
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInternal Coulombic assistance in intermolecular frustrated Lewis pair activation of dihydrogen†
Alicia
Rey Planells
 a,
Arturo
Espinosa Ferao
*a,
Rainer
Streubel
*b and
Antonio
Frontera
c
a,
Arturo
Espinosa Ferao
*a,
Rainer
Streubel
*b and
Antonio
Frontera
c
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Murcia, Spain. E-mail: artuesp@um.es
bInstitut für Anorganische Chemie, Rheinische Friedrich-Wilhelms-Universität, Bonn, Germany. E-mail: r.streubel@uni-bonn.de
cDepartament de Química, Universitat de les Illes Balears, Palma de Mallorca 07122, Spain
First published on 15th February 2024
Abstract
1,3-Zwitterions consisting of a phosphonium cation linked to a silicate centre through a one-atom bridge, X3P–E–SiY4 (3), are computationally studied. Their phosphonium acidic group together with a Lewis base constitute a frustrated Lewis pair (FLP) in the activation of H2, with the silicate side-arm providing Coulombic stabilization to the positive charge at the Lewis base.
The development of FLPs (frustrated Lewis pairs)1 has been a fundamental discovery in p-block chemistry. They are composed of Lewis acidic and basic centres that, generally, are not likely to form a binary adduct due to steric factors. This concept arose from the early finding that H2 (and other small molecules) can be reversibly activated by combinations of Lewis acids and bases with high steric demand, demonstrating capabilities exclusively attributed to transition metals until then.2 The acceptor-donor nature of the elements of groups 13 and 15 are classic examples of Lewis acids and bases, respectively. Therefore, it could be envisaged that such systems could activate small molecules3 through a synergistic action of both centres, provided that their direct interaction is sterically precluded.
It was not until 2006's discovery of p-(Mes2PH)C6F4-BH(C6F5)24 that this chemical frustration could be used for synthetic purposes, being capable of activating the H2 molecule under very mild conditions (25 °C, 1 atm). Thereafter, the scope of FLP activations was broadened including a variety of small molecules such as olefins,5 alkynes,6 and a number of element oxides, primarily carbon dioxide7 and nitrous oxide.8 Furthermore, FLP chemistry now includes unique stoichiometric transformations such as metal-free catalytic hydroaminations,9 polymerizations10 and even recent applications in bio-inorganic and materials chemistry, as well as in heterogeneous catalysis.11
Among the most commonly used Lewis bases are phosphines,12 amines,13 thioethers14 and carbenes,15 while polyfluorinated boranes16 and aluminium-based centres17 have been used as Lewis acidic counterparts in FLPs. In addition, Lewis acids based on carbon or silicon have been reported, the latter being able to act as Lewis acid (silylium–phosphine adducts)18 or base (silylium–silylene) FLPs.19 The phosphonium cation20 is a well-known Lewis acid in which the acidity is derived from a σ* orbital.21 1,3,5-Triphosphinine (1,3,5-triphosphabenzene) was reported to act as a kind of intramolecular FLP in the cleavage of the H2 molecule through resonance structures in which a positive charge is located at the phosphorus and a negative charge at the carbon.22 These cationic Lewis acids are able to promote dehydro-fluorination reactions, isomerization, hydrosilylation and hydrogenation of olefins, as well as dehydrocoupling of silanes with alcohols, acids, amines and thiols.23
1,3-Zwitterionic species24 could be good candidates for behaving as FLPs provided that the cationic and anionic centres could exhibit Lewis acid and basic properties, respectively. A recent example of a crypto-FLP bearing a P–O–Si framework with a basic phosphide centre and (neutral) electrophilic Si atom was reported.25 An interesting inverse combination of 1,3-zwitterionic species for the formation of an FLP, that has not yet been reported, is the use of a phosphonium cation as a Lewis acid, bearing a one-atom bridged negatively charged centre.
Herein, the formation of these types of zwitterions X3P–E–SiY4 (3) from stable σ4λ5-phosphorus derivatives X3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (1) and saturated SiY4 compounds (2) is explored (Scheme 1). Species 1 already have resonant structures with 1,2-dipolar structure. Nevertheless, this dipole is unable to activate small molecules. Species 3 could also enable a P-to-Si E-group transfer, giving rise to X3P (4) and E:→SiY4 (5). This scheme has been studied for different families varying the E bridge that contains group 14 (CH2, SiH2), 15 (NH, PH) or 16 (O or S) central atoms. Interestingly, 3 could provide the phosphonium acidic centre engaged in an intermolecular FLP with an external Lewis base (LB), the silicate side arm allowing vicarious stabilization via additional Coulombic interaction with the newly originated cationic centre resulting upon small molecule AB activation (Scheme 1).
E (1) and saturated SiY4 compounds (2) is explored (Scheme 1). Species 1 already have resonant structures with 1,2-dipolar structure. Nevertheless, this dipole is unable to activate small molecules. Species 3 could also enable a P-to-Si E-group transfer, giving rise to X3P (4) and E:→SiY4 (5). This scheme has been studied for different families varying the E bridge that contains group 14 (CH2, SiH2), 15 (NH, PH) or 16 (O or S) central atoms. Interestingly, 3 could provide the phosphonium acidic centre engaged in an intermolecular FLP with an external Lewis base (LB), the silicate side arm allowing vicarious stabilization via additional Coulombic interaction with the newly originated cationic centre resulting upon small molecule AB activation (Scheme 1).
 | ||
| Scheme 1 Studied potential interconversions (E = CH2, SiH2, NH, PH, O or S) and possible vicarious Coulombic stabilization of the silicate moiety upon small molecule FLP activation. | ||
The choice of substituents was first investigated, as they might play a key role in finding stable 1,3-zwitterionic dipoles 3 (Scheme 1). Electron donating substituents X at P (PXDo3) and electron acceptors Y at Si (SiYAcc4) are expected to favour the formation of zwitterionic structures 3 by stabilizing the separated electric charges. With this aim, simple model donor substituents Me, NH2 and SiMe3 (Tms), together with F and CF3 as acceptor substituents, were studied. As expected, the above-mentioned combination of substituents does indeed stabilize 1,3-dipolar species 3. Most of the energies for the dipole 3 formation process are highly exergonic (Table S1, ESI†), especially for CH2- and NH-bridged dipoles and most O-bridged ones with the highest exergonicity found for Tms3P–CH2–Si(CF3)4 (3Cf). As a general trend, the dipole formation is more exergonic as a function of the bridge “E” element group in the order tetrel ≥ pnictogen > chalcogen and decreasing for third (Si, P and S) compared to second-row elements (C, N and O). Indeed, S-bridged zwitterionic structures 3Sa–f are not formed as such, but van der Waals complexes 1·2 with only peripheral interactions (no direct S⋯Si bond) were found instead (Fig. S2, ESI†). Other exceptions are 3Sia,c and 3Pa whose formation is moderately endergonic. On the contrary, the opposite combinations of accepting substituents on P (PXAcc3) and donor substituents on Si (SiYDo4) destabilize the molecular 1,3-zwitterionic species 3, which were never located as minima.
The E-group transfer reaction from 1 to 2 by means of the alternative cleavage of the E–Si bond in 3 (Scheme 1) leads to a hypothetical species E:→SiY4 (5) which was never located as a minimum, as it rearranges furnishing a [1,2]Y-migration product Y–E–SiY3 (5′). Nevertheless, this E–Si bond-cleavage with rearrangement is generally endergonic (Table S1, ESI†) and leads to products (4+5′) that are much less stable than the starting materials (1+2), except for all SiH2-bridged zwitterions. In only a few exceptional cases (3Sia,c,e and 3Oe) such a cleavage is exergonic (Table S1, ESI†).
The intramolecular single electron transfer (SET) from the potentially reducing σ3λ3-P(III)-compound (4) to the P–E bond cleavage species E:→SiY4 (5) was also explored. However, although all species 5˙− were found to be stable (i.e. no rearrangement observed), the hypothetical (separated) ion pair 4˙+ + 5˙− turned out to be highly unstable (see Table S1, ESI†). Most likely, this is because the electronic effects of the substituents that favour formation of the zwitterion 3 are destabilizing the intramolecular SET from 4 to 5.
Once the appropriately substituted dipolar species 3 were shown to be stable compounds, the Coulombic interactions between the P and Si ionic centres were studied. As expected, the P⋯Si distances of dipolar species 3 with a third-row atom at the bridging E position are larger than those having second row atoms, due to their larger atomic size. (Table S2, ESI†). The Wiberg bond indices (WBI) for the P⋯Si (electrostatic) interaction are generally quite low, with only three non-linear molecules displaying values above 0.015 (3Sic, 3Pc and 3Pd), thus accounting for very weak interactions. Moreover, the atoms-in-molecules26 (AIM) methodology reveals the existence of bond critical points (BCP) corresponding to non-covalent interactions (NCI) between the X and Y substituents on P and Si, respectively (Fig. S1, ESI†), which satisfactorily explains the observed shortening of the P⋯Si distance in some cases.
In zwitterionic species 3 it is worth inspecting the nature of the E–Si bond formally arising from the interaction between the stable species X3PE (1) and SiY4 (2). To highlight the effect of the bridging unit “E”, a small subset of compounds 3 with the simplest substituents, namely Me3P–E–SiF4, was selected for this study. According to IUPAC, a dative bond is formed between two entities, one of them acting as a donor and the other as an acceptor of the pair of electrons to be shared in the complex formed. Compared to covalent bonds it presents “significant polarity, less resistance and greater length” and its signature is “that its minimum energy breakdown in the gas phase or in inert solvents follows the heterolytic bond cleavage path”. When compared to the covalent E-Si bond in analogous dipolar structures HE-SiF4, lacking the PX3 fragment, a clear elongation of the E–Si bond in 3 is observed (Table S3, ESI†), which may be indicative of a dative linkage. The elongation varies in the order SiH2 < PH < CH2 < NH < O ≪S, the latter agreeing with their van der Waals complex (1·2) nature.
The computed bond dissociation energies (BDE) show a clear preference for the heterolytic over the homolytic cleavage, and summation of the (Mulliken) electric charges over the atoms belonging to the SiY4 group ( ) shows a remarkable charge transfer and hence unveils its acceptor character (Table S3, ESI†), both facts pointing to a E:→Si dative bond nature.
) shows a remarkable charge transfer and hence unveils its acceptor character (Table S3, ESI†), both facts pointing to a E:→Si dative bond nature.
Another approach to analyse dative bonding arises from the representation of the Laplacian of the electron density along the bond path.27 The ∇2ρ function shows a pronounced sharp minimum, which is a valence shell charge concentration (VSCC) band corresponding to the electron donor atom of the ligand, VSCCC for 3Ca as a case in point (Fig. S3, ESI†), and another shoulder attributable to the associated acceptor Si atom (VSCCSi). Both lie within the basin of the donor atom, which constitutes a signature for dative bonds together with a small positive value of ∇2ρ at the BCP (2.88 e Å−5). For the rest of the E bridges, the two VSCC minima fall within the same basin (see ESI†) except for the bridge with S (3Sa), which constitutes a van der Waals complex, as already indicated.
Since the main interest of FLPs is their potential use in the activation of small molecules, the efficacy of the zwitterionic molecules 3 was checked for this purpose. As aforementioned, 3 can provide the phosphonium acidic centre acting as an intermolecular FLP with an external base, for which the two lighter trimethyl-pnictogens Me3Pn (Pn = N, P) 6Pn were used as models. As cases in point, two oxygen atom-bridged triphenylphosphonium silicates Ph3P–O–SiY4 (Y = F (3Og), Cl (3Oh)) were employed as zwitterionic species and their FLP behaviour checked for the activation of H2 (Scheme 2).
 | ||
| Scheme 2 Activation reaction of the H2 molecule by the dipole structures 3Og–j and Lewis bases 6Pn. | ||
The quantum chemical calculations provide evidence that the H2 molecule is activated by these complexes 3Og,h/6Pn, endergonically producing the corresponding ion pairs 7g,hPn by cleavage of the H–H bond with a moderate energy barrier (Fig. 1). The lowest barriers correspond to the FLPs engaging the amine 6N as a base (ca. 25 kcal mol−1) and the most stable final adducts are those having Cl as a substituent at silicon, 7hN and 7hP (Fig. 1). Altogether, 3Oh/6N resulted in the most favourable FLP combination for the activation of H2.
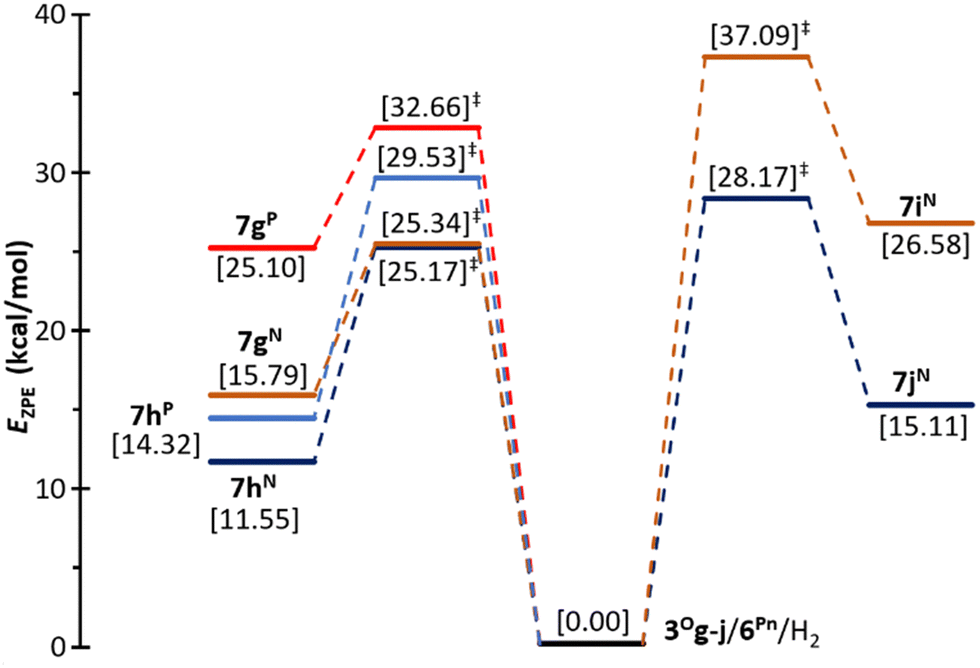 | ||
| Fig. 1 Computed (CCSD(T)/def2-TZVPP//PBEh-3c) zero-point corrected energy profile for the activation reaction of the H2 molecule by 6N,P and 3Og,h (left) and by 6N and 3Oi,j (right) according to Scheme 2. | ||
In these processes the approximation of H2 (and the Lewis base 6) occurred exclusively syn with respect to the silicate side-arm (Fig. 2), thus making profit of the vicarious Coulombic stabilization with the newly generated pnictogenium centre (Pn+). The beneficial effect of this Coulombic interaction is demonstrated by the fact that the analogous control reaction between triphenylphosphine oxide and trimethylamine did not produce the desired activation of the dihydrogen molecule, but 1,2-addition along the PO bond (Ph3P(H)–OH).
 | ||
| Fig. 2 Computed (PBEh-3c) TS for the formation of 7gN (left) and 7iN (right). | ||
In order to make the phosphonium centre more electrophilic, pentafluorophenyl (instead of phenyl) substituents were placed at P in zwitterions 3Oi,j (Scheme 2). As expected, the effect of increased electrophilicity causes the LUMO energy to decrease from −0.657/−0.834 eV in 3Og,h to −1.661/−1.894 eV in 3Oi,j, respectively, favouring the approximation of the hydridic H atom to the sigma hole of the phosphonium cation, which corresponds to the LUMO of mainly σ*(P–O) character (Fig. S8, ESI†). This is also reflected in the higher magnitude of the sigma-hole in 3Oi,j compared to 3Og,h (Fig. 3). However, this does not entail a decrease in the energy of the transition state with respect to the P-phenyl substituted homologues (Fig. 1), most likely because the sigma hole (anti) approximation (Fig. 3b) precludes the vicarious stabilization by Coulombic interaction between the silicate side arm and the newly originated cationic centre at the pnictogen atom of the Lewis base. This demonstrates that the stabilization caused by the Coulombic interaction of the silicate side-arm in the syn approximation to 3Og,h (Fig. 3a) is more effective than a Lewis acidic centre with a considerably lower LUMO in 3Oi,j. For the latter, no syn approximation pathway was found. Localization of the transition state for the formation of 7i,jP was not possible probably due to the larger size of the P atom in the Lewis base 6 and the scarce space left by the F-substituents to access the phosphonium acidic centre.
 | ||
| Fig. 3 Computed (B3LYP-D4/def2-TZVP//PBEh-3c) electrostatic potential plot mapped onto an electron density isosurface (0.01 a.u.) for (a) 3Og, (b) 3Oh, (c) 3Oi and (d) 3Oj. | ||
Conclusions
A series of zwitterionic species X3P–E–SiY4 (3) with group 14 (CH2, SiH2), 15 (NH, PH) or 16 (O or S) E-bridges were computationally studied. Only those having donor substituents at P and acceptor substituents at Si turned out to be stable. The E–Si linkage in 3 could be better described as a dative bond (E:→Si) on the basis of either the observed elongated bonds, significant charge transfer, predominant heterolytic over homolytic bond cleavage, or AIM-related criteria. In the presence of an external Lewis base Me3Pn (Pn = N, P), 1,3-dipolar compounds 3 display FLP reactivity in the activation of H2 which is facilitated by vicarious stabilization in the transition state due to the Coulombic interaction of the pendant silicate side-arm with the newly generated pnictogenium centre (Pn+) in the resulting ion pair 7.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Servicio de Cálculo Científico (SCC – University of Murcia) for the computational resources used. A. R. P. is indebted to an Erasmus Placement opportunity (3 months) and the University of Bonn Fellowship Programme (3 months) for financing short stays in Bonn. A. F. thanks MICIU/AEI of Spain (PID2020-115637GB-I00, FEDER funds) for financial support and the Alexander von Humboldt foundation for the J. C. Mutis award.Notes and references
- (a) G. Linti and H. Schnöckel, Coord. Chem. Rev., 2000, 206–207, 285 CrossRef CAS; (b) B. D. Ellis and C. L. B. Macdonald, Coord. Chem. Rev., 2007, 251, 936 CrossRef CAS; (c) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302 CrossRef CAS PubMed.
- S. Schulz, Coord. Chem. Rev., 2015, 297–298, 49 CrossRef CAS.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018 CrossRef CAS PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124 CrossRef CAS PubMed.
- (a) G. C. Welch and D. W. Stephan, J. Am. Chem. Soc., 2007, 129, 1880 CrossRef CAS PubMed; (b) P. Spies, G. Erker, K. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC.
- (a) L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164 CrossRef CAS PubMed; (b) L. Greb, C. G. Daniliuc, K. Bergander and J. Paradies, Angew. Chem., Int. Ed., 2013, 52, 5876 CrossRef CAS PubMed; (c) J. Paradies, Synlett, 2013, 777 CrossRef CAS; (d) J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552 CrossRef CAS PubMed; (e) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968 CrossRef CAS PubMed.
- (a) K. Chernichenko, A. Madarasz, I. Papai, M. Nieger, M. Leskela and T. Repo, Nat. Chem., 2013, 5, 718 CrossRef CAS PubMed; (b) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 8396 CrossRef CAS PubMed; (c) C. Jiang, O. Blacque and H. Berke, Organometallics, 2010, 29, 125 CrossRef CAS; (d) M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 659 Search PubMed.
- (a) C. M. Mamming, E. Otten, G. Kehr, R. Fröhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643 CrossRef PubMed; (b) A. Berkefeld, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2010, 132, 10660 CrossRef CAS PubMed; (c) A. E. Ashley, A. L. Thompson and D. O’Hare, Angew. Chem., Int. Ed., 2009, 48, 9839 CrossRef CAS PubMed.
- (a) E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918 CrossRef CAS PubMed; (b) R. C. Neu, E. Otten and D. W. Stephan, Angew. Chem., Int. Ed., 2009, 48, 9709 CrossRef CAS PubMed.
- (a) T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 12418 CrossRef CAS PubMed; (b) Y. Wang, Z. Liu, W. Guo, C. Zhang and X. Zhang, Macromolecules, 2023, 56, 4901 CrossRef CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- (a) Y. Guo and S. Li, Inorg. Chem., 2008, 47, 6212 CrossRef CAS PubMed; (b) M. Shi, L. H. Chen and C. Q. Li, J. Am. Chem. Soc., 2005, 127, 3790 CrossRef CAS PubMed.
- (a) T. A. Rokob, A. Hamza and I. Pápai, J. Am. Chem. Soc., 2009, 131(30), 10701 CrossRef CAS PubMed; (b) M. G. Gardiner and C. L. Raston, Coord. Chem. Rev., 1997, 166, 1 CrossRef CAS; (c) D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46 CrossRef CAS PubMed; (d) S. J. Geier, M. A. Dureen, E. Y. Ouyang and D. W. Stephan, Chem. – Eur. J., 2010, 16, 988 CrossRef CAS PubMed.
- (a) V. Sumerin, F. Schulz, M. Nieger, M. Leskel, T. Repo and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 6001 CrossRef CAS PubMed; (b) V. Sumerin, F. Schulz, M. Atsumi, C. Wang, M. Nieger, M. Leskel, T. Repo, P. Pyykk and B. Rieger, J. Am. Chem. Soc., 2008, 130, 14117 CrossRef CAS PubMed; (c) S. J. Geier, A. L. Gille, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2009, 48, 10466 CrossRef CAS PubMed; (d) G. Ers, H. Mehdi, I. Papai, T. A. Rokob, P. Kiraly, G. Tarkanyi and T. Soos, Angew. Chem., Int. Ed., 2010, 49, 6559 CrossRef PubMed . For imines, see: ; (e) P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050 CrossRef CAS PubMed . For aromatic N-heterocycles, see: ; (f) S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476 CrossRef CAS PubMed; (g) S. J. Geier, A. L. Gille, T. M. Gilbert and D. W. Stephan, Inorg. Chem., 2009, 48, 10466 CrossRef CAS PubMed; (h) S. J. Geier, P. A. Chase and D. W. Stephan, Chem. Commun., 2010, 46, 4884 RSC.
- M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594 CrossRef CAS.
- (a) D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS PubMed; (b) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS PubMed; M. Alcarazo, C. Gomez, S. Holle and R. Goddard, Angew. Chem., Int. Ed., 2010, 49, 5788 Search PubMed.
- W. Uhl, P. Wegener, M. Layh, A. Hepp and E. Würthwein, Z. Naturforsch. B, 2016, 71, 1043 CrossRef CAS.
- T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J. P. White, P. A. Hunt and A. E. Ashley, Chem. Commun., 2014, 50, 12753 RSC.
- A. Schafer, M. Reissmann, A. Schafer, M. Schmidtmann and T. Müller, Chem. – Eur. J., 2014, 20, 9381 CrossRef PubMed.
- N. Burford and P. J. Ragogna, J. Chem. Soc., Dalton Trans., 2002, 4307 RSC.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374 CrossRef CAS PubMed.
- L. E. Longobardi, C. A. Russell, M. Green, N. S. Townsend, K. Wang, A. J. Holmes, S. B. Duckett, J. E. McGrady and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 13453 CrossRef CAS PubMed.
- A. Bismuto, G. S. Nichol, F. Duarte, M. J. Cowley and S. P. Thomas, Angew. Chem., Int. Ed., 2020, 59, 12731 CrossRef CAS PubMed.
- R. Kunzmann, Y. Omatsu, G. Schnakenburg, A. Espinosa Ferao, T. Yanagisawa, N. Tokitoh and R. Streubel, Chem. Commun., 2020, 56, 3899 RSC.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083 CrossRef CAS.
- (a) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 CrossRef; (b) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS; (c) C. F. Matta and R. J. Boyd, in The Quantum Theory of Atoms in Molecules, ed. C. F. Matta and R. J. Boyd, Wiley-VCH, New York, 2007, p. 1 CrossRef.
- (a) M. W. Stanford, J. I. Schweizer, M. Menche, G. S. Nichol, M. C. Holthausen and M. J. Cowley, Angew. Chem., Int. Ed., 2019, 58, 1329 CrossRef CAS PubMed; (b) V. Nesterov, R. Baierl, F. Hanusch, A. Espinosa Ferao and S. Inoue, J. Am. Chem. Soc., 2019, 141, 14576 CrossRef CAS PubMed; (c) A. Espinosa Ferao, A. García Alcaraz, S. Zaragoza Noguera and R. Streubel, Inorg. Chem., 2020, 59, 12829 CrossRef CAS PubMed; (d) D. Biskup, G. Schnakenburg, R. T. Boeré, A. Espinosa Ferao and R. K. Streubel, Nat. Commun., 2023, 14, 6456 CrossRef CAS PubMed; (e) D. Biskup, G. Schnakenburg, A. Espinosa Ferao and R. Streubel, Dalton Trans., 2024, 53, 2517 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details, other theoretical results and computed structures. See DOI: https://doi.org/10.1039/d4nj00523f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |