
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of dithioglycol-functionalized periodic mesoporous organosilicas for the simultaneous removal of mercury ions and organic dyes from water†
Yuanjiang
Zhao
,
Yuhang
Liu
and
Liming
Jiang
 *
*
Key Laboratory of Macromolecular Synthesis and Functionalization of Ministry of Education, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310058, China. E-mail: cejlm@zju.edu.cn
First published on 30th January 2024
Abstract
A novel ethylene-bridged silsesquioxane was synthesized by a heat-induced thiol–ene addition reaction of 2-acetylthioethanethiol to 1,2-bis(triethoxysilyl)ethene. Upon deacetylation, the formed organosiloxane precursor bearing the dithiol moiety (–SCH2CH2SH) self-assembled into a sulfur-rich PMO material in the presence of the structure-directing agent under acidic conditions. The loading of sulfur-bound ligands in the PMO framework can be adjusted by co-condensing the precursor with tetraethyl orthosilicate. The structure of the resulting PMOs was characterized by powder X-ray diffraction, X-ray photoelectron spectroscopy, scanning electron microscopy, transmission electron microscopy, infrared spectroscopy, and nitrogen adsorption and desorption studies. The PMO prepared by using the thio-bis-silane precursor alone can be used for efficient and fast capture of mercury ions from water with a saturation Hg(II) uptake capacity of 1253 mg g−1 and recycled several times with negligible loss in the adsorption efficiency. This material also showed a high adsorption selectivity for Hg(II) over other competitive metal ions like Zn2+, Pb2+, Fe3+, Cu2+ and Mn2+. Remarkably, the new adsorbent proved to be effective for simultaneously removing Hg(II) and rhodamine B (RhB) from simulated sewage with removal rates of over 97% and 91% for both at 100 ppm, respectively, indicating its potential for the practical application in environmental remediation.
1. Introduction
Mercury contamination in water is a serious threat to the ecological environment and human health.1,2 Among various techniques known for the treatment of Hg(II)-polluted aqueous effluents, the adsorptive removal using porous materials is regarded as the most convenient and affordable method.3–5 In this context, a frequently adopted strategy is to integrate sulfur-bound ligands into solid carriers because of their strong affinity towards the Hg species based on the favorable soft–soft interactions. Such adsorbents include thiol-modified mesoporous silica,6–8 sulfurated mesoporous carbon,9 chalcogenide frameworks/aerogels,10,11 and others.12 In recent years, a number of thiol-/thioether-functionalized porous organics, such as covalent organic frameworks (COFs)13–17 and porous organic polymers (POPs),3,18–23 have been demonstrated to be outstanding in removing mercury ions from aqueous media, but their high-cost synthesis may present a hurdle to their practical applications.24Periodic mesoporous organosilicas (PMOs),25–27 featuring the unique organic–inorganic hybrid frameworks, have attracted increasing attention for various sorption-related applications due to their large surface area, suitable pore volume, excellent hydrothermal stability, and chemical/structural tunability.28–30 These materials are generally synthesized via a sol–gel self-assembly process of bridged alkoxysilanes of type (R′O)3Si–R–Si(OR′)3 with the structure-directing agents in aqueous solution under acidic or basic catalysis. One of the important features of PMOs is that the bridging organic (functional) groups are homogeneously and covalently incorporated in the pore walls at the molecular level, thus allowing a higher loading of organic matter and better prevention of channel blockage.28,29 Recently, Esquivel et al.31 reported the synthesis of a thiol-tagged silsesquioxane precursor by a thiol acid–ene click reaction and the corresponding ethylene bridged PMO material with well-ordered structure and high loading of sulfur groups. The uptake capacity of the material for Hg(II) in water reached 1183 mg g−1, exceeding the values reported for most of the POP/COF-based adsorbents.3,14,17,18,20,22 Bhaumik and coworkers developed nitrogen- and sulfur-rich highly ordered PMO materials containing thiadiazole and thiol moieties inside the pore-wall, which show a very fast and exceptionally high uptake capacity for mercury (2081 mg g−1).32 Ha et al. synthesized a series of sulphonic acid functionalized urea-bridged PMO materials and successfully applied them for the highly selective separation of cobalt(II) ions from artificial seawater.33 These studies highlight the great potential of the organosilica framework as a platform for the design of versatile adsorbent materials. However, PMOs presently are mostly explored for the single metal-ion capture,33–40 and no reports on the simultaneous removal of heavy metal and toxic organic species from the same water sample have been published so far.41 Thus, there are still opportunities to expand the use of PMOs as adsorbent materials for different kinds of co-existing pollutants.
This study reports here the preparation of a new PMO material containing a high density of thiol and thioether moieties in the framework. The employed protocol began with the synthesis of a bridged silsesquioxane through a heat-induced thiol–ene click reaction of 2-acetylthioethanethiol with 1,2-bis(triethoxysilyl)ethene (BTEE), followed by aminolysis to remove acetyl groups of intermediate (![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) ), and finally hydrolysis and condensation of the resulting organosilane (DT-BTEE) in the presence of the surfactant Pluronic P123 (Scheme 1). The structural features of the PMO particles could be tuned by the co-condensation of DT-BTEE with tetraethoxysilane. The introduction of more binding sites in the pendant alkyl chain (i.e. –SCH2CH2SH) was designed to facilitate the chelation of metal species in the PMO matrix. As expected, the dithiol-functionalized PMO obtained with DT-BTEE as a single organosilica source was capable of effectively and efficiently removing mercury from aqueous solution. This material was investigated in-depth in terms of its thermodynamic adsorption behavior, uptake kinetics, selectivity and recyclability. Also, we evaluated the simultaneous removal efficiency of the new adsorbent towards Hg(II) and coexisting organic dyes from simulated sewage using RhB as a model compound.
), and finally hydrolysis and condensation of the resulting organosilane (DT-BTEE) in the presence of the surfactant Pluronic P123 (Scheme 1). The structural features of the PMO particles could be tuned by the co-condensation of DT-BTEE with tetraethoxysilane. The introduction of more binding sites in the pendant alkyl chain (i.e. –SCH2CH2SH) was designed to facilitate the chelation of metal species in the PMO matrix. As expected, the dithiol-functionalized PMO obtained with DT-BTEE as a single organosilica source was capable of effectively and efficiently removing mercury from aqueous solution. This material was investigated in-depth in terms of its thermodynamic adsorption behavior, uptake kinetics, selectivity and recyclability. Also, we evaluated the simultaneous removal efficiency of the new adsorbent towards Hg(II) and coexisting organic dyes from simulated sewage using RhB as a model compound.
 | ||
| Scheme 1 Synthetic route of the thio-bis-silane precursor (DT-BTEE) and the corresponding dithiol-functionalized ethylene-bridged PMO materials (DT-PMOs). | ||
2. Experimental
2.1. Materials
Vinyltriethoxysilane (98%), tetraethoxysilane (TEOS, 98%), 1,2-dithioglycol, azobisisobutyronitrile (AIBN), and rhodamine B (RhB) were supplied by Shanghai Aladdin Bio-Chem Technology Co., Ltd (China). (PCy3)2Cl2Ru = CHPh was provided by Shanghai Haohong Scientific Co., Ltd., China. Pluronic P123 (EO20PO70EO20) was purchased from Aldrich, and all other chemicals were commercially available and used as received unless otherwise indicated.2.2. Characterization
Fourier transform infrared (FT-IR) spectra were recorded on a Nicolet is10 spectrophotometer (Bruker TENSOR II, Germany) in the range of 4000–400 cm−1 with 32 successive scans at a resolution of 2 cm−1. Before analysis, the samples were diluted in KBr and pressed to a 1 cm diameter pellet. Raman spectra were recorded on a Renishaw inVia-Reflex using a 50 mW, 532 nm laser and <0.65 cm−1 detector resolution. NMR spectra were acquired on a Bruker Avance III 400 spectrometer (400 MHz for 1H NMR and 101 MHz for 13C NMR). Chemical shifts are reported in parts per million (ppm) relative to those of tetramethylsilane (TMS). The powder X-ray diffraction (PXRD) patterns were collected on an X′Pert3 analytic diffractometer (PANalytical, Netherlands) at 40 kV and 40 mA using CuKα radiation (λ = 4.5419 Å). X-ray photoelectron spectroscopy (XPS) was performed on a K-Alpha+ photoelectron spectrometer (Thermo Fisher Scientific, USA) using monochromatic Al Kα X-ray as the excitation source (a working voltage of 15 kV and a filament current of 15 mA). Inductively coupled plasma–optical emission spectroscopy (ICP–OES) measurements were performed on an Agilent 730-ES instrument. UV-vis spectra were recorded on a Shimadzu UV-2600 spectrophotometer (Japan). Analyses of the morphology and chemical composition of the samples were conducted using a Hitachi S-4800 field-emission scanning electron microscope (FE-SEM) equipped with an energy dispersive X-ray (Oxford Instruments INCA EDX) system. The samples were coated with gold by sputtering before imaging. Transmission electron microscopy (TEM) images were acquired using a HT7700 transmission electron microscope (Hitachi, Japan) at an acceleration voltage of 100 kV. The samples were dispersed in ethanol and dropped on a lacey support film followed by drying under ambient conditions. Thermogravimetric analysis (TGA) was performed on a Q50 thermogravimetric analyzer (TA Instruments, USA) at a heating rate of 10 °C min−1 from room temperature to 800 °C under an airflow of 0.6 mL min−1. Nitrogen adsorption and desorption measurements were performed at 77 K on a BELSORP-max automated gas sorption apparatus (MicrotracBEL Corp., Japan). Before the measurement, the samples (20–30 mg) were degassed under dynamic vacuum (1 × 10−5 Pa) at 100 °C for 12 h. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) theory, and the pore size distribution was analyzed using the Barrett–Joyner–Halenda (BJH) model from desorption isotherm data.2.3. Synthesis of starting materials and the precursor thio-bis-silane
1,2-Bis(triethoxysilyl)ethene (BTEE) was synthesized by a previously reported procedure.42 Briefly, 54.3 mg of Grubbs′ first generation catalyst ((PCy3)2Cl2Ru = CHPh, 0.066 mmol) was added to 38.732 g of vinyltriethoxysilane (203.5 mmol) under a N2 atmosphere. The resulting mixture was stirred at room temperature for 1 h and later refluxed for 4 h. Unreacted vinyltriethoxysilane was collected by distillation. The bis-silane was separated under vacuum distillation as a colorless liquid (91% yield). The chemical structure of the product was confirmed by NMR spectroscopy (Fig. S1, ESI†). The synthesis and structural characterization of 2-acetylthioethanethiol (![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif) ) are detailed in the ESI† (Fig. S2).
) are detailed in the ESI† (Fig. S2).
The designed thio-bis-silane precursor, 1-(2-mercaptoethylthio)-1,2-bis(triethoxysilyl)ethane (DT-BTEE), was prepared according to the reported method with minor modifications outlined below.43 A mixture of AIBN (150 mg, 0.91 mmol), BTEE (8.28 g, 23.5 mmol), and 2-acetylthioethanethiol (; 3.52 g, 25.85 mmol) was shaken under a nitrogen atmosphere until complete dissolution of the initiator occurred. The solution was then stirred overnight at 70 °C. Purification by column chromatography using dichloromethane as the eluent resulted in 1-(2-thioacetylethylthio)-1,2-bis(triethoxysilyl)ethane () as a colorless liquid in 91% yield (10.4 g). Subsequent aminolysis of the thioacetyl groups (–SCOCH3 → –SH) was carried out by treatment with 9 mL of dry propylamine (109.5 mmol) at 0 °C for 2 min. Then, the excess propylamine was removed under reduced pressure to give a liquid containing DT-BTEE and propylacetamide (![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ) (see Fig. S3 and S4, ESI†). The product was directly used for the synthesis of PMOs without further purification.
) (see Fig. S3 and S4, ESI†). The product was directly used for the synthesis of PMOs without further purification.
2.4. Synthesis of dithiol-functionalized PMOs
The synthesis of dithiol-modified PMOs followed the literature method with a minor modification.44 In one of the optimized cases, 0.42 g of P123 and 2.65 g of KCl were added to a solution containing 2.1 mL of hydrochloric acid (36%) and 14.76 mL of deionized water and then the mixture was stirred overnight at 40 °C. After complete dissolution of the surfactant, the silane precursors DT-BTEE and TEOS were added at a predetermined molar ratio, and the solution was stirred at 40 °C for 24 h, followed by ageing overnight at 100 °C. The white solid was collected by centrifugation and washed thoroughly with deionized water and ethanol. To remove the residual surfactant template and propylacetamide, the as-prepared PMO material (0.5 g) was refluxed in 120 mL of acidified ethanol (EtOH/36% HCl 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for 24 h. After repeating this process twice, the product was filtered, washed with ethanol and dried in vacuo at 60 °C.
:1, v/v) for 24 h. After repeating this process twice, the product was filtered, washed with ethanol and dried in vacuo at 60 °C.
2.5. Adsorption experiments
Batch adsorption experiments were performed in acidic aqueous media with 1000 ppm Hg(II) ions. The pH was maintained around 3 to avoid precipitation of metal hydroxides. Typically, 20 mg of DT-PMOs was dispersed in 30 mL of 1000 ppm aqueous Hg(NO3)2 solution and stirred for 12 h at room temperature. After the separation of the adsorbent by centrifugation, the content of mercury ions in the supernatant was determined by means of ICP-OES. Each adsorption experiment was carried out in duplicate. The adsorption capacity (q, mg g−1) was calculated based on the expression31q = (C0 − Ct) × (V/m), where C0 represents the initial Hg(II) concentration in solution, Ct is the Hg(II) concentration in solution at time t, V is the volume of solution, and m is the mass of PMO. Also, the removal efficiency was calculated according to the formula R = 100 × (C0 − Ct)/C0.The organic dye adsorption experiments were conducted in the same way as in the case of mercury ion adsorption, but using UV-vis analysis to determine the change in the dye concentration in solution before and after adsorption.
2.6. Interpretation of the adsorption characteristics
The thermodynamic adsorption process was carried out as follows: 5 mg of the DT-PMO adsorbent was dispersed in 5 mL of Hg(NO3)2 solution (50–1000 ppm; for RhB 20–400 ppm) and stirred for 24 h at room temperature. After centrifugal separation, the supernatant was collected and the residual pollutant concentration was analyzed according to the method described above. The equilibrium adsorption data were fitted to the Langmuir and Freundlich models,45,46 shown in eqn (1) and (2), where q (mg g−1) and qmax (mg g−1) are the adsorption amount at equilibrium and the maximum adsorption capacity, respectively, Ce (mmol L−1) is the residual pollutant concentration at equilibrium, KL is the Langmuir constant, and KF and n are the Freundlich constants. | (1) |
| q = KFCe1/n | (2) |
In the kinetic studies, the mixtures of DT-PMO (40 mg) and 40 mL of solutions containing 1000 ppm Hg2+ and 400 ppm RhB were shaken at room temperature for 30–240 min. The influence of the contact time was elucidated using two linearized kinetic models, i.e., the pseudo-first order and pseudo-second order. The relevant formulas are presented in eqn (3) and (4), where qe is the amount of pollutant (Hg2+ or RhB) adsorbed per gram of sorbent at equilibrium, qt is the adsorption amount at particular time t; k1 and k2 represent the sorption rate constants estimated, respectively, by the pseudo first- and pseudo second-order models.47,48
| ln(qe − qt) = lnqe − k1t | (3) |
 | (4) |
2.7. Multi-metal adsorption
10 mg of DT-PMO was added into 10 mL of 10 ppm aqueous solution (pH 3) containing the following metal ions: Hg2+, Cu2+, Ca2+, Pb2+, Mg2+, Zn2+, Mn2+ and Fe3+ (each at 10 ppm). The rest of the procedure was the same as that for Hg(II) adsorption experiments (see Section 2.5).2.8. Simultaneous adsorption of Hg(II) and RhB from simulated sewage
For the multicomponent adsorption tests, a synthesized solvent was first prepared according to the OECD guidelines for the testing of chemicals.49 Specifically, MgSO4·7H2O (2 mg), CaCl2·2H2O (4 mg), NaCl (7 mg), K2HPO4 (28 mg), urea (30 mg), meat extract (110 mg) and peptone (160 mg) were dissolved in 1000 mL of tap water. Then, Hg(NO3)2 and RhB (10–100 ppm each) ware added into the synthesized solvent to yield the test water samples. To determine the adsorption capacity of DT-PMO for Hg(II) and RhB, 10 mg of the adsorbent was shaken with 10 mL of the simulated sewage for 24 h at room temperature. The rest of the procedure was the same as that for the single-pollutant analysis (see Section 2.5).2.9. Elution and recycling experiments
50 mg of the Hg(II)-adsorbed DT-PMO sample was dispersed in 30 mL of an aqueous solution of thiourea and hydrochloric acid (0.1 M each) and stirred for 24 h at room temperature. Afterward, the adsorbent was isolated by centrifugation and washed with deionized water until no detectable pollutants were observed. The elution of RhB from DT-PMO was similar to that described above for Hg(II) except for the use of an ethanol solution of 1 M HCl as the eluting agent. After drying, the regenerated adsorbent was used for the next adsorption test.3. Results and discussion
3.1. Synthesis and characterization of adsorbents
As delineated in Scheme 1, the dithiol-functionalized PMOs were obtained by self-assembly assisted co-condensation of the homemade organosilica precursor DT-BTEE and TEOS in various molar ratios. For the sake of clarity, the resultant materials are denoted as DT-PMOs or PMO-nT, where n (n = 100, 75, and 50) is the molar percent of DT-BTEE in the initial silane mixture. Performing the process with TEOS as the sole Si-source led to the generation of ordered mesoporous silica (OMS), which was used as the control sample for the adsorption measurements. All the materials presented irregularly shaped platelet morphology with the size of tens of nanometers to several microns, as can be seen from SEM images (Fig. S5, ESI†).The structural order of the above-mentioned four siliceous samples was assessed by powder X-ray diffraction measurements. From Fig. 1a, it was observed that the OMS particles showed three characteristic diffraction peaks in the low-angle region, which correspond to the (100), (110) and (200) reflections of a 2D hexagonal material with p6mm symmetry.50 These peaks shifted slightly towards a higher 2θ value for the hybrid PMOs, and the intensity gradually decreased upon increasing the organic moieties in the framework. Only one peak appeared in the XRD patterns of PMO-75T and PMO-100T, which is indexed to the (100) plane, indicating a markedly decreased long-range ordering compared to the pure silica OMS. Apparently, the ordering degree of mesostructures for the DT-PMOs was disturbed by the existence of pendant dithiol residues at the bridging groups. A similar phenomenon was also observed with the disulfide or tetrasulfide functionalized porous nanomaterials.38–40
 | ||
| Fig. 1 (a) XRD patterns, (b)–(e) N2 adsorption–desorption isotherms, and (f) the pore size distributions of the DT-PMO and OMS particles after surfactant extraction. The inset in (a) displays the locally magnified patterns of DT-PMOs. | ||
The N2 adsorption–desorption isotherms of OMS and PMO particles show the characteristic type IV isotherms with one-step capillary condensation at a relative pressure of around 0.4–0.5 (Fig. 1b–e), an indication of a well-developed mesoporous structure.51 An H2 hysteresis loop for these samples demonstrates the existence of complex porous structures. In the case of DT-PMOs, the BET surface areas (SBET), pore volumes (Vp), and pore diameters were 265–350 m2 g−1, 0.24–0.41 cm3 g−1, and 2.82–3.52 nm, respectively (Table 1). These values tend to increase with the molar fraction of TEOS in the reaction mixture, being in agreement with the literature.31 Moreover, the mesopores of DT-PMOs could be observed by TEM, but the contrast was relatively weak for PMO-75T and PMO-100T (Fig. 2) due to the disordered pore arrangement. Albeit having a smaller pore width compared to OMS particles, the pore size distribution of DT-PMOs was narrow, as illustrated in Fig. 1f.
| Samples | Molar ratio (DT-BTEE:TEOS) |
C%a | S%a | S BET (m2 g−1) | V p (cm3 g−1) | D p (nm) |
|---|---|---|---|---|---|---|
| a Weight% determined by elemental analysis. b S BET and Vp are BET surface areas and pore volumes obtained from N2 adsorption–desorption isotherms, respectively. c The pore diameter (Dp) denotes the peak values in the corresponding pore size distribution curves. | ||||||
| PMO-100T | 100:0 |
35.83 | 24.70 | 265.1 | 0.2415 | 2.824 |
| PMO-75T | 75:25 |
34.57 | 22.93 | 341.1 | 0.3638 | 2.912 |
| PMO-50T | 50:50 |
33.00 | 20.67 | 350.5 | 0.4089 | 3.522 |
| OMS | 0:100 |
— | — | 386.3 | 0.7583 | 6.819 |
 | ||
| Fig. 2 TEM images of (a) OMS, (b) PMO-50T, (c) PMO-75T, and (d) PMO-100T. | ||
The composition of DT-PMOs was characterized by FT-IR and Raman spectroscopies. The absorption peak at 2973 cm−1 is attributed to the asymmetrical stretching vibration of CH3 in the unhydrolyzed organosilane species (Fig. 3a). The presence of organic bridging moieties (–CH(SCH2CH2SH)CH2–) in the DT-PMOs was evidenced by the peaks at 2923 cm−1 and 2880 cm−1, which correspond to the asymmetrical stretching vibration of CH2 and CH groups.32 A weak S–H absorbance peak at 2562 cm−1 indicated that the thio-bis-silane DT-BTEE had condensed in the PMO matrices. In the Raman spectra (Fig. 3b), a peak at 2562 cm−1 consistent with the S–H vibration52 is clearly visible for all DT-PMOs, and the intensity is roughly proportional to the content of sulfur-bound groups in the frameworks (Fig. 3c). Besides, DT-PMOs show a band at ∼508 cm−1, characteristic of the vibration modes of S–S moieties,53 suggesting that some sulfhydryl groups formed disulfide linkages by air oxidation during the hydrolysis and co-condensation processes.31 In contrast, these peaks were not present in the FT-IR and Raman spectra of the pure silica particles. Elemental analysis revealed that the sulfur contents (w/w) for PMO-50T, PMO-75T and PMO-100T are 24.70%, 22.93%, and 20.67%, respectively (Table 1). As a representative example, Fig. S6 (ESI†) shows the TGA curve of PMO-100T, for which high thermal stability (up to 270 °C) was observed, allowing the material to be suitable for environmental applications.
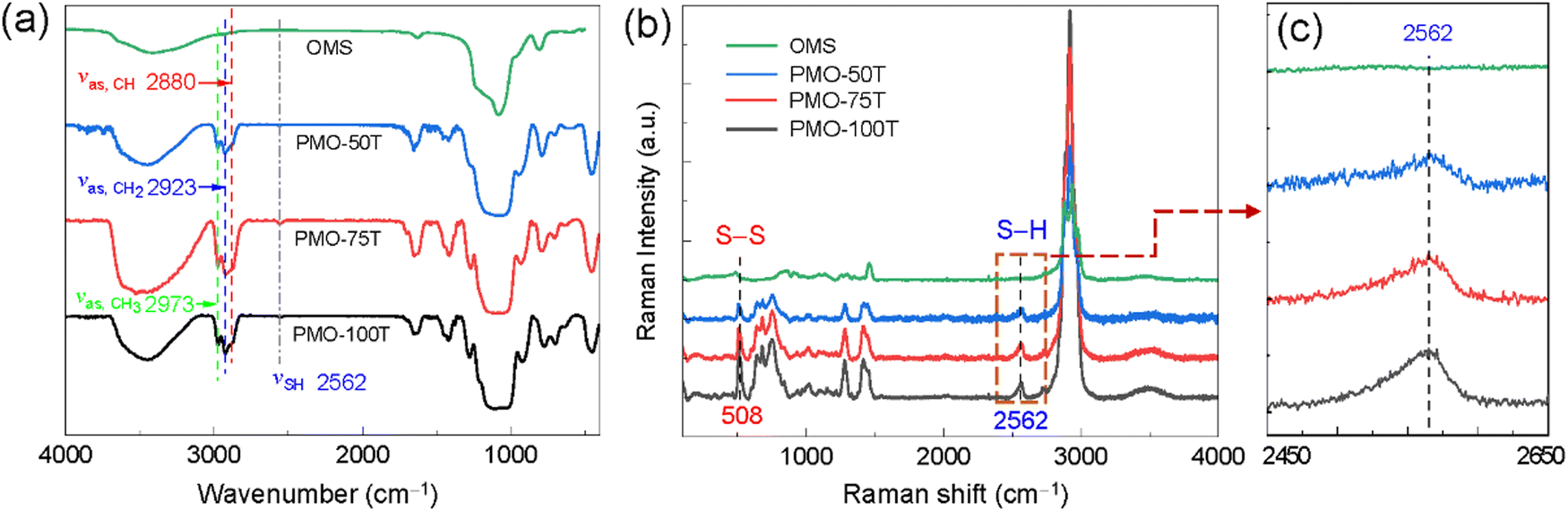 | ||
| Fig. 3 (a) FT-IR spectra of DT-PMOs and ordered mesoporous silica (OMS), (b) Raman spectra and (c) the corresponding locally magnified Raman spectra. | ||
3.2. Hg(II) adsorption capacity of DT-PMOs
The metal adsorption experiments were performed in an aqueous solution of 1000 ppm Hg(NO3)2 and different DT-PMOs at pH 3. As can be seen from Table 2, the PMO materials are very effective mercury adsorbents, whereas their silica counterpart (OMS) showed almost no adsorption of Hg(II) ions under the same conditions due to the lack of specific binding sites in the internal pores. The adsorption affinity of DT-PMOs for Hg(II) increases with increasing sulfur content in the framework. Despite less ordered pore structure and lower porosity (265.1 m2 g−1) in comparison with the other two samples, PMO-100T was the most efficient for Hg(II) removal, and its uptake capacity (1231 mg g−1) was higher than those of most reported thiol- and thioether-functionalized porous materials such as mesoporous silicas,6–8 COFs,13,14,17 POPs,3,18–20,22 PMOs,31,32,34 and MOFs.54–61 This result also signifies that the ordered alignment of channels in PMOs was not a requirement for efficient Hg(II) uptake. On the other hand, the calculated Stotal/Hg ratio is below 1 (0.79–0.72) in all cases, implying that partial binding sites were not exposed to mercury ions due to their embedding in the pore wall.32 Moreover, the highly selective adsorption of PMO-100T for Hg(II) was almost unaffected by commonly coexisting ions such as Zn2+, Pb2+, Fe3+, Ca2+ Mg2+, Cu2+ and Mn2+ (Fig. S7, ESI†).| Samples | S total (mmol g−1) | Hg(II) adsorption capacity (mg g−1) | Hg/Stotal |
|---|---|---|---|
| a Adsorbent dosage 20 mg, Hg(NO3)2 solution (1000 ppm, 30 mL), pH = 3; see Section 2.5 for details. b Calculated from elemental analysis. | |||
| PMO-100T | 7.72 | 1231.3 ± 6.9 | 0.79 |
| PMO-75T | 7.17 | 1067.4 ± 10.2 | 0.74 |
| PMO-50T | 6.45 | 925.7 ± 13.0 | 0.72 |
| OMS | 0 | 5.9 ± 4.2 | — |
Fig. 4 shows the SEM image of PMO-100T and the corresponding energy-dispersive X-ray (EDX) mapping after Hg(II) adsorption. It can be seen that the sulfur atoms and adsorbed mercury are evenly distributed throughout the matrix. From the XPS spectra shown in Fig. S8 (ESI†), it was observed that the binding energies of Si 2p, C 1s and O 1s were not influenced by the mercury uptake. Also, a slight shift to higher binding energies for the S 2p peak of Hg(II)-loaded PMO-100T was observed compared with the pristine adsorbent, which is attributed to the electron-donation of sulfur atoms to Hg(II) ions.60
 | ||
| Fig. 4 (a) SEM image and (b) EDX of PMO-100T after Hg(II) uptake as well as the corresponding EDX elemental maps of S (c) and Hg (d) showing the distribution of the adsorbed mercury in the PMO matrix. | ||
3.3. Adsorption isotherms and adsorption kinetics
To assess the sorption capacity of PMO-100T for Hg(II) from water solution, the thermodynamic binding properties were examined by dispersion in an aqueous solution of Hg(NO3)2with initial concentrations of 50–1000 ppm (Fig. 5a). With the increase of Hg(II) concentration, the adsorption capacity of PMO-100T increased rapidly and showed a steep sorption profile. The resulting fitting parameters for the Langmuir and Freundlich adsorption models are listed in Table S1 (ESI†). The Langmuir model (eqn (1)) was more appropriate for the thermodynamic binding data (correlation coefficient R2 = 0.9861), indicating monolayer adsorption.62 The calculated adsorption capacity (qm) of Hg(II) was 1253 mg g−1, close to the experimental value (1227.9 mg g−1). | ||
| Fig. 5 Adsorption isotherms (a) and adsorption kinetic curves (b) for adsorption of Hg(II) onto PMO-100T at room temperature (the initial concentration of Hg(II) ions = 1000 ppm). | ||
The rate of mercury removal by PMO-100T was investigated by detecting the adsorption capacity at different contact times at an initial Hg(II) concentration of 1000 ppm. It is evident from Fig. 5b that the adsorption equilibrium was achieved with the adsorbent within 90 min. Such an efficient removal capability should be relevant to the strong interaction between Hg(II) ions and a large number accessible active binding sites evenly located on the pore wall of the PMO matrix.32,35 Two typical kinetic models were employed to fit the experimental data, which can be seen in Table S2 (ESI†). The kinetic sorption was well fitted by the pseudo second-order kinetic model (R2 ≥ 0.9999), which suggests a chemisorption mechanism.61 The linearized plot for the pseudo-2nd order model is shown in the Fig. 5b inset.
3.4. Simultaneous separation of Hg(II) and RhB and adsorbent reusability
Apart from Hg(II), PMO-100DT also exhibited high adsorption performance for the water-soluble organic dye RhB (Fig. S9, ESI†). For example, the treatment of 400 ppm RhB solution with the adsorbent at a pH of 5 resulted in ∼67% removal of RhB with an uptake capacity of 268 mg g−1, which is superior to those of the recently reported dye adsorbents based on porous organic polymers.63 When the initial dye concentration was 100 ppm, the RhB removal rate reached 91.5% for PMO-100DT. The good adsorption efficiency can be attributed to the electrostatic interaction between the dye and adsorbent and hydrogen bonding as well as favorable structural properties of the mesoporous framework of PMOs.64The above results prompted us to further explore the potential of PMO-100T for the water-purification application in realistic scenarios. To this end, PMO-100T was dispersed in a simulated sewage sample containing 10–100 ppm Hg(II) and RhB followed by agitation for 24 h at room temperature. From Fig. 6a, it was found that the Hg(II) removal from water by the adsorbent was more than 97% at the initial concentration of 10–100 ppm, while the removal efficiency for RhB decreased slightly with decreasing concentration. Nonetheless, the removal rate higher than 91% for both pollutants revealed excellent simultaneous uptake ability of the new adsorbent.
 | ||
| Fig. 6 (a) Removal efficiency of PMO-100T for Hg(II) and RhB in the simulated sewage. Adsorption measurements were performed by dispersing the adsorbent (10 mg) in 10 mL of the water sample (see Section 2.8). (b) Hg(II) and RhB removal efficiency and recovery of PMO-100DT after 5 cycles of sequential adsorption–desorption steps (see Section 2.9). | ||
The reusability of PMO-100T was evaluated through the elution/regeneration process. The experimental data showed that the mixed solution of 0.1 M thiourea−0.1 M HCl can effectively elute the adsorbed Hg(II) ions from the adsorbent, while ethanol acidified with 1 M HCl is sufficient to extract RhB within the PMO matrix. Qualitative analysis of the collected filtrates revealed that the percentage desorption for both pollutants was more than 97%. After five elution–regeneration cycle operations, the removal efficiencies of PMO-100T decreased from 98.2% to 93.5% for the Hg(II) ions and from 95.5% to 90.1% for the RhB dye, respectively (Fig. 6b). The removal efficiency dropped by only ∼5% in both cases, and the structural integrity of the PMO adsorbent remained virtually unchanged for five cycles as evidenced by XRD measurements (Fig. S10, ESI†).
4. Conclusions
In summary, a new ethylene-bridged periodic mesoporous organosilica material has been prepared by the self-assembly of a home-made thio-bis-silane precursor in the presence of P123 under acidic conditions. The PMO material functionalized with thiol and thioether groups was capable of selectively capturing Hg(II) ions from aqueous media with a maximum uptake capacity of up to 1253 mg g−1, outperforming most existing Hg(II) adsorbents. The adsorption of Hg(II) on the PMO particles followed the Langmuir isotherm model and the pseudo-second-order kinetic model. The PMO adsorbent also showed good adsorption efficiency for the simultaneous removal of mercury ions and the amphoteric dye RhB from simulated sewage. When the initial concentration of each pollutant was 100 ppm, the removal rates for Hg(II) and RhB reached 97% and 91%, respectively. Moreover, the adsorbent can be regenerated by a gentle acidic treatment, and its adsorption properties were largely maintained even after five consecutive adsorption–desorption cycles. High uptake capacity, fast adsorption kinetics, good recyclability and cost-effective synthesis make this material a promising candidate for applications in environmental remediation.Author contributions
Yuanjiang Zhao: conceptualization, experiment, investigation, validation, visualization, writing – original draft; Yuhang Liu: validation, data curation; and Liming Jiang: conceptualization, funding acquisition, resources, writing – reviewing and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors greatly appreciate the financial support for this work from the National Key Research and Development Program (grant no. 2016YFA0200301).References
- S. Zhu, Z. Zhang and D. Žagar, Sci. Total Environ., 2018, 639, 538–549 CrossRef CAS PubMed.
- H. R. Rajabi, M. Shamsipur, M. M. Zahedi and M. Roushani, Chem. Eng. J., 2015, 259, 330–337 CrossRef CAS.
- B. Li, Y. Zhang, D. Ma, Z. Shi and S. Ma, Nat. Commun., 2014, 5, 5537 CrossRef CAS PubMed.
- N. Huang, L. Zhai, H. Xu and D. Jiang, J. Am. Chem. Soc., 2017, 139, 2428–2434 CrossRef CAS PubMed.
- M. B. Feng, P. Zhang, H. C. Zhou and V. K. Sharma, Chemosphere, 2018, 209, 783–800 CrossRef CAS PubMed.
- X. Feng, G. E. Fryxell, L. Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923–926 CrossRef CAS.
- V. Antochshuk, O. Olkhovyk, M. Jaroniec, I. S. Park and R. Ryoo, Langmuir, 2003, 19, 3031–3034 CrossRef CAS.
- M. Santhamoorthy, D. Thirumalai, K. Thirupathi and S.-C. Kim, Appl. Nanosci., 2023, 13, 6015–6024 CrossRef CAS.
- Y. Shin, G. E. Fryxell, W. Um, K. Parker, S. V. Mattigod and R. Skaggs, Adv. Funct. Mater., 2007, 17, 2897–2901 CrossRef CAS.
- S. Bag, P. N. Trikalitis, P. Chupas, G. S. Armatas and M. G. Kanatzidis, Science, 2007, 317, 490–493 CrossRef CAS PubMed.
- Y. Oh, C. D. Morris and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 14604–14608 CrossRef CAS PubMed.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS PubMed.
- S. Y. Ding, M. Dong, Y. W. Wang, Y. T. Chen, H. Z. Wang, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- N. Huang, L. P. Zhai, H. Xu and D. L. Jiang, J. Am. Chem. Soc., 2017, 139, 2428–2434 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Ear, C. W. Abney, Y. C. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Q. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- L. Merí-Bofí, S. Royuela, Z. Félix, M. L. Ruiz-González, J. L. Segura, R. Muñoz-Olivas and M. J. Mancheño, J. Mater. Chem. A, 2017, 5, 17973–17981 RSC.
- Y. Li, T. Hu, R. Chen, R. Xiang, Q. Wang, Y. Zeng and C. He, Chem. Eng. J., 2020, 398, 125566 CrossRef CAS.
- X. P. Li, C. Q. Bian, X. G. Meng and F. S. Xiao, J. Mater. Chem. A, 2016, 4, 5999–6005 RSC.
- B. Aguila, Q. Sun, J. A. Perman, L. D. Earl, C. W. Abney, R. Elzein, R. Schlaf and S. Q. Ma, Adv. Mater., 2017, 29, 1700665 CrossRef PubMed.
- Y. Fu, W. G. Yu, W. J. Zhang, Q. Huang, J. Yan, C. Y. Pan and G. P. Yu, Polym. Chem., 2018, 9, 4125–4131 RSC.
- S. Mondal, S. Chatterjee, S. Mondal and A. Bhaumik, ACS Sustainable Chem. Eng., 2019, 7, 7353–7361 CrossRef CAS.
- J. Cheng, Y. F. Li, L. Li, P. P. Lu, Q. Wang and C. Y. He, New J. Chem., 2019, 43, 7683–7693 RSC.
- D. Shetty, S. Boutros, A. Eskhan, A. Marie De Lena, T. Skorjanc, Z. Asfari, H. Traboulsi, J. Mazher, J. Raya, F. Banat and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 12898–12903 CrossRef CAS PubMed.
- A. Modak, P. Bhanja, M. Selvaraj and A. Bhaumik, Environ. Sci.: Nano, 2020, 7, 2887–2923 RSC.
- T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867–871 CrossRef CAS.
- S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611–9614 CrossRef CAS.
- B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302–3308 CrossRef CAS.
- N. Mizoshita, T. Taniab and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC.
- P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC.
- B. Karimi, N. Ganji, O. Pourshiani and W. R. Thiel, Prog. Mater. Sci., 2022, 125, 100896 CrossRef CAS.
- D. Esquivel, J. Ouwehand, M. Meledina, S. Turner, G. Van Tendeloo, F. J. Romero-Salguero, J. De Clercq and P. Van Der Voort, J. Hazard. Mater., 2017, 339, 368–377 CrossRef CAS PubMed.
- S. Das, S. Chatterjee, S. Mondal, A. Modak, B. K. Chandra, S. Das, G. D. Nessim, A. Majee and A. Bhaumik, Chem. Commun., 2020, 56, 3963–3966 RSC.
- S. H. Lee, S. S. Park, S. Parambadath and C. S. Ha, Microporous Mesoporous Mater., 2016, 226, 179–190 CrossRef CAS.
- E. De Canck, L. Lapeire, J. De Clercq, F. Verpoort and P. Van Der Voort, Langmuir, 2010, 26, 10076–10083 CrossRef CAS PubMed.
- M. A. O. Lourenço, P. Figueira, E. Pereira, J. R. B. Gomes, C. B. Lopes and P. Ferreira, Chem. Eng. J., 2017, 322, 263–274 CrossRef.
- O. Olkhovyk and M. Jaroniec, J. Am. Chem. Soc., 2005, 127, 60–61 CrossRef CAS PubMed.
- N. Velikova, Y. Vueva, Y. Ivanova, I. Salvado, M. Fernandes, P. Vassileva, R. Georgieva and A. Detcheva, J. Non-Cryst. Solids, 2013, 378, 89–95 CrossRef CAS.
- L. Zhang, W. Zhang, J. Shi, Z. Hua, Y. Li and J. Yan, Chem. Commun., 2003, 210–211 RSC.
- J. Liu, J. Yang, Q. Yang, G. Wang and Y. Li, Adv. Funct. Mater., 2005, 15, 1297–1302 CrossRef CAS.
- N. Hao, L. Han, Y. Yang, H. Wang, P. A. Webley and D. Zhao, Appl. Surf. Sci., 2010, 256, 5334–5342 CrossRef CAS.
- H. Li, D. Shen, H. Lu, F. Wu, X. Chen, R. Pleixats and J. Pan, Sep. Purif. Technol., 2021, 277, 119453 CrossRef CAS.
- C. Vercaemst, M. Ide, B. Allaert, N. Ledoux, F. Verpoort and P. Van Der Voort, Chem. Commun., 2007, 2261–2263 RSC.
- D. Esquivel, O. van den Berg, F. J. Romero-Salguero, F. Du Prez and P. Van Der Voort, Chem. Commun., 2013, 49, 2344–2346 RSC.
- D. Esquivel, J. Amaro-Gahete, N. Caballero-Casero, C. Jiménez-Sanchidrián, J. R. Ruiz, S. Rubio, P. Van Der Voort and F. J. Romero-Salguero, ACS Appl. Nano Mater., 2020, 3, 2373–2382 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- H. Freundlich, Z. Phys. Chem., 1906, 57, 385–470 CAS.
- C. Xiong, C. Xue, L. Huang, P. Hu, P. Fan, S. Wang, X. Zhou, Z. Yang, Y. Wang and H. Ji, J. Cleaner. Prod., 2021, 316, 128372 CrossRef CAS.
- Y. S. Ho and G. McKay, Process Biochem., 1999, 34, 451–465 CrossRef CAS.
- OECD guideline for the testing of chemicals in the Hershberger assay; OECD: Paris, 2001.
- Y. Han and J. Y. Ying, Angew. Chem., Int. Ed., 2005, 44, 288–292 CrossRef CAS PubMed.
- P. T. Tanev and T. J. Pinnavaia, Chem. Mater., 1996, 8, 2068–2079 CrossRef CAS.
- E. Cano-Serrano, J. Campos-Martin and J. Fierro, Chem. Commun., 2003, 246–247 RSC.
- S. J. Billinge, E. J. McKimmy, M. Shatnawi, H. Kim, V. Petkov, D. Wermeille and T. J. Pinnavaia, J. Am. Chem. Soc., 2005, 127, 8492–8498 CrossRef CAS PubMed.
- K.-K. Yee, N. Reimer, J. Liu, S.-Y. Cheng, S.-M. Yiu, J. Weber, N. Stock and Z. Xu, J. Am. Chem. Soc., 2013, 135, 7795–7798 CrossRef CAS PubMed.
- K. Leus, J. P. H. Perez, K. Folens, M. Meledina, G. Van Tendeloo, G. Du Laing and P. Van Der Voort, Faraday Discuss., 2017, 201, 145–161 RSC.
- J. He, K.-K. Yee, Z. Xu, M. Zeller, A. D. Hunter, S. S.-Y. Chui and C.-M. Che, Chem. Mater., 2011, 23, 2940–2947 CrossRef CAS.
- H. Saleem, U. Rafique and R. P. Davies, Microporous Mesoporous Mater., 2016, 221, 238–244 CrossRef CAS.
- G. Boix, J. Troyano, L. Garzon-Tovar, C. Camur, N. Bermejo, A. Yazdi, J. Piella, N. G. Bastus, V. F. Puntes, I. Imaz and D. Maspoch, ACS Appl. Mater. Interfaces, 2020, 12, 10554–10562 CrossRef CAS PubMed.
- C. Wang, S. Y. Tao, W. Wei, C. G. Meng, F. Y. Liu and M. Han, J. Mater. Chem., 2010, 20, 4635–4641 RSC.
- J. Li, X. D. Li, A. Alsaedi, T. Hayat and C. L. Chen, J. Colloid Interface Sci., 2018, 517, 61–71 CrossRef CAS PubMed.
- B. Zeng, G. Lin, J. Li, W. Wang and L. Zhang, Microporous Mesoporous Mater., 2022, 345, 112251 CrossRef CAS.
- I. M. Kenawy, M. A. H. Hafez, M. A. Ismail and M. A. Hashem, Int. J. Biol. Macromol., 2018, 107, 1538–1549 CrossRef CAS PubMed.
- A. Sanjabi, S. Azizian, M. Torabi, M. A. Zolfigol and M. Yarie, Microporous Mesoporous Mater., 2023, 348, 112367 CrossRef CAS.
- J. R. Deka, C. L. Liu, T. H. Wang, W. C. Chang and H. M. Kao, J. Hazard. Mater., 2014, 278, 539–550 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj04896a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |