
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the synthesis of aminal guanidine-based molecules: synthesis of cernumidine and analogues, and survey of its anti-inflammatory activity†
Rafael
Rippel
 a,
Flávia
Leitão
a,
Miglena K.
Georgieva
a,
Rafael
Mamede
a,
Clara S. B.
Gomes
a,
Catarina
Roma-Rodrigues
bc,
Alexandra R.
Fernandes
bc,
Ana
Lourenço
a,
Luísa M.
Ferreira
*a and
Paula S.
Branco
*a
a,
Flávia
Leitão
a,
Miglena K.
Georgieva
a,
Rafael
Mamede
a,
Clara S. B.
Gomes
a,
Catarina
Roma-Rodrigues
bc,
Alexandra R.
Fernandes
bc,
Ana
Lourenço
a,
Luísa M.
Ferreira
*a and
Paula S.
Branco
*a
aLAQV@REQUIMTE, Chemistry Department of Nova School of Science and Technology, Nova University of Lisbon, Campus de Caparica, 2829-516 Caparica, Portugal. E-mail: lpf@fct.unl.pt; paula.branco@fct.unl.pt
bAssociate Laboratory i4HB – Institute for Health and Bioeconomy, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal
cUCIBIO – Applied Molecular Biosciences Unit, Department of Life Sciences, NOVA School of Science and Technology, NOVA University Lisbon, 2819-516 Caparica, Portugal
First published on 26th February 2024
Abstract
A novel approach has been developed for the efficient synthesis of the unsymmetrical (2-aminopyrrolidin-1-yl)carboxamidine alkaloidal core found in cernumidine (1) and its analogs (20a, 20c, 20f, 20i–o). The key transformation in this process involves the utilization of the Curtius rearrangement, which plays a pivotal role in constructing the aminal moiety. One of the major challenges encountered during this synthesis was the instability of the free aminal core intermediate. Furthermore, a noteworthy observation during the synthesis was the racemization process that occurred during the isocyanate trapping by organometallic reagents. Detailed DFT calculations shed light on this phenomenon, revealing a neighboring coordination-induced mechanism. The resulting compounds were subjected to evaluation for their anti-inflammatory properties using lipopolysaccharide-stimulated human THP1 cells. Notably, compounds featuring the guanidine moiety and electron-donating groups exhibited significant anti-inflammatory activity. These findings suggest that these compounds hold promise as potential candidates for further development as anti-inflammatory agents.
Introduction
Cernumidine (1) is a natural alkaloid isolated in 2011 from the Brazilian specie Solanum cernuum Vell.1 Structurally, although the X-ray structure of cernumidine (1) showed the presence of both enantiomers, the isolated compound can be accurately characterized as a scalemic mixture. This conclusion arises from the clear dominance of the dextro isomer, which is evident through its measured optical rotation of [α]20D +10.9 (c 0.84, MeOH). Like many of the members of the genus Solanum,2 cernumidine (1) displays antioxidant3,4 and antitumor activity.1,5 The latter is probably owned to its ability to inhibit the production of interleukin-8 (IL-8), as showed in 2011 on colorectal carcinoma cells (HT-29).1 Given the well-known role of IL-8 in the inflammatory process,6–8 cancer progression9 and chemotherapy resistance,10–13 and more recently in COVID-19 treatment,7 the development of new IL-8 inhibitors can be a valuable strategy for treating inflammatory disorders and certain malignancies.Structurally, cernumidine (1) has a (2-aminopyrrolidin-1-yl)carboxamidine core acylated with isoferulic acid (3-hydroxy-4-methoxycinnamic acid) in an E configuration (Fig. 1). Another relevant feature of this molecule is the presence of an unusual aminal function. The aminal group, commonly referred as the N,N-analogue of acetals14 is usually obtained from the condensation of diamines with aldehydes. Generally, N,N-aminals can be found with both nitrogens being endocyclic or exocyclic (if N-protected), while cernumidine (1) has a hybrid system (only one nitrogen is contained within a cyclic system).
 | ||
| Fig. 1 Relevant compounds containing an aminal functionality. | ||
Although considered to be unstable,15–18 aminals can be found in several privileged structures with different applications, ranging from bioactive natural compounds,1,19,20 to organocatalysts,21,22 while also having useful synthetic applications (Fig. 1).23–29 As reminiscent of the proposed biosynthetic process1 (Scheme 1a), in 202030 we developed a racemic approach based on the decarboxylative coupling of L-arginine and L-proline derivatives for the racemic synthesis of cernumidine (1) and analogues (Scheme 1b). Following our previous efforts, we decided to propose a new enantioselective approach for the total synthesis of 1. In our retrosynthetic approach (Scheme 1c), we devised two convergent strategies for the enantioselective synthesis of 1 and its analogues. A pivotal step in both strategies involves the Curtius rearrangement of the acyl azide (2) to form the isocyanate (3), establishing the aminal core. In a forward perspective, isoferulic acrylamide (5) could be produced either through amide coupling with compound 4 (derived from isocyanate hydrolysis) or via a Heck reaction involving 6 and the corresponding haloarene. Compound 6, essential for this route, is prepared by trapping 3 with vinylmagnesium bromide. The acyl azide 2, a substrate for the Curtius rearrangement, can be readily obtained from L-proline.
 | ||
| Scheme 1 (a) Proposed biosynthesis of cernumidine. (b) Previous bio-inspired synthetic approach. (c) Retrosynthetic proposal for the enantioselective synthesis of cernumidine. | ||
Results and discussion
Anticipating the inherent instability of the unprotected aminal core, we made the strategic choice to start our investigation by synthesizing the aminal moiety through a Curtius rearrangement (Scheme 2a) of the acyl azide (7). Our approach began with the utilization of N,N′-di(Boc)-protected guanidine (8) as the initial substrate, and we tailored our methodology based on the established protocol developed by Verardo for the synthesis of carbamoyl azides.31 However, since all the methodologies described for the synthesis of acyl azides rely on the activation of the starting carboxylic acid (in the protocol used, a mixed anhydride), hydantoin derivative (9) was attained exclusively, resulting from an intramolecular cyclization. Therefore, N-Boc-L-proline was chosen as the starting material, which enabled the synthesis of 2 in excellent yield, followed by the Curtius rearrangement to yield the isocyanate 3 (Scheme 2b). With compound 3 in hand, various hydrolysis conditions were explored (see ESI,† Section S1.4 and Table S1 for details) to achieve the synthesis of the free aminal 4. Surprisingly, when the isocyanate hydrolysis was followed by an amidation reaction with benzoyl chloride, only trace amounts of the desired product (10a) were obtained. The resulting urea derivative (11), formed from the coupling of the desired free aminal (4) and the isocyanate, was identified. To the best of our knowledge, there are no precedents in the literature where the competing reaction between isocyanate hydrolysis and urea formation presents a challenge during isocyanate hydrolysis. | ||
| Scheme 2 (a) Attempt synthesis of acyl azide derivative from 8, (b) synthesis of acyl azide (2) from and N-Boc-L-proline, hydrolysis of isocyanate (3) and trapping of free-aminal (4), (c) preparation of compounds 10a–d. | ||
Given the unsuccessful hydrolysis of the isocyanate, we redirected our efforts towards its trapping using Grignard reagents and conducted a systematic investigation into the trapping of isocyanate (3) by various organometallic reagents. Table 1 outlines the results for the trapping of isocyanate (3) with PhMgBr (entries 1–6) to prepare the model substrate 10a. Carrying out the reaction either at room temperature (Rt) or 0 °C (Table 1, entries 1 and 2) gave 10a in moderate yields, which can be explained by the high number of secondary products observed when following the reaction by TLC.
| Entry | RMa | T (°C) | Time (h) | Yield (%) | eeb (%) |
|---|---|---|---|---|---|
| a All reaction were performed with 1.2 equiv. of the correspondent organometallic reagent. b Obtained by HPLC using a Phenomenex Lux 5 μm cellulose-2 column. c Not determined. | |||||
| 1 | PhMgBr | Rt | 0.25 | 48 | 0 |
| 2 | PhMgBr | 0 | 0.25 | 64 | 0 |
| 3 | PhMgBr | −41 | 1 | 87 | 30 |
| 4 | PhMgBr | −78 | 1 | 85 | 33 |
| 5 | PhMgBr | −41 | 2 | 11 | |
| 6 | PhMgBr | −41 | 3 | 4 | |
| 7 | VinylMgBr | 0 | 0.25 | 48 | |
| 8 | VinylMgBr | 0 | 0.5 | 3 | |
| 9 | VinylMgBr | −41 | 0.5 | 61 | |
| 10 | VinylMgBr | −41 | 1 | 66 | 64 |
| 11 | VinylMgBr | −78 | 0.5 | 60 | |
| 12 | VinylMgBr | −78 | 1 | 61 | |
| 13 | MeMgBr | −41 | 1 | 22 | 0 |
| 14 | BuLi | −41 | 1 | 18 | 0 |
| 15 | PhLi | −41 | 1 | 75 | 42 |
Being aware of the possibility of isocyanate exclusion during the Curtius rearrangement of amino acids derivatives,32 which would consequently lead to racemization, the chiral integrity of our substrates was verified through chiral-HPLC. Under the previously mentioned conditions, at Rt and 0 °C (Table 1, entries 1 and 2), compound 10a was obtained in a racemic form.
However, reducing the temperature (Table 1, entries 3 and 4) resulted in a significantly cleaner reaction, leading to both higher yields and enantiomeric excess (ee) (ca. 30%). Conversely, extending the reaction time (Table 1, entries 5 and 6) was observed to decrease the ee. Similar observations were made with the vinylic substrate (10b), where the ee remained consistent at both −78 °C and −41 °C (Table 1, entries 10 and 12). Notably, shorter reaction times did not produce any significant change in ee (Table 1, entries 9–11). Given that conducting the reaction at −41 °C provided no significant differences in the yield (Table 1, entries 3 and 4) and ee, this condition was deemed optimal for the Grignard addition and subsequently extended to other organometallic reagents (Table 1, entries 13–15). While complete racemization was observed for substrates 10c–d, compounds 10a and 10b were found in the form of scalemic mixtures (enantiomers in a ratio different than 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
In summary, upon analyzing the results, it becomes evident that complete racemization occurs when aliphatic substituents are transferred (10c–d). However, when substituents bearing conjugated π-systems are involved (10a and 10b), some level of chirality is maintained which is time and temperature dependent. These findings lead us to exclude the possibility of a mechanism similar to the one proposed by Baumman (isocyanate exclusion).32 If racemization were to occur during the Curtius rearrangement, all substrates would yield racemic mixtures regardless of the subsequent Grignard reagent used.
Therefore, two pathways for aminal racemization can be proposed based on an iminium-induced ring opening mechanism (Scheme 3). We can either postulate that iminium formation occurs via the amide or the pyrrolidine nitrogen. The former would lead to a zwitterionic species that reacts intramolecularly to reforge the aminal core (pathway A), while in the latter (pathway B) the aminal would be formed by an intermolecular reaction between an intimate ion pair. If pathway B were to happen, a contrary trend as the one observed for substrates (10a–d) would have been observed, given that more acidic amides would be more prone to elimination. Probably indicating that the proposed pathway A is more likely to happen.
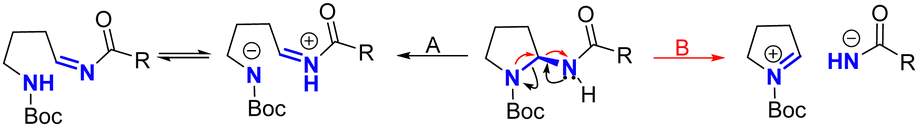 | ||
| Scheme 3 Proposed pathways for iminium-induced racemization. | ||
We can now rationalize at which point of the reaction it occurs: (i) during the reaction quenching by the proton source or (ii) during the organometallic addition process (as shown in Scheme 4a). The former hypothesis (Scheme 4a-i) assumes that the ring opening is the rate-limiting step of the racemization process, and that it occurs via an intramolecular hydrogen atom migration during the reaction quenching. To test this proposal, the reaction was carried out in an apolar solvent, such as toluene (32% ee), where it was expected that the proton transfer required for ring opening would be hindered or even suppressed when compared with THF.
 | ||
| Scheme 4 (a) Proposed mechanistic pathway for Grignard- or quenching-induced racemization (b) ee changes across different aminal scaffolds. | ||
Additionally, two experiments were conducted in an attempt to observe a decrease in the reaction rate either through a kinetic isotopic effect or by lowering the temperature. In the first experiment the reaction was quenched with MeOD while maintaining THF as the solvent (33% ee), while in the second experiment the reaction was performed at −78 °C (Table 1, entry 4). However, neither of these reactions showed significant changes in the ee from the control reaction. For our second hypothesis (Scheme 4a-ii) we proposed the intermediates (A and B) after the Grignard addition to the isocyanate. We believed that although the formation of A would be favored due the higher oxygen electronegativity, intermediate B would promote the ring opening reaction. We further explored other variables, such as the metallic ion (Table 1, entry 15) and scaffold changes (Scheme 4b) on the aminal racemization process. When performing the synthesis of 10a with phenyl lithium the ee increased to 42% (30% with PhMgBr), reflecting that the metallic ion of the organometallic reagent has an influence on the reaction outcome.
To evaluate the influence of scaffold changes on aminal racemization, analogues 12 and 13 were prepared (Scheme 4b). Compound 12 derive from L-pipecolic carboxylic acid and compound 13 from (S)-(−)-indoline-2-carboxylic acid. Both were synthesized using the same conditions applied for the synthesis of 10a–d and through the Curtius rearrangement of the respective acyl azide derivatives followed by trapping of the isocyanates with PhMgBr. Regarding the ee changes, HPLC data revealed that 13 completely preserved its stereochemistry [α]20D −10 (c 0.40, MeOH), whereas 12 fully racemized.
Driven by these results, we sought to investigate computationally the mechanism responsible for the loss of chiral integrity during the aminal synthesis.
Since experimentally the racemization seems to be induced by the organometallic reagent, this was our premise for the DFT calculations performed. The calculations focused on 3 aspects: (i) finding the thermodynamically stable complex(es) that are formed after Grignard addition, (ii) rationalize the reaction mechanism of the full reactional process, (iii) explain the origin of the chirality loss. We started by looking into the energies of the intermediates A and B (Scheme 4a) proposed after the addition of phenyl magnesium bromide to the isocyanate using 10a as a model case (Fig. 2). To our surprise, coordination with the amide nitrogen results in a more stable intermediate (B) compared to coordination with oxygen (A), and a third (C) and forth intermediates (D = IM1) were found. Intermediate C is formed when the MgBr species bonds with O1 (amide oxygen) and coordinates with Boc oxygen (O2), resulting in an 8-membered intermediate. Like in B, that suggests that coordination with the amide nitrogen (N1) is preferred, D is formed by the coordination between N1 (amide nitrogen) and O2 rendering the most stable intermediate. This outcome suggests the thermodynamic preference for a more stable 6-membered intermediate that can adopt a favorable boat conformation. It is noteworthy that IM1 will always represent the most stable intermediate found for every analyzed substrate. From IM1, the reaction proceeds through TS1 (ΔG = −8.31 kcal mol−1) to form a tri-coordinating complex with magnesium bromide between N1–N2–O2, initiating the ring-opening process and identified as the rate-limiting step of the reaction, with an energy barrier of 26.23 kcal mol−1 (Fig. 3).
 | ||
| Fig. 2 Optimized intermediates formed from the addition of phenyl magnesium bromide to isocyanate using PBE1PBE/6-311++G**//PBE1PBE/6-31+G** level of theory in solvent THF. Energies relative to the common reactants are indicated in kcal mol−1. | ||
 | ||
| Fig. 3 Optimized intermediates formed from the addition of phenyl magnesium bromide to isocyanate using PBE1PBE/6-311++G**//PBE1PBE/6-31+G** level of theory in solvent THF. Energies relative to the common reactants are indicated in kcal mol−1. | ||
After the ring opening, the formation of an imine (C1–N1) leads to the loss of the stereogenic center, and a di-coordinated magnesium complex is formed by the loss of coordination with N1 (IM2). From IM2, TSrot (ΔG = −15.27 kcal mol−1) is formed and the rotation over C1–C2 occurs to form IM2′. Next, the stereogenic center is reforged via TS2 through a 5-exo-trig cyclization by the (re)-attack of N1 to the imine. Finally leading to IM4, that after the reaction quenching afford the product with the inverse (R) configuration.
From these computational results, it seems that coordination with Boc carbonyl is what drives the pyrrolidine ring opening. Additionally, the low activation energy value associated to the conversion IM2 → TSrot (ΔG‡ = −3.23 kcal mol−1) indicates that rotation occurs easily once the endocyclic C–N bond is cleaved. As stated earlier, the synthesis of 10a showed an increase in the ee when PhLi was used (42% ee) compared to PhMgBr (30% ee). To understand the impact of the metallic ion on the racemization process, the energy profiles of the intermediates resulting from the addition of PhLi to the isocyanate were calculated. Similarly to the addition of magnesium bromide, four intermediates were discovered (see ESI,† Section S5), with the 6-membered complex of lithium (N1–Li–O2) (ΔG = −42.75 kcal mol−1) rendering the most stable intermediate. As the rate-limiting step (IM1 → TS1) has essentially the same activation energy for both lithium and magnesium (see ESI,† Section S5), the higher ee observed for the synthesis of 10a with phenyl lithium might be associated with the highest stability of IM1 (ΔG = −42.75 kcal mol−1) vs. (ΔG = −34.54 kcal mol−1) and the increase of almost 4-fold for the system rotation energy (IM2 → TSrot). The same rationale used for substrate 10a was applied for the calculations of compounds 12–13 (see ESI,† Section S5) to address the scaffold influence in the observed ee changes (Fig. 4).
 | ||
| Fig. 4 Calculated energy profile for the reaction of isocyanate and phenyl magnesium bromide for the synthesis of 10a, 12 and 13. Calculation performed using PBE1PBE/6-311++G**//PBE1PBE/6-31+G**level of theory in solvent THF. For 10a, IM1 = D. For 12, IM1 = C. | ||
Examining the reactional profile of indoline derivative (13), it is evident that ring closure (IM2 → TS1 – ΔG‡ = 7.49 kcal mol−1) is significantly more favorable than the expected pathway leading to the system rotation (IM2 → TSrot – ΔG‡ = 19.84 kcal mol−1). This preference can be attributed to the greater rigidity of the aromatic system, which is reflected in the highest TSrot value (ΔG = −5.37 kcal mol−1). This high TSrot value, makes it the limiting step for this substrate, and might help explain the observed retention of stereochemistry. For the pipecolic derivative 12, the low energy value for IM1(C) (ΔG = −40.52 kcal mol−1) is quite striking and contrary to the other derivatives is associated with the formation of an 8-membered ring intermediate (see ESI,† Section S5). This value leads to a Keq for IM1 that is 105 orders of magnitude higher than the IM1(D) formation of 13 (ΔG = 34.93 kcal mol−1). However, when compared with 10a (IM1 → TS1 – ΔG‡ = 26.23 kcal mol−1) the lower rate limiting step energy for 12 (IM1 → TS1 – ΔG‡ = 18.33 kcal mol−1) is what might justify the complete racemization observed for this derivative (Fig. 4). Based on the analysis of the studied systems (12 and 13), it appears that the ability to form stable complexes between the metallic species and the carbonyl oxygens is the determining factor for monocyclic systems. However, in the case of indoline, despite having a similar stable IM1(D) system compared to the pyrrolidine derivative, other additional factors may contribute to the absence of racemization. These factors include the reverse reaction preference (IM2 → TS1) and the high energy required for bond rotation. Regarding the analysis of substituent effects on enantiomeric excess, an examination of the energy profiles of derivatives 10a–c reveals a significant concern regarding the stability of the final product. This issue becomes particularly pertinent due to the lack of conclusive evidence in the energy profiles of these derivatives to explain the complete racemization of aliphatic aminals during the Grignard addition step. In the context of the racemic synthesis of 10d (conducted via BuLi, with the energy profile not calculated), it is deduced that aliphatic aminals exhibit inherent instability, leading to the formation of racemic adducts. This observation aligns with the observed trend wherein more basic aminals tend to be less stable.15–18
Next, we sought to confirm experimentally our computational results and validate the influence of the Boc group on the racemization process. We defined as a premise that the formation of the intermediates depicted in Scheme 5 occurs and that coordination with the Boc group is what leads to racemization.
 | ||
| Scheme 5 Study of the neighbouring group effect on aminal racemization (a) via ortho-directing groups at the phenyl ring and (b) N-protecting groups lacking hydrogen bond acceptors. | ||
Therefore, we predicted three scenarios: (i) higher reaction times would favor the formation of IM1 (six-membered intermediate) and ultimately lead to a lower ee, (ii) ortho-directing groups at the phenyl ring would further stabilize the intermediates (IM1) and transition states increasing the degree of racemization, (iii) N-protecting groups lacking hydrogen bond acceptors would inhibit the formation of IM1, thus decreasing or even preventing racemization entirely. To investigate the influence of time in the reaction outcome, the reaction time for the synthesis of 10a was increased (Table 1, entries 3, 5, 6 and ESI,† Section S2.14). As anticipated, the ee value decreased as the reaction time increased to 2 hours (11% ee) and 3 hours (4% ee). Afterwards, to evaluate the impact of ortho-coordinating groups on the phenyl ring, isocyanate (3) was trapped by 2-methoxyphenylmagnesium bromide to yield 10e in 53% yield with 5% ee (see ESI,† Section S2.18). In our pursuit to synthesize the corresponding isocyanates, we prepared several N-substituted-L-proline derivatives (14a–c) as outlined in Scheme 5b. Regrettably, all attempts to rearrange the acyl azides to the desired isocyanates proved unsuccessful. While the initial formation of the isocyanate was observed (FTIR 2200 cm−1), we suspected that subsequent exclusion of the isocyanate had taken place. The resulting mixtures were intractable and indicated a decomposition-like mechanism akin to the one described by Baumann,32 where the released isocyanate led to further decomposition of the intermediates. This phenomenon could be attributed to the higher basicity and nucleophilicity of the proline nitrogen in these substrates, which potentially diminished the stability of the aminal system during the rearrangement process. Although our investigation did not allow for a direct assessment of the influence of the Boc group, we were able to confirm two out of the three proposed scenarios. Based on these findings, we can conclude that increased reaction time and additional stabilization by o-directing groups promote the formation of IM1, resulting in a higher loss of ee during the Grignard addition. Furthermore, to assess the stability of aminal under the basic conditions generated after Grignard quenching, 10b (64% ee) was subjected to various conditions, including exposure to (i) MgBr2, (ii) a MeOH solution, and (iii) LiOH, all at room temperature. In all cases, there was no evidence of racemization occurring during the quenching process.
Following the envisioned approach, we then proceed with synthesis of 15via Heck reaction of 10b. We commenced our studies by analyzing the optimal parameters for the Heck reaction of the vinyl derivative (10b), using iodobenzene to achieve 15a as a model substrate (Scheme 6a). Starting with palladium acetate and PPh3 in acetonitrile under reflux yielded 15a in 25% in an incomplete reaction. Increasing the pressure within the system (sealed tube) increased the reaction yield to 50%, while also leading to the formation of the di-arylated product (15b). Next, despite resulting in only a slight increase in the reaction yield, changing the solvent to dioxane and carrying out the reaction at 110 °C resulted in complete reaction after 18 h (60% yield). Following the previous rationale, performing the reaction in dioxane in a sealed tube afforded 15a in 84% yield after 5 h. Stablished the optimal protocol for the Heck reaction, we analysed the scope of the reaction (Scheme 6b). The use of 4-bromo aniline, 4-bromo catechol and 1-bromo-3,4,5-trimethoxybenzene as coupling partners resulted only in unreacted starting material, probably due the coordinating character of nitrogen and oxygen with palladium in this type of reaction.33 Additionally, considering the enantiomeric stability of the indoline derivative (13) during the Grignard addition, the vinylic analogue (16) was prepared and subjected to the Heck reaction. Unfortunately, despite observing the formation of the desired product (17a) by NMR, the presence of the di-arylated adduct (17b) rendered the separation of both compounds 17a and 17b impracticable (Scheme 6c). Compound 15e was obtained with an ee of 20% and the mixture of both enantiomers were observed by X-ray analysis (see ESI,† Section S6).
 | ||
| Scheme 6 (a) Synthesis of compounds 15a–b through Heck reaction of 10b and iodobenzene. (b) Reaction scope of the Heck reaction of substrate 10b. (c) Heck reaction of derivative 16 with iodobenzene. | ||
In a parallel assay, we successfully achieved an ee of 82% for the cernumidine precursor 15. However, as the reaction was conducted without a chiral auxiliary, any enhancement in ee should be attributed to fluctuations in the enantiomeric excess during the synthesis of the starting material (10b). Given that most compounds presented a decrease in the ee comparatively to (10b), we sought to investigate the stability of 10b under the Heck conditions performed. For that, we started by analyzing the aminal stability under the reactional conditions in the absence of catalyst, in the presence of Pd(0) and Pd(II) species, and other metals (Table 2). The Heck reaction conditions at Rt, did not affect the ee (Table 2, entry 1). Although complete racemization of the aminal occurs at 110 °C (Table 2, entry 2), palladium displayed a protective-like effect that reduced the loss of chirality (Table 2, entries 3 and 4).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Solvent, T (°C) | Time (h) | ee (%) |
| 1 | Pd(OAc)2, PPh3 | Dioxane, Rt | 4 | 64 |
| 2 | No catalyst | Dioxane, 110 | 4 | 0 |
| 3 | Pd(OAc)2 | Dioxane, 110 | 4 | 44 |
| 4 | Pd(OAc)2, PPh3 | Dioxane, 110 | 4 | 44 |
| 5 | Cu(OAc)2 | Dioxane, 110 | 4 | 43 |
| 6 | ZnCl2 | Dioxane, 110 | 4 | 42 |

Similar observations were made with other metal complexes, such as Cu(OAc)2 and ZnCl2, under the same reaction conditions (Table 2, entries 5 and 6). Given the observed reduction in ee in the Heck coupling reaction, upcoming studies will focus on understanding the substrate's influence and identifying the optimal conditions. This investigation will encompass factors such as the catalyst, ligand, solvent, and temperature with the aim of minimizing the observed racemization.
Continuing with the designed approach, we then proceed with the removal of the Boc protecting group using standard Brønsted acid conditions (see ESI,† Section S1.8 and Table S3). In light of the high instability of N-free aminals (observed during the isocyanate (3) hydrolysis) and the fact that aminals are typically found within cyclic systems, we were interested in investigating whether the unprotected endocyclic nitrogen would yield a stable N,N-aminal. When compound 10a was subjected to TFA or HCl at different reaction conditions, we were able to verify the presence of desired product (18a) as its HCl or TFA salts in mixtures with benzamide as major the product (Scheme 7a).
 | ||
| Scheme 7 (a) Reactional conditions for the Boc removal of 10a and proposed structure of 18a and 18b. (b) X-ray structure of 19. | ||
Recently, alternative milder Boc removal methods compatible with acid labile groups have been reported. Among them, the use of Lewis acids34–39 has gained significant attention. Additionally, metal free methodologies have also been reported, such as the use of catalytic iodide,40 silica gel,41 and oxalyl chloride42 as a milder acidic approach. Alternatively, although requiring high temperatures and longer reaction times, when compared with the aforementioned methodologies, Ewing43 and Guillaumet44 also reported the use of inorganic bases. From the myriad of possibilities available, we decided to evaluate the efficiency of Lewis acids in the synthesis of 18b. Starting with iodine40 gave only unreacted starting material, while FeCl3,38 TMSOTf,45 and Sn(OTf)39 yielded benzamide. Regarding aminals, Ideguchi et al.46 reported the use of AlMe3 to selectively remove the Boc group, while maintaining the aminal core integrity in the total synthesis of neoxaline.
When applying these conditions, the use of AlMe3 allowed us the synthesis of 18b with a considerable reduction of benzamide formation (Scheme 7a). It is worth noting that during the monitoring of this reaction by TLC, no formation of benzamide was observed. We believe that the degradation into benzamide might occur during the work-up process, which involves quenching with a solution of sodium potassium tartrate (Rochelle Salt), followed by multiple sequential extractions with DCM to isolate the product from the aqueous phase. It is at this stage that the formation of benzamide becomes evident through TLC analysis. This led us to believe that contrary to the results observed under Brønsted acid conditions the use of AlMe3 rendered a stable aminal. In fact, when analyzing the stability of 18b in polar a solvent (e.g. MeOD) complete decomposition into benzamide and 1-pyrroline47 was observed after 4 h (see ESI,† Section S2.27). These results not only confirmed the aminal instability under acidic conditions, as also showed a clear difference in the stability between the ammonium salt (18a) and the free base (18b). The stability of 18b was initially thought to be related to the existence of a N-coordinating complex with AlMe3 (Scheme 7a). However, a complex-free structure was fully supported, for both pyrrolidine and indoline derivatives, by ICP-OES (inductively coupled plasma optical emission spectrometry – see ESI,† Section S4) and the X-ray structure (Scheme 7b) of the indoline derivative (19) (see ESI,† Section S6).
Interestingly, in natural products containing aminals, N-unprotected aminals are only observed when the respective nitrogen electron pair can be delocalized and when the unprotected nitrogen is contained within a cyclic core. This highlights the importance of the cyclic structure and adjacent conjugated systems in determining the stability of aminals and explain the divergent stability between compound 18a–b and 4 (attempted during the isocyanate hydrolysis).
Finally, to complete the synthesis of cernumidine (1) and analogues, different guanylating reagents (GR) and conditions were screened for the installation of the guanidine funtionality in 18a and 18b (Scheme 8a and see ESI,† Section S1.9, Table S4). Initial attempts to use cyanamide (GR1) for the synthesis of the guanidine moiety (in MeCN/H2O), proved unsuccessful due to the low electrophilicity of GR1 and the thermal instability of 18a, rendering only unreacted starting material (Rt) and benzamide (50 °C). To address any potential instability issue of 18a in water, organic solvents able to afford homogeneous reactions were screened (MeCN, DMA, MeOH and DMF), resulting only in unreacted starting material. To overcome the low basicity of GR1, which was initially expected to deprotonate 18a, inorganic and organic bases (NaHCO3, DABCO, DIPEA) were evaluated at room temperature. As expected, the low reactivity of cyanamide and instability of 18a under basic conditions resulted in only benzamide. As the conditions used were limited by stability constraints, a more reactive guanylating reagent was tested, N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (GR3), but only decomposition of 18a into benzamide was observed using. Considering 18a as an unsuitable substrate for the installation of the guanidine moiety, unprotected substrate 18b was evaluated. While no reaction occurred in the presence of cyanamide (GR1) at room temperature the desired guanylated product was obtained using pyrazole-1-carboxamidine reagents (GR2 and GR3). A direct correlation with the GR reactivity was observed, consistent with results reported by Matsueda,48 where GR3 (57%) was more reactive than GR2 (46%) due to the electron withdrawing effect by the carbamate group. Although the optimal conditions were achieved using GR3, GR2 was selected as the guanylating reagent to avoid further deprotection steps. Provided the optimal conditions, a set of N-Boc protected derivatives (10a, 10d, 15 and 15a–g) was subjected to Boc removal conditions, followed by the guanylation reaction with GR2, in a one-pot manner, to synthesize cernumidine (1) and its derivatives (Schemes 8b, 20a, 20c, 20f, 20j–o). To attain cernumidine a sample of 15 with 82% ee was used for the Boc deprotection followed by the guanylation reaction. The final compound cernumidine was obtained with a slight decrease on the ee (70%), [α]20D +12.4 (c 0.35, MeOH).
 | ||
| Scheme 8 (a) Guanylation reaction of 18a–b. (b) Scope of the guanilation reactions. | ||
To the other guanylated compounds synthesized the same HPLC method was unsuccessful in achieving any enantiomeric separation, making it impractical to draw any conclusions regarding ee changes during the Boc removal and guanylation steps. It is plausible to assume that the final ee will decrease relative to the N-Boc precursors. Additionally, when considering steric or electronic effects that could explain the achieved yields, no clear trend can be observed. This has led to the conclusion that the stability of the unprotect aminal likely controls the reaction yield and the ee.
Knowing that N-Boc derivatives (15) could maintain their chiral integrity after months in solution (MeOH), we decided to investigate the stability of cernumidine. Contrary to the analogous N-Boc derivatives, cernumidine (1) completely racemized after one week in MeOH (see ESI,† Section S2.1). Probably explaining the racemic entity found by X-ray when cernumidine was first isolated in 2011.1
Aware of the instability of pyrrolidine-based aminals, we decided to explore the enhanced stability provided by the indoline scaffold (13). Despite providing the N-deprotected adduct (19) in excellent yield, all our attempts to install the guanidine moiety resulted either in decomposition or unreacted starting material and 21 was not identified (Scheme 8b). In the absence of base all the GR tested failed to give the desired guanylated product (21). Similarly, the use of DIPEA did not change the reaction outcome. As in the N-functionalization of indoles, we used NaH to achieve the deprotonation of indoline which resulted in the formation of indole and benzamide as decomposition products (see ESI,† Section S1.9 and Table S5). Although the guanylation of indoline was anticipated to pose challenges, this outcome has prompted us to question how the aminal core of our substrates affects their nucleophilicity compared to the initial acid analogues. Therefore, we decided to use the N-Boc protection reaction as a model reaction to evaluate the nucleophilicity of 19 compared to (S)-(−)-indoline-2-carboxylic acid. The latter reacts with Boc2O in the presence of TEA to afford the N-protected product in 96% yield after 6 h.49 In contrast, when subjected to the same reaction conditions, 19 exhibited a conversion of 14% after 24 hours, which increased to 30% after 5 days of reaction.
These experimental results were supported by computational calculations (see ESI,† Section S5 and Fig. S5). L-Proline and (S)-(−)-indoline-2-carboxylic acid were found to be more nucleophilic, with a LUMO–HOMO gap value of 0.12 and 0.19 eV less, respectively, compared to their aminal analogues (18b and 19). These findings help to support that associated/adding to the instability of this core, aminals are less nucleophilic than their amino acid precursors.
Biological activity
Aware of the biological activity of cernumidine, we sought to evaluate the anti-inflammatory potential of cernumidine (1) and its guanylated analogues (20a, 20c and 20j–o), as well as the N-Boc precursors (10a, 10b, 10d and 15), as shown in Fig. 5. | ||
| Fig. 5 Anti-inflammatory activity of compounds in THP1 cells. (a) Tumor necrosis factor alpha (TNFα) mRNA relative expression. (b) Nuclear factor kappa B (NF-κB) protein relative expression. | ||
Compounds containing a guanidine group generally exhibited higher anti-inflammatory activity compared to their N-Boc precursors. However, this increased activity was not observed for nitro-substituted arenes (20l and 20m) and the mono-aryl cinnamic derivative (20c) when tested. Interestingly, the presence of a fluor substituent at the para position (20n) had a favorable impact on the anti-inflammatory activity. In terms of the effect of the guanidine group, Cernumidine (1) showed higher activity than its N-Boc precursor (15), indicating the importance of this functionality. As it was impractical to ascertain the enantiomeric excess, we cannot exclude the possibility that the observed activity results may stem from scalemic mixtures.
Experimental
For experimental part see please ESI.†Conclusions
Given the ever growing pursue by the pharmaceutical industry of new lead compounds, the preliminary results showcased here should be a valuable tool in the discovery of novel anti-inflammatory alternatives based on the (2-aminopyrrolidin-1-yl)carboxamidine alkaloidal core. Moreover, from a fundamental standpoint, the findings presented in this study highlight that while the enantioselective synthesis of cernumidine (1) can be accomplished, the instability of the aminal-guanidine core imposes limitations. This instability requires careful consideration of experimental conditions and appropriate storage conditions. Nevertheless, it may be feasible for other substrates with a (2-aminopyrrolidin-1-yl)carboxamidine alkaloidal core. This work is significant as it has the potential to pave the way for further studies into the mechanisms that dictate the equilibrium between the opening and closing equilibrium of hybrid aminal-imine compounds.Author contributions
The contributions of the authors to this work is as follows: Conceptualization, P. S. B. and L. M. F.; methodology, P. S. B. and L. M. F.; A. F.; formal analysis, A. L.; laboratory work, R. R., F. L., R. M. and C. R.-R.; X-ray analysis, C. S. B. G.; computational studies, M. K. G. and R. R.; writing—original draft preparation, P. S. B., L. M. F., R. R.; writing—review and editing, P. S. B., L. M. F. and A. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by national funds from FCT – Fundação para a Ciência e a Tecnologia, I.P., in the scope of the project, Associate Laboratory for Green Chemistry – LA/P/0008/2020 (DOI: 10.54499/LA/P/0008/2020), UIDP/50006/2020 (DOI: 10.54499/UIDP/50006/2020) and UIDB/50006/2020 (DOI: 10.54499/UIDB/50006/2020), through national funds and UIDP/04378/2020 (DOI 10.54499/UIDP/04378/2020) and UIDB/04378/2020 (DOI: 10.54499/UIDB/04378/2020) of the Research Unit on Applied Molecular Biosciences – UCIBIO and the project LA/P/0140/2020 (DOI: 10.54499/LA/P/0140/2020) of the Associate Laboratory Institute for Health and Bioeconomy – i4HB. RR thanks the fellowship SFRH/BD/136692/2018. C. S. B. Gomes acknowledges the XTAL – Macromolecular Crystallography group (UCIBIO and i4HB) for granting access to the X-ray diffractometer. X-Ray infrastructure was financed by FCT-MCTES through project RECI/BBBBEP/0124/2012.References
- L. C. Lopes, B. Roman, M. A. Medeiros, A. Mukhopadhyay, P. Utrilla, J. Galvez, S. G. Maurino, V. Moltiva, A. Lourenco and A. S. Feliciano, Cernumidine and isocernumidine, new type of cyclic guanidine alkaloids from Solanum cernuum, Tetrahedron Lett., 2011, 52, 6392–6395 CrossRef CAS.
- T. Kowalczyk, A. Merecz-Sadowska, P. Rijo, M. Mori, S. Hatziantoniou, K. Gorski, J. Szemraj, J. Piekarski, T. Sliwinski, M. Bijak and P. Sitarek, Hidden in Plants-A Review of the Anticancer Potential of the Solanaceae Family in In Vitro and In Vivo Studies, Cancers, 2022, 14, 42 CrossRef PubMed.
- M. A. Miranda, M. Lemos, K. A. Cowart, D. Rodenburg, J. D. McChesney, M. M. Radwan, N. Furtado and J. K. Bastos, Gastroprotective activity of the hydroethanolic extract and isolated compounds from the leaves of Solanum cernuum Vell, J. Ethnopharmacol., 2015, 172, 421–429 CrossRef PubMed.
- J. L. Damasceno, P. F. de Oliveira, M. A. Miranda, M. Lima, J. K. Bastos and D. C. Tavares, Antigenotoxic and Antioxidant Properties of Solanum cernuum and Its Alkaloid, Cernumidine, Biol. Pharm. Bull., 2016, 39, 920–926 CrossRef CAS PubMed.
- M. A. Miranda, A. Mondal, M. Sachdeva, H. Cabral, Y. Neto, I. Khan, M. Groppo, J. D. McChesney and J. K. Bastos, Chemosensitizing Effect of Cernumidine Extracted from Solanum cernuum on Bladder Cancer Cells in Vitro, Chem. Biodiversity, 2019, 16, e1900334 CrossRef CAS PubMed.
- S. Bernhard, S. Hug, A. E. P. Stratmann, M. Erber, L. Vidoni, C. L. Knapp, B. D. Thomass, M. Fauler, B. Nilsson, K. N. Ekdahl, K. Fohr, C. K. Braun, L. Wohlgemuth, M. Huber-Lang and D. A. C. Messerer, Interleukin 8 Elicits Rapid Physiological Changes in Neutrophils That Are Altered by Inflammatory Conditions, J. Innate Immun., 2021, 13, 225–241 CrossRef CAS PubMed.
- M. C. Cesta, M. Zippoli, C. Marsiglia, E. M. Gavioli, F. Mantelli, M. Allegretti and R. A. Balk, The Role of Interleukin-8 in Lung Inflammation and Injury: Implications for the Management of COVID-19 and Hyperinflammatory Acute Respiratory Distress Syndrome, Front. Pharmacol., 2022, 12, 808797 CrossRef PubMed.
- B. S. Qazi, K. Tang and A. Qazi, Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis, Int. J. Inflammation, 2011, 908468 Search PubMed.
- K. Fousek, L. A. Horn and C. Palena, Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression, Pharmacol. Ther., 2021, 219, 107692 CrossRef CAS PubMed.
- H. Jiang, J. Cui, H. Chu, T. Xu, M. Xie, X. Jing, J. Xu, J. Zhou and Y. Shu, Targeting IL8 as a sequential therapy strategy to overcome chemotherapy resistance in advanced gastric cancer, Nature, 2022, 8, 235 CAS.
- Y. Pu, Q. Li, Y. Wang, L. Xu, Q. Qiao, Y. Guo and C. Guo, pERK-mediated IL8 secretion can enhance the migration, invasion, and cisplatin resistance of CD10-positive oral cancer cells, BMC Cancer, 2021, 21, 1283 CrossRef CAS PubMed.
- S. Limpakan, P. Wongsirisin, S. Yodkeeree, B. Chakrabandhu, W. Chongruksut and P. Limtrakul, Interleukin-8 associated with chemosensitivity and poor chemotherapeutic response to gastric cancer, J. Gastrointest. Oncol., 2019, 10, 1120–1132 CrossRef PubMed.
- S. Y. Park, J. Han, J. B. Kim, M. G. Yang, Y. J. Kim, H. J. Lim, S. Y. An and J. H. Kim, Interleukin-8 is related to poor chemotherapeutic response and tumourigenicity in hepatocellular carcinoma, Eur. J. Cancer, 2014, 50, 341–350 CrossRef CAS PubMed.
- L. Duhamel, Amino, Nitroso, Nitro Compounds, Their Derivatives Supplement F: Part 2, Wiley, 1982, vol. 2, ch. 20, pp. 849–907 Search PubMed.
- J. A. Carmona, C. Rodriguez-Franco, J. Lopez-Serrano, A. Ros, J. Iglesias-Siguenza, R. Fernandez, J. M. Lassaletta and V. Hornillos, Atroposelective Transfer Hydrogenation of Biaryl Aminals via Dynamic Kinetic Resolution. Synthesis of Axially Chiral Diamines, ACS Catal., 2021, 11, 4117–4124 CrossRef CAS.
- E. Sawatzky, A. Drakopoulos, M. Rolz, C. Sotriffer, B. Engels and M. Decker, Experimental and theoretical investigations into the stability of cyclic aminals, Beilstein J. Org. Chem., 2016, 12, 2280–2292 CrossRef CAS PubMed.
- H. Brederec, G. Simchen, H. Hoffmann, P. Horn and R. Wahl, Stability of aminal esters and their reactions with aromatic aldehydes, Angew. Chem., Int. Ed. Engl., 1967, 6, 356–357 CrossRef.
- A. Greenberg, N. Molinaro and M. Lang, Structure and dynamics of troger base and simple derivatives in acidic media, J. Org. Chem., 1984, 49, 1127–1130 CrossRef CAS.
- I. Bosque, J. C. Gonzalez-Gomez, M. I. Loza and J. Brea, Natural Tetraponerines: A General Synthesis and Antiproliferative Activity, J. Org. Chem., 2014, 79, 3982–3991 CrossRef CAS PubMed.
- M. Kuramoto, N. Miyake, Y. Ishimaru, N. Ono and H. Uno, Cylindradines A and B: Novel Bromopyrrole Alkaloids from the Marine Sponge Axinella cylindratus, Org. Lett., 2008, 10, 5465–5468 CrossRef CAS PubMed.
- O. V. Runarsson, J. Artacho and K. Warnmark, The 125th Anniversary of the Troger's Base Molecule: Synthesis and Applications of Troger's Base Analogues, Eur. J. Org. Chem., 2012, 7015–7041 CrossRef CAS.
- A. Quintard, S. Belot, E. Marchal and A. Alexakis, Aminal-Pyrrolidine Organocatalysts - Highly Efficient and Modular Catalysts for alpha-Functionalization of Carbonyl Compounds, Eur. J. Org. Chem., 2010, 927–936 CrossRef CAS.
- J. F. Bai, H. Sasagawa, T. Yurino, T. Kano and K. Maruoka, In situ generation of N-Boc-protected alkenyl imines: controlling the E/Z geometry of alkenyl moieties in the Mukaiyama-Mannich reaction, Chem. Commun., 2017, 53, 8203–8206 RSC.
- J. G. Pereira, J. P. M. Antonio, R. Mendonca, R. F. A. Gomes and C. A. M. Afonso, Rediscovering aminal chemistry: copper(ii) catalysed formation under mild conditions, Green Chem., 2020, 22, 7484–7490 RSC.
- T. Kano, Y. Aota, D. Asakawa and K. Maruoka, Brønsted acid-catalyzed Mannich reaction through dual activation of aldehydes and N-Boc-imines, Chem. Commun., 2015, 51, 16472–16474 RSC.
- K. Yasumoto, T. Kano and K. Maruoka, One-Pot Synthesis of Less Accessible N-Boc-Propargylic Amines through BF3 -Catalyzed Alkynylation and Allylation Using Boronic Esters, Org. Lett., 2019, 21, 3214–3217 CrossRef CAS PubMed.
- M. Kohr and U. Kazmaier, C-H Functionalization of Peptides via Cyclic Aminal Intermediates, Org. Lett., 2021, 23, 5947–5951 CrossRef CAS PubMed.
- T. D. Harris and G. P. Roth, Ortho-Lithiation via a Carbonyl Synthon, J. Org. Chem., 1979, 44, 2004–2007 CrossRef CAS.
- S. G. Ouellet, J. B. Tuttle and D. W. C. MacMillan, Enantioselective organocatalytic hydride reduction, J. Am. Chem. Soc., 2005, 127, 32–33 CrossRef CAS PubMed.
- R. Rippel, L. Pinheiro, M. Lopes, A. Lourenco, L. M. Ferreira and P. S. Branco, Synthetic Approaches to a Challenging and Unusual Structure-An Amino-Pyrrolidine Guanine Core, Molecules, 2020, 25, 17 CrossRef PubMed.
- G. Verardo, E. Bombardella, C. D. Venneri and P. Strazzolini, A Convenient Synthesis of Unsymmetrically Substituted Ureas via Carbamoyl Azides of alpha-N-Protected Amino Acids, Eur. J. Org. Chem., 2009, 6239–6244 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Interrupted Curtius Rearrangements of Quaternary Proline Derivatives: A Flow Route to Acyclic Ketones and Unsaturated Pyrrolidines, J. Org. Chem., 2021, 86, 14199–14206 CrossRef CAS PubMed.
- I. P. Beletskaya and A. V. Cheprakov, The Heck reaction as a sharpening stone of palladium catalysis, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed.
- J. S. Yadav, B. V. S. Reddy, K. S. Reddy and K. B. Reddy, Indium-mediated facile cleavage of the t-butoxycarbonyl group from di-t-butylimidodicarbonate, Tetrahedron Lett., 2002, 43, 1549–1551 CrossRef CAS.
- R. S. Giri, S. Roy, G. Dolai, S. R. Manne and B. Mandal, FeCl3-Mediated Boc Deprotection: Mild Facile Boc-Chemistry in Solution and on Resin, ChemistrySelect, 2020, 5, 2050–2056 CrossRef CAS.
- R. Frank and M. Schutkowski, Extremely mild reagent for Boc deprotection applicable to the synthesis of peptides with thioamide linkages, Chem. Commun., 1996, 2509–2510 RSC.
- A. M. Felix, Cleavage of protecting groups with boron tribromide, J. Org. Chem., 1974, 39, 1427–1429 CrossRef CAS.
- J. M. Lopez-Soria, S. J. Perez, J. N. Hernandez, M. A. Ramirez, V. S. Martin and J. I. Padron, A practical, catalytic and selective deprotection of a Boc group in N,N '-diprotected amines using iron(III)-catalysis, RSC Adv., 2015, 5, 6647–6651 RSC.
- D. S. Bose, K. K. Kumar and A. V. N. Reddy, A new protocol for selective deprotection of N-tert-butoxycarbonyl protective group (t-boc) with Sn(OTf)(2), Synth. Commun., 2003, 33, 445–450 CrossRef CAS.
- G. P. Kumar, D. Rambabu, M. V. B. Rao and M. Pal, Iodine-Mediated Neutral and Selective N-Boc Deprotection, J. Chem., 2013, 5 Search PubMed.
- T. Apelqvist and D. Wensbo, Selective removal of the N-BOC protective group using silica gel at low pressure, Tetrahedron Lett., 1996, 37, 1471–1472 CrossRef CAS.
- N. George, S. Ofori, S. Parkin and S. G. Awuah, Mild deprotection of theN-tert-butyloxycarbonyl (N-Boc) group using oxalyl chloride, RSC Adv., 2020, 10, 24017–24026 RSC.
- N. J. Tom, W. M. Simon, H. N. Frost and M. Ewing, Deprotection of a primary Boc group under basic conditions, Tetrahedron Lett., 2004, 45, 905–906 CrossRef CAS.
- S. El Kazzouli, J. Koubachi, S. Berteina-Raboin, A. Mouaddib and G. Guillaumet, A mild and selective method for the N-Boc deprotection by sodium carbonate, Tetrahedron Lett., 2006, 47, 8575–8577 CrossRef CAS.
- Z. J. Liu, N. Yasuda, M. Simeone and R. A. Reamer, N-Boc Deprotection and Isolation Method for Water-Soluble Zwitterionic Compounds, J. Org. Chem., 2014, 79, 11792–11796 CrossRef CAS PubMed.
- T. Ideguchi, T. Yamada, T. Shirahata, T. Hirose, A. Sugawara, Y. Kobayashi, S. Omura and T. Sunazuka, Asymmetric Total Synthesis of Neoxaline, J. Am. Chem. Soc., 2013, 135, 12568–12571 CrossRef CAS PubMed.
- R. Alam and G. A. Molander, Photoredox-catalyzed Direct Reductive Amination of Aldehydes without an External Hydrogen/Hydride Source, Org. Lett., 2018, 20, 2680–2684 CrossRef CAS PubMed.
- M. S. Bernatowicz, Y. L. Wu and G. R. Matsueda, Urethane protected derivatives of 1-guanylpyrazole for the mild and efficient preparation of guanidines, Tetrahedron Lett., 1993, 34, 3389–3392 CrossRef CAS.
- S. L. Gwaltney, H. S. Jae, D. M. Kalvin, L. G. H. L. Sham, Q. Li, A. K. Claiborne, L. Wang, K. J. Barr and K. W. Woods, Oxazoline antiproliferative agents, US6228868B1, 2001.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2306654 and 2306655. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj05406c |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |