
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, reactivity and coordination behaviour of a ferrocene phosphinostibine and intramolecular interactions in its P(V) and Sb(V) derivatives†
Jakub
Antala
 ,
Jiří
Schulz
,
Ivana
Císařová
and
Petr
Štěpnička
*
,
Jiří
Schulz
,
Ivana
Císařová
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, Prague 128 40, Czech Republic. E-mail: stepnic@natur.cuni.cz
First published on 16th February 2024
Abstract
Compared to their P,N-analogues, compounds combining P and Sb substituents remain less common. This contribution describes the synthesis of a new ferrocene phosphinostibine, Cy2PfcSbPh2 (3; fc = ferrocene-1,1′-diyl, Cy = cyclohexyl), and its derivatives modified at the phosphine and stibine moieties, viz., Cy2P(E)fcSbPh2 (E = BH3, O, S, AuCl), Cy2P(E)fcSbCl2Ph2 (E = BH3, S, AuCl) and stiboranes Cy2P(E)fcSb(O2C6Cl4)Ph2 (E = void, BH3, O, S and AuCl). The increased Lewis acidity of the Sb atom in the catecholatostiboranes Cy2PfcSb(O2C6Cl4)Ph2 and Cy2P(O)fcSb(O2C6Cl4)Ph2 resulted in intramolecular P → Sb and O → Sb dative interactions, which were not detected for the respective parent stibines or even in Cy2P(S)fcSb(O2C6Cl4)Ph2. While the P → Sb interaction in Cy2PfcSb(O2C6Cl4)Ph2 was stronger than that in the “all-phenyl” analogue Ph2PfcSb(O2C6Cl4)Ph2 due to the electron-donating effect of the cyclohexyl groups, the bonding situations in Cy2P(O)fcSb(O2C6Cl4)Ph2 and Ph2P(O)fcSb(O2C6Cl4)Ph2, where the substituent effect was only indirect, remained very similar according to DFT calculations. A coordination study with 3 resulted in isolation of phosphine ([AuCl(3-κP)]), P,Sb-bridging ([(μ(P,Sb)-3)(AuCl)2]), and P,Sb-chelate ([(arene)MCl(3-κ2P,Sb)]X, where (arene)M/X = (η6-p-cymene)Ru(II)/PF6, (η5-C5Me5)Rh(III)/Cl, and (η5-C5Me5)Rh(III)/PF6, and [MCl2(3-κ2P,Sb)], where M = Pd and Pt) complexes. For some of these compounds, structure determination revealed structural distortions suggesting weak intramolecular Cl–Sb interactions, which were confirmed by theoretical methods.
Introduction
Stibine ligands have long been overlooked in favour of the ubiquitous phosphines.1 This situation was possibly influenced by the simplified notion that stibines are simple phosphine analogues, albeit less stable (due to the weaker Sb–C bonds) and weaker coordinating ligands. Although many parallels can be found between the chemistries of phosphine and stibine ligands, stibines exhibit additional peculiar features that make them attractive, such as changed stereochemical profiles (typically lower steric demands than for the corresponding phosphines), increased Lewis acidity, and facile conversion into Lewis acidic stiboranes.2 In the chemistry of ferrocene ligands,3 compounds bearing stibine substituents were long limited to (diphenylstibino)ferrocene derivatives bearing donor groups at position 2 of the ferrocene scaffold, which were studied for possible Sb-donor interactions.4,5Recently, we synthesised 1,1′-bis(diphenylstibino)ferrocene (1 in Scheme 1),6 which is a direct analogue of the widely studied 1,1′-bis(diphenyphosphino)ferrocene (dppf),7 and explored the reactivity and coordination behaviour of this compound.6 In the following research, we focused on the analogous mixed-donor derivative 1-(diphenylphoshino)-1′-(diphenylstibino)ferrocene (2) and its P(V)- and Sb(V)-derivates.8 These compounds were studied mainly because of possible intramolecular dative interactions between the P- and Sb-substituents, which were indeed identified in stiboranes resulting from oxidation of the stibine moiety. Unfortunately, studies on the coordination behaviour of 2 were hampered by the tendency of this compound to form disordered structures, in which the phosphine and stibine groups alternated in their positions.
 | ||
| Scheme 1 Structures of dppf and compounds 1–3 (Cy = cyclohexyl). | ||
To circumvent this problem, we synthesised desymmetrised derivative 3 bearing cyclohexyl (Cy) substituents at the phosphorus atom. This compound allowed us to not only study the coordination properties of this hybrid P,Sb-ligand but also elucidate the influence of the phosphine substituents on the interactions between the functional groups in 3 and its oxidised derivatives. In particular, we report here the synthesis of 3 and analogous compounds with P(V) or Sb(V) substituents, detailed experimental and theoretical characterisation of these compounds focused on interactions between the pnictogen substituents and a study of the coordination behaviour of 3 as a new, hybrid P,Sb-ligand.9
Results and discussion
Preparation and reactivity of compound 3
Phosphinostibine 3 was obtained by sequential lithiation/functionalisation of 1,1′-dibromoferrocene10,11 (Scheme 2) and was isolated as an air-stable orange solid in approximately 40% yield over the two reaction steps after final crystallisation. Attempted synthesis of 3 using a “one-pot” approach (i.e., without isolation of intermediate 4) proved unsuitable because the isolated crude product contained substantial amounts of monosubstituted side products FcSbPh2 and FcPCy2 (Fc = ferrocenyl), which could not be efficiently removed by chromatography and crystallisation. | ||
| Scheme 2 Synthesis of 3 and further modifications to the phosphine moiety (Cy = cyclohexyl, tht = tetrahydrothiophene) | ||
To illustrate the different reactivities of the two pnictogen substituents, compound 3 was treated with elemental sulfur (1 equiv.) and BH3·SMe2 (1.5 equiv.). Under such conditions, only the phosphine moiety reacted to give phosphine sulfide 3S and adduct 3·BH3. Synthesis of the corresponding phosphine oxide 3O could not be similarly performed (e.g., with H2O2 as the usual oxidant) because of unwanted reactions at the stibine moiety. Alternatively, this compound was obtained similarly to 3 through lithiation of precursor 4O, which had a preinstalled phosphine oxide moiety, followed by subsequent reaction with ClSbPh2. The reaction between 3 and [AuCl(tht)] (1 equiv., tht = tetrahydrothiophene) afforded a phosphine complex [AuCl(3-κP)] (5) as the sole product, in line with our previous observations.8
All the compounds were characterised by a combination of multinuclear NMR spectroscopy, electrospray ionisation (ESI) mass spectrometry, and elemental analysis, and in most cases, the solid-state structures were established by single-crystal X-ray diffraction analysis. The NMR spectra showed signals of the asymmetrically 1,1′-disubstituted ferrocene unit and characteristic signals of the cyclohexyl12 and phenyl substituents at the pnictogen groups (especially in the 13C{1H} NMR spectra). The manipulation of the phosphine substituent was reflected in the 31P{1H} NMR spectra (Table 1). Of particular note are the similarities in the 31P NMR shifts determined for 3E (E = void, BH3, O, and S) and 5 to those for analogous “simple” compounds such as (dicyclohexylphosphino)ferrocene (δP −6.0) and its P-oxide (δP 46.8),13 1,1′-bis(dicyclohexylphosphinothioyl)ferrocene (fc(P(S)Cy2)2; fc = ferrocene-1,1′-diyl,14δP 57.3), 1-(dicyclohexylphosphino)-1′-methylferrocene–borane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (Cy2PfcMe·BH3; δP 24.1),15 and the complex [{μ(P,P′)-fc(PCy2)2}(AuCl)2] (δP 41.2; all values in CDCl3),16 which ruled out any significant interactions between the stibine moiety and the phosphorus substituents in all cases.
:1) (Cy2PfcMe·BH3; δP 24.1),15 and the complex [{μ(P,P′)-fc(PCy2)2}(AuCl)2] (δP 41.2; all values in CDCl3),16 which ruled out any significant interactions between the stibine moiety and the phosphorus substituents in all cases.
The molecular structures of 3, 3·BH3, 3O, and 3S (Fig. 1 and Table 2) were generally similar. The ferrocene units adopted their usual geometry with practically negligible tilting of the cyclopentadienyl rings (<5°) and similar open 1,3′ conformations17 (cf. the τ angles with the ideal value of 144°). Even the substituents at the pnictogen atoms were similarly positioned, with one pointing away from the ferrocene unit and one directed to the side. The Sb–C bonds (Sb–C(fc) < Sb–C(Ph)) varied rather marginally in the entire series in contrast to the P–C bonds, which were affected by transformations of the phosphine moiety. Specifically, the introduction of the fourth substituent to the phosphorus atom resulted in a decrease in the P–C distance (more in the P-chalcogenides 3O and 3S than in 3·BH3). A similar trend could be found for tricyclohexylphosphine and its derivatives, which also had comparable P–E bond lengths.18 The C–Sb–C angles were smaller19 than the C–P–C angles, which further opened upon addition of the fourth substituent. All cyclohexyl rings adopted a chair conformation with the ring puckering parameter θ20 deviating by no more than 3° from the ideal value of 0/180° in the entire series, and the pivotal P–C bonds occupied equatorial positions.
 | ||
| Fig. 1 Molecular structures of 3, 3·BH3, 3O and 3S (for displacement ellipsoid plots, see the ESI†). | ||
| Parametera | 3 | 3·BH3 | 3O | 3S |
|---|---|---|---|---|
| a Definitions: Cp1 and Cp2 are the cyclopentadienyl rings C(1–5) and C(6–10), respectively, and Cg1 and Cg2 represent their centroids. τ is the torsion angle C1–Cg1–Cg2–C6. b Not applicable. | ||||
| E | Void | B | O | S |
| Sb–C1 | 2.125(3) | 2.131(2) | 2.150(2) | 2.129(2) |
| Sb–C23 | 2.146(3) | 2.153(2) | 2.156(2) | 2.155(2) |
| Sb–C29 | 2.160(4) | 2.157(3) | 2.160(2) | 2.154(2) |
| P–C6 | 1.828(3) | 1.800(2) | 1.787(2) | 1.793(1) |
| P–C11 | 1.856(3) | 1.839(2) | 1.824(2) | 1.834(1) |
| P–C17 | 1.868(3) | 1.839(3) | 1.821(2) | 1.842(1) |
| ∠Cp1,Cp2 | 2.8(2) | 1.7(1) | 3.61(9) | 3.15(8) |
| τ | −139.9(2) | −141.7(2) | −142.6(1) | −137.6(1) |
| P–E | —b | 1.932(3) | 1.491(1) | 1.9635(5) |
Notably, the individual molecules of 3O assembled into chains via non-bonding contacts between the stibine Sb and phosphoryl oxygen from adjacent molecule (Sb⋯O = 3.033(1) Å; Fig. S3, ESI†). No such interactions were observed in the structures of 3 and 3S.
Further experiments focused on compounds oxidised at the stibine moiety, namely, on stiboranes. Unfortunately, direct oxidation of 3 with thionyl chloride or sulfuryl chloride failed to provide the targeted phosphino-stiborane due to the concurrent oxidation of the phosphine moiety. Reaction of 3 with SOCl2 produced the doubly oxidised product Cy2P(S)fcSbCl2Ph2 (6S; 56% in the reaction mixture), which arose from unwanted thionation of the phosphine moiety with in situ generated sulfur,21 and two additional unidentified products. The analogous reaction employing SO2Cl2 resulted in a mixture of several products dominated by an unidentified species showing a broad 31P NMR resonance at δP 67.6 (≈84%).
Conversely, oxidation of 3·BH3 and 3S, whose phosphine moieties were efficiently protected, smoothly proceeded, producing the respective dichlorostiboranes 6·BH3 and 6S (Scheme 3). Even so, oxidation of Au(I) complex 5 afforded an analogous complex with a “terminal” stiborane substituent, [AuCl(Cy2PfcSbCl2Ph2-κP)] (7), in nearly quantitative yield (96%, Scheme 3). Notably, while oxidation of 3S and 5 could be achieved equally well with SOCl2 and SO2Cl2, oxidation of 3·BH3 had to be performed with SOCl2 because a similar reaction of 3·BH3 with SO2Cl2 as the halogenating agent produced 6·BH3 contaminated by ≈25% of another compound giving rise to a 31P NMR signal at δP 8.4. The side product, presumably the boron-halogenated compound 6·BH2Cl,22 could not be simply separated because all dichlorostiboranes readily decomposed on silica gel (most likely via hydrolysis at the column) and had to be purified by crystallisation, which proved inefficient for this particular mixture.
 | ||
| Scheme 3 Oxidation of 3O, 3·BH3 and Au(I) complex 5. | ||
Compounds 6·BH3, 6S and 7 were characterised similarly to their precursors. Oxidation of the stibine moiety was mainly reflected in the 13C NMR spectra, which showed that the signals due to Sb-bound aromatic rings (C5H4 and Ph) shifted to a lower field than those of the respective precursor. In contrast, the 31P NMR shifts (Table 1) only slightly differed from those of the stibine analogues, confirming the absence of any significant Sb–S interactions in 3S, which is indeed in line with the results of crystal structure determination (Fig. 2 and Table 3).
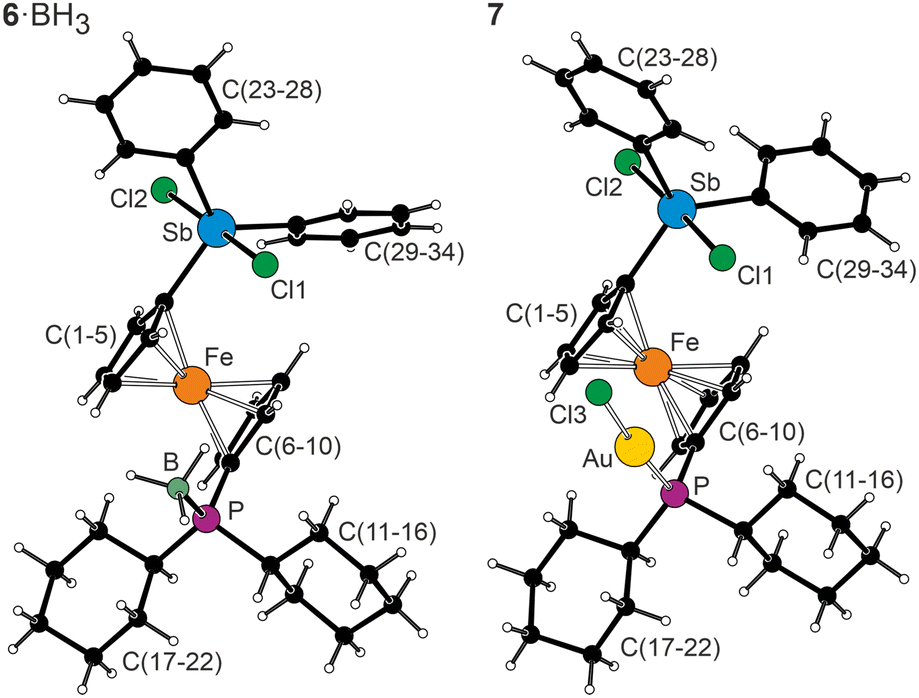 | ||
| Fig. 2 Structures of 6·BH3 and 7 (molecule 1). Additional structure diagrams (including that of 6S, which is isostructural with 6·BH3) are available in the ESI.† | ||
| Parametera | 6·BH3 | 6S | 7 (mol 1) | 7 (mol 2) |
|---|---|---|---|---|
| a The parameters are defined similarly as for the parent stibines (see footnote to Table 1). b Value affected by disorder. | ||||
| E | B | S | Au | Au |
| Sb–C1 | 2.083(1) | 2.078(2) | 2.094(6) | 2.090(6) |
| Sb–C23 | 2.104(2) | —b | 2.123(5) | —b |
| Sb–C29 | 2.122(2) | 2.121(2) | 2.137(6) | —b |
| Sb–Cl1 | 2.4586(4) | 2.4721(5) | 2.442(1) | 2.462(2) |
| Sb–Cl2 | 2.4651(4) | 2.4540(5) | 2.476(1) | 2.462(2) |
| P–C6 | 1.805(1) | 1.802(2) | 1.806(6) | 1.805(5) |
| P–C11 | 1.839(2) | 1.837(2) | 1.840(5) | 1.835(5) |
| P–C17 | 1.839(1) | 1.840(2) | 1.841(6) | 1.832(5) |
| ∠Cp1,Cp2 | 3.35(9) | 3.3(1) | 1.5(4) | 3.5(3) |
| τ | 141.5(1) | −141.7(2) | 124.8(4) | −152.8(4) |
| P–E | 1.940(2) | 1.9642(6) | 2.229(1) | 2.237(1) |
While the transformation of the stibine moiety left the PCy2·BH3 moiety and conformation of the ferrocene unit in the structure of 6·BH3 virtually unchanged, the arrangement around the Sb atom was changed to trigonal bipyramidal with the Cl atoms located at axial positions (Cl1–Sb–Cl2 177.25(2)°), and the Sb–C bonds were shortened by ≈0.04 Å (compared to 3·BH3). The τ5 index23 (0.91) indicated only a minor distortion (the ideal trigonal bipyramid would yield τ5 = 1.00), likely due to variations in the equatorial angles (C–Sb–C = 116.45(6)–122.47(6)°).
Similar features were observed in the structure of 6S, where one of the phenyl rings was partly disordered, and complex 7, which crystallised with two independent molecules per asymmetric unit (one molecule showed disorder of the phenyl rings; in this case, the conformation of the ferrocene unit differed between the two molecules). The P–Au–Cl units in 7 were linear (molecule 1/2: 175.29(6)/177.32(5)°), and the interatomic distances therein (Au–P 2.229(1)/2.237(1) Å, Au–Cl 2.298(1)/2.299(2) Å) were comparable to the values determined for [AuCl(Cy2PfcCN-κP)] (Au–P 2.2319(7) Å, Au–Cl 2.2850(7) Å).24
Synthesis and structural characterisation of catecholatostiboranes
To further enhance the Lewis acidity of the antimony atom and thus elicit possible dative P/P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E → Sb interactions, additional Sb(V) derivatives were prepared via oxidation of compounds with free stibine groups with o-chloranil (3,4,5,6-tetrachloro-1,2-benzoquinone). Even in this case, direct oxidation of free phosphine 3 was rather difficult due to the poor selectivity25 (Scheme 4). The reaction of 3 with 1 equiv. o-chloranil produced a mixture containing unreacted 3, the targeted stiborane 8, and the corresponding phosphine oxide 8O in an approximately 3:3:4 ratio. This suggested that both stibine and phosphine moieties were oxidised with o-chloranil but that the phosphorane was hydrolysed more easily to give phosphine oxide 8O.8 Indeed, a reaction performed using dried reagents allowed isolation of the unstable intermediate Cy2P(O2C6Cl4)fcSbPh2(O2C6Cl4) (9), which cleanly converted into 8O when treated with water. When water was present in the reaction mixture (introduced as a wet solvent), oxidation of 3 produced 8O as the sole product in good yield (85%).
E → Sb interactions, additional Sb(V) derivatives were prepared via oxidation of compounds with free stibine groups with o-chloranil (3,4,5,6-tetrachloro-1,2-benzoquinone). Even in this case, direct oxidation of free phosphine 3 was rather difficult due to the poor selectivity25 (Scheme 4). The reaction of 3 with 1 equiv. o-chloranil produced a mixture containing unreacted 3, the targeted stiborane 8, and the corresponding phosphine oxide 8O in an approximately 3:3:4 ratio. This suggested that both stibine and phosphine moieties were oxidised with o-chloranil but that the phosphorane was hydrolysed more easily to give phosphine oxide 8O.8 Indeed, a reaction performed using dried reagents allowed isolation of the unstable intermediate Cy2P(O2C6Cl4)fcSbPh2(O2C6Cl4) (9), which cleanly converted into 8O when treated with water. When water was present in the reaction mixture (introduced as a wet solvent), oxidation of 3 produced 8O as the sole product in good yield (85%).
 | ||
| Scheme 4 Oxidation of 3 with o-chloranil. | ||
Oxidation of adduct 3·BH3, phosphine chalcogenides 3O and 3S, and model Au(I) complex 6 proceeded without any complications and afforded corresponding stiboranes 8·BH3, 8O, 8S, and 10 in good yields after crystallisation (Scheme 5). This ultimately enabled an alternative route towards 8 based on deprotection26 of borane adduct 8·BH3 with 1,4-diazabicyclo[2.2.2]octane (dabco) in warm THF (Scheme 5) to be devised, which avoided problems associated with product isolation (the compounds decomposed on silica gel, which substantially complicated isolation of individual products from their mixtures, e.g., when 8 and 8O were both present).
 | ||
| Scheme 5 Synthesis of catecholatostiboranes. | ||
Initial NMR characterisation of the catecholatostiboranes already indicated differences between compounds 8/8O and 8·BH3/8S/10. While the 31P{1H} NMR signals of the latter compounds were observed at positions similar to those of the signals of the respective precursors (i.e., 3·BH3, 3S and 5), the 31P{1H} NMR signals of 8 and 8O significantly shifted to a lower field (Table 1), thus suggesting the presence of P → Sb and O → Sb interactions. For 8, the dative interaction was further indicated by splitting of the signals due to Cipso carbons in the Sb-bound C5H4 and Ph rings into 31P-coupled doublets (even one of the cyclopentadienyl CH groups showed such interactions), which was not observed in the 13C{1H} NMR spectra of the other compounds.
No similar interactions were obviously possible in the compounds where the phosphorus substituent was “blocked” by the formation of a Lewis adduct and coordination (8·BH3 and 10) and was not even suggested for phosphine sulfide 8S. This trend was clearly manifested in the crystal structures of 8, 8O, 8S and 8·BH3 (Fig. 3 and Table 4).
 | ||
| Fig. 3 Molecular structures of 8·C6H14 (molecule 1), 8O (molecule 1), 8·BH3 and 8S (complete structure diagrams are available in the ESI†). | ||
| Parametera | 8·C6H14 (molecule 1) | 8·C6H14 (molecule 2) | 8O (molecule 1) | 8O (molecule 2) | 8·BH3 | 8S |
|---|---|---|---|---|---|---|
| a The parameters are defined the same as those for the parent stibines (see footnote to Table 1). n.a. = not applicable. b Parameter affected by disorder of the phenyl ring. | ||||||
| E | Void | Void | O | O | B | S |
| P/E⋯Sb | 2.8126(9) | 2.8288(9) | 2.255(2) | 2.187(2) | n.a. | n.a. |
| Sb–C1 | 2.146(3) | 2.127(3) | 2.113(3) | 2.117(3) | 2.079(2) | 2.073(2) |
| Sb–C23 | 2.132(3) | 2.134(4) | 2.142(3) | 2.150(3) | 2.099(2) | 2.097(2) |
| Sb–C29 | 2.134(3) | 2.142(3) | 2.132(2) | 2.143(3) | 2.137(2) | 2.135(2) |
| Sb–O1 | 2.084(2) | 2.081(2) | 2.079(2) | 2.076(2) | 2.020(1) | 2.111(2) |
| Sb–O2 | 2.122(2) | 2.119(3) | 2.075(2) | 2.089(2) | 2.110(1) | 2.016(1) |
| P–C6 | 1.806(3) | 1.798(4) | 1.795(3) | 1.779(3) | 1.805(2) | 1.806(2) |
| P–C11 | 1.844(3) | 1.847(3) | 1.811(3) | 1.808(4) | 1.844(2) | 1.838(2) |
| P–C17 | 1.857(3) | 1.852(3) | 1.821(3) | —b | 1.843(2) | 1.837(2) |
| ∠Cp1,Cp2 | 5.7(2) | 4.8(2) | 3.2(2) | 1.3(2) | 2.23(9) | 2.2(1) |
| τ | 17.9(2) | −16.4(2) | 22.4(2) | −28.2(2) | 142.8(1) | 143.4(1) |
| P–E | n.a. | n.a. | 1.519(2) | 1.519(2) | 1.927(2) | 1.9562(7) |
The crystal structure of solvated 8 contained two practically identical, albeit crystallographically independent, stiborane molecules (Fig. S23, ESI†), whose arrangement implied a strong P–Sb interaction. The P–Sb separation (≈2.81 and 2.83 Å in the two molecules) was shorter than that in the analogous compound with phenyl substituents, Ph2PfcSbPh2(O2C6Cl4) (8Ph, 3.0987(6) Å), in line with the occurrence of an interaction enhanced by the increased electron-donating ability of the phosphine group (N.B. the P–Sb distance falls between the sum of the covalent radii and the sum of the van der Waals radii, 2.46 and 3.86 Å,27 being closer to the former). The phosphorus atom thus completed an octahedral coordination sphere around the Sb atom, where the interligand cis-angles ranged from approximately 78–104° (in both molecules), with the smallest angle associated with the catecholate ligand (O1–Sb–O2) and the widest angle for C1–Sb–C29 at the adjacent position. The Sb atom was displaced from the “equatorial” plane defined by C1, C29, O1 and O2 towards C23 by approximately 0.2 Å, and the P–Sb–C23 axis was slightly bent (169.97(1)/170.2(1)° in molecule 1/2). Compared to the structure of the parent stibine 3, the ferrocene substituents in 8 were rotated closer, allowing for the P–Sb interaction (cf. τ angles in Tables 2 and 3).
The structure of 8O was generally similar except that the PO⋯Sb distances were more similar to the values reported for Ph2P(O)fcSbPh2(O2C6Cl4) (2.256(1) Å; N.B. the influence of the phosphine substituents was now only indirect). The interligand angles within the octahedron surrounding the Sb atoms were approximately 77–106°, with the largest value occurring for the C23–Sb–C29 angle (cf. O–Sb–C23 = 168.18(8)/171.48(9)°). By featuring a longer “bridge” between the cyclopentadienyl rings, the ferrocene units in 8O exhibited more relaxed conformations (with larger τ) and lower tilt angles.
Compounds 8·BH3 and 8S crystallised isostructurally and their molecules adopted extended structures with substituents at the ferrocene unit in remote positions (the τ values were similar to those of 3·BH3 and 3S). While the arrangements of the phosphorus substituents in 8·BH3 and 8S were similar to those in the respective parent compounds, the ψ-tetrahedral stibine groups were converted into pentacoordinate stiborane moieties with a severely distorted arrangement, as indicated by the τ5 indices (τ5 = 0.55 for 8·BH3 and 0.56 for 8S), which were practically halfway between the values expected for an ideal trigonal bipyramid (1.0) and an ideal square pyramid (0.0).23 However, the bond distances did not significantly differ from those determined for Ph3Sb(O2C6Cl4).28
Analysis of the bonding situation and electrochemistry
The nature and strength of the intramolecular interactions in stiboranes 8 and 8O were evaluated through topological analysis of the electron density using the Atoms in Molecules (AIM) theory29 (Fig. 4). Compound 8S, for which no PS → Sb interaction was detected, was included in the calculations for comparison.
 | ||
| Fig. 4 Contour plots of the electron density ρ (top) and Laplacian of the electron density ∇2ρ (middle; positive – full red lines, negative – dashed blue lines) in the plane defined by the pivotal C, P and Sb (for 8) or by chalcogen atoms (O or S), P and Sb (for 8O and 8S). The yellow and blue dots indicate the positions of the critical points, and the brown lines indicate the P–Sb and E–Sb bond paths. (bottom) Selected intrinsic bond orbitals (IBOs) of stiboranes 8, 8O and 8S. The values in parentheses indicate the fraction of bonding electrons assigned to the individual atoms (lp = lone pair). | ||
The experimentally proven presence or absence of intramolecular interactions already matched the calculated energy differences between the (hypothetical) “open” and “closed” forms of the investigated species (Table 5). While the closed (interacting) forms of stiboranes 8 and 8O were strongly favoured in both vacuum and chloroform (the solvent effects were approximated by the PCM model30), the difference in energy between the open and closed forms of 8S was practically negligible, and the former arrangement was even slightly preferred when solvent effects were considered.
| Compound | Vacuum | Chloroform |
|---|---|---|
| a Calculated at the PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory. Solvent effects were approximated using the PCM model (see Experimental section). | ||
| 8 | –27 | –29 |
| 8O | –47 | –41 |
| 8S | –3 | +0.4 |
The values of the calculated real space functions (Table 6) were mostly in agreement with the values recently reported8 for analogous stiboranes bearing only phenyl substituents (i.e., compounds derived from phosphinostibine 2). The low electron densities (ρbcp) and positive values for their Laplacians (∇2ρbcp) at the bond critical points (bcps) corresponded to values typical for donor–acceptor “complexes” involving heavy elements with diffuse valence shells (such as antimony).31 A closer inspection of the calculated local properties revealed that the values previously found for stiboranes Ph2P(E)fcSbPh2(O2C6Cl4) (E = O or S) were practically identical to those estimated for chalcogenides 8O and 8S described here. However, significant differences were noted for Ph2PfcSbPh2(O2C6Cl4) (8Ph) and 8. The most significant difference was the change in the ratio of the potential to the kinetic energy density (|Vbcp|/Gbcp) at the bcps. The magnitude of this parameter indicates whether the bonding has a prevalently covalent (|Vbcp|/Gbcp > 2) or ionic (|Vbcp|/Gbcp < 1) character,32 while values falling in the intermediate region (2 > |Vbcp|/Gbcp > 1) are characteristic of dative interactions, as in the case of all chalcogenide derivatives R2P(E)fcSbPh2(O2C6Cl4) (R = Cy and Ph) and phenyl-substituted phosphinostiborane 8Ph previously studied.
| Compd | Bond | Bond length [Å] | ρ bcp [e Å−3] | ∇2ρbcp [e Å−5] | H bcp [a.u.] | |Vbcp|/Gbcp [a.u.] | G bcp/ρbcp [a.u.] | H bcp/ρbcp [a.u.] | |
|---|---|---|---|---|---|---|---|---|---|
| Experimental | Calculated | ||||||||
| a Calculated at the PBE0(d3)/def2-TZVP:sdd(Sb,Fe) level of theory. b Hypothetical closed isomer. c Not available. | |||||||||
| 8 | P⋯Sb | 2.8126(9)/2.8288(9) | 2.859 | 0.047 | 0.026 | −1.17 × 10−2 | 2.10 | 0.22 | −0.25 |
| 8O | O⋯Sb | 2.255(2)/2.187(2) | 2.298 | 0.054 | 0.170 | −1.20 × 10−2 | 1.44 | 0.51 | −0.22 |
| 8S | S⋯Sb | —c | 2.898 | 0.035 | 0.044 | −0.60 × 10−2 | 1.51 | 0.34 | −0.17 |
In addition, the value calculated for 8 (|Vbcp|/Gbcp = 2.10) suggested an increase in the covalent character of the P → Sb dative interaction in this compound. The increased covalent nature was further implied by the higher energy density at the bcp and by the more negative total energy density-to-electron density ratio, Hbcp/ρbcp. An inspection of the Laplacian profiles along the P → Sb bond path in 8 and 8Ph (see the ESI,† Fig. S24) revealed a slightly greater accumulated charge density in the region of the phosphorus lone electron pair for 8. In addition, the corresponding valence shell charge concentration (VSCC) was shifted closer to the bcp, which otherwise lies in the charge-depleted region. The higher covalency of the P → Sb interaction in 8 compared to that in its phenyl analogue 8Ph was also indicated by the calculated Mayer bond orders (MBOs: 0.56 in 8 and 0.49 in 8Ph) and Wiberg bond indices (WBIs: 0.30 and 0.22, respectively). In contrast, the low values of both the MBO and WBI (Table 7) found for 8O implicated that the electrostatic contribution was the dominant component of the PO → Sb interaction, as also reflected by the relatively high value of the kinetic energy density-to-electron density ratio Gbcp/ρbcp. This indeed corresponded with the higher polarisation of the PO bond towards P(+)–O(−).33
| Compound | MBO | WBI | ||
|---|---|---|---|---|
| P/E⋯Sb | PE |
P/E⋯Sb | PE |
|
| a Calculated at the PBE0(d3)/def2-TZVP:sdd(Sb,Fe) level of theory. n.a. = not applicable. | ||||
| 8 | 0.56 | n.a. | 0.30 | n.a. |
| 8O | 0.07 | 1.27 | 0.19 | 2.05 |
| 8S | 0.55 | 1.36 | 0.24 | 1.80 |
The conclusions obtained from the topological analysis were further supported by intrinsic bond orbital (IBO) analysis.34 The IBOs corresponding to the interaction between the antimony and respective donor atoms are also shown in Fig. 4. The charge distribution between bonded atoms reflects the different degrees of electron sharing (for an ideal covalent bond, it would be exactly 1.0/1.0). The greater charge localisation at the acceptor atom indicated higher covalency of the P → Sb interaction in 8 [P(1.59)/Sb(0.31)] compared not only to its chalcogenide derivatives 8O [O(1.77)/Sb(0.15)] and 8S [S(1.69)/Sb(0.18)] but also to the cognate compound 8Ph [P(1.68)/Sb(0.20)]. This increase reflected an increase in the donation ability of the phosphine group upon introduction of cyclohexyl substituents, which was also suggested by the substantially higher methyl cation affinity (MCAs)35 estimated for the phosphine group in 3 (P: 695, Sb: 552 kJ mol−1) as compared to the phenyl analogue 2 (P: 549, Sb: 675 kJ mol−1;8 calculated at the PBE0(d3)/def2-TZVP:sdd(Fe,Sb) level of theory in vacuum).
Considering that all the compounds contain the redox-active ferrocene unit, whose redox potential reflects the electronic influence of the substituents and can thus be used as a reporter group at the molecular level, stibines 3, 3·BH3, 3O and 3S and the corresponding catecholatostiboranes were studied by cyclic voltammetry on a glassy carbon disc electrode in CH2Cl2 containing Bu4N[PF6] as a supporting electrolyte. Attention was given to the primary electrochemical oxidations, which were assumed to occur at the ferrocene unit. For 3 and 3O as the model compounds, this was supported by DFT calculations showing that the highest occupied molecular orbitals (HOMOs) were localised predominantly at the ferrocene unit, though with a significant contribution from the phosphorus orbitals for phosphine 3. Conversely, the HOMOs of stiboranes 8 and 8O were localised mainly at the tetrachlorocatecholate units (see the ESI,† Fig. S25).
The initial oxidation of the phosphine chalcogenides and borane adducts (Fig. 5 and Fig. S28–S30, ESI†) was reversible, even though additional irreversible redox transitions could be detected at more positive potentials for most compounds (these redox changes were not investigated further). In contrast, the oxidation of 3 and 8 was only quasireversible, appearing virtually reversible at relatively fast scan rates (100 mV s−1 or higher) but losing reversibility when the scan rate was decreased, most likely due to associated chemical reactions that consumed the electrogenerated species, as is typical for (uncoordinated) ferrocene phosphines (cf. the HOMO).36
 | ||
| Fig. 5 Cyclic voltammograms for 3, 3O and 3S and the corresponding stiboranes 8, 8O and 8S, showing the first oxidative wave (glassy carbon disc electrode, 0.1 M Bu4N[PF6] in CH2Cl2, scan rate 100 mV s−1). | ||
The redox potentials of the first oxidation were 0.06 V for 3, 0.23 V for 3·BH3, 0.22 V for 3O, and 0.23 V for 3S relative to the ferrocene/ferrocenium (ref. 37). This trend roughly corresponded with the electronic nature of the substituents, as suggested by the Hammett σp constants (0.19 for PPh2, 0.53 for P(O)Ph2, and 0.47 for P(S)Ph2).38,39
Oxidation of the stibine moiety resulted in a shift towards more positive potentials (E°′ = 0.18 V for 8, 0.30 V for 8·BH3, 0.36 V for 8O, and 0.32 V for 8S), which indicated that the electron density at the ferrocene unit decreased. Notably, the differences in the stibine-stiborane pairs were substantially smaller for the borane adducts and phosphine sulfides, for which no interactions were detected between the P- and Sb-substituents (ΔE = 0.07 and 0.08 V), than for the compounds showing dative P → Sb and O → Sb interactions (ΔE = 0.12 and 0.14 V). This could be tentatively ascribed to the intramolecular interactions reducing the electron density at the P-substituents, thereby decreasing the ability of the phosphorus groups to buffer the decrease in electron density associated with stibine-to-stiborane conversion.
Coordination study with phosphinostibine 3
One of the original aims of the present work was to study the coordination behaviour of 3 towards selected metals. For this purpose, we chose soft, late transition metal ions with different coordination geometries. As stated above, compound 3 reacted with 1 molar equiv. of [AuCl(tht)] to produce [AuCl(3-κP)] (5), in which only the more basic phosphine group coordinated (vide supra). A similar behaviour was reported for 2,8 while 1 produced the ionic, ligand-redistribution product [Au(1-κ2Sb,Sb′)2][AuCl2],6 and dppf gave rise to the coordination polymer [AuCl(dppf)n]40 or polynuclear species41 under similar conditions. In contrast, the reaction between 3 and 2 equiv. of the Au(I) precursor (Scheme 6) afforded the symmetrical, ligand-bridged digold complex [(μ(P,Sb)-3)(AuCl)2] (11; δP 40.7), similar to compounds resulting from dppf,421 and 2. | ||
| Scheme 6 Preparation of complex 11 (tht = tetrahydrothiophene). | ||
The salient feature of the solid-state structure of 11 (Fig. 6) was the intramolecular aurophilic interaction43 (Au⋯Au = 3.1371(4) Å, Cl–Au–Au–Cl = −101.69(2)°) between the linear LAuCl units, which controlled the conformation of the ferrocene unit (τ = −79.4(1)°, tilt angle: 5.0(1)°). A similar arrangement was noted in [(μ(Sb,Sb′)-1)(AuCl)2] (Au⋯Au = 2.9878(5) Å) but not in either the polymorphs or solvatomorphs of the analogous dppf complex.42,44 Parameters pertaining to the individual “LAuCl” moieties were unexceptional in view of those reported for [AuCl(FcSbPh2-κSb)]8 and [AuCl(Cy2PfcCN-κP)].24
 | ||
| Fig. 6 Molecular structure of 11. Selected distances and angles (in Å and deg.): Au1–Sb 2.4967(5), Au1–Cl1 2.2980(6), Sb–Au1–Cl 173.29(2), Au2–P 2.2349(5), Au2–Cl2 2.3019(5), and P–Au2–Cl2 171.58(3). The Au⋯Au interaction is indicated by a red dashed line. | ||
Reaction of 1 with [RuCl(μ-Cl)(η6-p-cymene)]2 (Ru:3 = 2:1) in dichloromethane produced a mixture of the anticipated bridged complex [(μ(P,Sb)-3){RuCl2(η6-p-cymene)}2] (≈95%; δP 18.4) and a minor unidentified species (≈5%; δP 17.6). Unfortunately, repeated attempts to isolate the bridged complex failed due to decomposition of the reaction mixture upon crystallisation or prolonged standing. A product mixture was also obtained when the ligand amount was reduced to one molar equivalent per Ru. Gratifyingly, addition of Na[PF6] to the reaction mixture as a halide scavenger resulted in the formation of P,Sb-chelate complex 12 (Scheme 7), which could be separated from a minor, Ru-containing impurity by chromatography and isolated in 56% yield (in a partly solvated form).
 | ||
| Scheme 7 Preparation of (arene)M complexes 12 and 13. | ||
Analogous reactions employing the isoelectronic precursor [RhCl(μ-Cl)(η5-C5Me5)]2 did not yield any ligand-bridged dirhodium complex but instead spontaneously produced P,Sb-chelate complex 13a (Scheme 7) irrespective of the Rh:3 molar ratio (1:1 or 2:1), presumably due to the greater polarising power of the harder, formally trivalent Rh atom. Similar compound 13b with a hexafluorophosphate counteranion was obtained when Na[PF6] was added to a mixture containing [RhCl(μ-Cl)(η5-C5Me5)]2 and 3 (Rh:3 = 1:1). Even in this case, the behaviour of 3 differed from that of dppf and 1, which produced ligand-bridged dinuclear complexes at the M:L 2:1 ratio, while the formation of chelate complexes with these ligands required halide abstraction (i.e., did not spontaneously proceed, such as with 3 and the Rh precursor).
Due to the asymmetry of their coordination sphere, the metal ions in 12 and 13a,b were stereogenic. As the result, eight distinct 1H and 13C NMR resonances were observed due to the diastereotopic ferrocene CH groups, and similar NMR responses were noted for the cyclohexyl and phenyl groups. The 31P{1H} NMR signal of 12 was observed as a singlet at δP 40.8 (coordination shift: δP = 48.2 ppm), and those of 13a and 13b were observed as doublets at δP 40.5 and 40.2 (δP = 48.0 and 47.7 ppm), respectively, due to coupling with 103Rh (I = ½, monoisotopic; 1JRhP = 136 Hz for both complexes).
The cations in the structures of 12·C2H4Cl2, 13a·4CHCl3 and 13b·C2H4Cl2 (Fig. 7 and Table 8) adopted similar piano stool geometries, which were slightly asymmetric due to dissimilar M–ligand distances (M–Sb > M–Cl/P) and varying steric demands of the donor moieties, which could be illustrated by the CgM–M–donor angles decreasing in the sequence CgM–M–P (131–132°) > CgM–M–Sb (125–128°) > CgM–M–Cl (118–123°; CgM stands for the centroid of the π-bound arene ligand). The individual M-donor distances were similar to the values reported for analogous complexes featuring 1 and dppf as chelating ligands.6,45 Similar values (within a few degrees) were also found for the bite angles (Sb–M–P), which, however, seemed to be affected by the packing forces (cf. the values for 13a·4CHCl3 and 13b·C2H4Cl2). The ferrocene units in chelating 3 were eclipsed (|τ| < 7°) and showed negligible tilting.
 | ||
| Fig. 7 Views of the complex cations in the structures of 12·C2H4Cl2 and 13b·C2H4Cl2 (for additional structure diagrams, see the ESI†). | ||
| Parametera | 12·C2H4Cl2 | 13a·4CHCl3 | 13b·C2H4Cl2 |
|---|---|---|---|
| a CgM is the centroid of the π-coordinated aromatic ring (C(35–40) for the Ru complex and C(35–39) for both Rh complexes). Other parameters are defined the same as those for the free ligand (see footnote to Table 2). | |||
| M | Ru | Rh | Rh |
| M–CgM | 1.763(1) | 1.859(1) | 1.8625(9) |
| M–Sb | 2.6074(5) | 2.5787(4) | 2.5913(4) |
| M–P | 2.3882(6) | 2.3725(7) | 2.3700(5) |
| M–Cl | 2.4026(8) | 2.3998(7) | 2.3967(9) |
| Sb–M–P | 91.74(2) | 94.53(2) | 92.94(1) |
| Cl–M–Sb | 79.47(2) | 81.74(2) | 82.21(2) |
| Cl–M–P | 89.28(2) | 89.18(2) | 91.20(2) |
| ∠Cp1,Cp2 | 1.6(2) | 2.1(1) | 2.3(1) |
| τ | 3.8(2) | −6.5(1) | −3.0(1) |
In contrast to experiments previously mentioned, reactions of 3 with [MCl2(cod)] (M = Pd, Pt; cod = cycloocta-1,5-diene), as MCl2 synthetic proxies, proceeded uneventfully to give the expected P,Sb-chelate complexes 14 and 15 (Scheme 8) in approximately 70% yields after crystallisation.
 | ||
| Scheme 8 Synthesis of Pd(II) and Pt(II) dichloride complexes 14 and 15 (cod = cycloocta-1,5-diene) | ||
The chelate structures of 14 and 15, corroborated by structure determination (vide infra), were suggested by a low-field shift in the 31P{1H} NMR resonances, which were observed as singlets at δP 55.4 and 21.6, respectively, with the latter flanked by a pair of satellites due to the 195Pt isotopomer (I = ½, abundance: 34%; 1JPtP = 3608 Hz), and by shifts in the 13C{1H} NMR signals due to the Sb-substituted rings. Compound 14 was further characterised by splitting of the 13C{1H} NMR signal due to the ferrocene Cipso-Sb into doublets by 31P (δC 65.48, JPC = 6 Hz).
Complexes [MCl2(3-κ2P,Sb)] (M = Pd and Pt) (Fig. 8) crystallised with similar structures but were not isostructural due to differences in solvation. The coordination environments of the Pd(II) and Pt(II) ions in these compounds were square planar, as expected, but were distorted with varying interligand angles and with the Cl2 atom displaced (by 0.367(1) Å and 0.165(1) Å in solvated 14 and 15, respectively; vide infra) from the plane of the remaining atoms in the coordination sphere, {M, Cl1, P, Sb}, which, in turn, were coplanar within ≈0.04 Å. Even for 14 and 15, the M-donor distances were similar to the values reported for similar complexes with the symmetrical ligands 1 and dppf. Similarly, the ligand bite angles (100° for 14, and 98° for 15), which were the largest among the interligand angles, did not differ from those in [PdCl2(dppf-κ2P,P′)] (98–99° for different solvates),46 [PdCl2(Cy2PfcPCy2-κ2P,P′)] (102°),16 [PdCl2(Ph2PfcPCy2-κ2P,P′)] (101°),15 and [PtCl2(dppf-κ2P,P′)] (99°).47 The ferrocene units adopted an intermediate conformation with τ = 28.2(2)° [16.6(1)°] (values for 14 [15]) and tilt angles of 5.1(1)° [4.1(1)°].
 | ||
| Fig. 8 View of the complex molecule in the structure of 14·1.5CH2Cl2 (the structure diagram of 15·1.5CH2Cl2 is available in the ESI†). Selected distances and angles (in Å and deg.) for 14·1.5CH2Cl2 (M = Pd) [for 15·CH2Cl2 in parentheses (M = Pt)]: M–Sb 2.4977(3) [2.5085(4)], M–P 2.2607(6) [2.2524(5)], M–Cl1 2.3380(6) [2.3441(5)], M–Cl2 2.3653(6) [2.3762(5)], Sb–M–P 100.44(2) [97.82(2)], Cl1–M–Cl2 90.53(2) [87.60(2)], Cl1–M–P 90.03(2) [92.21(2)], and Cl2–M–Sb 79.38(2) [82.13(2)]. | ||
Although the structures of platinum metal complexes 12–15 were rather unexceptional, closer inspection of these complexes revealed minor structural distortions suggesting the presence of Cl → Sb interactions (most significantly in 12 and 14; Fig. 9). In particular, the chloride ligands in the structures of the (arene)M complexes were slightly, albeit notably, bent towards the stibine group (Fig. 9), while in 14 and 15, the chloride ligand adjacent to the stibine substituent (Cl2) was forced from the coordination plane towards Sb with concomitant closure of the Sb–M–Cl2 angle. The Sb⋯Cl distances were 3.2063(8) Å in solvated complex 12, 3.2605(6)/3.2827(7) Å in 13a/13b, and 3.2103(5)/3.1074(6) Å in 14/15. In all cases, these distances were shorter than the sum of the van der Waals radii (Sb–Cl, 3.81 Å).27b
 | ||
| Fig. 9 Views of the complex cations in the structures of 12·C2H4Cl2 and 13a·4CHCl3 in the direction perpendicular to the π-arene ligand (top) and of the complex molecules in the structures of 14·1.5CH2Cl2 and 15·1.5CH2Cl2 in the direction perpendicular to the coordination plane. Selected angles (in deg.) for 12·C2H4Cl2: P–Ru–Cl 89.28(2), P–Ru–Sb 91.74(2), and Cl–Ru–Sb 79.47(2), and for 13a·4CHCl3 [13b·C2H4Cl2]: P–Rh–Cl 89.18(2) [91.20(2)], P–Rh–Sb 94.53(2) [92.94(1)], and Cl–Rh–Sb 81.74(2) [82.21(2)]. | ||
To rule out a mere steric influence of the pnictogen substituents, the presumed Sb⋯Cl interaction was visualised using the noncovalent interaction analysis (NCI),48 which is used to distinguish between repulsive and attractive weak contacts based on the reduced density gradient (RDG) plots. The attractive and repulsive contacts are discerned based on the magnitude of the sign(λ2)ρ product, so that positive values correspond to repulsive contacts and negative values indicate attractive interactions. Indeed, the NCI analysis for model compounds 12 and 14 revealed regions of significantly negative sign(λ2)ρ values between the Sb and Cl atoms (see Fig. S26, ESI†), indicating weak attractive interactions in both complexes.
Conclusions
In summary, this study further illustrates the remarkable properties of derivatives functionalised with phosphine and stibine groups arising from the different reactivities of their functional substituents, which mainly reflects the differing chemistries of pentavalent phosphorus and antimony. Particularly attractive is the possibility of independently altering the properties of the pnictogen groups in derivatives that combine phosphorus and antimony substituents, which is relatively easily achieved by changing their substituents and the oxidation state of their “central” atom. When properly combined, these manipulations can result in intramolecular P → Sb or PE → Sb dative interactions between the Lewis basic phosphine or phosphine chalcogenide moieties and Lewis acidic stiboranes. As demonstrated here via a combination of experimental and theoretical methods, the P → Sb interactions can be further fine-tuned through substituents at the phosphorus atom (viz., the change observed for 8-type compounds upon replacement of the phenyl substituents at the phosphorus atom by cyclohexyl groups; similar changes in the corresponding P-chalcogenides had only a minimal impact).
Additionally, the coordination behaviour of the mixed derivatives is different from that of the analogous symmetrical ligands. In P,Sb-donors such as 2 and 3, the phosphine group acts as the primary donor site for soft metal ions, while the stibine moiety remains accessible for further modifications or coordination if additional metal ions or vacant coordination sites are available. In the case of Au(I) ions, the behaviour of 3 parallels that of distibine 1, which differs from that of dppf. An entirely different reactivity is observed towards the (C5Me5)Rh(III)Cl2 fragment, where 3 favours the formation of cationic P,Sb-chelate complexes, whereas 1 and dppf form primarily symmetrical, ligand-bridged dinuclear complexes, and the formation of cationic chelates requires the presence of halide scavengers (which naturally facilitate reactions producing the P,Sb-chelate complexes even from 3). Some complexes exhibit short Cl⋯Sb contacts in their structures, suggesting additional (weak) Sb–Cl interactions, in which the lone pair electron density at a terminal chloride ligand is partly donated to the Sb atom, whose Lewis acidity is enhanced by coordination to a metal centre.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the financial support from the Czech Science Foundation (project no. 21-02316S). Theoretical calculations were performed using the resources provided by the e-INFRA CZ project (ID: 90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.References
- Phosphorus(III) Ligands, in Homogeneous Catalysis, ed. P. C. J. Kamer and P. W. N. M. van Leeuwen, Wiley, Chichester, 2012 Search PubMed.
- (a) N. R. Champness and W. Levason, Coord. Chem. Rev., 1994, 133, 115 CrossRef CAS; (b) W. Levason and G. Reid, Coord. Chem. Rev, 2006, 250, 2565 CrossRef CAS; (c) H. Werner, Angew. Chem., Int. Ed., 2014, 43, 938 CrossRef PubMed; (d) S. L. Benjamin and G. Reid, Coord. Chem. Rev, 2015, 297–298, 168 CrossRef CAS; (e) V. K. Greenacre, W. Levason and G. Reid, Coord. Chem. Rev, 2021, 432, 213698 CrossRef CAS; (f) J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699 CrossRef CAS PubMed; (g) W. M. Hollingsworth and E. A. Hill, J. Coord. Chem., 2022, 75, 1436 CrossRef CAS; (h) J. L. Wardell, Arsenic, Antimony and Bismuth, in Comprehensive Organometallic Chemistry II, ed. E. W. Abel, F. G. A. Stone and G. Wilkinson, Elsevier, Oxford, 1995, vol. 2, ch. 8, pp. 321–347 Search PubMed; (i) W. Levason and G. Reid, Acyclic Arsine, Stibine, and Bismuthine Ligands, in Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Oxford, 2003, vol. 1, ch. 1.16, pp. 377–389 Search PubMed.
- (a) A. Togni and T. Hayashi, ed., Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, VCH, Weinheim, 1995 Search PubMed; (b) P. Štěpnička, ed., Ferrocenes: Ligands, Materials and Biomolecules, Wiley, Chichester, 2008 Search PubMed; (c) R. C. J. Atkinson, V. C. Gibson and N. J. Long, Chem. Soc. Rev., 2004, 33, 313 RSC; (d) R. Gómez Arrayás, J. Adrio and J. C. Carretero, Angew. Chem., Int. Ed., 2006, 45, 7674 CrossRef PubMed; (e) L. Cunningham, A. Benson and P. J. Guiry, Org. Biomol. Chem., 2020, 18, 9329 RSC; (f) P. Štěpnička, Dalton Trans., 2022, 51, 8085 RSC.
- (a) P. Sharma, J. G. Lopez, C. Ortega, N. Rosas, A. Cabrera, C. Alvarez, A. Toscano and E. Reyes, Inorg. Chem. Commun., 2006, 9, 82 CrossRef CAS; (b) J. Vázquez, P. Sharma, A. Cabrera, A. Toscano, S. Hernández, J. Pérez and R. Gutiérrez, J. Organomet. Chem., 2007, 692, 3486 CrossRef; (c) D. Pérez, P. Sharma, N. Rosas, A. Carbrera, J. L. Arias, F. del Rio-Portilla, J. Vazquez, R. Gutierrez and A. Toscano, J. Organomet. Chem., 2008, 693, 3357 CrossRef; (d) A. M. Ortiz, P. Sharma, D. Pérez, N. Rosas, A. Cabrera, L. Velasco, A. Toscano and S. Hernández, J. Organomet. Chem., 2009, 694, 2037 CrossRef CAS; (e) D. Pérez, P. Sharma, A. Cabrera, N. Rosas, I. Arellano, A. Toscano and S. Hernández, Polyhedron, 2009, 28, 3115 CrossRef; (f) D. Pérez, P. Sharma, A. M. Ortiz, A. Cabrera, S. Hernández, A. Toscano and R. Gutiérrez, Z. Naturforsch., 2012, 67b, 36 CrossRef; (g) D. Perez, C. Herrera, M. Sharma, R. Gutierrez, S. Hernández, A. Toscano and P. Sharma, J. Organomet. Chem., 2013, 743, 97 CrossRef CAS.
- An earlier report mentioned triferrocenylstibine, albeit
without any synthetic or characterisation details: A. E. Ermoshkin, N. P. Makarenko and K. I. Sakodynskii, J. Chromatogr., 1984, 290, 377 CrossRef CAS . Furthermore, a sterically stabilised distibene, Fc*SbSbFc* (Fc* = 2,5-bis(3,5-di-t-butylphenyl)ferrocenyl), was reported in: M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. – Asian J., 2013, 8, 690 CrossRef PubMed.
- J. Schulz, J. Antala, I. Císařová and P. Štěpnička, Dalton Trans., 2023, 52, 1198 RSC.
- (a) K.-S. Gan and T. S. A. Hor, 1,1′-Bis(diphenylphosphino)ferrocene. Coordination Chemistry, Organic Syntheses, and Catalysis, in Ferrocenes: Homogeneous Catalysis, Organic Synthesis Materials Science, ed. A. Togni and T. Hayashi, Wiley-VCH, Weinheim, Germany, 1995, ch. 1, pp. 3–104 Search PubMed; (b) S. W. Chien and T. S. A. Hor, The Coordination and Homogeneous Catalytic Chemistry of 1,1′-Bis(diphenylphosphino)ferrocene and its Chalcogenide Derivatives, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpnička, Wiley, Chichester, UK, 2008, ch. 2, pp. 33–116 Search PubMed; (c) T. J. Colacot and S. Parisel, Synthesis, Coordination Chemistry and Catalytic Use of dppf Analogs, in Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Štěpničkas, Wiley, Chichester, U.K., 2008, ch. 3, pp. 117–140 Search PubMed; (d) G. Bandoli and A. Dolmella, Coord. Chem. Rev., 2000, 209, 161 CrossRef CAS; (e) D. J. Young, S. W. Chien and T. S. A. Hor, Dalton Trans., 2012, 41, 12655 RSC; (f) S. Dey and R. Pietschnig, Coord. Chem. Rev., 2021, 437, 213850 CrossRef CAS.
- J. Schulz, J. Antala, D. Rezazgui, I. Císařová and P. Štěpnička, Inorg. Chem., 2023, 62, 14028 CrossRef CAS PubMed.
- For representative examples of P,Sb-donors, see: (a) J. W. Dawson and L. M. Venanzi, J. Am. Chem. Soc., 1968, 90, 7229 CrossRef CAS; (b) B. R. Higginson, C. A. McAuliffe and L. M. Venanzi, Inorg. Chim. Acta, 1971, 5, 37 CrossRef CAS; (c) W. Levason and C. A. McAuliffe, Inorg. Chim. Acta, 1974, 11, 33 CrossRef CAS; (d) W. Levason and C. A. McAuliffe, Inorg. Chim. Acta, 1976, 16, 167 CrossRef CAS; (e) H. H. Karsch and E. Witt, J. Organomet. Chem., 1977, 529, 151 CrossRef; (f) W. Levason, K. G. Smith, C. A. McAuliffe, F. P. McCullough, R. D. Sedgwick and S. G. Murray, J. Chem. Soc., Dalton Trans., 1979, 1718 RSC; (g) L. R. Gray, A. L. Hale, W. Levason, F. P. McCullough and M. Webster, J. Chem. Soc., Dalton Trans., 1983, 2573 RSC; (h) T. Kaufmann, R. Joußen, N. Klas and A. Vahrenhorts, Chem. Ber., 1983, 116, 473 CrossRef; (i) J. R. Black, W. Levason, M. D. Spicer and M. Webster, J. Chem. Soc., Dalton Trans., 1993, 3129 RSC; (j) M. Manger, J. Wolf, M. Laubender, M. Teichert, D. Stalke and H. Werner, Chem. – Eur. J., 1997, 3, 1442 CrossRef CAS; (k) M. Manger, O. Gevert and H. Werner, Chem. Ber., 1997, 130, 1529 CrossRef CAS; (l) H. C. Jewiss, W. Levason, M. D. Spicer and M. Webster, Inorg. Chem., 1987, 26, 2102 CrossRef CAS; (m) S. Yasuike, S. Kawara, S. Okajima, H. Seki, K. Yamaguchi and J. Kurita, Tetrahedron Lett., 2004, 45, 9135 CrossRef CAS; (n) C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2011, 50, 7369 CrossRef CAS PubMed; (o) I.-S. Ke and F. P. Gabbaï, Inorg. Chem., 2013, 52, 7145 CrossRef CAS PubMed; (p) J. S. Jones, C. R. Wade and F. P. Gabbaï, Angew. Chem., Int. Ed., 2014, 53, 8876 CrossRef CAS PubMed; (q) I.-S. Ke, J. S. Jones and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 53, 2633 CrossRef PubMed; (r) H. Yang and F. P. Gabbaï, J. Am. Chem. Soc., 2015, 137, 13425 CrossRef CAS PubMed; (s) B. A. Chalmers, M. Bühl, K. S. A. Arachchige, A. M. Z. Slawin and P. Kilian, Chem. – Eur. J., 2015, 21, 7520 CrossRef CAS PubMed; (t) B. A. Chalmers, C. B. E. Meigh, P. S. Nejman, M. Bühl, T. Lébl, J. D. Woollins, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2016, 55, 7117 CrossRef CAS PubMed; (u) J. S. Jones and F. P. Gabbaï, Chem. – Eur. J., 2017, 23, 1136 CrossRef CAS PubMed; (v) D. You and F. P. Gabbaï, J. Am. Chem. Soc., 2017, 139, 6843 CrossRef CAS PubMed; (w) J. S. Jones, C. R. Wade, M. Yang and F. P. Gabbaï, Dalton Trans., 2017, 46, 5598 RSC; (x) S. Sen, I.-S. Ke and F. P. Gabbaï, Organometallics, 2017, 36, 4224 CrossRef CAS; (y) S. Furan, E. Hupf, J. Boidol, J. Brünig, E. Lork, S. Mebs and J. Beckmann, Dalton Trans., 2019, 48, 4504 RSC; (z) M. Piesch, F. P. Gabbaï and M. Scheer, Z. Anorg. Allg. Chem., 2021, 647, 266 CrossRef CAS.
- (a) L.-L. Lai and T.-Y. Dong, J. Chem. Soc., Chem. Commun., 1994, 2347 RSC; (b) I. R. Butler and R. L. Davies, Synthesis, 1996, 1350 CrossRef CAS.
- The synthesis of 4 was performed as previously reported: P. Štěpnička and I. Císařová, Dalton Trans., 2013, 42, 3373 RSC.
- J. Schraml, M. Čapka and V. Blechta, Magn. Res. Chem., 1992, 30, 544 CrossRef CAS.
- C. Baillie, L. Zhang and J. Xiao, J. Org. Chem., 2004, 69, 7779 CrossRef CAS PubMed.
- K. D. Reichl, C. L. Mandell, O. D. Henn, W. G. Dougherty, W. S. Kassel and C. Nataro, J. Organomet. Chem., 2011, 696, 3882 CrossRef CAS.
- P. Vosáhlo, I. Císařová and P. Štěpnička, J. Organomet. Chem., 2018, 860, 14 CrossRef.
- L. E. Hagopian, A. N. Campbell, J. A. Golen, A. L. Rheingold and C. Nataro, J. Organomet. Chem., 2006, 691, 4890 CrossRef CAS.
- S. I. Kirin, H.-B. Kraatz and N. Metzler-Nolte, Chem. Soc. Rev., 2006, 35, 348 RSC.
- (a) J. A. Davies, S. Dutremez and A. A. Pinkerton, Inorg. Chem., 1991, 30, 2380 CrossRef CAS; (b) N. P. Taylor, J. A. Gonzalez, G. S. Nichol, A. García-Domínguez, A. G. Leach and G. C. Lloyd-Jones, J. Org. Chem., 2022, 87, 721 CrossRef CAS PubMed; (c) Y. J. Tan, C. I. Yeo, N. R. Halcovitch, M. M. Jotani and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2017, 73, 493 CrossRef CAS PubMed.
- The C–Sb–C angles in 3 and 3·BH3 (93.6(1)–96.5(1)° and 95.0(1)–96.8(1)°) vary significantly less than those in 3O (90.71(6)–99.60(6)°) and 3S (92.75(6)–99.42(6)°).
- D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354 CrossRef CAS.
- The oxidation of a stibine SbR3 with SOCl2 produces stiborane SbCl2R3 and “SO”, which disproportionates into SO2 and S: P. W. Schenk and R. Steudel, Angew. Chem., Int. Ed. Engl., 1965, 4, 402 CrossRef.
- Compare the 31P NMR chemical shift with the value reported for Ph2PC(O)fcPCy2·BH2Cl (δP 8.1): P. Vosáhlo, I. Císařová and P. Štěpnička, New J. Chem., 2022, 46, 21536 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- O. Bárta, I. Císařová, J. Schulz and P. Štěpnička, New J. Chem., 2019, 43, 11258 RSC.
- Recently, it was shown that selective oxidation of the stibine moiety in phosphinostibines can be achieved with 3,5-di-tert-butyl-o-benzoquinone: B. Zhou, S. Bedajna and F. P. Gabbaï, Chem. Commun., 2024, 60, 192 RSC.
- (a) H. Brisset, Y. Gourdel, P. Pellon and M. Le Corre, Tetrahedron Lett., 1993, 34, 4523 CrossRef CAS; (b) J. M. Brunel, B. Faure and M. Maffei, Coord. Chem. Rev., 1998, 178–180, 665 CrossRef CAS.
- (a) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC; (b) M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- R. R. Holmes, R. O. Day, V. Chandrasekhar and J. M. Holmes, Inorg. Chem., 1987, 26, 163 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS.
- (a) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed; (b) G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- (a) D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627 CrossRef; (b) P. Macchi, D. M. Proserpio and A. Sironi, J. Am. Chem. Soc., 1998, 120, 13429 CrossRef CAS; (c) P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238, 383 CrossRef; (d) D. Stalke, Chem. – Eur. J., 2011, 17, 9264 CrossRef CAS PubMed.
- (a) R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360 CrossRef CAS PubMed; (b) E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- (a) D. B. Chesnut, J. Am. Chem. Soc., 1999, 121, 2335 CrossRef CAS; (b) K. Yamada and N. Koga, J. Comput. Chem., 2013, 34, 149 CrossRef CAS PubMed; (c) B. Lindquist-Kleissler, J. S. Wenger and T. C. Johnstone, Inorg. Chem., 2021, 60, 1846 CrossRef CAS PubMed.
- (a) G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834 CrossRef CAS PubMed; (b) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518 CrossRef CAS PubMed.
- (a) C. Lindner, B. Maryasin, F. Richter and H. Zipse, J. Phys. Org. Chem., 2010, 23, 1036 CrossRef CAS; (b) C. Lindner, R. Tandon, B. Maryasin, E. Larionov and H. Zipse, Beilstein J. Org. Chem., 2012, 8, 1406 CrossRef CAS PubMed.
- (a) J. C. Kotz and C. L. Nivert, J. Organomet. Chem., 1973, 52, 387 CrossRef CAS; (b) B. Corain, B. Longato, G. Favero, G. Ajo, G. Pilloni, U. Russo and F. R. Kreissl, Inorg. Chim. Acta, 1989, 157, 259 CrossRef CAS; (c) G. Pilloni, B. Longato and B. Corain, J. Organomet. Chem., 1991, 420, 57 CrossRef CAS; (d) J. Podlaha, P. Štěpnička, J. Ludvík and I. Císařová, Organometallics, 1996, 15, 543 CrossRef CAS; (e) C. Nataro, A. N. Campbell, M. A. Ferguson, C. D. Incarvito and A. L. Rheingold, J. Organomet. Chem., 2003, 673, 47 CrossRef CAS.
- (a) G. Gritzner and J. Kůta, Pure Appl. Chem., 1984, 56, 461 CrossRef; (b) R. R. Gagné, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854 CrossRef.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- The difference in the redox potentials determined for (diphenylphosphino)ferrocene and the corresponding phosphine oxide was approximately 120 mV in MeCN, see ref. 36d.
- (a) A. Houlton, D. M. P. Mingos, D. M. Murphy, D. J. Williams, L.-T. Phang and T. S. A. Hor, J. Chem. Soc., Dalton Trans., 1993, 3629 RSC; (b) L.-T. Phang, T. S. A. Hor, Z.-Y. Zhou and T. C. W. Mak, J. Organomet. Chem., 1994, 469, 253 CrossRef CAS.
- A. Houlton, D. M. P. Mingos, D. M. Murphy and D. J. Williams, Acta Crystallogr., Sect. C: Struct, Chem, 1995, 51, 30 CrossRef.
- D. T. Hill, G. R. Girard, F. L. McCabe, R. K. Johnson, P. D. Stupik, J. H. Zhang, W. M. Reiff and D. S. Eggleston, Inorg. Chem., 1989, 28, 3529 CrossRef CAS.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- (a) F. Canales, M. C. Gimeno, P. G. Jones, A. Laguna and C. Sarroca, Inorg. Chem., 1997, 36, 5206 CrossRef CAS; (b) O. Crespo, M. C. Gimeno, P. G. Jones and A. Laguna, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, 1433 CrossRef PubMed; (c) N. Meyer, F. Mohr and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m168 CrossRef CAS PubMed.
- (a) S. B. Jensen, S. J. Rodger and M. D. Spicer, J. Organomet. Chem., 1998, 556, 151 CrossRef CAS; (b) J.-F. Ma and Y. Yamamoto, J. Organomet. Chem., 1999, 574, 148 CrossRef CAS.
- (a) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158 CrossRef CAS; (b) I. R. Butler, W. R. Cullen, T. J. Kim, S. J. Rettig and J. Trotter, Organometallics, 1984, 4, 972 CrossRef.
- (a) D. A. Clemente, G. Pilloni, B. Corain, B. Longato and M. Tiripicchio-Camellini, Inorg. Chim. Acta, 1986, 115, L9 CrossRef CAS; (b) A. Muller, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m210 CrossRef CAS; (c) G. M. de Lima, C. A. L. Filgueiras, M. T. S. Giotto and Y. P. Mascarenhas, Trans. Met. Chem., 1995, 20, 380 CrossRef CAS.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed; (b) E. Pastorczak and C. Corminboeuf, J. Chem. Phys., 2017, 146, 120901 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental details, additional structure diagrams, result from the DFT calculations, additional cyclic voltammograms, copies of the NMR spectra and Cartesian coordinates for the DFT-optimised structures. CCDC 2320741–2320757. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj00349g |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |