DOI:
10.1039/D4NJ00235K
(Paper)
New J. Chem., 2024,
48, 8844-8859
Piperidine and piperazine analogs in action: zinc(II)-mediated formation of amidines†
Received
15th January 2024
, Accepted 15th April 2024
First published on 16th April 2024
Abstract
The catalytic activity of zinc(II) compounds was exploited in the nucleophilic addition of amines to nitriles to form amidines. The reaction systems comprised a Zn(II) starting material [Zn(quin)2(H2O)] (quin− = quinaldinate, an anion of quinaldinic acid), secondary cyclic amine, acetonitrile, and in some cases, methanol. Amines with additional heteroatoms in the ring (thiomorpholine, piperazine) or ring substituents (piperidine derivatives, piperazine derivatives) with different steric and electronic effects were used. The aim of the study was to determine how the nature of the amine affects the formation of amidines. Different types of Zn(II) products were obtained: mono- or diamine complexes, amidine complexes, and also an ionic compound with a protonated amine. In one case, the amidine itself crystallized. A Zn(II) complex with acetamidine was also obtained. Acetamidine was formed from acetonitrile and ammonia, which most likely originated from the hydrolysis of acetonitrile under harsh reaction conditions. The hydrolysis could terminate at the acetamide step, which was confirmed by the isolation of a cocrystal containing acetamide. In the case of piperazine (pz), a polymeric compound with the composition [Zn(quin)2(pz)]n was isolated regardless of the reaction conditions. The same coordination polymer was also observed to form in 1-methylpiperazine, 1-ethylpiperazine, and 1-acetylpiperazine reaction systems, containing piperazine as an inherent impurity. This was unambiguously confirmed by the crystal structure of a cocrystal, [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN, which formed in the 1-acetylpiperazine (1-Acpz) reaction system.
Introduction
One of the most important goals in chemistry is to develop methods for sustainable, efficient, and selective synthesis, wherein metal catalysis is an effective approach to achieve this goal. However, most of the metals in common use, such as Pd, Rh, Ru, and Ir, pose challenges due to their limited availability, high cost, or toxicity. Current research is therefore focused on replacing these metals with cheaper and less toxic alternatives.1 In particular, zinc(II) compounds have attracted significant research interest for several reasons. Firstly, zinc is more abundant in the Earth's crust than the aforementioned metals.2 More importantly, zinc(II) plays a key role in many vital biological processes and is essential to all organisms.3 For this reason, zinc(II) is considered to be relatively nontoxic.4 The widespread biological use of Zn(II) can be attributed to its unique properties, which arise mainly due to its d10 electron configuration. Zinc(II) ions show no stereochemical preferences due to ligand-field stabilization effects. This flexibility allows them to adopt a wide range of coordination numbers and geometries,5 affording low activation barriers, which are crucial for efficient catalysis.6 Zinc(II) catalysts have great potential, as demonstrated by their numerous applications in organic chemistry.1,7 The main role of Zn(II) catalysts lies in their ability to act as Lewis acids. Simple Zn(II) salts are often used in catalytic reactions, while coordination compounds with more intricate ligands are used only selectively.8,9 Zn(II) catalysts have the ability to facilitate the formation of chemical bonds, e.g., C–C, C–N, and C–O bonds.10,11 One example is the conversion of carbon dioxide into cyclic carbonates, which is an emerging area of research because of the widespread use of fossil fuels.12 Despite not being a redox-active metal, zinc(II) species can catalyze redox reactions in the presence of suitable reducing or oxidizing agents.13 Zn(II) catalysts can also be used in polymerization reactions; for example, in the ring-opening polymerization of lactide, which produces one of the most commercially important biodegradable polymers, polylactic acid.14,15
It comes as no surprise that Zn(II) species have also been shown to catalyze the nucleophilic addition of amines to nitriles to form amidines,16,17 where no amidine products were observed in reactions without Zn(II) species.17 Our initial reaction system comprised a Zn(II) starting material, a secondary cyclic amine (either piperidine or pyrrolidine), and acetonitrile as a solvent. The starting compound was [Zn(quin)2(H2O)], where quin− stands for quinaldinate, an anion of quinaldinic acid (IUPAC name: quinoline-2-carboxylic acid). Quinaldinate was chosen to limit the chemical space because it normally exhibits strong N,O-bidentate chelating coordination to transition metal ions,18,19 whereas water served as a ligand that could be substituted. The reactions were conducted under harsh conditions in an autoclave or under ambient conditions with a solvent system comprising of acetonitrile and methanol. The reactions resulted in the formation of either neutral amidine complexes with a [Zn(quin)2(amidine)] composition or amidinium salts.17 To test the generality of the reaction, acetonitrile was replaced with propionitrile or benzonitrile. For both nitriles, the formation of amidine was confirmed with no substantial differences in reactivity.20 The influence of the Zn(II) starting material was also investigated. Quinaldinic acid was replaced by structurally similar picolinic acid or pyrazinoic acid. The choice of Zn(II) precursor was shown to have no effect on the amidine formation.21 In the final study, presented herein, the range of secondary cyclic amines was expanded. Scheme 1 shows the structural formulas and abbreviations of thiomorpholine, piperidine analogs, and piperazine analogs used in this study. To the best of our knowledge, among the chosen amines only piperazine reacted with acetonitrile to produce an amidine, which coordinated to rhenium(I).22 The aim of this study was to determine how the nature of the amine, in particular the ring substituents, affects the reaction between the amine and acetonitrile (Scheme 2). The inductive effects of the substituents entail changes in the nucleophilicity of the non-substituted (free) nitrogen atom and, thus, in its reactivity. Our reactions afforded a series of products that encompass a salt with a protonated amine, the amidine itself (in one instance only), and Zn(II) complexes, either with the reacting amines, amidines, or acetamidine. The amidines were named by appending the suffix am to the amine abbreviation, whereas acetamidine was named solely am. All new compounds under this investigation were fully characterized using infrared (IR) and nuclear magnetic resonance (NMR) spectroscopies, elemental analyses, mass spectrometry (MS), and single-crystal X-ray diffraction analysis (XRD). A complete list of products is given in Table 1.
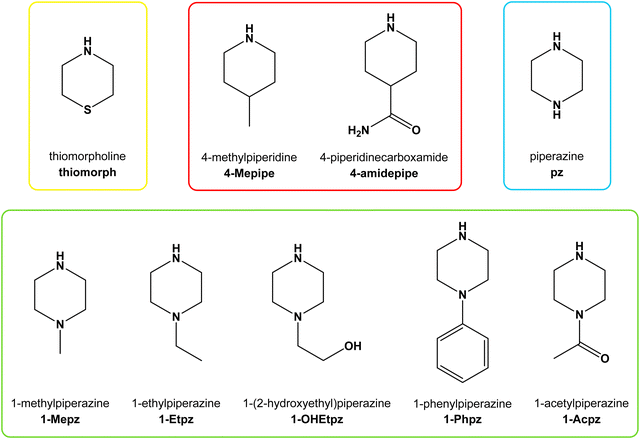 |
| | Scheme 1 Structural formulas, names, and abbreviations of the amines used in this study. Similar amines are grouped in boxes. | |
 |
| | Scheme 2 Nucleophilic addition of piperazine analogs to acetonitrile to form amidines. See Scheme 1 for R. | |
Table 1 Novel compounds, prepared in the course of this study
|
4-MepipeH+ stands for protonated 4-methylpiperidine.
|
| [Zn(quin)2(thiomorph)2] |
1
|
| [Zn(quin)2(thiomorpham)]·CH3OH·CH3CN |
2
|
| [Zn(quin)2(4-Mepipe)2] |
3
|
| 4-MepipeH[Zn(quin)3]a |
4
|
| [Zn(quin)2(4-Mepipeam)]·CH3CN |
5
|
| [Zn(quin)2(4-amidepipe)2]·2CH3CN |
6
|
| [Zn(quin)2(4-amidepipeam)] |
7
|
| 4-amidepipeam |
8
|
| [Zn(quin)2(pz)]n |
9
|
| [Zn(quin)2(1-Mepz)] |
10
|
| [Zn(quin)2(1-Mepz)2]·0.5CH3CN |
11
|
| [Zn(quin)2(1-Mepzam)] |
12
|
| [Zn(quin)2(1-Etpz)2]·0.5CH3CN |
13
|
| [Zn(quin)2(1-Etpzam)]·H2O |
14
|
| [Zn(quin)2(1-OHEtpz)2]·2CH3CN |
15
|
| [Zn(quin)2(1-OHEtpzam)] |
16
|
| [Zn(quin)2(1-Phpz)2]·CH3CN |
17
|
| [Zn(quin)2(1-Phpz)2]·2acetamide |
18 (polymorphs 18a and 18b) |
| [Zn(quin)2(1-Phpzam)]·0.5(1-Phpz)·CH3CN |
19
|
| [Zn(quin)2(1-Acpz)2] |
20
|
| [Zn(quin)2(1-Acpz)2]·2CH3CN |
21 (polymorphs 21a and 21b) |
| [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN |
22
|
| [Zn(quin)2(am)]·1.3CH3CN·0.2H2O |
23
|
Experimental
General
All reagents were purchased from commercial sources. Acetonitrile was dried over molecular sieves before use.23 The starting materials, [Zn(quin)2(H2O)] and [Zn(quin)2(NH3)], were prepared according to a previously published procedure.24 For ligands added in large excess compared to Zn(II), only the mass or volume is given. IR spectra were recorded on a Bruker Alpha II FT-IR spectrophotometer with an attenuated total reflection (ATR) module in the range of 4000–400 cm−1. The spectra are shown as obtained, without corrections. Band intensities are given as follows: w = weak, m = medium, s = strong, vs = very strong, and vvs = very very strong. 1H NMR spectra were recorded with a Bruker Avance III 500 MHz or Bruker Avance NEO 600 MHz instruments. Deuterated dimethyl sulfoxide ((CD3)2SO) with 0.03% tetramethylsilane (TMS) standard was used as a solvent. Trifluoroacetic acid (TFA) was added to increase the solubility of 1, 6, and 9. In each case, the TFA addition resulted in the dissociation of the complex. The resonances in the spectra can, therefore, be attributed to the protonated species. The residual solvent peak of (CD3)2SO at 2.50 ppm or the TMS standard at 0.00 ppm was used as a reference for the chemical shifts.25 Chemical shifts (δ) are given in ppm and coupling constants (J) in Hz. Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, dt = doublet of triplet, td = triplet of doublet, q = quartet, m = multiplet, and br = broad signal. Spectra were processed using the MestReNova program (version 11.0.4).261H NMR spectra of 4, 7, 8 and 9 form part of the ESI,† whereas the others may be obtained from the authors upon request. High-resolution mass spectra were recorded on an Agilent 6224 Accurate Mass TOF LC/MS system. Elemental analyses of carbon, nitrogen, and hydrogen were performed on a PerkinElmer 2400 II analyzer. For some compounds which crystallized with solvent molecules of crystallization, the elemental analysis data are not provided. The solvates were usually unstable outside the mother liquor, and partial or complete loss of solvent molecules was observed. The yields for unstable solvates are therefore reported as the mass of solid remaining after approximately 15 min of air drying.
X-ray structure analysis
Single-crystal XRD data were collected on an Agilent SuperNova diffractometer equipped with a molybdenum (Mo-Kα, λ = 0.71073 Å) or copper (Cu-Kα, λ = 1.54184 Å) X-ray source at low temperature (150 K). Each crystal was mounted on the tip of a glass fiber using silicone grease and then mounted on the head of a goniometer. Data processing was performed using CrysAlis PRO.27 The crystal structures were solved in the Olex2 software,28 using methods implemented in ShelXT.29 The structures were refined using least squares methods in ShelXL.30 Anisotropic displacement parameters were determined for all non-hydrogen atoms. Where possible, hydrogens on heteroatoms were located using residual electron density and refined with isotropic displacement parameters. The remaining hydrogen atoms were added in calculated positions. For 2, 12, 16, and 19, the solvent molecules were crystallographically disordered and could not be modeled. The solvent contribution to the scattering factors was thus accounted for using a solvent mask function. Platon31 was used to analyze the data, and the images were prepared using Mercury.32 All crystal structures were deposited at the Cambridge Crystallographic Data Centre (CCDC) and assigned the deposition numbers: 2181834 (1), 2181835 (2), 2181836 (3), 2181837 (5), 2181838 (6), 2181839 (8), 2181840 (10), 2181841 (11), 2181842 (12), 2181843 (13), 2181844 (14), 2181845 (15), 2181846 (16), 2181847 (17), 2181848 (18a), 2181849 (18b), 2181850 (19), 2181851 (20), 2181852 (21a), 2181853 (21b), 2181854 (22), and 2181855 (23). Crystallographic data for 1–23 are collected in Tables S1–S7 (ESI†).
Preparation of [Zn(quin)2(thiomorph)2] (1).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and thiomorpholine (0.5 mL) was stirred for 3 days at room temperature in a closed flask. White crystalline solid, [Zn(quin)2(thiomorph)2] (1), was filtered off. Yield: 104 mg, 72%. Single crystals were prepared as follows. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and thiomorpholine (0.5 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Unreacted starting material was filtered off (78 mg) and the filtrate was stored at 4 °C. Single crystals of 1 were filtered off on the following day. IR (ATR, cm−1): 3191m, 3054w, 2953w, 2919m, 1638vs, 1598m, 1568s, 1509m, 1484w, 1464m, 1446w, 1432w, 1417w, 1366s, 1351s, 1299w, 1284s, 1273s, 1229s, 1207m, 1176m, 1156m, 1137m, 1113m, 1089m, 1029w, 1002m, 993m, 963m, 950s, 896m, 861s, 821s, 810s, 796s, 777vvs, 746m, 692w, 664m, 635s, 600s, 556w, 523m, 499m, 429w, 403m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.76 (3H, br s), 8.64 (2H, d, J = 8.4 Hz), 8.24 (2H, d, J = 8.5 Hz), 8.18 (2H, d, J = 8.5 Hz), 8.14–8.13 (2H, m), 7.93–7.90 (2H, m), 7.80–7.77 (2H, m), 3.32–3.29 (8H, m), 2.85–2.84 (8H, m) ppm. Note. Due to poor solubility of 1 in (CD3)2SO, trifluoroacetic acid was added, which has a resonance at 14.14 ppm. The ratio of the integrals agrees with the proposed composition of the complex. Elemental analysis calcd for C28H30N4O4S2Zn (%): C, 54.59; H, 4.91; N, 9.09. Found (%): C, 54.77; H, 4.75; N, 9.31. ESI-HRMS m/z calcd for [C24H22N3O4SZn]+ = [Zn(quin)2(thiomorph) + H]+: 512.0617, found: 512.0594.
Preparation of [Zn(quin)2(thiomorpham)]·CH3OH·CH3CN (2).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and thiomorpholine (1 mL) was stirred for 3 days at room temperature in a closed flask. Afterwards, the reaction mixture was filtered and the filtrate was stored at 4 °C. Single crystals of [Zn(quin)2(thiomorpham)]·CH3CN·CH3OH (2) were filtered off on the following day. Yield: 57 mg. IR (ATR, cm−1): 3426w, 3282w, 3061w, 2908w, 1637s, 1585s, 1571s, 1507m, 1460s, 1435m, 1341s, 1284m, 1254m, 1237m, 1218s, 1194m, 1181s, 1161m, 1144m, 1063m, 1022m, 968w, 953m, 897s, 874m, 853m, 828m, 802vs, 773vvs, 740m, 638s, 602s, 561m, 544m, 522s, 497s, 405m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.79 (2H, d, J = 8.2 Hz, quin−), 8.52 (2H, d, J = 8.2 Hz, quin−), 8.36 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.1 Hz, quin−), 8.00–7.98 (2H, m, quin−), 7.84–7.81 (2H, m, quin−), 7.15 (1H, br s, thiomorpham), 3.58–3.57 (4H, m, thiomorpham), 2.36–2.32 (4H, br m, thiomorpham), 1.95 (3H, s, thiomorpham) ppm. ESI-HRMS m/z calcd for [C26H25N4O4SZn]+ = [Zn(quin)2(thiomorpham) + H]+: 553.0882, found: 553.0876.
Preparation of [Zn(quin)2(4-Mepipe)2] (3).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 4-methylpiperidine (1 mL) was stirred for 3 days at room temperature in a closed flask. Afterwards, the reaction mixture was filtered and the filtrate was stored at 4 °C. Single crystals of [Zn(quin)2(4-Mepipe)2] (3) were filtered off. Yield: 45 mg, 32%. IR (ATR, cm−1): 3168w, 3048w, 2953m, 2938m, 2919m, 2897m, 2861m, 1635vvs, 1600s, 1568s, 1509m, 1465s, 1448s, 1429m, 1363vs, 1290m, 1268m, 1217w, 1202m, 1178s, 1153m, 1109m, 1090w, 1045m, 1020m, 991w, 977m, 961m, 946m, 893s, 851s, 805s, 795s, 771vvs, 737m, 636s, 626s, 600s, 553w, 523m, 499s, 478w, 427w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.96 (2H, d, J = 7.9 Hz, quin−), 8.79 (2H, d, J = 8.3 Hz, quin−), 8.41 (2H, d, J = 8.3 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.01 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 2.84 (4H, dt, J = 12.3, 2.8 Hz, 4-Mepipe), 2.37 (4H, td, J = 12.3, 2.8 Hz, 4-Mepipe), 1.46–1.42 (4H, m, 4-Mepipe), 1.36–1.28 (2H, m, 4-Mepipe), 0.98–0.91 (4H, m, 4-Mepipe), 0.78 (6H, d, J = 6.6 Hz, 4-Mepipe) ppm. Elemental analysis calcd for C32H38N4O4Zn (%): C, 63.21; H, 6.30; N, 9.21. Found (%): C, 63.21; H, 6.64; N, 9.29. ESI-HRMS m/z calcd for [C26H26N3O4Zn]+ = [Zn(quin)2(4-Mepipe) + H]+: 508.1209, found: 508.1216.
Preparation of 4-MepipeH[Zn(quin)3] (4).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 4-methylpiperidine (0.5 mL) was stirred for 3 days at room temperature in a closed flask. Unreacted starting material was filtered off (a small amount) and the filtrate was stored at 4 °C. After a longer period of time, a small amount of small crystals of 4-MepipeH[Zn(quin)3] (4) was obtained. They were not suitable for X-ray diffraction analysis on a single crystal. The identity of the compound was determined by IR and 1H NMR spectroscopy. The reaction was not reproducible. IR (ATR, cm−1): 3055w, 3007w, 2958m, 2928m, 2851w, 2732w, 2506w, 1651s, 1628s, 1558s, 1509m, 1459s, 1430w, 1358vs, 1342s, 1299m, 1259m, 1218m, 1207m, 1175s, 1151m, 1108w, 1076w, 1041w, 1021w, 993w, 965m, 951w, 896s, 856s, 801vs, 775vvs, 753s, 742m, 631s, 601s, 552w, 523m, 499s, 483w, 447w, 413m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.59 (3H, d, J = 8.5 Hz, quin−), 8.55 (3H, d, J = 8.5 Hz, quin−), 8.38 (2H, br s, 4-MepipeH+), 8.20 (3H, d, J = 8.5 Hz, quin−), 8.05 (3H, d, J = 7.8 Hz, quin−), 7.75–7.73 (3H, m, quin−), 7.67–7.65 (3H, m, quin−), 3.20 (2H, dt, J = 12.6, 2.8 Hz, 4-MepipeH+), 2.79 (2H, td, J = 12.6, 2.8 Hz, 4-MepipeH+), 1.71–1.67 (2H, m, 4-MepipeH+), 1.61–1.53 (1H, m, 4-MepipeH+), 1.24–1.17 (2H, m, 4-MepipeH+), 0.88 (3H, d, J = 6.6 Hz, 4-MepipeH+) ppm.
Preparation of [Zn(quin)2(4-Mepipeam)]·CH3CN (5).
Procedure A. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 4-methylpiperidine (1 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered and the filtrate was stored at 4 °C. Single crystals of [Zn(quin)2(4-Mepipeam)]·CH3CN (5) were filtered off on the following day. Yield: 74 mg. Procedure B. Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 4-methylpiperidine (0.5 mL) was stirred for 3 days at room temperature in a closed flask. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of 5 were filtered off on the following day. Yield: 61 mg. IR (ATR, cm−1): 3288m, 3059w, 3024w, 2948w, 2925w, 2870w, 2249w, 1638s, 1584s, 1570s, 1506m, 1484m, 1459s, 1433s, 1341vs, 1257m, 1218m, 1180s, 1146m, 1115m, 1084m, 1052m, 1021w, 969m, 958m, 896s, 875m, 852m, 826m, 801vvs, 780s, 770vvs, 735s, 637s, 628s, 602s, 560m, 523m, 497s, 481m, 428w, 404m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 8.1 Hz, quin−), 8.49 (2H, d, J = 8.4 Hz, quin−), 8.36 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.0 Hz, quin−), 7.99–7.96 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 6.96 (1H, br s, 4-Mepipeam), 3.87–3.70 (2H, br m, 4-Mepipeam), 2.72–2.62 (2H, br m, 4-Mepipeam), 1.90 (3H, s, 4-Mepipeam), 1.50–1.41 (2H + 1H, m, 4-Mepipeam), 0.74 (3H, d, J = 6.2 Hz, 4-Mepipeam), 0.69–0.61 (2H, br m, 4-Mepipeam) ppm. Elemental analysis calcd for C28H28N4O4Zn = [Zn(quin)2(4-Mepipeam)] (%): C, 61.15; H, 5.13; N, 10.19. Found (%): C, 60.69; H, 5.54; N, 10.18. ESI-HRMS m/z calcd for [C28H29N4O4Zn]+ = [Zn(quin)2(4-Mepipeam) + H]+: 549.1475, found: 549.1474.
Preparation of [Zn(quin)2(4-amidepipe)2]·2CH3CN (6).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 4-piperidinecarboxamide (100 mg). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Single crystals of [Zn(quin)2(4-amidepipe)2]·2CH3CN (6) were filtered off. Yield: 152 mg. IR (ATR, cm−1): 3261m, 3151m, 3067m, 2967w, 2928w, 2858w, 2252w, 1687m, 1628vvs, 1597s, 1568m, 1557m, 1510m, 1464s, 1449m, 1429w, 1371vs, 1346s, 1300m, 1273m, 1254m, 1213w, 1179m, 1158s, 1121w, 1091m, 1025m, 1003m, 961m, 920m, 893s, 861m, 803s, 777vs, 750m, 718m, 672m, 637s, 602s, 580m, 521m, 503s, 472m, 427m, 404m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.81 (2H, d, J = 8.4 Hz), 8.57 (2H, br s), 8.36 (2H, d, J = 8.5 Hz), 8.29 (2H, d, J = 8.5 Hz), 8.21 (2H, d, J = 8.0 Hz), 8.01–7.98 (2H, m), 7.86–7.83 (2H, m), 7.43 (2H, br s), 6.94 (2H, br s), 3.32 (4H, dt, J = 12.8, 3.3 Hz), 2.96–2.89 (4H, m), 2.47–2.42 (2H, m), 1.92–1.89 (4H, m), 1.79–1.72 (4H, m) ppm. Note. Due to poor solubility of 6 in (CD3)2SO, TFA was added, which has a resonance at 14.61 ppm. The ratio of the integrals agrees with the proposed composition of the complex. ESI-HRMS m/z calcd for [C26H25N4O5Zn]+ = [Zn(quin)2(4-amidepipe) + H]+: 537.1111, found: 537.1113.
Preparation of [Zn(quin)2(4-amidepipeam)] (7).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 4-piperidinecarboxamide (100 mg) was stirred for 3 days at room temperature in a closed flask. White crystalline solid, [Zn(quin)2(4-amidepipeam)] (7), was filtered off. Yield: 63 mg, 47%. The filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. After a few days, large crystals of 4-carboxamidopiperidinoacetamidine (4-amidepipeam, 8) grew from the solution. We were unable to obtain single crystals of 7 suitable for X-ray diffraction analysis. The identity of 7 was determined by IR, 1H NMR spectroscopy and mass spectrometry. IR (ATR, cm−1): 3437w, 3297m, 3177w, 3067w, 2955w, 2929w, 2250w, 1684m, 1640vvs, 1584s, 1568s, 1509m, 1494m, 1460s, 1448m, 1434m, 1402m, 1369s, 1349s, 1340s, 1267m, 1214m, 1177s, 1154m, 1117m, 1082w, 1028m, 995m, 982w, 963w, 920w, 899s, 877m, 857s, 828m, 803vs, 771vs, 744m, 733m, 701m, 637s, 602s, 577m, 561m, 544m, 522m, 497s, 485s, 444w, 406m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 7.2 Hz, quin−), 8.51 (2H, d, J = 6.6 Hz, quin−), 8.35 (2H, d, J = 8.0 Hz, quin−), 8.21 (2H, d, J = 7.2 Hz, quin−), 8.00–7.96 (2H, br m, quin−), 7.82–7.80 (2H, m, quin−), 7.20 (1H, br s, 4-amidepipeam), 7.08 (1H, br s, 4-amidepipeam), 6.75 (1H, br s, 4-amidepipeam), 3.91–3.77 (2H, br m, 4-amidepipeam), 2.80–2.72 (2H, br m, 4-amidepipeam), 2.27–2.22 (1H, m, 4-amidepipeam), 1.93 (3H, s, 4-amidepipeam), 1.60–1.55 (2H, br m, 4-amidepipeam), 1.30–1.22 (2H, br m, 4-amidepipeam) ppm. ESI-HRMS m/z calcd for [C28H28N5O5Zn]+ = [Zn(quin)2(4-amidepipeam) + H]+: 578.1376, found: 578.1374. IR for 4-amidepipeam (8) (ATR, cm−1): 3349m, 3165m, 2945m, 2925m, 2854w, 1665s, 1622vs, 1585s, 1544s, 1476m, 1444s, 1416vs, 1370s, 1343s, 1289m, 1271m, 1254m, 1239m, 1166s, 1148s, 1122s, 1032m, 997m, 975w, 944m, 928m, 915m, 816m, 777s, 745m, 664vvs, 584s, 568s, 545s, 523m, 469m, 448m. 1H NMR for 4-amidepipeam (8) ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 7.24 (1H, br s, 4-amidepipeam), 6.74 (1H, br s, 4-amidepipeam), 6.62 (1H, br s, 4-amidepipeam), 4.02–3.96 (2H, br m, 4-amidepipeam), 2.64 (2H, td, J = 13.0, 2.5 Hz, 4-amidepipeam), 2.27–2.22 (1H, m, 4-amidepipeam), 1.98 (3H, s, 4-amidepipeam), 1.64–1.61 (2H, m, 4-amidepipeam), 1.45–1.38 (2H, m, 4-amidepipeam) ppm. ESI-HRMS m/z calcd for [C8H16N3O]+ = [4-amidepipeam + H]+: 170.1288, found: 170.1287.
Preparation of [Zn(quin)2(pz)]n (9).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and piperazine (40 mg, 0.46 mmol). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. White solid was filtered off and air-dried. Yield: 105 mg, 91%. The same product was obtained at ambient conditions. Despite numerous tries, we were unable to obtain single crystals of 9. The composition of [Zn(quin)2(pz)] was confirmed by 1H NMR spectroscopy. The polymeric structure is assumed due to extremely low solubility of the compound. IR (ATR, cm−1): 3245m, 3117w, 3087w, 3054w, 2960w, 2936w, 1639vs, 1594m, 1564m, 1509w, 1473w, 1458s, 1430w, 1361vs, 1344s, 1326s, 1295m, 1257m, 1208w, 1175m, 1145s, 1118s, 1021m, 978w, 958w, 894m, 884m, 862s, 800s, 782vvs, 637s, 616s, 601s, 554w, 522m, 503s, 416m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 9.09 (3H, br s), 8.79 (2H, d, J = 8.5 Hz), 8.34 (2H, d, J = 8.5 Hz), 8.27 (2H, d, J = 8.5 Hz), 8.20 (2H, d, J = 8.0 Hz), 8.00–7.97 (2H, m), 7.85–7.82 (2H, m), 3.36 (8H, s) ppm. Note. Due to poor solubility of 9 in (CD3)2SO, TFA was added, which has a resonance at 15.30 ppm. The ratio of the integrals agrees with the proposed composition of the complex.
Preparation of [Zn(quin)2(1-Mepz)] (10).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 1-methylpiperazine (1 mL) was stirred for 3 days at room temperature in a closed flask. White crystalline solid, [Zn(quin)2(1-Mepz)] (10), was filtered off. Yield: 100 mg, 84%. Single crystals were prepared as follows. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 1-methylpiperazine (0.5 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. White solid (either unreacted starting material or [Zn(quin)2(NH3)]) was filtered off (40–50 mg) and the filtrate was stored at 4 °C. Single crystals of 10 grew from the solution. Sometimes, a small amount of [Zn(quin)2(pz)]n (9) was obtained instead of 10. Piperazine is an impurity present in 1-methylpiperazine. IR (ATR, cm−1): 3082w, 3050w, 3012w, 2943w, 2925w, 2847w, 2797w, 1645vs, 1596m, 1570m, 1559m, 1509w, 1460m, 1451m, 1434w, 1361s, 1343s, 1289m, 1274w, 1254m, 1208m, 1176s, 1147s, 1116w, 1098m, 1067m, 1040w, 1025m, 1003m, 963m, 910w, 897m, 886m, 874s, 864m, 855s, 809s, 800s, 776vvs, 741s, 657w, 636s, 604s, 559w, 522m, 502m, 495m, 426w, 403s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.87 (2H, d, J = 8.3 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.0 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 2.63 (4H, t, J = 4.8 Hz, 1-Mepz), 2.20–2.11 (4H, br m, 1-Mepz), 2.05 (3H, s, 1-Mepz) ppm. Elemental analysis calcd for C25H24N4O4Zn (%): C, 58.89; H, 4.74; N, 10.99. Found (%): C, 58.66; H, 4.79; N, 11.04. ESI-HRMS m/z calcd for [C25H25N4O4Zn]+ = [Zn(quin)2(1-Mepz) + H]+: 509.1162, found: 509.1161.
Preparation of [Zn(quin)2(1-Mepz)2]·0.5CH3CN (11).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 1-methylpiperazine (2 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered and the filtrate was stored at 4 °C. A small amount of single crystals of [Zn(quin)2(1-Mepz)2]·0.5CH3CN (11) grew from the solution. The reaction was not reproducible. IR (ATR, cm−1): 3205w, 3162w, 3048w, 2961w, 2931m, 2837w, 2790w, 2761w, 2737w, 2249w, 1634vs, 1600s, 1567s, 1508m, 1462m, 1442s, 1363vs, 1335m, 1286m, 1191m, 1181m, 1145s, 1106m, 1038m, 1022s, 998s, 963m, 906w, 892m, 856s, 807s, 794s, 772vvs, 740m, 638m, 627s, 600m, 554w, 522m, 497s, 483w, 425w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.89 (2H, d, J = 8.4 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 2.64 (8H, t, J = 4.9 Hz, 1-Mepz), 2.20–2.13 (8H, br m, 1-Mepz), 2.07 (6H, s, 1-Mepz) ppm. Elemental analysis calcd for C30H36N6O4Zn = [Zn(quin)2(1-Mepz)2] (%): C, 59.07; H, 5.95; N, 13.78. Found (%): C, 58.85; H, 5.71; N, 13.84. ESI-HRMS m/z calcd for [C25H25N4O4Zn]+ = [Zn(quin)2(1-Mepz) + H]+: 509.1162, found: 509.1162.
Preparation of [Zn(quin)2(1-Mepzam)] (12).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 1-methylpiperazine (1 mL) was stirred for 3 days at room temperature in a closed flask. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of [Zn(quin)2(1-Mepzam)] (12) were filtered off on the following day. Yield: 64 mg. The product is a solvate. The crystals are not stable when taken out from the mother liquor. The solvent molecules are in a crystallographic disorder which could not be modelled. As a result, the identity of the solvent and its quantity are unknown. Furthermore, crystals of two different unit cells were obtained. The parameters for the first cell are: triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.6790(4), b = 13.2115(8), c = 15.0392(9) Å, α = 80.519(5), β = 80.889(5), γ = 78.091(5)°, V = 1460.24(15) Å3, and for the second: triclinic P, a = 7.9372(2), b = 11.9685(4), c = 15.1081(4) Å, α = 79.394(3), β = 79.461(2), γ = 82.938(2)°, V = 1380.97(7) Å3. The better quality data, the second one, were deposited with the CCDC. IR (ATR, cm−1): 3474w, 3273m, 2940w, 2849w, 2800w, 1640vvs, 1586s, 1564s, 1510m, 1483m, 1462s, 1433m, 1365s, 1356s, 1344vs, 1286s, 1267m, 1240m, 1220m, 1180m, 1152m, 1127s, 1114m, 1089w, 1044m, 1008s, 971m, 943w, 917w, 899s, 875w, 852s, 800vs, 774vvs, 743m, 639s, 631s, 603s, 562m, 521s, 493s, 483s, 430m, 405s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 8.0 Hz, quin−), 8.51 (2H, d, J = 8.4 Hz, quin−), 8.36 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.0 Hz, quin−), 8.00–7.97 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 7.10 (1H, br s, 1-Mepzam), 3.26–3.25 (4H, m, 1-Mepzam), 2.08–2.06 (4H + 3H, br m, 1-Mepzam), 1.92 (3H, s, 1-Mepzam) ppm. ESI-HRMS m/z calcd for [C27H28N5O4Zn]+ = [Zn(quin)2(1-Mepzam) + H]+: 550.1427, found: 550.1420.
, a = 7.6790(4), b = 13.2115(8), c = 15.0392(9) Å, α = 80.519(5), β = 80.889(5), γ = 78.091(5)°, V = 1460.24(15) Å3, and for the second: triclinic P, a = 7.9372(2), b = 11.9685(4), c = 15.1081(4) Å, α = 79.394(3), β = 79.461(2), γ = 82.938(2)°, V = 1380.97(7) Å3. The better quality data, the second one, were deposited with the CCDC. IR (ATR, cm−1): 3474w, 3273m, 2940w, 2849w, 2800w, 1640vvs, 1586s, 1564s, 1510m, 1483m, 1462s, 1433m, 1365s, 1356s, 1344vs, 1286s, 1267m, 1240m, 1220m, 1180m, 1152m, 1127s, 1114m, 1089w, 1044m, 1008s, 971m, 943w, 917w, 899s, 875w, 852s, 800vs, 774vvs, 743m, 639s, 631s, 603s, 562m, 521s, 493s, 483s, 430m, 405s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 8.0 Hz, quin−), 8.51 (2H, d, J = 8.4 Hz, quin−), 8.36 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.0 Hz, quin−), 8.00–7.97 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 7.10 (1H, br s, 1-Mepzam), 3.26–3.25 (4H, m, 1-Mepzam), 2.08–2.06 (4H + 3H, br m, 1-Mepzam), 1.92 (3H, s, 1-Mepzam) ppm. ESI-HRMS m/z calcd for [C27H28N5O4Zn]+ = [Zn(quin)2(1-Mepzam) + H]+: 550.1427, found: 550.1420.
Preparation of [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 1-ethylpiperazine (2 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered and the filtrate was stored at 4 °C. Single crystals of [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13) were filtered off on the following day. Yield: 87 mg. Reactions in an autoclave with a smaller amount of ligand (0.5 or 1 mL) produced a complex with water or ammonia as the first precipitate, whereas 13 or [Zn(quin)2(pz)]n (9) crystallized from the filtrate. Piperazine is an impurity in 1-ethylpiperazine. IR (ATR, cm−1): 3161w, 3061w, 2971w, 2930m, 2816w, 2768w, 1634vvs, 1600s, 1567s, 1508m, 1464s, 1441m, 1364vs, 1306m, 1284m, 1271w, 1257w, 1206m, 1178s, 1148m, 1104m, 1056w, 1022s, 963w, 939s, 917w, 893m, 855s, 807s, 795s, 774vvs, 739m, 635s, 627m, 600m, 554w, 522m, 499m, 481w, 457w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.92 (2H, d, J = 8.4 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 2.64 (8H, t, J = 4.9 Hz, 1-Etpz), 2.25–2.18 (12H, m, 1-Etpz), 0.92 (6H, t, J = 7.2 Hz, 1-Etpz) ppm. Elemental analysis calcd for C32H40N6O4Zn = [Zn(quin)2(1-Etpz)2] (%): C, 60.23; H, 6.32; N, 13.17. Found (%): C, 60.03; H, 6.33; N, 13.36. ESI-HRMS m/z calcd for [C26H27N4O4Zn]+ = [Zn(quin)2(1-Etpz) + H]+: 523.1318, found: 523.1317.
Preparation of [Zn(quin)2(1-Etpzam)]·H2O (14).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 1-ethylpiperazine (2 mL) was stirred for 3 days at room temperature in a closed flask. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of [Zn(quin)2(1-Etpzam)]·H2O (14) were filtered off on the following day. Yield: 71 mg, 52%. IR (ATR, cm−1): 3395m, 3287m, 3068w, 2975w, 2812m, 2768w, 1635vs, 1583s, 1563s, 1509m, 1489m, 1460s, 1429m, 1366s, 1345s, 1307s, 1290m, 1265m, 1239m, 1220m, 1182s, 1146s, 1125s, 1093w, 1076w, 1051m, 1021m, 1004s, 969m, 898m, 876m, 853s, 834m, 800s, 774vvs, 740s, 638s, 602s, 560m, 522s, 491s, 403s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 8.0 Hz, quin−), 8.50 (2H, d, J = 8.4 Hz, quin−), 8.36 (2H, d, J = 8.4 Hz, quin−), 8.21 (2H, d, J = 8.0 Hz, quin−), 7.99–7.97 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 7.11 (1H, br s, 1-Etpzam), 3.27–3.25 (4H, m, 1-Etpzam), 2.20 (2H, q, J = 7.2 Hz, 1-Etpzam), 2.12–2.10 (4H, m, 1-Etpzam), 1.92 (3H, s, 1-Etpzam), 0.91 (3H, t, J = 7.2 Hz, 1-Etpzam) ppm. Elemental analysis calcd for C28H31N5O5Zn = [Zn(quin)2(1-Etpzam)]·H2O (%): C, 57.69; H, 5.36; N, 12.01. Found (%): C, 58.01; H, 5.55; N, 12.17. ESI-HRMS m/z calcd for [C28H30N5O4Zn]+ = [Zn(quin)2(1-Etpzam) + H]+: 564.1584, found: 564.1578.
Preparation of [Zn(quin)2(1-OHEtpz)2]·2CH3CN (15).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 1-(2-hydroxyethyl)piperazine (800 mg) was heated in a round-bottom flask under reflux for 6 h. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Reaction mixture was filtered and the filtrate was stored at 4 °C. Single crystals of [Zn(quin)2(1-OHEtpz)2]·2CH3CN (15) were filtered off on the following day. Yield: 109 mg. IR (ATR, cm−1): 3230w, 3056w, 2940m, 2875w, 2813m, 2246w, 1627vs, 1564s, 1508m, 1461s, 1442m, 1431m, 1365vs, 1346s, 1322m, 1306m, 1278m, 1268m, 1216w, 1176s, 1153m, 1133m, 1104m, 1087m, 1065s, 1040m, 1025m, 1011m, 996s, 963m, 926m, 894s, 869m, 850s, 800vs, 774vvs, 635s, 600s, 557m, 522m, 497m, 451w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.89 (2H, d, J = 7.3 Hz, quin−), 8.79 (2H, d, J = 8.1 Hz, quin−), 8.41 (2H, d, J = 8.1 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.00 (2H, br m, quin−), 7.83–7.81 (2H, m, quin−), 4.31 (2H, br s, 1-OHEtpz), 3.44 (4H, t, J = 6.4 Hz, 1-OHEtpz), 2.64 (8H, t, J = 4.9 Hz, 1-OHEtpz), 2.31–2.25 (12H, m, 1-OHEtpz) ppm. ESI-HRMS m/z calcd for [C26H27N4O5Zn]+ = [Zn(quin)2(1-OHEtpz) + H]+: 539.1267, found: 539.1263.
Preparation of [Zn(quin)2(1-OHEtpzam)] (16).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 1-(2-hydroxyethyl)piperazine (500 mg) was stirred for 3 days at room temperature in a closed flask. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of [Zn(quin)2(1-OHEtpzam)] (16) were filtered off on the following day. Yield: 74 mg. IR (ATR, cm−1): 3394m, 3294m, 3066w, 2950w, 2909w, 2859w, 2799w, 2751w, 2249w, 1639vs, 1591s, 1569s, 1505m, 1486m, 1459s, 1433m, 1376s, 1356s, 1343vs, 1303s, 1288s, 1255m, 1241m, 1216m, 1184s, 1173m, 1152s, 1144s, 1083m, 1073m, 1064m, 1051m, 1010s, 966m, 943w, 917w, 897s, 852m, 802vvs, 776vvs, 743m, 654w, 638s, 602s, 560m, 522s, 497s, 482m, 405m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 7.8 Hz, quin−), 8.54 (2H, d, J = 7.8 Hz, quin−), 8.36 (2H, d, J = 8.2 Hz, quin−), 8.21 (2H, d, J = 7.8 Hz, quin−), 8.00–7.97 (2H, m, quin−), 7.83–7.81 (2H, m, quin−), 7.13 (1H, br s, 1-OHEtpzam), 4.38 (1H, t, J = 5.3 Hz, 1-OHEtpzam), 3.43–3.41 (2H, m, 1-OHEtpzam), 3.26–3.25 (4H, br m, 1-OHEtpzam), 2.26 (2H, t, J = 6.1 Hz, 1-OHEtpzam), 2.19–2.17 (4H, br m, 1-OHEtpzam), 1.92 (3H, s, 1-OHEtpzam) ppm. Elemental analysis calcd for C28H29N5O5Zn = [Zn(quin)2(1-OHEtpzam)] (%): C, 57.89; H, 5.03; N, 12.06. Found (%): C, 57.46; H, 5.17; N, 12.16. ESI-HRMS m/z calcd for [C28H30N5O5Zn]+ = [Zn(quin)2(1-OHEtpzam) + H]+: 580.1533, found: 580.1532.
Preparation of [Zn(quin)2(1-Phpz)2]·CH3CN (17).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 1-phenylpiperazine (0.5 mL) was stirred for 3 days at room temperature in a closed flask. White crystalline solid, [Zn(quin)2(1-Phpz)2]·CH3CN (17), was filtered off. Yield: 134 mg. Single crystals were prepared as follows. A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 1-phenylpiperazine (0.5 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Unreacted starting material was filtered off (74 mg) and the filtrate was stored at 4 °C. Single crystals of 17 grew from the solution. IR (ATR, cm−1): 3163w, 3057w, 2959w, 2929m, 2831w, 2252w, 1629vs, 1598vs, 1567s, 1495s, 1463s, 1440m, 1378s, 1361vs, 1337s, 1243s, 1191m, 1175s, 1158s, 1109m, 1067m, 1045m, 1021s, 992s, 962m, 922vs, 893s, 855s, 806s, 794s, 775vvs, 755vs, 693vs, 653m, 635s, 627s, 602s, 555w, 522s, 498s, 485w, 445w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.88 (2H, d, J = 8.7 Hz, quin−), 8.79 (2H, d, J = 8.4 Hz, quin−), 8.41 (2H, d, J = 8.4 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 8.03–8.00 (2H, m, quin−), 7.83–7.80 (2H, m, quin−), 7.20–7.16 (4H, m, 1-Phpz), 6.89–6.87 (4H, m, 1-Phpz), 6.76–6.73 (2H, m, 1-Phpz), 3.02–2.99 (8H, m, 1-Phpz), 2.82–2.80 (8H, m, 1-Phpz) ppm. Elemental analysis calcd for C40H40N6O4Zn = [Zn(quin)2(1-Phpz)2] (%): C, 65.44; H, 5.49; N, 11.45. Found (%): C, 65.19; H, 5.63; N, 11.53. ESI-HRMS m/z calcd for [C30H27N4O4Zn]+ = [Zn(quin)2(1-Phpz) + H]+: 571.1318, found: 571.1308.
Preparation of [Zn(quin)2(1-Phpz)2]·2acetamide (18).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (2 mL) and 1-phenylpiperazine (2 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of [Zn(quin)2(1-Phpz)2]·2acetamide grew from the solution. The compound crystalized in two polymorphic modifications, which were labelled 18a and 18b (the label 18 pertains to the compound). Yield: 91 mg, 46%. IR for the first polymorph (ATR, cm−1): 3359m, 3222m, 3170m, 3052w, 3021w, 2965w, 2937w, 2884w, 2820m, 1673s, 1641vs, 1597vs, 1577m, 1564s, 1508m, 1491s, 1459s, 1444m, 1416m, 1365vs, 1347s, 1331s, 1322s, 1297m, 1283m, 1268m, 1257s, 1239vs, 1189m, 1175m, 1152s, 1106s, 1077w, 1055m, 1030s, 1021s, 994m, 962m, 934m, 917s, 893s, 855m, 808s, 798s, 775vs, 760vvs, 745s, 694vs, 652s, 634s, 599s, 587s, 530s, 501m, 481m, 460s, 422w, 403m. IR for the second polymorph (ATR, cm−1): 3420w, 3297w, 3234m, 3062w, 2977w, 2959w, 2928w, 2855w, 2826w, 1728w, 1669vvs, 1630s, 1597s, 1569m, 1557m, 1497s, 1461s, 1443m, 1433m, 1372vs, 1347s, 1336s, 1300w, 1270m, 1244s, 1208m, 1196m, 1181m, 1156s, 1112m, 1044m, 1016s, 990m, 964w, 922s, 894s, 858m, 812s, 802s, 770vvs, 745m, 699s, 636s, 601s, 584s, 559s, 528s, 521s, 500s, 483m, 447s. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.83–8.79 (4H, m, quin−), 8.41–8.40 (2H, m, quin−), 8.20 (2H, d, J = 7.1 Hz, quin−), 8.04–7.98 (2H, br m, quin−), 7.84–7.80 (2H, br m, quin−), 7.27 (2H, br s, acetamide), 7.21–7.17 (4H, m, 1-Phpz), 6.91–6.88 (4H, m, 1-Phpz), 6.76–6.74 (2H, m, 1-Phpz), 6.67 (2H, br s, acetamide), 3.03–3.01 (8H, m, 1-Phpz), 2.83–2.81 (8H, m, 1-Phpz), 1.75 (6H, 2s, acetamide) ppm. Elemental analysis calcd for C44H50N8O6Zn = [Zn(quin)2(1-Phpz)2]·2acetamide (%): C, 62.01; H, 5.91; N, 13.15. Found (%): C, 62.16; H, 6.27; N, 13.17. ESI-HRMS m/z calcd for [C30H27N4O4Zn]+ = [Zn(quin)2(1-Phpz) + H]+: 571.1318, found: 571.1291.
Preparation of [Zn(quin)2(1-Phpzam)]·0.5(1-Phpz)·CH3CN (19).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (5 mL), methanol (5 mL) and 1-phenylpiperazine (1 mL) was stirred for 3 days at room temperature in a closed flask. The reaction mixture was filtered and the filtrate was concentrated under reduced pressure on a rotary evaporator. A glass vial with diethyl ether was carefully inserted into the Erlenmeyer flask with the concentrate. Single crystals of [Zn(quin)2(1-Phpzam)]·0.5(1-Phpz)·CH3CN (19) were filtered off on the following day. Yield: 77 mg. IR (ATR, cm−1): 3281m, 3062w, 3005w, 2824m, 2250w, 1639s, 1598s, 1579s, 1494s, 1460s, 1427m, 1365s, 1354s, 1345vs, 1293m, 1279m, 1254m, 1228s, 1182s, 1150s, 1106m, 1070m, 1033m, 1021m, 1005m, 967m, 944w, 920w, 898s, 888m, 854m, 836m, 802vs, 773vvs, 759s, 747s, 694s, 651m, 637s, 602s, 571m, 562m, 522s, 493m, 441w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.78 (2H, d, J = 8.1 Hz, quin−), 8.55 (2H, d, J = 8.3 Hz, quin−), 8.37 (2H, d, J = 8.3 Hz, quin−), 8.20 (2H, d, J = 8.1 Hz, quin−), 7.97–7.95 (2H, m, quin−), 7.80–7.77 (2H, m, quin−), 7.21–7.17 (3H, m, 1-Phpzam + 1-Phpz), 6.90–6.88 (1H, m, 1-Phpz), 6.86–6.84 (2H, br m, 1-Phpzam), 6.80–6.77 (1H, m, 1-Phpzam), 6.76–6.73 (0.5H, m, 1-Phpz), 3.45–3.43 (4H, m, 1-Phpzam), 3.02–3.00 (2H, m, 1-Phpz), 2.96–2.94 (4H, br m, 1-Phpzam), 2.82–2.80 (2H, m, 1-Phpz), 1.99 (3H, s, 1-Phpzam) ppm. ESI-HRMS m/z calcd for [C32H30N5O4Zn]+ = [Zn(quin)2(1-Phpzam) + H]+: 612.1584, found: 612.1576.
Preparation of [Zn(quin)2(1-Acpz)2]·2CH3CN (21).
Reaction mixture containing [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (10 mL) and 1-acetylpiperazine (1.00 g) was stirred for 3 days at room temperature in a closed flask. White crystalline solid, [Zn(quin)2(1-Acpz)2]·2CH3CN (21), was filtered off. Yield: 95 mg. The compound crystalized in two polymorphic modifications, which were labelled 21a and 21b (the label 21 pertains to the compound). For the preparation of single crystals of 21a and 21b see below. IR (ATR, cm−1): 3202m, 3057w, 2968w, 2935w, 2867w, 2249w, 1631vvs, 1598m, 1566m, 1511w, 1429s, 1385s, 1366s, 1322m, 1297m, 1263s, 1219w, 1195w, 1177m, 1157m, 1104m, 1051m, 1032w, 1020m, 987s, 958s, 911w, 894m, 850s, 802vs, 778vs, 748m, 636s, 599m, 578s, 524m, 501s, 473w, 451w. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 600 MHz): δ 8.84 (2H, d, J = 8.2 Hz, quin−), 8.79 (2H, d, J = 7.9 Hz, quin−), 8.41 (2H, d, J = 7.9 Hz, quin−), 8.20 (2H, d, J = 8.0 Hz, quin−), 8.03–7.99 (2H, br m, quin−), 7.83–7.81 (2H, m, quin−), 3.34–3.30 (8H, m, 1-Acpz, overlapped with the peak for H2O), 2.66–2.64 (4H, m, 1-Acpz), 2.59–2.58 (4H, m, 1-Acpz), 1.95 (6H, s, 1-Acpz) ppm. Elemental analysis calcd for C32H36N6O6Zn = [Zn(quin)2(1-Acpz)2] (%): C, 57.71; H, 5.45; N, 12.62. Found (%): C, 57.56; H, 5.63; N, 12.38. ESI-HRMS m/z calcd for [C26H25N4O5Zn]+ = [Zn(quin)2(1-Acpz) + H]+: 537.1111, found: 537.1112.
Preparation of [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22).
A Teflon container was filled with [Zn(quin)2(H2O)] (100 mg, 0.23 mmol), acetonitrile (7 mL) and 1-acetylpiperazine (1.50 g). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Unreacted starting material was filtered off (40 mg) and the filtrate was stored at 4 °C. After a few days, a mixture of crystals of [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22), [Zn(quin)2(1-Acpz)2]·2CH3CN (21a and 21b) and [Zn(quin)2(1-Acpz)2] (20) was obtained. The reaction was not reproducible. Piperazine is an impurity present in 1-acetylpiperazine. IR (ATR, cm−1): 3206s, 3063w, 2987w, 2962w, 2928w, 2866w, 2247w, 1652s, 1619vvs, 1568s, 1510m, 1462s, 1426s, 1377s, 1359vs, 1349s, 1319m, 1301m, 1267s, 1221w, 1208m, 1194w, 1178m, 1157m, 1143m, 1105m, 1095m, 1049m, 1022s, 988s, 963s, 916w, 892s, 866s, 851s, 800s, 778vs, 748m, 723w, 635s, 600s, 579s, 556w, 523m, 496m, 470m, 405m.
Preparation of [Zn(quin)2(am)]·1.3CH3CN·0.2H2O (23).
Procedure A. A Teflon container was filled with [Zn(quin)2(H2O)] (50 mg, 0.12 mmol), acetonitrile (5 mL) and methanol (2 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Unreacted starting material was filtered off (a small amount) and the filtrate was stored at 4 °C. After a few days, single crystals of [Zn(quin)2(am)]·1.3CH3CN·0.2H2O (23) were obtained. Yield: 25 mg. Compound 23 also formed in similar reaction systems of 4-piperidinecarboxamide, piperazine, 1-methylpiperazine, 1-ethylpiperazine, 1-(2-hydroxyethyl)piperazine, 1-phenylpiperazine and 1-acetylpiperazine. IR (ATR, cm−1): 3448w, 3267m, 3193m, 2251w, 1640vs, 1596s, 1561s, 1508m, 1460s, 1435m, 1365s, 1353s, 1342s, 1273s, 1219m, 1176m, 1152m, 1141m, 1112w, 1030w, 1021w, 988w, 968m, 918w, 898m, 874m, 854m, 802vs, 776vvs, 737m, 637s, 629m, 602s, 580m, 559m, 522s, 488s, 405m. 1H NMR ((CD3)2SO with 0.03% v/v TMS, 500 MHz): δ 8.78 (2H, d, J = 7.5 Hz, quin−), 8.54 (2H, d, J = 8.5 Hz, quin−), 8.35 (2H, d, J = 8.1 Hz, quin−), 8.22 (2H, d, J = 7.5 Hz, quin−), 8.02–7.99 (2H, m, quin−), 7.84–7.82 (2H, m, quin−), 7.28 (1H, br s, am), 6.68 (1H, br s, am), 1.97 (3H, br s, am) ppm. Elemental analysis calcd for C22H18N4O4Zn = [Zn(quin)2(am)] (%): C, 56.49; H, 3.88; N, 11.98. Found (%): C, 56.31; H, 4.32; N, 12.30. ESI-HRMS m/z calcd for [C22H19N4O4Zn]+ = [Zn(quin)2(am) + H]+: 467.0692, found: 467.0691.
Reaction of [Zn(quin)2(NH3)] with acetonitrile.
A Teflon container was filled with [Zn(quin)2(NH3)] (50 mg, 0.12 mmol), acetonitrile (5 mL) and methanol (2 mL). The container was closed and inserted into a steel autoclave, which was heated for 24 h at 105 °C. Afterwards, the reaction mixture was allowed to cool slowly to room temperature. Unreacted starting material was filtered off (19 mg) and the filtrate was stored at 4 °C. After a few days, single crystals of [Zn(quin)2(am)]·1.3CH3CN·0.2H2O (23) were obtained. Yield: 17 mg.
Synthetic considerations
The reaction system, comprising [Zn(quin)2(H2O)] as a Zn(II) starting material, a secondary amine, acetonitrile, and in some cases, methanol, was the same as the one employed in our previous studies.17 The beneficial factors concerning the formation of amidines, recognized in previous studies,17,33 were taken into account. A pool of reacting amines was broadened to include thiomorpholine, piperidine analogs, and piperazine and its analogs. The studied amines all share a common feature: all are secondary cyclic amines with an additional functional group that has a potential steric or electronic impact on the reactivity.
Firstly, the reaction systems without methanol will be described. The typical products isolated from these systems speak of the straightforward substitution of coordinated water in [Zn(quin)2(H2O)] with one or two molecules of the reacting amine. The fact that more diamine than monoamine complexes have been isolated suggests the transient nature of monoamine complexes.17 Of the nine amines used in this study, only one, namely 1-methylpiperazine, afforded a monoamine complex, [Zn(quin)2(1-Mepz)] (10). Interestingly, this monoamine complex formed under both mild (ambient temperature) or harsh reaction conditions (heating at 105 °C in an autoclave). Nevertheless, the autoclave reaction with an increased amount of amine predictably produced the corresponding diamine complex, [Zn(quin)2(1-Mepz)2], found in compound 11. As will be shown, the 1-methylpiperazine reaction system is more complicated because piperazine was found to be an inherent impurity, not only in 1-methylpiperazine, but in several other substituted piperazines. Irrespective of the conditions, the piperazine reactions produced a highly insoluble compound labeled as compound 9. Unfortunately, numerous attempts to obtain single crystals of 9 remained fruitless. The composition of 9, [Zn(quin)2(pz)]n, was determined using 1H NMR and IR spectroscopies. Compound 9 was assumed to be polymeric in nature due to its extremely low solubility. The same compound was obtained in small amounts (on the 10 mg scale) from the reaction systems of two other amines, 1-methylpiperazine and 1-ethylpiperazine. The precipitation of [Zn(quin)2(pz)]n (9) from the latter two reaction systems suggested that both amines contained piperazine as an impurity. The MS analysis of a sample of 1-ethylpiperazine showed a m/z peak at 87.0920, which is characteristic of piperazine. Furthermore, the NMR spectra of 1-ethylpiperazine undoubtedly revealed piperazine resonances at 2.59 ppm (1H) and at 46.9 ppm (13C). From the integral of the 2.59 ppm resonance, the piperazine content in 1-ethylpiperazine was estimated to be ca. 0.5%. With a quantitative conversion of piperazine to its Zn(II) complex, followed by its complete precipitation, the amount of isolated solid was sufficient to be unambiguously identified by IR spectroscopy as [Zn(quin)2(pz)]n (9). Further confirmation about the presence of piperazine in its analogs was obtained from the X-ray structure analysis of [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22), a compound which formed in the reaction mixture of 1-acetylpiperazine. The cocrystal contains two complex species, a diamine complex with 1-acetylpiperazine and a polymeric complex with piperazine. The amount of 22 was small, in line with the estimated piperazine content in 1-acetylpiperazine. The X-ray structure analysis of 22 further supports the polymeric nature of piperazine complex 9.
Very rarely, protonation of the reacting amine was observed, as exemplified by the room-temperature reaction of [Zn(quin)2(H2O)] with 4-methylpiperidine, which produced 4-MepipeH[Zn(quin)3] (4). The composition of 4 implies that even under ambient conditions, the rupture of quinaldinate–Zn(II) bonds took place. As observed previously,17 neither a source of the acid nor a resulting Zn(II) complex with fewer than two quinaldinates per metal ion were identified.
When methanol was introduced to the reaction mixture, the reaction took a different course. With the exception of 1-acetylpiperazine and piperazine, all the amines reacted with acetonitrile to form the corresponding amidine, which coordinated to Zn(II). The composition of the amidine complexes was [Zn(quin)2(amidine)]. As the only amine, 4-piperidinecarboxamide produced aside from the Zn(II) amidine complex also the amidine itself, i.e., 4-carboxamidopiperidinoacetamidine (8), which we conveniently named 4-amidepipeam. This compound has not been prepared previously. The largest of the reacting amines, 1-phenylpiperazine, also yielded the amidine complex. The bulky phenyl moiety did not intervene with the formation of amidine and its subsequent coordination to Zn(II). The failure of 1-acetylpiperazine to produce amidine can be explained in terms of the lowered nucleophilicity of its non-substituted nitrogen atom. A negative inductive effect of the acetyl moiety makes this nitrogen less reactive.34 For the piperazine system, a plausible explanation lies in the low solubility of [Zn(quin)2(pz)]n (9) and its prompt precipitation. Consequently, piperazine eliminated the Zn(II) species, which is necessary for the catalysis, from the solution. Nevertheless, both the 1-acetylpiperazine and piperazine reaction systems produced a complex with acetamidine (am) [Zn(quin)2(am)], as found in compound 23. It is to be noted that the acetamidine complex 23 formed in the reaction systems of several other amines (see the Experimental). Acetamidine is known to form in a manner similar to that of amidines, with the only difference being that the reacting amine is ammonia (Scheme 3). A few other transition metal ions are known to catalyze this reaction. By analogy to our system, the in situ formed acetamidine coordinated to the transition metal ion used.35–43 The reaction described in Scheme 3 implies the presence of ammonia in the reaction systems. The formation of ammonia has been observed previously.24 In some instances, an ammine complex with the [Zn(quin)2(NH3)] composition was obtained. Ammonia could result from the decomposition of amines or the hydrolysis of acetonitrile under solvothermal conditions. A blank experiment when the [Zn(quin)2(H2O)]–methanol–acetonitrile mixture was heated at 105 °C in an autoclave without any other reagents supports the latter. This mixture produced a Zn(II) acetamidine complex reproducibly and in good yield. Unfortunately, the amount of water in the mixture could not be controlled as it was introduced not only via the zinc(II) starting material, but also with methanol. Thus, the extent of acetonitrile that underwent hydrolysis remained variable from one batch to another. In another synthetic strategy to prepare the acetamidine complex [Zn(quin)2(am)], the Zn(II) ammine complex [Zn(quin)2(NH3)] was used as a source of ammonia. The ammine complex was reacted under the same conditions with a mixture of acetonitrile and methanol. Taking into account that 38% of the starting material remained undissolved, the conversion of the consumed [Zn(quin)2(NH3)] to the acetamidine complex [Zn(quin)2(am)] was ca. 50%. In spite of numerous attempts, the yield could not be improved. To introduce larger amounts of ammonia in the reaction mixture, a simple ammonium salt and a base were used. Thus, ammonium chloride and triethylamine were added in an up to ten-fold excess to the [Zn(quin)2(H2O)]–methanol–acetonitrile mixture. Yet, 24-h heating at 105 °C in autoclaves produced a complex with ammonia or a complex with acetamidine or both. It appears that with a larger amount of ammonia, more [Zn(quin)2(NH3)] formed. With larger amounts of the complex with ammonia, its crystallization was more likely to take place. This being the case, the concentration of Zn(II) species in the solution was lowered to such a level that only negligible amounts or no acetamidine complex was isolated. Acetonitrile hydrolysis can stop halfway and produce acetamide. Its formation was undoubtedly confirmed for the 1-phenylpiperazine-[Zn(quin)2(H2O)]–methanol–acetonitrile mixture, which was heated in an autoclave at 105 °C (see the preparation of 18). Namely, a 1-phenylpiperazine diamine complex formed a cocrystal with acetamide, which interestingly crystallized in two polymorphic forms.
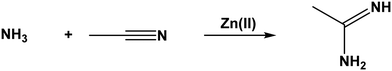 |
| | Scheme 3 The reaction between ammonia and acetonitrile leads to the formation of acetamidine. | |
Crystal structures
The crystal structures of the following compounds with coordinated amines were determined: [Zn(quin)2(thiomorph)2] (1), [Zn(quin)2(4-Mepipe)2] (3), [Zn(quin)2(4-amidepipe)2]·2CH3CN (6), [Zn(quin)2(1-Mepz)] (10), [Zn(quin)2(1-Mepz)2]·0.5CH3CN (11), [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13), [Zn(quin)2(1-OHEtpz)2]·2CH3CN (15), [Zn(quin)2(1-Phpz)2]·CH3CN (17), [Zn(quin)2(1-Phpz)2]·2acetamide (18a and 18b), [Zn(quin)2(1-Acpz)2] (20), [Zn(quin)2(1-Acpz)2]·2CH3CN (21a and 21b), and [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22). The quinaldinate ligand is bound in the usual N,O-bidentate chelating manner, whereas the amine ligands exhibit monodentate coordination via their non-substituted nitrogen donor atom. The only exception of monodentate coordination was observed in 22, in which the piperazine is coordinated in a bidentate bridging manner. The formation of a diamine complex was observed for all the amines used. The Zn(II) ion is surrounded by two quinaldinates and two amine ligands that adopt a trans configuration. The formation of cis isomers was not observed. In our previous studies, the cis isomer was obtained only as part of a cocrystal [Zn(quin)2(pipe)]·cis-[Zn(quin)2(pipe)2].17 The diamine complexes are centrosymmetric, with the N4O2 donor set occupying the vertices of a distorted octahedron. Their geometric parameters are similar, as shown in Table 2. The Zn(II)-quinaldinate and Zn(II)–amine bond lengths are comparable to those of known complexes.17 In the diamine complexes, the bonds between the Zn(II) ion and the amine ligand span a 2.1848(13)–2.2488(10) Å interval. The ORTEP drawing of [Zn(quin)2(1-Mepz)2] of 11 is shown in Fig. 1, while the ORTEP drawings of all other diamine complexes are found in the ESI† (Fig. S1–S7). In a monoamine complex, [Zn(quin)2(1-Mepz)] (10), the Zn(II) ion is five-coordinated with two quinaldinates and one 1-methylpiperazine ligand. The ORTEP drawing of the monoamine complex is shown in Fig. 1. Several geometric parameters of 10 are significantly shorter than those of the diamine complexes. The difference in the lengths of the Zn(II)–quinaldinate bonds is, on average, smaller than 0.1 Å, but more pronounced is the difference in the Zn(II)–amine bond, e.g., 2.0444(16) Å in 10 compared to 2.1915(12)–2.2135(12) Å in 11. The N3O2 donor set of the monoamine complex occupies the vertices of a distorted square pyramid, as shown by the τ parameter of 0.28.44 In 10, the aromatic planes of the quinaldinates form a dihedral angle of 53.84(4)°.
Table 2 Relevant geometric parameters [Å] of amine complexes
| Compound |
Donors |
Zn–N(amine) |
Zn–O(quin−) |
Zn–N(quin−) |
|
Pertains to [Zn(quin)2(1-Acpz)2].
Pertains to [Zn(quin)2(pz)]n.
|
| [Zn(quin)2(thiomorph)2] (1) |
N4O2 |
2.2205(11) |
2.0611(9) |
2.2612(10) |
| [Zn(quin)2(4-Mepipe)2] (3) |
N4O2 |
2.1848(13), 2.2153(13) |
2.0694(10), 2.0576(10) |
2.2746(12), 2.2521(13) |
| [Zn(quin)2(4-amidepipe)2]·2CH3CN (6) |
N4O2 |
2.2186(15) |
2.0331(11) |
2.2422(15) |
| [Zn(quin)2(1-Mepz)] (10) |
N3O2 |
2.0444(16) |
2.0105(13), 1.9960(13) |
2.1822(15), 2.1645(15) |
| [Zn(quin)2(1-Mepz)2]·0.5CH3CN (11) |
N4O2 |
2.2083(12), 2.2135(12), 2.1915(12), 2.2177(12) |
2.0406(10), 2.0461(10), 2.0806(10), 2.0520(10) |
2.2809(12), 2.2719(12), 2.2312(12), 2.2348(11) |
| [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13) |
N4O2 |
2.1868(14), 2.2208(15) |
2.0677(12), 2.0441(12) |
2.2316(14), 2.2799(14) |
| [Zn(quin)2(1-OHEtpz)2]·2CH3CN (15) |
N4O2 |
2.2214(14), 2.2044(14) |
2.0391(12), 2.0701(12) |
2.2346(13), 2.2629(13) |
| [Zn(quin)2(1-Phpz)2]·CH3CN (17) |
N4O2 |
2.1914(11), 2.2115(11) |
2.0754(9), 2.0378(9) |
2.2415(11), 2.3050(11) |
| [Zn(quin)2(1-Phpz)2]·2acetamide (18a) |
N4O2 |
2.1894(12) |
2.0545(9) |
2.2665(12) |
| [Zn(quin)2(1-Phpz)2]·2acetamide (18b) |
N4O2 |
2.2228(10) |
2.0492(8) |
2.2368(9) |
| [Zn(quin)2(1-Acpz)2] (20) |
N4O2 |
2.2488(10) |
2.0290(9) |
2.2484(10) |
| [Zn(quin)2(1-Acpz)2]·2CH3CN (21a) |
N4O2 |
2.1953(16), 2.1887(16) |
2.0529(13), 2.0639(13) |
2.2741(14), 2.2479(14) |
| [Zn(quin)2(1-Acpz)2]·2CH3CN (21b) |
N4O2 |
2.223(2) |
2.0432(15) |
2.2423(18) |
| [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22) |
N4O2a |
2.2254(15) |
2.0453(11) |
2.2417(14) |
| N4O2b |
2.2417(14) |
2.0544(13) |
2.2038(15) |
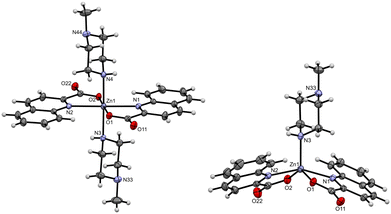 |
| | Fig. 1 ORTEP diagram of [Zn(quin)2(1-Mepz)2] found in 11 (left) and [Zn(quin)2(1-Mepz)] (10) (right). The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
Of the structures listed above, a few should be highlighted. The cocrystal [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22) is of interest in terms of its composition and structure as it consists of a six-coordinated diamine complex and one-dimensional (1D) coordination polymer. The coordination environment of Zn(II) in the six-coordinated complex is the same as described above and comprises two N,O-bidentate chelating quinaldinates and two 1-acetylpiperazine ligands in a trans orientation. The coordination environment of Zn(II) in the 1D polymer also comprises two bidentate chelating quinaldinates and two piperazines in a trans arrangement. The piperazine ligand has utilized in coordination both nitrogen atoms, thereby adopting a bidentate bridging fashion. The N4O2 donors occupy the vertices of a distorted octahedron. Fig. 2 shows the repeating unit and a chain segment in [Zn(quin)2(pz)]n. The shortest Zn⋯Zn contacts within the polymeric chain are 7.3460(2) Å. [Zn(quin)2(1-Acpz)2] molecules are hydrogen bonded into supramolecular chains. Interestingly, the same distance between the Zn(II) ions, 7.3460(2) Å, is observed within these supramolecular chains. In the structure of 22, the [Zn(quin)2(1-Acpz)2] complex molecules and the 1D coordination polymer are linked by hydrogen bonds, forming supramolecular layers in which the chains of a coordination polymer alternate with the supramolecular chains of the hydrogen-bonded complex molecules (Fig. S22, ESI†).
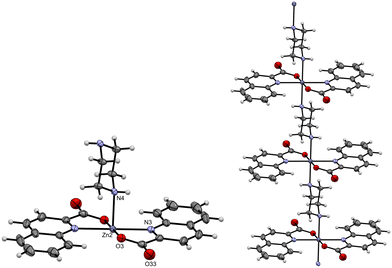 |
| | Fig. 2 ORTEP diagram of the repeating unit (left) and a short chain segment (right) of the polymeric part in [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22). The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
The connectivity motifs of the polymorphs are described in more detail. In our study, polymorphism was observed for two compounds, [Zn(quin)2(1-Phpz)2]·2acetamide and [Zn(quin)2(1-Acpz)2]·2CH3CN. This was not surprising, as around 50% of all compounds crystallize in multiple polymorphic forms.45 [Zn(quin)2(1-Phpz)2]·2acetamide crystallized in two polymorphic modifications, in triclinic (18a) and monoclinic (18b) forms. In both, the composition of the complex molecules is the same. Each complex crystallizes with two acetamide molecules per formula unit. The similarity in the complex molecules of both polymorphs was checked by superposition, which was achieved in the Mercury software package (Fig. S13, ESI†).32 The geometric parameters of the complex molecules are very similar, as shown by the value of the root mean square deviation, 0.3471. The superposition of the complexes from both polymorphs reveals that the largest difference is in the twist of the phenyl rings. Therefore, the maximum distance between two equivalent atoms is 0.8221 Å. The two polymorphs differ significantly in hydrogen bonding (Table S8, ESI†) and, consequently, in their connectivity motifs (Fig. 3). The greatest differences are observed in the hydrogen bonds involving the amine group of the acetamide. In 18a, this group is involved in the hydrogen bonds, whereas in 18b, its interaction with the carboxylate is longer than the sum of the van der Waals radii (N⋯O = 3.0704(17) Å).46 In 18a, both hydrogen atoms of the amine group of the acetamide are involved in the hydrogen bonding; one hydrogen atom of the amine group forms a hydrogen bond with the non-coordinated carboxylate oxygen of the quinaldinate and the other with the carbonyl group of the adjacent acetamide molecule. The acetamide carbonyl is engaged in another hydrogen bond with the amine N–H of an adjacent complex molecule, thus leading to the formation of supramolecular layers. The linkage of two acetamide molecules via two N–H⋯O hydrogen bonds represents a characteristic acetamide⋯acetamide homosynthon.47 Interestingly, 18b lacks such a homosynthon. Instead, a hydrogen bond between the carbonyl oxygen of acetamide and the amine group of 1-phenylpiperazine is observed, linking the complex species and two acetamide molecules. The polymorphism with acetamide molecules engaged in different hydrogen bonding patterns is not unprecedented, as exemplified by polymorphs of gallic acid and acetamide cocrystals.48 Characteristic acetamide⋯acetamide homosynthons were observed in two polymorphs, while they were absent in the other two.48
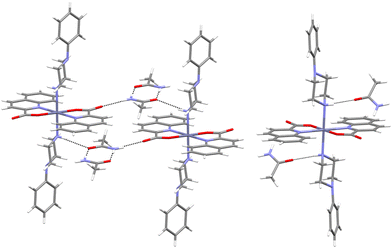 |
| | Fig. 3 Hydrogen bonding between [Zn(quin)2(1-Phpz)2] and acetamide in polymorphs 18a (left) and 18b (right). | |
Crystallization in several forms was also observed for the complex with 1-acetylpiperazine. [Zn(quin)2(1-Acpz)2] crystallized without solvent (20) or with two molecules of acetonitrile per formula unit. The latter compound crystallized in two polymorphic modifications, in triclinic (21a) and monoclinic (21b) forms. In 20, the acetyl group participates in hydrogen bonding, and supramolecular layers are observed (Fig. S20, ESI†). Each complex molecule is surrounded by six others from the same layer. The asymmetric unit in 20 contains one-half of the complex molecule. The same is true for 21b, while the asymmetric unit in 21a contains two halves of two complex molecules. The similarity was again checked by overlay (Fig. S14, ESI†). The superposition of the complexes from 21a and 21b shows that the largest difference lies in the orientation of the acetyl substituent in the piperazine ligand. The difference in connectivity patterns in the pair is not so obvious since the same type of hydrogen bonding is observed. The N–H group of the amine is hydrogen bonded to the non-coordinated carboxylate oxygen atoms of the adjacent molecule, forming infinite supramolecular chains. Sections of the two chains are shown in Fig. 4. Interestingly, the acetyl group – a hydrogen bond acceptor – is not involved in the hydrogen bonding. Close examination reveals that the difference between the chains lies in the orientation of the complex molecules (Fig. S21, ESI†). In 21a, the molecules constituting the chains are arranged in two different directions; in contrast, the molecules in 21b are fully aligned. The dissimilarity in the chains leads to different packing motifs. The packing of the chains in 21a is dense, with π⋯π stacking interactions between adjacent chains (arene⋯arene type, centroid⋯centroid distance = 3.6568(12) Å, interplanar angle = 0.00(9)°, shift distance = 0.899 Å). In the structure of 21b, there are no π⋯π stacking interactions with centroid⋯centroid distances less than 4.0 Å.49
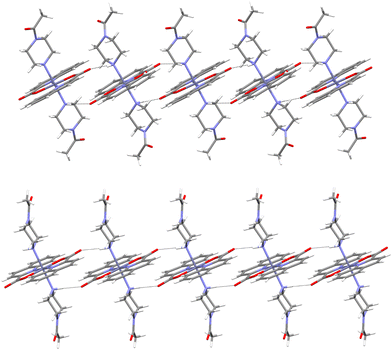 |
| | Fig. 4 Hydrogen bonding in polymorphs 21a (top) and 21b (bottom). Acetonitrile molecules are not shown for clarity. | |
Several compounds with coordinated amidines were obtained: [Zn(quin)2(thiomorpham)]·CH3CN·CH3OH (2), [Zn(quin)2(4-Mepipeam)]·CH3CN (5), [Zn(quin)2(1-Mepzam)] (12), [Zn(quin)2(1-Etpzam)]·H2O (14), [Zn(quin)2(1-OHEtpzam)] (16), [Zn(quin)2(1-Phpzam)]·0.5(1-Phpz)·CH3CN (19), and [Zn(quin)2(am)]·1.3CH3CN·0.2H2O (23). The relevant geometrical parameters of these compounds are summarized in Table 3. In all, the coordination environment of Zn(II) consists of two bidentate chelating quinaldinates and a monodentate amidine bound via an imine nitrogen. The τ parameters range from 0.41 to 0.70, suggesting that the N3O2 donors describe a polyhedron that can be regarded as an intermediate between a square pyramid and a trigonal bipyramid.44 The dihedral angle between the quinaldinates ranges from 57.56(3)° to 74.01(5)°. The Zn(II)–amidine bond, which is in the range of 1.967(2)–2.0016(13) Å, is generally much shorter than the Zn(II)–amine bond in diamine complexes (2.1848(13)–2.2488(10) Å) and slightly shorter than the corresponding bond in five-coordinated amine species (2.0444(16) Å). The Zn(II)–quinaldinate bonds are comparable to those in the monoamine complex 10. Due to the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond, amidines can exist in two configurations (as depicted in Scheme 4), referred to as E or Z. The literature suggests that the E isomers are the kinetically more favorable products.50 In our case, all amidines are in the E configuration, with the exception of acetamidine in 23. Fig. 5 shows ORTEP drawings of [Zn(quin)2(1-OHEtpzam)] (16), which features the E conformation and [Zn(quin)2(am)] in 23 with the Z conformation. The ORTEP drawings of all the other amidine complexes are shown in the ESI† (Fig. S8–S12). The absence of Z isomers can be ascribed to steric interactions between the bulky amine residues of the axial amidine and the quinaldinate ligands located in the equatorial plane.22 Interestingly, all the amidine complexes, including acetamidine complex 23, feature NH(imine)⋯COO− intermolecular interactions (Table S9, ESI†). The ORTEP drawing of the amidine itself, 4-carboxamidopiperidinoacetamidine (8), is shown in Fig. 6. In the structure of 8, each amidine molecule is hydrogen bonded via amine⋯carbonyl and amine⋯imine interactions to four neighboring molecules, which results in the formation of stacked supramolecular layers (Fig. S17, ESI†). The bonds in 8 are very similar to the ones in complexes with coordinated amidines (2, 5, 12, 14, 16, 19, and 23). For example, the length of the CN(imine) bond is 1.291(2) Å in the coordinated thiomorpham in [Zn(quin)2(thiomorpham)]·CH3CN·CH3OH (2) and 1.286(2) Å in the ‘free’ 4-amidepipeam (8). The same observation applies to the remaining bonding pattern of the sp2 hybridized carbon. It is noted that the amidine coordination to Zn(II) imparts slight changes in the angles involving this carbon atom.
N double bond, amidines can exist in two configurations (as depicted in Scheme 4), referred to as E or Z. The literature suggests that the E isomers are the kinetically more favorable products.50 In our case, all amidines are in the E configuration, with the exception of acetamidine in 23. Fig. 5 shows ORTEP drawings of [Zn(quin)2(1-OHEtpzam)] (16), which features the E conformation and [Zn(quin)2(am)] in 23 with the Z conformation. The ORTEP drawings of all the other amidine complexes are shown in the ESI† (Fig. S8–S12). The absence of Z isomers can be ascribed to steric interactions between the bulky amine residues of the axial amidine and the quinaldinate ligands located in the equatorial plane.22 Interestingly, all the amidine complexes, including acetamidine complex 23, feature NH(imine)⋯COO− intermolecular interactions (Table S9, ESI†). The ORTEP drawing of the amidine itself, 4-carboxamidopiperidinoacetamidine (8), is shown in Fig. 6. In the structure of 8, each amidine molecule is hydrogen bonded via amine⋯carbonyl and amine⋯imine interactions to four neighboring molecules, which results in the formation of stacked supramolecular layers (Fig. S17, ESI†). The bonds in 8 are very similar to the ones in complexes with coordinated amidines (2, 5, 12, 14, 16, 19, and 23). For example, the length of the CN(imine) bond is 1.291(2) Å in the coordinated thiomorpham in [Zn(quin)2(thiomorpham)]·CH3CN·CH3OH (2) and 1.286(2) Å in the ‘free’ 4-amidepipeam (8). The same observation applies to the remaining bonding pattern of the sp2 hybridized carbon. It is noted that the amidine coordination to Zn(II) imparts slight changes in the angles involving this carbon atom.
Table 3 Relevant geometric parameters [Å, °] of amidine complexes
| Compound |
Zn–N(amidine) |
Zn–O(quin−) |
Zn–N(quin−) |
τ
5
44
|
Dihedral anglea |
|
Angle between the quinaldinate aromatic planes.
|
| [Zn(quin)2(thiomorpham)]·CH3CN·CH3OH (2) |
1.9760(13) |
2.0218(10), 2.0063(10) |
2.1331(11), 2.1903(11) |
0.41 |
73.71(3) |
| [Zn(quin)2(4-Mepipeam)]·CH3CN (5) |
2.0016(13) |
2.0225(10), 2.0032(10) |
2.1738(11), 2.1533(11) |
0.63 |
60.96(3) |
| [Zn(quin)2(1-Mepzam)] (12) |
1.9953(16) |
2.0108(13), 2.0070(13) |
2.1537(14), 2.1890(14) |
0.65 |
63.46(4) |
| [Zn(quin)2(1-Etpzam)]·H2O (14) |
1.998(3) |
2.005(2), 2.0008(19) |
2.181(2), 2.159(2) |
0.70 |
58.98(6) |
| [Zn(quin)2(1-OHEtpzam)] (16) |
1.967(2) |
2.0023(17), 2.0163(16) |
2.2066(17), 2.1384(16) |
0.56 |
74.01(5) |
| [Zn(quin)2(1-Phpzam)]·0.5(1-Phpz)·CH3CN (19) |
1.9971(12) |
2.0028(10), 2.0011(10) |
2.1375(11), 2.1805(11) |
0.47 |
57.56(3) |
| [Zn(quin)2(am)]·1.3CH3CN·0.2H2O (23) |
1.9759(19) |
2.0128(15), 2.0058(14) |
2.1481(18), 2.1981(16) |
0.57 |
65.62(5) |
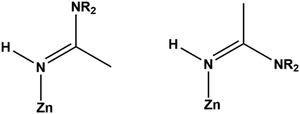 |
| | Scheme 4 Structural formulas of the E (left) and Z (right) isomers of coordinated amidine, where NR2 represents the amine residue. | |
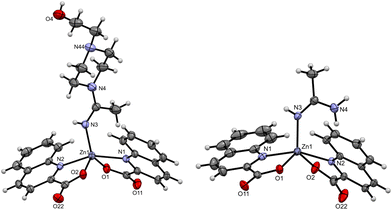 |
| | Fig. 5 ORTEP diagram of [Zn(quin)2(1-OHEtpzam)] (16) (left) and [Zn(quin)2(am)] in 23 (right). The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
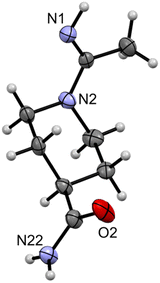 |
| | Fig. 6 ORTEP diagram of 4-carboxamidopiperidinoacetamidine (8). The displacement ellipsoids are shown at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radii. | |
In the solid state, intricate intermolecular bonding patterns are observed. Complex species are linked together by hydrogen bonds and form infinite supramolecular chains or layers. In general, the connectivity pattern is largely dependent on the amine moiety. Compounds containing amines with substituents that are not hydrogen bond donors/acceptors, e.g., thiomorpholine, 4-methylpiperidine, 1-methylpiperazine, 1-ethylpiperazine, and 1-phenylpiperazine, usually form only one type of hydrogen bond, NH⋯COO−. The amine or amidine ligands act as hydrogen bond donors via their N–H groups, whereas the non-coordinated carboxylate oxygen atoms act as acceptors. Each complex molecule is, therefore, hydrogen bonded to two adjacent molecules (Fig. S15 and S16, ESI†). Thus, infinite supramolecular chains were observed for 2, 3, 5, 10, 11, 12, 13, 17, and 19. Moreover, these chains can be arranged to form channels that can accommodate solvent molecules of crystallization. These channels allow the solvent molecules to escape when the crystals are removed from the mother liquor, which explains the observed instability of some solvates. The same type of hydrogen bond, NH⋯COO−, is observed in 1, but the connectivity is different. Each molecule is hydrogen bonded to four adjacent molecules, forming 2D supramolecular layers. Another exception is 14, where the presence of solvent molecules capable of forming hydrogen bonds alters the mode of the hydrogen bonding. The presence of substituents on the amine rings, which serve as hydrogen bond donors/acceptors, e.g., in 4-piperidinecarboxamide, 1-(2-hydroxyethyl)piperazine, and 1-acetylpiperazine, ensures the formation of supramolecular layers as the substituents participate in hydrogen bonding (Tables S8 and S9, ESI†). Such layers can be observed in 6, 15, 16, 20, and 23. Selected examples of intramolecular connectivities are shown in the ESI† (Fig. S15–S18, S20, and S23).
IR spectroscopy
The IR spectra of the Zn(II) complexes presented in this study feature a large number of absorption bands. The spectra are presented in the ESI,† but the following discussion is focused on the most prominent bands for each group of compounds. The coordinated quinaldinate leaves a characteristic imprint over the spectra of its complexes. The most informative and intense bands can be attributed to the asymmetric and symmetric C–O stretching vibrations of the COO− group, labeled as νas(COO−) and νs(COO−). For example, in the spectrum of [Zn(quin)2(thiomorph)2] (1), bands are observed at 1638, 1366, and 1351 cm−1. Ideally, the νas(COO−) − νs(COO−) difference should give information on the carboxylate binding mode.51,52 For compound 1, this difference amounts to 287 cm−1, corresponding to the monodentate binding mode of the carboxylate. Very similar values can be calculated from the spectra of compounds 2–7 and 9–23, which all exhibit monodentate carboxylate coordination.
The spectra of the amine complexes share a single absorption band of weak intensity in the region of the stretching N–H vibration as a common feature, with the band spanning a region of ca. 100 cm−1. Thus, the piperazine in [Zn(quin)2(pz)]n (9) absorbs at 3245 cm−1, whereas the 1-ethylpiperazine in [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13) exhibits a band at 3161 cm−1. For some amine complexes, with [Zn(quin)2(4-amidepipe)2]·2CH3CN (6) being such an example, no unequivocal assignment was possible. Namely, the 4-piperidinecarboxamide complex 6 possesses two groups with N–H bonds, amine and amide groups. In the spectrum of 6, two bands can be observed at 3261 and 3151 cm−1. Interestingly, the spectrum of [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22), which contains two different amine complexes, features a single ν(N–H) band at 3206 cm−1. The spectrum of 4-MepipeH[Zn(quin)3] (4), a compound that contains a protonated amine, is very similar to the spectra of related compounds featuring protonated piperidine and pyrrolidine.17 All the compounds display a very broad absorption band of medium intensity at ca. 2800 cm−1, which can be attributed to the stretching of the N–H bonds in the NH2+ group engaged in hydrogen bonding. Furthermore, the νas(COO−) and δ(NH2+) absorption bands could not be unambiguously assigned. In the spectrum of 4, these bands are observed at 1651 and 1628 cm−1. It should be noted that in the absence of X-ray structure analysis, the identification of 4 is based on its IR and NMR spectra.
As a rule, the amidine complexes spectra feature two characteristic bands, wherein the high-frequency band at ca. 3290 cm−1 can be attributed to N–H stretching and the low-frequency band at ca. 1585 cm−1 to CN stretching. For example, the absorptions of [Zn(quin)2(thiomorpham)]·CH3OH·CH3CN (2) can be observed at 3282 and 1585 cm−1, and for [Zn(quin)2(4-Mepipeam)]·CH3CN (5) at 3288 and 1584 cm−1. However, the spectrum of [Zn(quin)2(4-amidepipeam)] (7) is, owing to the interference with the –CONH2 substituent, more complicated. For 7, two bands, one at 3437 cm−1 and the other at 3297 cm−1, can be observed in the ν(N–H) region. In the absence of X-ray structural data, the identification of 7 is heavily dependent on its IR and NMR spectra. The spectrum of the acetamidine compound 23, features three bands in the ν(N–H) region, at 3448, 3267, and 3193 cm−1. The bands at 3448 and 3193 cm−1 can be tentatively assigned to NH2 group vibrations, whereas the 3267 cm−1 band is more likely to arise from the imine moiety. For 23, interestingly, two bands, at 1596 and 1561 cm−1, can be observed in the ν(CN) region.
1H NMR spectroscopy
The 1H NMR spectra of the (CD3)2SO solutions confirmed the proposed compositions of the products. In cases where single crystals suitable for X-ray diffraction analysis could not be obtained, NMR spectroscopy was indispensable as an identification method. The monoamine, diamine, and amidine complexes with the compositions [Zn(quin)2(amine)], [Zn(quin)2(amine)2], and [Zn(quin)2(amidine)], respectively, all feature two quinaldinate ligands which exhibit 1H NMR resonances in the range of 8.96–7.77 ppm. The corresponding resonances in 4-MepipeH[Zn(quin)3] (4), which contains a coordination anion with three bidentate chelating quinaldinates, are observed at lower chemical shifts, namely 8.59–7.65 ppm. The opposite is true for the amine resonances, which shift downfield upon protonation. The protonated amine in 4 exhibits ring resonances at 3.20, 2.79, 1.71–1.67, 1.61–1.53, and 1.24–1.17 ppm, while the spectrum of the same amine in a coordinated form in [Zn(quin)2(4-Mepipe)2] (3) features resonances at 2.84, 2.37, 1.46–1.42, 1.36–1.28, and 0.98–0.91 ppm. The spectra of amidine complexes share common features. The amidine ligands are comprised of two parts, one originating from the acetonitrile and the other from the amine residue. The imine hydrogen resonates in the range of 7.15–6.96 ppm, e.g., for [Zn(quin)2(4-Mepipeam)]·CH3CN (5) at 6.96 ppm. The resonance for the methyl group, which originates from acetonitrile, appears in the spectra of all the compounds at around 1.95 ppm, e.g., for 5 at 1.90 ppm. This resonance is located at 1.98 ppm for the amidine itself, 4-amidepipeam (8). The resonances for amine residues in amidines are shifted compared to those of the coordinated amine. For example, the 1-ethylpiperazine resonances in the spectrum of [Zn(quin)2(1-Etpz)2]·0.5CH3CN (13) can be observed at 2.64 and 2.25–2.18 ppm. The corresponding resonances of the amine residue in [Zn(quin)2(1-Etpzam)]·H2O (14) can be observed at 3.27–3.25, 2.20, and 2.12–2.10 ppm, where the greatest difference is observed for the first listed resonance. The resonances for the substituents remain at virtually identical chemical shifts. In spectrum of [Zn(quin)2(1-Phpz)2]·2acetamide (18) the resonances for acetamide are clearly visible. Two broad singlets can be observed at 7.27 and 6.67 ppm, which can be assigned to the two protons of the NH2 group, and a resonance can be observed for the methyl group at 1.75 ppm. Finally, the spectrum of the acetamidine compound 23, is described. Two overlapping broad singlets are observed at 7.28 and 6.68 ppm, which can be attributed to the exchangeable protons of amino and imino groups. The resonance for the methyl group of acetamidine can be observed at 1.97 ppm. The spectrum is consistent with the literature data reporting 7.37 ppm for the imino proton, 7.24 and 6.95 ppm for the amino protons, and 1.93 ppm for the methyl group.40
Conclusions
Zn(II) ions act as efficient catalysts for the nucleophilic addition of cyclic amines to the triple bond in acetonitrile, which was used as a solvent in the reaction systems presented herein. The Zn(II) compound used as a starting material was [Zn(quin)2(H2O)]. The products of the simple water substitution included the monoamine complex with the composition [Zn(quin)2(amine)] and the trans isomer of the diamine complex [Zn(quin)2(amine)2]. It should be noted that the monoamine complex formed only in the reaction system of 1-methylpiperazine. Protonation of the amine was observed in one case only, resulting in the isolation of the ionic compound 4-MepipeH[Zn(quin)3] (4). In the presence of zinc(II) compounds, all the amines, except for piperazine and 1-acetylpiperazine, reacted with acetonitrile under ambient conditions in methanol to form amidine. Only in one case did the amidine, 4-amidepipeam (8), crystallize in its free form, indicating a tendency of the in situ formed amidines to coordinate to Zn(II). Neutral complexes with the composition [Zn(quin)2(amidine)] were therefore isolated. The amidine of 1-phenylpiperazine with a bulky substituent showed that steric factors do not interfere with amidine formation. The same cannot be said for the electronic effect of 1-acetylpiperazine. The negative inductive effect of the acetyl group makes the nitrogen atom less nucleophilic and consequently less reactive. In the case of piperazine, a possible explanation lies in the low solubility of [Zn(quin)2(pz)]n, which precipitated regardless of the reaction conditions. [Zn(quin)2(pz)]n also formed in the reaction systems of 1-methylpiperazine, 1-ethylpiperazine, and 1-acetylpiperazine, which all contained piperazine as an impurity. Its presence was undoubtedly confirmed by the crystal structure of [Zn(quin)2(1-Acpz)2]·[Zn(quin)2(pz)]n·4CH3CN (22). Interestingly, the reaction systems of several amines yielded under robust conditions, a complex with a coordinated acetamidine [Zn(quin)2(am)]. Acetamidine was formed by the reaction of ammonia with acetonitrile. A rational synthetic route was sought, and it was found that the hydrolysis of acetonitrile is the source of the ammonia. This process may end at the acetamide stage, the formation of which was confirmed by [Zn(quin)2(1-Phpz)2]·2acetamide, which crystallized in two polymorphic forms.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the Slovenian Research and Innovation Agency (Junior Researcher Grant for N. P. and Research Core Funding grants P1-0134 and P1-0230) is gratefully acknowledged. The authors acknowledge the support of the Centre for Research Infrastructure at the University of Ljubljana, Faculty of Chemistry and Chemical Technology, which is part of the Network of Research and Infrastructural Centres UL (MRIC UL) and is financially supported by the Slovenian Research and Innovation Agency (Infrastructure program no. I0-0022).
References
-
S. Enthaler and X.-F. Wu, in Zinc Catalysis: Applications in Organic Synthesis, ed. S. Enthaler and X.-F. Wu, Wiley-VCH, Weinheim, Germany, 2015 Search PubMed.
-
J. Burgess and R. H. Prince, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley, Hoboken, NJ, 2nd edn, 2005 Search PubMed.
-
N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Butterworth-Heinemann, Oxford, UK, 2nd edn, 1997 Search PubMed.
- G. J. Fosmire, Am. J. Clin. Nutr., 1990, 51, 225–227 CrossRef CAS PubMed.
-
F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley, New York, 6th edn, 1999 Search PubMed.
- H. Vahrenkamp, Dalton Trans., 2007, 4751–4759 RSC.
- S. Enthaler, ACS Catal., 2013, 3, 150–158 CrossRef CAS.
- K. Keerthi Krishnan, S. M. Ujwaldev, S. Saranya, G. Anilkumar and M. Beller, Adv. Synth. Catal., 2019, 361, 382–404 CrossRef CAS.
- D. Nakatake, Y. Yokote, Y. Matsushima, R. Yazaki and T. Ohshima, Green Chem., 2016, 18, 1524–1530 RSC.
- B. M. Trost and V. S. C. Yeh, Angew. Chem., Int. Ed., 2002, 41, 861–863 CrossRef CAS PubMed.
- X.-F. Wu and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef CAS.
- K. Naveen, H. Ji, T. S. Kim, D. Kim and D.-H. Cho, Appl. Catal., B, 2021, 280, 119395 CrossRef CAS.
- X.-F. Wu, Chem. – Asian J., 2012, 7, 2502–2509 CrossRef CAS PubMed.
- C. A. Wheaton and P. G. Hayes, Chem. Commun., 2010, 46, 8404–8406 RSC.
- I. D’Auria, V. Ferrara, C. Tedesco, W. Kretschmer, R. Kempe and C. Pellecchia, ACS Appl. Polym. Mater., 2021, 3, 4035–4043 CrossRef.
- S. Roy, S. Bhattacharya and S. Chattopadhyay, J. Coord. Chem., 2016, 69, 112–122 CrossRef CAS.
- N. Podjed, B. Modec, M. M. Alcaide and J. López-Serrano, RSC Adv., 2020, 10, 18200–18221 RSC.
-
C. D. Johnson, in Comprehensive Heterocyclic Chemistry II, ed. A. McKillop, Pergamon Press, Oxford, UK, 1996, vol. 5 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- N. Podjed and B. Modec, New J. Chem., 2022, 46, 23225–23238 RSC.
- N. Podjed and B. Modec, J. Mol. Struct., 2023, 1284, 135457 CrossRef CAS.
- P. Abhayawardhana, P. A. Marzilli, T. Perera, F. R. Fronczek and L. G. Marzilli, Inorg. Chem., 2012, 51, 7271–7283 CrossRef CAS PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- N. Podjed, P. Stare, R. Cerc Korošec, M. M. Alcaide, J. López-Serrano and B. Modec, New J. Chem., 2020, 44, 387–400 RSC.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed.
- M. R. Willcott, J. Am. Chem. Soc., 2009, 131, 13180 CrossRef CAS.
-
Agilent, CrysAlis PRO, Agilent Technologies Ltd, Yarnton, Oxfordshire, England, 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- G. Rousselet, P. Capdevielle and M. Maumy, Tetrahedron Lett., 1993, 34, 6395–6398 CrossRef CAS.
-
P. Vollhardt and N. Schore, Organic Chemistry: Structure and Function, W. H. Freeman and Company, New York, 8th edn, 2018 Search PubMed.
- N. C. Stephenson, J. Inorg. Nucl. Chem., 1962, 24, 801–808 CrossRef.
- R. Norrestam, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 873–876 CrossRef.
- D. Cornacchia, R. Z. Pellicani, F. P. Intini, C. Pacifico and G. Natile, Inorg. Chem., 2009, 48, 10800–10810 CrossRef CAS PubMed.
- M. A. A. F. de. C. T. Carrondo and V. Felix, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 2451–2453 CrossRef.
- S. G. Feng, P. S. White and J. L. Templeton, Organometallics, 1993, 12, 1765–1774 CrossRef CAS.
- B. Longato, G. Bandoli, A. Mucci and L. Schenetti, Eur. J. Inorg. Chem., 2001, 3021–3029 CrossRef CAS.
- T. Perera, F. R. Fronczek, P. A. Marzilli and L. G. Marzilli, Inorg. Chem., 2010, 49, 7035–7045 CrossRef CAS PubMed.
- W. C. Corbin, G. S. Nichol and Z. Zheng, Inorg. Chem., 2016, 55, 9505–9508 CrossRef CAS PubMed.
- E. P. Shestakova, Y. S. Varshavsky, V. N. Khrustalev and S. N. Smirnov, J. Organomet. Chem., 2013, 735, 47–51 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- G. P. Stahly, Cryst. Growth Des., 2007, 7, 1007–1026 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- R. Kaur and T. N. Guru Row, Cryst. Growth Des., 2012, 12, 2744–2747 CrossRef CAS.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- U. Belluco, F. Benetollo, R. Bertani, G. Bombieri, R. A. Michelin, M. Mozzon, A. J. L. Pombeiro and F. C. Guedes da Silva, Inorg. Chim. Acta, 2002, 330, 229–239 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds. Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, Hoboken, NJ, 6th edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for 1–23 (Tables S1–S7), ORTEP drawings of zinc(II) complexes with amines (Fig. S1–S7), ORTEP drawings of zinc(II) complexes with amidines (Fig. S8–S12), overlays of complex molecules in polymorphs 18a/18b and 21a/21b (Fig. S13 and S14), intermolecular interactions (Tables S8, S9 and Fig. S15–S23), IR spectra of 1 to 23 (Fig. S24–S46), 1H NMR spectra of 4, 7, 8 and 9 (Fig. S47–S50). CCDC 2181834 (1), 2181835 (2), 2181836 (3), 2181837 (5), 2181838 (6), 2181839 (8), 2181840 (10), 2181841 (11), 2181842 (12), 2181843 (13), 2181844 (14), 2181845 (15), 2181846 (16), 2181847 (17), 2181848 (18a), 2181849 (18b), 2181850 (19), 2181851 (20), 2181852 (21a), 2181853 (21b), 2181854 (22), and 2181855 (23). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj00235k |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Janez
Košmrlj
,
Janez
Košmrlj









