
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCreation of a facile heterojunction in Co/ZnO–TiO2 for the photocatalytic degradation of alizarin S†
Aditi
Prabhu
a,
Preetha Chandrasekharan
Meenu
a and
Sounak
Roy
 *ab
*ab
aDepartment of Chemistry, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad, 500078, India. E-mail: sounak.roy@hyderabad.bits-pilani.ac.in
bMaterials Center for Sustainable Energy & Environment, Birla Institute of Technology and Science Pilani, Hyderabad Campus, Hyderabad, 500078, India
First published on 13th May 2024
Abstract
Advanced oxidative process is an efficient method to photocatalytically degrade the effluent pollution by organic dyes with a suitable catalyst. Photocatalysts with a heterojunction not only improve the excitation efficiency of the catalyst in the presence of light by broadening the wavelength range of absorption but also reduce the rate of recombination. ZnO and TiO2 were successfully synthesized in this study via solution combustion synthesis. Wurtzite ZnO was grown along with anatase TiO2 to create a heterojunction of ZnO–TiO2, and Co loading was carried out to improve the rate of electron transfer between the heterojunction for enhanced photocatalytic activity. The photogenerated electrons are injected from the conduction band of ZnO to TiO2via Co to create ˙O2− and holes from TiO2 to form OH˙, which help to degrade alizarin S. The detailed photocatalytic experiment confirmed Co/ZnO–TiO2 as an extremely efficient catalyst, which shows the complete degradation of the dye alizarin S in just 40 minutes under UV light illumination.
1. Introduction
Organic dyes released from various industries such as textile, leather, plastic, food, drug and cosmetic industries have harmful effects on the environment as well as humans and usually tend to be carcinogenic and mutagenic due to their toxic nature and high resistance to decomposition. Alizarin red S (1,2-dihydroxy-9,10-anthraquinonesulfonic acid sodium salt; ARS; mordant red 3) is such a water-soluble bright red-coloured anthraquinone dye, which is extensively employed as a pH indicator, biological stain in chemical analyses, and during the dyeing process in textile industries.1,2 The robust stability of this particular dye is attributed to its intricate aromatic structure, which prevents its degradation over time. Moreover, the non-biodegradable nature in aerobic and anaerobic environments makes its release detrimental for the aquatic ecosystem and also poses a direct threat to human beings and animals.3–7 Thus, the high chemical resistance of alizarin Red S makes it highly challenging to be removed from water bodies.Various methods have been adopted for the removal of dyes. One such process is advanced oxidative process (AOP),8,9 which involves a strong oxidizing species such as OH˙ and ˙O2− radicals that break down the dye molecule into smaller and less harmful substances. The photocatalytic advanced oxidative process generates active free radicals OH˙ and ˙O2− primarily by the oxidation of water with the help of a suitable semiconductor catalyst.10–12 The photocatalytic oxidation involves various fundamental processes including charge carrier formation, separation, and transportation. One of the crucial steps is the creation and recombination of the electron–hole pair.13 Electron–hole pair formation occurs under UV light illumination corresponding to the semiconductor photocatalytic material's band gap. When exposed to light, electrons (e−) from the valence band (VB) of the semiconductor are excited to the conduction band (CB), leading to the generation of positive holes (h+) in the VB. The resulting e−–h+ pairs, through a series of redox reactions with the solvent water or gaseous oxygen, yield active oxygen species such as hydroxyl radicals ˙OH and superoxide radical anions O2•−, contributing to the mineralization of dyes.14–18
Among the photocatalysts studied, TiO2 and ZnO are the most widely researched materials for dye degradation.16,19–24 Both TiO2 and ZnO as photocatalysts can give rise to photoinduced electron/hole (e−/h+) pairs and commence a chain of chemical reactions, thus decomposing the pollutants. What distinguishes these two materials is their stability, non-toxicity, cost-effectiveness, and high reactivity.25 However, currently, the semiconductor photocatalysis field faces a significant challenge–the low efficiency of photocatalysis. This issue arises from the swift recombination of e−–h+ pairs generated during the process and the inherent difficulty in achieving a balance between broad light absorption and ample redox ability. It is widely acknowledged that in a one-step excitation system utilizing a single photocatalyst and satisfying multiple performance criteria simultaneously is nearly impossible. Consequently, the concept of composite photocatalysts has emerged, combining the strengths of different materials. TiO2–ZnO mixed oxides are one of the popular choices of heterojunction photocatalysts due to the separation efficiency of electrons and holes.26–28 The formation of a heterojunction can enhance the photogeneration process and cause electron transfer from the ZnO CB to the TiO2 CB and thus hole transfer from the TiO2 VB to the ZnO VB, giving rise to higher electron–hole transfer separation and lifetime, as has been reported by many studies.29,30 In composite photocatalytic systems, the rational design of heterojunctions stands out as a core strategy. Among different type of heterojunctions, the popular types are semiconductor–semiconductor heterojunction and semiconductor–metal heterojunction. However, most often, both the types are limited by visible light responses.
For broadening the wavelength range of absorption by photocatalysis, a multicomponent heterojunction can be invoked, where the two semiconductor bands create heterojunctions with metallic bands.31 Herein, we studied the photocatalytic degradation of alizarin S dye utilizing the nano-composite heterojunction of TiO2 and Co/ZnO. The primary objective for the particular design of a heterojunction is to improve the excitation efficiency of the catalyst in the presence of light by broadening the wavelength range of absorption. Electrons from the CB of ZnO will easily flow into the Co due to a higher CB (or Fermi level) compared to Co, facilitating rapid electron transfer. This process will be faster than electron–hole recombination between the VB and CB of ZnO, allowing ample electrons to be stored in the metal. Likewise, holes from the VB of TiO2 will readily flow into Co, outpacing electron–hole recombination.
2. Materials and methods
2.1 Synthesis
TiO2 and ZnO were prepared by a single-step solution combustion method that includes oxidisers (metal precursors) and organic fuel glycine.23,32–34 For the preparation of ZnO, stoichiometric amounts of Zn(NO3)2·6H2O (S D Fine-Chem Ltd, LR) and glycine (Sigma Aldrich) were homogeneously mixed in 50 mL DI water. The solution was then placed in a muffle furnace preheated at 450 °C. After the complete evaporation of water, the fuel ignited with a sudden increase in temperature of about 1000 °C and a puffy, voluminous solid product was obtained, which was ground to a fine powder and collected.35,36 TiO2 was also prepared following the same procedure of solution combustion synthesis. Initially, titanium isopropoxide (99%, Sigma Aldrich) was completely hydrolysed with water and dissolved in concentrated HNO3 to prepare the titanyl nitrate precursor. Stochiometric amounts of titanyl nitrate and glycine were introduced in a preheated muffle furnace. The resulting voluminous TiO2 powder was ground finely using a mortar and pestle.25,37The heterojunction oxide of ZnO–TiO2 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio was prepared by the reflux method. A desired amount of combustion-synthesized TiO2 was dispersed in 62.5 mL of 0.01 M Zn(CH3COO)2/methanol solution. The mixture was vigorously stirred and subjected to 30 minutes of ultrasonication in order to prepare a homogenous mixture. Then, 32.5 mL of 0.03 M NaOH was added dropwise into the mixture while stirring. Subsequently, the mixture was subjected to reflux at 80 °C and 600 rpm for a duration of 6 hours. The resulting ZnO–TiO2 was filtered and dried overnight in an oven at 70 °C.38
:1 stoichiometric ratio was prepared by the reflux method. A desired amount of combustion-synthesized TiO2 was dispersed in 62.5 mL of 0.01 M Zn(CH3COO)2/methanol solution. The mixture was vigorously stirred and subjected to 30 minutes of ultrasonication in order to prepare a homogenous mixture. Then, 32.5 mL of 0.03 M NaOH was added dropwise into the mixture while stirring. Subsequently, the mixture was subjected to reflux at 80 °C and 600 rpm for a duration of 6 hours. The resulting ZnO–TiO2 was filtered and dried overnight in an oven at 70 °C.38
The synthesis of Co-supported ZnO–TiO2 heterojunction oxide involved loading 3, 5 and 7 wt% Co onto ZnO using the incipient wet-impregnation method. Initially, a precise amount of Co(NO3)2·6H2O was added to ZnO synthesized via combustion and stirred to achieve a uniform mixture. Subsequently, a minute quantity of 0.1 M NaBH4 was gradually added until the solution turned greyish in colour. The mixture was then filtered and dried in an oven for 24 hours. Approximately 90 mg of the prepared Co/ZnO was dispersed in methanol and adjusted to 100 mL in a volumetric flask. Similarly, 90 mg of combustion-synthesized TiO2 was dispersed in 62.5 mL of the aforementioned solution and thoroughly stirred. This mixture underwent ultrasonication for 30 minutes, followed by the addition of 32.5 mL of 0.03 M NaOH dropwise while stirring. The resulting solution was refluxed for 6 hours at 80 °C and 600 rpm. The resulting Co/ZnO–TiO2 was filtered, dried, and ground.39
2.2 Characterisation
The structural characterization of the synthesized ZnO, TiO2, ZnO–TiO2 and Co/ZnO–TiO2 was carried out with a Rigaku Ultima IV X-ray diffractometer (XRD) with Cu Kα radiation (λ = 1.5418 Å) at a scan rate of 1° min−1 with a step size of 0.01°. The average nanocrystal diameters (D) were estimated using Scherrer's formula: D = 0.9λ/Bcosθ, where B is the full-width at half-maximum, λ is the wavelength of the radiation, and θ is the corresponding angle. Energy-dispersive X-ray fluorescence (ED-XRF) was used to study the actual atomic ratio of the synthesized heterojunction and Co-doped heterojunction with the help of an Epsilon 1 PANanalytical instrument. A Microtrac BEL Corp mini-II surface area analyser was used to study the surface area of the synthesized materials. The samples were degassed in vacuum at 200 °C for 2 h prior to the N2 sorption measurements at 77 K. The BET equation was used to estimate the specific surface area. The surface morphology of the synthesized materials was studied using a high-resolution field emission scanning electron microscope (FE-SEM, FEI-ApreoS) at 30 kV acceleration. The surface composition, elemental oxidation state and bonding were explicitly studied by X-ray photoelectron spectra (XPS) recorded on a Thermo Scientific K-Alpha surface-analysis spectrometer housing Al Kα as the X-ray source (1486.6 eV). The instrument was operated at 72 W. The base pressure in the analysing chamber was maintained at 5 × 10−9 mbar. The data profiles were subjected to a nonlinear least-squares curve fitting program with a Gaussian–Lorentzian production function and processed with Avantage software. The binding energy (B.E.) of all XPS data was calibrated versus the standard C 1s peak at 284.85 eV. The electronic properties and band gap of the synthesized materials were studied using solid state UV by a JASCO V-670 UV-visible spectrophotometer, and photoluminescence (PL) spectra were obtained using a JASCO FP-6300 on the synthesized material to determine the recombination rate. The concentration of the key reaction species, surface HO˙, was measured by converting coumarin to umbelliferone under photocatalytic conditions. 15 mg of all the four samples were dispersed in an aqueous solution of coumarin (0.1 mM) in a quartz cuvette and was exposed to UV light irradiation for 120 min. Further, 0.5 g of KCl was added into the light-irradiated suspension and then the suspension was kept in the dark for 12 hours. After the sedimentation of the catalysts, the fluorescence of the clear solution was measured using an excitation wavelength of umbelliferone at 332 nm.
2.3 Photocatalytic degradation of alizarin S
Photodegradation studies were carried out using a cylindrical annular batch photoreactor fitted with a medium pressure mercury vapor lamp of 125 W, which consisted of a double-walled borosilicate immersion well for water circulation around the lamp to maintain the reaction at room temperature. All degradations were performed in an open system to provide enough oxygen for the oxidative degradation of the dye. For a particular batch reaction, the catalyst loading was 25 mg for 100 mL of 10 ppm alizarin Red S. During irradiation, 2 mL of the suspension was collected at regular time intervals, and the progress of photodegradation was monitored by observing the disappearance of the absorption peak of alizarin Red S at 425 nm in the UV-visible absorption spectra recorded using a JASCO V-650 UV-visible spectrophotometer. The photocatalytic degradation studies were repeated three times to provide the error bars.3. Results and discussion
3.1 Bulk, surface and electronic characterisation
The crystallinity and structural information of the synthesised TiO2, ZnO, ZnO–TiO2, and 5% Co/ZnO–TiO2 obtained from the XRD patterns are shown in Fig. 1a. The XRD patterns of the rest of the materials (3%Co/ZnO–TiO2 and 7%Co/ZnO–TiO2) are plotted in Fig. S1a (ESI†). The ZnO synthesized by the solution-combustion method crystallised in a phase-pure wurzite structure with space group P63mc (JCPDS card No. 75-0576). The dominant diffraction peaks at 2θ = 32.8°, 35.3°, and 37.3° corresponding to (100), (002) and (101), respectively, matched well with the literature.40 The XRD pattern of TiO2 is in good agreement with standard anatase TiO2 (JCPDS card No. 21-1272), and there was no impurity corresponding to the rutile or brookite phases.18,41,42 The anatase TiO2 crystallised in a tetragonal crystal system with space group I41/amd. In case of the heterojunction of ZnO–TiO2, the presence of both wurzite ZnO and anatase TiO2 were confirmed with a predominant phase of TiO2. However, in the case of 5% Co/ZnO–TiO2, it has been observed that the peaks of ZnO were predominant than TiO2 and the characteristic peaks of crystalline Co were obscured. A similar phenomenon was also observed for 3% Co/ZnO–TiO2 and 7% Co/ZnO–TiO2. The actual concentration of all the elements including Co was confirmed using XRF and is tabulated in ESI† Table S1. | ||
| Fig. 1 (a) XRD patterns and (b) BET adsorption isotherms of ZnO, TiO2, ZnO–TiO2, 5% Co/ZnO–TiO2. | ||
The specific surface area of the synthesized materials was studied using N2 adsorption–desorption isotherms, as shown in Fig. 1b and Fig. S1b (ESI†). ZnO exhibited a type II isotherm with a surface area of 12.24 m2 g−1, while TiO2, ZnO–TiO2, and 5%Co/ZnO–TiO2 exhibited type IV isotherm with a surface area of 101.7, 104.6 and 100.5 m2 g−1, respectively, indicating their mesoporous nature. The total pore volume was 0.129, 0.156, 0.190, and 0.201 cm3 g−1 for ZnO, TiO2, ZnO–TiO2 and 5%Co/ZnO–TiO2, respectively. 3% and 7% Co/ZnO–TiO2 also exhibited type IV isotherm with a surface area of 145.0 and 137.1 m2 g−1, respectively. Their corresponding pore volume was 0.266 and 0.262 cm3 g−1, respectively. Further, the surface morphology was investigated by FE-SEM at different magnifications and is shown in Fig. 2. TiO2 exhibited a diffused structure with pores on its surface, whereas ZnO had an agglomerated sphere-like morphology. Heterojunction ZnO–TiO2 also exhibited agglomerated spherical ZnO particles deposited on the surface of diffused TiO2. 5% Co/ZnO–TiO2 showed the presence of Co on the diffused ZnO–TiO2 surface. The EDAX mapping in Fig. S2 and S3 (ESI†) confirmed the high distribution of Co, Zn and Ti in the matrix. The HR-TEM images of 5% Co/ZnO–TiO2 shown in Fig. 3a also confirmed the diffused structure of ZnO–TiO2 with Co on its surface. The interplanar distances of 0.23 and 0.24 nm calculated from the fringes shown in Fig. 3(b) and (c) correspond to the (004) plane of anatase TiO2 and the (101) plane of wurtzite ZnO, respectively. The selected area electron diffraction (SAED) pattern in Fig. 3d also corroborates the same.
 | ||
| Fig. 2 FE-SEM images of (a) and (b) TiO2 (c) and (d) ZnO (e) and (f) ZnO–TiO2, (g) and (h) 5% Co/ZnO–TiO2. | ||
 | ||
| Fig. 3 (a–c) HR-TEM images and (d) SAED patterns of 5% Co/ZnO–TiO2. | ||
Further, XPS measurements were used to investigate the surface composition and the chemical state of the synthesised of ZnO, TiO2, ZnO–TiO2 and Co/ZnO–TiO2. Fig. S4a (ESI†) shows the XPS survey spectra containing all the individual elements of Ti, Zn, O, and Co in the synthesized composites. The high-resolution Zn 2p spectra plotted in Fig. 4a exhibit two characteristic peaks from Zn 2p3/2 and Zn 2p1/2 at 1021.5 and 1044.6 eV, respectively, indicating the presence of Zn2+ in the synthesized materials.43–45 The Ti 2p XPS spectra in Fig. 4b also display two peaks at 458.5 and 468.1 eV corresponding to Ti 2p3/2 and Ti 2p1/2 from Ti4+ in the synthesized oxides.38 The Co 2p spectra of 3% Co/ZnO–TiO2, 5% Co/ZnO–TiO2 and 7% Co/ZnO–TiO2 in Fig. S4b (ESI†) exhibit two main peaks corresponding to the spin orbit coupling of Co 2p3/2 and 2p1/2. The deconvoluted spectra of the as-prepared materials exhibited primarily metallic Co (at 778.4 and 794.8 eV) with a minor occurrence of Co2+ (at 780.9 and 798.3 eV). The distribution of metallic to ionic Co is shown in Fig. S4b (ESI†). Three satellite peaks at 784.6, 788.3 and 802.1 eV were also observed in the Co 2p spectra.46,47Fig. 4c shows the high-resolution O 1s core level spectra of the oxide samples. The spectra consist of three deconvoluted peaks. The primary peak at 529.9 eV (designated as OL) corresponds to the lattice oxygen in the synthesized oxides.38,48 The peaks at higher binding energy at 531.3 and 532.6 eV correspond to the surface oxygen (OS) and hydroxyl species attached to the oxygen vacancy sites (OO–H). It was interesting to see that the peaks corresponding to the surface oxygen species as well as the oxygen vacancy sites are significantly prominent in ZnO–TiO2 and 5% Co/ZnO–TiO2 compared to ZnO and TiO2.
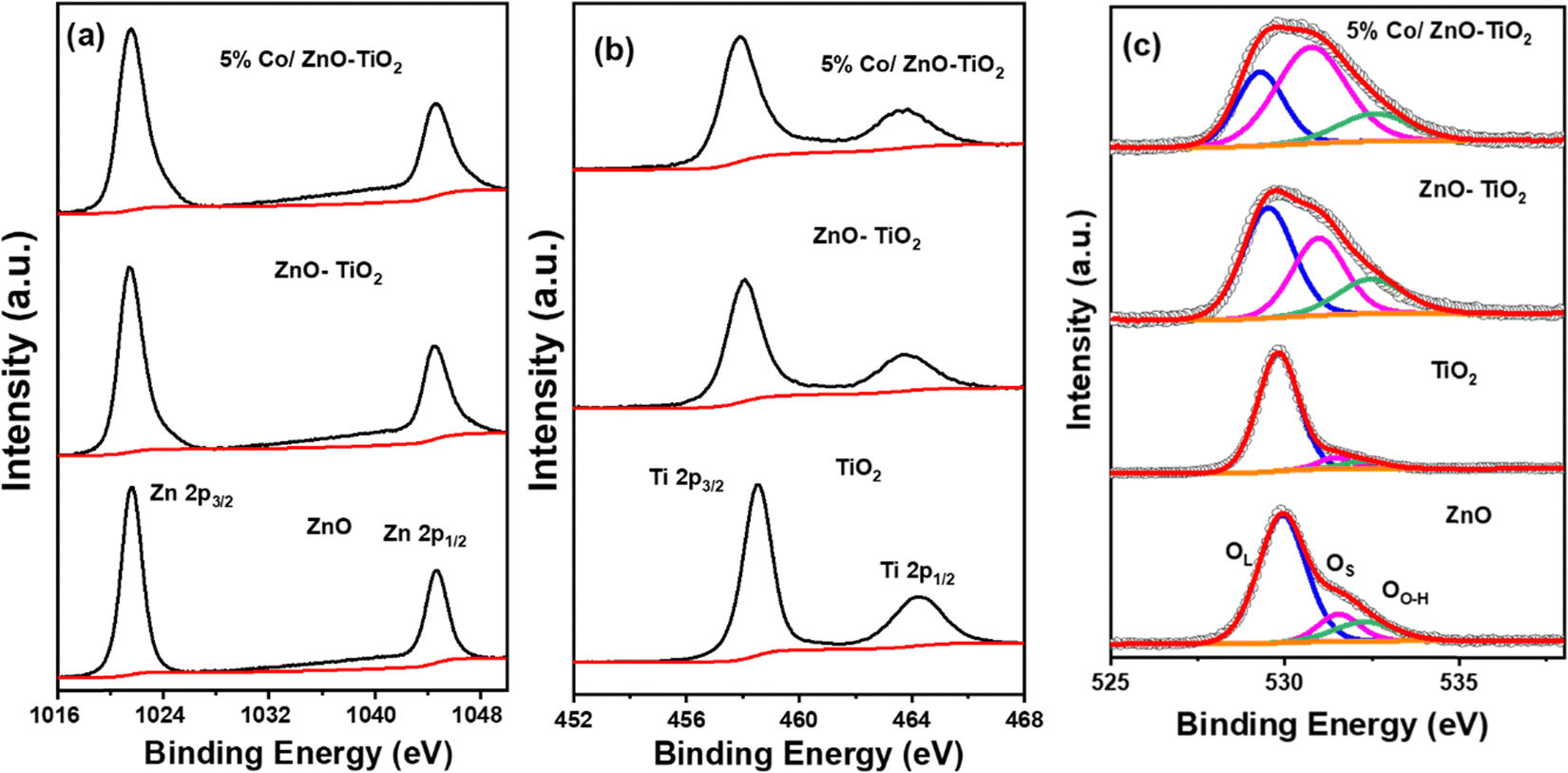 | ||
| Fig. 4 (a) Zn 2p, (b) Ti 2p, and (c) O 1s core level spectra of ZnO, TiO2, ZnO–TiO2, and 5% Co/ZnO–TiO2. | ||
While TiO2 is an indirect semiconductor, ZnO is a direct semiconductor oxide. To explore the optical properties of the synthesized pristine and heterojunction nanomaterials, room temperature UV-visible absorption spectrum was obtained in the wavelength range of 200–800 nm. As shown in Fig. 5a, ZnO exhibits maximum absorbance at 361 nm, while TiO2 shows the maxima at 341 nm. In case of heterojunction ZnO–TiO2 and 5% Co/ZnO–TiO2, the maximum absorbance was observed at 350 nm. This can effectively extend the photoresponses of the system, leading to an improvement of solar light utilization. Further, the band gap of all the synthesized nanomaterials was calculated via the Kubelka–Munk method using the equation  , where R represents the % reflectance and F(R) is the molar extinction coefficient. The plot of (F(R) × hν)2vs. energy in eV in Fig. 5b was adopted for ZnO as it is a direct semiconductor, whereas the plot of (F(R) × hν)1/2vs. energy in Fig. 5c was adopted for indirect semiconductor TiO2. ZnO and TiO2 exhibited a band gap of 3.19 and 3.16 eV, respectively. Heterojunction oxide ZnO–TiO2 is regarded as a type II semiconductor with indirect band gap.49,50 Accordingly, the band gap was calculated and the composite ZnO–TiO2 exhibited a reduced band gap of 2.99 eV, whereas 5% Co/ZnO–TiO2 showed the lowest band gap of 1.4 eV among the four materials. Evidently, the heterojunction indeed reduced the optical band gap of the materials, and the presence of Co helped in the further reduction of the band gap.51,52 The absorbance of incident irradiation over TiO2 occurs due to the electronic transition from O 2p to Ti 3d, whereas for ZnO, the absorbance corresponds to the electronic transition between the O 2p valence band and the Zn 4s conduction band.53,54 The reduction in the band gap for the heterojunction can be attributed to the settling of the TiO2 particles over the ZnO structure, which is known to scatter the incident light.34,35 The lowering of band gap can also be attributed to the microstates originated from the oxygen vacancies formed in the heterojunction oxide, as evident from the XPS studies.28,55
, where R represents the % reflectance and F(R) is the molar extinction coefficient. The plot of (F(R) × hν)2vs. energy in eV in Fig. 5b was adopted for ZnO as it is a direct semiconductor, whereas the plot of (F(R) × hν)1/2vs. energy in Fig. 5c was adopted for indirect semiconductor TiO2. ZnO and TiO2 exhibited a band gap of 3.19 and 3.16 eV, respectively. Heterojunction oxide ZnO–TiO2 is regarded as a type II semiconductor with indirect band gap.49,50 Accordingly, the band gap was calculated and the composite ZnO–TiO2 exhibited a reduced band gap of 2.99 eV, whereas 5% Co/ZnO–TiO2 showed the lowest band gap of 1.4 eV among the four materials. Evidently, the heterojunction indeed reduced the optical band gap of the materials, and the presence of Co helped in the further reduction of the band gap.51,52 The absorbance of incident irradiation over TiO2 occurs due to the electronic transition from O 2p to Ti 3d, whereas for ZnO, the absorbance corresponds to the electronic transition between the O 2p valence band and the Zn 4s conduction band.53,54 The reduction in the band gap for the heterojunction can be attributed to the settling of the TiO2 particles over the ZnO structure, which is known to scatter the incident light.34,35 The lowering of band gap can also be attributed to the microstates originated from the oxygen vacancies formed in the heterojunction oxide, as evident from the XPS studies.28,55
 | ||
| Fig. 5 (a) UV-visible spectra and band gap elucidation of (b) ZnO, (c) TiO2, ZnO–TiO2 and 5% Co/ZnO–TiO2. | ||
The photocatalytic activity of a catalyst is also largely determined by the efficiency of the photogenerated electron–hole separation. Since photoluminescence originates from the recombination of charge carriers, the PL spectra are highly informative to study the recombination or trapping of charge carriers. Fig. 6a shows the PL spectra for the pristine ZnO, TiO2, and the nanocomposite of ZnO–TiO2 and 5% Co/ZnO–TiO2 exhibiting emission in the range of 350–600 nm. As observed from the PL spectra, the highest intensity corresponds to TiO2, followed by ZnO. As compared to pristine TiO2 and ZnO, the nano-composites exhibited a fairly low PL intensity. The lower PL intensity vis-à-vis the lower recombination rate of the photogenerated charge carriers indicated a longer separated carrier lifetime, which in turn promotes the photocatalytic activity of the nanocomposite heterojunction material. The enhanced life-time of the exciton over 5% Co/ZnO–TiO2 is primarily due to the formed heterojunction, which promotes efficient charge transfer, thereby increasing the quantum efficiency of the photocatalytic reaction.56,57
 | ||
| Fig. 6 (a) PL spectra of ZnO, TiO2, ZnO–TiO2 and 5% Co/ZnO–TiO2. (b) Concentration of umbelliferone with respect to light irradiation time in the presence of ZnO, TiO2, ZnO–TiO2, and 5% Co/ZnO–TiO2. | ||
Thus, the lower recombination rate of excitons might have helped the long-lived h+ to produce more OH˙, which is the key for the degradation mechanism. Therefore, we carried out an experiment to detect the photogenerated OH˙ species under UV light irradiation over the synthesized materials with the help of coumarin as a fluorescent probe. Coumarin reacts with photogenerated OH˙ to produce 7-OH coumarin (umbelliferone), which is fluorescent in nature and can be probed with PL against the time of irradiation. All the four materials of 15 mg weight were added to 3.5 mL of 0.1 mM coumarin solution in a quartz cuvette, and UV light was irradiated for 120 min. After irradiation, 0.5 g of KCl was added into the suspension and kept in the dark for 12 hours to get a clear solution. The formed umbelliferone was measured using PL spectroscopy. The generation of umbelliferone against time over the synthesized materials is plotted in Fig. 6b, and it is evident from the data that the concentration of 7-OH coumarin is the highest over 5% Co/ZnO–TiO2. Therefore, it can be inferred that 5% Co/ZnO–TiO2 showed the least rate of electron–hole recombination, which could be beneficial for the heterojunction nanocomposite material for catalytic performances.
3.2 Photocatalytic degradation of alizarin S
The photocatalytic activity of the synthesized materials was determined by conducting experiments for the degradation of alizarin S dye under UV light illumination, and the concentration of the degraded dye was measured from the standard graph derived from the UV-Vis absorption spectrum of the dye (Fig. S5, ESI†). It should be noted that the UV-Vis spectrum of alizarin S contains multiple peaks involving the excitation from n or π to π*, and the transitions being associated with a significant charge-transfer phenomenon rationalizes the intense colour of alizarin S.Adsorption plays a significant role in the catalytic reaction mechanism irrespective of the photocatalysis. Efficient adsorption provides higher retention time and better interaction between the reactants and the catalyst. At first, the dark adsorption of alizarin S at room temperature was performed for an hour, and the corresponding UV-visible spectra are shown in Fig. S6 (ESI†). The relative concentration C/C0 derived from the UV-visible spectra in the dark for 60 min is plotted in Fig. 7a and Fig. S8a (ESI†). Interestingly, ZnO adsorbed alizarin S better than the synthesized TiO2. Among the synthesized materials, 5% Co/ZnO–TiO2 exhibited the best adsorption behaviour. However, the effective adsorption equilibrium was reached by ∼30 min over all the four materials in the dark.
 | ||
| Fig. 7 (a) Adsorption of alizarin S over ZnO, TiO2, ZnO–TiO2, and 5% Co/ZnO–TiO2 in dark. (b) alizarin S photodegradation over ZnO, TiO2, ZnO–TiO2, 5% Co/ZnO–TiO2 and also in the absence of any catalyst. (c) Photocatalytic degradation of alizarin S over P25 TiO2 and 5% Co/ZnO. (d) Degradation kinetics of alizarin S following the pseudo first-order reaction kinetics. | ||
Therefore, the optimized time of 30 minutes for effective adsorption equilibrium was considered for the photocatalytic degradation of alizarin S over ZnO, TiO2, ZnO–TiO2, and Co/ZnO–TiO2 catalysts. The corresponding UV-visible spectra in the wavelength range of 200–700 nm is shown in Fig. S7 (ESI†), and the relative concentration C/C0 derived from the UV-visible spectra under the exposure of light is plotted in Fig. 7b. Evidently, in the absence of any catalyst, the degradation was only 20% after 2 hours of irradiation. On irradiating the dye in the presence of TiO2 and ZnO, the percentage of degradation increased to 60 and 80%, respectively. It must be noted that the higher adsorption capability of ZnO coupled with the lower optical band gap and reduced electron–hole recombination facilitated ZnO to outperform TiO2. The presence of heterojunction ZnO–TiO2 exhibited a further increase in the photodegradation of alizarin S to 90%. The enhanced photocatalytic activity of the ZnO–TiO2 heterojunction compared to the single oxides can be explained via the established mechanism of type II heterojunction between ZnO and TiO2.58 The photocatalytic performance of Co/ZnO–TiO2 catalysts with varying Co concentrations is illustrated in Fig. S8b (ESI†). Notably, 5% Co/ZnO–TiO2 exhibited the highest catalytic efficiency, achieving the complete degradation of alizarin S within just 30 minutes of irradiation. In contrast, 3% Co/ZnO–TiO2 and 7% Co/ZnO–TiO2 required over 90 minutes for complete degradation. These findings indicate that optimal 5 wt% of Co doping in ZnO–TiO2 enhances the photocatalytic activity by improving the efficiency of the electron–hole pair separation. Further, 5% Co–ZnO and P25 TiO2 were also compared with 5% Co/ZnO–TiO2 (Fig. 7c). While P25 TiO2 and 5% Co–ZnO achieved only 49.9% and 71.5% photocatalytic degradation of alizarin S within 120 minutes, respectively, 5% Co/ZnO–TiO2 achieved complete degradation within just 30 minutes of irradiation, confirming the superiority of the composite catalyst. The wider band gap of P25 TiO2, less adsorption capacity and low OH˙ species on the catalyst surface might have diminished its photocatalytic performance towards alizarin S. Additionally, the quantum yield (QY) efficiency of the optimized 5% Co–ZnO/TiO2 catalyst was calculated using the following equation
 C0/C and irradiation time (t) was also plotted in Fig. 7d. Here, C0 is the initial concentration of the dye and C is the concentration of the dye at time (t). The linear relationship between ln(C0/C) and t demonstrated that the photocatalytic degradation of alizarin S followed pseudo first-order kinetics, and the slope (k) is the reaction rate constant (min−1). The rate constant was calculated to be 0.020 min−1, 0.013 min−1, 0.004 min−1, and 0.007 min−1 for 5% Co/ZnO–TiO2, ZnO–TiO2, TiO2 and ZnO, respectively.
C0/C and irradiation time (t) was also plotted in Fig. 7d. Here, C0 is the initial concentration of the dye and C is the concentration of the dye at time (t). The linear relationship between ln(C0/C) and t demonstrated that the photocatalytic degradation of alizarin S followed pseudo first-order kinetics, and the slope (k) is the reaction rate constant (min−1). The rate constant was calculated to be 0.020 min−1, 0.013 min−1, 0.004 min−1, and 0.007 min−1 for 5% Co/ZnO–TiO2, ZnO–TiO2, TiO2 and ZnO, respectively.
The degradation of the dye alizarin S over the optimized 5% Co/ZnO–TiO2 was further assessed by identifying the intermediates formed during the reaction. Samples were analysed using the HR-MS technique, and the spectra are presented in Fig. S9 (ESI†), while a plausible degradation pathway of alizarin red S is depicted in Fig. 8. The mass spectra of alizarin red S, taken after 5 minutes of degradation, revealed major peaks at m/z = 338.34, corresponding to the radical formation of alizarin S (labelled as “a” in Fig. 8). After 10 minutes of degradation, peaks with m/z = 288.2 and 261.1, corresponding to intermediates “b” and “d,” respectively, were observed. At the 15th minute, a peak with m/z value = 165.0, corresponding to intermediate “c,” was detected. Further degradation was observed after 30 minutes, with the peaks at m/z = 141.9 and 182.9 corresponding to intermediates “e” and “f,” respectively.
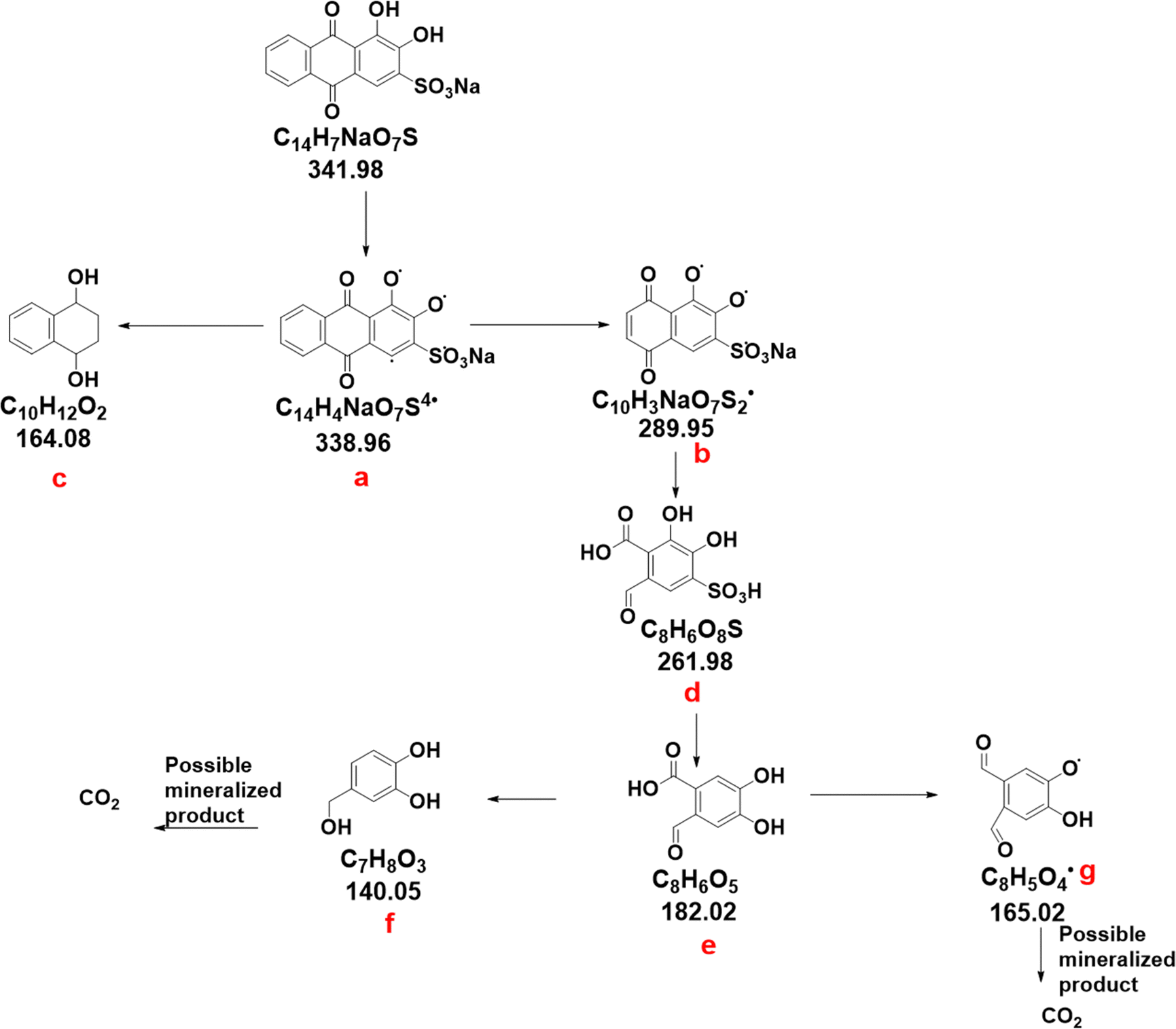 | ||
| Fig. 8 The intermediates observed in HR-MS during the photocatalytic degradation of alizarin S using 5% Co/ZnO–TiO2 catalyst. | ||
To verify the catalyst's reproducibility, we conducted three cycles of degradation studies with the exhausted 5% Co/ZnO–TiO2 catalyst (Fig. 9a). The first cycle achieved approximately 90.9% degradation of alizarin S, followed by 84.1% and 71.0% in subsequent cycles. The decline in the degradation can be attributed to active site blockage from dye intermediate adsorption and loss of catalyst particles during collection and rinsing.59 After completing the three cycles of photocatalytic degradation, the exhausted catalyst was dried and the powder XRD data was collected to assess the structural stability (Fig. 9b). XRD patterns indicate no structural difference in the exhausted 5%Co/ZnO–TiO2 compared to the as-prepared material, except for the reduced crystallinity.
 | ||
| Fig. 9 (a) Cycle study of 5% Co/ZnO–TiO2. (b) XRD pattern of the exhausted catalyst. | ||
To understand the catalytic reactivity in terms of the band structure, we evaluated the position of the VB maxima from the onset of the VB XPS spectra, as shown in Fig. S10 (ESI†). The BE of the on-set edge divulges the energy gap between the VB maxima and the Fermi level, and for all practical purposes, the valence band spectra obtained from XPS coincides with the density of states. The VB maxima position of TiO2 and ZnO in vacuum with respect to the eV scale was found to be 2.48 eV and 2.30 eV, respectively. Therefore, the CB minima levels were calculated using the equation ECB = EVB + Eg and were found to be −0.65 and −0.89 eV, respectively, over TiO2 and Co/ZnO. The enhanced photocatalytic activity of the Co/ZnO–TiO2 heterojunction compared to the single oxides can be explained via the established mechanism of type II heterojunction between Co/ZnO and TiO2. Fig. 10 illustrates the scheme of the photodegradation mechanism of alizarin S. On light illumination, the electrons in the CB of TiO2 easily flow into Co (electron transfer I) through the Schottky barrier because the CB (or the Fermi level) of TiO2 is higher than that of the Co. This process of electron transfer I is faster than the electron–hole recombination between the VB and the CB of TiO2, as we have evidenced from the PL studies. Consequently, holes with a strong oxidation power in the VB of TiO2 can form OH˙, which helps in oxidizing alizarin S. Conversely, as the energy level of Co is above the VB of ZnO, the holes in the VB of ZnO also flow easily into the metal (electron transfer II), which is faster than the electron–hole recombination between the VB and CB of ZnO. The electrons accumulated in the CB of ZnO can react with oxygen molecules, which are absorbed on the surface and produce the superoxide radical (O2−•). Hence, simultaneous electron transfers I and II (illustrated as vectorial electron transfer from TiO2 → Co → ZnO in Fig. 10) can take place upon light irradiation. In these processes, Co within multicomponent heterojunction systems serves as a storage or recombination center for electrons in the CB of TiO2 and holes in the VB of ZnO, thereby enhancing the interfacial charge transfer and facilitating the complete separation of holes in the VB of TiO2 and electrons in the CB of ZnO. As a result, the multicomponent heterojunction systems greatly enhance the photocatalytic activity compared with a single semiconductor or semiconductor heterojunctions. The superior behaviour of 5% Co/ZnO–TiO2 can be rationalized by the following features: (i) lowest optical band gap of the nano-composite to increase the photo-efficacy, as observed in the DRS studies, (ii) lowest recombination rate of the exciton pair due to the heterojunction, as evident from the PL spectra, and (iii) the highest concentration of photogenerated OH˙, as observed from the reaction with coumarin. The existence of Co acted as a medium for faster and more efficient transfer of electrons (and holes) between the semiconductors.57,60 The combinatorial effect of superior charge transition between the multicomponent heterojunction due to Co and the long-lived key species O2−˙ and OH˙ formed due to h+ made 5% Co/ZnO–TiO2 an exceptional photocatalyst for the degradation of a dye like alizarin S.
 | ||
| Fig. 10 Schematic illustration of the mechanism of photodegradation of alizarin S using the multicomponent 5% Co/ZnO–TiO2 catalyst. | ||
To corroborate the photodegradation mechanism of alizarin S over the multicomponent heterojunction catalyst, we further studied the photocatalysis using hole scavengers such as methanol and electron scavenger such as H2O2, as depicted in Fig. 11. Methanol, acting as a hole scavenger, can intercept photogenerated holes before they react with water, forming oxidized products instead of OH˙. Apparently, the results demonstrated a significant reduction in the degradation efficacy in the presence of the hole scavenger methanol. In the presence of methanol, ˙OH radicals react with it, abstracting a hydrogen atom to form formaldehyde (HCHO) in the presence of oxygen, as shown in the equation below. Consequently, the availability of ˙OH radicals for reacting with adsorbed molecules decreases, leading to a reduction in the degradation percentage.61 This outcome confirms the indispensable role of ˙OH in the photocatalytic degradation of alizarin S.
| ˙OH + CH3OH → ˙CH2OH + H2O |
| ˙CH2OH + O2 → HCHO + HO2˙ |
 | ||
| Fig. 11 Photocatalytic degradation of 5% Co/ZnO–TiO2 in the presence of holes and electron scavengers. | ||
It has been reported that the utilization of inorganic oxidants, such as H2O2, enhances the quantum efficiencies by either hindering electron–hole pair recombination through scavenging conduction band electrons at the semiconductor surface or by providing an additional oxygen atom as an electron acceptor to generate the superoxide radical ion.62 In the case of the electron scavenger H2O2, its addition has been found to increase the degradation of alizarin S. When H2O2 is introduced into the system, it can trap electrons, thereby preventing their recombination with holes, consequently promoting the formation of ˙OH on the photocatalyst's surface.61,63,64
| O2−˙ + H2O2 → ˙OH + OH− + O2 |
| e− + H2O2 → ˙OH + OH− |
4. Conclusion
ZnO and TiO2 were synthesized successfully by the solution-combustion method, whereas a simple reflux method was adopted for the synthesis of the nano-composites. Among the materials, 5% Co/ZnO–TiO2 exhibited an appreciable photocatalytic degradation of alizarin S. Upon light illumination, the electrons in the conduction band and h+ in the VB through a series of redox reactions with the solvent water result in active oxygen species like hydroxyl radicals OH˙ that help in mineralizing alizarin S. The (i) optimum band gap, (ii) the higher flux of holes in the valence band, (iii) the lower recombination rate of excitons, and (iv) the high rate of formation of OH˙ species on the catalyst surface were found to be the crucial factors for the high photocatalytic efficacy of the material.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
Authors thank BITS Pilani Hyderabad Campus for the financial aid for this work.References
- F. M. Machado, S. A. Carmalin, E. C. Lima, S. L. P. Dias, L. D. T. Prola, C. Saucier, I. M. Jauris, I. Zanella and S. B. Fagan, J. Phys. Chem. C, 2016, 120, 18296–18306 CrossRef CAS
.
- H. Puchtler, S. N. Meloan, M. S. Terry and S. Terry, J. Histochem. Cytochem., 1969, 17, 110–124 CrossRef CAS PubMed
- A. Amat, C. Miliani, A. Romani and S. Fantacci, Phys. Chem. Chem. Phys., 2015, 17, 6374–6382 RSC
- S. Sood, S. K. Mehta, A. Umar and S. K. Kansal, New J. Chem., 2014, 38, 3127–3136 RSC
- M. Baruah, A. Supong, P. Chandra, B. Rituparna and K. Chubaakum, Nanotechnol. Environ. Eng., 2020, 5, 1–13 CrossRef
- S. Akshatha, S. Sreenivasa, L. Parashuram, V. U. Kumar, S. C. Sharma, H. Nagabhushana, K. Sandeep and T. Maiyalagan, J. Environ. Chem. Eng., 2019, 7, 103053–103065 CrossRef CAS
- L. Jiang, J. Li, K. Wang, G. Zhang, Y. Li and X. Wu, Appl. Catal., B, 2020, 260, 118181–118191 CrossRef CAS
- A. B. Albadarin and C. Mangwandi, J. Environ. Manage., 2015, 164, 86–93 CrossRef CAS PubMed
- Z. Wang, K. Wang, Y. Li, L. Jiang and G. Zhang, Appl. Surf. Sci., 2019, 498, 143850–143860 CrossRef CAS
- S. Roy, J. Phys. Chem. C, 2020, 124, 28345–28358 CrossRef CAS
- K. Mamun, R. Asw and K. Fahmida, Appl. Water Sci., 2017, 7, 1569–1578 CrossRef
- M. A. Rauf and S. S. Ashraf, Chem. Eng. J., 2009, 151, 10–18 CrossRef CAS
- Q. Guo, C. Zhou, Z. Ma and X. Yang, Adv. Mater., 2019, 31, 1901997–1902023 CrossRef CAS PubMed
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS
- H. Kumari, S. Suman, R. Ranga and S. Chahal, Water, Air, Soil Pollut., 2023, 234, 349–375 CrossRef CAS PubMed
- S. Challagulla, K. Tarafder, R. Ganesan and S. Roy, Sci. Rep., 2018, 7, 8783–8794 CrossRef PubMed
- A. L. Linsebigler, G. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS
- S. Challagulla, K. Tarafder, R. Ganesan and S. Roy, J. Phys. Chem. C, 2017, 121, 27406–27416 CrossRef CAS
- H. Lachheb, E. Puzenat, A. Houas, M. Ksibi, E. Elaloui, C. Guillard and J. Herrmann, Appl. Catal., B, 2002, 39, 75–90 CrossRef CAS
- M. Vautier, C. Guillard and J. Herrmann, J. Catal., 2001, 201, 46–59 CrossRef CAS
- S. Payra, S. Challagulla, Y. Bobde, C. Chakraborty, B. Ghosh and S. Roy, J. Hazard. Mater., 2019, 373, 377–388 CrossRef CAS PubMed
- D. A. Kuznetsov, B. Han, Y. Yu, R. R. Rao, J. Hwang, Y. Román-Leshkov and Y. Shao-Horn, Joule, 2018, 2, 225–244 CrossRef CAS
- S. Challagulla and S. Roy, J. Mater. Res., 2017, 32, 2764–2772 CrossRef CAS
- S. Challagulla, R. Nagarjuna, R. Ganesan and S. Roy, ACS Sustainable Chem. Eng., 2016, 4, 974–982 CrossRef CAS
- B. Soman, S. Challagulla, S. Payra, S. Dinda and S. Roy, Res. Chem. Intermed., 2018, 44, 2261–2273 CrossRef CAS
- G. K. Upadhyay, J. K. Rajput, T. K. Pathak, V. Kumar and L. P. Purohit, Vacuum, 2019, 160, 154–163 CrossRef CAS
- N. Bai, X. Liu, Z. Li, X. Ke, K. Zhang and Q. Wu, J. Sol-Gel Sci. Technol., 2021, 99, 92–100 CrossRef CAS
- H. Mousa, J. Alenezi Fares, I. M. A. Mohamed, A. S. Yasin, A.-F. M. Hashem and A. Abdal-hay, J. Alloys Compd., 2021, 886, 161169–161181 CrossRef CAS
- S. Hernández, D. Hidalgo, A. Sacco, A. Chiodoni, A. Lamberti, V. Cauda, E. Tresso and G. Saracco, Phys. Chem. Chem. Phys., 2015, 17, 7775–7786 RSC
- B. Bethi, S. H. Sonawane, B. A. Bhanvase and S. P. Gumfekar, Chem. Eng. Process., 2016, 109, 178–189 CrossRef CAS
- K. Wang, Y. Li, J. Li and G. Zhang, Appl. Catal., B, 2020, 263, 117730–117740 CrossRef CAS
- W. Wen and J. Wu, RSC Adv., 2014, 4, 58090–58100 RSC
- K. Rajeshwar and N. R. De Tacconi, Chem. Soc. Rev., 2009, 38, 1984–1998 RSC
- A. Varma, A. S. Mukasyan, A. S. Rogachev and K. V. Manukyan, Chem. Rev., 2016, 116, 14493–14586 CrossRef CAS PubMed
- S. Payra, S. K. Ganeshan, S. Challagulla and S. Roy, Adv. Powder Technol., 2020, 31, 510–520 CrossRef CAS
- S. Shraavan, S. Challagulla, S. Banerjee and S. Roy, Bull. Mater. Sci., 2017, 40, 1415–1420 CrossRef CAS
- S. Challagulla, R. Nagarjuna, R. Ganesan and S. Roy, Nano-Struct. Nano-Objects, 2017, 12, 147–156 CrossRef CAS
- C. Sun, Q. Xu, Y. Xie, Y. Ling and Y. Hou, J. Mater. Chem. A, 2018, 6, 8289–8298 RSC
- K. Xu, Z. Liu, S. Qi, Z. Yin, S. Deng, M. Zhang and Z. Sun, RSC Adv., 2020, 10, 34702–34711 RSC
- S. Payra and S. Roy, J. Phys. Chem. C, 2021, 125, 8497–8507 CrossRef CAS
- P. Ganji and S. Roy, Energy Fuels, 2019, 33, 5293–5303 CrossRef CAS
- S. Roy, S. Payra, S. Challagulla, R. Arora, S. Roy and C. Chakraborty, ACS Omega, 2018, 3, 17778–17788 CrossRef CAS PubMed
- M. Claros, M. Setka, Y. P. Jimenez and S. Vallejos, Nanomaterials, 2020, 10, 471–487 CrossRef CAS PubMed
- A. Abdalla, S. Bereznev, N. Spalatu, O. Volobujeva and N. Sleptsuk, Sci. Rep., 2019, 9, 17443–17453 CrossRef PubMed
- D. Xu, D. Fan and W. Shen, Nanoscale Res. Lett., 2013, 8, 46–55 CrossRef PubMed
- B. Eren and A. R. Head, J. Phys. Chem. C, 2020, 124, 3557–3563 CrossRef CAS
- R. Riva, H. Miessner, R. Vitali and G. Del Piero, Appl. Catal., A, 2000, 196, 111–123 CrossRef CAS
- P. C. Meenu and S. Roy, ACS Appl. Mater. Interfaces, 2023, 15, 36154–36166 CrossRef PubMed
- K. Xu, Z. Liu, S. Qi, Z. Yin, S. Deng, M. Zhang and Z. Sun, RSC Adv., 2020, 10, 34702–34711 RSC
- W. Sun, S. Meng, S. Zhang, X. Zheng, X. Ye, X. Fu and S. Chen, J. Phys. Chem. C, 2018, 122, 15409–15420 CrossRef CAS
- X. Ma, L. Xue, X. Li, M. Yang and Y. Yan, Ceram. Int., 2015, 41, 11927–11935 CrossRef CAS
- I. Tunc, M. Bruns, H. Gliemann and P. Koelsch, Surf. Interface Anal., 2010, 42, 835–841 CrossRef CAS
- A. Mohamad Azmin, M. Hassan Syafiq, M. Yakoob Kamil, M. Taib Mohamad Fariz, F. Badrudin Wafi, O. Hassan Hasdinir and M. Yahya Azhan Zu, J. King Saud Univ., Eng. Sci., 2017, 29, 278–283 Search PubMed
- J. J. Reinosa, C. María, Á. Docio and V. Z. Ramírez, Ceram. Int., 2018, 44, 2827–2834 CrossRef CAS
- A. Das, P. M. Kumar, M. Bhagavathiachari and R. G. Nair, Appl. Surf. Sci., 2020, 534, 147321–147355 CrossRef CAS
- K. Jug and V. A. Tikhomirov, J. Phys. Chem. A, 2009, 113, 11651–11655 CrossRef CAS PubMed
- Y. Lu, Y. Lin, D. Wang, L. Wang, T. Xie and T. Jiang, Nano Res., 2011, 4, 1144–1152 CrossRef CAS
- Z. Wang, X. Ning, Y. Feng, R. Zhang, Y. He, H. Zhao, J. Chen, P. Du and X. Lu, Anal. Chem., 2022, 94, 8539–8546 CrossRef CAS PubMed
- X. Cui, Y. Wang, G. Jiang, Z. Zhao, C. Xu, A. Duan, J. Liu, Y. Wei and W. Bai, J. Mater. Chem. A, 2014, 2, 20939–20946 RSC
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC
- A. N. Ökte, M. S. Resat and Y. Inel, Toxicol. Environ. Chem., 2001, 79, 171–178 CrossRef
- T.-L. T. Le, T.-H. T. Le, K. Nguyen Van, H. Van Bui, T. G. Le and V. Vo, J. Sci.: Adv. Mater. Devices, 2021, 6, 516–527 CAS
- O. Makota, E. Dutková, J. Briančin, J. Bednarcik, M. Lisnichuk, I. Yevchuk and I. Melnyk, Molecules, 2024, 29, 1190–1208 CrossRef CAS PubMed
- D. Saha, M. M. Desipio, T. J. Hoinkis, E. J. Smeltz, R. Thorpe, D. K. Hensley, S. G. Fischer-Drowos and J. Chen, J. Environ. Chem. Eng., 2018, 6, 4927–4936 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: XRF, EDX mapping, XPS survey spectra, VB spectra, UV-visible spectra in dark condition and light condition, degradation studies, HR-MS are provided. See DOI: https://doi.org/10.1039/d4nj00407h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |