
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeoxygenation of heterocyclic N-oxides employing iodide and formic acid as a sustainable reductant†
Alicia Elvira
Cruz-Jiménez‡
a,
Paola Alejandra
Argumedo-Castrejón‡
a,
Jeferson B.
Mateus-Ruiz‡
a,
Victor A.
Lucas-Rosales‡
 b,
Octavio Adrián
Valle-González
a,
J. Oscar C.
Jiménez-Halla
b and
J. Armando
Luján-Montelongo
*a
b,
Octavio Adrián
Valle-González
a,
J. Oscar C.
Jiménez-Halla
b and
J. Armando
Luján-Montelongo
*a
aDepartamento de Química, Centro de Investigación y de Estudios Avanzados (Cinvestav) Av. Instituto Politécnico Nacional 2508 San Pedro Zacatenco, 07360 Ciudad de México, Mexico. E-mail: jalujanm@cinvestav.mx
bDepartamento de Química, División de Ciencias Naturales y Exactas, Sede Noria Alta, Universidad de Guanajuato, Noria Alta s/n, C.P, 36050, Guanajuato, Gto., Mexico
First published on 9th May 2024
Abstract
We present a novel deoxygenation method for heterocyclic N-oxides utilizing iodide as a catalyst. Iodide acts as a reducing catalyst that is regenerated by formic acid, which also serves as a Brønsted activator and solvent. The method demonstrates high efficiency and excellent selectivity in the reduction of a variety of heterocyclic N-oxides and tertiary amines. Our computational DFT investigation revealed that the reduction mechanism entails a direct interaction between iodide and the oxygen of the N-oxide within a Mg2+/formic acid framework, resulting in the formation of the N-heterocycle and the release of a hypoiodite unit. Additionally, a molecular mechanism for the regeneration of iodide from hypoiodite, facilitated by formic acid, is suggested. This method provides an environmentally friendly approach for the deoxygenation of N-oxides and related species.
Pyridines and quinolines are fundamental heterocyclic structures, extensively found in natural products,1 and playing pivotal roles across various fields. Their significance ranges from their inherent presence in biologically active compounds2 to applications in medicinal chemistry (Fig. 1a),3 and materials science.4 In consequence, compounds containing N-pyridinic moieties are among the most commonly used in the manufacture of drug candidates.5 Unfortunately, the direct functionalization of pyridines and their derivatives through direct electrophilic substitutions imposes a challenge due to poor chemoselectivity and a lower π-system energy compared to benzene.6 A compelling strategy for effective pyridine functionalization involves the use of N-activated pyridines,7 among which N-oxide derivatives (Fig. 1b), with remarkable availability and reactivity, enable functionalization through various strategies.8 Therefore, after the functionalization of pyridine/quinoline N-oxides, regeneration of the N-heterocycle through deoxygenation would be needed. Among the reductant systems used for heterocyclic N-oxide deoxygenation (NOD) many are based on transition metals (TM)9 including Mo, Pd, In, Ir, Ru, Sm, etc., while other TM-free based methods are found in lesser extent.10
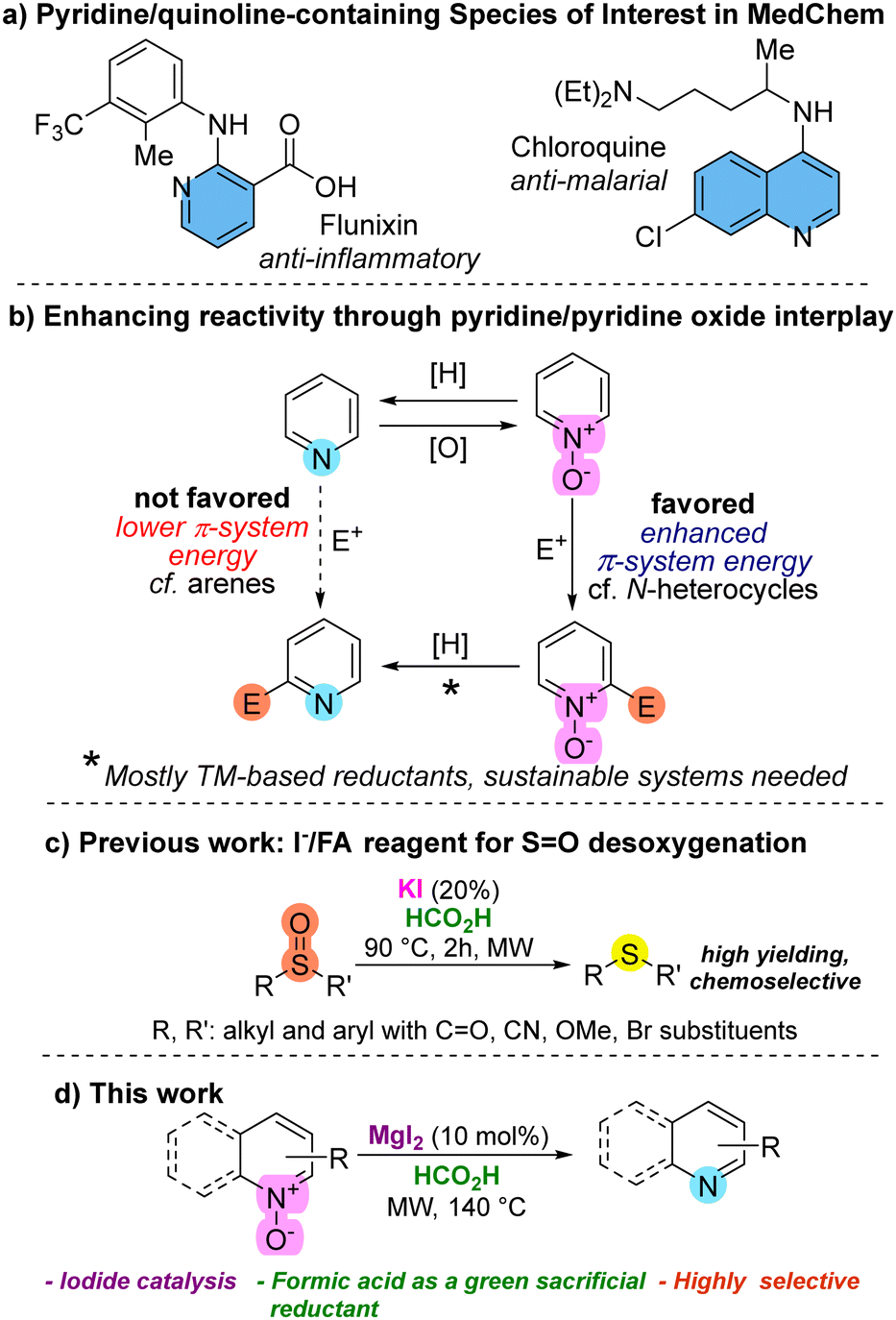 | ||
| Fig. 1 (a) Examples of relevant N-oxide pyridine species in medicinal chemistry (MedChem). (b) Pyridine N-oxide features enhanced reactivity compared to pyridine. (c) I−/FA reagent for the efficient reduction of sulfoxides (shown) and methyl sulfinates (not shown). (d) MgI2 as a convenient iodide source for a TM-free reduction based on the I−/FA reagent. | ||
Iodine-based deoxygenations of N-functionalities have emerged as alternatives,11,12 particularly since some methods employ green sacrificial reductants.12 In the case of iodide-based methodologies for NODs, iodide is often used as a sacrificial reductant, in stoichiometric or superstoichiometric quantities, and often paired with metals, including TMs.13
The search for cost-effective, efficient, and environmentally friendly methods for the selective reduction of heterocyclic N-oxides is crucial. Formic acid (FA) has emerged as a prominent reducing agent.14 Furthermore, its production from biomass or CO2 valorization adds a sustainable appeal.15 Recently, we introduced the I−/FA as a green reagent for reducing methylsulfinates and sulfoxides (Fig. 1c).16 This method utilizes iodide as the catalytic reductant, which is regenerated by formic acid. Additionally, formic acid serves both as the medium/solvent and in the elaboration of an activating Brønsted template. Under the optimal conditions, we observed exceptional reducing efficiency, coupled with remarkable orthogonality to various reducible functional groups.
Following our research program, which focuses on the development of TM-free16,17 and iodine/iodide-based synthetic methods,16,18 we present a practical method that enables the deoxygenation of pyridine N-oxides (and related species such as N-oxides derived from quinolines, isoquinolines and tertiary amines) using the I−/FA reagent. Our experimentation began exposing quinoline N-oxide (1a) to KI (10% mol) in formic medium, under MW irradiation, delivering 40% yield of quinoline (2a) (entry 1, Table 1). As expected, iodide was essential for the reduction as the absence of KI or its replacement by potassium salts such as K2SO4 and K2HPO4 delivered no reduced product (entries 2–4). Other iodide sources such as TBAI, NaI, KIO3, ZnI2, I2, and MgI2 were also evaluated, with the last two (entries 9 and 10) exhibiting superior performance compared to KI. Although KI, I2 and MgI2 delivered comparable results in terms of yield, MgI2 delivered better reproducibility and chemoselectivity, prompting us to proceed with it for subsequent experiments. We had prior evidence indicating that iodide demonstrated superior performance compared to other halides in similar reductions.16 This was reinforced for the N–O reduction through additional experiments employing magnesium chloride and bromide (entries 16 and 17), delivering low conversion. No contribution from Mg2+ was observed, further supporting the role of iodide as the reducing entity in MgI2 (cf. entries 10 and 11). From tuning of reaction conditions, we identified 10% mol of MgI2 and 3 h of reaction time as optimal (entry 14). An additional experiment conducted with conventional heating underscored the necessity of microwave activation (cf. entries 13 and 18).19,20
|
|
||||
|---|---|---|---|---|
| Entry | Halide source [I−] | (% mol) | Time (h) | Yield (%)a |
| a Determined by 1H-NMR. b Isolated yields. c Reaction performed using a conventional heating reactor at 140 °C (Anton-Paar Monowave® 50). | ||||
| 1 | KI | 10 | 1 | 40 |
| 2 | — | — | 1 | — |
| 3 | K2SO4 | 10 | 1 | — |
| 4 | K2HPO4 | 10 | 1 | — |
| 5 | TBAI | 10 | 1 | 39 |
| 6 | NaI | 10 | 1 | 37 |
| 7 | KIO3 | 10 | 1 | 37 |
| 8 | ZnI2 | 5 | 1 | 14 |
| 9 | I2 | 5 | 1 | 42 |
| 10 | MgI2 | 5 | 1 | 46 |
| 11 | MgSO4 | 5 | 1 | — |
| 12 | MgI2 | 5 | 3 | 57 |
| 13 | MgI2 | 10 | 1 | 47 |
| 14 | MgI 2 | 10 | 3 | 91/91 |
| 15 | MgI2 | 10 | 4 | 95/85b |
| 16 | MgCl2 | 10 | 3 | 2 |
| 17 | MgBr2 | 10 | 3 | 2 |
| 18c | MgI2 | 10 | 3 | 65/49b |
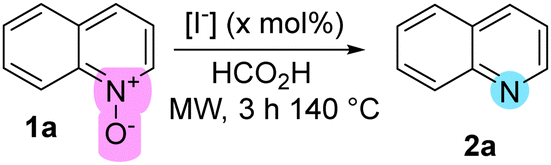
With the optimal conditions at hand, diverse N-oxides with varying substituents were submitted to the optimal deoxygenation conditions. Isoquinoline, benzo[h]quinoline, and phenazine N-oxides (1b–1d) were successfully deoxygenated with comparable yields to those obtained using TM-based technologies (Table 2).9
Interestingly, sterically hindered 2,6-methylated pyridine N-oxides (1e, 1f) underwent deoxygenation without any detrimental effects, yielding the corresponding pyridines with excellent yields. Furthermore, complete orthogonality was observed for carbonyl-containing N-oxides, such as 4-acetylpyridine N-oxide (1h) and 4-benzoylpyridine N-oxide (1i). These results significantly support a wider range of conditions under which the I−/FA reductive reagent is effective (90–140 °C), without affecting carbonyl groups.21 Additionally, a cyano-substituted substrate (1j) and 6-MeO-quinoline N-oxide (1m) underwent chemoselective N-deoxygenation, producing the corresponding heterocycles with excellent yields.
Under standard conditions, 3-nitroquinoline N-oxide (1k) underwent reduction of both the N-oxide and nitro groups, accompanied by formylation to yield product 2ka. In contrast, 2-aminopyridine N-oxide (1g) was deoxygenated with excellent yield and without formylation, a result attributed to the significant electronic differences between positions 2 and 3 on the pyridine ring.22 After adjusting the reaction conditions, 2k was successfully obtained with the nitro group remaining unaffected (see ESI†). As for 4-nitroquinoline N-oxide (1l), a Nef-type hydrolytic removal of the nitro group was impossible to avoid, delivering quinolin-4-(H)-one (2l) as the sole product.23 All attempts to adjust the conditions to obtain the nitro-containing deoxygenated product were unsuccessful (see ESI†). Finally, N-oxides of tertiary amines such as 1n and 1o were obtained with particularly good yields. Importantly, in all successful experimental trials, the reactor vial was pressurized during the reaction process and maintained a high pressure (>10 psi) after cooling to room temperature, which is consistent with the release of CO2 as byproduct.24 Upon completion of the reductions, the reaction mixtures typically developed a brownish hue, indicative of the presence of molecular iodine.
To get insight in the reaction mechanism for the N–O reduction, we performed DFT calculations (see ESI,† for further details) employing pyridine N-oxide as a model and considering the optimal conditions above discussed. First, we explored the interaction between MgI2 and solvent molecules (Fig. 2). In a recent previous work, we demonstrated that the coordination modes of the Mg metal center vary based on the σ-donor capability of the Lewis base.25 Consequently, FA acts as a ligand and magnesium can coordinate up to four units, through the carbonyl O atom, resulting in Mg–O–C angles of approximately 120°. Contrary to what one might expect, FA-ligands are monodentate and not bidentate. The formation of the lower coordination modes A-1 and A-2 is spontaneous, while the penta- and hexa-coordinated species, A-3 and A-4, corresponding to sp3d and sp3d2 hybridizations at the Mg center, exist in equilibrium with the tetrahedral A-2 mode. The NOD mechanistic pathway and the subsequent iodide regeneration begins with a double substitution of iodide for two FA-ligands within the Mg coordination sphere (A-4 → I-1 → I-2, Fig. 3). The formation of the [Mg2+(OCHOH)6]2I− complex (I-2) requires ΔGA-4 → I−2 = +4.2 kcal mol−1. Subsequently, the spontaneous addition of a pyridine N-oxide (C5H5NO) unit forms I-3 (ΔGI-2 → I-3 = −4.5 kcal mol−1). This addition is deemed favorable due to the hydrogen bonding between the oxygen in pyridine N-oxide and protons from formic acid units. Despite the pKa values of pyridine N-oxide (0.79)26 and formic acid (3.5)27 suggesting that proton transfer (PT) between these Brønsted species might not be favored, our findings show that this process is both barrierless and exergonic (ΔGI-3 → I-4 = −3.5 kcal mol−1). Thus, it seems that an effect of increased acidity of FA protons, through Mg-FA coordination, facilitates the PT process to access I-4 effortlessly. Next, a nucleophilic substitution (SN2-type) takes place, centered on the oxygen atom viaTS-1 (ΔG‡I-4 → TS-1 = +32.5 kcal mol−1, in agreement with T = 140 °C), marking the rate-determining step and resulting in the formation of the pyridine-hypoiodous acid pair in I-5. Remarkably, formic acid acts as a Brønsted-activator enabling the electrophilicity of the pyridine N-oxide, so the nucleophilic attack of iodide is feasible. From I-5, a hydrogen transfer process was determined viaTS-2A, with an energy barrier of ΔG‡I-5 → TS-2A = +20.1 kcal mol−1, leading to the production of hydroiodic acid and carbon dioxide (P-A) through a net exothermic process. Then, hydroiodic acid spontaneously transfers its proton to the pyridine supported by a H2O unit.
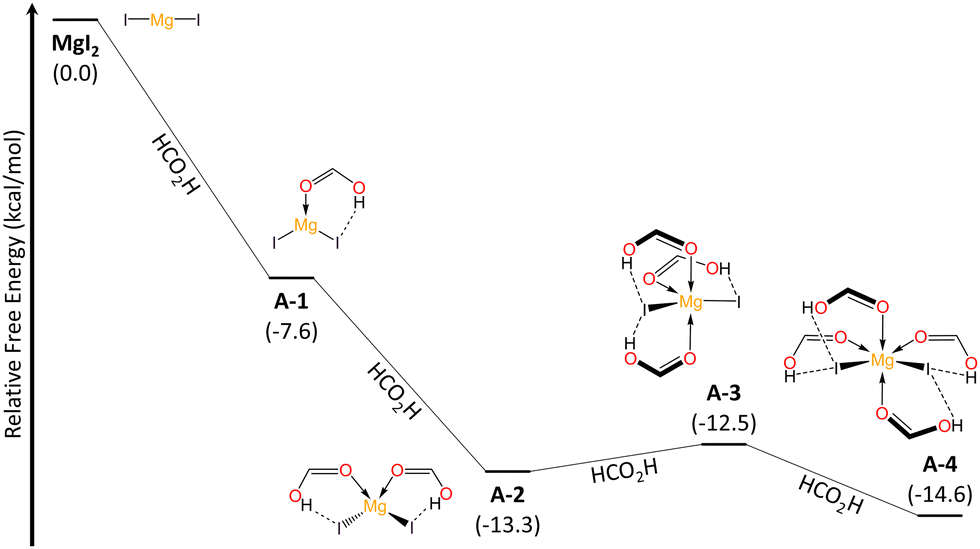 | ||
| Fig. 2 Relative Gibbs free energy (kcal mol−1) profile for the progressive coordination of formic acid ligands to MgI2 at the (SMD:HCO2H)ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVP level and 298.15 K. | ||
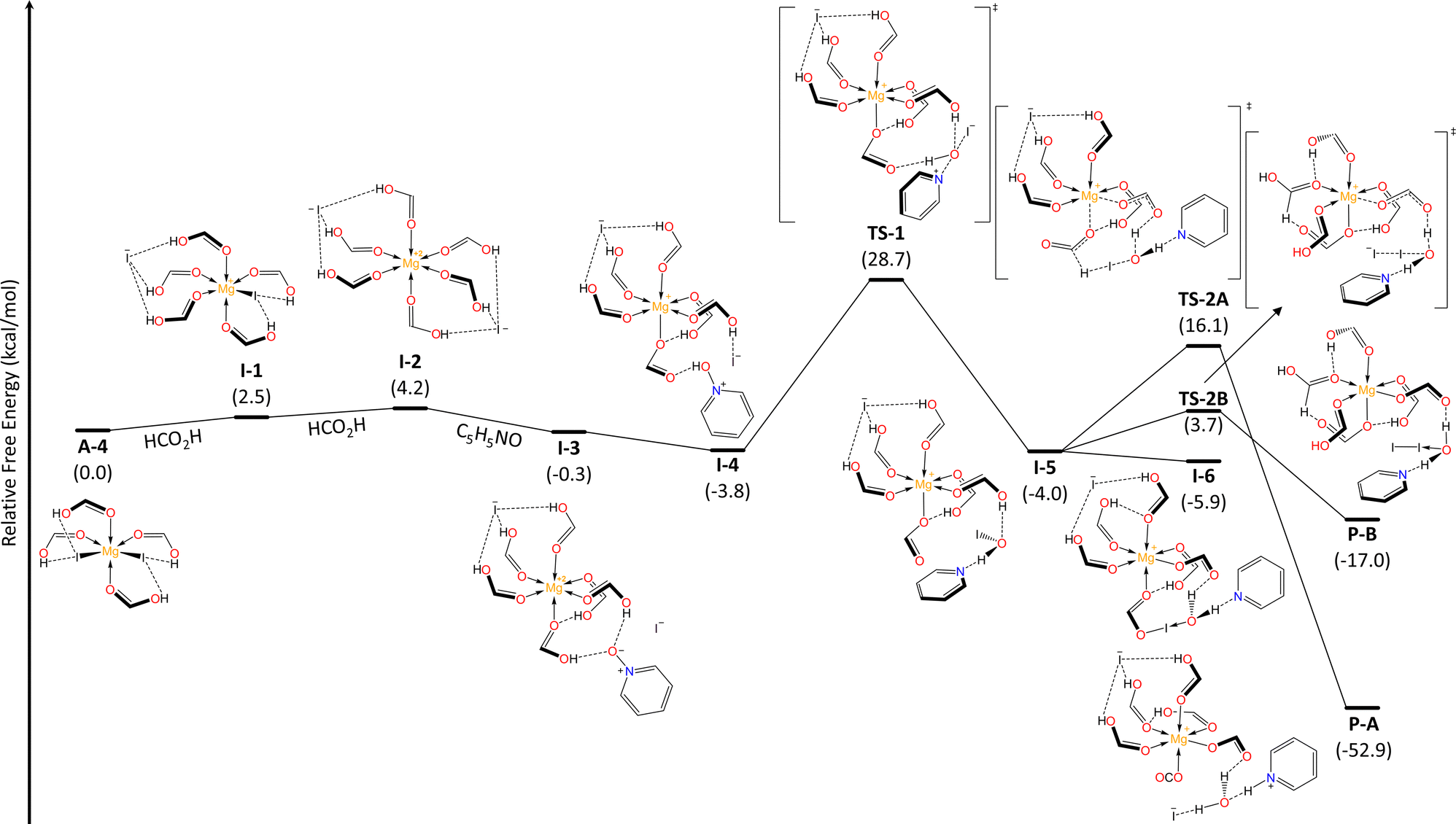 | ||
| Fig. 3 Relative Gibbs free energy (kcal mol−1) profile for the pyridine N-oxide deoxygenation using FA-coordinated MgI2 (A-4) at the (SMD:HCO2H)ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVP level and 298.15 K. | ||
We identified an equilibrium between I-5 and a formyl hypoiodite species (I-6, ΔGI-5 → I-6 = −1.9 kcal mol−1). Finding a transition state connecting these minima proved to be difficult. Starting from I-6, I-5 can also lead to the formation of molecular iodine (I2, P-B) through iodide addition, with an energy barrier of ΔG‡I-6 → TS-2B = +9.6 kcal mol−1. These equilibria towards non-iodide productive species determines the molar percentage required for the iodide reactant, as amounts of MgI2 lower than standard significantly affect the conversion rate (cf. entries 12,14, Table 1). However, our thermochemical data suggest that iodine (P-B) can be converted into P-A by accessing I-5 and then proceeding through TS-2A, requiring an energy investment of ΔG‡P-B → TS-2A = +33.1 kcal mol−1.
Conclusions
We developed a methodology with a sustainable basis for the deoxygenation of pyridine N-oxides, as formic acid is employed as activator, solvent, and stoichiometric reductant, using MgI2 as a convenient iodide source, which serves as a catalytic reductant. This method proved to be compatible with various potentially reducible functional groups, including carbonyl, cyano, benzyl, and to some extent, the nitro group. The same system also allowed for the deoxygenation of tertiary amines. The mechanistic study through DFT suggests that the N-oxide deoxygenation occurs via an SN2-type mechanism. Formic acid performs Brønsted-activation, increasing its acidity through Mg2+ coordination (Lewis to Brønsted acid relay). Also, the molar concentration of MgI2 must be such to overcome non-iodide productive oxidized iodine species, such as I2. The regeneration of iodide mainly occurs through the reduction of hypoiodous acid by hydrogen transfer delivering a net exothermic process. To regenerate iodide from molecular iodine, the pathway involves the reversion towards hypoiodite followed by hydrogen transfer, requiring +33.1 kcal mol−1 to complete the reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from CONACYT/CONAHCYT (Mexico) through a Ciencia de Frontera grant (CF-2019-51493). A. C.-J. would like to express gratitude to CONACYT/CONAHCYT for the PhD fellowship 708711 and V.A.L.-R. also acknowledges CONAHCYT for financial support through a MSc fellowship (1312084). P. A. A.-C. and J. B. M.-R. thank CONAHCYT for undergraduate and postdoctoral fellowships, respectively, within grant CF-2019-51493. The authors would like to acknowledge the computing time supported by CONACYT (Grant CB-2015-252356), and additional resources provided by LANCAD and CONACYT/CONAHCYT at the super-computer hybrid cluster Xiuhcoatl at the General Coordination of Information and Communication Technologies (CGSTIC) of CINVESTAV (https://clusterhibrido.cinvestav.mx). Special thanks are extended to Teresa Cortéz, Géiser Cuellar, Ma. Luisa Rodríguez and Víctor González for assistance in spectral acquisition.Notes and references
- M. F. Grundon, Nat. Prod. Rep., 1984, 1, 195–200 RSC.
- Selected literature: (a) T. Tahir, M. Ashfaq, M. Saleem, M. Rafiq, M. I. Shahzad, K. Kotwica-Mojzych and M. Mojzych, Molecules, 2021, 26, 4872 CrossRef CAS PubMed; (b) B. S. Matada, R. Pattanashettar and N. G. Yernale, Bioorg. Med. Chem., 2021, 32, 115973 CrossRef CAS PubMed; (c) H. William, K. Kutterer, A. Crombie and J. Clemens, Prog. Heterocycl. Chem., 2009, 6, 289–332 Search PubMed; (d) A. Y. Khan and G. Suresh Kumar, Biophys. Rev., 2015, 7, 407–420 CrossRef CAS PubMed.
- Selected literature: (a) R. Kaur and K. Kumar, Eur. J. Med. Chem., 2021, 215, 113220 CrossRef CAS PubMed; (b) P. E. Alford. Six-Membered Ring Systems: Pyridines and Benzo Derivatives. in Progress in Heterocyclic Chemistry, vol. 22, ed. G. Gribble and J. A. Joule, Elsevier, 2011, pp. 349–391 Search PubMed; (c) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed; (d) A. Basnet, P. Thapa, R. Karki, Y. Na, Y. Jahng, B. S. Jeong, T. C. Jeong, C. S. Lee and E. S. Lee, Bioorg. Med. Chem., 2007, 15, 4351–4359 CrossRef CAS PubMed; (e) G. Bentzinger, E. Pair, J. Guillon, M. Marchivie, C. Mullié, P. Agnamey, A. Dassonville-Klimpt and P. Sonnet, Tetrahedron, 2020, 76, 131088 CrossRef CAS.
- Selected literature: (a) S. Varshney and N. Mishra. Pyridine-based polymers and derivatives: Synthesis and applications. in Recent Developments in the Synthesis and Applications of Pyridines, ed P. E. Singh, Elsevier, 2022, pp. 43–69 Search PubMed; (b) K. Kumar, A. Karmakar, F.-R. Chen, J.-H. Jou, S. Ghosh, S. Banik and S. Kumar, Phys. Chem. Chem. Phys., 2023, 25, 19648–19659 RSC; (c) C. Verma, K. Y. Rhee, M. A. Quraishi and E. E. Ebenso, J. Taiwan Inst. Chem. Eng., 2020, 117, 265–277 CrossRef CAS.
- (a) J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC; (b) S. De, A. Kumar, S. K. Shah, S. Kazi, N. Sarkar, S. Banerjee and S. Dey, RSC Adv., 2022, 12, 15385–15406 RSC.
- A. Katritzky and R. TaylorHeteroaromatics Containing One Six-Membered Ring. In Advances in Heterocyclic Chemistry, Academic Press, 1990, vol. 47, pp. 277–323 Search PubMed.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charrette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- (a) D. Wang, L. Désaubry, G. Li, M. Huang and S. Zheng, Adv. Synth. Catal., 2021, 363, 2–39 CrossRef CAS; (b) R. Tomar, A. Kumar, A. Dalal, D. Bhattacharya, P. Singh and S. Arulananda Babu, Asian J. Org. Chem., 2022, 11, e202200311 CrossRef CAS; (c) J. Zhong, Y. Long, X. Yan, S. He, R. Ye, H. Xiang and X. Zhou, Org. Lett., 2019, 21, 9790–9794 CrossRef CAS PubMed ; For a recent umpolung strategy for the electrophilic functionalization of N-oxides see: ; (d) D. I. Bugaenko, M. A. Yurovskaya and A. V. Karchava, Org. Lett., 2021, 23, 6099–6104 CrossRef CAS PubMed.
- Selected literature: (a) J. Li, S. Liu, T. L. Lohr and T. J. Marks, ChemCatChem, 2019, 11, 4139–4146 CrossRef CAS; (b) R. Rubio-Presa, M. A. Fernández-Rodríguez, M. R. Pedrosa, F. J. Arnáiz and R. Sanz, Adv. Synth. Catal., 2017, 359, 1752–1757 CrossRef CAS; (c) Y. Kuwahara, Y. Yoshimura, K. Haematsu and H. Yamashita, J. Am. Chem. Soc., 2018, 140, 9203–9210 CrossRef CAS PubMed; (d) K. D. Kim and J. Lee, Org. Lett., 2018, 20, 7712–7716 CrossRef CAS PubMed; (e) Y. Handa, J. Inanaga and M. Yamaguchi, J. Chem. Soc., Chem. Commun., 1989, 298–299 RSC; (f) J. A. Fuentes and M. L. Clarke, Synlett, 2008, 2579–2582 CAS; (g) B. W. Yoo and J. W. Choi, Synth. Commun., 2009, 39, 3550–3554 CrossRef CAS.
- Selected literature: (a) Y. Kawazoe and M. Tachibana, Chem. Pharm. Bull., 1965, 13, 1103–1107 CrossRef CAS PubMed; (b) S. R. Ram, K. P. Chary and D. S. Iyengar, Synth. Commun., 2000, 30, 3511–3515 CrossRef CAS; (c) H. P. Kokatla, P. F. Thomson, S. Bae, V. R. Doddi and M. K. Lakshman, J. Org. Chem., 2011, 76, 7842–7848 CrossRef CAS PubMed; (d) H. Suzuki, N. Sato and A. Osuna, Chem. Lett., 1980, 9, 459–460 CrossRef; (e) A. Romero and H. Cerecetto, Eur. J. Org. Chem., 2020, 1853–1865 CrossRef CAS.
- D. S. Raghuvanshi, N. Verma and A. Gupta, ChemistrySelect, 2019, 4, 2075–2078 CrossRef CAS.
- T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Green Chem., 2016, 18, 2966–2970 RSC.
- Selected literature: (a) T. Morita, K. Kuroda, Y. Okamoto and H. Sakurai, Chem. Lett., 1981, 10, 921–924 CrossRef; (b) R. Balicki, Chem. Ber., 1990, 123, 647–648 CrossRef CAS; (c) B. W. Yoo and M. C. Park, Synth. Commun., 2008, 38, 1646–1650 CrossRef CAS; (d) B. W. Yoo, J. W. Choi and S. Park, Bull. Korean Chem. Soc., 2008, 29, 909–910 CrossRef CAS; (e) M. M. Khodaei, A. Alizadeh and H. A. Hezarkhani, J. Iran. Chem. Soc., 2018, 15, 1843–1849 CrossRef CAS; (f) M. Boruah and D. Konwar, Synlett, 2001, 795–796 CrossRef CAS.
- Selected literature: (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (b) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed; (c) L.-H. Gong, Y.-Y. Cai, X.-H. Li, X. Y.-N. Zhang, J. Su and J.-S. Chen, J. Green Chem., 2014, 16, 3746–3751 RSC.
- (a) D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross. The Biofine Process – Production of Levulinic Acid, Furfural, and Formic Acid from Lignocellulosic Feedstocks. in Biorefineries-Industrial Processes and Products: Status Quo and Future Directions. ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley, 2005, pp. 139–164 Search PubMed; (b) K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- J. A. Luján-Montelongo, L. J. García de la Cuesta, A. E. Cruz-Jiménez, P. Hernández and A. Vela, Green Chem., 2023, 25, 7963–7970 RSC.
- O. A. Valle-González, Á. I. Salazar-Bello and J. A. Luján-Montelongo, Org. Biomol. Chem., 2023, 21, 2894–2898 RSC.
- J. A. Luján-Montelongo, J. B. Mateus-Ruiz and R. M. Valdez-García, Eur. J. Org. Chem., 2023, 26, e202201156 CrossRef.
- (a) G. B. Dudley, R. Richert and A. E. Stiegman, Chem. Sci., 2015, 6, 2144–2152 RSC; (b) J. Zhou, W. Xu, Z. You, Z. Wang, Y. Luo, L. Gao, C. Yin, R. Peng and L. Lan, Sci. Rep., 2016, 6, 25149 CrossRef CAS PubMed.
- A Biotage® Initiator+ microwave reactor and 0.5-2 mL reaction tubes were used in the N-O deoxygenation reactions. The radiation absorption parameter was set to VERY HIGH, with the apparatus usually recording a pressure increase (10-20 Psi).
- The sulfinyl reduction with I−/FA usually is carried out at 90 °C (see reference 16) whereas heterocyclic N-oxides required 140 °C. For selected literature featuring C-O bond reductions involving iodide: (a) L. D. Hicks, J. K. Han and A. J. Fry, Tetrahedron Lett., 2000, 41, 7817–7820 CrossRef CAS; (b) M. Allukian, G. Han, L. Hicks and A. J. Fry, ARKIVOC, 2002, 76–79 Search PubMed; (c) J. E. Milne, T. Storz, J. T. Colyer, O. R. Thiel, M. D. Seran, R. D. Larsen and J. A. Murry, J. Org. Chem., 2011, 76, 9519–9524 CrossRef CAS PubMed; (d) L.-Z. Yuan, D. Renko, I. Khelifi, O. Provot, J.-D. Brion, A. Hamze and M. Alami, Org. Lett., 2016, 18, 3238–3241 CrossRef CAS PubMed; (e) L.-Z. Yuan, G. Zhao, A. Hamze, M. Alami and O. Provot, Adv. Synth. Catal., 2017, 359, 2682–2691 CrossRef CAS.
- Recently, a formylation of primary and secondary amines under comparable conditions was demonstrated: H. Liang, T. Zhao, J. Ou, J. Liu and X. Hu, ACS Sustainable Chem. Eng., 2023, 11, 14317–14322 CrossRef CAS.
- J. Xu, X. Miao, L. Liu, Y. Wang and W. Yang, ChemSusChem, 2021, 14, 5311–5319 CrossRef CAS PubMed.
- N. D. Rode, A. Arcadi, M. Chiarini and F. Marinelli, Synthesis, 2017, 49, 2501–2512 CrossRef CAS.
- L. I. Lugo-Fuentes, V. A. Lucas-Rosales, J. A. Sandoval-Mendoza, R. Shang, J. P. Martínez and J. O. C. Jimenez-Halla, Chem. – Eur. J., 2024, 30, e202304130 CrossRef CAS PubMed.
- (a) L. Chmurzyński, Anal. Chim. Acta, 1996, 321, 237–244 CrossRef; (b) P. Mech, M. Bogunia, A. Nowacki and M. Makowski, J. Phys. Chem. A, 2020, 124, 538–551 CrossRef CAS PubMed.
- (a) M. H. Kim, C. S. Kim, H. W. Lee and K. Kim, J. Chem. Soc., Faraday Trans., 1996, 92, 4951–4956 RSC; (b) X.-x. Wang, H. Fu, D.-m. Du, Z.-y. Zhou, A.-g. Zhang, C.-f. Su and K.-s. Ma, Chem. Phys. Lett., 2008, 460, 339–342 CrossRef CAS; (c) F. R. Dutra, C. de S. Silva and R. Custodio, J. Phys. Chem. A, 2021, 125, 65–73 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the calculated reaction mechanism. See DOI: https://doi.org/10.1039/d4nj00913d |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |