
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct pseudomorphic transformation of silica from rice husk into organo-functionalized MCM-41†
Lilia
Sennoun
 a,
Chun-Cheng
Lee
a,
Yohan
Fretel
a,
Margaux
Clavié
b,
Gilles
Subra
b,
Yoann
Ladner
b,
Alfredo
Napoli
c,
Anne
Galarneau
a,
Peter
Hesemann
a and
Ahmad
Mehdi
*a
a,
Chun-Cheng
Lee
a,
Yohan
Fretel
a,
Margaux
Clavié
b,
Gilles
Subra
b,
Yoann
Ladner
b,
Alfredo
Napoli
c,
Anne
Galarneau
a,
Peter
Hesemann
a and
Ahmad
Mehdi
*a
aICGM, Univ Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: ahmad.mehdi@umontpellier.fr
bIBMM, Univ Montpellier, CNRS, ENSCM, Montpellier, France
cCIRAD-PERSYST-UPR BioWooEB, Montpellier, France
First published on 15th March 2024
Abstract
To reduce the cost of silica-based materials, an agricultural waste, rice husk (RH), was transformed into higher valuable products such as MCM-41 and organo-functionalized MCM-41 in the frame of circular economy. RH is the biomass waste containing the highest amount of silica (214 g kg−1). Direct calcination of RH led to silica with low surface area (10 m2 g−1), which could not be transformed into MCM-41. After optimization, silica of 310 m2 g−1 was produced by washing RH with HCl 0.1 mol L−1 at 25 °C and calcination at 600 °C. Such silica was successfully transformed in one-step synthesis at 115 °C for 24 h into highly ordered MCM-41 (800 m2 g−1) and organo-functionalized MCM-41 (510–720 m2 g−1) including aminopropyl and amide derivatives of amino acids (leucine, serine and tyrosine). The corresponding organo-triethoxysilanes alone or in mixture were directly added in the synthesis with silica from RH, octadecyltrimethylammonium and NaOH solution. Surfactant removal was successfully performed with EtOH/NH4NO3 solution at 60 °C. The mesopore diameter of the materials were homogeneous and varied from 3.8 to 4.2 nm depending on the organic functions. Thanks to the use of the concept of pseudomorphic transformation, the particle size of the materials ranged from 1 to 100 μm. As proof of concept, the 50–100 μm fraction (30% of the volume) was successfully used to fill columns to run size exclusion chromatography for protein separation. The largest protein, bovine serum albumine (BSA) was excluded from all materials as its kinetic diameter (d = 7.2 nm) was larger than the mesopore diameter. The smallest proteins: carbonic anhydrase (d = 4.2 nm), myoglobin (d = 3.8 nm) and lysozyme (d = 3.4 nm) were retained by the materials. Their retention factor increased as the kinetic diameter decreased, as the mesopore diameter decreased and as the hydrophobicity of the materials increased. The best separation of proteins was obtained with MCM-41 functionalized with both leucine and serine amide derivatives.
1. Introduction
Rice, as the second most produced cereal in the world, generates large number of biomass wastes such as rice straw and rice husks. Rice husk (RH) consists of the protective envelope of the rice grain during its growth, which is removed during the milling process. Globally, 970 millions tons of rice per year are produced in the world, leading to 272 millions tons of RH per year,1 with the major contributors being Asia, North America and Latin America. In Europe, eight countries contribute to this production, with a total quantity of 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons, with half generated by Italy. France (Camargue region) produces annually 16500 tons.2 It is mostly used as a fuel for thermal power generation, and also in building materials or steel production sectors.2 RH has attracted much attention in recent years as a source of biogenic silica nanoparticles.3 Besides being a bio-based material, this silica is also a material of circular economy, as it is a by-product of agricultural activities that will gain much higher value thanks to its applications. The production of this bio-circular silica will be launched by the end of 2024 by Solvay in Europe (Livorno, Italy) as an alternative to silica nanoparticles coming from sand for the tires industry.4 This more sustainable source of silica will lead to a 50% reduction in CO2 emissions per ton of silica. Based on dry matter, RH consists of 80 wt% organics (33 wt% cellulose, 20 wt% hemicellulose, 22 wt% lignin, 4 wt% proteins, oils) and 20 wt% inorganic components, mostly silica5 (87–97 wt%) with some traces of metallic impurities (3–13 wt%).6–8 Various researchers have developed methods to reach high-purity silica from different sources of RH (see for example a recent review on the best synthesis to produce silica nanoparticles from RH and its applications).3
000 tons, with half generated by Italy. France (Camargue region) produces annually 16500 tons.2 It is mostly used as a fuel for thermal power generation, and also in building materials or steel production sectors.2 RH has attracted much attention in recent years as a source of biogenic silica nanoparticles.3 Besides being a bio-based material, this silica is also a material of circular economy, as it is a by-product of agricultural activities that will gain much higher value thanks to its applications. The production of this bio-circular silica will be launched by the end of 2024 by Solvay in Europe (Livorno, Italy) as an alternative to silica nanoparticles coming from sand for the tires industry.4 This more sustainable source of silica will lead to a 50% reduction in CO2 emissions per ton of silica. Based on dry matter, RH consists of 80 wt% organics (33 wt% cellulose, 20 wt% hemicellulose, 22 wt% lignin, 4 wt% proteins, oils) and 20 wt% inorganic components, mostly silica5 (87–97 wt%) with some traces of metallic impurities (3–13 wt%).6–8 Various researchers have developed methods to reach high-purity silica from different sources of RH (see for example a recent review on the best synthesis to produce silica nanoparticles from RH and its applications).3
First strategies have concerned the dissolution of silica contained in biomass ashes in basic solutions (NaOH or Na2CO3) to form a sodium silicate solution, which was then used as a precursor for the synthesis of silica-based materials as ordered mesoporous silica materials such as MCM-41 and SBA-15.1,9,10 However, the yield in silica is low and rarely given. More recent strategies used acidic pretreatments of RH to remove metallic impurities (especially K+ responsible of silica crystallization and hence of the low resulting surface area) followed by controlled calcination protocols to remove organic components. Silica with specific surface area of 200–350 m2 g−1 and a broad distribution of mesopore diameter around 6 nm have been produced with high yield (Table 1).11–19 However, these procedures are barely detailed in literature. To reach larger mesopore diameter, a multiple-steps synthesis was applied consisting in the pyrolysis of the acid leached (HCl) RH, followed by silica extraction in NaOH to form a sodium silicate solution, followed by the formation of a gel by adding HCl. After drying and crushing silica gels feature surface area of 350 m2 g−1 and homogeneous mesopores of 10 nm diameter with a good yield in silica of 90%.20 To reach higher surface area (800 m2 g−1), this silica was transformed into MCM-41 in presence of surfactant and acetic acid in basic medium. Surfactant removal was performed in MeOH/HCl solution under reflux.21 These syntheses were realized mainly to synthesize silica nanoparticles.3
| RH origin | Pretreatment | Acid washing | Calcination | S BET m2 g−1 | Ref. |
|---|---|---|---|---|---|
| China | Dried 105 °C | HCl 0.7 mol L−1 1 h RT | Put 0.5 h in oven already at 600 °C | 210 | 11 |
| H2SO4 0.7 mol L−1 1 h RT | 240 | ||||
| Malaysia | C12SO4Na2/H2O | HCl 0.5 mol L−1 0.5 h 60 °C | 600 °C 2 h | 218 | 12 |
| Dried 110 °C | H2SO4 0.5 mol L−1 60 °C 0.5 h | 208 | |||
| Venezuela | HCl 4 mol L−1 24 h | 350 °C 3 h, 550 °C 2 h, 700 °C 3 h, grinding 12 h | 234 | 13 | |
| China | HCl 1 mol L−1 2.5 h RT stirring | 600 °C 2 h, grinding 10 min | 248 | 14 | |
| India | H2O | H2SO4 1 mol L−1 | 700 °C 6 h | 220 | 15 |
| Egypt | H2O | Citric acid (5 wt%) | 310 °C 1 h, 400 °C 2 h, 510 °C 5 h, 600 °C 0.5 h | 313 | 16 |
| Dried 110 °C | 50 °C 3 h and 80 °C 1 h | ||||
| Milling | |||||
| Turkey | H2O | Boiling 2 h HCl (3% v/v) | 600 °C 4 h (10 °C min−1) | 312 | 17 |
| Dried 110 °C | |||||
| China | H2O | HCl 8 wt%, 120 °C 4 h, H2O pH 7, dried 110 °C 3 h | 300 °C 0.5 h N2 | 352 | 18 |
| Dried 110 °C | 610 °C 3 h O2 | ||||
| Pulverized 10–60 Mesh | |||||
| France | H2O sieving | HNO3 100 °C 1 h, H2O pH 7, dried 100 °C 12 h | 700 °C (5 °C min−1) | 330 | 19 |
| France | H2O | HCl 0.1 mol L−1 25 °C 2 h (3 times) | 600 °C 6 h (2 °C min−1) | 312 | This work |
| Senegal | H2O pH 7 | 307 | |||
Our goal is in opposite to maintain large particle size (50–100 μm) of silica-based materials to use them in columns for various flow applications and to develop high surface area silica materials (800–1000 m2 g−1) in the minimum of synthesis steps. For this purpose, the pseudomorphic transformation of silica particles into MCM-41 is the best option.22–24 A pseudomorph has the outward appearance of the parent material.24 The pseudomorphic transformation consists to adapt the rate of dissolution of the parent silica to the rate of formation or crystallization of another silica-based material leading to the conservation of the initial particle morphology. Only one study reports the direct synthesis of unfunctionalized MCM-41 from RH silica by pseudomorphic transformation.25 Particles of 100–300 μm of MCM-41 were obtained featuring a hierarchical structure of macropores (1–1.5 μm) coming from the parent silica and homogeneous mesopores of 3.4 nm after 48 h reaction at 120 °C leading to a surface area of 920 m2 g−1.25 No application was proposed.
To be used in various applications mesoporous silica need to be functionalized, for example by organic species. Functionalization of ordered mesoporous silica, such as MCM-41 or SBA-15 materials, produce organic–inorganic hybrid silica materials20,21,26–32 of high surface area for a great variety of applications, as biotechnology,27,33 catalysis and biocatalysis,26,27 environmental remediation,28,29 biosensors,30 proteins separation,34 high-performance liquid chromatography (HPLC)22etc. Two different pathways exist for mesoporous silica functionalization: post-grafting method with organo-trialkoxylsilanes,22,32,35,36 or direct synthesis by co-condensation of tetraalkylorthosilicate with organo-trialkoxylsilanes followed by surfactants extraction with solvents.37–39 The co-condensation method offers several advantages over the post-grafting method. It is simpler, involving a reduction of synthesis steps, no calcination is needed, and allows a uniform incorporation of functional groups throughout the material.40,41 This method provides a higher degree of uniformity of functional group distribution and surface chemistry.33,38,39,42
In the present study, we combine the pseudomorphic transformation of silica from RH into MCM-41 together with the co-condensation with organo-triethoxysilanes to reach in one-step organo-functionalized MCM-41 suitable for flow applications. First the optimization of RH acidic pretreatment and calcination protocol were performed. The resulting silica from RH was then transformed by pseudomorphic transformation in a one-step procedure into MCM-41 or organo-functionalized MCM-41 by self-assembly with octadecyltrimethylammonium surfactants in basic medium. Surfactant was extracted with an ethanol/NH4NO3 solution. Purely siliceous MCM-41 and MCM-41 functionalized with aminopropyl and amino acid derivatives of tyrosine, serine, leucine were obtained and tested in protein separations.
2. Experimental
2.1 Material preparation
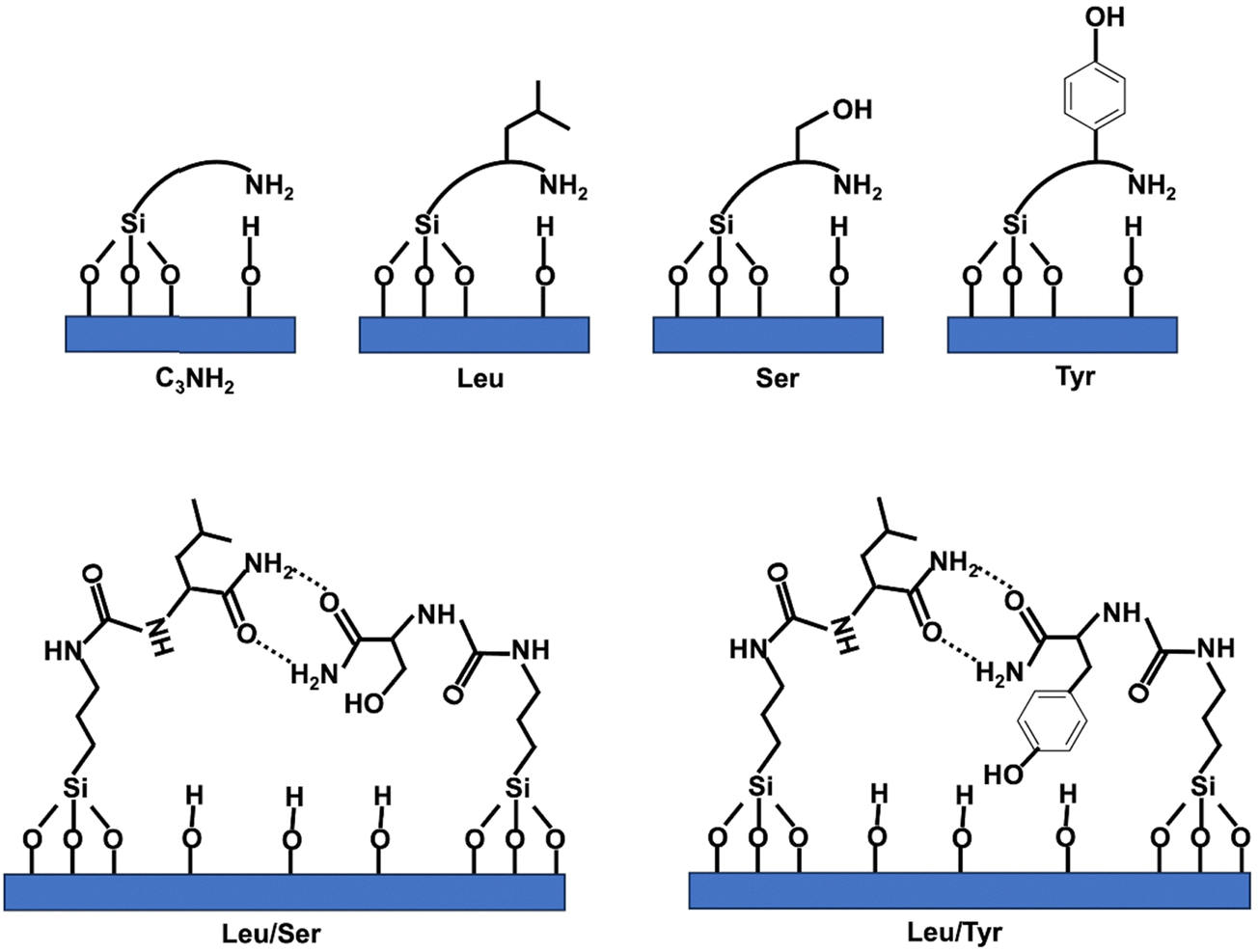
Two sources of rice husks (RH) were used: RH from France was provided by SARL Balleconcept, Arles, France, and RH from Senegal by CIRAD, Montpellier, France. Different chemicals were used in the material preparations: octadecyltrimethylammonium bromide (C18TAB, Aldrich, 392.50 g mol−1), sodium hydroxide (NaOH, ≥98.0%, Anlar Normapur), hydrochloric acid (HCl, ACS Reagents, 37%), ethanol (EtOH, Normapur, 96%), 3-aminopropyl triethoxysilane (APTES, Aldrich, 221.37 g mol−1) and deionized water (pH = 6.8). Three amide derivatives of amino-acid (AA) (L-tyrosine (Tyr), L-serine (Ser), L-leucine (Leu)) triethoxysilanes (EtO3Si-R), named Si-Tyr-NH2, Si-Ser-NH2, Si-Leu-NH2 (Scheme 1), were synthesized accordingly to the previously described protocol.32 The molar weights are 427.57, 351.48, 377.56 g mol−1, respectively. Acetonitrile (ACN) was purchased from Sigma-Aldrich. Lysozyme from chicken egg white, Lys ≥99% (16.6 kDa, hydrodynamic diameter (Dh) 3.4 nm (globular protein), isoelectic point (pI) of 9.7), bovine myoglobin (17.1 kDa, Dh = 3.8 nm, pI = 6.2), carbonic anhydrase (AC) from bovine erythrocytes (30 kDa, Dh = 4.2 nm, pI = 6.1), bovine serum albumin (BSA) (67 kDa, Dh = 7.2 nm (globular protein), pI = 4.5) were supplied from Sigma-Aldrich.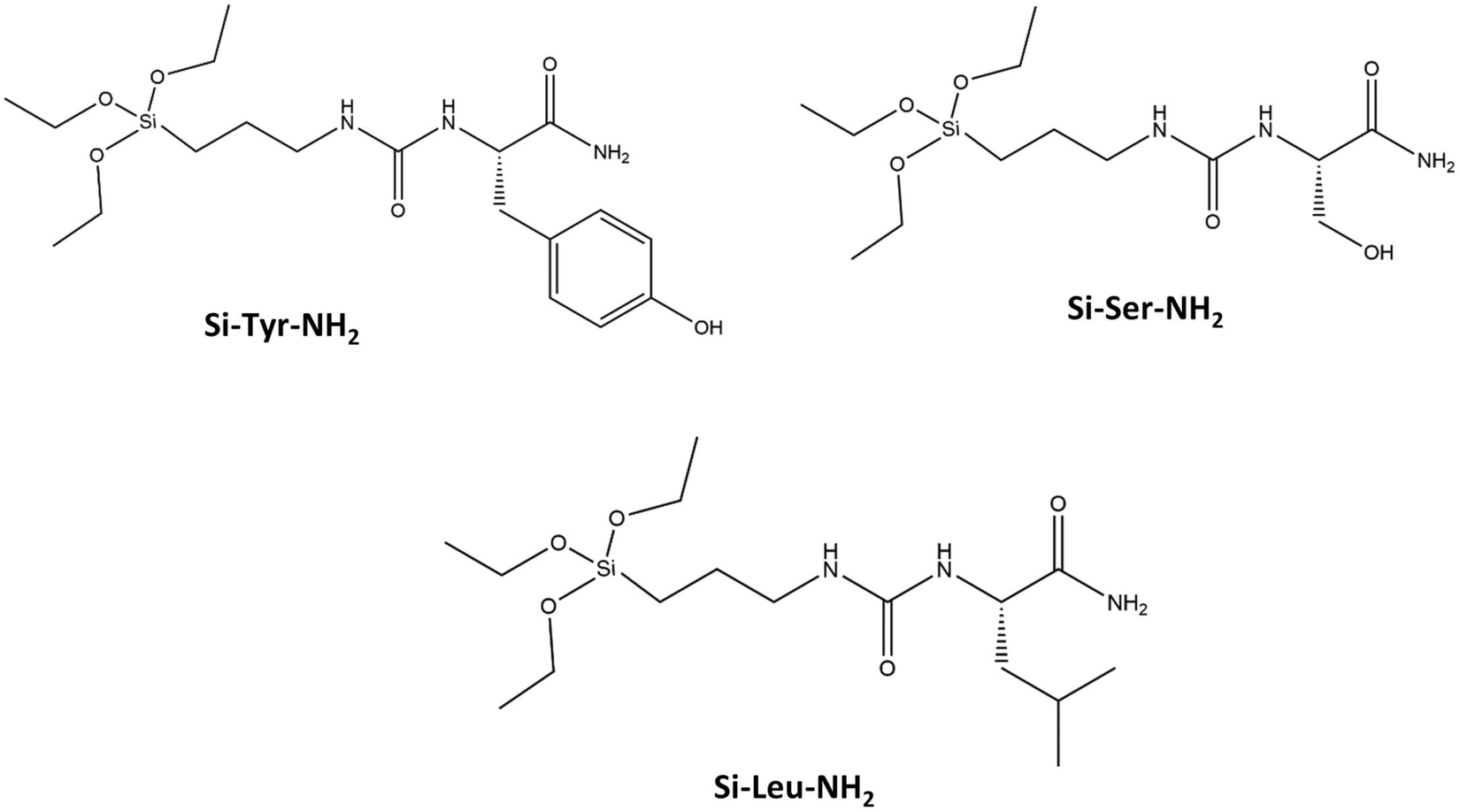 | ||
| Scheme 1 Representation of silylated amide derivatives of amino acids. | ||
:0.1 C18TAB:0.25 NaOH:50 H2O, corresponding to 15.0 g H2O, 0.167 g NaOH, 0.654 g C18TAB, 1.0 g SiO2. The mixture is poured into a Teflon autoclave and placed in an oven at 115 °C for 24 h. The autoclave is then put in an ice bath to cool down and stop the reaction. The slurry is washed with 40 mL of water and centrifuged (4–5 times) until to reach pH 7. Finally, the sample is dried in an oven for 12 h at 90 °C and a white powder of as-synthesized MCM-41 is obtained. To remove the surfactant, 1 g MCM-41 is dispersed into 150 mL of EtOH containing 0.3 g of NH4NO3 and the mixture is mechanically stirred (500 rpm) with an endless screw for 15 min at 60 °C.43 The amount of NH4NO3 corresponds to an excess of NH4+/CTMA+ = 2 molar ratio. Surfactant-extracted MCM-41 is recovered by centrifugation, washed with around 40 mL of ethanol and centrifuged again. The treatment is repeated 3 times. The material is then dried at 80 °C for 12 h.
:1/3 Si-Leu-NH2:1/3 Si-Ser-NH2): (1) in 1/39 molar ratio corresponding to the addition of 60.9 mg Si-Tyr-NH2, 50.1 mg Si-Ser-NH2, 53.8 mg Si-Leu-NH2 or (2) in 1/19 molar ratio corresponding to the addition of 125 mg Si-Tyr-NH2, 102.8 mg Si-Ser-NH2, 110.4 mg Si-Leu-NH2. The resulting materials are named: MCM-41@3AA(1/39), MCM-41@3AA(1/19). The surfactant was extracted by the previous protocol and the resulting materials were dried at 80 °C for 12 h.
2.2 Characterization
X-ray diffraction (XRD) is performed using an X’pert Pro diffractometer (PanAnalytical) with Ni-filtered Cu radiation (λ = 1.5418 Å). The diffracted intensity is detected using an ultrafast X’celerator detector. The material is deposited on a plexiglass sample holder and measurements are performed at room temperature (25 °C) between 2θ = 1 and 7° using rapid scans (analysis time = 12 min). Thermogravimetric analysis (TGA) is performed with a PerkinElmer STA6000 Simultaneous Thermal analyzer using a Teflon crucible. The samples are heated under air from 30 to 900 °C (heating rate of 10 °C min−1). Elemental analysis (EA) (%C, %H, %N) is performed with a Elementar Vario Micro Cube. Nitrogen sorption isotherms at 77 K are determined using a gas analyzer Micromeritics instrument – Tristar II Plus. Prior measurements, all samples are outgassed at least 6 h at 120 °C. The specific surface area (SBET) and the CBET parameter are determined using the Brunauer–Emmett–Teller (BET) equation following with the Rouquerol criteria.44,45CBET parameter is a constant varying with the adsorbent–adsorbate interactions and is related to the differential heat of adsorption of the first layer of adsorbate to the heat of adsorption of the following layers assimilated to the heat of condensation. The pore diameter is calculated using the Barret–Joyner–Halenda (BJH)46 (DBJH) and Broekhoff–de Boer (BdB)47,48 (DBdB) methods, as comparison. Transmission electron microscopy (TEM) analyses are performed on a JEOL 1400 microscope from the MEA platform, Univ. Montpellier. The samples are dispersed in ethanol and drop-coated on Lacey carbon grids before observation. Scanning electron microscopy (SEM) analyses are performed on a ZEISS EVO HD 15 microscope with Secondary electron detector and 5-segment backscatter detector under high vacuum. Energy Dispersive Spectroscopy (EDS) chemical analyses (in atomic%) were performed on a FEI Quanta 200F (15 kV) apparatus.2.3 Chromatographic applications
Protein stock sample solution was prepared in buffer pH 7 0.1 mol L−1 at 5 mg mL−1. Buffer pH 7 0.1 mol L−1 was prepared by dissolving 545 mg Na2HPO4 and 735 mg NaH2PO4 in 100 mL deionized water with a volumetric flask. Dilutions at 1 mg mL−1 of proteins were done in buffer pH 7 in a 200 μL Eppendorf tube and stored at −20 °C. Aliquot (250 μL) of proteins (lysozyme, myoglobin, AC, BSA) were added in 1 mL of mobile phase and flow through the columns at a flow rate of 1 mL min−1. The mobile phase used for isocratic elution was a mixture of water/ACN 98/2 (v/v). The pH of distilled water is 6.5. Liquid chromatographic separations were carried out using a Dionex UltiMate 3000 instrument (Thermo Fisher, Waltham, USA) equipped with a UV detection system and an oven to control the temperature of the column. Data were acquired by Chromeleon software (Version 7.0, Thermo Fisher Scientific). Injected sample volumes were 20 μL. Detection was performed at 214 nm. The temperature was maintained at 30 °C. Chromatographic columns 10 cm length, 2.1 mm internal diameter were filled with 150 mg of materials. MCM-41 and organo-functionalized MCM-41 particles were sieved into the fraction 50–100 μm. The washing of the columns after used was performed with ACN/H2O 85/15 (v/v) for reuse. The retention factor k was determined after three repetitions. k is calculated using the retention time tr and the dead time to pass the dead volume through the column t0 by the formula: k = (tr − t0)/t0.3. Results and discussion
3.1 Production of high surface area silica from RH
As-received raw RH (France) is first washed with water and then treated with different acidic protocols: HCl 0.1 or 1 mol L−1, at 25 or 100 °C, under stirring or in static. The first water washings allow to remove dust. The supernatant solution becomes less turbid after 3 washings (Fig. S1, ESI†). Acidic washing allows to remove metallic impurities,16 all supernatant solutions are totally clear. The final wash with water removes acid to reach a neutral pH. SEM images show that RH has two different aspects: a smooth inside, which is in contact with the rice grain and the outside, which is rough (“toothed” like an “egg box”) (Fig. 1 and Fig. S2, ESI†) to protect rice again harmful insects. Differences appear between the chemical composition of the inner and outer parts of raw RH. EDS analysis shows the presence of elements (Mg, S, K, Ca, Mn, Na, Cl) in such small quantities (<1%) that it was impossible to precisely quantify them due to the limit of detectability of the EDS method. However, in literature it was already reported that the first water washing removed few metal impurities, and that it is mainly the acidic treatment, which allowed for a higher removal of metal impurities due to cellulose and hemicellulose hydrolysis.16 | ||
| Fig. 1 SEM images of (a)–(c) RH treated with HCl 0.1 mol L−1 showing (a) the inside part of RH, (b) the outside part of RH and (c) the interface; (d) image and (e), (f) SEM images of SiO2 particles from RH after HCl 0.1 mol L−1 treatment and calcination at 600 °C 6 h. | ||
EDS analysis shows that Si is mainly in the outer part of raw RH, as previously reported in literature.19 The composition of RH is followed after water and HCl treatments by EDS analysis. After the first water washings, Si disappears from the inside part and remains only in the outside part, and hence after HCl washing (Fig. S3, ESI†). SEM images shows that HCl washing does not change the external surface morphology of RH (Fig. S2, ESI†), but affects strongly the interface between the inside and the outside part of RH by creating macropores of 5–10 μm (Fig. 1), certainly due to some cellulose and hemicellulose acidic hydrolysis.49 Indeed, acidic treatments of biomass are known to destroy amorphous cellulosic domains. In plants, cellulose fibers features generally diameter of 13–22 μm,50 which is in accordance with the diameter of the macropores observed by SEM in HCl treated RH. After calcination, RH is transformed into white needles of silica of ∼3 mm length and 0.4 mm diameter (Fig. 1), which are friable. In SEM silica from RH appears as particles of approximately 50–100 μm with macropores of 5–10 μm (Fig. 1). These particles are built by an aggregation of silica nanoparticles of ∼70 nm (Fig. 1), which were shown to be themselves formed by an aggregation of smaller silica nanoparticles of ∼5–10 nm.19
The silica materials from RH resulting of the different acidic pretreatments (HCl 0.1 or 1 mol L−1, at 25 or 100 °C, under stirring or in static) and calcination protocols (at 600 or 700 °C for 6 h, 2 °C min−1) were analyzed by XRD and nitrogen sorption isotherms at 77 K. All silica from RH is amorphous (Fig. 2). All nitrogen isotherms of silica from RH are of the type IV characteristic of mesoporous materials (Fig. 2) with a broad pore size distribution extending from 5 to 15 nm with an average diameter of 6.5 nm. The shape of the hysteresis is characteristic of spaces in between nanoparticles of ∼5–10 nm. Some small differences appear in the specific surface area of silica from RH depending on the acidic treatments performed and on the calcination temperatures. The specific surface areas vary from 255 to 312 m2 g−1 (Fig. 3). The highest specific surface area is obtained for the treatment with HCl 0.1 mol L−1 at 25 °C under shaking and a calcination at 600 °C for 6 h (2 °C min−1).
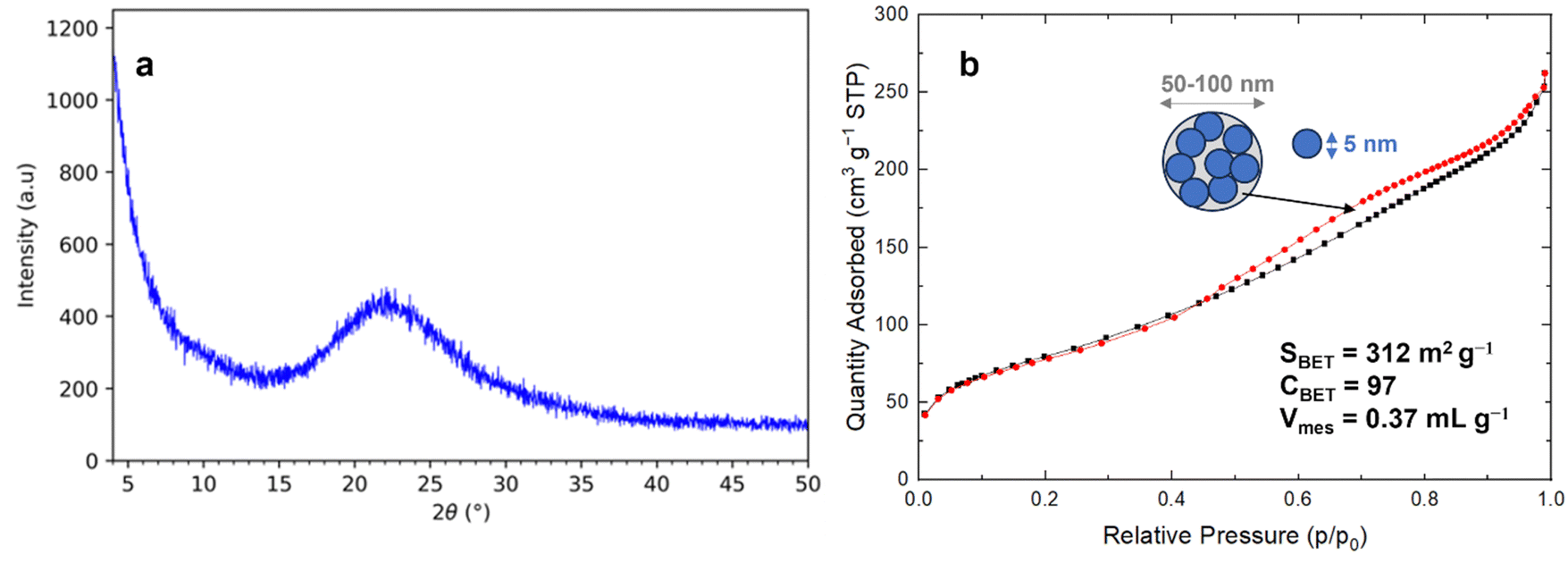 | ||
| Fig. 2 (a) XRD pattern and (b) N2 sorption isotherm at 77 K of silica from RH (France) treated with 0.1 mol L−1 HCl at 25 °C with shaking followed by calcination at 600 °C for 6 h (2 °C min−1). | ||
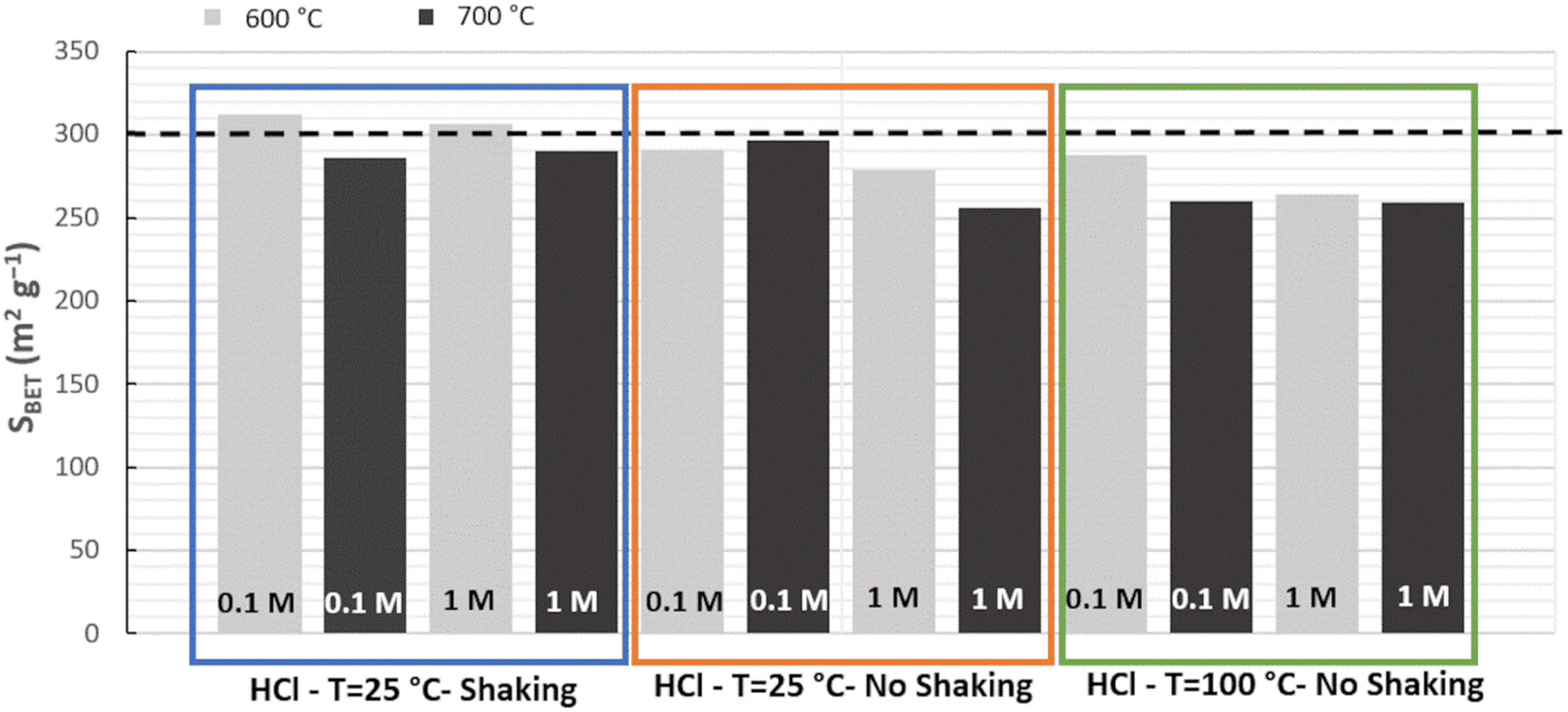 | ||
| Fig. 3 Specific surface area (SBET) of silica from RH (France) depending on the different HCl pretreatments: HCl 0.1 or 1 mol L−1 (noted 0.1 or 1 M in the figure) at 25 or 100 °C under shaking or in static, and calcination temperatures (grey) 600 °C or (black) 700 °C for 6 h (2 °C min−1). | ||
The same study was also performed with another source of RH coming from Senegal. The same result was reached, confirming that the best protocol is effectively HCl 0.1 mol L−1 at 25 °C under shaking followed by a calcination at 600 °C. The highest surface area for silica from RH from Senegal was 307 m2 g−1. These values of specific surface area are among the highest found in the literature for silica from RH of different origins (Table 1). Using this best pretreatment protocol, raw RH yield to 12 and 15 wt% of silica for RH from France and Senegal, respectively. This suggests that RH from Senegal contains more Si than the one coming from France due to differences in climate and soil.5
A scale-up was performed with 50 g of raw RH (France) using the best protocol: HCl 0.1 mol L−1 at 25 °C under shaking and calcination at 600 °C for 6 h (2 °C min−1). After the first water washings, the weight of RH decreased to 46 g to reach at the end, after HCl treatment and water neutralization, 45 g (Fig. S1, ESI†). The removal of dust during the water washing is responsible for the larger weight loss. TGA of raw and treated RH show two steps: the first between 25 and 200 °C corresponds to the weight loss of hydration water and the second between 200 and 500 °C is related to the weight loss of organic matter. The remaining mass corresponds to the inorganic oxides weight, mainly due to silica, especially after HCl treatment (Table 2). Hence, TGA show that raw RH, H2O washed RH and HCl treated RH contain 16.7, 11.7, 12.6 wt% of inorganics, respectively, corresponding on dry basis matter to 18.2, 13.0, 13.4 wt%. RH from France is hence able to produce 13 wt% of pure silica. After calcination at 600 °C, the silica from RH obtained from the 50 g scale-up features a specific surface area of 298 m2 g−1, in good agreement with the surface obtained for the 10 g batch (312 m2 g−1).
| RH | H2O (wt%) | Organic (wt%) | Inorganic oxides (wt%) |
|---|---|---|---|
| Raw | 8.3 | 75.2 | 16.7 |
| H2O washed | 9.8 | 78.5 | 11.7 |
| HCl washed | 6.1 | 81.2 | 12.6 |
3.2 Direct synthesis of MCM-41 from silica from RH
The transformation of raw RH (untreated with HCl) directly calcined at 600 °C, corresponding to RH ashes (RHA) produced by RH carbonization plants, into MCM-41 by pseudomorphic transformation did not succeed, as the surface area of such silica is too low (∼10 m2 g−1). Indeed, pseudomorphic transformation requires silica with sufficient surface area to reach the transformation of the entire particle, overwise as it was previously demonstrated for silica with low surface area (16 m2 g−1), only external surface is transformed into a rough surface.51 Silica from RH (France) with surface area ∼310 m2 g−1 obtained following the best pretreatment protocol (Section 3.1) was successfully transformed into MCM-41 by pseudomorphic transformation (Fig. 4).24,25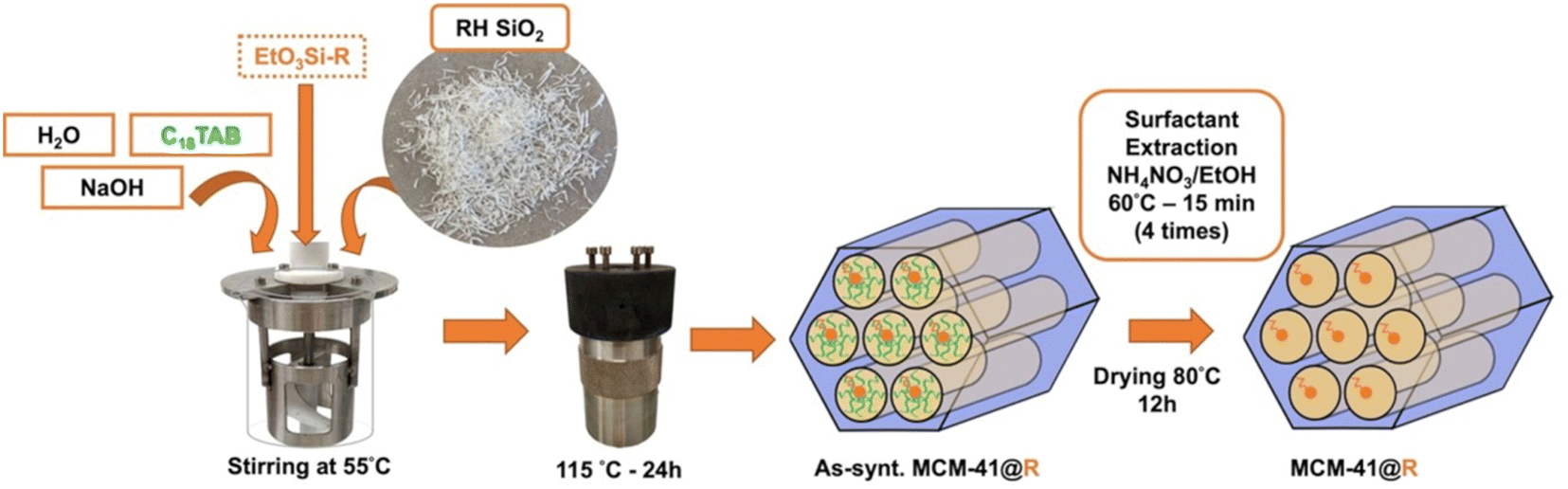 | ||
| Fig. 4 Schematic representation of the synthesis of organo(R)-functionalized MCM-41 and purely siliceous MCM-41 by pseudomorphic transformation by adding or not the corresponding triethoxy organosilane (EtO3Si–R), respectively. R = aminopropyl, amide derivates of amino acids (Leu, Ser, Tyr). | ||
XRD patterns of as-synthesized MCM-41 and extracted MCM-41 (Fig. 4) reveal the hexagonal organization of the mesopores of MCM-41 material (Fig. 5).52 After surfactant removal, the d100 reflection (cell parameter a = 5.29 nm (Table 3)) does not shift indicating that the extraction procedure do not modify the unit cell, no contraction of the hexagonal network occurs during solvent extraction,43 in contrary to calcination processes.53,54 The highly ordered hexagonal organization of the mesopores is confirmed by TEM images (Fig. 5).
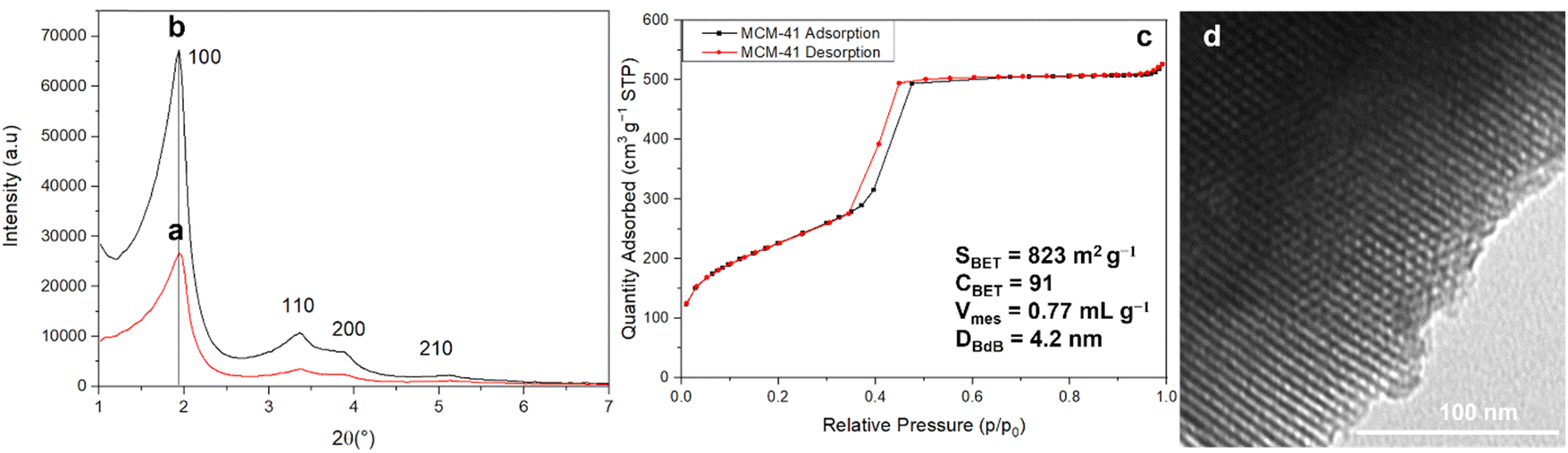 | ||
| Fig. 5 XRD patterns of (a) as-synthesized MCM-41 and (b) surfactant extracted MCM-41, (c) N2 sorption isotherm at 77 K and (d) TEM image of surfactant extracted MCM-41. | ||
| Materials | d 100 nm | a nm | S BET m2 g−1 | C BET | V mes mL g−1 | D BJH nm | D BdB nm | t BJH nm | t BdB nm |
|---|---|---|---|---|---|---|---|---|---|
|
a
d-Spacing (d100) is measured by XRD with the formula λ = 2d100sinθ.
b Cell parameter: a = 2d100/√3.
c Wall thickness: t = a − 0.95 D with D = DBJH or DBdB.
|
|||||||||
| MCM-41 | 4.58 | 5.29 | 823 | 91 | 0.77 | 3.3 | 4.2 | 2.2 | 1.3 |
| MCM-41@NH2(1/39) | 4.50 | 5.20 | 514 | 71 | 0.49 | 3.1 | 4.1 | 2.3 | 1.3 |
| MCM-41@1AA-Tyr(1/39) | 4.52 | 5.22 | 721 | 89 | 0.65 | 3.1 | 4.0 | 2.3 | 1.4 |
| MCM-41@1AA-Leu(1/39) | 4.57 | 5.27 | 719 | 71 | 0.65 | 3.1 | 4.1 | 2.3 | 1.3 |
| MCM-41@1AA-Ser(1/39) | 4.57 | 5.28 | 696 | 86 | 0.65 | 3.1 | 4.2 | 2.3 | 1.3 |
| MCM-41@3AA(1/39) | 4.50 | 5.19 | 680 | 75 | 0.60 | 3.0 | 4.1 | 2.3 | 1.3 |
| MCM-41@3AA(1/19) | 4.51 | 5.21 | 606 | 59 | 0.52 | 2.8 | 3.8 | 2.6 | 1.6 |
N2 sorption isotherm at 77 K of extracted MCM-41 is of type IV characteristic of mesoporous materials with narrow pore size distribution typical of MCM-41 materials (Fig. 5).54 The presence of a sharp sorption step (inflexion at p/p0 = 0.407) indicates that the mesopores are homogeneous in diameter, with a high specific surface area of 823 m2 g−1 and a high pore volume of 0.77 mL g−1 (Table 3). The CBET parameter of 91 is classical for MCM-4124 and shows that nitrogen interacts strongly with the silica surface. Hence, it was demonstrated that to calculate the mesopore diameters, the BdB method47 should be applied rather than the BJH method, which underestimated pore diameters by 20%.48 However, for organic-functionalized silica (as in the following), leading to lower CBET values, N2 molecules interact less strongly with the surface, and it was demonstrated that mesopore diameter values were in between those determined by BdB and BJH methods.55 Hence, mesopores diameter were expressed by both methods in Table 3 for an easier comparison with literature results.25 The mesopore diameter of extracted MCM-41 calculated by the BdB method is 4.2 nm, leading to wall thickness tBdB between mesopores (t = a − 0.95)48 of 1.3 nm (Table 3). These values allow to calculate the geometrical surface area S of MCM-41 materials by the following equation (eqn (1)):56
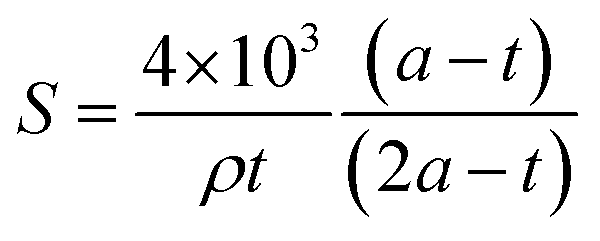 | (1) |
The geometrical surface area is 781 m2 g−1, which is close to SBET result (Table 3). This formula (eqn (1)) could not be applied to organo-functionalized MCM-41 due to the change of wall density by the presence of organic matter.
Highly ordered MCM-41 was obtained after only 24 h of reaction. This was not the case for the previous pseudomorphic transformation of silica from RH into MCM-41 described by Alyosef et al.25 A poorly ordered MCM-41 was obtained after 24 h (SBET = 718 m2 g−1, V = 0.51 mL g−1, DBJH = 2.8 nm (inflexion: p/p0 = 0.35)). After 3 days reaction, a poorly ordered MCM-41 was obtained featuring larger surface area (SBET = 1120 m2 g−1, V = 0.80 mL g−1, DBJH = 2.8 nm) and additional spherical nanoparticles (300–700 nm) on the external surface of the particles. After 6 days reaction, “worm-like” fibers with a hexagonal section of 3–5 μm further protruded out of the particles, as already observed for pseudomorphic transformation of spherical silica particles into MCM-41 when materials were left too long in the mother solution.24 These “worm-like” fibers of MCM-41 contributed to the obtention of a highly ordered MCM-41 (SBET = 1210 m2 g−1, V = 1.00 mL g−1, DBJH = 2.8 nm). The kinetic of our pseudomophic transformation was faster although Alyosef et al.25 were using a silica from RH of similar surface area (313 m2 g−1) with similar molar ratio of reactants. The difference comes from RH acidic pretreatment, which was for Alyosef et al.25 hot citric acid and led to smaller macropores (0.2–5 μm)25 in silica particles in comparison to the macropores of 5–10 μm obtained in the present study (Fig. 1). Hence, the presence of larger macropores in the parent silica favors the diffusion of the reactants (C18TAB and NaOH) leading to a faster pseudomorphic transformation. Textural characterizations of MCM-41 obtained by Alyosef et al.25 were performed on calcined MCM-41 materials, no surfactant extraction by solvents was attempted.
3.3 Direct synthesis of NH2 functionalized MCM-41 from silica from RH
Aminopropyl triethoxysilane (APTES) was added with silica from RH during the pseudomorphic transformation into MCM-41 in a molar ratio of APTES/SiO2 = 1/39, corresponding to a mixture of molar ratio: 1 SiO2:0.1 C18TAB:0.25 NaOH:50 H2O:0.0256 APTES. Surfactant was extracted with NH4NO3/EtOH solution. The material is named MCM-41@NH2(1/39). Starting from 1 g of silica from RH, the maximum weight expected for extracted MCM-41@NH2(1/39) should be 1.037 g (dry matter). The mass found for as-synthesized and extracted MCM-41@NH2(1/39) was 1.42 and 0.91 g, respectively (Table S1, ESI†), which is higher than for purely siliceous MCM-41 (1.14 and 0.70 g, respectively), indicating the incorporation of organic functions.
XRD patterns of as-synthesized and extracted MCM-41@NH2(1/39) (Fig. 6) show highly ordered hexagonal structures as purely siliceous MCM-41 (Fig. 5). The extraction does not affect the cell parameter, which remains at a = 5.2 nm (Table 3). TEM images of extracted MCM-41@NH2(1/39) confirms that the highly ordered hexagonal structure is preserved during the surface modification with APTES. N2 sorption isotherm at 77 K of extracted MCM-41@NH2(1/39) shows a decrease of the specific surface area (from 823 to 514 m2 g−1) and pore volume (from 0.77 to 0.49 mL g−1) in comparison to MCM-41 (Fig. 6 and Table 3). The CBET parameter decreases (CBET = 71) demonstrating the presence of organic matter at the surface of the mesopores.36 The N2 adsorption step is less sharp than the one of MCM-41 suggesting a slightly broader pore size distribution due the presence of organic chains at the surface of the pores. The inflexion of the N2 adsorption step is occurring at a slightly lower relative pressure p/p0 = 0.397 instead of p/p0 = 0.407 for MCM-41, indicating that the mesopore diameter is slightly smaller than in MCM-41 due to the surface functionalization with NH2 groups. It is difficult to precisely determine the exact value of pore diameter for organo-functionalized materials based on nitrogen adsorption. For mesoporous materials with surface totally covered by octyl-grafted chains (CBET = 20), it was suggested that the more accurate value of pore diameter was the average between pore diameter obtained by BdB and BJH methods.55 For extracted MCM-41@NH2(1/39), DBdB is 4.1 nm and DBJH is 3.14 nm giving an average value of D = 3.62 nm. Hence mesopore diameter of extracted MCM-41@NH2(1/39) should be between 3.6 < D < 4.1 nm.
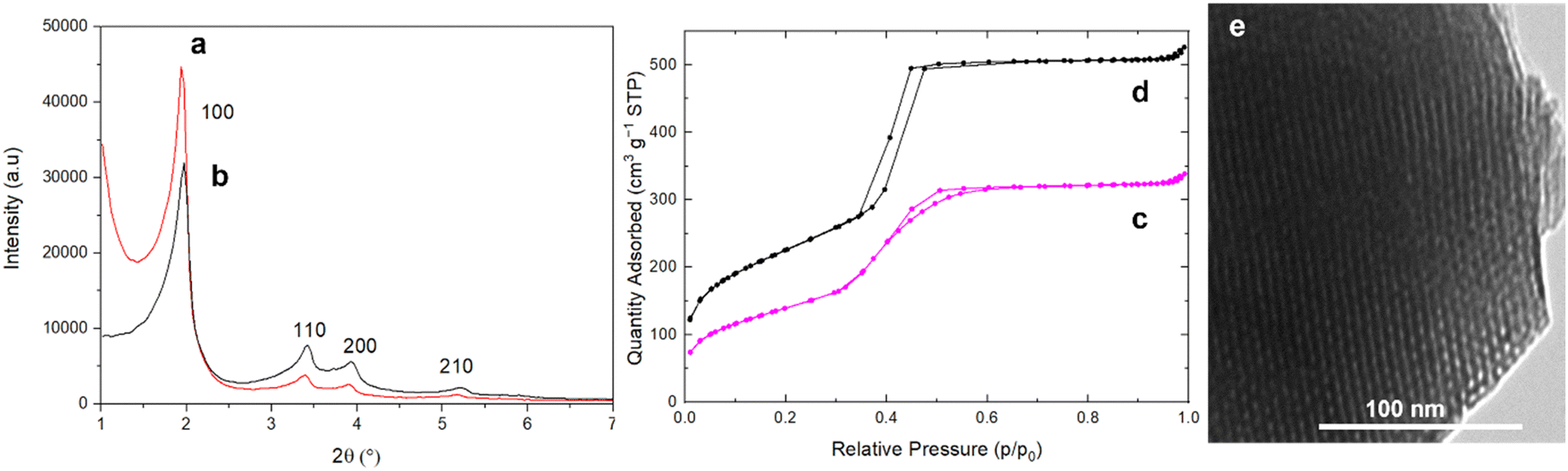 | ||
| Fig. 6 XRD patterns of (a) as-synthesized and (b) extracted MCM-41@NH2(1/39), N2 sorption isotherms at 77 K of extracted (c) MCM-41@NH2(1/39) and (d) MCM-41 for comparison. (e) TEM image of extracted MCM-41@NH2(1/39). | ||
3.4 Direct synthesis of amino-acid functionalized MCM-41 from RH
Amide derivates of amino acids (AA) triethoxysilane Si-Tyr-NH2, Si-Ser-NH2, Si-Leu-NH2 (Scheme 1) are added alone or in mixture (1/3, 1/3, 1/3) with silica from RH during the pseudomorphic transformation into MCM-41, corresponding to a mixture of molar ratio: 1 SiO2:0.1 C18TAB:0.25 NaOH:50 H2O:0.0256 AA or 0.0526 AA for molar ratio of AA/SiO2 = 1/39 or 1/19, respectively. Surfactants are extracted with NH4NO3/EtOH. The resulting materials are named MCM-41@1AA-Tyr(1/39), MCM-41@1AA-Ser(1/39), MCM-41@1AA-Leu(1/39), MCM-41@3AA(1/39), MCM-41@3AA(1/19). Starting from 1 g of silica from RH, 1.36–1.41 g of as-synthesized MCM-41@1AA or 3AA(1/39) are obtained and 1.58 g for MCM-41@3AA(1/19) (Table S1, ESI†). After surfactant extraction the weight decreases to 0.85–0.90 g for all materials. The resulting masses are closed to the one of as-synthesized and extracted MCM-41@NH2(1/39). XRD patterns and TEM images of AA-functionalized MCM-41 materials (Fig. 7 and Fig. S4, ESI†) show highly ordered hexagonal structures with cell parameters of 5.19–5.28 nm as aminopropyl functionalized MCM-41 (Fig. 6 and Table 3).
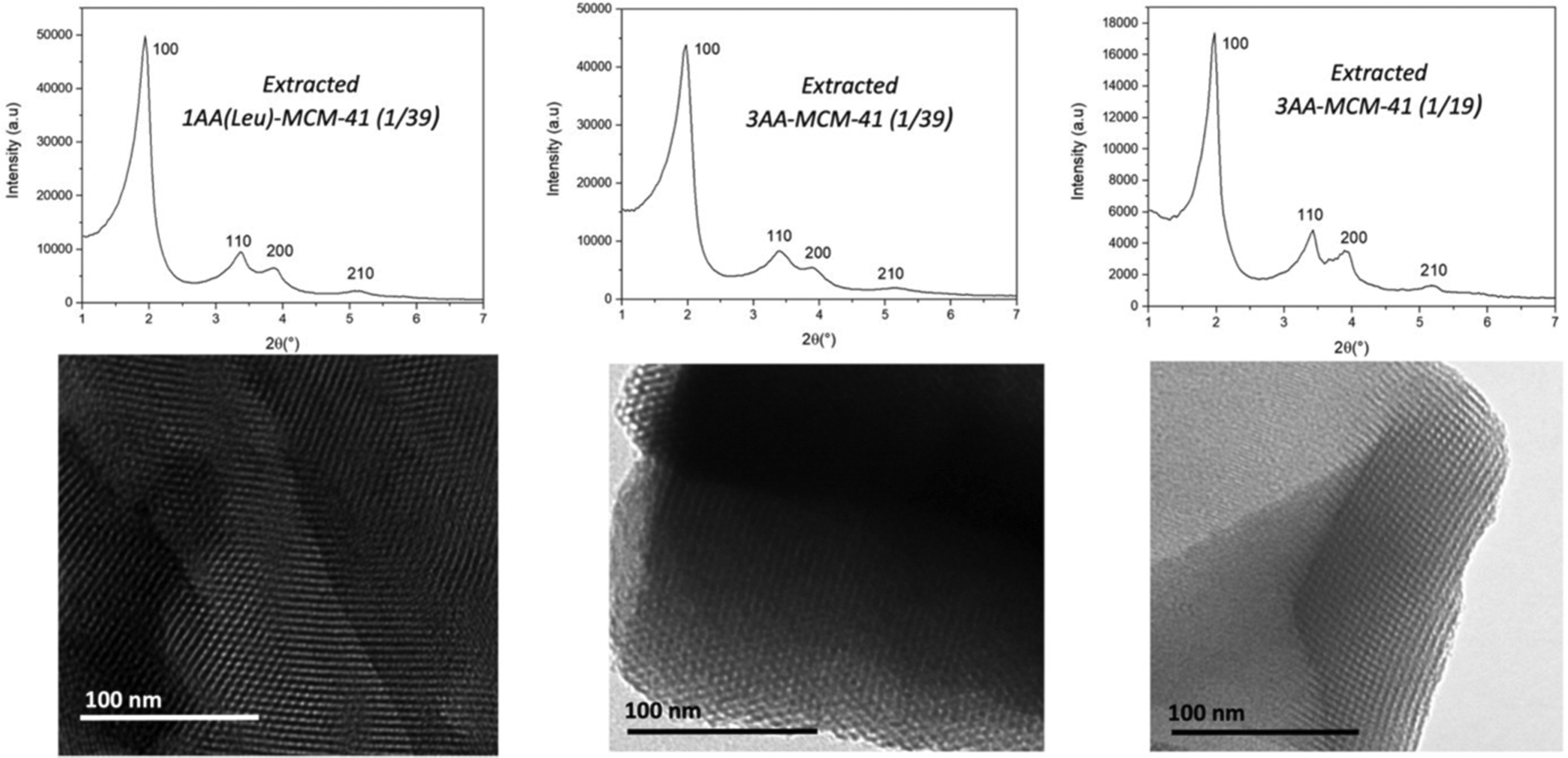 | ||
| Fig. 7 XRD patterns and TEM of extracted AA-functionalized MCM-41 with 1 AA (Leu) or a mixture of 3 AA (Leu, Tyr, Ser) in molar ratio 1/3, 1/3, 1/3 for the molar ratio AA based-triethoxysilane/SiO2 = 1/19 or 1/39. | ||
N2 sorption isotherms at 77 K of all extracted 1AA- and 3AA-MCM-41(1/39) are equivalent and exhibit similar shape as the one of MCM-41, with a straight adsorption step. Slightly lower pore volumes (0.60–0.65 mL g−1) and surface areas (680–720 m2 g−1) are obtained due to the presence of organic functions (Fig. 8 and Table 3). The pore volumes and surface areas are larger than the one of aminoproyl-functionalized MCM-41 and the shape of the isotherm is different. N2 sorption isotherm at 77 K of extracted 3AA-MCM-41(1/19) shows the lowest surface area and pore volume due to the increase of organosilane amount used in the synthesis, but remain larger than the surface area and pore volume of aminoproyl-functionalized MCM-41. This suggests a lower density of organic functions with AA-triethoxysilane at the surface of the materials in comparison to APTES.
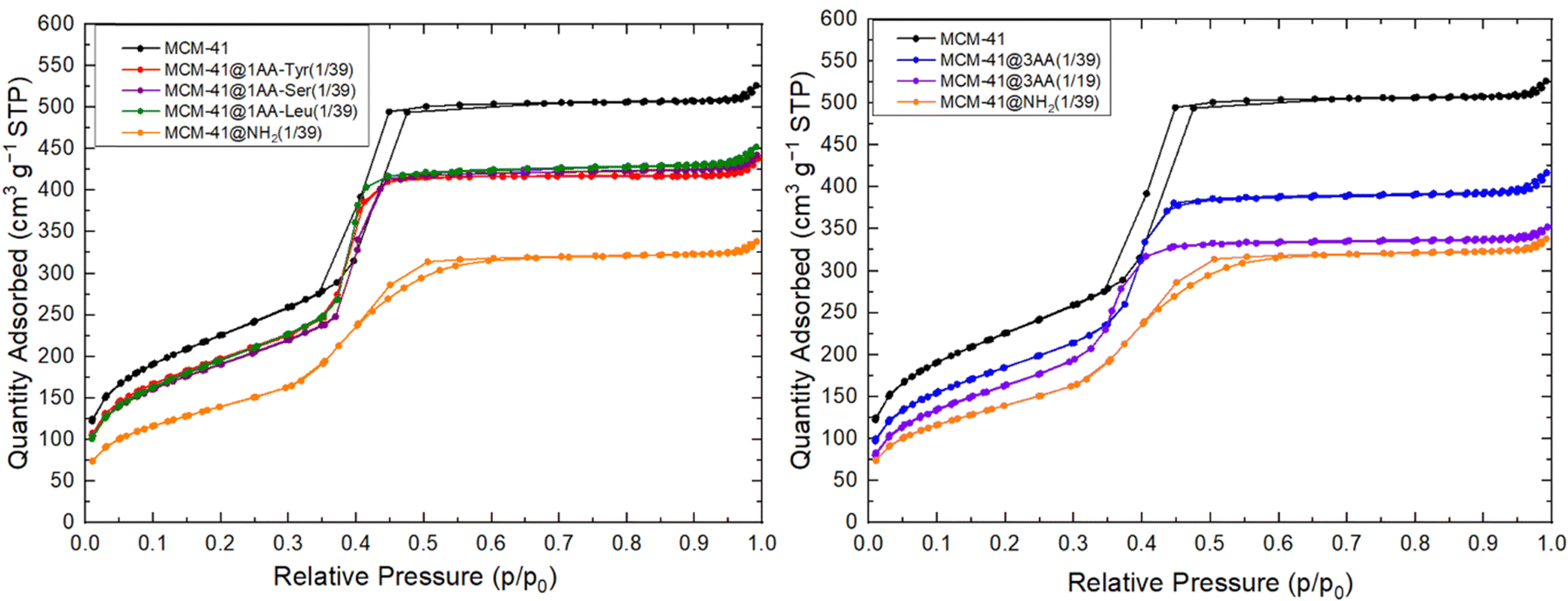 | ||
| Fig. 8 Nitrogen sorption isotherms at 77 K of extracted AA-functionalized MCM-41 with 1 AA or a mixture of 3 AA in molar ratio 1/3, 1/3, 1/3 for the molar ratio organo triethoxysilane/SiO2 = 1/19 or 1/39. Comparison with nitrogen sorption isotherm at 77 K of surfactant extracted MCM-41 and aminopropyl functionalized MCM-41. | ||
The CBET parameter of MCM-41@1AA-Leu(1/39) has a CBET parameter close to the one of MCM-41@NH2(1/39) (CBET = 71) demonstrating clearly the presence of organic matter at the surface of the pore. MCM-41@1AA-Tyr(1/39) and MCM-41@1AA-Ser(1/39) have CBET parameter (CBET = 86–89) closed to the one of MCM-41 (CBET = 91) (Table 3), indicating either that very few organic functions were grafted on the surface of the mesopores or that their amino group strongly interact with the surface silanols and that their OH groups interact with N2 as silanol does (Scheme 2).
 | ||
| Scheme 2 Schematic representation of aminopropyl- and AA-functionalized MCM-41 to explain CBET parameter values of nitrogen sorption isotherms at 77 K. | ||
For MCM-41@3AA(1/39) featuring the 3 AA, the CBET parameter are close to the one of MCM-41@1AA-Leu(1/39) (CBET = 75) indicating that OH functions of Ser and Tyr are not pointing to the interior of the pores. This suggests that when using AA mixture in the synthesis, the different AA are closed and interact between them for example by hydrogen bond between the active groups of the amide function of two different AA (Scheme 2). Similar interactions may be drawn between Tyr and Leu or Ser and Tyr. For larger amount of grafted AA, MCM-41@3AA(1/19) features a lower CBET (CBET = 59) indicating that a larger amount of organic functions are present at the surface of the pores.
3.5 Quantification of the organic matter in MCM-41 and organo-functionalized MCM-41
In term of yield, 1 g of silica from RH leads to 1.14 g of as-synthesized MCM-41 (Table S1, ESI†). TGA of as-synthesized MCM-41 shows a sharp step from 150 to 330 °C corresponding to 36.7 wt% followed by a continuous weight loss above 330 °C (Fig. S5, ESI†). Similar TGA curve was previously observed for MCM-41 with the combustion and decomposition of organic species identified by weight loss between 150–400 °C and the stage above 400 °C was attributed to water losses via condensation of silanol groups to form siloxande bonds.54 The weight loss from as-synthesized MCM-41 from RH between 150 and 400 °C is 60.8 wt%. Hence, the yield in octadecyltrimethyl ammonium (C18TMA+) is 86 wt% and the yield in silica is 69 wt%. This result is in agreement with previous pseudomorphic transformations for other kinds of silica with yields in surfactant and silica around 92–98 wt% and 57–66 wt%, respectively.24 This corresponds to 0.69 g of silica and 0.45 g of C18TMA+, for a total mass of 1.14 g. Hence, as-synthesized MCM-41 features in first approximation an empirical formula of C18TMA0.12SiO2.Surfactant of as-synthesized MCM-41 (1.14 g) was extracted with EtOH/NH4NO3 solution at 60 °C, which leads to a final weight of extracted-MCM-41 of 0.70 g (Table S1, ESI†). This surfactant extraction starts by a cationic exchange between C18TMA+ and NH4+.43 TGA curve of extracted MCM-41 with this protocol of extraction was not shown previously in literature.43 TGA of extracted MCM-41 (Fig. 9) features three distinct stages of weight loss: 4.3 wt% between 25 and 150 due to the water desorption, 2.6 wt% between 150 and 400 °C, which could be attributed to some residual C18TMA+ in comparison to TGA of MCM-41 (Fig. S5, ESI†), 5.5 wt% of organics between 400 and 650 °C, followed by a slight progressive decrease of 1.4 wt% until 900 °C due to dehydroxylation of silica. At 900 °C the remaining mass corresponds to 85 wt% of dehydroxylated silica. To understand what could be the organic species decomposing between 400 and 650 °C, a comparison of TGA and elemental analysis (EA) results (Table S2, ESI†) was performed.
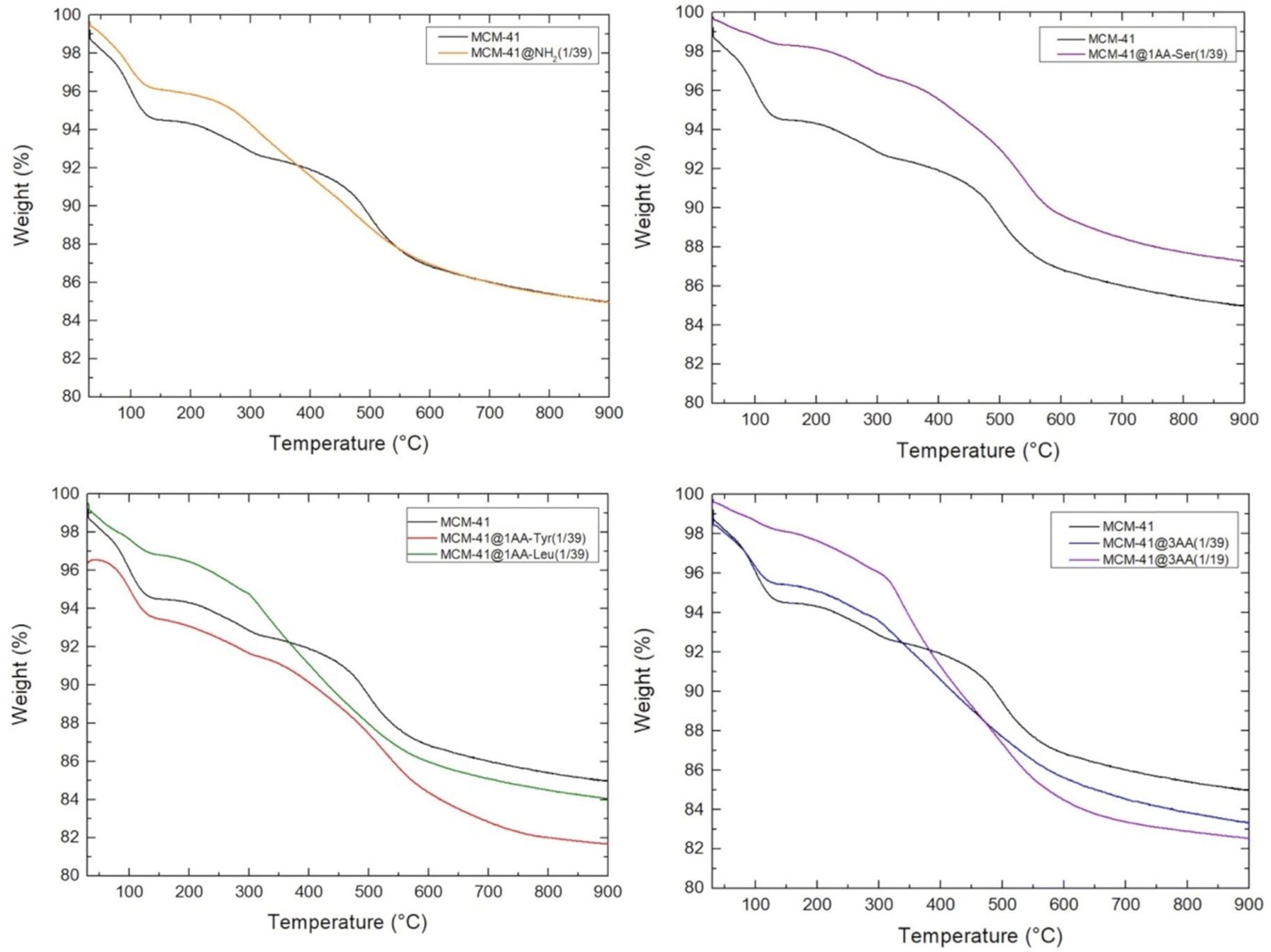 | ||
| Fig. 9 TGA curves of extracted MCM-41 and extracted MCM-41 functionalized with aminopropyl (NH2) or AA groups (1 AA or a mixture of 3 AA in ratio 1/3, 1/3, 1/3) for molar ratio organosilane/SiO2 = 1/19 or 1/39. AA = amide derivatives of Tyr, Leu, Ser. | ||
By EA (Table S2, ESI†), the amount of C is 5.15 wt%, and corresponds for 100 g of material to 0.429 mol C. In C18TMA+ surfactant, there is 21 C. Hence, EA would correspond to 0.020 mol of C18TMA+, and consequently 0.020 mol of N. However, the amount of N found by EA is 1.71 wt%, which corresponds for 100 g of material to 0.122 mol N. Therefore, there is another source of N, which could be NH4+ coming from the cation exchange with C18TMA+ and would amount for 0.102 mol of NH4+. Furthermore, 0.20 mol of C18TMA+ should lead to a weight loss in TGA of 6.4 wt%, which is not possible as the maximum weight loss for C18TMA+ between 150–400 °C is 2.6 wt%. Hence, there is also another source of C, which could be some ethoxy groups grafted on silanols at the surface of the silica coming from the surfactant extraction in EtOH at 60 °C.
By TGA, residual C18TMA+ corresponds to a weight loss of 2.6 wt%, hence for 100 g of material there is 0.0083 mol of C18TMA+, so 0.0083 mol of N corresponding to 0.116 g of N and 0.1746 mol C corresponding to 2.096 g of C. By EA, the total amount of N is 0.122 mol, so by difference, the amount of N of NH4+ should be 0.1138 mol which would correspond to a weight loss of NH3 in TGA of 1.935 g. By EA, the total amount of C is 5.15 g of C, so by difference with the amount of C of C18TMA+ (2.096 g of C), there should be 3.054 g of C coming from 0.1272 mol of grafted ethoxy groups, which would correspond to a weight loss of ethylene (thermal decomposition of ethyl group) in TGA of 3.563 g. The weight loss corresponding to the degradation of NH4+ and of grafted ethoxy groups should be 5.498 g. This value is in agreement with the weight loss (5.51 wt%) experimentally observed in TGA between 400–650 °C. Hence, extracted-MCM-41 is mainly under the form NH4-MCM-41 with an empirical formula of (NH4)0.080C18TMA0.0059(EtO)0.090SiO2. This formula corresponds to 95% of surfactant extraction, which is in agreement with what was previously observed in literature with 97% of surfactant extraction for purely siliceous MCM-41 after 3 extractions with EtOH/NH4NO3 solution.43
In literature, it was also shown that the first extraction was leading to 66% of surfactant extraction for purely siliceous MCM-41, whereas 100% surfactant extraction was obtained after 15 min for Al-MCM-41 with Si/Al = 1.25.43 This method of extraction was more efficient for Al-MCM-41 and the efficiency increased with the increase of Al content in MCM-41. The fact that it is more difficult to extract surfactants in purely siliceous MCM-41 in comparison to Al-MCM-41 is surprising, as stronger electrostatic interactions are expected between positively charged ammonium cations and negatively charged aluminosilicates in comparison to silanols. However, it was previously shown by EPR that purely siliceous MCM-41 contained a certain percentage of surfactants in strong interaction with the surface, which increased with the chain length of the surfactant (or the mesopore diameter) and was of 50 and 68% for C16TMA+, C18TMA+, respectively.57 It was concluded that in MCM-41, featuring hexagonal mesopore section52,58 the surfactant in strong interactions was localized on the hydrophilic sides of the hexagon, and the ones in weak interactions in the hydrophobic corners. We can propose a scheme (Fig. 10) to explain the difficulty to extract surfactants from purely siliceous MCM-41. During MCM-41 formation, silicates surround the quaternary ammonium head groups of the surfactants leading to the formation of some silicate shell around the head groups. These silicate/surfactant species arrange into micelles, extend to their maximum diameter and then length perpendicularly and in the same times organize into hexagonal arrays.59 The head group ((CH3)N+(CH2–)) of the surfactants protrudes inside the walls of MCM-41 forming some “cavities” of ∼0.4–0.5 nm diameter (Fig. 10). The total length of C18TMA+ is 2.601 nm,60 and the C17 chain inside the pore would then exhibit 2.14 nm length, in accordance with a mesopore diameter DBdB of 4.2 nm (Table 3). We propose that during surfactants extraction, NH4+ cations replace the surfactant head groups and get trapped in those cavities.
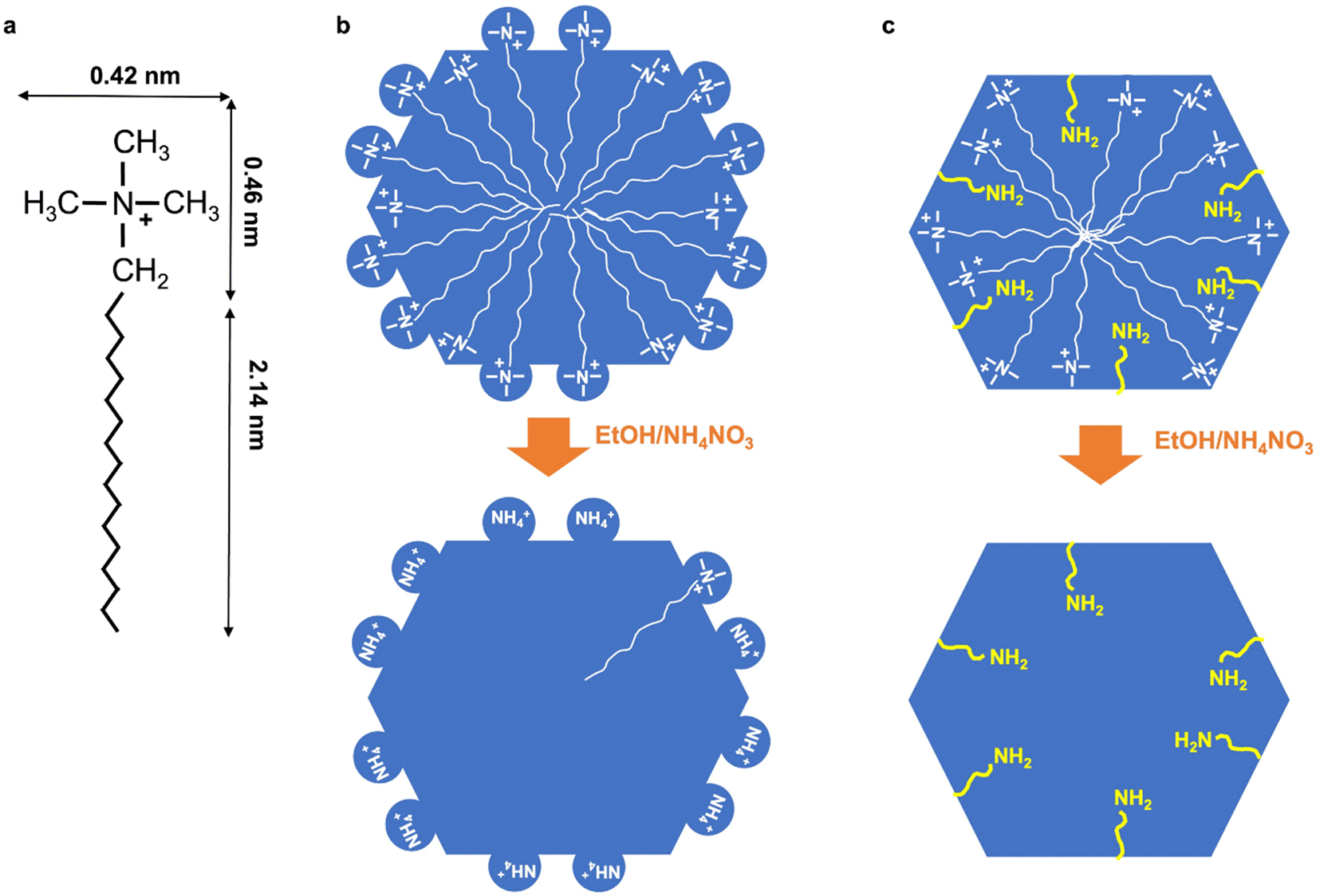 | ||
| Fig. 10 (a) Dimension of C18TMA+ surfactant. Schematic representation of the pores of as-synthesized and extracted (b) purely siliceous MCM-41 and (c) amino-functionalized MCM-41 obtained by co-condensation with amino-functionalized triethoxysilane (as aminopropyl and AA functions). The same scenario as (c) is expected for aluminum rich Al-MCM-41 (Si/Al = 1.25) (without yellow organic chains). | ||
Twenty years ago, there was a strong debate about the surface roughness of MCM-41. The existence of strong heterogeneities inside the pore walls was suspected, which was qualified as pore walls not totally smooth, with a surface micro-roughness leading to a masked microporosity, with significant micropore filling for adsorbate of kinetic diameter <0.5 nm.61 In the scheme we proposed, cavities with low depth could effectively account for a masked microporosity for nitrogen adsorption62 and hyperpolarized 129Xe NMR63,64 as it is the case for SBA-15 materials synthesized at 100–110 °C. Some models simulating XRD and TEM data revealed the possible existence of some cavities in MCM-41 walls.52 Adding alumina in the synthesis could prevent the formation of such cavities, as silica environment around the head of surfactant will become more rigid with shorter Si–O–Al bonds, less flexible, with less possible curvature of silico–aluminate bonds. The depth of the cavities might decrease with the increase of Al content. The same scenario could be proposed when amino-functionalized triethoxysilane is added in the synthesis of MCM-41 (see below).
TGA of extracted amino-functionalized (aminopropyl- and AA-functionalized MCM-41) (Fig. 9) does not exhibit the defined weight loss between 400–650 °C observed for extracted MCM-41. Hence, this could indicate that no cavities trapping NH4+ are formed during the pseudomorphic transformation of MCM-41 when amino-functionalized triethoxysilane are added. Amino-functionalized alkoxysilane are very efficient for silica grafting as it does not require the addition of catalysts in contrary to other alkoxysilanes.36 Thanks to its amino group and its bendable alkyl chain, it is a self-catalyst for its condensation and grafting. By promoting silica condensation, it should avoid the formation of a flexible layer of silicates surrounded quaternary ammonium head group, avoiding the formation of cavities in the walls of MCM-41 and leading to smooth pore walls (Fig. 10).
As previously performed for extracted MCM-41, TGA (Fig. 9 and Scheme 3) and EA (Table S2, ESI†) were compared. The quantification of organic functions in organo-functionalized MCM-41 is complex due to the possible presence of lot of organic groups: remaining C18TMA+, presence of NH4+, ethoxy grafted on silanols and the degree of co-condensation of organo-ethoxysilane with silica forming even mono-, bi-, tridentate organo-silanes. The best accordance between TGA and EA is reported in Table 4 and Scheme 3. MCM-41@NH2(1/39) is most probably composed of half bidentate and half monodentate organosilanes with the presence of remaining C18TMA+ and NH4+, which are however in lower amount than in extracted MCM-41. AA-functionalized MCM-41 materials do not exhibit remaining C18TMA+. MCM-41@1AA-Leu(1/39) is composed of 62% of tridentate and 38% of bidentate organosilanes. MCM-41@1AA-Ser(1/39) and MCM-41@1AA-Tyr(1/39) exhibit only tridentate organosilane with additional ethoxy groups grafted on the surface and NH4+ species, respectively.
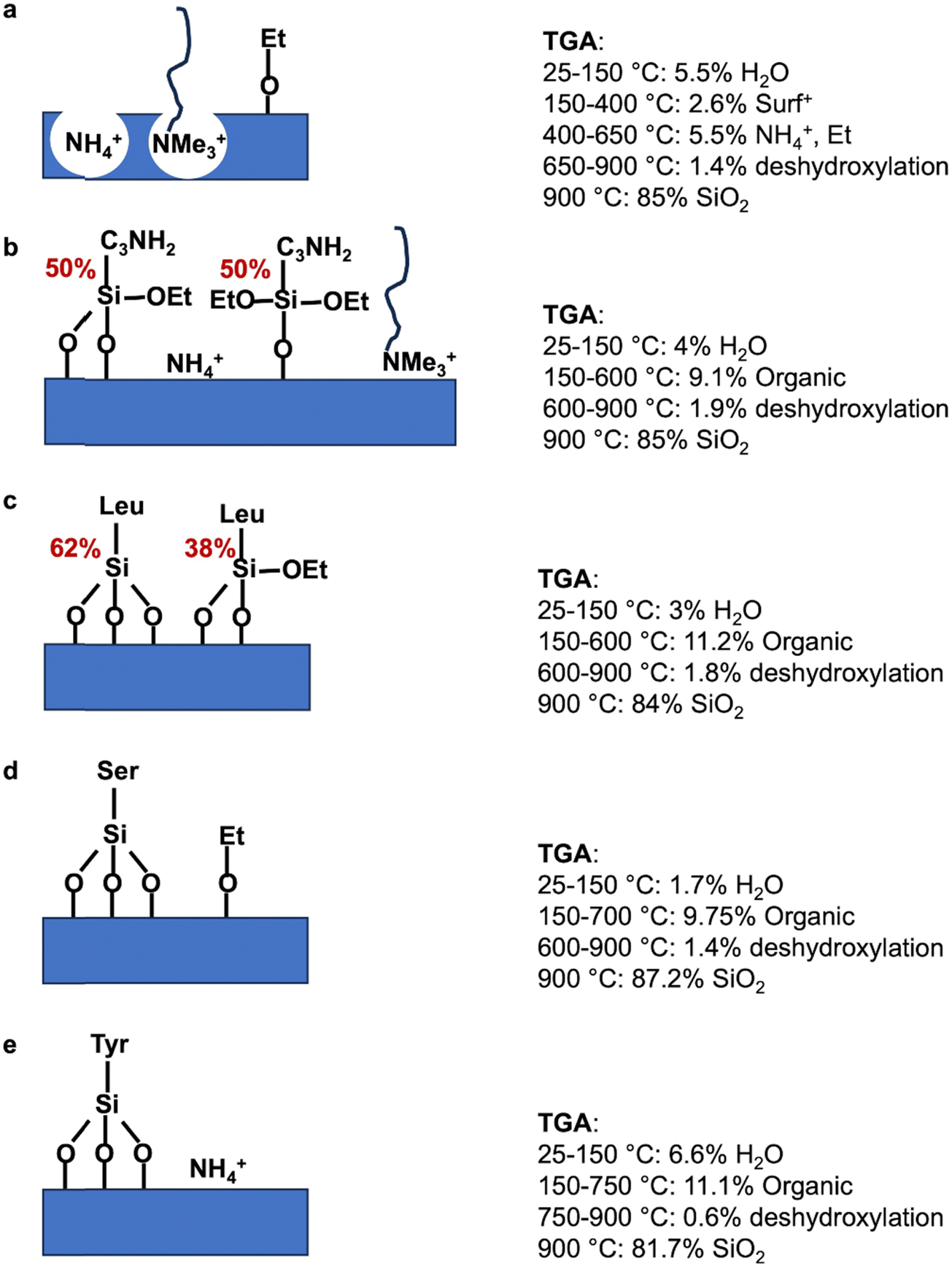 | ||
| Scheme 3 Schematic representation of the species present at the surface of the mesopores of extracted materials: (a) MCM-41, (b) MCM-41@NH2(1/39), (c) MCM-41@1AA-Leu(1/39), (d) MCM-41@1AA-Ser(1/39), (e) MCM-41@1AA-Tyr(1/39) with TGA results of the different materials. | ||
| Materials 100 g | Organic function mol | SiOEta mol | OEtb mol | NH4+ mol | C18TMA+ mol |
|---|---|---|---|---|---|
| a In TGA, with the increase of temperature, SiOEt groups react with surface silanols to complete tridentate grafting and EtOH is released. b In TGA, degradation of grafted ethoxy groups give ethylene. | |||||
| MCM-41 | 0 | 0 | 0.1272 | 0.1138 | 0.0083 |
| MCM-41@NH2(1/39) | 0.0522 | 0.0780 | 0 | 0.0772 | 0.0056 |
| MCM-41@1AA-Leu(1/39) | 0.0476 | 0.0183 | 0 | 0 | 0 |
| MCM-41@1AA-Ser(1/39) | 0.0414 | 0 | 0.0733 | 0 | 0 |
| MCM-41@1AA-Tyr(1/39) | 0.0411 | 0 | 0 | 0.0151 | 0 |
By subtracting hydration water and organic functions (Table 4 and Scheme 3) from the yields of extracted materials (Table S1, ESI†), it is possible to calculate the yield of silica during the pseudomorphic transformation. For MCM-41@NH2(1/39), MCM-41@1AA-Leu(1/39), MCM-41@1AA-Ser(1/39) and MCM-41@1AA-Tyr(1/39), the yields in silica are 76, 74, 72, 71%, respectively. These results are in good agreement with the yield of silica in pseudomorphic transformation of MCM-41 (70%). The empirical formula corresponding to the resulting materials are given in Table 5.
| Materials | Empirical formula |
|---|---|
| MCM-41 | SiO2(C18TMA)0.0059 (NH4)0.080 (OEt)0.090 |
| MCM-41@NH2(1/39) | SiO2(C3NH2)0.037 (C18TMA)0.0039 (NH4)0.054 (SiOEt)0.078 |
| MCM-41@1AA-Leu(1/39) | SiO2(Leu)0.034 (SiOEt)0.013 |
| MCM-41@1AA-Ser(1/39) | SiO2(Ser)0.029 (OEt)0.050 |
| MCM-41@1AA-Tyr(1/39) | SiO2(Tyr)0.030 (NH4)0.011 |
| MCM-41@3AA(1/39) | SiO2(Leu)0.0144 (Ser)0.0144 (Tyr)0.0043 (NH4)0.01152 |
| MCM-41@3AA(1/19) | SiO2(Leu)0.020 (Ser)0.020 (OEt)0.044 |
For the material MCM-41@3AA(1/39) synthesized with the mixture of 3 AA (1/3, 1/3, 1/3), TGA shows that the weight loss of hydration water (25–150 °C) is 4.57%, the weight of organics between 150–650 °C is 10.18%, and the remaining 83.3% is SiO2 (Fig. 9). EA gives 5.08% C and 2.16% N (Table S2, ESI†). The best accordance between TGA and EA is obtained by a total grafting as tridentate species for Leu and Ser-based organo-triethoxysilanes in equal quantity (0.020 mol for 100 g material) and 3 times less for Tyr (0.006 mol Tyr for 100 g materials). Leu and Ser should preferentially form bi-complex as presented in Scheme 2. NH4+ (0.016 mol NH4+ for 100 g material) should be added to fullfill the amount of N found by EA and the total weight loss by TGA. This would correspond to an empirical formula of SiO2(Leu)0.0144 (Ser)0.0144 (Tyr)0.0043 (NH4)0.01152 (Table 5). By removing the weight of organics (AA and NH4+) and hydration water from the final weight of the sample (Table S1, ESI†), the weight of silica is 0.72 g when the synthesis is performed with 1 g of SiO2 from RH. This yields to a transformation of 72% of silica from RH into MCM-41@3AA(1/39), which is in agreement with the yield of silica found in pseudomorphic transformation.
The shape of TGA of MCM-41@3AA(1/19) is closed to the one of MCM-41@3AA(1/39), although the double of AA triethoxysilanes have been used. All triethoxy silane does not participate to the formation of the material. For the material MCM-41@3AA(1/19) synthesized with the mixture of 3 AA (1/3, 1/3, 1/3), TGA shows that the weight loss of hydration water (25–150 °C) is 2%, the weight of organics between 150–600 °C is 13.8%, the dehydroxylation (600–900 °C) is 1.85% and the remaining weight 82.5% is SiO2 (Fig. 9). EA gives 7.05% C and 2.31% N (Table S2, ESI†). The best accordance between TGA and EA is obtained for bidentate species of Leu and Ser-based AA-triethoxysilanes in equal quantity (0.0275 mol for 100 g material) with no presence of Tyr and 0.0604 mol of (Si)OEt. For larger amount of AA, AA derivates from Tyr should be preferentially excluded due to steric hindrance in the bi-complex formation in comparison to the other AA (Scheme 2). This would correspond to an empirical formula of SiO2(Leu)0.020(Ser)0.020(OEt)0.044 (Table 5). By removing the weight of organics (AA and (Si)OEt) and hydration water from the final weight of the sample (Table S1, ESI†), the weight of silica is 0.72 g when the synthesis is performed with 1 g of SiO2 from RH. This yields to a transformation of 72% of silica from RH into MCM-41@3AA(1/19), which is in agreement with the yield of silica found in pseudomorphic transformation.
3.6 Organo-functionalized MCM-41 for flow applications
The pseudomorphic transformation of silica from RH into MCM-41 and organo-functionalized MCM-41 did not preserve the millimetric needles morphology. Powders were obtained composed of microparticles of 1–100 μm, mainly around 50 μm from SEM observations (Fig. 11 and Fig. S7, ESI†). Sieving the powders allows to recover 30% in volume of particles of 50–100 μm suitable to fill columns and perform flow applications with low pressure drop. Aminopropyl-fonctionalized MCM-41 (MCM-41@NH2) is an interesting material that could be used for biomolecules immobilization, such as enzymes through glutaraldehyde coupling for biocatalysis applications,35 or could be directly used as basic catalyst as in Knoevenagel reactions,65 for example. | ||
| Fig. 11 SEM images of MCM-41, MCM-41@NH2(1/39) and MCM-41@3AA(1/39). | ||
AA-functionalized MCM-41 materials could be used for proteins separation as it was performed with silica capillaries grafted with similar AA-triethoxysilanes in capillary electrophoresis.66 Therefore, AA-functionalized MCM-41 were tested in proteins separation by size exclusion chromatography (Fig. 12 and Fig. S8, ESI†) using mobile phase of water/ACN 98/2 (v/v) and a mixture of proteins of different hydrodynamic diameters (Dh): BSA (Dh = 7.2 nm), carbonic anhydrase (AC) (Dh = 4.2 nm), myoglobin (Dh = 3.8 nm), lysozyme (Dh = 3.4 nm).
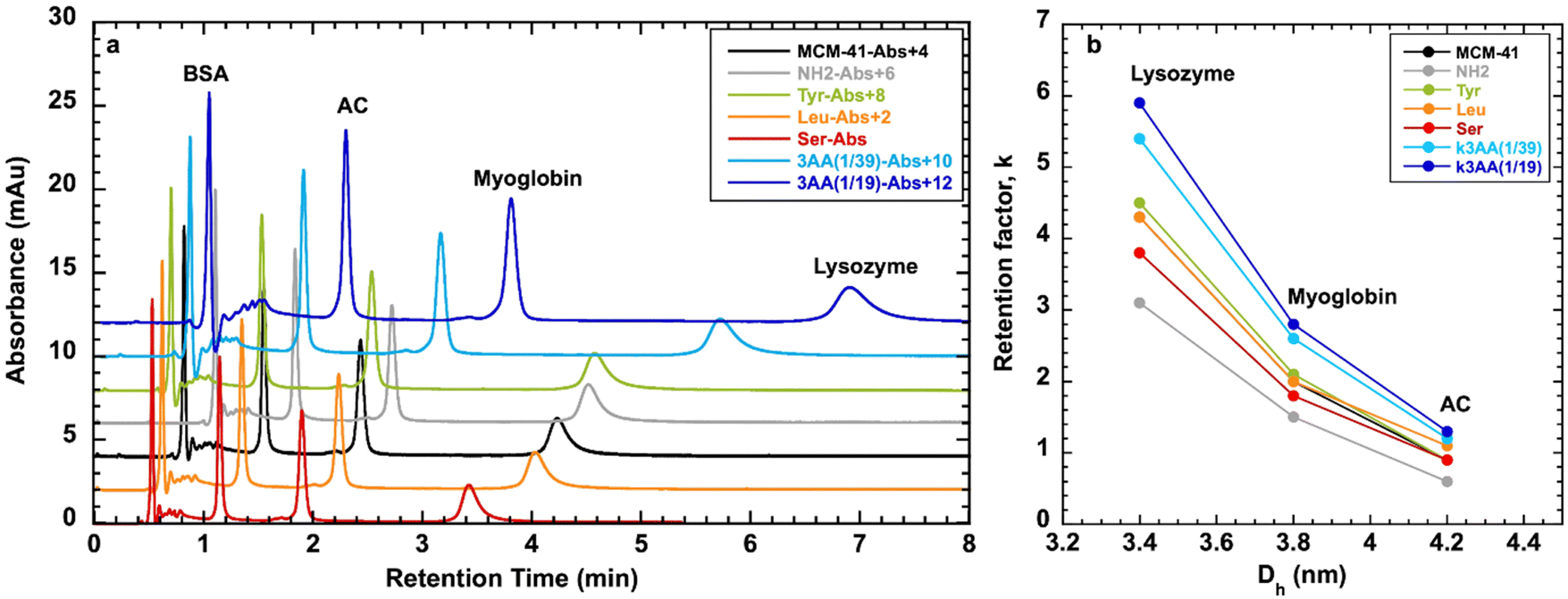 | ||
| Fig. 12 (a) Chromatograms of proteins separations with columns filled of organo-functionalized MCM-41 as a function of retention time (each absorbance was up of 2 mAu for a better observation). (b) Retention factors as a function of hydraulic diameter of proteins. Conditions: particles 50–100 μm, mobile phase H2O/ACN 98/2 (v/v), flow rate 1 mL min−1. | ||
BSA with the largest hydrodynamic diameter was not retained by the materials and get out the column with the dead volume, as Dh (7.2 nm) is larger than the mesopore diameter (∼4 nm) of MCM-41 type materials. AA-functionalized MCM-41, as well as MCM-41 and MCM-41@NH2 retain the proteins with hydrodynamic diameter (3.4 < Dh < 4.2 nm) smaller or equal to the mesopore diameter of the materials (3.8 < DBDB < 4.2 nm) (Table 3). Owing to to this result, the BdB method to determine mesopore diameter is more appropriate than the BJH method for AA-functionalized MCM-41 as DBJH gave mesopore diameter values of 2.8–3.1 nm, too small to accommodate the proteins.
The retention factor of the proteins in the materials increases with the decrease of the hydrodynamic diameter of the proteins (Fig. 12). The selectivity between proteins is similar for all materials: α1 = kLys/kAC ∼ 4.5 and α2 = kLys/kMyo ∼ 2 (Table S3, ESI†). For AC with Dh = 4.2 nm, the retention factors are similar (k ∼ 1) for all the materials (Fig. S8, ESI†), indicating that AC is adsorbed at the entrance of the mesopores. For Lysozyme with Dh = 3.4 nm, except for two materials MCM-41@NH2(1/39) and MCM-41@3AA(1/39), retention factor increases with the decrease of mesopore diameter (Fig. S8, ESI†), indicating that the protein is adsorbed inside the mesopores and get more trapped when the mesopores are smaller. For Myoglobin with Dh = 3.8 nm, similar trend as Lysozyme is observed.
Protein adsorption on solid surface is driven by a combination of different contributions such as mesopore diameter, but also electrostatic forces, hydrophobic effect and rearrangement of the tertiary structure at the surface of the material.67 The hydrophobicity nature of the surface of the materials can be deduced from the value of CBET parameter obtained by nitrogen adsorption (Table 3). Indeed, for a hydrophilic surface as hydroxylated silica CBET = 100, and CBET decreases when the surface hydrophobicity increases to reach an optimum of CBET = 20 for highly hydrophobic surface, such as totally grafted silica with octyldimethyl silane.36 Hydrophobicity could have also an impact of the retention factor. The analysis of the retention factors as a function of CBET parameter (Fig. S8, ESI†) showed that a more hydrophobic surface (lower CBET parameter) led to an increase of retention factor, except for MCM-41@NH2(1/39).
The exception found with MCM-41@NH2(1/39) with lower retention times for proteins than for the other materials is certainly coming from electrostatic forces leading to repulsion. Indeed the conjugate acid of an amine has a pKa of ∼9.5 and propylamino group is therefore positively charge at pH 6.5 and is under the form NH3+, whereas the the conjugate acid of an amide has a pKa of ∼ −0.5 and therefore amide derivatives of AA (Leu, Ser, Tyr) at pH 6.5 are majoritary under their neutral form (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, NH2) or in less extend under zwitterionic structure (C–O−, NH2+). The charge of the proteins at pH 6.5 is given by their isoelectric point (pI). The proteins, such as AC (pI = 6.1) and Myoglobin (pI = 6.2) are almost neutral at pH 6.5, only slightly negatively charged. BSA (pI = 4.5) is negatively charged and Lyzosyme (pI = 9.7) is positively charged. Lysozyme and MCM-41@NH2(1/39) should show strong repulsion and it is effectively what is observed with the lowest retention factor for Lysozyme adsorbed in MCM-41@NH2(1/39) in comparison to the other materials (Fig. S8, ESI†). However, lowest retention factors are also obtained for AC and Myoglobin adsorbed on MCM-41@NH2(1/39). Some positive charge present on these proteins should lead to repulsion, although the global charge is neutral or slightly negative.
O, NH2) or in less extend under zwitterionic structure (C–O−, NH2+). The charge of the proteins at pH 6.5 is given by their isoelectric point (pI). The proteins, such as AC (pI = 6.1) and Myoglobin (pI = 6.2) are almost neutral at pH 6.5, only slightly negatively charged. BSA (pI = 4.5) is negatively charged and Lyzosyme (pI = 9.7) is positively charged. Lysozyme and MCM-41@NH2(1/39) should show strong repulsion and it is effectively what is observed with the lowest retention factor for Lysozyme adsorbed in MCM-41@NH2(1/39) in comparison to the other materials (Fig. S8, ESI†). However, lowest retention factors are also obtained for AC and Myoglobin adsorbed on MCM-41@NH2(1/39). Some positive charge present on these proteins should lead to repulsion, although the global charge is neutral or slightly negative.
These observations mean that electrostatic interactions are not the driving force for the initial phase of interaction, but that mesopore diameter of the materials are the main driving force followed by the hydrophobic effect. Hydrophobicity is a usual factor that explains that proteins adsorb on hydrophobic surfaces under unfavorable electrostatic conditions, i.e. when surface and protein bear the same charge sign or are both neutral (Table S4, ESI†).67
In conclusion, the highest retention factors for proteins (k = 1.4 for AC, k = 3 for myoglobin, k = 6 for lysozyme) were obtained with MCM-41@3AA(1/19) synthesized with the highest amount of AA, featuring the smallest mesopore diameter (DBDB = 3.8 nm) and the highest hydrophobic character (CBET = 59). Similar retention times were obtained with MCM-41@3AA(1/19) for the separation of a mixture of two proteins including BSA (Fig. S9, ESI†), showing that proteins adsorb individually with no synergy between them. Silica from RH does not retain any proteins (Fig. S9, ESI†). This demonstrates the importance to perform the transformation of non-ordered silica into MCM-41 with homogeneous mesopores diameter and to functionalize the materials with AA for a more efficient protein separations.
4. Conclusion
Pure silica featuring a surface area of ∼300 m2 g−1 with mesopore diameter of ∼6.5 nm and macropores diameter of 5–10 μm was produced from rice husk (RH) thanks to optimize HCl 0.1 mol L−1 pretreatment at 25 °C and a calcination at 600 °C for 6 h. This silica from RH was successfully transformed into highly ordered MCM-41 and organo-functionalized (aminopropyl and amide derivates of amino acids (AA)) MCM-41 by direct pseudomorphic transformation in 24 h at 115 °C. Surfactants were successfully removed from AA-functionalized MCM-41 by NH4NO3/EtOH solution at 60 °C. The materials featured homogeneous cylindrical mesopores of ∼4 nm. The fraction of 50–100 μm particles size of the materials were used to fill columns and successfully used in flow applications, such as proteins separation. The MCM-41 functionalized with the largest amount of three AA (leucine, serine, tyrosine) featured the highest retention times for the separation of four proteins: BSA, carbonic anhydrase, myoglobin, lyzosyme. BSA was not retained by the material, as its hydrodynamic diameter (7.2 nm) was larger than the mesopore diameter. The retention factor increased mainly with the decrease of the hydrodynamic diameter of the protein (3.4–4.2 nm), and also in a less extend with the decrease of the mesopore diameter of the materials and the increase of its surface hydrophobicity. Such AA-functionalized MCM-41 could be used for the extraction and concentration of small proteins from body fluids. Also, aminopropyl-functionalized MCM-41 could be used in other applications, such as basic catalysis and biocatalysis after enzymes coupling with glutaraldehyde.This approach of valorizing a biogenic silica source highlights the technical advantages of these materials compared to mineral silica or commercial molecular precursors. The use of this bio-sourced silica can be extended to other materials such as functional nanoparticles, mixed oxides and zeolites in different domains i.e. health, energy and environment. Furthermore, it opens up new perspectives regarding the increasing integration of environmental assessments, particularly carbon footprint considerations.
Author contributions
Conceptualization: A. M., Y. L., G. S., A. G.; supervision: A. M., P. H., A. G.; project administration: A. M., P. H.; investigation: L. S., Y. F., C.-C. L., M. C., A. N.; methodology: A. M., A. G., Y. L.; formal analysis: L. S., Y. L., A. G., A. M.; validation: A. G., A. M.; writing – original draft: L. S., A. G.; writing – review & editing: L. S., A. G., A. M., M. C., G. S., Y. L., A. N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from Algerian Governement for the Algerian grant of excellence 2019 of Lilia Sennoun. The authors gratefully thank Bruno Lacrotte from Balle Concept (https://www.balleconcept.com) for providing them the rice husk generously.References
- A. G. Gebretatios, A. R. Kadiri Kanakka Pillantakath, T. Witoon, J.-W. Lim, F. Banat and C. K. Cheng, Chemosphere, 2023, 310, 136843 CrossRef CAS PubMed.
- K. Y. Foo and B. H. Hameed, Adv. Colloid Interface Sci., 2009, 152, 39–47 CrossRef CAS PubMed.
- A. Rodriguez-Otero, V. Vargas, A. Galarneau, J. Castillo, J. H. Christensen and B. Bouyssiere, Processes, 2023, 11, 3373 CrossRef CAS.
- Reducing the carbon footprint of tires with… rice! https://www.solvay.com/en/article/reducing-carbon-footprint-tires, (accessed January 2, 2024).
- T.-H. Liou, Carbon, 2004, 42, 785–794 CrossRef CAS.
- W. Nakbanpote, B. A. Goodman and P. Thiravetyan, Colloids Surf., A, 2007, 304, 7–13 CrossRef CAS.
- S. Naeem, N. Zahra and U. Zafar, Bangladesh J. Sci. Ind. Res., 2011, 45, 367–370 CrossRef.
- Q. Feng, Q. Lin, F. Gong, S. Sugita and M. Shoya, J. Colloid Interface Sci., 2004, 278, 1–8 CrossRef CAS PubMed.
- S. Xu, C. Zhou, H. Fang, W. Zhu, J. Shi and G. Liu, Environ. Res., 2023, 231, 116070 CrossRef CAS PubMed.
- N. K. Renuka, A. K. Praveen and K. Anas, Mater. Lett., 2013, 109, 70–73 CrossRef CAS.
- P. Chen, W. Gu, W. Fang, X. Ji and R. Bie, Environ. Prog. Sustain. Energy, 2017, 36, 830–837 CrossRef CAS.
- R. A. Bakar, R. Yahya and S. N. Gan, Procedia Chem., 2016, 19, 189–195 CrossRef.
- J. Castillo, V. Vargas, G. Gonzalez, W. Ruiz and B. Bouyssiere, J. Dispers. Sci. Technol., 2022, 43, 873–879 CrossRef CAS.
- W. Xu, J. Wei, J. Chen, B. Zhang, P. Xu, J. Ren and Q. Yu, Materials, 2018, 11, 1697 CrossRef PubMed.
- R. S. Bakdash, I. H. Aljundi, C. Basheer and I. Abdulazeez, Sci. Rep., 2020, 10, 19526 CrossRef CAS PubMed.
- H. A. Alyosef, A. Eilert, J. Welscher, S. S. Ibrahim, R. Denecke, W. Schwieger and D. Enke, Part. Sci. Technol., 2013, 31, 524–532 CrossRef CAS.
- N. Yalçin and V. Sevinç, Ceram. Int., 2001, 27, 219–224 CrossRef.
- S. Gu, J. Zhou, C. Yu, Z. Luo, Q. Wang and Z. Shi, Ind. Crops Prod., 2015, 65, 1–6 CrossRef CAS.
- B. Russo, J. Causse, C. Rey, J. Lautru, D. Rebiscoul and A. Ayral, Sustain. Mater. Technol., 2023, 36, e00601 CAS.
- T.-H. Liou and C.-C. Yang, Mater. Sci. Eng., B, 2011, 176, 521–529 CrossRef CAS.
- H. Purwaningsih, S. Raharjo, V. M. Pratiwi, D. Susanti and A. Purniawan, Mater. Sci. Forum, 2019, 964, 88–96 Search PubMed.
- T. Martin, A. Galarneau, F. Di Renzo, D. Brunel, F. Fajula, S. Heinisch, G. Crétier and J.-L. Rocca, Chem. Mater., 2004, 16, 1725–1731 CrossRef CAS.
- A. Galarneau, J. Iapichella, K. Bonhomme, F. Di Renzo, P. Kooyman, O. Terasaki and F. Fajula, Adv. Funct. Mater., 2006, 16, 1657–1667 CrossRef CAS.
- T. Martin, A. Galarneau, F. Di Renzo, F. Fajula and D. Plee, Angew. Chem., Int. Ed., 2002, 41, 2590–2592 CrossRef CAS PubMed.
- H. A. Alyosef, H. Uhlig, T. Münster, G. Kloess, W.-D. Einicke, R. Gläser and D. Enke, Chem. Eng. Trans., 2014, 37, 667–672 Search PubMed.
- G. Martínez-Edo, A. Balmori, I. Pontón, A. Martí del Rio and D. Sánchez-García, Catalysts, 2018, 8, 617 CrossRef.
- S. Bhattacharyya, G. Lelong and M.-L. Saboungi, J. Exp. Nanosci., 2006, 1, 375–395 CrossRef CAS.
- Y. Liu, C. Li, A. Peyravi, Z. Sun, G. Zhang, K. Rahmani, S. Zheng and Z. Hashisho, J. Hazard. Mater., 2021, 408, 124911 CrossRef CAS PubMed.
- V. Rizzi, J. Gubitosa, P. Fini, S. Nuzzo and P. Cosma, Sustain. Mater. Technol., 2020, 26, e00231 CAS.
- S. Yusan, M. M. Rahman, N. Mohamad, T. M. Arrif, A. Zubaidi, A. Latif, M. Aznan and W. S. B. Wan Nik, J. Anal. Methods Chem., 2018, 2018, 2687341 Search PubMed.
- M. Kruk, M. Jaroniec and A. Sayari, Langmuir, 1997, 13, 6267–6273 CrossRef CAS.
- R. Gutiérrez-Climente, M. Clavié, J. Gouyon, G. Ngo, Y. Ladner, P. Etienne, P. Dumy, P. Martineau, M. Pugnière, C. Perrin, G. Subra and A. Mehdi, Molecules, 2021, 26, 6085 CrossRef PubMed.
- K. Bouchmella, Q. Lion, C. Gervais and M. B. Cardoso, ACS Omega, 2023, 8, 12154–12164 CrossRef CAS PubMed.
- Z. Tavakoli, B. Rasekh, F. Yazdian, A. Maghsoudi, M. Soleimani and J. Mohammadnejad, Int. J. Biol. Macromol., 2019, 135, 600–608 CrossRef CAS PubMed.
- W. Sebai, S. Ahmad, M.-P. Belleville, A. Boccheciampe, P. Chaurand, C. Levard, N. Brun, A. Galarneau and J. Sanchez-Marcano, Front. Chem. Eng., 2022, 4, 823877 CrossRef.
- T. Martin, A. Galarneau, D. Brunel, V. Izard, V. Hulea, A. C. Blanc, S. Abramson, F. Di Renzo and F. Fajula, Stud. Surf. Sci. Catal., 2001, 135, 178 CrossRef.
- T. Yokoi, H. Yoshitake and T. Tatsumi, J. Mater. Chem., 2004, 14, 951–957 RSC.
- R. Mouawia, A. Mehdi, C. Reyé and R. J. P. Corriu, J. Mater. Chem., 2008, 18, 4193–4203 RSC.
- R. Mouawia, A. Mehdi, C. Reyé and R. J. P. Corriu, New J. Chem., 2006, 30, 1077–1082 RSC.
- M. Colilla and M. Vallet-Regí, Compr. Biomater. II, 2017, 4, 644–685 CAS.
- A.-M. Putz, M. Ciopec, A. Negrea, O. Grad, C. Ianăşi, O. I. Ivankov, M. Milanović, I. Stijepović and L. Almásy, Materials, 2021, 14, 628 CrossRef CAS PubMed.
- M. Mahmoodi, A. Behzad-Behbahani, S. Sharifzadeh, S. S. Abolmaali and A. Tamaddon, IET Nanobiotechnol., 2017, 11, 995–1004 CrossRef PubMed.
- N. Lang and A. Tuel, Chem. Mater., 2004, 16, 1961–1966 CrossRef CAS.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 160, 49–56 CrossRef CAS.
- L. Desmurs, A. Galarneau, C. Cammarano, V. Hulea, C. Vaulot, H. Nouali, B. Lebeau, T. J. Daou, C. Vieira Soares, G. Maurin, M. Haranczyk, I. Batonneau-Gener and A. Sachse, ChemNanoMat, 2022, 8, e202200051 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- J. C. P. Broekhoff and J. H. de Boer, J. Catal., 1968, 10, 368–376 CrossRef CAS.
- A. Galarneau, D. Desplantier, R. Dutartre and F. Di Renzo, Microporous Mesoporous Mater., 1999, 27, 297–308 CrossRef CAS.
- J. Chun and J. H. Lee, Sustainability, 2020, 12, 10683 CrossRef CAS.
- J. D. P. de Amorim, K. C. de Souza, C. R. Duarte, I. da Silva Duarte, F. de Assis Sales Ribeiro, G. S. Silva, P. M. A. de Farias, A. Stingl, A. F. S. Costa, G. M. Vinhas and L. A. Sarubbo, Environ. Chem. Lett., 2020, 18, 851–869 CrossRef.
- A. Galarneau, N. Calin, J. Iapichella, M. Barrande, R. Denoyel, B. Coasne and F. Fajula, Chem. Mater., 2009, 21, 1884–1892 CrossRef CAS.
- S. Schacht, M. Janicke and F. Schüth, Microporous Mesoporous Mater., 1998, 22, 485–493 CrossRef CAS.
- A. Galarneau, D. Desplantier-Giscard, F. Di Renzo and F. Fajula, Catal. Today, 2001, 68, 191–200 CrossRef CAS.
- C.-Y. Chen, S. L. Burkett, H.-X. Li and M. E. Davis, Microporous Mater., 1993, 2, 27–34 CrossRef CAS.
- L. Guillemot, T. Biben, A. Galarneau, G. Vigier and É. Charlaix, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19557–19562 CrossRef CAS PubMed.
- F. Di Renzo, D. Desplantier, A. Galarneau and F. Fajula, Catal. Today, 2001, 66, 75–79 CrossRef CAS.
- M. F. Ottaviani, A. Galarneau, D. Desplantier-Giscard, F. Di Renzo and F. Fajula, Microporous Mesoporous Mater., 2001, 44–45, 1–8 CrossRef CAS.
- V. Alfredsson, M. Keung, A. Monnier, G. D. Stucky, K. K. Unger and F. Schüth, J. Chem. Soc., Chem. Commun., 1994, 921–922 RSC.
- A. Galarneau, F. Di Renzo, F. Fajula, L. Mollo, B. Fubini and M. F. Ottaviani, J. Colloid Interface Sci., 1998, 201, 105–117 CrossRef CAS.
- O. Yoshida and M. Okamoto, J. Polym. Eng., 2006, 26, 919–940 CAS.
- Á. Berenguer-Murcia, D. Cazorla-Amorós and Á. Linares-Solano, Adsorpt. Sci. Technol., 2011, 29, 443–455 CrossRef.
- A. Galarneau, H. Cambon, F. Di Renzo and F. Fajula, Langmuir, 2001, 17, 8328–8335 CrossRef CAS.
- A. Nossov, E. Haddad, F. Guenneau, A. Galarneau, F. Di Renzo, F. Fajula and A. Gédéon, J. Phys. Chem. B, 2003, 107, 12456–12460 CrossRef CAS.
- A. Galarneau, M. Nader, F. Guenneau, F. Di Renzo and A. Gédéon, J. Phys. Chem. C, 2007, 111, 8268–8277 CrossRef CAS.
- A. El Kadib, R. Chimenton, A. Sachse, F. Fajula, A. Galarneau and B. Coq, Angew. Chem., Int. Ed., 2009, 48, 4969–4972 CrossRef CAS PubMed.
- J. Gouyon, M. Clavié, R. Gutiérrez-Climente, G. Ngo, P. Dumy, P. Etienne, P. Martineau, M. Pugnière, A. Mehdi, G. Subra, C. Perrin and Y. Ladner, Electrophoresis, 2023, elps.202300168 Search PubMed.
- A. Assarsson, I. Nasir, M. Lundqvist and C. Cabaleiro-Lago, RSC Adv., 2016, 6, 35868–35874 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj00715h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |