
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical investigation of the effect of radical substituents on the open-shell character of polycyclic aromatic hydrocarbons†
Tomohito
Shinozuka
a,
Daiki
Shimizu
 *a and
Kenji
Matsuda
*ab
*a and
Kenji
Matsuda
*ab
aDepartment of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan. E-mail: dshimizu@sbchem.kyoto-u.ac.jp; kmatsuda@sbchem.kyoto-u.ac.jp
bFukui Institute for Fundamental Chemistry (FIFC), Kyoto University, Sakyo-ku, Kyoto 606-8103, Japan
First published on 10th April 2024
Abstract
The open-shell character y of a molecule correlates with its physical properties, such as singlet fission and nonlinear optical responses. In this study, we investigated the effect of various radical substituents on the y of polyaromatic hydrocarbons (PAHs) using occupation numbers of natural orbitals calculated at the LC-UBLYP(μ=0.33)/6-311G(d,p) level of theory. It was found that radical substituents significantly altered the y of PAHs compared to non-radical substituents. Specifically, substituting methyl-type radicals such as the dicyanomethyl radical decreased the y value, while substituting allyl-2-yl-type radicals such as nitronyl nitroxide increased it. In addition, we also evaluated the correlation between the y and the HOMO–LUMO gaps and between the y and the spin-state energy gaps.
1. Introduction
For closed-shell molecules, the ground electronic state can be described by a single electronic configuration in which two electrons occupy the orbitals up to the HOMO, and none of the electrons occupy the orbitals starting from the LUMO. However, for open-shell molecules with quasi-degenerate frontier orbitals, it is necessary to consider additional electronic configurations with excited electrons in the LUMO or orbitals above it. Dissociation of a hydrogen molecule is the simplest example to show how to describe the partially-bond-dissociated state (Fig. 1). The electronic state Ψ of a hydrogen molecule with arbitrary bond lengths can be described using two electronic configurations with minimal basis: the ground state configuration ϕg and doubly excited configuration ϕe.1| Ψ = c1ϕg − c2ϕe where c12 + c22 = 1 | (1) |
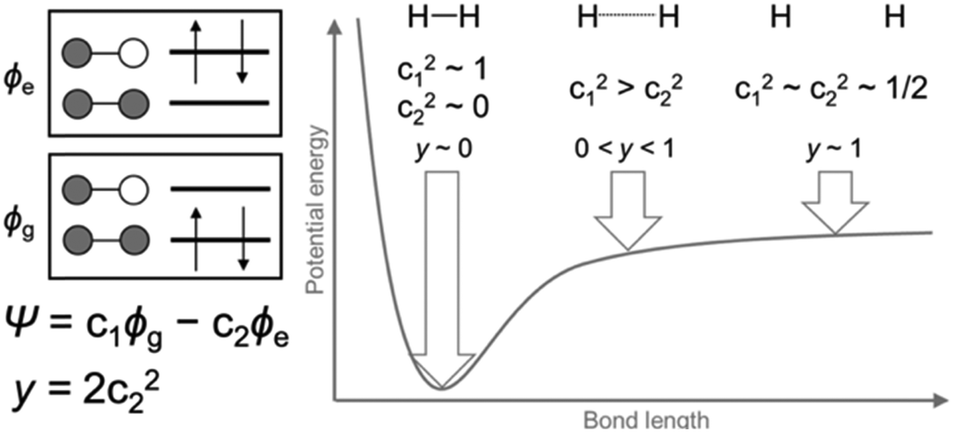 | ||
| Fig. 1 Dissociation of a hydrogen molecule and open-shell character y. | ||
Nakano et al. have revealed the correlation between the open-shell character y and molecular properties such as singlet fission4 and nonlinear optical responses,5 providing a guideline for designing functional molecules. The y strongly depends on the size and shape of molecules.6 The y values have been reported for a variety of polycyclic aromatic hydrocarbons (PAHs) such as oligoacenes,7 bisphenalenyls,8 zethrenes,9 anthenes,10 and rylene ribbons.11
It has also been reported that y can be controlled without significantly changing the molecular framework (Fig. 2). For example, introducing donor/acceptor substituents or placing the molecule in an external electric field can reduce y values.12 Haley et al. have reported that y can be increased by introducing electron-rich thiophene moieties or by extending the conjugation system.13
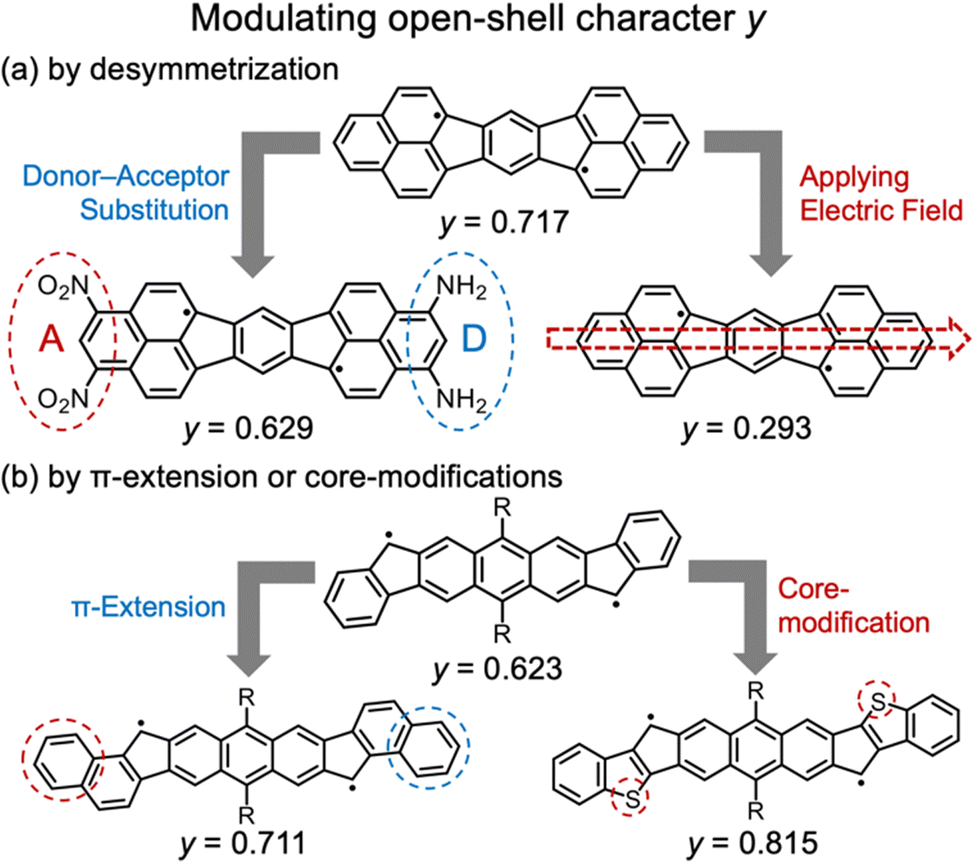 | ||
| Fig. 2 Reported guidelines for modulating y values: (a) desymmetrization12 and (b) π-extension or core-modification.13 | ||
In this study, we focused on the introduction of radical substituents with unpaired electrons as a new attempt to control y and evaluated their effects using computational chemistry. We found that the radical substitution can significantly change y compared to conventional closed-shell structural modifications, such as donor–acceptor substitution. It has been reported that conjugated radical substituents affect the excited-state dynamics of acene derivatives,14–20 which have been utilized for enhancing the photostability of pentacene.17–20 We have also reported that a radical substituent significantly enhances the conductance of a molecular wire, which was proved by single molecular measurements.21
In this work, PAHs with various open-shell characters were selected as model molecules. The y of a molecule is generally evaluated by computational means. Although an experimental method for assessing y has been proposed,22 it is not widely applied at present due to practical difficulties. In addition to the estimation of y, we also evaluated HOMO–LUMO gaps and the energy differences between spin states, which are generally considered to correlate with y.
2. Computational methodology
2.1 Model structures
We examined the y of various molecules by combinations of core structures and substituents linked by saturated (–CH2CH2–) or conjugated (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) linkers. These linkers were selected to make the PAH and substituent coplanar as much as possible. The core structures are PAHs with a wide range of open-shell characters from y0 = 0.000 (anthracene) to y0 = 1.000 (triangulene). We surveyed various substituents on them, such as spin-localized/delocalized radical substituents and donor/acceptor or π-conjugated non-radical substituents. Core structures and substituents reported in this work are summarized in Fig. 3.
C–) linkers. These linkers were selected to make the PAH and substituent coplanar as much as possible. The core structures are PAHs with a wide range of open-shell characters from y0 = 0.000 (anthracene) to y0 = 1.000 (triangulene). We surveyed various substituents on them, such as spin-localized/delocalized radical substituents and donor/acceptor or π-conjugated non-radical substituents. Core structures and substituents reported in this work are summarized in Fig. 3.
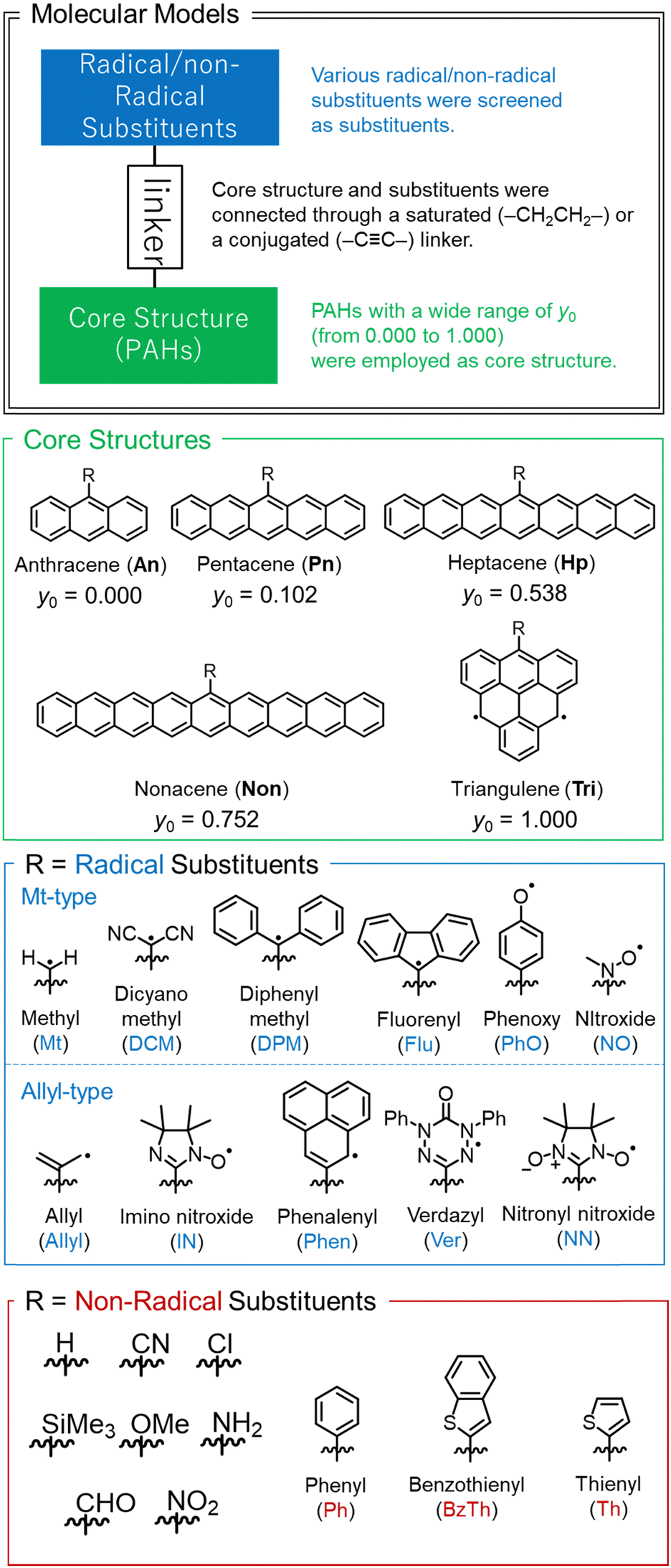 | ||
| Fig. 3 Summary of molecules examined in this work. The values of y0 shown in the 2nd panel are for PAHs with “R = H” listed in the 4th panel. | ||
2.2 Calculation methods
In this study, we examined the diradical character of an even-electron system without radical substituents. For an odd-electron system with one radical substituent, we analyzed the triradical character to evaluate the effect of radical substituents on inducing an extra open-shell character.Open-shell character y was evaluated using occupation numbers n of natural orbitals of the LUMO or orbitals above it (LUMO+i; i = 0, 1, 2…).23
| yi = nLUMO+i | (2) |
We also evaluated the HOMO–LUMO gap of even-electron systems without radical substituents at the LC-RBLYP(μ=0.33)/6-311G(d,p) level and those of radical mono-substituted systems at the LC-ROBLYP(μ=0.33)/6-311G(d,p) level. Not only the HOMO–LUMO gap, but the singlet–triplet energy gap (ΔEST) is also known to correlate with open-shell character y. The relationship between the ΔEST and y is expressed by the following equation:32
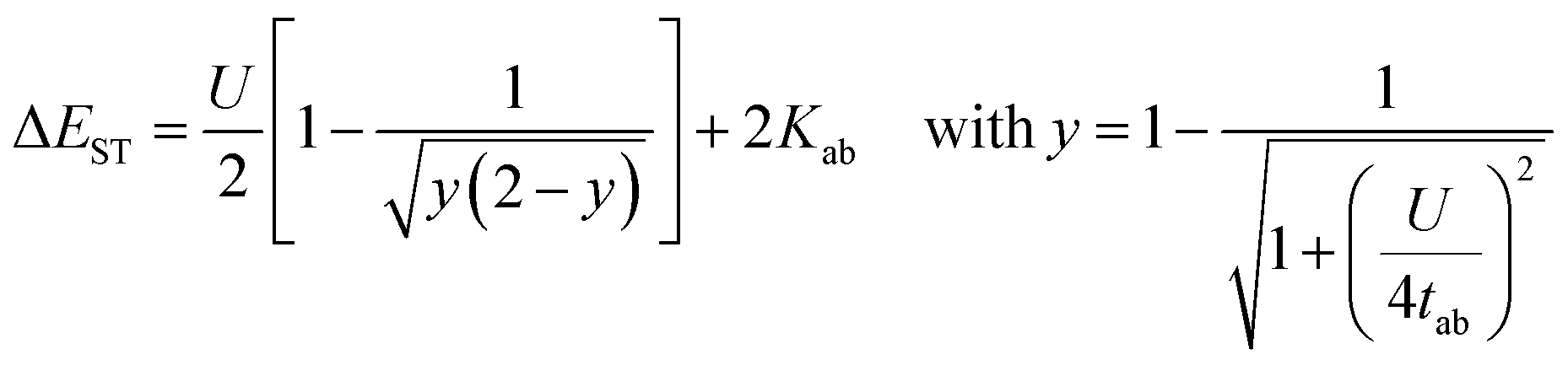 | (3) |
To evaluate the energy difference between the spin states, the ΔEST of even-electron systems with non-radical substituents and the doublet–quartet gap (ΔEDQ) of radical substituted systems were calculated. The energy gaps between spin states were estimated by the RAS-SF33 method using the cc-pVTZ34 basis set (RAS-SF/cc-pVTZ) implemented in the Q-Chem 5.4 package.35
3. Results and discussion
3.1 Effect of radical substituents on the open-shell character y of the pentacene
First, we discuss the substituent effect on the pentacene core, which has a non-zero diradical character (y0 = 0.102). In Fig. 4, substituents are classified into three groups: the “Sat-Radical” group with radical substituents via saturated hydrocarbon linkers, the “Non-Radical” group with non-radical substituents via conjugated linkers, and the “Radical” group with radical substituents via conjugated linkers.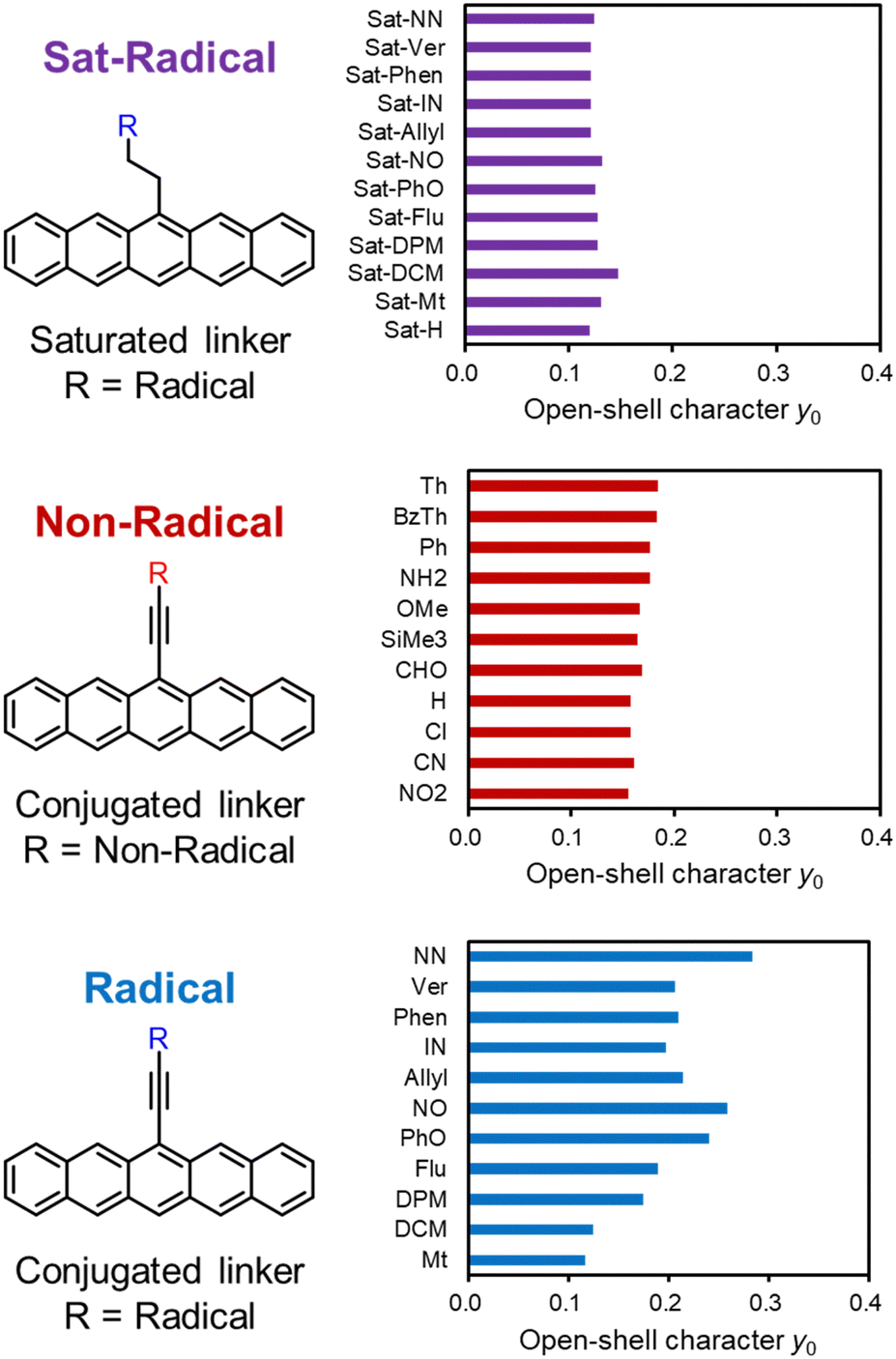 | ||
| Fig. 4 Calculated structures and y0 values of pentacene derivatives with Sat-Radical (purple bars), Non-Radical (red bars), and Radical (blue bars) substituents. | ||
For the Sat-Radical group, the triradical character y0 was not affected by the substituents (y0 = 0.1268 ± 0.008) as expected from the non-conjugated nature (Fig. 4 top and Table S8, ESI†). Importantly, the triradical character is comparable to the diradical character of pristine pentacene (y0 = 0.102, Fig. 3). The triradical character can be divided into one pure radical of the substituents and diradical character of the core. Therefore, triradical character of radical-substituted species can be interpreted as the induced diradical character of the core unit.
For the Non-Radical group, the effect of substituents on the open-shell (diradical) character y0 is also small regardless of whether the substituent is electron donating/withdrawing, π-conjugated, or heteroatoms (y0 = 0.1683 ± 0.010, Fig. 4 middle and Table S6, ESI†). The minor substitution effect will be discussed later.
Non-conjugated radical substituents and conjugated non-radical substituents do not affect the open-shell character. In the case of the conjugated radical substituents, however, we found that the y0 value widely ranges from 0.117 to 0.284, unlike the other two groups (Fig. 4 bottom, Table S7, ESI†). The radical substituents have a significant effect on the open-shell properties of PAHs when they interact with the PAH π-conjugation.
3.2 Relationship between y0 and HOMO–LUMO gap in substituted pentacene
The open-shell character y0 is generally correlated with the HOMO–LUMO (H–L) gaps of molecules, and this was also observed in this study. Fig. 5 shows the scatter plots of y0 and H–L gaps for the three groups of pentacene derivatives (see Tables S6–S8 for details, ESI†). It should be noted that the restricted methods (RDFT or RODFT) were used to qualitatively evaluate the HOMO–LUMO gaps free from spin polarization.36 As shown in Fig. 5, the H–L gaps and y0 are well-correlated; the Sat-Radical and Non-Radical groups do not change the H–L gaps, and substituents in the conjugated Radical group significantly alter the H–L gap.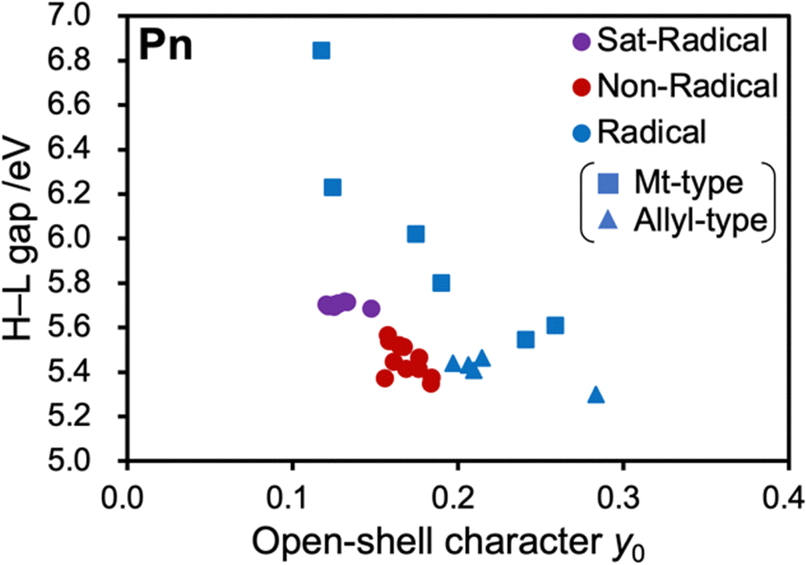 | ||
| Fig. 5 Scatter plot of HOMO–LUMO gaps and y0 of the three groups of pentacene derivatives shown in Fig. 4. The plots of the Radical groups are distinguished by square and triangle icons, which indicate methyl-type radical substituted pentacenes and allyl-type radical substituted pentacenes, respectively. | ||
To disclose how the radical substituents change the H–L gap of PAHs, the radical substituents are divided into two types in terms of conjugational topology: methyl-type (squares) and allyl-2-ly-type (triangles, hereafter called allyl-type), shown in Fig. 5. The plot shows a trend that the methyl-type radicals tend to decrease y0 and the allyl-type radicals tend to increase y0.
3.3 Interpretation of the effects of radicals based on Hückel molecular orbitals
We examined the effects of the two types of radical substituents (methyl-type and allyl-type) based on the simple Hückel molecular orbitals. Fig. 6 shows the orbital interactions between methyl/allyl radical substituents and pentacene (see Table S1 for details, ESI†).37 First, the SOMO of the methyl radical strongly interacts with both frontier orbitals of pentacene, resulting in an expansion of the H–L gap. On the other hand, in the case of the allyl radical, the SOMO of the radical moiety cannot interact with the frontier orbitals of pentacene because the orbital coefficient on the carbon atom directly bonding to pentacene is zero. Instead, the HOMO and LUMO of the radical moiety have significant orbital coefficients on the carbon atom. These orbitals interact with the frontier orbitals of the pentacene moiety, leading to the narrower H–L gap and larger y0. In summary, the increase or decrease in y0 observed for the radical substituents can be understood from the conjugational topology of the radical moiety, and it is a fundamental property of radical substituents. The same trend was observed for the molecular orbitals obtained by DFT calculations (Fig. S1 and Table S2, ESI†).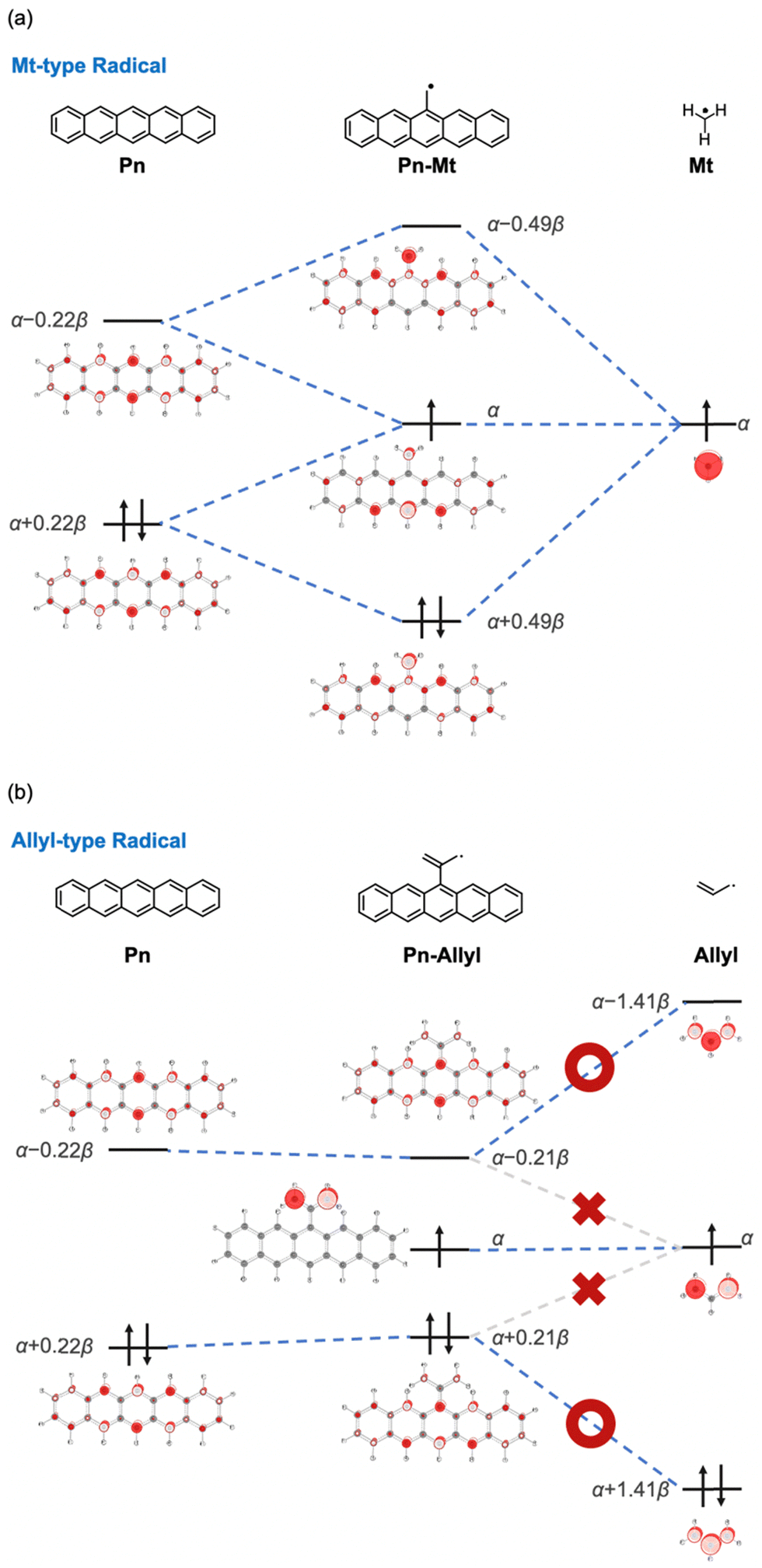 | ||
| Fig. 6 Orbital interactions between PAHs and (a) methyl radicals or (b) allyl-2-yl radicals expressed by simple Hückel molecular orbitals (α represents the Coulomb integral and β represents the resonance integral). | ||
3.4 Influence of radical substituents on different PAHs
To discuss the generality of the substituent effect, we move on to the calculation results of other core structures (Fig. 7 and Tables S3–S17, ESI†). Similar to pentacene, non-conjugated radical and conjugated non-radical substituents have a negligible impact, but the conjugated radical substituents significantly alter the open-shell character except for anthracene. The sensitivity of y0 differs from anthracene (y0 = 0.012–0.083) to triangulene (y0 = 0.143–1.000), suggesting a trend that a more prominent open-shell character leads to higher sensitivity of y0 to substitution. Other than anthracene, the methyl radical (Mt) substituent gives the smallest y0, and the nitronyl nitroxide (NN) substituent gives the largest y0 among all examined substituents. | ||
| Fig. 7 Relationship of HOMO–LUMO gaps and y0 of the Sat-Radical/Non-Radical/Radical groups of the PAHs shown in Fig. 3. Horizontal blue and orange arrows represent the decrease and increase in y0 induced by introducing Mt and NN radical substituents, respectively. All calculated data in these plots are summarized in Tables S3–S17 (ESI†). | ||
The larger the y0 value of the original PAH, the larger the decrease in y0 due to Mt, and the smaller the y0 value of the original PAH, the larger the increase in y0 due to NN. This is also interpreted by the orbital interaction between PAHs and radicals. The methyl-type radicals cause orbital interactions with their SOMO. Hence, a larger y0 of PAH (with a smaller H–L gap or energetically close frontier orbitals to the SOMO of radical units) is more favorable for orbital interaction. On the other hand, the allyl-type substituents interact with the HOMO and LUMO of the radical moiety, and so a smaller y0 of PAH (with larger H–L gaps or energetically close frontier orbitals to the HOMO and LUMO of radical units) is favorable for orbital interaction. These trends are also found in the simple Hückel calculation results (Table S1, ESI†).
3.5 Effect of radical substituents on the magnetic properties of molecules
Finally, Fig. 8 shows the correlation between y0 and the singlet–triplet (S–T) gap for the Non-Radical groups and the doublet–quartet (D–Q) gap for the Radical group for each PAH as a measurable relationship of molecular properties and y0. Fig. 8 reveals the correlation between the H–L gap and y0 shown in Fig. 7, indicating that radical substituents can significantly change the magnetic properties of open-shell molecules. Correlated with y0, the Mt substituted PAHs show the largest D–Q gap, and the NN substituted counterparts show the smallest D–Q gap. The D–Q gaps of the Mt- and NN-substituted PAHs are at least different by 1 eV, including the case of anthracene. The 1 eV change corresponds to a difference of more than 104 K in thermal energy, clearly showing how significant the effect of radical substituents is on the magnetic properties of molecules.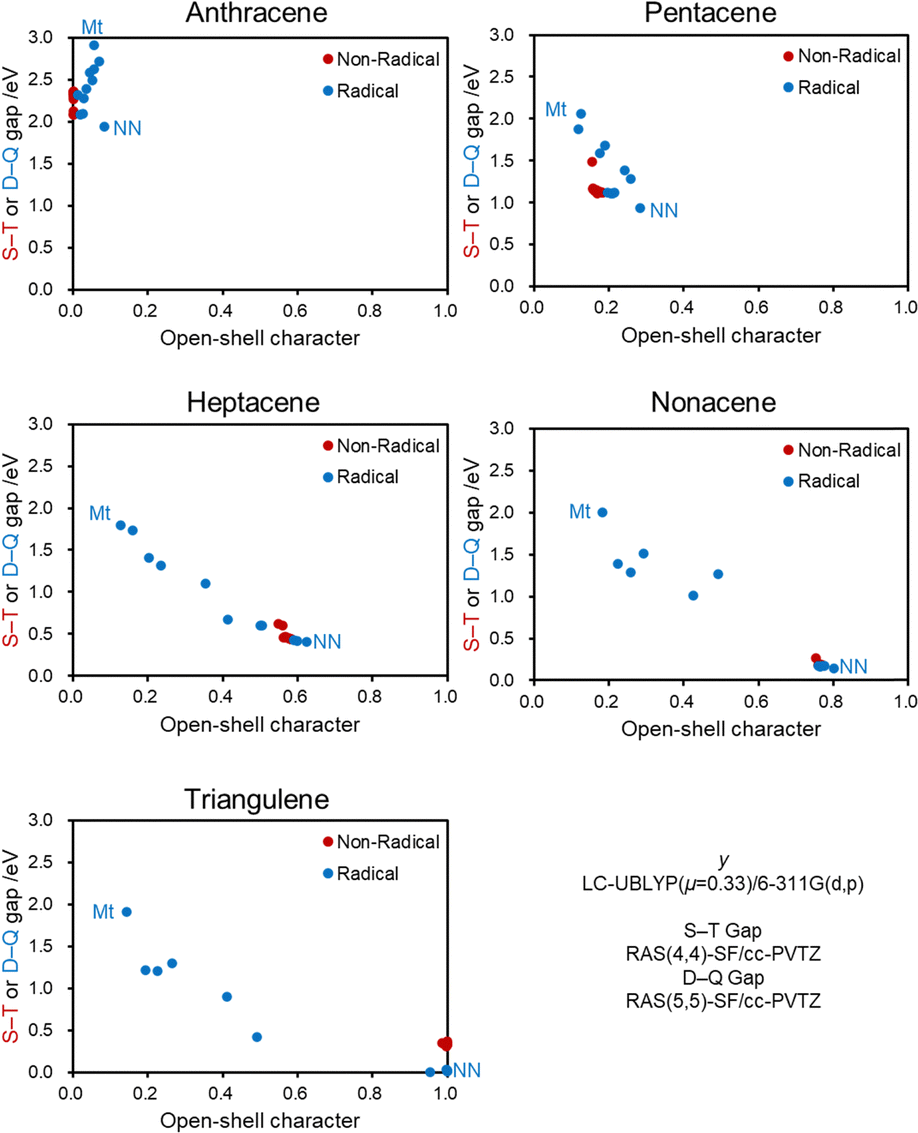 | ||
| Fig. 8 Relationship between the singlet–triplet (S–T)/doublet–quartet (D–Q) gaps and y0 of the Non-Radical/Radical groups of the PAHs shown in Fig. 3. All calculated data in these plots are summarized in Tables S3–S17 (ESI†). | ||
4. Conclusions
In summary, we have evaluated the effect of radical substituents on the open-shell character y0, which correlates with various molecular properties. We found that radical substituents change the y0 more significantly than non-radical substituents and that the influence of the radical substituents can be understood in terms of orbital interactions. Finally, we also predicted the significant substitution effect on measurable magnetic properties.This study demonstrated the power of radical substituents in controlling the open-shell character and related properties of a molecule. We are also working on the correlation between open-shell character and nonlinear optical response, which is not well understood for three-spin systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Transformative Research Areas (A) “Condensed Conjugation” (JSPS KAKENHI Grant Number JP20H05868) from MEXT, Japan. This work was also supported by a Grant-in-Aid for Young Scientists (JSPS KAKENHI Grant Number JP20K15259) and a Grant-in-Aid for Scientific Research (B) (JSPS KAKENHI Grant Numbers JP19H02788 and JP23H01948) from JSPS, Japan. We acknowledge financial support from the Iketani Science and Technology Foundation (Grant Number 0351047-A). TS acknowledges the establishment of university fellowships towards the creation of science technology innovation (JST Grant Number JPMJFS2123) from MEXT, Japan. The computational resource was provided by the SuperComputer System, Institute for Chemical Research, Kyoto University.Notes and references
- A. Szabo and N. S. Ostlund, Modern Quantum Chemistry, Dover Publication Inc., 1996 Search PubMed.
- E. F. Hayes and A. K. Q. Siu, Electronic structure of the open forms of three-membered rings, J. Am. Chem. Soc., 1971, 93, 2090 CrossRef CAS.
- L. Salem and C. Rowland, The Electronic Properties of Diradicals, Angew. Chem., Int. Ed. Engl., 1972, 11, 92 CrossRef CAS.
- T. Minami and M. Nakano, Diradical Character View of Singlet Fission, J. Phys. Chem. Lett., 2012, 3, 145 CrossRef CAS.
- M. Nakano and B. Champagne, Theoretical Design of Open-Shell Singlet Molecular Systems for Nonlinear Optics, J. Phys. Chem. Lett., 2015, 6, 3236 CrossRef CAS.
- M. Nakano, Open-Shell-Character-Based Molecular Design Principles: Applications to Nonlinear Optics and Singlet Fission, Chem. Rec., 2017, 17, 27 CrossRef CAS PubMed.
- H. Yamada and H. Hayashi, Synthesis of oligoacenes using precursors for evaluation of their electronic structures, Photochem. Photobiol. Sci., 2022, 21, 1511 CrossRef CAS PubMed.
- K. Nakasuji and T. Kubo, Multi-Stage Amphoteric Redox Hydrocarbons Based on a Phenalenyl Radical, Bull. Chem. Soc. Jpn., 2004, 77, 1791 CrossRef CAS.
- P. Hu and J. Wu, Modern zethrene chemistry, Can. J. Chem., 2017, 95, 223 CrossRef CAS.
- A. Konishi, Y. Hirao, J. Kurata, T. Kubo, M. Nakano and K. Kamada, Anthenes: Model systems for understanding the edge state of graphene nanoribbons, Pure Appl. Chem., 2014, 86, 497 CAS.
- W. Zeng, H. Phan, T. S. Herng, T. Y. Gopalakrishna, N. Aratani, Z. Zeng, H. Yamada, J. Ding and J. Wu, Rylene Ribbons with Unusual Diradical Character, Chem, 2017, 2, 81 CAS.
- M. Nakano, T. Minami, K. Yoneda, S. Muhammad, R. Kishi, Y. Shigeta, T. Kubo, L. Rougier, B. Champagne, K. Kamada and K. Ohta, Giant Enhancement of the Second Hyperpolarizabilities of Open-Shell Singlet Polyaromatic Diphenalenyl Diradicaloids by an External Electric Field and Donor–Acceptor Substitution, J. Phys. Chem. Lett., 2011, 2, 1094 CrossRef CAS.
- J. J. Dressler and M. M. Haley, Learning how to fine-tune diradical properties by structure refinement, J. Phys. Org. Chem., 2020, 33, 4114 CrossRef.
- Y. Teki, S. Miyamoto, K. Iimura, M. Nakatsuji and Y. Miura, Intramolecular Spin Alignment Utilizing the Excited Molecular Field between the Triplet (S = 1) Excited State and the Dangling Stable Radicals (S = 1/2) as Studied by Time-Resolved Electron Spin Resonance:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Observation of the Excited Quartet (S = 3/2) and Quintet (S = 2) States on the Purely Organic π-Conjugated Spin Systems, J. Am. Chem. Soc., 2000, 122, 984 CrossRef CAS.
Observation of the Excited Quartet (S = 3/2) and Quintet (S = 2) States on the Purely Organic π-Conjugated Spin Systems, J. Am. Chem. Soc., 2000, 122, 984 CrossRef CAS. - Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, π-Topology and Spin Alignment Utilizing the Excited Molecular Field: Observation of the Excited High-Spin Quartet (S = 3/2) and Quintet (S = 2) States on Purely Organic π-Conjugated Spin Systems, J. Am. Chem. Soc., 2001, 123, 294 CrossRef CAS PubMed.
- Y. Teki, T. Toichi and S. Nakajima, π Topology and Spin Alignment in Unique Photoexcited Triplet and Quintet States Arising from Four Unpaired Electrons of an Organic Spin System, Chem. – Eur. J., 2006, 12, 2329 CrossRef CAS PubMed.
- Y. Kawanaka, A. Shimizu, T. Shinada, R. Tanaka and Y. Teki, Using Stable Radicals To Protect Pentacene Derivatives from Photodegradation, Angew. Chem., Int. Ed., 2013, 52, 6643 CrossRef CAS PubMed.
- A. Ito, A. Shimizu, N. Kishida, Y. Kawanaka, D. Kosumi, H. Hashimoto and Y. Teki, Excited-State Dynamics of Pentacene Derivatives with Stable Radical Substituents, Angew. Chem., Int. Ed., 2014, 53, 6715 CrossRef CAS PubMed.
- A. Shimizu, A. Ito and Y. Teki, Photostability enhancement of the pentacene derivative having two nitronyl nitroxide radical substituents, Chem. Commun., 2016, 52, 2889 RSC.
- N. Minami, K. Yoshida, K. Maeguchi, K. Kato, A. Shimizu, G. Kashima, M. Fujiwara, C. Uragami, H. Hashimoto and Y. Teki, π-Topology and ultrafast excited-state dynamics of remarkably photochemically stabilized pentacene derivatives with radical substituents, Phys. Chem. Chem. Phys., 2022, 24, 13514 RSC.
- R. Yasui, D. Shimizu and K. Matsuda, Large Enhancement of Single Molecular Conductance of Molecular Wire through a Radical Substituent, Chem. – Eur. J., 2022, 28, e202104242 CrossRef CAS PubMed.
- K. Kamada, K. Ohta, A. Shimizu, T. Kubo, R. Kishi, H. Takahashi, E. Botek, B. Champagne and M. Nakano, Singlet Diradical Character from Experiment, J. Phys. Chem. Lett., 2010, 1, 937 CrossRef CAS.
- K. Yamaguchi, Self-Consistent Field: Theory and Applications, Elsevier, 1990, pp. 727–828 Search PubMed.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, A long-range correction scheme for generalized-gradient-approximation exchange functionals, J. Chem. Phys., 2001, 115, 3540 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- R. Kishi, S. Bonness, K. Yoneda, H. Takahashi, M. Nakano, E. Botek, B. Champagne, T. Kubo, K. Kamada, K. Ohta and T. Tsuneda, Long-range corrected density functional theory study on static second hyperpolarizabilities of singlet diradical systems, J. Chem. Phys., 2010, 132, 094107 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Thiophene and its sulfur inhibit indenoindenodibenzothiophene diradicals from low-energy lying thermal triplets, Nat. Chem., 2018, 10, 1134 CrossRef CAS PubMed.
- D. Casanova and M. Head-Gordon, Restricted active space spin-flip configuration interaction approach: theory, implementation and examples, Phys. Chem. Chem. Phys., 2009, 11, 9779 RSC.
- T. H. Dunning, Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen, J. Chem. Phys., 1989, 90, 1007 CrossRef CAS.
- M. Head-Gordon, et al., Advances in molecular quantum chemistry contained in the Q-Chem 5.4 program package, Mol. Phys., 2015, 113, 184 CrossRef.
- The H–L gaps calculated by UDFT method showed less correlation with y0. The plots of UDFT H–L gaps vs. y0 are shown in Fig. S2 in ESI†.
- Y. Carissan, D. Hagebaum-Reignier, D. N. Goudard and S. Humbel, Hückel molecular orbitals were obtained with the HuLis program, 2008, https://www.hulis.free.fr Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nj00555d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |