
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of rigidity on the emission of quadrupolar strongly polarized dyes†
Bartosz
Szymański
ad,
Smruti Ranjan
Sahoo
b,
Rashid
Valiev
c,
Olena
Vakuliuk
a,
Piotr
Łaski
d,
Katarzyna N.
Jarzembska
d,
Radosław
Kamiński
 d,
Glib
Baryshnikov
*bf,
Mohammad B.
Teimouri
*ae and
Daniel T.
Gryko
*a
d,
Glib
Baryshnikov
*bf,
Mohammad B.
Teimouri
*ae and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bLaboratory of Organic Electronics, Department of Science and Technology, Linköping University, SE-60174, Norrköping, Sweden. E-mail: glib.baryshnikov@liu.se
cDepartment of Chemistry, University of Helsinki, FI-00014, Helsinki, Finland
dDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland. E-mail: rkaminski85@uw.edu.pl
eFaculty of Chemistry, Kharazmi University, 15719-14911 Tehran, Iran. E-mail: teimouri@khu.ac.ir
fDepartment of Chemistry and Nanomaterials Science, Bohdan Khmelnytsky National University, 18031 Cherkasy, Ukraine
First published on 4th January 2024
Abstract
Hybrid dyes comprising 1,4-dihydropyrrolo[3,2-b]pyrrole and two strongly electron-withdrawing benzoxadiazole substituents, differing in their level of planarization, offer an insightful window into the interplay between internal conversion and intersystem crossing as relaxation pathways from the excited state. The emission intensity is strong for non-fused systems whilst it is minimal for fused dyes. Computational evaluation has revealed that internal conversion, and not inter-system crossing, is the main non-radiative process for planar, fused quadrupolar dyes.
Introduction
Centrosymmetric dyes with acceptor–donor–acceptor (A–D–A) and donor–acceptor–donor (D–A–D) architectures,1–5 which form emissive CT states, can display solvatofluorochromism, caused by excited-state symmetry breaking (ES-SB).6–10 Many new quadrupolar,11–14 centrosymmetric systems have been reported over the last few years, but there has been no consensus on the influence of restricted rotation (rigidity) on the emission properties, ES-SB and thermodynamics of intramolecular charge transfer (ICT).15In this regard we hypothesized that it could be possible to directly study the influence of restricted rotation on the fate of quadrupolar dye molecules in the excited state by bridging strongly electron-donating and electron-deficient moieties. Assessing this hypothesis requires two dyes possessing identical, quadrupolar scaffolds except for their degrees of rigidity. Designing the quadrupolar architectures, we considered several candidates for the electron-rich scaffold, but we eventually decided upon 1,4-dihydropyrrolo[3,2-b]pyrrole (DHPP) due to the combination of the following properties: (1) exceptionally high-lying HOMO; (2) straightforward and modular synthesis; (3) possibility of vast structural modification.16–18 The combination of these features has been utilized several times, predominantly to decipher the fluorescence of nitro-aromatics.19,20 Simultaneously, we reasoned that incorporation of 2,1,3-benzoxadiazole21–24 at the strongly conjugated positions 2 and 5 of DHPP represents an optimal choice as an electron-acceptor. The versatility and generality of the multicomponent reaction affording tetraarylpyrrolo[3,2-b]pyrroles (TAPPs)25 enabled us to assemble the required quadrupolar dye 1 in just one step (Scheme S1 (ESI†) and Fig. 1). Subsequently the benzo[c][1,2,5]oxadiazole-4-carbaldehyde (2) was condensed with 2-bromo-4-octadecylaniline (3) affording TAPP 5, which was subjected to intramolecular direct arylation,26 giving almost planar quadrupolar centrosymmetric architecture 6 (Scheme 1).
 | ||
| Fig. 1 Structural formula of centrosymmetric TAPP 1. | ||
 | ||
| Scheme 1 Synthesis of dye 6. | ||
The photophysical properties of dyes 1 and 6 were studied in solvents of various polarity and in the polycrystalline state (Table 1 and Fig. 2). The presence of the strongly electron-withdrawing benzoxadiazole substituents shifts the absorption and emission about 1900–2300 cm−1 bathochromically with respect to the analogous TAPP possessing two 4-nitrophenyl substituents at positions 2 and 5,19 and makes TAPP 1 susceptible to changes in solvent polarity. Dye 1 exhibits strong fluorescence in non-polar solvents, characterized by a relatively low Stokes shift (Table 1). However, the fluorescence is quenched in polar solvents due to the presence of specific non-radiative relaxation pathways, namely charge-transfer-type transitions, and vibrational relaxation of the polar excited state. Solvents such as toluene, tetrahydrofuran (THF) or dichloromethane (DCM) effectively stabilize this state, resulting in larger Stokes shifts (Fig. 2).
| Dye | Solvent or powder | λ maxabs/nm (ε 10−4/M−1 cm−1) | λ maxem/nm | Φ f1 | Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 /cm−1 |
|---|---|---|---|---|---|
| a Reference: rhodamine 6G in EtOH (Φf1 = 0.95). b Measured with an integrating sphere. c Reference: oxazine 1 in MeOH (Φf1 = 0.11). | |||||
| 1 | Cyclohexane | 499 (29) | 553 | 0.77a | 2000 |
| Toluene | 506 (25) | 590 | 0.67a | 2800 | |
| THF | 501 (25) | 623 | 0.44a | 3900 | |
| DCM | 504 (23) | 673 | 0.08a | 5000 | |
| Cryst. powd. | 518 | 582 | 0.20b | 2100 | |
| 6 | Toluene | 354, 452, 580, 626 | 683, 724 (sh) | 0.11c | 1330 |
| THF | 351, 448, 576, 637 | 714 | 0.03c | 1700 | |
| Cryst. powd. | 678 | 743 | <0.01b | 1300 | |
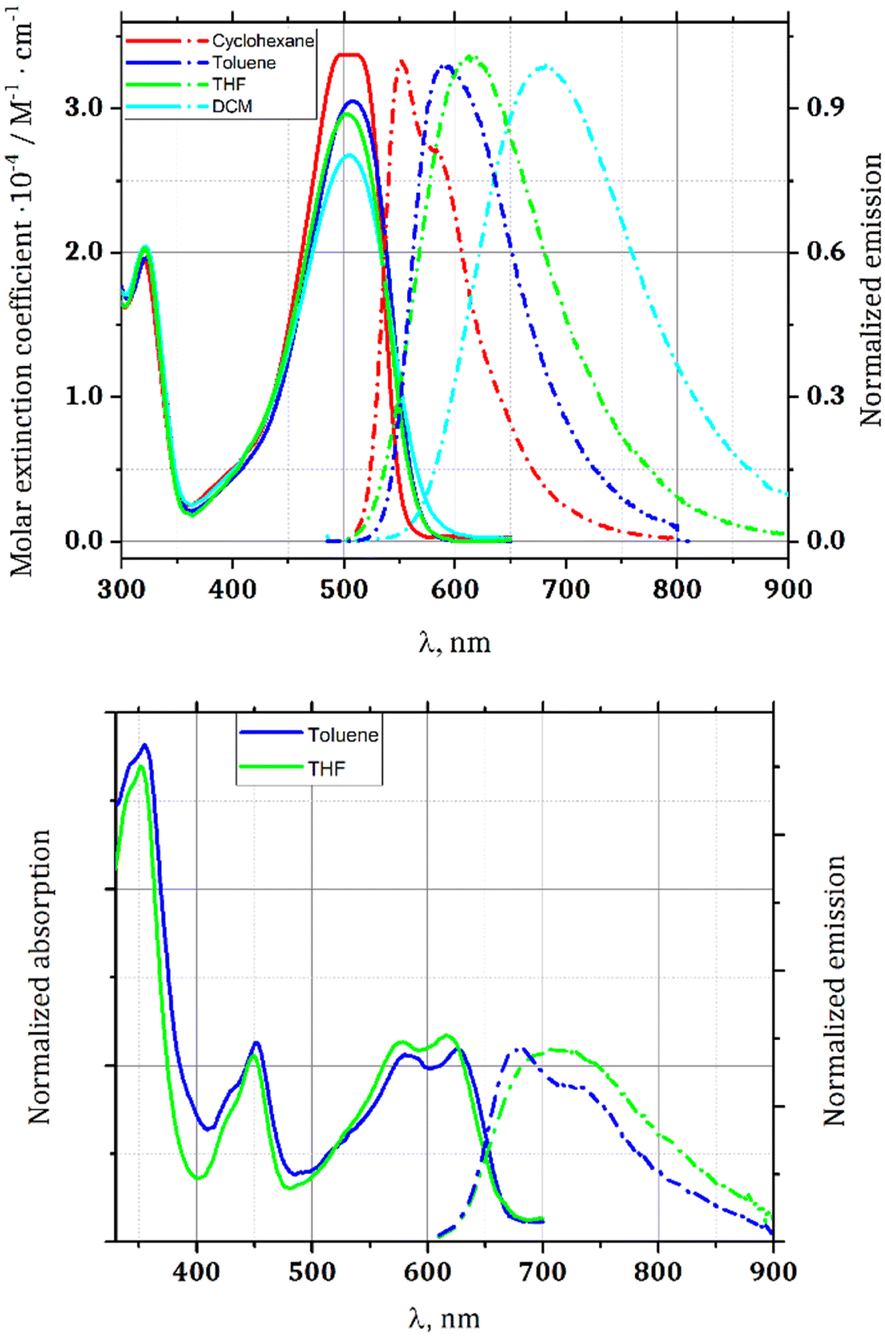 | ||
| Fig. 2 Absorption (solid lines) and emission (dot-dashed lines) spectra of TAPP 1 (upper) and 6 (lower). | ||
On the other hand, rigid and planar (vide infra) dye 6 shows remarkably different characteristics. Predominantly, it has extremely low solubility in the majority of solvents. Careful experiments have proven that in many solvents, it does not form real solutions but rather an ultrafine suspension, which cannot pass through a 0.45 μm syringe filter. Consequently, its photophysical studies were conducted only in toluene and in THF. The large size and planarity of the chromophore inclined us to investigate the possibility to form aggregates. The measurements of the dependence of the absorption on the concentration, performed in toluene, have proven that there is no aggregation within the measured concentration range.
The emission maximum for compound 6 is bathochromically shifted to 683 nm in toluene. Noteworthily, 6 possesses a more rigid chromophore compared to 1 and thus in the excited state it is less exposed to the vibronic relaxation pathways. Hence, compound 6 deactivates with the geometry close to the 1FC* state with preserved vibronic structure in the emission spectra (in toluene), while for compound 1 the detailed vibrational structure is lost and the emission spectrum appears as a broad band for most of the cases.
Compound 6 exhibits weak fluorescence, getting even lower in more polar THF, whereas solvatofluorochromism is weaker, but it remains observable (Table 1). It is worth noting that Stokes shifts are significantly lower for compound 6. They also do not correlate with solvent polarity. For compound 1 they are, however, enormous in polar solvents, providing bathochromic shift of emission (Table 1). Here, planarization causes a strikingly different effect to when nitro groups play the role of electron-withdrawing substituents.27
TAPP 1 crystallizes in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group with two halves of two symmetry-inequivalent molecules in the asymmetric unit (Fig. 3). The C4–C5–C7–N1 and C7–N1–C10–C11 torsion angles are 26.4(5)° and 33.0(4)°, respectively. The distance between C4 and C11 carbon atoms (which are bound in 6) is 3.098(4) Å in the crystal structure of 1. The main influence on the stability of the crystal structure stems from van der Waals interactions between the long alkyl chains, which results in a layered architecture of the crystal (Fig. 3). There is a hydrogen bond between two TAPP molecules namely C38–H38⋯N2 (Fig. S11, ESI†).
space group with two halves of two symmetry-inequivalent molecules in the asymmetric unit (Fig. 3). The C4–C5–C7–N1 and C7–N1–C10–C11 torsion angles are 26.4(5)° and 33.0(4)°, respectively. The distance between C4 and C11 carbon atoms (which are bound in 6) is 3.098(4) Å in the crystal structure of 1. The main influence on the stability of the crystal structure stems from van der Waals interactions between the long alkyl chains, which results in a layered architecture of the crystal (Fig. 3). There is a hydrogen bond between two TAPP molecules namely C38–H38⋯N2 (Fig. S11, ESI†).
 | ||
| Fig. 3 Molecular structure of 1 (upper). Torsion angles and the distance between C4 and C11 in the 1 crystal structure. Note that ellipsoids correspond to 50% probability of atom location. Alkyl chains and hydrogen atoms are omitted for clarification. Packing in the crystal of 1 (lower). There are some intermolecular interactions between aryl rings, which probably stabilize the structure. | ||
Having the single crystal structure in hand, we performed solid-state photophysical measurements. As far as the solid-state emission is concerned, both examined quadrupolar dyes, 1 and 6, exhibit strikingly different properties. In the case of TAPP 1, there are two emission maxima of a single crystal at room temperature, located around 460 nm and 580 nm, and the excited-state lifetime is estimated to be 3.800(9) ns (Fig. S7, ESI†). For dye 6, owing to the fused benzene rings when compared to 1, two emission maxima are notably red-shifted to about 550 nm and 745 nm. As expected, the emission decay of 6 is much faster than that of 1. The respective room-temperature lifetime is estimated to be ca. 1 ns, which is at the accuracy limit of the spectroscopic setup used (Fig. S7, ESI†). It should also be noted that temperature has a greater effect on the emission decay of 6. When lowering the temperature to 100 K, the emission maximum gets red-shifted ≈35 nm, to ca. 780 nm with almost no effect on emission lifetime. In the case of dye 1 no significant temperature-dependent spectral changes are observed.
Fluorescence quantum yields in the solid state are markedly different for both compounds. Whereas 1 exhibits a 20% quantum yield, which is considered moderate, dye 6 is almost not fluorescent at all. This corresponds well with results obtained from measurements in various solvents.
The TD DFT optimized excited states (S1) of both 1 and 6 do not significantly differ from their corresponding ground state (S0) geometries. Apparently, when going from the S0 to the S1 state, it is observed that the dihedral angles change in the range 0.2°–7.8° and 0.5°–4.5° between the benzoxiadiazolyl group and DHPP and the benzoalkyl group and DHPP, respectively for non-fused dye 1; whereas these values are calculated to be nearly zero for fused dye 6, i.e. 0.02°–0.13° and 0.02°–0.34°, supporting the rigidity and planarity of dye 6 in the excited state.
Computational calculations using the TD-DFT/PCM/B3LYP-37/6-31G(d,p) formalism were in good agreement with the experimental photophysical properties of dyes 1 and 6 (Table 1). The main absorption and emission bands correspond to the intramolecular HOMO → LUMO charge-transfer transition (Fig. 4).
 | ||
| Fig. 4 Calculated optimized ground (S0) state structures of dye 1 (a) and 6 (b) with ethyl chains and isosurfaces for the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). The calculated EHOMO/ELUMO values for dye 1 (a) and 6 (b) are −5.55/−1.99 eV and −5.41/−2.21 eV, respectively. Both S0 → S1 optical absorption and S1 → S0 emission correspond to HOMO → LUMO electronic transition. | ||
The calculated S1 → S0 radiative rates (kr) of dye 1 in different solvents are approximately the same (2.0 × 108 s−1), while for fused dye 6 the radiative rate is approximately three times smaller (0.7–0.8 × 108 s−1). We found two triplet states (T1 and T2) energetically lower than the S1 state for both dyes. The spin–orbit coupling matrix elements (SOCMEs) between S1 and T1 states for both 1 and 6, however, are negligibly small (less than 0.05 cm−1) while the S1–T1 energy gap is considerable (0.7–0.9 eV for 1 and near 0.6 eV for 6) which results in a calculated rate of S1 → T1 intersystem crossing  of 102–103 s−1 order of magnitude (Table 2 and Table S8, ESI†), i.e. uncompetitive with the radiative rate. At the same time, the S1–T2 gap is much smaller for both 1 and 6, while SOCME between the S1 and T2 states for 1 is large (near 0.4 cm−1). This results in
of 102–103 s−1 order of magnitude (Table 2 and Table S8, ESI†), i.e. uncompetitive with the radiative rate. At the same time, the S1–T2 gap is much smaller for both 1 and 6, while SOCME between the S1 and T2 states for 1 is large (near 0.4 cm−1). This results in  for 1 being in region of 0.36–2.0 × 108 s−1, i.e. it is comparable with the radiative rate constant. However, for compound 6 the SOCME between the S1 and T2 states is predicted to be zero, which corresponds to a zero rate constant for
for 1 being in region of 0.36–2.0 × 108 s−1, i.e. it is comparable with the radiative rate constant. However, for compound 6 the SOCME between the S1 and T2 states is predicted to be zero, which corresponds to a zero rate constant for  . This means that fluorescence quenching for dye 6 originates from S1–S0 internal conversion (IC). Indeed, our calculations predict kIC for compound 6 to increase with an increase of solvent polarity from 4.3 × 108 s−1 in cyclohexane to 7.5 × 108 s−1 in DCM. For compound 1, internal conversion also significantly contributes to fluorescence quenching and demonstrates a similar solvent polarity dependence as for fused dye 6. The calculated values of fluorescence quantum yield (ϕtheorf1) estimated as a relation between radiative rate and the sum of radiative and all non-radiative rate constants finally demonstrates a good agreement with the experimental measurements (Table 2 and Table S8, ESI†).
. This means that fluorescence quenching for dye 6 originates from S1–S0 internal conversion (IC). Indeed, our calculations predict kIC for compound 6 to increase with an increase of solvent polarity from 4.3 × 108 s−1 in cyclohexane to 7.5 × 108 s−1 in DCM. For compound 1, internal conversion also significantly contributes to fluorescence quenching and demonstrates a similar solvent polarity dependence as for fused dye 6. The calculated values of fluorescence quantum yield (ϕtheorf1) estimated as a relation between radiative rate and the sum of radiative and all non-radiative rate constants finally demonstrates a good agreement with the experimental measurements (Table 2 and Table S8, ESI†).
| Dye | Solvent | E S1/eV | E T1/eV | E T2/eV | SOCMES1T1/cm−1 | SOCMES1T2/cm−1 |
|
k r a × 108/s−1 | k IC b × 108/s−1 | ϕ theorf1(ϕexpf1) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cyclohexane | 1.99 | 1.11 | 1.62 | 0.04 | 0.37 | 2.06 | 2.053 | 1.0 | 0.61 (0.77) |
| Toluene | 1.97 | 1.10 | 1.61 | 0.03 | 0.37 | 1.35 | 2.053 | 1.0 | 0.58 (0.67) | |
| THF | 1.83 | 1.05 | 1.57 | 0.03 | 0.39 | 12.8 | 2.027 | 2.3 | 0.32 (0.44) | |
| DCM | 1.81 | 1.05 | 1.56 | 0.03 | 0.39 | 13.7 | 2.023 | 2.4 | 0.31 (0.08) | |
| 6 | Cyclohexane | 1.64 | 1.02 | 1.53 | 0.02 | 0.00 | 16.2 | 0.738 | 4.3 | 0.14 |
| Toluene | 1.62 | 1.01 | 1.52 | 0.02 | 0.00 | 17.9 | 0.752 | 4.7 | 0.13 (0.11) | |
| THF | 1.53 | 0.98 | 1.48 | 0.01 | 0.00 | 27.8 | 0.805 | 7.0 | 0.10 (0.03) | |
| DCM | 1.52 | 0.98 | 1.48 | 0.01 | 0.00 | 29.1 | 0.810 | 7.5 | 0.09 (< 0.01) | |

Conclusions
In conclusion, by planarization of the geometry of highly polarized, centrosymmetric dyes, it is possible to gain control over the fate of the molecular excited state. Furthermore, our observations indicate that while balanced donor–acceptor coupling ensures fast radiative deactivation and slow ISC essential for large fluorescence quantum yields, planarization changes this balance towards IC. Planarization reduces spin–orbit coupling matrix elements between the singlet and triplet excited states to essentially zero, shifting the main mechanism of non-radiative deactivation from S1 to S0 from intersystem crossing to internal conversion. These mechanistic paradigms set important design principles for molecular photonics and electronics.Author contributions
Conceptualization: D. T. G. and M. B. T.; investigation: B. S., O. V., M. B. T., P. Ł., S. R. S., R. V.; supervision: D. T. G., G. B., R. K., K. N. J.; visualization: D. T. G., B. S., P. Ł.; writing – original draft: B. S., D. T. G., O. V., G. B., P. Ł., R. K., K. N. J.; writing – review and editing: D. T. G., O. V., R. K., K. N. J., G. B.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Polish National Science Center, Poland (grants OPUS 2020/37/B/ST4/00017 and 2019/33/B/ST4/03144), and by grant no. 324139 of the Research Council of Norway (AG). G. B. thank the Ministry of Education and Science of Ukraine for support (Project No. 0121U107533). We thank K. Durka (Warsaw, Poland) for his help with solid-state quantum-yield measurements. The X-ray diffraction experiments were carried out at the Department of Physics, University of Warsaw, on a Rigaku Oxford Diffraction SuperNova diffractometer, which was co-financed by the European Union within the European Regional Development Fund (program no. POIG.02.01.00-14.122/09). The quantum-chemical calculations were performed with computational resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS 2023/5-77) at the National Supercomputer Centre (NSC) at Linköping University partially funded by the Swedish Research Council through grant agreement no. 2022-06725. G. B. thanks the support by the Swedish Research Council through starting grant no. 2020-04600. G. B. and S. R. S. are also thankful for support from Olle Engkvists Stiftelse (Sweden), project number 212-0136.Notes and references
- Y. Feng, P. J. Das, R. M. Young, P. J. Brown, J. E. Hornick, J. A. Weber, J. S. W. Seale, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2022, 144, 16841–16854 CrossRef CAS PubMed.
- R. Feng, N. Sato, T. Yasuda, H. Furuta and S. Shimizu, Chem. Commun., 2020, 56, 2975–2978 RSC.
- S. Shimizu, A. Murayama, T. Haruyama, T. Iino, S. Mori, H. Furuta and N. Kobayashi, Chem. – Eur. J., 2015, 21, 12996–13003 CrossRef CAS PubMed.
- T. M. Cooper, J. E. Haley, D. M. Krein, A. R. Burke, J. E. Slagle, A. Mikhailov and A. Rebane, J. Phys. Chem. A, 2017, 121, 5442–5449 CrossRef CAS PubMed.
- C. L. Anderson, T. Zhang, M. Qi, Z. Chen, C. Yang, S. J. Teat, N. S. Settineri, E. A. Dailing, A. Garzón-Ruiz, A. Navarro, Y. Lv and Y. Liu, J. Am. Chem. Soc., 2023, 145, 5474–5485 CrossRef CAS PubMed.
- Z. Szakács and E. Vauthey, J. Phys. Chem. Lett., 2021, 12, 4067–4071 CrossRef PubMed.
- B. Dereka, A. Rosspeintner, Z. Li, R. Liska and E. Vauthey, J. Am. Chem. Soc., 2016, 138, 4643–4649 CrossRef CAS PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 CrossRef CAS PubMed.
- T. Kim, J. Kim, H. Mori, S. Park, M. Lim, A. Osuka and D. Kim, Phys. Chem. Chem. Phys., 2017, 19, 13970–13977 RSC.
- T. Beppu, K. Tomiguchi, A. Masuhara, Y.-J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332–7335 CrossRef CAS PubMed.
- (a) Y. Hong, F. Schlosser, W. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2022, 144, 15539–15548 CrossRef CAS PubMed; (b) R. H. Kim, J. S. Park, K.-S. Lee, K. Zojer and J.-L. Brédas, Int. J. Quantum Chem., 2017, e25441 CrossRef; (c) Z. Szakács, F. Glöcklhofer, F. Plasser and E. Vauthey, Phys. Chem. Chem. Phys., 2021, 23, 15150–15158 RSC; (d) J. A. Clark, D. Kusy, O. Vakuliuk, M. Krzeszewski, K. J. Kochanowski, B. Koszarna, O. O'Mari, D. Jacquemin, T. Gryko and V. I. Vullev, Chem. Sci., 2023, 14, 13537–13550 RSC.
- E. Vauthey, J. Phys. Chem. Lett., 2022, 13, 2064–2071 CrossRef CAS PubMed.
- M. Söderberg, B. Dereka, A. Marrocchi, B. Carlotti and E. Vauthey, J. Phys. Chem. Lett., 2019, 10, 2944–2948 CrossRef PubMed.
- C. W. Stark, M. Rammo, A. Trummal, M. Uudsemaa, J. Pahapill, M.-M. Sildoja, S. Tshepelevitsh, I. Leito, D. C. Young, B. Szymański, O. Vakuliuk, D. T. Gryko and A. Rebane, Angew. Chem., Int. Ed., 2022, 61, e202212581 CrossRef CAS PubMed.
- N. I. Nijegorodov and W. S. Downey, J. Phys. Chem., 1994, 98, 5639–5643 CrossRef CAS.
- M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334–2345 CrossRef CAS PubMed.
- G. Sanil, B. Koszarna, Y. M. Poronik, O. Vakuliuk, B. Szymański, D. Kusy and D. T. Gryko, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2022, vol. 138, pp. 335–409.
- M. Krzeszewski, Ł. Dobrzycki, A. L. Sobolewski, M. K. Cyrański and D. T. Gryko, Angew. Chem., Int. Ed., 2021, 60, 14998–15005 CrossRef CAS PubMed.
- Y. M. Poronik, G. V. Baryshnikov, I. Deperasińska, E. M. Espinoza, J. A. Clark, H. Ågren, D. T. Gryko and V. I. Vullev, Commun. Chem., 2020, 3, 190 CrossRef CAS PubMed.
- K. Skonieczny, I. Papadopoulos, D. Thiel, K. Gutkowski, P. Haines, P. M. McCosker, A. D. Laurent, P. A. Keller, T. Clark, D. Jacquemin, D. M. Guldi and D. T. Gryko, Angew. Chem., Int. Ed., 2020, 59, 16104–16113 CrossRef CAS PubMed.
- T. Oyama, L. Mendive-Tapia, V. Cowell, A. Kopp, M. Vendrell and L. Ackermann, Chem. Sci., 2023, 14, 5728–5733 RSC.
- F. de Moliner, I. Biazruchka, K. Konsewicz, S. Benson, S. Singh, J.-S. Lee and M. Vendrell, Front. Chem. Sci. Eng., 2022, 16, 128–135 CrossRef CAS.
- P. K. Mehta, K. Ryu, C. K. Kim and K.-H. Lee, New J. Chem., 2022, 46, 7003–7013 RSC.
- S. Benson, F. de Moliner, A. Fernandez, E. Kuru, N. L. Asiimwe, J.-S. Lee, L. Hamilton, D. Sieger, I. R. Bravo, A. M. Elliot, Y. Feng and M. Vendrell, Nat. Commun., 2021, 12, 2369 CrossRef CAS PubMed.
- (a) M. Tasior, O. Vakuliuk, D. Koga, B. Koszarna, K. Górski, M. Grzybowski, Ł. Kielesiński, M. Krzeszewski and D. T. Gryko, J. Org. Chem., 2020, 85, 13529–13543 CrossRef CAS PubMed; (b) A. Janiga, E. Glodkowska-Mrowka, T. Stoklosa and D. T. Gryko, Asian J. Org. Chem., 2013, 2, 411–415 CrossRef CAS.
- W. Hagui, H. Doucet and J.-F. Soulé, Chem, 2019, 5, 2006–2078 CAS.
- G. Łukasiewicz, H. G. Ryu, A. Mikhaylov, C. Azarias, M. Banasiewicz, B. Kozankiewicz, K. H. Ahn, D. Jacquemin, A. Rebane and D. T. Gryko, Chem. – Asian J., 2017, 12, 1736–1748 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, compound characterization, X-ray structural data, supporting spectroscopic plots, computational details, computed spectroscopic characteristics, Cartesian coordinates for optimized structures and spectral data for all new compounds. CCDC 2288872. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj05467e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |