ||
| A5T250P280 | 280 | 81 | C/T | 10 | C/T + m | 44 | ||
| A8T250P120 | 8 | 250 | 120 | m | C/T + m | 39 | ||
| A8T250P200 | 200 | 78 | C/T | 5 | C/T + M | 34 | ||
| A8T250P280 | 280 | 153 | m | C/T + m | 38 | |||
| A10T250P120 | 10 | 250 | 120 | 98 | m | C/T | 40 | |
| A10T250P200 | 200 | 63 | m | C/T + M | 30 | |||
| A10T250P280 | 280 | 81 | m | C/T + M | 38 | |||
| A10T300P120 | 10 | 300 | 120 | 139 | M | C/T | 51 | |
| A10T300P200 | 200 | 150 | C/T | 7 | C/T | 54 | ||
| A10T300P280 | 280 | 95 | C/T | 41 | ||||
Several syntheses were also performed with the other acid contents (8 and 10 mL) at different pressures (120, 200 and 280 bar) (samples [A8/A10]-T250-[P120/P200/P280]). However, only a few experiments led to crystallized powders at the end of the reaction without the monoclinic phase.
The experiments leading to crystallized powders include the two A5 experiments (A5T250-P200 and -P280) and the A8T250P200 one. The latter exhibits weak diffraction peaks, indicating a low crystallinity or formation of really small nano-crystallites (Fig. 5). Even though a positive impact of the pressure was observed for the A5-powders, the reverse effect was observed with the A8-powders (Fig. 6). Indeed, with the highest pressure, monoclinic phase diffraction peaks were observed without cubic/tetragonal structures, while the desired structure was obtained at 200 bar (A8T250P200). This outcome leads to the hypothesis that, even if the pressure helps the powder crystallization, it may also promote the monoclinic zirconia formation (with inhomogeneous yttrium incorporation).
After a thermal treatment of the powders at 400 °C for 4 h, all the powders exhibit a cubic/tetragonal phase with crystallites whose dimensions are between 8 and 10 nm. By firing the powders at 1150 °C (Fig. 4–6), the induced crystallite growth allows to check for the presence of other crystalline phases and the stability of the cubic phase that has been formed.
The structural analysis of the sintered powders is in line with the previous theory on the destabilization of the desired cubic structure: for each acid content tested, all the powders synthesized at 120 bar (-P120 powders) are monoclinic-free, while monoclinic peaks were observed for the highest pressures. For A5-powders, only monoclinic traces were observed on the -P200 and -P280 powder diffractograms. This calcination step allows us to show that this set of experiments can lead to the synthesis of YSZ powders at 250 °C. The obtained powders contain 30–55 nm-sized crystallites after the 1150 °C calcination step. According to these experiments, both A5T250-P200 and -P280 experiments were retained, as the powders obtained are crystallized in the correct phase.
During the T250 series of experiments, it was observed that the A10T250 powders were mostly crystallized in the monoclinic phase. The results were attributed to a degradation of the gel by nitric acid during the maturation phase, leading to the destabilization of the cubic/tetragonal phase. This monoclinic phase was also observed after the thermal treatments (Fig. 6). However, an increase in the temperature led to a crystalline powder at 200 bar (A10T300P200). The A10T300-powders exhibit an opposite behavior in comparison to the powders synthesized at 250 °C, with an absence of crystallinity for the high-pressure synthesis (-P280) and with a destabilization of the cubic/tetragonal phase at the lowest pressure (-P120). The high acid amount combined with high pressure and high-temperature conditions may result in a drastic increase in the reactivity of nitric acid molecules. This high reactivity may be the reason for the gel degradation for the A10T300P280 sample. The crystallization at the intermediate pressure again illustrates the dual effect of the pressure on the reaction.
The A10T300P200 powders were kept for their crystallized structure at the reactor outlet, even though a small quantity of monoclinic phase is observed with a small diffraction peak. Moreover, the powders exhibit a fine microstructure, with a high specific surface area of 150 m2 g−1 and 7 nm diameter crystallites. Finally, its cubic/tetragonal structure is stable after the 1150 °C thermal treatment and no more monoclinic structure was observed (Fig. 7).
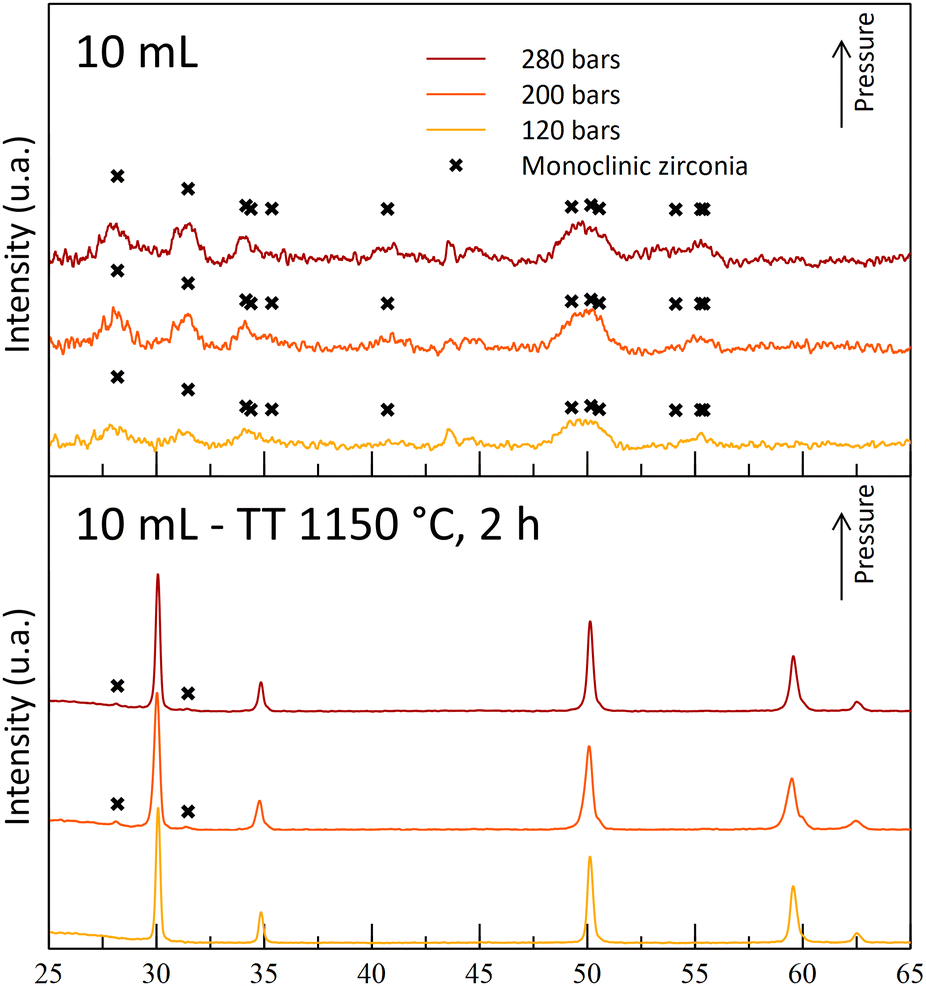 | ||
| Fig. 7 Diffractograms of powders synthesized with 10 mL nitric acid at 250 °C and at different operating pressures before and after thermal treatments (400 °C, 4 h + 1150 °C, 2 h). | ||
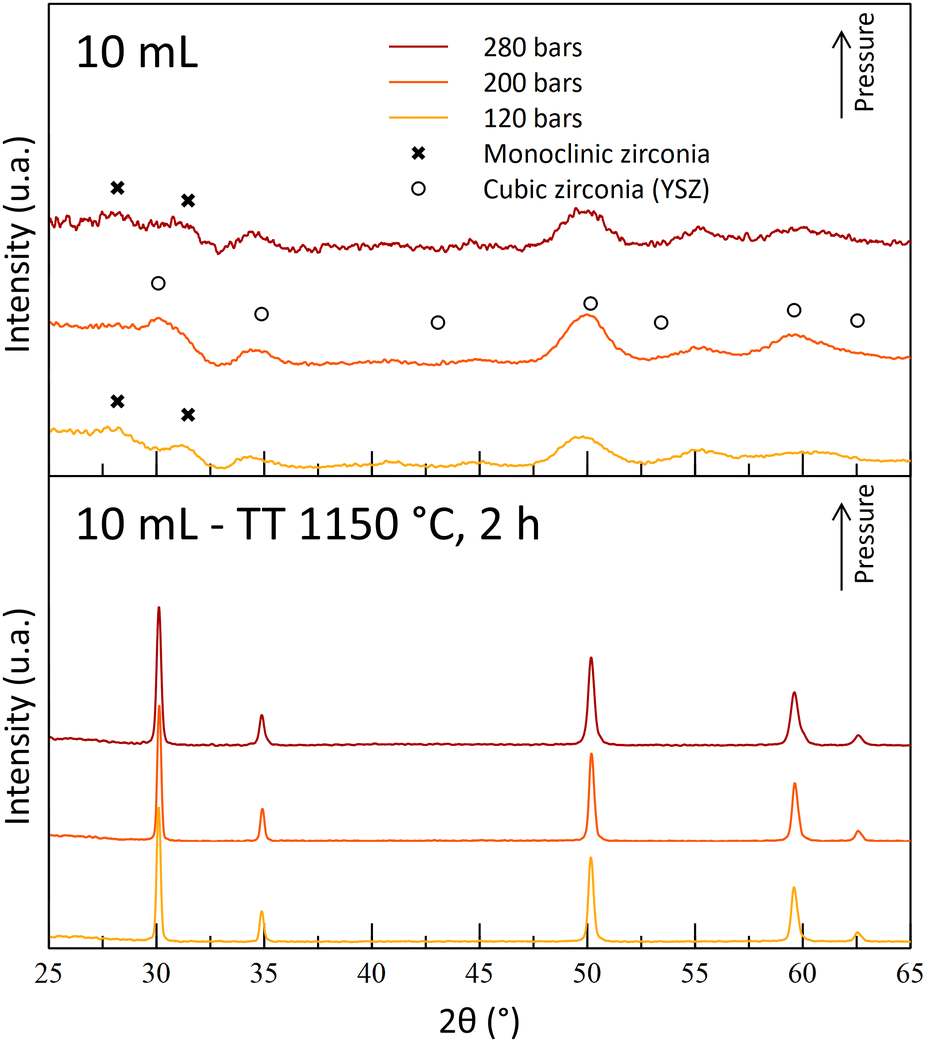 | ||
| Fig. 8 Diffractograms of powders synthesized with 10 mL nitric acid at 300 °C and at different operating pressures before and after thermal treatments (400 °C for 4 h + 1150 °C for 2 h). | ||
Thermal treatments and microstructure evolution
After the study of the influence of operating parameters on crystalline phase stabilization, the optimized parameters involve the supercritical treatment of the starting solution containing 10 mL nitric acid at 300 °C and 200 bar (A10T300P200 powder). The powders exhibit a specific surface area of 150 m2 g−1 with 7 nm crystallites at the reactor outlet. The porosimetry analysis by nitrogen adsorption/desorption is shown in Fig. 9. The isotherms and the pore size plot reveal the presence of micropores and mesopores with a diffuse size distribution. The pore size distribution exhibits two weak maxima around 6 nm and 20 nm. In all the pore size distribution plots, the high-intensity fine peaks appearing around 2 nm are spurious. These artifacts are the result of the gap between the adsorption and desorption branches of the isotherms.40 The BET-specific surface area was compared to the theoretical geometrical surface of the crystallites. The calculation of the latter was based on the assumption that crystallites were spherical. The surface area for the untreated powder was shown to be close to the geometrical surface that was calculated, which is about 170 m2 g−1 for spherical YSZ crystallites. This comparison emphasizes a low particle aggregation of the powders, which is an important argument for the supercritical route, as the powders do not require a size reduction by milling or an aggregate breaking step. A particle size analysis showed that the grain size distribution is centered around 400 nm, with a few wider particles reaching 100 μm (Fig. 9). However, the granulometry of the particles was measured without the presence of a dispersing agent. A lower particle size distribution may be obtained with a dispersant combined with ultrasound. SEM images highlight the fineness of the particles, with a low agglomeration (Fig. 10). Some areas appear denser but still consist of small primary particles with small pores in between.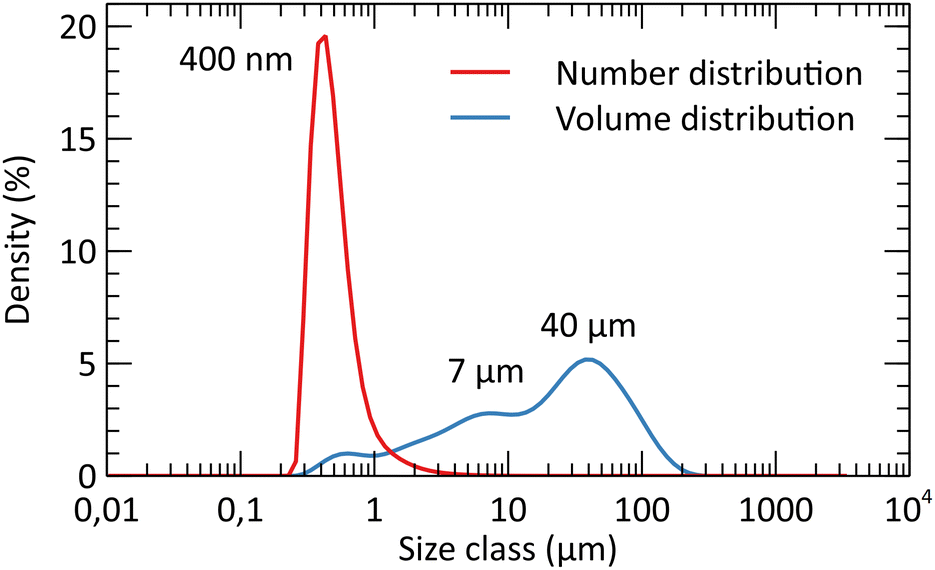 | ||
| Fig. 9 Particle size analysis in water of the selected powders (A10T300P200) after the 400 °C thermal treatment (4 h). | ||
 | ||
| Fig. 10 SEM images of the A10T300P200 powder after (a) a 400 °C thermal treatment for 4 h and (b) after an additional treatment at 1150 °C for 2 h. | ||
The thermal behavior of the powder was studied by measuring the crystallite size and the specific surface area evolution through a series of thermal treatments (Fig. 11). It was shown that the surface area is decreasing significantly beyond 450 °C, while the mean crystallite size really starts to increase beyond 650 °C. This gap between the collapsing surface area and the crystallite growth shows that the sintering of the crystallites starts at low temperatures. The crystallites growth is starting only at higher temperatures. The collapse of the specific surface area is due to the disappearance of the open micropores, with the centering of the pore size toward wider pores, around 20 nm. Based on the SEM analysis, after the 1150 °C thermal treatment, the powder still exhibits a much-dispersed structure (Fig. 12). The intra-particle porosity loss does not seem to affect the appearance of the powder on a moderately close-up view.
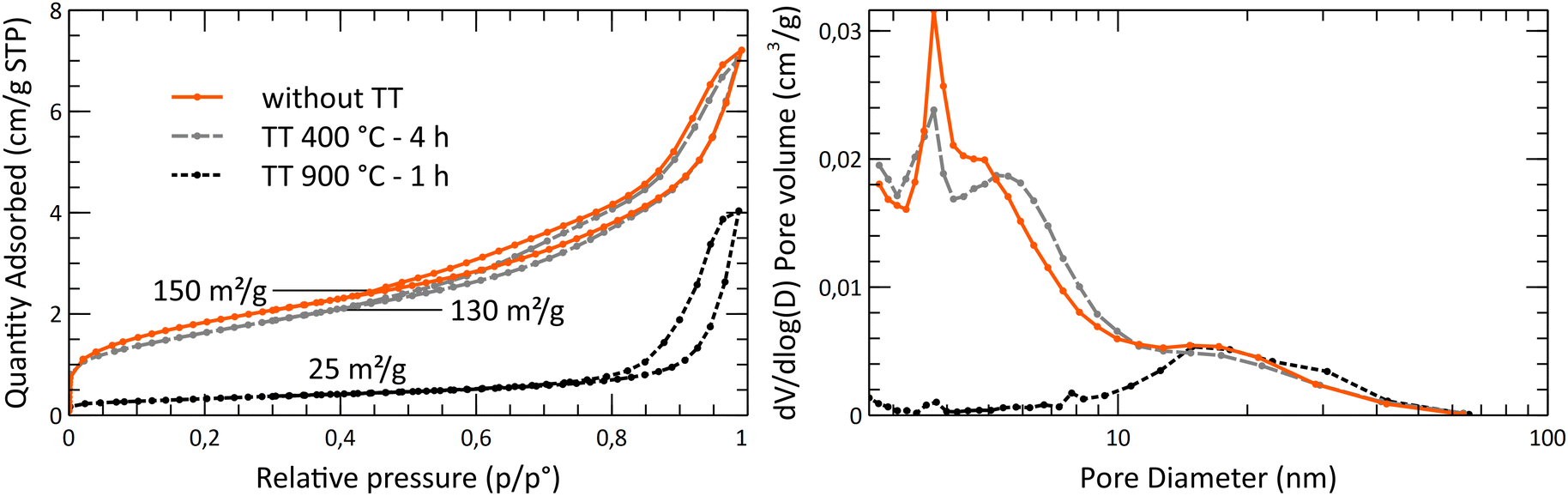 | ||
| Fig. 11 Nitrogen adsorption and desorption isotherms and BJH pore size distribution of the A10T300P200 powders. The measurement was carried out on the powder as synthesized after a thermal treatment at 400 °C for 4 h and after a second thermal treatment at 900 °C for 1 h. BJH calculations are based on the desorption branch of the isotherms. | ||
 | ||
| Fig. 12 Evolution of specific surface area and crystallite size as a function of calcination temperature. The experimental values are compared to a geometrical curve that represents the combined evolution of surface area and diameter if the crystallites were independent of each other. | ||
Pellet shaping and sintering
The thermal behavior of the powders was also studied to select a temperature for the pre-treatment of the powders before processing. Indeed, the small dimension of the powders prevents them from being shaped by compaction into a final material (pellet breaking). Therefore, a two-step sintering was performed on the pellet.Thereby, the A10T300P200 powders were briefly calcined at 900 °C under air for 1 h before being shaped into a pellet. The obtained density of the pellet was close to the theoretical density of YSZ (5.93 g cm−3). The apparent density of the pellet calculated from the caliper measurement was estimated to be 92% of the theoretical value, while the helium pycnometer measurement of the sintered pellet led to a 99% skeletal relative density value. The differences between those two values may be the result of the uncertainty of pellet volume measurement for the former, or by the presence of a residual porosity at the pellet surface for the latter. The high density obtained shows that the powders may be suitable for dense electrolyte processing.
Conclusion
The purpose of this work is to adapt a supercritical CO2-assisted process for fine YSZ particle generation. This route enabled the synthesis of dried crystallized powders at a temperature as low as 250 °C during the supercritical treatment. A parametric study was conducted to optimize the operating parameters in order to obtain the cubic YSZ structure without a monoclinic phase. It was shown that the pressure has a dual effect on the crystal formation: an increasing pressure promotes both the powder crystallization and destabilizes the cubic structure into a monoclinic structure. The nitric acid content in the starting solution also shows contradictory effects: on the one hand, a high acid content degrades the pentane gel, leading to monoclinic zirconia formation, and on the other hand, it also allows the full solubilization of the reactants. After studying these two parameters, the impact of the temperature was investigated, revealing that an increase in the temperature from 250 °C to 300 °C improves powder crystallization, as expected.Finally, the supercritical treatment was performed at 300 °C and 200 bar. The obtained powder exhibited a high specific surface area of around 150 m2 g−1, small crystallites below 10 nm with low agglomeration, and a fully stabilized cubic phase. This fine morphology may be interesting for a catalyst substrate.
The sintering behavior of the obtained powders was then investigated, following the evolution of the microstructure up to 1150 °C. The sintering of the powders starts at low temperatures, and a highly dense material was obtained. In addition to this work, measurements of ionic conductivity with temperature by EIS of the pellets will be performed. These measurements will enable us to check whether the powders are suitable for integration into a solid oxide cell. A study of the ionic conduction of the material would be an interesting addition to this work. The evaluation of the synthesized material as a dense electrolyte or as a catalyst support with a great catalyst dispersion would be a considerable step forward in the evaluation of the benefits of supercritical CO2 for ceramic material synthesis.
Author contributions
Loan Avédikian: conceptualization, methodology, investigation, data curation, formal analysis, writing – original draft. Thomas David: resource, formal analysis, investigation, writing – review & editing. Julien Vulliet: resource, methodology, investigation, writing – review & editing. Audrey Hertz: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Pierre Venditti and Alexandre Gaillard (CEA/ISEC/LPSD) for the support for supercritical processes, Pascal Antonucci (CEA/ISEC/LFSM) for the X-ray diffraction analysis, Yves Faucherand (CEA/LITEN/LCAE) for the SEM sample preparation, Adrien Gerenton (CEA/ISEC/LFCM) for the characterizations using gas porosimeter, and Benoit Meilleray (CEA/ISEC/LPSD) for the grain size analysis.References
- H. N. Ralph and G. Wilfing, Ullmann's Encycl. Ind. Chem., 2010, 39, 753–776 Search PubMed.
- M. N. Tsampas, F. M. Sapountzi and P. Vernoux, Catal. Sci. Technol., 2015, 5, 4884–4900 RSC.
- P. Mercera, J. Van Ommen, E. Doesburg, A. Burggraaf and J. Ross, Appl. Catal., 1991, 71, 363–391 CrossRef CAS.
- M. Laguna-Bercero, S. Skinner and J. Kilner, J. Power Sources, 2009, 192, 126–131 CrossRef CAS.
- P. Temluxame, P. Puengjinda, S. Peng-ont, W. Ngampuengpis, N. Sirimungkalakul, T. Jiwanuruk, T. Sornchamni and P. Kim-Lohsoontorn, Int. J. Hydrogen Energy, 2021, 46, 24568–24580 CrossRef CAS.
- Y.-M. Chiang, D. P. Birnie and W. D. Kingery, Physical ceramics: principles for ceramic science and engineering, John Wiley & Sons, Inc., New York, 1997 Search PubMed.
- O. Yamamoto, Electrochim. Acta, 2000, 45, 2423–2435 CrossRef CAS.
- S. Badwal, Solid State Ionics, 1992, 52, 23–32 CrossRef CAS.
- A. Hauch, S. D. Ebbesen, S. H. Jensen and M. Mogensen, J. Mater. Chem., 2008, 18, 2331–2340 RSC.
- N. H. Menzler, F. Tietz, S. Uhlenbruck, H. P. Buchkremer and D. Stöver, J. Mater. Sci., 2010, 45, 3109–3135 CrossRef CAS.
- B. Yu, W. Zhang, J. Chen, J. Xu and S. Wang, Sci. China, Ser. B: Chem., 2008, 51, 289–304 CrossRef CAS.
- C. Suciu, A. C. Hoffmann and P. Kosinski, J. Mater. Process. Technol., 2008, 202, 316–320 CrossRef CAS.
- X. Xu, Y. Feng, Z. Zhong, X. Guo, Z. Zhang, J. Li, S. Zhao, S. Wu and H. Sun, J. Rare Earths, 2023, 41, 1385–1391 CrossRef CAS.
- A. Zarkov, A. Stanulis, J. Sakaliuniene, S. Butkute, B. Abakeviciene, T. Salkus, S. Tautkus, A. F. Orliukas, S. Tamulevicius and A. Kareiva, J. Sol-Gel Sci. Technol., 2015, 76, 309–319 CrossRef CAS.
- T. He, Q. He and N. Wang, J. Alloys Compd., 2005, 396, 309–315 CrossRef CAS.
- S. Buinachev, M. Mashkovtsev, A. Dankova, N. Zhirenkina and K. Kharisova, Powder Technol., 2022, 399, 117201 CrossRef CAS.
- M. Hajizadeh-Oghaz, R. S. Razavi and M. L. Estarki, Bull. Mater. Sci., 2014, 37, 969–973 CrossRef CAS.
- S. Bursich, S. Morelli, G. Bolelli, G. Cavazzini, E. Rossi, F. G. Mecca, S. Petruzzi, E. Bemporad and L. Lusvarghi, Surf. Coat. Technol., 2024, 476, 130270 CrossRef CAS.
- A. Hauch, K. Brodersen, M. Chen and M. B. Mogensen, Solid State Ionics, 2016, 293, 27–36 CrossRef CAS.
- O. Durá, M. L. De La Torre, L. Vázquez, J. Chaboy, R. Boada, A. Rivera-Calzada, J. Santamaria and C. Leon, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 184301 CrossRef.
- X. Kuang, G. Carotenuto and L. Nicolais, Adv. Perform. Mater., 1997, 4, 257–274 CrossRef CAS.
- P. Colomban, Ceramics, 2020, 3, 312–339 CrossRef CAS.
- Y. Mansilla, M. Arce, C. G. Oliver, H. Troiani and A. Serquis, Mater. Today: Proc., 2019, 14, 92–95 CAS.
- C. Erkey, J. Supercrit. Fluids, 2009, 47, 517–522 CrossRef CAS.
- B. Shri Prakash, S. Senthil Kumar and S. T. Aruna, Renewable Sustainable Energy Rev., 2014, 36, 149–179 CrossRef CAS.
- A. Chafidz, T. Jauhary, M. Kaavessina, S. Sumarno and F. H. Latief, Commun. Sci. Technol., 2018, 3, 57–63 CrossRef.
- S. B. Barim, E. Uzunlar, S. E. Bozbag and C. Erkey, J. Electrochem. Soc., 2020, 167, 054510 CrossRef CAS.
- S. Bozbag, D. Sanli and C. Erkey, J. Mater. Sci., 2012, 47, 3469–3492 CrossRef CAS.
- L. Henry, J. Roger, Y. Le Petitcorps, C. Aymonier and L. Maillé, J. Supercrit. Fluids, 2018, 141, 113–119 CrossRef CAS.
- S. Bozbag and C. Erkey, J. Supercrit. Fluids, 2012, 62, 1–31 CrossRef CAS.
- A. Hertz, Y.-M. Corre, S. Sarrade, C. Guizard, A. Julbe, J.-C. Ruiz and B. Fournel, J. Eur. Ceram. Soc., 2010, 30, 1691–1698 CrossRef CAS.
- A. Hertz, M. Drobek, J. C. Ruiz, S. Sarrade, C. Guizard and A. Julbe, Chem. Eng. J., 2013, 228, 622–630 CrossRef CAS.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- F. T. L. Muniz, M. R. Miranda, C. Morilla dos Santos and J. M. Sasaki, Acta Crystallogr., Sect. A: Found. Adv., 2016, 72, 385–390 CrossRef CAS PubMed.
- K. He, N. Chen, C. Wang, L. Wei and J. Chen, Cryst. Res. Technol., 2018, 53, 1700157 CrossRef.
- T. Adschiri, Y. Hakuta and K. Arai, Ind. Eng. Chem. Res., 2000, 39, 4901–4907 CrossRef CAS.
- X. Zhang, S. Heinonen and E. Levänen, RSC Adv., 2014, 4, 61137–61152 RSC.
- I. Ushiki, R. Fujimitsu and S. Takishima, J. Supercrit. Fluids, 2020, 164, 104909 CrossRef CAS.
- Y. Zhao, Y. Shimoyama, T. Momose and Y. Shimogaki, J. Supercrit. Fluids, 2017, 120, 209–217 CrossRef CAS.
- W. Lai, S. Yang, Y. Jiang, F. Zhao, Z. Li, B. Zaman, M. Fayaz, X. Li and Y. Chen, Adsorption, 2020, 26, 633–644 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |