
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-fused polycyclic indoles via a Pd-catalyzed multicomponent cascade reaction consisting of an amide-directed [3+1+1] annulation reaction of 3-diazo oxindole and isocyanides†
Pooja
Soam
a,
Debasish
Mandal
 ab and
Vikas
Tyagi
*ab
ab and
Vikas
Tyagi
*ab
aSchool of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala-147004, Punjab, India. E-mail: vikas.tyagi@thapar.edu
bCentre of Excellence for Emerging Materials, Thapar Institute of Engineering and Technology, Patiala-147004, Punjab, India
First published on 5th January 2024
Abstract
Herein, we report a Pd-catalyzed multicomponent cascade reaction of 3-diazo oxindole and isocyanides to synthesize N-fused polycyclic indoles consisting of an in situ generated amide-assisted regioselective [3+1+1] annulation reaction. Additionally, a mechanism is proposed and investigated using DFT that strongly favors the role of amide in assisting the annulation reaction, which is further confirmed using control experiments.
Introduction
N-Fused polycyclic indoles have attracted great attention in synthetic chemistry due to their ubiquitous presence in several natural products and pharmaceutical agents (Fig. 1). They display a wide range of biological activities such as anticancer, anti-inflammatory, antimicrobial, antiatherogenic, antihypertensive, etc. Moreover, indole-fused heterocycles are imperative structural units of various agrochemicals, plastics, and dyes that are integrated into everyday life.1a–g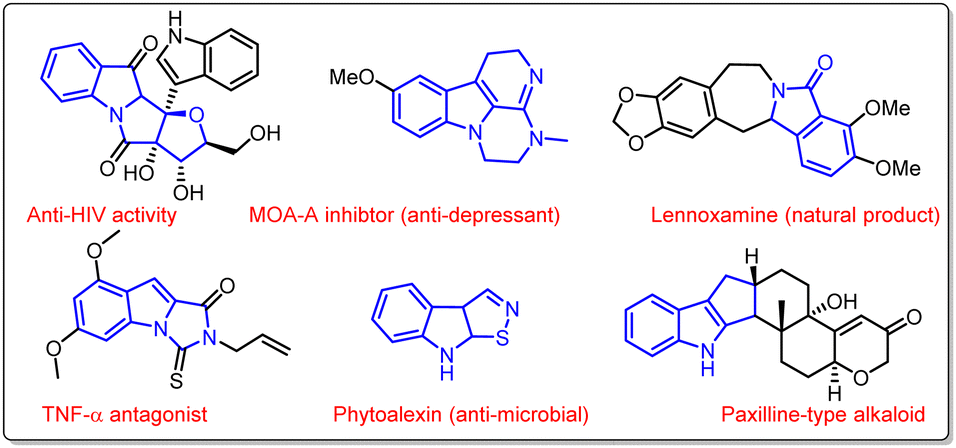 | ||
| Fig. 1 Biologically important indole-fused heterocycles. | ||
In this context, various combinations of polycyclic indole-fused heterocycles have been designed and synthesized in the past.2a–e However, the production of these moieties is very challenging due to the requirement of multistep synthesis, which is associated with their disadvantages. So, the development of novel and efficient methodologies to synthesize indole-fused heterocyclic scaffolds is highly admirable in synthetic chemistry. Throughout the significant progress made in this area, different methods have been developed to synthesize tricyclic oxazolo[3,2-a]indole moieties. In this context, Zhu et al. synthesized oxazolo[3,2-a]indolones via a visible light-induced aerobic dearomative reaction of N-substituted indoles in the presence of a Ru-catalyst (Scheme 1a).3 Next, a metal- and solvent-free approach to synthesize oxazolo[3,2-a]indolones using propargylic alcohols and indole derivatives was developed by Oparina's group (Scheme 1b).4 Recently, Wang and co-workers reported a cascade approach to synthesize oxazolo[3,2-a]indolones along with indolo [2,3-b] indoles in the presence of a rhodium catalyst at 130 °C (Scheme 1c).5 However, most of these approaches are associated with certain limitations such as complex starting materials, longer reaction time, lower efficiency, and substrate scope.
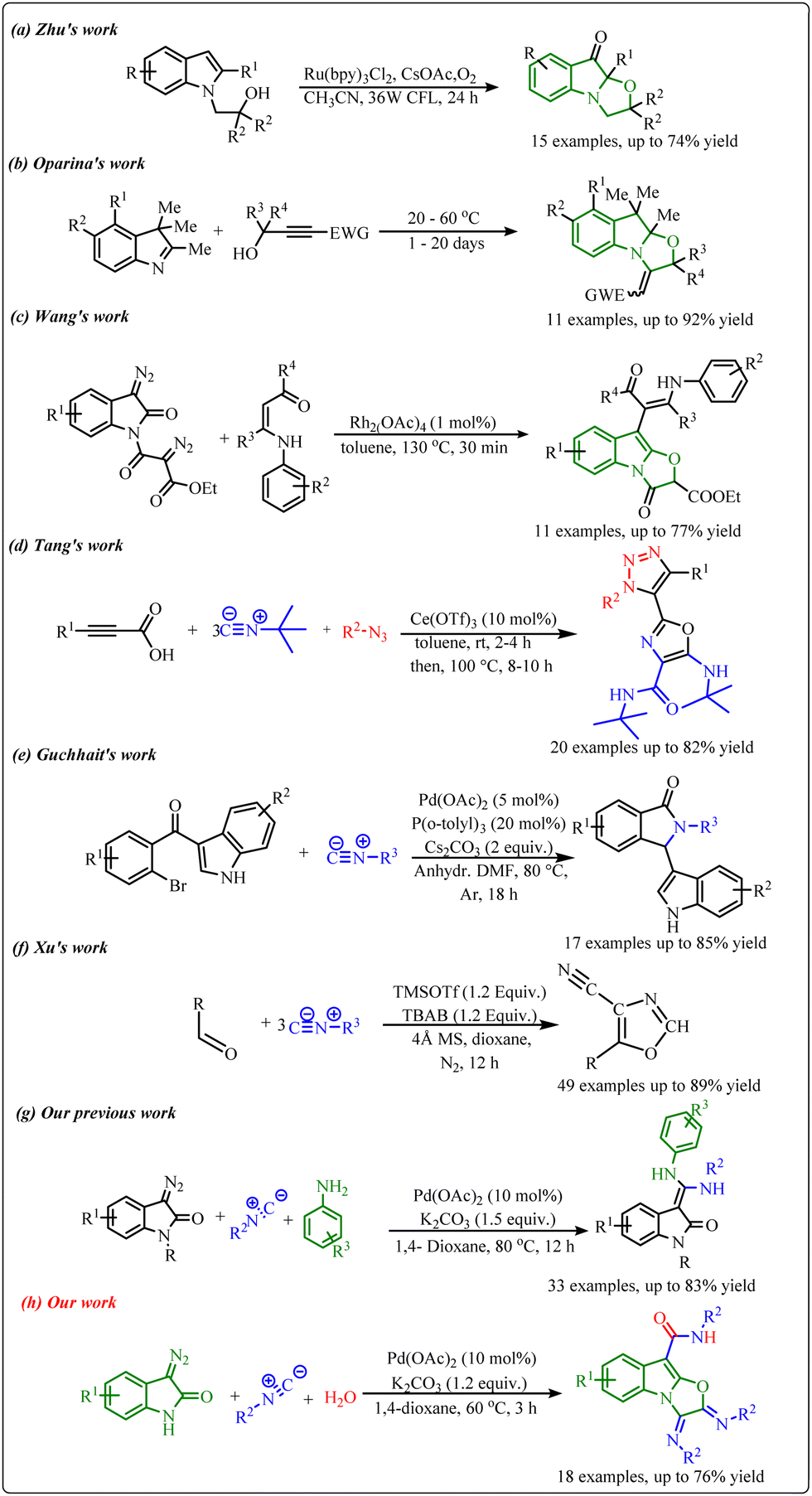 | ||
| Scheme 1 (a)–(c) Recent approaches to synthesizing tricyclic oxazolo[3,2-a]indole derivatives, (d)–(f) recent isocyanide insertion approaches, (g) our previous work, and (h) recent work. | ||
On the other hand, cascade or tandem reactions have been found to be very advantageous to improve synthetic efficiency in modern organic chemistry.6a–e Furthermore, the cascade reaction involves multiple bond-forming reactions in one pot, representing an attractive strategy towards the facile synthesis of novel molecular frameworks with less wastage of resources. In particular, transition metal-catalyzed one-pot cascade reactions are being used to generate novel fused heterocycles.7a–c Interestingly, isocyanides have been considered important substrates for transition metal-catalyzed one-pot cascade reactions since they can react both as a nucleophile or an electrophile in chemical reactions.8a–e In this context, several cascade reactions using isocyanides have been developed in the past. Recently, Tang's group reported a cerium triflate catalyzed triple isocyanide insertion approach to develop a library of oxazole derivatives.9 Next, Guchhait et al. developed a cascade C–H activation methodology to construct bicyclic indolyl motifs via intramolecular isocyanide insertion using Pd-catalysts.10 Another efficient approach towards consecutive triple isocyanide insertion into aldehydes to synthesize a broad library of 4-cyano oxazole derivatives was reported by Xu and co-workers. (Scheme 1d–f)11 Also, we have developed a Pd-catalysed one-pot cascade reaction to access benzoxazine-fused 1,2,3-triazoles using N-aryl-α-(tosylhydrazone)acetamides with isocyanide.12 In continuation of our efforts to develop novel cascade reactions by the use of isocyanide and diazo compounds (Scheme 1g),13 herein, we have developed a Pd-catalysed multicomponent cascade reaction of 3-diazo oxindole, isocyanide, and H2O to generate tricyclic oxazolo[3,2-a]indole scaffolds (Scheme 1h).
Results and discussion
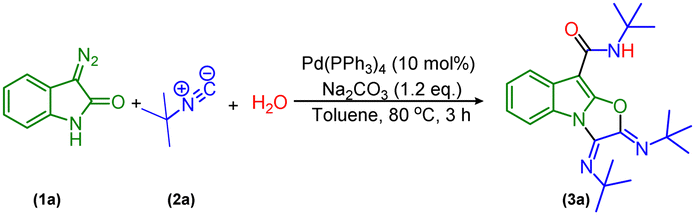
Initially, we envisioned the synthesis of indole-3-acetamide via the formation of indole-based ketenimine intermediates by the reaction of isocyanide and 3-diazo oxindole as previously reported by our group and others. To actualize our hypothesis, we set up a reaction of tert-butyl isocyanide, water, and 3-diazo oxindole using 10 mol% of Pd(PPh3)4 and 1.2 equivalent of Na2CO3 in toluene as a solvent at 80 °C, and obtained the product in a 30% isolated yield. However, characterization techniques such as 1H-NMR, 13C-NMR, HRMS, and X-ray confirm the unprecedented formation of a tricyclic oxazolo[3,2-a]indole derivative (Scheme 2).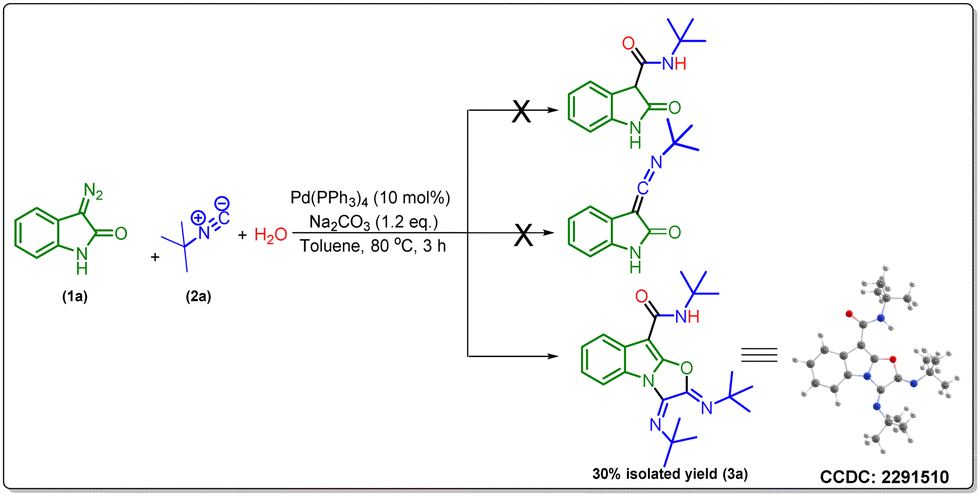 | ||
| Scheme 2 Possible structures under a Pd-catalyzed reaction. | ||
Drawing encouragement from these findings, next, we decided to optimize the reaction conditions to improve the yield of the multicomponent cascade reaction by selecting 3-diazo oxindole (1a), tert-butyl isocyanide (2a), and H2O as model substrates. Initially, we screened different transition metal catalysts such as Cu(OAc)2 and Rh2(OAc)4 in addition to Pd(PPh3)4, and unexpectedly the reaction worked only with the Pd-catalyst (Table 1, entries 1–3). Afterward, we examined different palladium-based catalysts like Pd(OAc)2, Pd(TFA)2, Pd(OPiv)2, PdCl2, PdCl2(NCC6H5)2, Pd(DPPF)Cl2, Pd2(dba)3etc. and obtained product (3a) in a maximum yield, i.e. 56% when 10 mol% of Pd(OAc)2 was added as a catalyst (Table 1, entry 4–6) and (Table S2, entries 1–4, ESI†). However, on decreasing the amount of Pd(OAc)2 from 10 mol% to 5 mol%, the yield of the model reaction got decreased (Table S2, entry 5, ESI†). Also, it must be noticed that the reaction did not work in the absence of a palladium catalyst (Table 1, entry 7). Next, we screened different bases and found that the reaction provided a maximum yield in the case of K2CO3 as compared to other bases such as Na2CO3, Cs2CO3, KOt-Bu, NEt3 and DBU (Table 1, entries 8–12). Also, it was observed that in the absence of a base, the yield of product (3a) decreased, up to a 27% yield (Table S2, entry 6, ESI†). Furthermore, toluene was replaced by various solvents like 1,4-dioxane, CH3CN, DMF, DMSO, and THF; however, 1,4-dioxane was found to be the best solvent (Table 1, entry 13) and (Table S2, entries 7–10, ESI†). Besides, there was no improvement in the outcome of the model reaction observed while adding different additives like AgOAc and CsOAc (Table S2, entries 11 and 12, ESI†). Moreover, temperature variation significantly affected the outcome of the model reaction. In particular, decreasing the temperature from 80 °C to 60 °C increases the yield of 3a to 71% (Table 1, entry 14). Then, the yield of 3a was reduced by further decreasing the temperature to 50 °C (Table 1, entry 15). The reaction was almost diminished below 50 °C and the product was observed only in trace amounts at 40 °C (Table 1, entries 16 and 17). Subsequently, increasing the equivalent of water slightly decreased the yield to 60% (Table 1, entry 18). Noticeably, in the absence of water, only a 20% yield of product 3a was observed, which suggests the requirement of water in an equimolar amount in this transformation (Table 1, entry 19). Also, the molar ratio of the substrates was varied; however, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.2:1 of 3-diazo oxindole, tert-butyl isocyanide, and water, respectively, remained the best ratio (Table S2, entries 13–16, ESI†).
:3.2:1 of 3-diazo oxindole, tert-butyl isocyanide, and water, respectively, remained the best ratio (Table S2, entries 13–16, ESI†).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yieldb (%) |
| a Reaction conditions: 3-diazo oxindole 1a (1.0 equiv., 0.31 mmol, and 50 mg), tert-butyl isocyanide 2a (3.2 equiv., 113 μL, and 0.99 mmol), H2O (1.0 equiv., 5.6 μL), Pd-catalyst (10 mol%), base (1.2 equiv.), and solvent (2 ml), for 3 h. b Isolated yield. | ||
| 1 | No deviation | 30 |
| 2 | Cu(OAc)2 instead of Pd(PPh3)4 | — |
| 3 | Rh2(OAc)4 instead of Pd(PPh3)4 | — |
| 4 | Pd(OAc)2 instead of Pd(PPh3)4 | 56 |
| 5 | Pd(TFA)2 instead of Pd(PPh3)4 | 38 |
| 6 | Pd(OPiv)2 instead of Pd(PPh3)4 | 31 |
| 7 | No catalyst | — |
| 8 | Cs2CO3 instead of Na2CO3 | 51 |
| 9 | K2CO3 instead of Na2CO3 | 59 |
| 10 | KOt-Bu instead of Na2CO3 | 35 |
| 11 | NEt3 instead of Na2CO3 | 43 |
| 12 | DBU instead of Na2CO3 | 39 |
| 13 | 1,4-Dioxane instead of toluene | 64 |
| 14 | 60 °C | 71 |
| 15 | 50 °C | 36 |
| 16 | 40 °C | Trace |
| 17 | rt | — |
| 18 | 2 equiv. of H2O | 60 |
| 19 | No H2O | 20 |
After having the optimized reaction conditions in hand, we evaluated the feasibility of the reaction with different isocyanides as well as 3-diazo oxindole as revealed in Scheme 3.
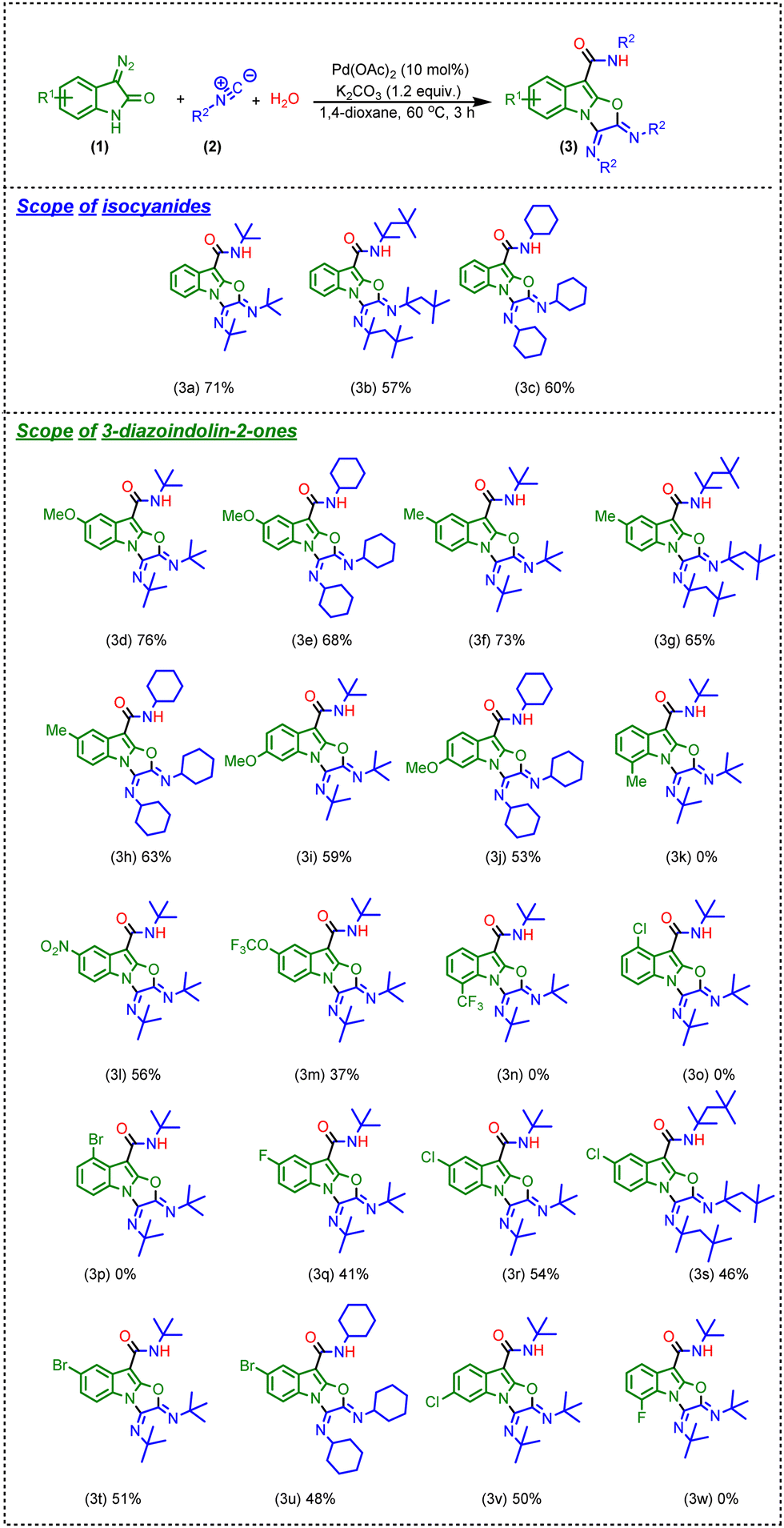 | ||
| Scheme 3 The substrate scope of the Pd-catalyzed multicomponent cascade reaction. Reaction conditions: 3-diazo oxindoles (1) (1.0 equiv.), isocyanides (2) (3.2 equiv.), H2O (3) (1.0 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (1.2 equiv.), and 1,4-dioxane (2 mL), for 3 h, at 60 °C. | ||
Firstly, we have replaced tert-butyl isocyanide with 1,1,3,3-tetramethyl butyl isocyanide or cyclohexyl isocyanide, and as a result, the corresponding products (3b and 3c) were obtained in slightly lower yields in comparison of the product 3a (Scheme 3). These results represent the structural role of isocyanides; as the steric hindrance increases, the yield of the reaction decreases. Besides, we have tried reactions with other isocyanides like p-methoxy phenyl isocyanide and p-toluene sulphonyl methyl-isocyanide; however, no product formation was observed with these isocyanides. Next, we have examined the effect of various electron-donating, electron-withdrawing, and halide substituents at different positions of 3-diazo oxindole. Initially, a range of electron-donating groups at different positions of the phenyl ring of 3-diazo oxindole were tested. Noticeably, in the case of 5-OMe-substituted 3-diazo oxindole, the reaction provided products (3d) and (3e) in 76% and 68% yields, respectively, whereas the reaction provided moderate to good yields of the desired products when 5-CH3-substituted 3-diazo oxindole was employed with tert-butyl isocyanide, 1,1,3,3-tetramethyl butyl isocyanide, and cyclohexyl isocyanide, respectively (3f–h, Scheme 3). Also, 6-OMe-substituted 3-diazo oxindole provided products 3i and 3j in slightly lower yields, i.e., 59 and 53% (Scheme 3). It should be noted that the yield diminished when the electron-donating group, i.e. –CH3, was incorporated at the 7-position of 3-diazo oxindole and it might be due to hindrance enhancement near the fringe where the reaction occurred (3k, Scheme 3). This abatement of yield indicated the significance of the electronic and steric effects of the substituents at different positions of 3-diazo oxindole. Next, electron-withdrawing groups i.e., 5-nitro and 5-OCF3 substituted 3-diazo oxindole were used in the reaction and the products (3l and 3m) were attained in 56% and 37% isolated yield (Scheme 3), which slightly lower in comparison to 3-diazo oxindole having an electron-donating group at the same position. However, there was no product formation observed when 7-CF3-containing 3-diazo oxindole was employed in the reaction (3n, Scheme 3). Afterward, we examined the effect of halides at different positions of 3-diazo oxindoles and found that in the case of 4-Cl and 4-Br substitutions, the yields almost diminished (3o and 3p, Scheme 3). Next, 5-F-substituted 3-diazo oxindole provides product 3q in a lower yield as compared to 3-diazo oxindole having electron-withdrawing and -donating groups. Next, 5-Cl- and 5-Br-substituted 3-diazo oxindoles were employed in the reaction, and the corresponding products (3r to 3u) were obtained in lower yields, i.e., 46 to 54% as compared to electron-donating and electron-withdrawing substitutions at 3-diazo oxindole. However, the reaction provided the product in a moderate yield when 6-Cl-substituted 3-diazo oxindole was used in the reaction (3v, Scheme 3). Again, halide substitution at the 7-position of 3-diazo oxindole did not work (3w, Scheme 3).
Besides, a Hammet study was performed to get further information about the effect of substituents on the rate of the reaction.14 In this view, 3-diazo oxindole having substituents like 5-OMe, 5-Me, 5-Cl and 5-NO2 along with unsubstituted 3-diazo oxindole was employed in the multicomponent reaction (Scheme 4). The best linear fit (R2 = 0.91) was achieved using the standard σp parameters vs. logkX/kH, providing a ρ value (slope value) of –0.28. The value of ρ is less than 1, which indicates that a positive charge is built up during the rate-determining step. Also, the positive slope value suggests that the rate of reaction increases in the presence of the electron-donating groups.
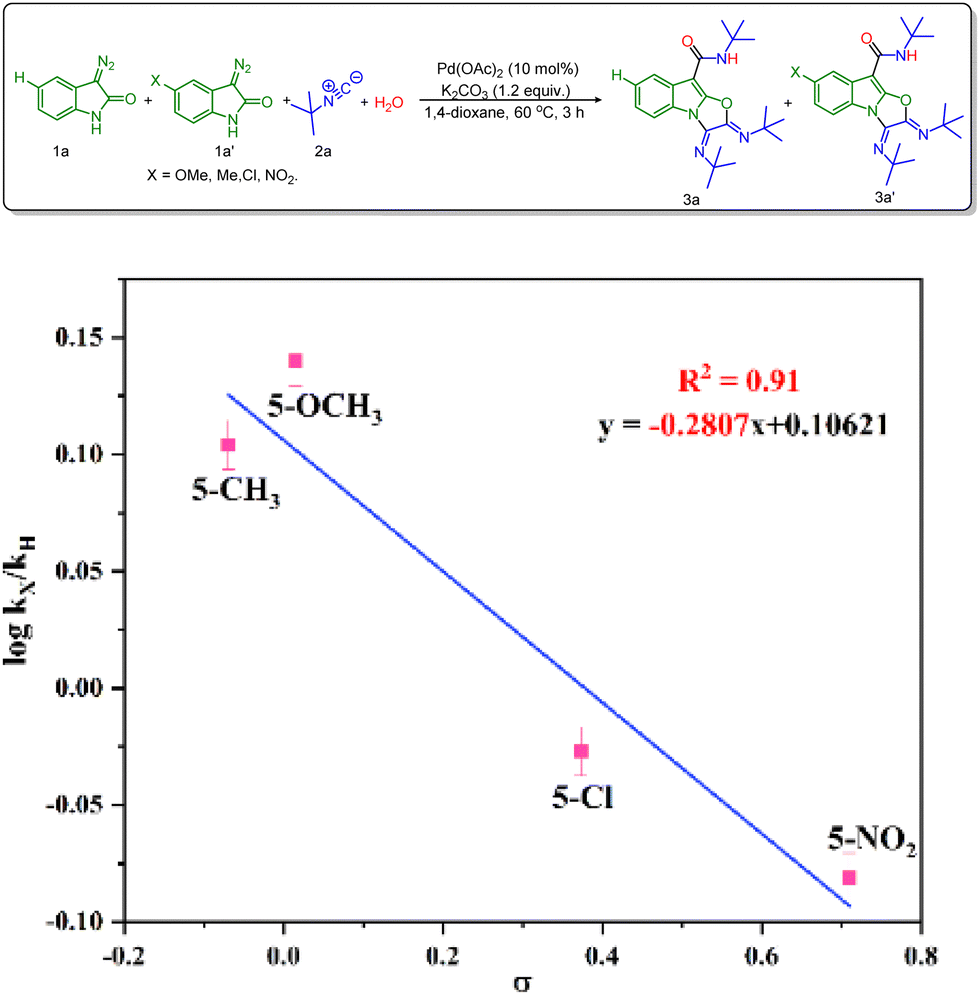 | ||
| Scheme 4 Hammett plot for the reaction of isocyanide with electronically varied 3-diazo oxindoles.a aReaction conditions: 3-diazo oxindole 1a (1.0 equiv., 0.31 mmol), substituted 3-diazo oxindole 1a′ (1.0 equiv., 0.31 mmol), tert-butyl isocyanide 2a (3.2 equiv., 0.99 mmol), H2O (1.0 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (1.2 equiv.), and 1,4-dioxane (2 mL), for 3 h, at 60 °C. | ||
Next, we have presented a plausible mechanism for the synthesis of tricyclic oxazolo[3,2-a]indole scaffolds as shown in Fig. 2.15 Initially, tert-butyl isocyanide reacts with Pd(OAc)2 to generate complex (I).12,16a–d Then, tert-butyl isocyanide acts as a neutral ligand in complex (I), and palladium remains in its divalent oxidation state. Complex (I) then linked with the 3-diazo oxindole (1a) substrate to generate square planar complex (II) that served as our starting complex. In this context, Fig. 2 shows the overall mechanism's steps, and Fig. 3(a) displays the associated DFT-computed potential free energy profiles in relation to the energies of the initial complex (I + 1a). Furthermore, Fig. 3(b) depicts the optimized transition states computed at the B3LYP/6-31+G(d,p) level of theory.
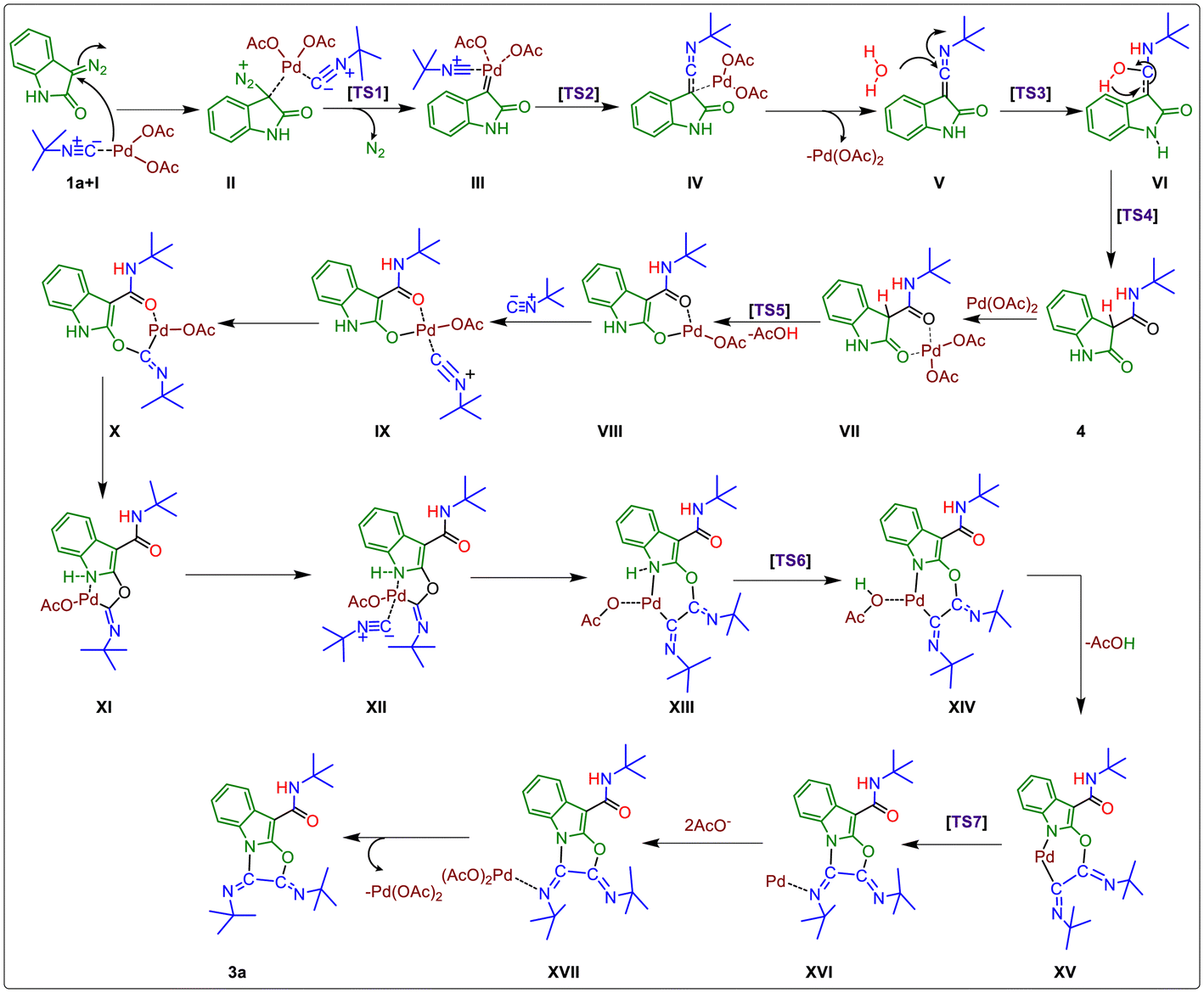 | ||
| Fig. 2 The plausible mechanism for Pd-catalyzed multicomponent cascade reaction. | ||
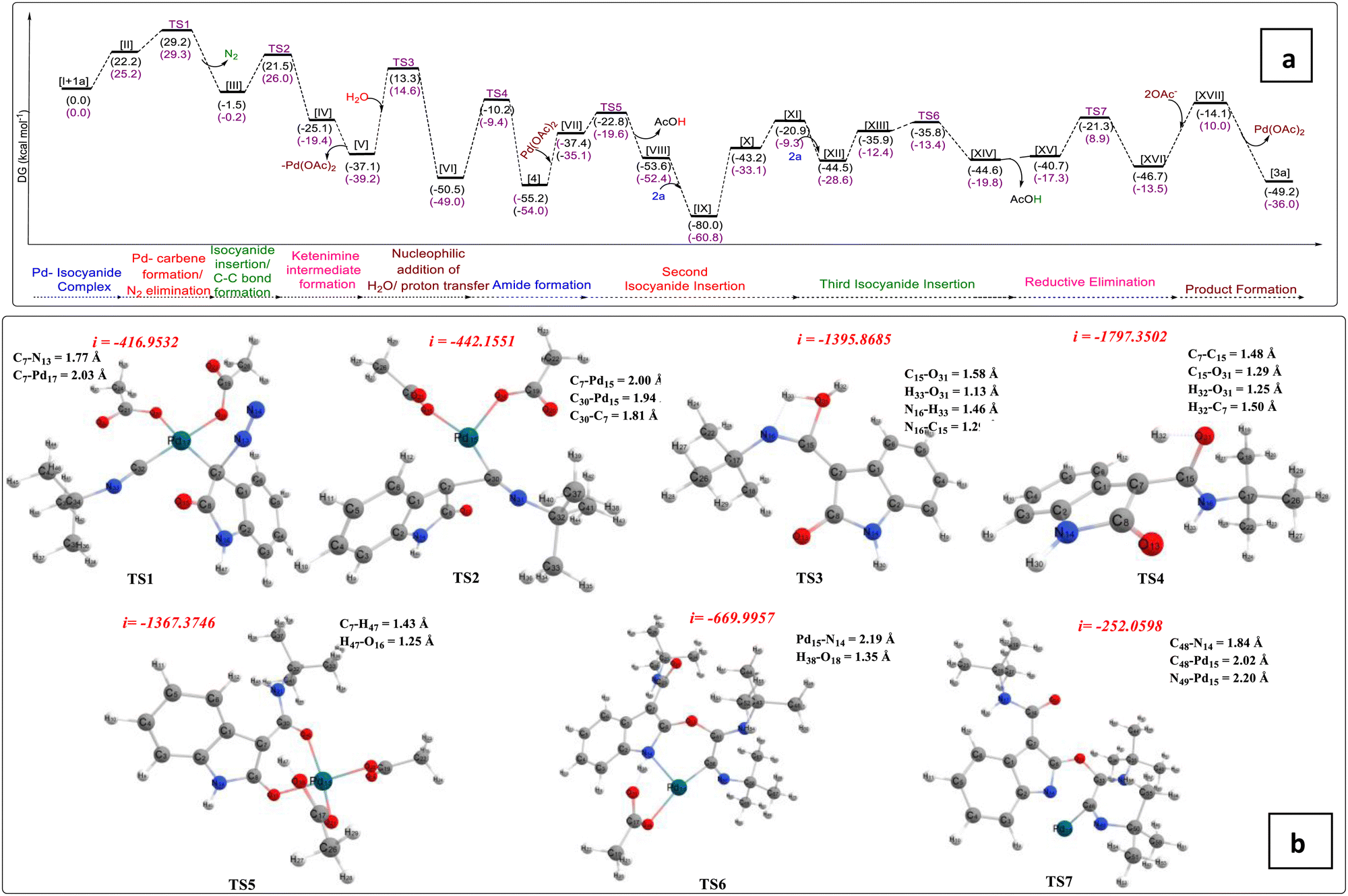 | ||
| Fig. 3 (a) The potential energy profiles computed at the M06/6-31++G(2d,2p)/B3LYP/6-31+G(d,p) level of theory related to the plausible mechanism represented in Fig. 2. (b) Optimized transition states computed at the B3LYP/6-31+G(d,p) level of theory. | ||
Furthermore, the elimination of N2 from complex (II) took place and generated Pd–carbene complex (III) viaTS1 (Fig. 3(b)) with the free energy of activation ΔG‡ = 29.2 kcal mol−1. The next step is the migration of isocyanide into the Pd–carbene complex through the three-membered ring transition state, TS2 (Fig. 3(b)), which has a 21.5 kcal mol−1 free energy of activation and produces Pd-ketenimine complex (IV), which readily releases Pd(OAc)2 to form highly unstable ketenimine intermediate (V). Furthermore, H2O is connected to the central electrophilic carbon of the ketenimine intermediate (V) with simultaneous proton transfer to the nitrogen of the isocyanide unit viaTS3 (Fig. 3(b)) with ΔG‡ = 13.3 kcal mol−1 to form intermediate VI, which is stabilized by –37.2 kcal mol−1 from TS3 (Fig. 3(b)).
After that, the tautomerization step takes place via proton transfer to the C-3 carbon of VI from the OH group to form amide intermediate (N-(tert-butyl)-2-oxoindoline-3-carboxamide) 4, through TS4 (Fig. 3(b)) with a barrier of 40.3 kcal mol−1.17a,b Afterward, N-(tert-butyl)-2-oxoindoline-3-carboxamide (4) interacts with palladium acetate to form six-membered complex VII, which releases the proton from the C-3 position and acetate group from the palladium catalyst as acetic acid to form complex VIIIvia transition state TS5 (Fig. 3(b)) with ΔG‡ = 14.6 kcal mol−1. During this step, the oxidation state of Pd remains unchanged, which is further coordinated with a second tert-butyl isocyanide to form a highly stabilized (–80.0 kcal mol−1) tetravalent Pd-containing intermediate IX. Then, the amidic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group at the C2-position gets activated and an isocyanide unit is inserted into the Pd–O bond to form seven-membered complex X, which readily converts to five-membered palladacyclic intermediate complex XI having an energy of 22.3 kcal mol−1, higher than complex X. Next, the third tert-butyl isocyanide proceeds to link to the Pd atom of complex (XI) to form stable tetravalent complex XII. Again, this isocyanide is inserted into the Pd–C bond and regains its six-membered intermediate to form intermediate XIII, having a higher energy of 8.6 kcal mol−1 from complex XII. The acetate group present at the palladacyclic intermediate (XIII) abstracts the N–H proton via transition state TS6 (Fig. 3(b)), which is located at the point of –35.8 kcal mol−1 in the PES and form another complex XIV, which possesses 44.6 kcal mol−1 less energy from the starting complex. Afterward, complex XIV releases AcOH and forms another complex XV having slightly higher energy (3.8 kcal mol−1) than complex XIII. Finally, C–N bond formation takes place via transition state, TS7 (Fig. 3(b)), having ΔG‡ = 19.4 kcal mol−1 from complex XV and forms complex XVI. Afterward, reductive elimination of palladium occurs in the form of palladium acetate to give the final product (3a) through intermediate XVII.
O group at the C2-position gets activated and an isocyanide unit is inserted into the Pd–O bond to form seven-membered complex X, which readily converts to five-membered palladacyclic intermediate complex XI having an energy of 22.3 kcal mol−1, higher than complex X. Next, the third tert-butyl isocyanide proceeds to link to the Pd atom of complex (XI) to form stable tetravalent complex XII. Again, this isocyanide is inserted into the Pd–C bond and regains its six-membered intermediate to form intermediate XIII, having a higher energy of 8.6 kcal mol−1 from complex XII. The acetate group present at the palladacyclic intermediate (XIII) abstracts the N–H proton via transition state TS6 (Fig. 3(b)), which is located at the point of –35.8 kcal mol−1 in the PES and form another complex XIV, which possesses 44.6 kcal mol−1 less energy from the starting complex. Afterward, complex XIV releases AcOH and forms another complex XV having slightly higher energy (3.8 kcal mol−1) than complex XIII. Finally, C–N bond formation takes place via transition state, TS7 (Fig. 3(b)), having ΔG‡ = 19.4 kcal mol−1 from complex XV and forms complex XVI. Afterward, reductive elimination of palladium occurs in the form of palladium acetate to give the final product (3a) through intermediate XVII.
Finally, several control studies were carried out to confirm the role of amide in this synthesis. We set up a reaction using tert-butyl 3-diazo-2-oxoindoline-1-carboxylate (1p) with tert-butyl isocyanide under standard reaction conditions (Scheme 5i) and got product N-(tert-butyl)-2-oxoindoline-3-carboxamide (4) in a 78% yield. In continuation, we performed a reaction of N-(tert-butyl)-2-oxoindoline-3-carboxamide (4) with tert-butyl isocyanide under the same reaction conditions. Delightfully, we got the desired product (3a) in a good yield (Scheme 5ii), which confirmed the formation of N-(tert-butyl)-2-oxoindoline-3-carboxamide (4) as an intermediate in this transformation. To further confirm the role of in situ generated amide, we have tried the reaction with three different oxindole derivatives such as ethyl 2-oxoindoline-3-carboxylate (5), indolin-2-one (6), and 3-hydroxy-3-(2-oxopropyl)indolin-2-one (7). Surprisingly there was no reaction with these substrates {Scheme 5(iii–v)}. So, these control experiments suggest that the amide group at the C-3 position of oxindole is required to assist the reaction for further steps.8,18 Next, we tried a reaction with D2O in place of H2O, and product formation was confirmed with HRMS analysis. HRMS data of the crude reaction mixture showed a peak at 398.2648, equivalent to a deuterated amide-based product. Hence, this experiment suggests the participation of H2O molecules in the formation of amide {Scheme 5(vi)}.
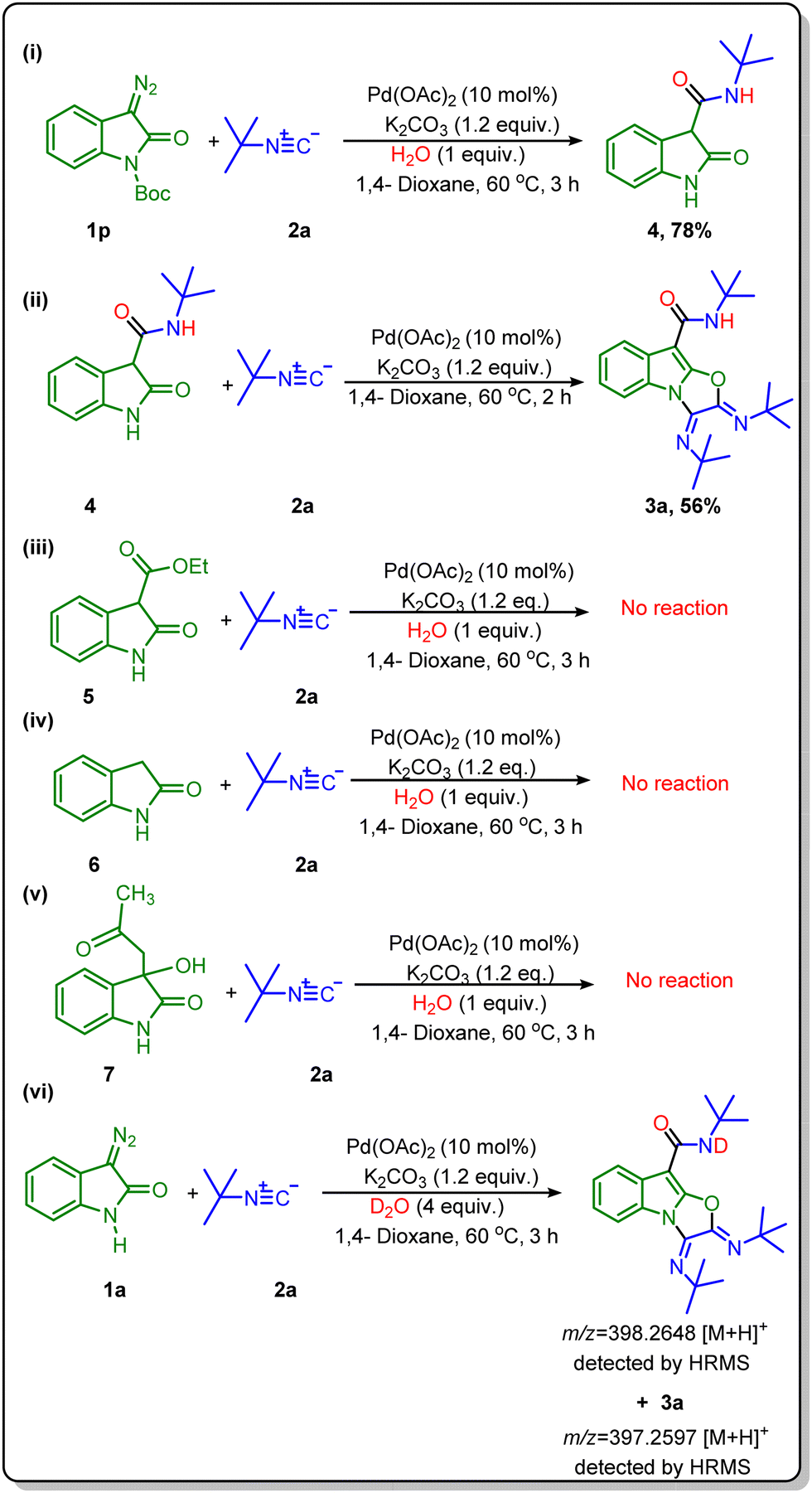 | ||
| Scheme 5 Control experiments. Reaction conditions for (i)–(v): substrates 1p/4/5/6 or 7 (1 equiv.), isocyanides (2) (3.2 equiv.), H2O (1.0 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (1.2 equiv.), and 1,4-dioxane (2 mL), for 3 h, at 60 °C. (vi) 1a (1 equiv.), isocyanides (2) (3.2 equiv.), D2O (4.0 equiv.), Pd(OAc)2 (10 mol%), K2CO3 (1.2 equiv.), and 1,4-dioxane (2 mL), for 3 h, at 60 °C. | ||
Conclusions
In summary, we have developed a Pd-catalysed and amide-assisted multicomponent reaction of 3-diazo oxindole, isocyanide, and water to generate oxazole-fused indole scaffolds. The functional group tolerance at different positions of 3-diazo oxindole along with different isocyanides was tested and the corresponding products were obtained in 37–76% isolated yields. Besides, a DFT study was performed to get an overview of the proposed mechanism, which recommended that the reaction involved amide formation at the C-3 position via isocyanide insertion, which subsequently assisted a [3+1+1] annulation reaction to furnish oxazole-fused indoles. Also, the role of in situ-generated amide to assist the annulation reaction was confirmed by setting up different control experiments.Procedure and characterization
General procedure to synthesize (2Z,3E)-N-(tert-butyl)-2,3-bis(tert-butylimino)-2,3-dihydrooxazolo[3,2-a]indole-9-carboxamide (3a–3q)
In a glass tube equipped with a stirrer bar, 3-diazo oxindole (1.0 equiv., 50 mg, 0.31 mmol), H2O (1.0 equiv., 5.6 μL, 0.31 mmol), Pd(OAc)2 (10 mol%, 7 mg), K2CO3 (1.5 equiv., 65 mg, 0.47 mmol) and 2 mL of 1,4-dioxane as a solvent were added. Then, the resulting reaction mixture was stirred at 60 °C in a preheated oil bath and tert-butyl isocyanide (3.2 equiv., 116 μL, 0.99 mmol) was added slowly with the help of a syringe. After completion of the reaction as indicated by TLC, work-up of the reaction mixture was done using ethyl acetate and distilled water in a 2:1 ratio followed by evaporation of volatiles under a reduced pressure. Next, the crude mixture was purified by column chromatography using ethyl acetate in hexane as an eluent affording the corresponding (2Z,3E)-N-(tert-butyl)-2,3-bis(tert-butylimino)-2,3-dihydrooxazolo[3,2-a]indole-9 carboxamide (3a–3w) in 37–76% yields.
Characterization data
:hexane v/v 1:19) afforded 3a; yellow solid, yield = 71% (88 mg, 0.223 mmol), 1H NMR (400 MHz, CDCl3): 8.20 (d, J = 7.2 Hz, 1H), 7.89 (d, J = 7.6 Hz, 1H), 7.27–7.18 (m, 2H), 5.70 (s, 1H), 1.52 (s, 9H), 1.50 (s, 9H), 1.47 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.3, 148.8, 136.5, 133.0, 128.9, 127.4, 124.2, 123.1, 121.9, 112.6, 87.8, 57.6, 55.9, 51.4, 30.0, 29.5, 29.4 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H33N4O2 397.2604; found 397.2601.
:hexane v/v 1:19) afforded 3b: yellow solid, yield = 57% (101 mg, 0.179 mmol), 1H NMR (400 MHz, CDCl3): 8.24 (d, J = 8 Hz, 1H), 7.94 (d, J = 8 Hz, 1H), 7.28–7.19 (m, 2H), 5.62 (s, 1H), 2.01 (s, 2H), 1.92 (s, 4H), 1.58 (s, 6H), 1.55 (s, 6H), 1.51 (s, 6H), 1.03 (s, 9H), 1.00 (s, 9H), 1.00 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.3, 148.4, 136.2, 131.8, 129.0, 127.5, 124.2, 123.1, 122.1, 112.5, 87.7, 61.4, 60.0, 55.3, 54.3, 54.1, 51.1, 32.2, 32.1, 32.0, 31.9, 31.7, 31.6, 31.0, 30.3, 30.2 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C35H57N4O2 565.4487; found 565.4487.
:hexane v/v 1:19) afforded 3c: yellow solid, yield = 60% (89 mg, 0.188 mmol), 1H NMR (400 MHz, CDCl3): 8.11 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.27–7.19 (m, 2H), 5.78 (d, J = 8.0 Hz, 1H), 4.78–4.71 (m, 1H), 4.09–3.97 (m, 2H), 2.00–1.26 (m, 30H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 160.7, 148.1, 138.2, 133.3, 127.6, 126.1, 123.1, 122.1, 120.5, 111.6, 86.7, 56.3, 55.3, 46.2, 32.9, 32.1, 32.0, 24.8, 24.7, 24.6, 23.4, 23.3, 22.7 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H39N4O2 475.3069; found 475.3073.
:hexane v/v 1:19) afforded 3d: yellow solid, yield = 76% (86 mg, 0.201 mmol), 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 2.4 Hz, 1H), 7.74 (s, 1H), 6.80 (dd, J = 8.8, 2.8 Hz, 1H), 5.70 (s, 1H), 3.86 (s, 3H), 1.50 (s, 9H), 1.49 (s, 9H), 1.46 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.6, 157.1, 149.2, 136.4, 132.8, 130.0, 121.8, 113.3, 112.2, 104.5, 87.8, 57.6, 55.8, 51.4, 30.0, 29.5, 29.4 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H35N4O3 427.2709; found 427.2712.
:hexane v/v 1:19) afforded 3e: yellow solid, yield = 68% (137 mg, 0.272 mmol), 1H NMR (400 MHz, CDCl3): 7.75–7.73 (m, 2H), 6.81 (dd, J = 8.4, 2.4 Hz, 1H), 5.74 (d, J = 8.0 Hz, 1H), 4.76–4.69 (m, 1H), 3.86 (s, 3H), 2.00–1.27 (m, 30H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.0, 157.1, 149.4, 139.2, 134.3, 130.0, 121.7, 113.5, 112.1, 104.7, 87.7, 57.4, 56.4, 55.7, 47.3, 34.0, 33.2, 33.1, 29.8, 25.85, 25.77, 25.72, 24.5, 24.4, 23.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H41N4O3 505.3179; found 505.3179.
:hexane v/v 1:19) afforded 3f: yellow solid, yield = 73% (86.5 mg, 0.211 mmol), 1H NMR (400 MHz, CDCl3): 8.04 (s, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.01 (dd, J = 1.2, 8.4 Hz, 1H), 5.69 (s, 1H), 2.41 (s, 3H), 1.51 (s, 9H), 1.49 (s, 9H), 1.46 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.6, 148.9, 136.6, 133.9, 132.9, 129.0, 125.6, 124.2, 122.1, 112.2, 87.6, 57.6, 55.8, 51.4, 30.0, 29.5, 29.3, 21.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H35N4O2 411.2760; found 411.2755.
:hexane v/v 1:19) afforded 3g: yellow solid, yield = 65% (108 mg, 0.188 mmol), 1H NMR (400 MHz, CDCl3): 8.08 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.02 (dd, J = 8.0 Hz, 1.2 Hz, 1H), 5.61 (s, 1H), 2.42 (s, 3H), 2.00 (s, 2H), 1.92 (s, 2H), 1.91 (s, 2H), 1.57 (s, 6H), 1.54 (s, 6H), 1.51 (s, 6H), 1.02 (s, 9H), 1.00 (s, 9H), 0.99 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.5, 148.6, 136.3, 133.9, 131.7, 129.2, 125.6, 124.2, 122.2, 112.1, 87.4, 61.4, 59.9, 55.3, 54.4, 54.1, 51.0, 32.2, 32.1, 32.0, 31.9, 31.7, 31.6, 31.0, 30.3, 30.1, 29.8, 21.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C36H59N4O2 579.4638; found 579.4632.
:hexane v/v 1:19) afforded 3h: yellow solid, yield = 63% (88.8 mg, 0.182 mmol), 1H NMR (400 MHz, CDCl3): 7.95 (s, 1H), 7.73 (d, 1H), 7.03 (dd, J = 8.4, 1.2 Hz, 1H), 5.75 (d, J = 7.6 Hz, 1H), 4.77–4.71 (m, 1H), 4.08–3.96 (m, 2H), 2.43 (s, 3H), 1.82–1.24 (m, 30H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 160.8, 148.1, 138.3, 133.3, 132.8, 127.8, 124.3, 123.2, 120.8, 111.3, 86.4, 56.3, 55.3, 46.2, 32.9, 32.1, 32.0, 28.6, 24.74, 24.67, 24.6, 23.4, 23.3, 22.7, 20.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H41N4O2 489.3230; found 489.3224.
:hexane v/v 1:19) afforded 3i: yellow solid, yield = 59% (66.5 mg, 0.156 mmol), 1H NMR (400 MHz, CDCl3): δ 8.06 (d, J = 8.8 Hz, 1H), 7.49 (d, J = 2.4 Hz, 1H), 6.86 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 5.67 (s, 1H), 3.83 (s, 3H), 1.51 (s, 9H), 1.49 (s, 9H), 1.46 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 162.4, 156.7, 147.9, 136.8, 133.1, 128.1, 122.5, 122.4, 111.6, 98.2, 87.7, 57.5, 55.9, 55.7, 51.3, 30.0, 29.5, 29.4 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H35N4O3 427.2709; found 427.2714.
:hexane v/v 1:19) afforded 3j: yellow solid, yield = 53% (70 mg, 0.140 mmol), 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 2.4 Hz, 1H), 6.88 (dd, J = 8.8 Hz, 2.4 Hz), 4.77–4.71 (m, 1H), 4.08–3.95 (m, 1H), 3.85 (s, 3H), 2.00–1.24 (m, 30H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 170.8, 161.9, 156.9, 148.2, 139.5, 134.5, 128.0, 122.3, 122.2, 111.7, 98.3, 87.6, 57.3, 56.4, 55.8, 47.3, 34.0, 33.2, 29.8, 25.9, 25.8, 25.7, 24.5, 24.4, 23.8, 23.1 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H40N4O3 505.3179; found 505.3177.
:hexane v/v 1:19) afforded 3l: yellow solid, yield = 56% (60.5 mg, 0.137 mmol), 1H NMR (400 MHz, CDCl3): δ 9.15 (d, J = 2.0 Hz, 1H), 8.13 (td, J = 8.8, 2.0 Hz, 1H), 7.96 (d, J = 8.8 Hz, 1H), 5.68 (s, 1H), 1.53 (s, 9H), 1.51 (s, 9H), 1.47 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 161.2, 149.7, 144.8, 135.0, 132.3, 130.3, 129.2, 119.0, 118.5, 112.4, 88.7, 58.2, 56.6, 51.7, 30.0, 29.34, 29.27, 28.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H32N5O4 442.2454; found 442.2463.
:hexane v/v 1:19) afforded 3m: yellow solid, yield = 37% (36.5 mg, 0.076 mmol), 1H NMR (700 MHz, CDCl3): 8.17 (d, J = 1.4 Hz, 1H), 7.91 (d, J = 9.1 Hz, 1H), 7.11 (dd, J = 8.4, 1.4 Hz, 1H), 5.70 (s, 1H), 1.55 (s, 9H), 1.54 (s, 9H), 1.50 (s, 9H) ppm. 13C{1H} NMR (175 MHz, CDCl3): δ 161.8, 149.4, 145.97, 145.96, 135.7, 132.5, 129.9, 125.6, 121.37, 119.91, 116.7, 115.1, 113.1, 88.0, 57.8, 56.0, 51.4, 29.9, 29.3, 29.2 ppm. 19F{1H} NMR (376 MHz, CDCl3): −57.86 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H32F3N4O3 481.2427; found 481.2430.
:hexane v/v 1:19) afforded 3q: yellow solid, yield = 41% (48 mg, 0.116 mmol), 1H NMR (700 MHz, CDCl3): δ 7.96 (dd, J = 9.8 Hz, 2.8 Hz, 1H), 7.84 (q, J = 4.2 Hz, 1H), 6.95 (td, J = 8.4 Hz, 2.1 Hz, 1H), 5.69 (s, 1H), 1.55 (s, 9H), 1.53 (s, 9H), 1.50 (s, 9H) ppm. 13C{1H} NMR (175 MHz, CDCl3): δ 162.0, 160.9, 159.5, 149.4, 135.9, 132.6, 130.2, 130.1, 123.7, 113.3, 113.2, 110.6, 110.5, 108.6, 108.4, 87.9, 57.7, 56.0, 51.4, 29.9, 29.4, 29.3 ppm. 19F{1H} NMR (376 MHz, CDCl3): −117.66 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H32FN4O2 415.2509; found 415.2513.
:hexane v/v 1:19) afforded 3r; yellow solid, yield = 54% (60 mg, 0.140 mmol), 1H NMR (400 MHz, CDCl3): 8.25 (d, J = 2.0 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.16 (dd, J = 2.0, 8.0 Hz, 1H), 5.66 (s, 1H), 1.51 (s, 9H), 1.50 (s, 9H), 1.46 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 161.9, 149.2, 135.9, 132.6, 130.2, 130.0, 125.7, 123.3, 121.9, 113.4, 87.6, 57.8, 56.1, 51.5, 30.0, 29.4, 29.3 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H32ClN4O2 431.2214; found 431.2212.
:hexane v/v 1:19) afforded 3s; yellow solid, yield = 46% (71 mg, 0.119 mmol), 1H NMR (400 MHz, CDCl3): δ 8.28 (d, J = 4.0 Hz, 1H), δ 7.83 (d, J = 8.4 Hz, 1H), 7.18 (dd, J = 8.4, 2.0 Hz, 1H), 5.58 (s, 1H), 1.99 (s, 2H), 1.91 (s, 4H), 1.57 (s, 6H), 1.54 (s, 6H), 1.50 (s, 6H), 1.02 (s, 9H), 1.00 (s, 9H), 0.99 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 161.8, 148.9, 135.5, 131.4, 130.3, 129.9, 125.7, 123.4, 122.0, 113.3, 87.5, 61.6, 60.2, 55.4, 54.3, 54.1, 51.0, 32.14, 32.09, 32.05, 31.9, 31.7, 31.6, 31.0, 30.2, 30.1 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C35H56ClN4O2 599.4092; found 599.4091.
:hexane v/v 1:19) afforded 3t: yellow solid, yield = 51% (51 mg, 0.107 mmol), 1H NMR (400 MHz, CDCl3): 8.41 (d, J = 2.0 Hz, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.30 (dd, J = 2.0, 8.0 Hz, 2H), 5.66 (s, 1H), 1.51 (s, 9H), 1.50 (s, 9H), 1.46 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 161.9, 149.1, 135.8, 132.6, 130.6, 126.1, 126.0, 124.8, 117.7, 113.8, 87.4, 57.8, 56.1, 51.5, 30.0, 29.4, 29.3 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H32BrN4O2 475.1709; found 475.1709.
:hexane v/v 1:19) afforded 3u: yellow solid, yield = 48% (56 mg, 0.101 mmol), 1H NMR (400 MHz, CDCl3): δ 8.32 (d, J = 2.0 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.31 (dd, J = 8.4 Hz, 2.0 Hz, 1H), 5.71 (d, J = 7.6 Hz, 1H), 4.76–4.70 (m, 1H), 4.08–3.96 (m, 1H), 2.00–1.24 (m, 30H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 161.3, 149.3, 138.7, 134.1, 130.5, 126.1, 125.8, 124.7, 117.7, 114.0, 87.4, 57.6, 56.6, 47.5, 33.9, 33.2, 33.1, 29.8, 25.81, 25.76, 25.7, 24.5, 24.4, 23.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H38BrN4O2 553.2178; found 553.2180.
:hexane v/v 1:19) afforded 3v:; yellow solid, yield = 50% (56 mg, 0.129 mmol), 1H NMR (700 MHz, CDCl3): δ 8.17 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.26 (dd, J = 8.4, 1.4 Hz, 1H), 5.71 (s, 1H), 1.55 (s, 9H), 1.53 (s, 9H), 1.50 (s, 9H) ppm. 13C{1H} NMR (175 MHz, CDCl3): δ 161.9, 148.6, 135.9, 132.5, 128.7, 124.50, 122.8, 112.1, 87.8, 57.8, 56.1, 51.4, 29.9, 29.3, 29.2 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C23H33N4O2 431.2213; found 431.2214.
:hexane v/v 1:9) afforded 4; off-white solid, yield = 78% (70 mg, 0.301 mmol), 1H NMR (400 MHz, CDCl3): δ 8.25 (s, 1H), 7.69 (d, J = 7.2 Hz, 1H), 7.33 (s, 1H), 7.24 (t, J = 6.8 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 6.87 (d, J = 7.2, 1H), 4.18 (s, 1H), 1.37 (s, 9H) ppm. 13C{1H} NMR (100 MHz, CDCl3): δ 175.7, 162.5, 140.5, 128.6, 127.4, 124.4, 123.2, 109.8, 51.7, 51.4, 28.8 ppm. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C13H16N2O2Na 255.1109; found 255.1110.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are thankful to the Science and Engineering Research Board (SERB), India (CRG/2021/002077) and Centre of Excellence for Emerging Materials, Thapar Institute of Engineering and Technology, Patiala (TIET/CEEMS/Regular/2022/042) for financial support. Also, we acknowledge the DST-FIST grant (SR/FST/CS-II/2018/69) at the School of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala and Single Crystal X-ray Diffraction (SCXRD) facility at the Department of Chemistry, Panjab University, Chandigarh for providing the X-ray crystallography data.References
- (a) Y. M. Khetmalis, M. Shivani, S. Murugesan and K. V. G. Chandra Sekhar, Oxindole and its derivatives: a review on recent progress in biological activities, Biomed. Pharmacother., 2021, 141, 111842 Search PubMed; (b) S. Ravi Suman Rudrangi, V. Kumar Bontha, V. Reddy Manda and S. Bethi, Oxindoles and their pharmaceutical significance-an overview, Asian J. Res. Chem., 2011, 4, 335–338 Search PubMed; (c) L. Yin, Q. Hu, J. Emmerich, M. M. C. Lo, E. Metzger, A. Ali and R. W. Hartmann, Novel pyridyl-or isoquinolinyl-substituted indolines and indoles as potent and selective aldosterone synthase inhibitors, J. Med. Chem., 2014, 57, 5179–5189 Search PubMed; (d) N. Monakhova, S. Ryabova and V. Makarov, Synthesis and some biological properties of Pyrrolo[1,2-a]indoles, J. Heterocycl. Chem., 2016, 53, 685–709 Search PubMed; (e) C. Bunders, J. Cavanagh and C. Melander, Flustramine inspired synthesis and biological evaluation of pyrroloindoline triazole amides as novel inhibitors of bacterial biofilms, Org. Biomol. Chem., 2011, 9, 5476–5481 Search PubMed; (f) L. M. Gaste, P. A. Wyman, E. S. Ellis, A. M. Brown and T. J. Young, 5-HT4receptor antagonists: oxazolo, oxazino and oxazepino[3,2-a]indole derivatives, Bioorg. Med. Chem. Lett., 1994, 4, 667–668 CrossRef; (g) W. C. Sumpter, THE CHEMISTRY OF ISATIN, Chem. Rev., 1944, 34, 393–434 Search PubMed.
- (a) L. Y. Mei, Y. Wei, X. Y. Tang and M. Shi, Catalyst-Dependent Stereodivergent and Regioselective Synthesis of Indole-Fused Heterocycles through Formal Cycloadditions of Indolyl-Allenes, J. Am. Chem. Soc., 2015, 137, 8131–8137 CrossRef CAS PubMed; (b) G. Pan, L. Lu, W. Zhuang and Q. Huang, Synthesis of Indole-Fused Six-, Seven-, or Eight-Membered N, O-Heterocycles via Rhodium-Catalyzed NH-Indole-Directed C–H Acetoxylation/Hydrolysis/Annulation, J. Org. Chem., 2021, 86, 16753–16763 CrossRef CAS PubMed; (c) X. Fang, S. Gao, Z. Wu, H. Yao and A. Lin, Pd(II)-Catalyzed oxidative dearomatization of indoles: substrate-controlled synthesis of indolines and indolones, Org. Chem. Front., 2017, 4, 292–296 RSC; (d) Y. Huang, Y. Yang, H. Song, Y. Liu and Q. Wang, Synthesis of Structurally Diverse 2,3-Fused Indoles via Microwave-Assisted AgSbF6-Catalysed Intramolecular Difunctionalization of o-Alkynylanilines, Sci. Rep., 2015, 5, 13516 CrossRef PubMed; (e) K. F. Suzdalev, S. V. Den’Kina and V. V. Tkachev, Formation of the same pyrimido[1,2-a]indoles from 1-(oxiran-2-ylmethyl)-1H-indole or [1,3]oxazolo[3,2-a]indole derivatives in its reactions with aromatic amines, Tetrahedron, 2013, 69, 8785–8789 CrossRef CAS.
- M. Zhang, Y. Duan, W. Li, Y. Cheng and C. Zhu, Visible-light-induced aerobic dearomative reaction of indole derivatives: access to heterocycle fused or spirocyclo indolones, Chem. Commun., 2016, 52, 4761–4763 RSC.
- L. A. Oparina, N. A. Kolyvanov, I. A. Ushakov, A. G. Mal’kina, A. V. Vashchenko and B. A. Trofimov, Metal-and Solvent-free Synthesis of Functionalized Dihydrooxazolo[3,2-a]indoles by One-Pot Tandem Assembly of 3H-Indoles and Propargylic Alcohols, Synthesis, 2019, 1445–1454 CAS.
- Y. Zhao, X. Niu, H. Yang, J. Yang, Z. Wang and Q. Wang, Substrate-directed divergent synthesis of fused indole polycycles through Rh(II)-catalyzed cascade reactions of bis (diazo) indolin-2-ones, Chem. Commun., 2022, 58, 8576–8579 RSC.
- (a) M. G. Ciulla, S. Zimmermann and K. Kumar, Cascade reaction based synthetic strategies targeting biologically intriguing indole polycycles, Org. Biomol. Chem., 2019, 17, 413–431 Search PubMed; (b) X. Ma and W. Zhang, Recent developments in one-pot stepwise synthesis (OPSS) of small molecules, Iscience, 2022, 25, 105005 CrossRef CAS PubMed; (c) A. Baccalini, G. Faita, G. Zanoni and D. Maiti, Transition Metal Promoted Cascade Heterocycle Synthesis through C−H Functionalization, Chem. – Eur. J., 2020, 26, 9749–9783 CrossRef CAS PubMed; (d) I. Alahyen, L. Benhamou, V. Dalla, C. Taillier and S. Comesse, 20 Years of Forging N-Heterocycles from Acrylamides through Domino/Cascade Reactions, Synthesis, 2021, 3409–3439 CAS; (e) R. Connon and P. J. Guiry, Recent advances in the development of one-pot/multistep syntheses of 3,4-annulated indoles, Tetrahedron Lett., 2020, 61, 151696 CrossRef CAS.
- (a) F. L. Hong and L. W. Ye, Transition metal-catalyzed tandem reactions of ynamides for divergent N-heterocycle synthesis, Acc. Chem. Res., 2020, 53, 2003–2019 CrossRef CAS PubMed; (b) A. Cabré, X. Verdaguer and A. Riera, Multi-component syntheses of heterocycles by transition-metal catalysis, Chem. Rev., 2022, 122, 269–339 Search PubMed; (c) Y. Xia and J. Wang, Transition-Metal-Catalyzed Cross-Coupling with Ketones or Aldehydes via N-Tosylhydrazones, J. Am. Chem. Soc., 2020, 142, 10592–10605 CrossRef CAS PubMed.
- (a) G. Qiu, Q. Wang and J. Zhu, Palladium-Catalyzed Three-Component Reaction of Propargyl Carbonates, Isocyanides, and Alcohols or Water: Switchable Synthesis of Pyrroles and Its Bicyclic Analogues, Org. Lett., 2017, 19, 270–273 CrossRef CAS PubMed; (b) Z. Tashrifi, M. Mohammadi Khanaposhtani, F. Gholami, B. Larijani and M. Mahdavi, Rapid Access to Fused Tetracyclic N-Heterocycles via Amino-to-Alkyl 1,5-Palladium Migration Coupled with Intramolecular C(sp3)–C(sp2) Coupling, Adv. Synth. Catal., 2023, 365, 926–947 CrossRef CAS; (c) J. W. Collet, T. R. Roose, B. Weijers, B. U. W. Maes, E. Ruijter and R. V. A. Orru, Recent advances in palladium-catalyzed isocyanide insertions, Molecules, 2020, 25, 4906 CrossRef CAS PubMed; (d) F. Zhou, K. Ding and Q. Cai, Palladium-Catalyzed Amidation of N-Tosylhydrazones with Isocyanides, Chem. – Eur. J., 2011, 17, 12268–12271 CrossRef CAS PubMed; (e) Y. Tian, L. Tian, X. He, C. Li, X. Jia and J. Li, Indium(III) chloride-catalyzed isocyanide insertion reaction to construct complex spirooxindole, Org. Lett., 2015, 17, 4874–4877 CrossRef CAS PubMed.
- M. Cao, Y. L. Fang, Y. C. Wang, X. J. Xu, Z. W. Xi and S. Tang, Ce(OTf)3-Catalyzed Multicomponent Reaction of Alkynyl Carboxylic Acids, tert-Butyl Isocyanide, and Azides for the Assembly of Triazole–Oxazole Derivatives, ACS Comb. Sci., 2020, 22, 268–273 CrossRef CAS PubMed.
- S. Sisodiya, A. Acharya, M. Nagpure, N. Roy, S. K. Giri, H. R. Yadav, A. R. Choudhury and S. K. Guchhait, A cascade reaction of indolyl-migratory isocyanide insertion, scaffold rearrangement and redox-neutral event with isocyanide as a C(sp3)H–N synthon efficiently constructs indolylisoindolinones, Chem. Commun., 2022, 58, 11827–11830 RSC.
- S. Lu, C. H. Ding and B. Xu, Triple-Consecutive Isocyanide Insertions with Aldehydes: Synthesis of 4-Cyanooxazoles, Org. Lett., 2023, 25, 849–854 CrossRef CAS PubMed.
- P. Soam, H. Gaba, D. Mandal and V. Tyagi, A Pd-catalyzed one-pot cascade consisting of C–C/C–O/N–N bond formation to access benzoxazine fused 1,2,3-triazoles, Org. Biomol. Chem., 2021, 19, 9936–9945 Search PubMed.
- P. Soam, D. Mandal and V. Tyagi, Divergent and Selective Synthesis of 3-Alkylidene Oxindoles Using Pd-Catalyzed Multicomponent Reaction, J. Org. Chem., 2023, 88, 11023–11035 CrossRef CAS PubMed.
- (a) B. Vol, D. H. McDANIEL and H. C. Brown, An extended table of Hammett substitutent constants based on the ionization of substituted benzoic acids, J. Org. Chem., 1958, 23, 420–427 CrossRef; (b) H. M. Yau, R. S. Haines and J. B. Harper, A Robust, “One-Pot” Method for Acquiring Kinetic Data for Hammett Plots Used to Demonstrate Transmission of Substituent Effects in Reactions of Aromatic Ethyl Esters, J. Chem. Educ., 2015, 92, 538–542 CrossRef CAS; (c) S. V. Khansole and Y. B. Vibhute, Kinetic of iodination of phenol and substituted phenols by pyridinium iodochloride in methanol, J. Iran. Chem. Res., 2009, 2, 151–156 Search PubMed.
- X. Zhang, L. W. Chung and Y. D. Wu, New mechanistic insights on the selectivity of transition-metal-catalyzed organic reactions: the role of computational chemistry, Acc. Chem. Res., 2016, 49, 1302–1310 CrossRef CAS PubMed.
- (a) Y. Xia, D. Qiu and J. Wang, Transition-metal-catalyzed cross-couplings through carbene migratory insertion, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed; (b) Y. Liu, Z. Luo, J. Z. Zhang and F. Xia, DFT calculations on the mechanism of transition-metal-catalyzed reaction of diazo compounds with phenols: O–H insertion versus C–H insertion, J. Phys. Chem. A, 2016, 120, 6485–6492 Search PubMed; (c) X. Bi, Z. Liu, S. Cao, J. Wu, G. Zanoni and P. Sivaguru, Palladium(II)-catalyzed cross-coupling of diazo compounds and isocyanides to access ketenimines, ACS Catal., 2020, 10, 12881–12887 CrossRef; (d) X. Hui Zhang, Z. Yuan Geng and K. Tai Wang, Theoretical studies on the mechanism of palladium(II)-catalysed ortho-carboxylation of acetanilide with CO, J. Chem. Sci., 2014, 126, 265–272 CrossRef.
- (a) O. Bakulina, A. Inyutina, D. Dar’in and M. Krasavin, Multicomponent reactions involving diazo reagents: a 5 year update, Molecules, 2021, 26, 6563 Search PubMed; (b) H. Gu, Z. Qiu, Z. Zhang, J. Li and B. Yan, A mechanistic study of Pd(OAc)2-catalyzed intramolecular C–H functionalization reaction involving CO/isonitrile insertion, Dalton Trans., 2015, 44, 9839–9846 Search PubMed.
- (a) F. Chen, C. Zhu and H. Jiang, [3+1+1] Annulation Reaction of Benzo-1,2-Quinones, Aldehydes and Hydroxylamine Hydrochloride: Access to Benzoxazoles with Inorganic Nitrogen Source, Adv. Synth. Catal., 2021, 363, 2124–2132 CrossRef CAS; (b) W. Q. Zhu, Y. C. Fang, W. Y. Han, F. Li, M. G. Yang and Y. Z. Chen, Palladium-catalyzed [2+2+1] annulation: access to chromone-fused cyclopentanones with cyclopropenone as the CO source, Org. Chem. Front., 2021, 8, 3082–3090 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystal data of compound 3a, copies of 1H NMR and 13C NMR data of the compounds (3a–3q, 4), computational details, optimized transition states and optimized Cartesian coordinates and electronic energies. CCDC 2291510. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04886a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |