
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization, and anticancer potential of pyrene-appended Schiff base tin(IV) complexes: experimental and computational insights†
Anup
Paul‡
 *a,
Rais Ahmad
Khan‡
b,
Gouse M.
Shaik
c,
Jilani P.
Shaik
c,
Dmytro S.
Nesterov
a,
M. Fátima C.
Guedes da Silva
ad and
Armando J. L.
Pombeiro
a
*a,
Rais Ahmad
Khan‡
b,
Gouse M.
Shaik
c,
Jilani P.
Shaik
c,
Dmytro S.
Nesterov
a,
M. Fátima C.
Guedes da Silva
ad and
Armando J. L.
Pombeiro
a
aCentro de Química Estrutural, Institute of Molecular Sciences, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: anuppaul@tecnico.ulisboa.pt
bDepartment of Chemistry, King Saud University, Riyadh-11451, Kingdom of Saudi Arabia
cDepartment of Biochemistry, King Saud University, Riyadh-11451, Kingdom of Saudi Arabia
dDepartamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 12th December 2023
Abstract
In this study, we report the synthesis and comprehensive characterization of two novel pyrene-appended Schiff base organotin(IV) compounds, trimethylstannyl (E)-4-((pyren-1-ylmethylene)amino)benzoate (1) and triphenylstannyl (E)-4-((pyren-1-ylmethylene)amino)benzoate (2). They were synthesized through the condensation of trimethylstannyl 4-aminobenzoate or triphenylstannyl 4-aminobenzoate with 1-pyrenealdehyde in toluene. The characterization included elemental analysis, infrared spectroscopy, 1H, 13C, and 119Sn NMR, and ESI-MS. The crystal structure of compound 1 was elucidated through single crystal X-ray diffraction analysis. The interaction of compounds 1 and 2 with calf thymus DNA (CT DNA) was investigated using absorption and fluorescence spectroscopic techniques. The calculated binding constants (Kb) unveiled a notably stronger binding propensity of compound 2 compared to 1. Furthermore, the in vitro cytotoxicity assays of these compounds were performed against human lung (A549) cancer cell lines, which revealed concentration-dependent cytotoxic effects, with 2 exhibiting a higher cytotoxicity than 1. Moreover, the scratch assay demonstrated the ability of both compounds to inhibit cancer cell migration, with 2 displaying stronger inhibitory effects. Furthermore, gene expression studies showed that both compounds downregulated the expression of MMP-2 and TGF-β genes, indicating their potential in modulating critical signaling pathways in cancer cells. Molecular docking simulations provided insights into the binding interactions of these compounds with MMP-2 and TGF-β, further supporting their potential as inhibitors of cancer-related proteins. Overall, our findings highlight the promising anticancer properties of these novel pyrene-appended Schiff base compounds, with 2 showing particular potency. This research provides an insight into the development of potential chemotherapeutic agents targeting cancer cell proliferation, migration, and signalling pathways.
Introduction
The rising incidence of cancer continues to be a global health crisis, necessitating an urgent need for the development of innovative and effective anticancer drugs. Metal-based compounds, exemplified by platinum-based drugs like cisplatin, carboplatin, nedaplatin, and oxaliplatin, have shown remarkable efficacy in cancer treatment.1–3 However, the limitations of platinum drugs, including severe side effects and drug resistance, demand the exploration of alternative metal-based anticancer agents.4,5 Organotin compounds have emerged as promising scaffolds due to their diverse biological properties, including antimicrobial, anti-HCV, antiparasitic, and anti-inflammatory properties.6–8 Moreover, organotin compounds have demonstrated potential as cancer chemotherapeutic agents, attributed to their ability to induce apoptosis and their lipophilic nature.9–17 This has prompted significant research efforts towards the design of organotin(IV) complexes as potential anticancer agents, aiming to enhance their therapeutic efficacy through selective cytotoxicity against cancer cells.11–18Schiff base organotin(IV) complexes have garnered significant attention within the realm of organotin compounds due to their remarkable biological activities. In particular, Schiff base organotin(IV) carboxylates have demonstrated potent antitumor properties, at times even surpassing the efficacy of cisplatin, a widely used platinum-based anticancer drug.19–24 The integration of the Schiff base unit into organotin(IV) carboxylate complexes has proven to be a pivotal development, leading to compounds with enhanced biological properties and increased selectivity. This innovative approach offers promising avenues for targeted therapeutic interventions.20,25,26 The specific alkyl/aryl groups and ligands attached to the tin atom in organotin(IV) compounds play a profound role in their anticancer activity.25,27 Notably, triorganotin(IV) complexes, characterized by their higher lipophilicity, have shown superior biological activities when compared to their di- and mono-organotin(IV) counterparts.25,28,29 This observation has generated substantial interest in exploring triorganotin(IV) carboxylates as potential anticancer agents.12,15–18,30,31 Beyond the choice of organotin compound and its structure, structural modifications to the ligand and the nature of its substituents have been found to significantly influence the antiproliferative activity of organotin(IV) carboxylates. This opens the door to fine-tuning the biological properties of these compounds. Researchers have been able to optimize the design of organotin(IV) complexes to achieve enhanced anticancer effects, ensuring that the compounds selectively target cancer cells, while minimizing damage to healthy tissues. Such advances in compound design and selection hold promise for developing more effective and less toxic treatments for cancer, addressing a critical need in oncology.
Pyrene, a polycyclic aromatic hydrocarbon, holds significant biological importance and has garnered attention across scientific fields.32,33 When incorporated into Schiff base organotin(IV) complexes, the pyrene moiety can greatly enhance their biological activities.33,34 The unique properties of pyrene, such as its aromaticity and hydrophobicity, facilitate improved cellular uptake and interactions with target biomolecules, leading to enhanced therapeutic efficacy. Therefore, the incorporation of the pyrene moiety into Schiff base organotin(IV) complexes represents an innovative approach to develop metal-based anticancer therapeutics with superior biological activities.
Motivated by these considerations, this study focuses on the synthesis, characterization, and biological evaluation of two novel pyrene-tagged Schiff base triorganotin(IV) complexes 1 and 2. By harnessing the unique properties of these complexes, we aim to enhance their biological activity, paving the way for the development of innovative metal-based anticancer drugs. Furthermore, in silico studies have been performed to investigate the interaction of these compounds with gene expression proteins, shedding light on the underlying mechanism of action.
Experimental section
Materials and methods
The following chemicals were obtained from Sigma Aldrich Chemical Co. and used as such: 4-aminobenzoic acid, 1-pyrenecarbaldehyde, Me3SnOH, and Ph3SnOH. Precursors trimethylstannyl 4-aminobenzoate (TM4M) and triphenylstannyl 4-aminobenzoate (TP4M) were prepared using a modified version of the procedure mentioned earlier.34 The IR spectra were obtained with a Bruker Alpha FTIR spectrometer. Tetramethylsilane [Si(CH3)4] was used as the internal reference for the 1H (300 MHz) and 13C (75.45 MHz) NMR spectra, and [Sn(CH3)4] for 119Sn (112 MHz) NMR spectra, acquired at room temperature (RT) on a Bruker 300 MHz spectrometer. Bruker Micro TOF QII and THERMO Finnigan LCQ Advantage Max ion trap mass spectrometers were used for collecting the electrospray ionization mass spectrometric data (ESI-MS) in MeOH.The absorption and fluorescence experiments were performed to evaluate the DNA binding propensity of complexes 1 and 2 by adopting the standard protocol35,36 and protocols previously adopted by our laboratory.37,38
Cell culture
The human lung cancer (A549) cell lines were obtained from ATCC (American-type culture collection). These cells were cultured in DMEM (Dulbecco's modified Eagle's medium) augmented with 10% FBS (foetal bovine serum) and penicillin and streptomycin in 5% CO2. Cells were passaged with 0.1% trypsin solution every 48 h.MTT assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dye reduction assay was adopted to analyze the cytotoxic effect of the organotin complexes 1 and 2 against A549 cell lines.39 The standard protocol was adopted and briefly described below.Around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells were seeded in 96 well plates and incubated overnight. The stock solutions of complexes 1 and 2 (1 mM) were prepared in DMSO. Then, from the stock solutions of the complexes, serial dilution was done for a volume of 1 mL to get the desired concentration. The final percentage of DMSO in the sample dilution is from 0.01% to 0.5%.
000 cells were seeded in 96 well plates and incubated overnight. The stock solutions of complexes 1 and 2 (1 mM) were prepared in DMSO. Then, from the stock solutions of the complexes, serial dilution was done for a volume of 1 mL to get the desired concentration. The final percentage of DMSO in the sample dilution is from 0.01% to 0.5%.
Then these cells were heated with 0.1% DMSO (control) and 0.1, 1 and 5 μM organotin compound 1 or 2. Upon treatment for 24 h, cell viability was analyzed using 100 μL of the MTT dye (0.1 mg mL per stock) and by incubating for another 4 h at 37 °C in the dark. The quantification of formazan was carried out by measuring absorbance at 570 nm in a microplate reader. The normalized value to control has been plotted. Images of control and treated cancer cells after 0 and 24 h were taken using a Leica DFC 450 microscope at 10× magnification.
Scratch plate assay
The scratch plate assay carried out to analyze the metastasis of the human lung A549 cancer cells upon treatment with organotin compounds 1 and 2 following the standard protocol40 is briefly described below.On the 24-well cell culture plate, the A549 cancer cells were sown. The cells were allowed to grow confluently as monolayers. Using a sterile pipette tip, a scratch was made to the monolayer. The cells were washed with DMEM to remove detached cells. After washing the cells, they were incubated with control 0.1% DMSO and 0.01 μM organotin compounds 1 and 2. The images of cell migration were taken after 0, 24 and 48 h. The images were converted to grey scale images and processed using the J software for comparison.41
Expression of MMP2 and TGF-β genes
The total cellular RNA from both control and treated (organotin compounds 1 and 2) A549 human lung cancer cells was extracted by adopting standard protocols using Trizol (Invitrogen Life Technologies). In the reverse transcription for cDNA synthesis, 2 μg of total RNA was utilized. Furthermore, this cDNA was utilized for the quantification of various genes by real-time PCR (Applied Biosystems 7500 real-time PCR system) using specific primers.Theoretical studies
The ORCA 5.0.4 package42,43 was used for all DFT calculations. The B3LYP functional with the ZORA-def2-TZVP basis set44 and SARC/J auxiliary basis set45,46 was used for all atoms except Sn, for which the SARC-def2-TZVP basis set45 was applied. AutoAux keyword47 was used to generate other auxiliary basis sets in all cases. The zero-order regular approximation (ZORA)48 was used because of the presence of a heavy element. The solvent medium (water) was accounted for using the conductor-like polarizable continuum model (C-PCM).49 The SCF optimization convergence criteria settled with the VeryTightSCF keyword, geometry optimization criteria with the TightOPT keyword, and integration grids of high density (Defgrid3 keyword) were employed. Analysis of bond critical points and non-covalent interaction indexes was performed using the Multiwfn 3.8 program.50 Dispersion correction was introduced through the D4 keyword (Grimme's atom pairwise approach).51 Visualization of the molecular orbitals was performed using the Avogadro 1.2 program.52,53 Cartesian coordinates of the optimized structures are provided in Tables S2 and S3 (ESI†). Complexes 1 and 2 were optimized using the ORCA 5.0.4 package as discussed above. Gene expression proteins PDB Id: 1rtg and PDB Id: 1VJY were downloaded in the PDB format from the protein data bank (www.rcsb.org). The protein was then processed and refined utilizing the Protein Preparation Wizard in the Discovery Studio tutorial. All the water molecules were deleted from the proteins and active sites were marked for docking. Hex 8.0.0. was used for docking using the protocols described previously.54,55Synthesis and characterization
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1352 ν(C–O), 772 ν(Sn–C), 552 ν(Sn–O). 1H NMR (300 MHz, CDCl3) [s, 1H, (C(H)N)], 9.08 [d, J = 9.4 Hz, 1H, H1 and H1′], 8.78 [d, J = 8.2 Hz, 1H, H2 and H2′], 8.41–8.05 (m, 9H, Py-H), 7.40 (d, J = 8.3 Hz, 2H), 0.7 [s, 9H, Sn-CH3, 2J 119Sn-1H = 54 Hz]. 13C NMR (CDCl3, 75.45 MHz, δC, ppm): 159.98 (–CO), 156.32 (C(H)N), 133.77, 131.56, 131.26, 130.81, 130.57, 129.31, 128.07, 127.49, 127.08, 126.35, 126.11, 125.08, 124.59, 122.41, 120.73, 113.78, −2.24. 119Sn NMR (CDCl3, 112 MHz): 137.32 ppm.
:1) after 5 days. Isolated yield = 0.140 g (39%). Anal. calcd for C42H29NO2Sn: C, 72.23; H, 4.19; N, 2.01; found C, 72.31; H, 4.23; N, 2.08. ATR-IR (cm−1): 1601 ν(CN), 1330 ν(C–O), 845 ν(Sn–C), 548 ν(Sn–O). 1H NMR (CDCl3, 300 MHz, δH, ppm) [s, 1H, (C(H)N)], (8.78–7.38) [m, 28H, Py-H and Ar-H]. 13C NMR (CDCl3, 75.45 MHz, δC, ppm): 160.28 [CO], 156.82 [(C)HN], 138.47, 137.32, 136.92, 136.70, 133.89, 132.09, 131.15, 130.83, 130.42, 130.10, 129.34, 128.95, 127.99, 127.39, 127.07, 126.34, 126.14, 125.09, 122.35, 120.68. 119Sn NMR (CDCl3, 112 MHz): −111.85 ppm.
N), 1352 ν(C–O), 772 ν(Sn–C), 552 ν(Sn–O). 1H NMR (300 MHz, CDCl3) [s, 1H, (C(H)N)], 9.08 [d, J = 9.4 Hz, 1H, H1 and H1′], 8.78 [d, J = 8.2 Hz, 1H, H2 and H2′], 8.41–8.05 (m, 9H, Py-H), 7.40 (d, J = 8.3 Hz, 2H), 0.7 [s, 9H, Sn-CH3, 2J 119Sn-1H = 54 Hz]. 13C NMR (CDCl3, 75.45 MHz, δC, ppm): 159.98 (–CO), 156.32 (C(H)N), 133.77, 131.56, 131.26, 130.81, 130.57, 129.31, 128.07, 127.49, 127.08, 126.35, 126.11, 125.08, 124.59, 122.41, 120.73, 113.78, −2.24. 119Sn NMR (CDCl3, 112 MHz): 137.32 ppm.
:1) after 5 days. Isolated yield = 0.140 g (39%). Anal. calcd for C42H29NO2Sn: C, 72.23; H, 4.19; N, 2.01; found C, 72.31; H, 4.23; N, 2.08. ATR-IR (cm−1): 1601 ν(CN), 1330 ν(C–O), 845 ν(Sn–C), 548 ν(Sn–O). 1H NMR (CDCl3, 300 MHz, δH, ppm) [s, 1H, (C(H)N)], (8.78–7.38) [m, 28H, Py-H and Ar-H]. 13C NMR (CDCl3, 75.45 MHz, δC, ppm): 160.28 [CO], 156.82 [(C)HN], 138.47, 137.32, 136.92, 136.70, 133.89, 132.09, 131.15, 130.83, 130.42, 130.10, 129.34, 128.95, 127.99, 127.39, 127.07, 126.34, 126.14, 125.09, 122.35, 120.68. 119Sn NMR (CDCl3, 112 MHz): −111.85 ppm.
Crystal structure determination
A crystal of 1 was immersed in cryo-oil, mounted on a nylon loop, and measured at room temperature. Intensity data were collected on a Bruker APEX-II PHOTON 100 diffractometer.56 Retrieved cell parameters were refined using Bruker SMART and SAINT software programs, respectively; SADAB was used for absorption corrections. The structure was solved by direct methods using the SHELXS-97 package and refined with SHELXL2018/3.57 WinGX System-Version 2018.3 was used to perform the calculations.58 All nonhydrogen atoms were refined anisotropically. The hydrogen atoms bonded to carbon were included in the model at geometrically calculated positions and refined using a riding model. CCDC no. 2296017. Crystallographic data and structure refinement details of 1 are presented in Table S1 (ESI†).Results and discussion
Synthesis and characterisation
For the synthesis of the target pyrene-appended Schiff base compounds, we condensed trimethylstannyl 4-aminobenzoate or triphenylstannyl 4-aminobenzoate with 1-pyrenealdehyde in toluene. Scheme 1 illustrates the synthetic route to prepare compounds 1 and 2.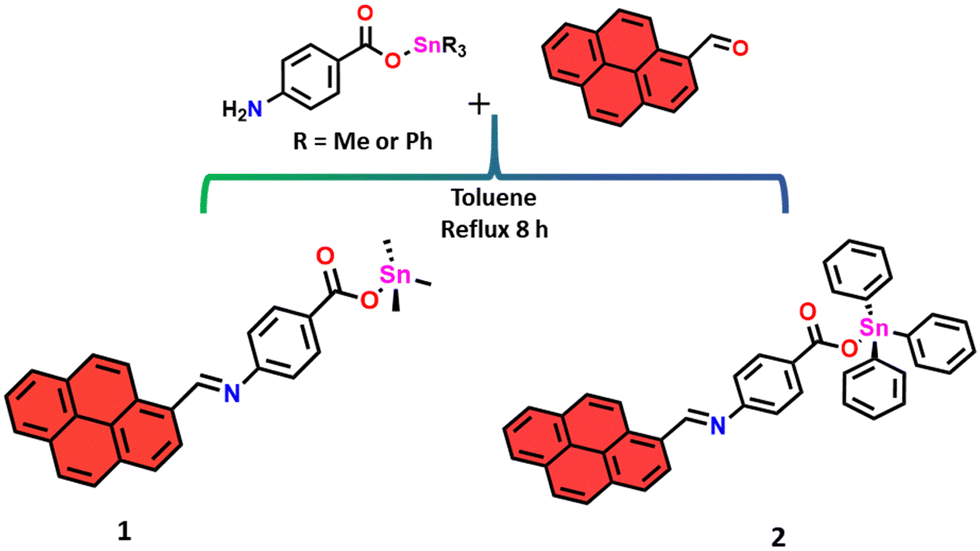 | ||
| Scheme 1 Synthesis of compounds 1 and 2. | ||
They were characterized by elemental analysis, infrared spectroscopy, 1H, 13C, and 119Sn NMR, and ESI-MS. The structure of 1 was elucidated through single-crystal X-ray diffraction analysis. Attempts to obtain X-ray-quality crystals of compound 2 were unsuccessful in our hands.
The ATR-IR spectra of 1 and 2 (Fig. S1, ESI†) reveal crucial information about their successful synthesis. Notably, a distinct peak at around ∼1600 cm−1, attributed to the ν(CN) stretching vibration,21,59,60 confirms the presence of the Schiff bases. The bands at approximately 1350 and 550 cm−1 correspond to the C–O and the Sn–C stretching, respectively, further supporting the characterization of the compounds.21,59,60
As depicted in Fig. S2 and S3 (ESI†), the 1H NMR spectra of compounds 1 and 2 exhibit a distinctive singlet at approximately δ 9.5, attributed to the –C(H)N– protons. The resonances at approximately δ 0.7 in 1 were attributed to the –SnMe3 protons, while compound 2 displays multiplets in the range of 9.09–7.39 ppm, indicative of the –SnPh3 protons. The 13C NMR spectra (Fig. S4 and S5, ESI†) further support the proposed structures. In the 119Sn NMR spectrum, compound 1 exhibits a resonance at +137.32 ppm (Fig. S6, ESI†), while compound 2 displays a peak at −111.85 ppm (Fig. S7, ESI†). The detected signals fall within the typical range observed for four-coordinate tin atoms in trialkyltin (153–99 ppm) and triphenyltin (−40 to −120 ppm) species featuring an Sn–O bond.31,61–63 This indicates that the polymeric structure observed for complex 1 in the solid state (see below) dissociates in solution, leading to the formation of monomeric species. In the coordination of carboxylate ligands to organotin compounds, there is a common occurrence of anisobidentate behavior, involving the formation of an additional secondary Sn⋯O contact in the solid state. However, the 119Sn NMR chemical shift in complex 1 suggests that the carboxylate-SnMe3 linkage is monodentate in solution.31,63 This points to the preservation of a robust ligand–metal bonding interaction in the dissolved state, providing insights into the dynamic nature of the complex in different environments. Furthermore, the 2J(1H–119Sn) coupling constant was successfully measured for compound 1, providing an insight into its coordination environment; the observed 2J(1H–119Sn) value of 54 Hz in 1 confirms the Sn tetracoordination in solution.61,62 Due to the complexity of the spectrum of compound 2, it was not possible to determine the 2J(1H–119Sn) coupling constant. The positive ion ESI mass spectra display prominent peaks at m/z = 514.10 (calcd 513.08) for compound 1, corresponding to [1 + H]+ (Fig. S8, ESI†). In the case of compound 2, peaks at m/z = 716.73 (calcd. 718.1) were observed, corresponding to [2 + H2O + H]+ (Fig. S9, ESI†).
Crystallographic analysis of compound 1
The asymmetric unit of 1 contains a tin cation, three methyl groups and an O-bound Schiff base ligand. Symmetry expansion reveals a 1D polymeric structure spreading along the crystallographic b-axis with the O-carboxylate atoms bridge chelating the metal atoms. In this way, every metal presents a trigonal bipyramid geometry with the oxygen atoms in the axial sites, with Sn⋯O distances of 2.125(6) and 2.643(7) Å and an O–Sn–O angle of 174.1(2)°; the methyl groups in the equatorial plane show Sn⋯C lengths between 2.113(10) and 2.125(10) Å with C–Sn–C angles of 113.8(5)°, 119.0(5)° and 124.3(5)° (Fig. 1 and 2). The carboxylate group stands in the plane of the phenyl moiety; the pyrene and the phenyl fractions make an angle of 55.7°. The intrachain Sn⋯Sn distance is 5.297(1) Å, while the shortest interchain distance is 6.456(1) Å.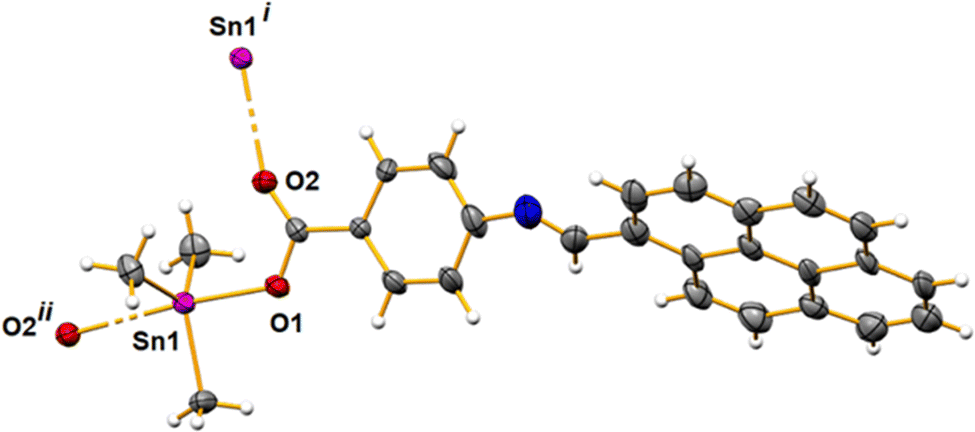 | ||
| Fig. 1 An ellipsoid plot of polymer 1 drawn at a 30% probability level, with a partial atom labeling scheme. Symmetry codes used to generate equivalent atoms: (i) −x, −1/2 + y, 1.5 − z; (ii) −x, 1/2 + y, 1.5 − z. | ||
 | ||
| Fig. 2 Ball and stick representation of the 1D structure of polymer 1 running along the crystallographic b-axis. | ||
In vitro DNA binding studies
 | ||
| Fig. 3 Absorption spectra at fixed concentration of the organotin complexes (a) 1 and (b) 2 (80 μM) and upon increasing the concentration of the CT DNA from 0–100 μM in phosphate buffer, pH 7.4 at 25 °C. | ||
Steady-state fluorescence study
We employed fluorescence spectroscopy to further analyze the binding interaction of the organotin 1 and 2 complexes with CT DNA. The CT DNA is weakly emissive and the organotin complex emits significant fluorescence when excited at 260 nm. The organotin 1 and 2 complexes were titrated with increasing concentration of the CT DNA from 0-100 μM keeping the concentration of organotin complexes constant (80 μM) in phosphate buffer at pH 7.4 at 25 °C (Fig. 4). The spectra depicted in Fig. 4 showed the quenching of the fluorescence intensity of organotin 1 and 2 complexes upon concomitant increase of the CT DNA concentration, thus ascertaining CT-DNA–organotin complex formation (Fig. 4(a) for 1 and Fig. 4(b) for 2). However, the fluorescence intensity is not decimated and thus the complete intercalation is ruled out, but partial intercalation might be possible with electrostatic interaction along the phosphate ends of the CT DNA by the Sn(IV) cation, which opens up the grooves for binding with the complex.65,66 | ||
| Fig. 4 Fluorescence spectra of the organotin complexes (a) 1 and (b) 2 (at a fixed concentration of 80 μM) and upon increasing the concentration of the DNA from 0-100 μM in phosphate buffer, at pH 7.4 at 25 °C. | ||
To quantify and ascertain the mechanism of interaction of DNA with organotin complexes, we further evaluated the Stern–Volmer constant ‘Ksv’ (Fig. S11a for 1 and S11b for 2, ESI†) and biomolecular quenching constant ‘kq’, using equations given below:66,67
| F0/F = 1+ Ksv [CT DNA] |
| kq = Ksv/τ0 |
Furthermore, we have evaluated the binding constant Kb for the organotin complexes 1 and 2 with CT DNA, using the below equation:
log(F0 − F/F) = logKb + n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) log[CT DNA] log[CT DNA] |
The Kb values obtained were found to be 0.20 × 103 M−1 (1) and 0.23 × 103 M−1 (2) (Fig. S11(c) and (d) for 1 and 2, ESI† respectively). These results are consistent with the findings obtained from UV-vis spectral studies.
In vitro cytotoxic activity
The anticancer properties of benzoate-derived organotin(IV) complexes have been extensively studied by our research group.70 Building upon our previous work, we conducted in vitro cytotoxicity assays of the organotin(IV) compounds 1 and 2 against human lung (A549) cancer cell lines to assess their potential as chemotherapeutic agents (Fig. 5). The MTT assay was employed, with 0.1% DMSO serving as the control. Both synthesized organotin(IV) compounds 1 and 2 demonstrated a concentration-dependent cytotoxic effect. Notably, compound 2 exhibited a higher cytotoxicity compared to compound 1. Fig. 3 presents images of treated and untreated A549 cancer cells with compounds 1 and 2 at a concentration of 5 μM, along with the corresponding concentration-dependent results graph (0.1, 1 and 5 μM). The higher cytotoxicity of compound 2 can be attributed to the presence of the triphenyl group, which enhances the structural activity relationship (SAR). The planarity of the phenyl groups facilitates interaction with DNA base pairs through intercalation, ultimately leading to apoptosis.71,72 The metal centre “Sn” of the tin(IV) compounds possesses a well-established affinity for the phosphodiester group of the DNA backbone, thus disrupting DNA repair in the presence of the enzyme phosphodiesterase.73 Moreover, tin compounds have been known to interact with glycoproteins and other cellular proteins, which are potential biomarkers of cancer.74,75 The encouraging outcomes displayed by compound 2 against A549 cancer cells have motivated us to delve further into our research, prompting us to conduct a scratch assay for evaluating cancer cell migration. | ||
| Fig. 5 Images of A549 cancer cells upon treatment with compounds 1 (5 μM) and 2 (5 μM) after 0 h and 24 h and untreated cells (0.1% DMSO, as control). Bar graph displays concentration dependent cytotoxicity of the two compounds 1 and 2 along with DMSO as control. Data are represented as means of three independent experiments. Bar shows means ± standard error. * denotes significant difference at p ≤ 0.05 with untreated control. | ||
Scratch assay: cell migration of A549 cells
In cancer progression, one of the most visible features is cell–cell interaction or cell migration.41 Thus, to study this phenomenon, we performed a scratch assay, a robust and frequently used technique (Fig. 6). The study was carried out against the A549 cancer cell monolayer with passage or scratch, untreated (control) and treated with 1 and 2 (0.1 μM), and the cells were observed at 0 h, 24 h and 48 h (Fig. 6). The control image displays cell–cell interaction and thus migration is evident with the appearance of cells in the passage or scratch marked (yellow lines gap). Treating the A549 cells with 1 and 2 stopped the migration. However, compound 2 produced more potential results even after 48 h. Thus, the scratch assay also supports the potential ability of 2 as compared to 1 to restrict cell migration. Agreeing on the disruption of the cytoskeleton rearrangement in the migration of the cells, it is fair to interpret that the organotin compounds have hampered this process.76,77 | ||
| Fig. 6 Images showing the A549 lung cancer cell migration or interaction: untreated (0.1% DMSO) as control and treated with compounds 1 and 2 after an interval of 0 h, 24 h and 48 h, respectively. | ||
Up/down regulation of MMP-2 and TGF-β genes
The versatile TGF-β (pleiotropic cytokine, transforming growth factor-β) plays a crucial role in regulating diverse cellular processes viz, cell proliferation, angiogenesis, metastasis, and immunosuppression.78 A biphasic effect on tumour growth is exhibited by TGF-β; it impedes carcinogenesis as well as early tumour suppression. However, in advanced aggressive-stage tumours, growth factor accelerates tumour progression.78 Thus, we investigated the effect of organotin compounds 1 and 2 on mRNA expression in lung cancer A549 cells (Fig. 7). TGF-β and MMPs work bidirectionally to activate each other. Our results indicated that A549 cells treated with 2 significantly reduced the expression of MMP-2 and TGF-β mRNA levels (figure). 1 did not show much difference in expression levels of MMP-2 and TGF-β mRNA. | ||
| Fig. 7 Histogram representing the TGF-β and MMP-2 gene expression upon treatment with compounds 1 and 2. | ||
The stability of the complexes was evaluated in a buffer solution (phosphate buffer of pH 7.4 with 0.1% DMSO) using absorption spectroscopy. Spectra were collected at 0, 24 and 48 h. Although a decrease in absorbance was noted, there were no significant changes observed in the spectra, confirming the stability of the compounds in the solutions (Fig. S12, ESI†).
Theoretical calculations
The geometries of 1 and 2 were optimized at the DFT level employing the ZORA relativistic approximation and C-PCM model to account for the solvent (water) effects. The optimized structures of 1 and 2 are depicted in Fig. S10, ESI.† In both cases, the frontier HOMO and LUMO orbitals are strongly delocalized over the pyrene-carboxylate Schiff base ligand (Fig. 8). The HOMO–LUMO gaps of 1 and 2 are almost equal, being 3.227 and 3.203 eV, respectively. The carboxylate group is monodentately coordinated in both 1 and 2 with the Sn–O bond distances of 2.102 and 2.098 Å, respectively. The analysis of the electron density in terms of the QTAIM theory79,80 and Reduced Density Gradient81 suggested the presence of a very weak attractive interaction between the uncoordinated oxygen atom of the carboxylate group and the tin atom in 1, distanced at 2.970 Å (Fig. 9). However, the analysis of the Laplacian of electron density did not reveal a bond critical point between this pair of atoms (Fig. 6). With the methyl groups replaced by phenyl groups (complex 2), the d(Sn⋯O) distances for coordinated and uncoordinated oxygen atoms become shorter, 2.098 and 2.784 Å, respectively. This shortening can be explained by the appearance of weak C–H⋯O hydrogen bonds [d(H⋯O) = 2.591 and 2.706 Å] between the uncoordinated oxygen atom and aromatic H atoms of the phenyl groups. The electron densities ρ(rBCP) at the (3, –1) bond critical points between O and H atoms (Fig. S13, ESI†) constitute 0.80 × 10−2 and 0.66 10−2 a.u. According to the model developed by Emamian and Lu,82 these ρ(rBCP) values correspond to binding energies of −1.03 and −0.74 kcal mol−1, respectively.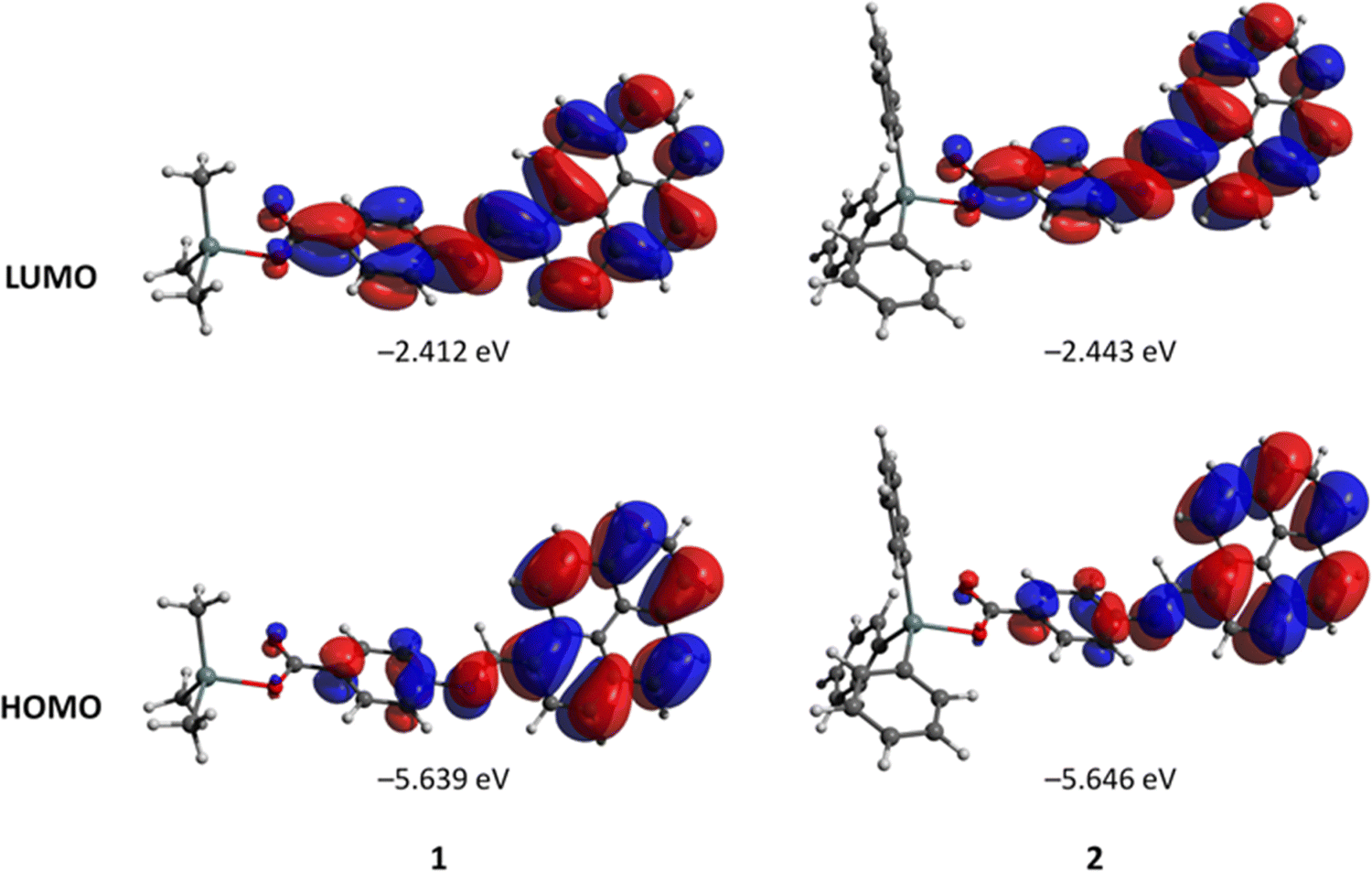 | ||
| Fig. 8 Plots and energies of the frontier molecular orbitals (shown at the 0.02 a.u. isosurface) of 1 and 2, calculated at the B3LYP/ZORA-def2-TZVP(SARC-ZORA-TZVP) level and based on atom coordinates optimized at the same level. Colour scheme: Sn, light grey; O, red; N, blue; C, grey; H, white. | ||
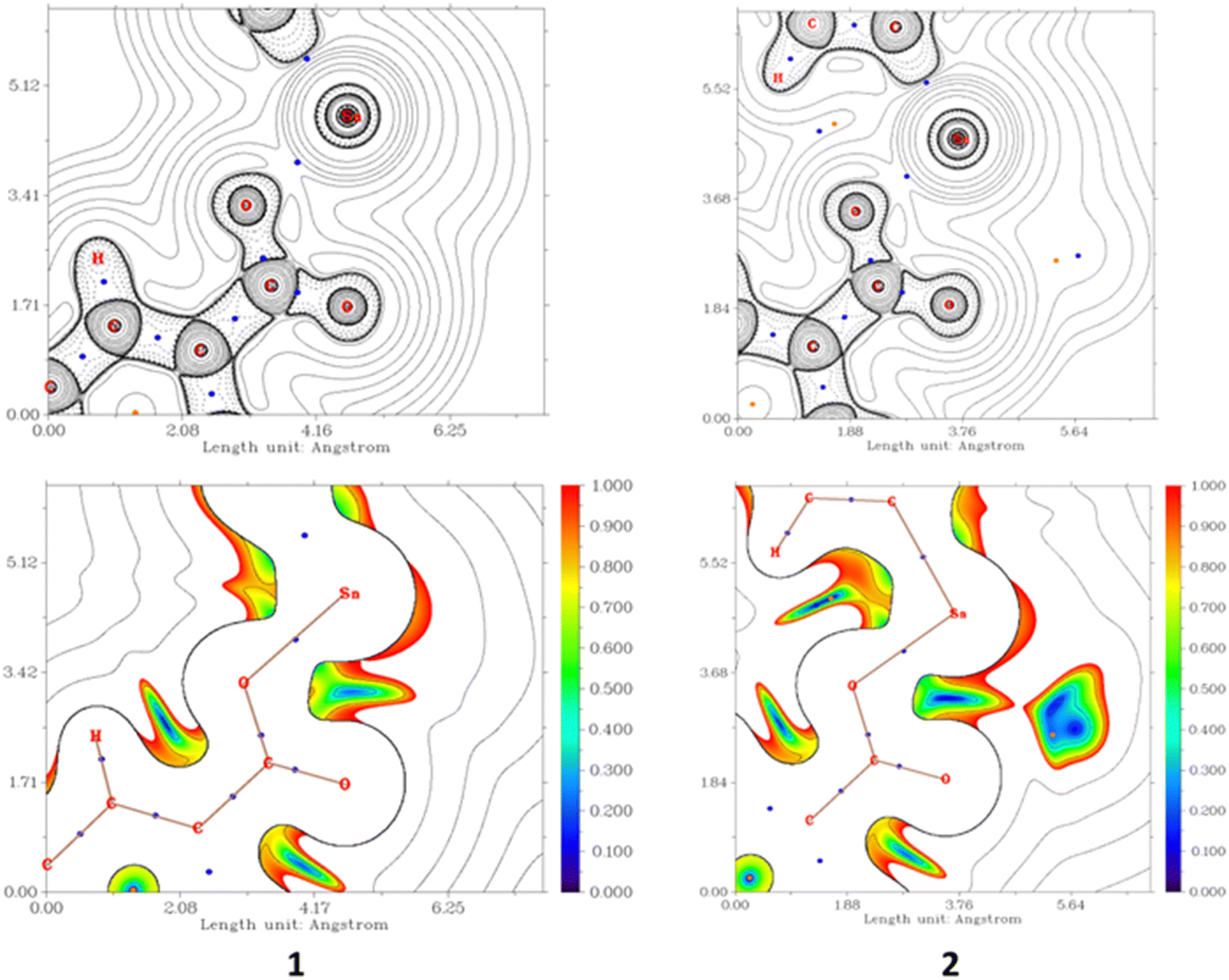 | ||
| Fig. 9 2D plots of the Laplacian of the electron density, ∇2ρ(r) (top), and RDG function for the same projections (Fig. S12, ESI†), showing weak contacts between Sn and uncoordinated O atoms in 1 and 2. | ||
Molecular docking studies
The docking analysis of the tin(IV) compounds 1 and 2 with MMP-2 (PDB ID: 1rtg) provides valuable insights into their binding interactions and potential implications for anticancer activity (Fig. 10(a) and 11(a)). These findings align with the observed differences in their cytotoxic effects in A549 lung cancer cell lines. Both compounds 1 and 2 displayed favourable binding energies (−269.2 kcal mol−1 for 1 and −295.05 kcal mol−1 for 2), suggesting their potential as inhibitors of MMP-2 activity. However, compound 2 exhibited somewhat superior binding characteristics compared to compound 1. From the analysis of the 2D diagram (Fig. 10(b)), we observed that compound 1 exhibited specific molecular interactions within the active site pocket of the receptor. Notably, it formed a hydrogen bond with the Asp569 residue at a distance of 2.45 Å. Additionally, compound 1 established favourable interactions, including π–alkyl bonds with Val565, Ala584, and Arg567, π–anion bonds with Asp521 and a C–H bond with Gln566. | ||
| Fig. 10 Docking representation of 1 with 1rtg (a). 2D diagram representing the links between 1 and protein, showing its interaction with amino acids aspartic acid (Asp521, 561), arginine (Arg567), alanine (Ala584), glutamine (Gln566) and valine (Val565) (b). | ||
 | ||
| Fig. 11 Docking representation of 2 with 1rtg (a). 2D diagram representing the links between 2 and protein, showing its interactions with amino acids aspartic acid (Asp521, 564, 569), arginine (Arg567), alanine (Ala584), lysine (587) and valine (Val565) (b). | ||
On the other hand, docking analysis of compound 2 (Fig. 11(b)) revealed intriguing interactions within the active site pocket. Compound 2 demonstrated π–anionic and π–cationic interactions with amino acid residues such as Asp521, Asp564, and Asp587. Furthermore, Arg567 formed π–Sigma and π–alkyl interactions with Ala584 and Val565.
Overall, these findings provide valuable insights into the molecular interactions of both compounds within the active site pocket of MMP-2. Such interactions contribute to the stabilization of the complexes and indicate the potential inhibitory activity of these compounds towards MMP-2.
In addition to investigating the interactions with MMP-2, we also performed docking simulations to explore the binding interactions of compounds 1 and 2 with TGF-β (PDB ID: 1VJY) (Fig. 12 and 13). In the docking study with TGF-beta, both compounds 1 and 2 demonstrated binding interactions with the receptor-binding pocket of TGF-β, albeit with differing affinities and modes of interaction. The binding energy of 1 was found to be −289.2 kcal mol−1, while for 2 it was −331.3 kcal mol−1.
 | ||
| Fig. 12 Docking representation of 2 with 1VJY (a). 2D diagram representing the links between 2 and protein, showing its interactions with amino acids glycine (Gly214), valine (Val219), threonine (Thr375), aspartic acid (Asp435), isoleucine (Ile211), leucine (Leu340), alanine (Ala350), lysine (Lys232, 337, 376) and arginine (Arg377) (b). | ||
 | ||
| Fig. 13 Docking representation of 1 with 1VJY (a). 2D diagram representing the links between 1 and protein, showing the interaction of 1 with amino acids aspartic acid (Asp521, 564, 569), arginine (Arg567), alanine (Ala584), lysine (587) and valine (Val565) (b). | ||
Compound 2 displayed stronger binding interactions with key residues involved in TGF-beta receptor recognition, such as Gly214 (hydrogen bond interaction), Val219 and Thr375 (π–Sigma), Asp435 (π–anionic), Ile211, Leu340, Ala350, Lys232, 337, 376 and Arg377 (π–alkyl) (Fig. 10(b)). These interactions correlated well with the significant reduction in TGF-beta gene expression observed in our gene expression study. The favourable binding affinity of compound 2 suggests its potential as a modulator of TGF-β signalling, possibly inhibiting TGF-β binding to its receptors and downstream signalling pathways.
Although compound 1 also exhibited binding interactions with the receptor-binding pocket of TGF-β, they were comparatively weaker. This weaker binding affinity may explain its less pronounced effect on reducing TGF-β gene expression in our study. It showed binding interactions with residues involved in TGF-β receptor recognition, such as Lys337 (hydrogen bonding interaction) and Val219 (π–alkyl) (Fig. 12). Nonetheless, it is worth noting that even moderate interactions with TGF-β could still have functional implications, and further investigations are warranted to understand the precise mechanisms by which compound 1 affects TGF-β signalling.
The docking results provide valuable insights into the binding interactions of tin(IV) compounds 1 and 2 with MMP-2 and TGF-β, which complement our gene expression study results. In the docking study, compound 2 exhibited stronger binding interactions with the active site residues of MMP-2. These interactions suggest that compound 2 has the potential to inhibit the activity of MMP-2, a protein known to play a crucial role in cancer progression and metastasis. By blocking MMP-2 activity, compound 2 may disrupt the degradation of extracellular matrix components and inhibit cancer cell invasion and migration.
Furthermore, compound 2 demonstrated favourable binding interactions with key residues involved in TGF-beta receptor recognition. These interactions support the potential of compound 2 to modulate TGF-beta signalling, as evidenced by the reduced expression of the TGF-beta gene observed in the experimental study. Modulating TGF-β signaling pathways can have significant implications for cancer progression, as TGF-β is known to regulate cell proliferation, migration, and immune evasion.
Compound 2 demonstrated superior cytotoxic activity in A549 lung cancer cell lines, which is correlated to its ability to significantly reduce the expression of MMP-2 and TGF-beta genes. The docking study provides a molecular rationale for these observed effects, shedding light on the potential interactions and mechanisms of action of compound 2 with MMP-2 and TGF-β. Compound 2 may disrupt multiple key processes involved in cancer development and progression, including tumour growth, invasion, metastasis, and immune evasion.
On the other hand, compound 1 (Fig. 13), although showing some anticancer activity, demonstrated weaker effects on reducing the expression of MMP-2 and TGF-β genes. The docking results provide insights into the potential reasons for these observations, suggesting that compound 1 may have weaker binding interactions with the active sites of MMP-2 and TGF-β, leading to a reduced impact on their activities. Thus the results obtained from docking studies are in accord with gene expressions.
Conclusions
We successfully synthesized and characterized two novel pyrene-appended Schiff base triorganotin(IV) compounds with three methyl or phenyl ligands designated as 1 and 2, respectively.The cytotoxicity assays against human lung (A549) cancer cell lines revealed that both compounds exhibited concentration-dependent cytotoxic effects, with compound 2 displaying higher cytotoxicity compared to 1. Absorption and fluorescence spectroscopy revealed a strong interaction of organotin complexes 1 and 2 with CT DNA. The results obtained from the spectroscopic studies suggested electrostatic interaction and groove binding propensity, emphasizing the preferential selectivity of Sn(IV) for DNA phosphate ends. This higher cytotoxicity of compound 2 can be attributed to the presence of the triphenyl group, which is known to enhance the structure activity relationship (SAR) by facilitating interactions with DNA base pairs through intercalation. Furthermore, scratch assays demonstrated that both compounds effectively inhibited cancer cell migration, with compound 2 exhibiting more potent inhibitory effects even after 48 h. This inhibition of cell migration is attributed to the disruption of cytoskeleton rearrangement by the organotin compounds.
To gain insights into the molecular mechanisms underlying their anticancer activity, we performed gene expression studies and molecular docking simulations. Both compounds showed binding interactions with MMP-2 and TGF-β, key proteins implicated in cancer progression. Compound 2 exhibited stronger binding characteristics with such proteins, providing a molecular rationale for its superior cytotoxic and antimigratory effects. These interactions suggest that compound 2 has the potential to disrupt multiple critical processes involved in cancer development and progression. These findings underscore the potential of these organotin compounds as candidates for further development as chemotherapeutic agents for cancer treatment. Further investigations are warranted to elucidate the precise mechanisms of action and to evaluate their in vivo efficacy, moving us closer to the development of promising anticancer drugs.
Author contributions
Anup Paul: conceptualization, formal analysis, investigation, methodology, project administration, supervision, writing – original draft, reviewing and editing; Rais Ahmad Khan: conceptualization, biological analysis, investigation, writing – original draft, reviewing and editing; Gouse Mohiddin Shaik: biological analysis, investigation, writing – original draft; Jilani. P. Shaik: biological analysis, investigation; Dmytro S. Nesterov: part of theoretical analysis, writing – original draft; M. Fátima C. Guedes da Silva: formal analysis, reviewing and editing; and Armando Pombeiro: writing – reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. P. is grateful to the FCT and IST, Portugal, for providing financial support through “DL/57/2017” (contract no. IST-ID/197/2019). R. A. K. gratefully acknowledges the Researchers Supporting Project (project number, RSP2023R400), King Saud University, Riyadh, KSA. This work has also been partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal, through projects UIDB/00100/2020, UIDP/00100/2020, and LA/P/0056/2020 of Centro de Química Estrutural. D. S. acknowledges FCT for providing financial support (contract no. IST-ID/086/2018). The authors also acknowledge the Portuguese NMR Network (IST-UL Centre) for providing access to the NMR facility and the IST Node of the Portuguese Network of Mass-spectrometry for ESI-MS measurements.References
- F. T. Vieira, G. M. de Lima, J. R. da, S. Maia, N. L. Speziali, J. D. Ardisson, L. Rodrigues, A. Correa and O. B. Romero, Eur. J. Med. Chem., 2010, 45, 883–889 CrossRef CAS PubMed
.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Theranostics, 2022, 12, 2115–2132 CrossRef CAS PubMed
- S. Alassadi, M. J. Pisani and N. J. Wheate, Dalton Trans., 2022, 51, 10835–10846 RSC
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC
- R. L. Lucaciu, A. C. Hangan, B. Sevastre and L. S. Oprean, Molecules, 2022, 27, 6485 CrossRef CAS PubMed
- T. S. B. Baul, Appl. Organomet. Chem., 2008, 22, 195–204 CrossRef CAS
- O. Hayat, N. Ullah, M. Sirajuddin, M. A. Giardini, J. V. Nguyen, K. R. Francisco, L. J. Liu, Y. U. Sun, S. Maurya, D. McGrosso, D. J. Gonzalez, C. R. Caffrey, A. Debnath and J. L. Siqueira-Neto, Pathogens, 2022, 11, 1424 CrossRef CAS PubMed
- S. N. Syed Annuar, N. F. Kamaludin, N. Awang and K. M. Chan, Front. Chem., 2021, 9, 1–15 Search PubMed
- T. S. Basu Baul, A. Paul, L. Pellerito, M. Scopelliti, A. Duthie, D. De Vos, R. P. Verma and U. Englert, J. Inorg. Biochem., 2012, 107, 119–128 CrossRef CAS PubMed
- S. Hazra, A. Paul, G. Sharma, B. Koch, M. F. C. G. da Silva and A. J. L. Pombeiro, J. Inorg. Biochem., 2016, 162, 83–95 CrossRef CAS PubMed
- W. Jiang, Q. Qin, X. Xiao and Y. Tan, J. Inorg. Biochem., 2022, 232, 111808 CrossRef CAS PubMed
- S. A. Patra, G. Sahu, P. Das Pattanayak, T. Sasamori and R. Dinda, Inorg. Chem., 2022, 61, 16914–16928 CrossRef CAS
- J. Wang, H. Chen, Q. Song, X. Liu, C. Li, H. Wang, C. Li and M. Hong, J. Inorg. Biochem., 2022, 236, 111983 CrossRef CAS PubMed
- I. J. Gómez, K. Ovejero-Paredes, J. M. Méndez-Arriaga, N. Pizúrová, M. Filice, L. Zajíčková, S. Prashar and S. Gómez-Ruiz, Chem. – Eur. J., 2023, 29, e202301845 CrossRef PubMed
- A. Paul, R. A. Khan, G. M. Shaik, J. P. Shaik, A. K. Rai, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Appl. Organomet. Chem., 2023, 37, e7232 CrossRef CAS
- S. Ramzan, S. Rahim, S. T. Hussain, K. B. Holt, J. K. Cockcroft, N. Muhammad, Z. Ur-Rehman, A. Nawaz and S. Shujah, Appl. Organomet. Chem., 2023, 37, e7161 CrossRef CAS
- N. N. Riaz, M. M. Ahmed, M. Kashif, M. Sajid, M. Ali and K. Mahmood, RSC Adv., 2023, 13, 10768–10789 RSC
- A. Paul, S. Hazra, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2020, 930–941 CrossRef CAS
- S. Yu, C. Li, S. Fan, J. Wang, L. Liang and M. Hong, J. Mol. Struct., 2022, 1257, 132585 CrossRef CAS
- Y. M. Ahmed and G. G. Mohamed, Inorg. Chem. Commun., 2022, 144, 109864 CrossRef CAS
- S. Sharma, N. Agnihotri, K. Kumar, S. Sihag, V. Randhawa, R. Kaur, R. Singh and V. Kaur, Appl. Organomet. Chem., 2022, 36, 1–20 Search PubMed
- S. Saroya, S. Asija, N. Kumar, Y. Deswal and J. Devi, J. Indian Chem. Soc., 2022, 99, 100379 CrossRef CAS
- S. Ramzan, S. Shujah, K. B. Holt, Z. ur Rehman, S. T. Hussain, J. K. Cockcroft, N. Malkani, N. Muhammad and A. Kauser, J. Organomet. Chem., 2023, 990, 122671 CrossRef CAS
- H. L. Singh, N. Dhingra and S. Bhanuka, J. Mol. Struct., 2023, 1287, 135670 CrossRef CAS
- S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 235–249 CrossRef CAS
- R. Wang, J. Zhang, G. Cui and L. Tian, Main Group Met. Chem., 2022, 45, 242–254 CrossRef CAS
- C. Pettinari, F. Marchetti, R. Pettinari, A. Cingolani, A. Drozdov and S. Troyanov, J. Chem. Soc., Dalton Trans., 2002, 188–194 RSC
- J. Liu, Y. Lin, M. Liu, S. Wang, Y. Li, X. Liu and L. Tian, Appl. Organomet. Chem., 2019, 33, 1–10 Search PubMed
- M. Gielen, M. Bouâlam, A. Meriem, B. Mahieu, M. Biesemans and R. Willem, Heteroat. Chem., 1992, 3, 449–452 CrossRef CAS
- T. Ali, N. Muhammad, Z. Ali, A. Samad, M. Ibrahim, M. Ikram, S. Rehman, S. Shujah, G. S. Khan, A. Wadood, S. Ali and C. Schulzke, J. Mol. Struct., 2021, 1234, 130190 CrossRef CAS
- P. Debnath, K. S. Singh, T. S. Devi, S. S. Singh, R. J. Butcher, L. Sieroń and W. Maniukiewicz, Inorg. Chim. Acta, 2020, 510, 119736 CrossRef CAS
- R. Kollock, H. Frank, A. Seidel, W. Meinl and H. Glatt, Toxicology, 2008, 245, 65–75 CrossRef CAS PubMed
- B. Babu, T. A. Ali, T. Ochappan, J. Mack, T. Nyokong and M. G. Sethuraman, Photodiagn. Photodyn. Ther., 2021, 33, 102102 CrossRef CAS PubMed
- D. Tzimopoulos, I. Sanidas, A. C. Varvogli, A. Czapik, M. Gdaniec, E. Nikolakaki and P. D. Akrivos, J. Inorg. Biochem., 2010, 104, 423–430 CrossRef CAS PubMed
- M. E. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS
- B. J. R. Lakowicz and G. Weber, Biochemistry, 1973, 12, 4161–4170 CrossRef PubMed
- S. Tabassum, R. A. Khan, F. Arjmand, A. S. Juvekar and S. M. Zingde, Eur. J. Med. Chem., 2010, 45, 4797–4806 CrossRef CAS PubMed
- S. Tabassum, R. A. Khan, F. Arjmand, M. Aziz, A. S. Juvekar and S. M. Zingde, Carbohydr. Res., 2011, 346, 2886–2895 CrossRef CAS PubMed
- A. H. Cory, T. C. Owen, J. A. Barltrop and J. G. Cory, Cancer Commun., 1991, 3, 207–212 CrossRef CAS PubMed
- C. C. Liang, A. Y. Park and J. L. Guan, Nat. Protoc., 2007, 2, 329–333 CrossRef CAS PubMed
- S. M. Hartig, Curr. Protoc. Mol. Biol., 2013, 1–12 Search PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, 1–15 Search PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- D. A. Pantazis and F. Neese, Theor. Chem. Acc., 2012, 131, 1–7 Search PubMed
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC
- S. Bai and M. Barbatti, J. Chem. Theory Comput., 2019, 15, 1503–1513 CrossRef CAS PubMed
- C. Van Wüllen, J. Chem. Phys., 1998, 109, 392–399 CrossRef
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 34112 CrossRef PubMed
- Avogadro: an open-source molecular builder and visualization tool. Version 1.2. https://avogadro.cc/
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek, G. R. Hutchison and G. R. Avogadro, J. Cheminf., 2012, 4, 17 CAS
- A. Paul, P. Singh, M. L. Kuznetsov, A. Karmakar, M. F. C. Guedes da Silva, B. Koch and A. J. L. Pombeiro, Dalton Trans., 2021, 50, 3701–3716 RSC
- A. Paul, S. Anbu, G. Sharma, M. L. Kuznetsov, B. Koch, M. F. C. Guedes Da Silva and A. J. L. Pombeiro, Dalton Trans., 2015, 44, 19983–19996 RSC
- D. Bandyopadhyay, J. C. Granados, J. D. Short and B. K. Banik, Oncol. Lett., 2012, 3, 45–49 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
- T. S. Basu Baul, S. Basu, D. De Vos and A. Linden, Invest. New Drugs, 2009, 27, 419–431 CrossRef CAS PubMed
- T. S. Basu Baul, I. Longkumer, A. Duthie, P. Singh, B. Koch and M. F. C. Guedes Da Silva, Dalton Trans., 2018, 47, 1993–2008 RSC
- M. Nath and S. Goyal, Main Group Met. Chem., 1996, 75–102 CAS
- U. Sair and A. Thakur, Mater. Today: Proc., 2021, 50, 1862–1866 Search PubMed
- T. S. Basu Baul, M. R. Addepalli, A. Duthie, P. Singh, B. Koch, H. Gildenast, U. Englert, I. Rojas-León and H. Höpfl, Appl. Organomet. Chem., 2021, 35, e6080 CrossRef CAS
- V. Barba, E. Vega, R. Luna, H. Höpfl, H. I. Beltrán and L. S. Zamudio-Rivera, J. Organomet. Chem., 2007, 692, 731–739 CrossRef CAS
- R. A. Khan, S. Yadav, Z. Hussain, F. Arjmand and S. Tabassum, Dalton Trans., 2014, 43, 2534–2548 RSC
- S. I. Parvin, M. K. Mandal, P. Gopi, S. Singh, M. R. Khan, P. Pandya, M. M. Islam and H. A. R. Gazi, J. Biomol. Struct. Dyn., 2023, 41, 10944–10956 CrossRef CAS PubMed
- I. Hussain, S. Fatima, S. Siddiqui, S. Ahmed and M. Tabish, Spectrochim. Acta, Part A, 2023, 41, 10944–10956 Search PubMed
- F. A. Qais, K. M. Abdullah, M. M. Alam, I. Naseem and I. Ahmad, Int. J. Biol. Macromol., 2017, 97, 392–402 CrossRef PubMed
- C. Albrecht, Anal. Bioanal. Chem., 2008, 390, 1223–1224 CrossRef CAS
- S. Tabassum, R. A. Khan, F. Arjmand, S. Sen, J. Kayal, A. S. Juvekar and S. M. Zingde, J. Organomet. Chem., 2011, 696, 1600–1608 CrossRef CAS
- E. N. M. Yusof, T. B. S. A. Ravoof and A. J. Page, Polyhedron, 2021, 198, 115069 CrossRef CAS
- R. A. Haque, M. A. Salam and M. A. Arafath, J. Coord. Chem., 2015, 68, 2953–2967 CrossRef CAS
- D. M. Templeton, Anal. Bioanal. Chem., 2003, 375, 1062–1066 CrossRef CAS PubMed
- C. Pellerito, L. Nagy, L. Pellerito and A. Szorcsik, J. Organomet. Chem., 2006, 691, 1733–1747 CrossRef CAS
- J. S. Casas, E. E. Castellano, M. D. Couce, J. Ellena, A. Sánchez, J. L. Sánchez, J. Sordo and C. Taboada, Inorg. Chem., 2004, 43, 1957–1963 CrossRef CAS PubMed
- F. Xu, C. Piett, S. Farkas, M. Qazzaz and N. I. Syed, Mol. Brain, 2013, 6, 1–15 CrossRef PubMed
- R. J. Cooper and N. Spitzer, Neurotoxicology, 2015, 48, 231–238 CrossRef CAS PubMed
- E. S. Kim, Y. W. Sohn and A. Moon, Cancer Lett., 2007, 252, 147–156 CrossRef CAS PubMed
-
C. F. Matta and R. J. Boyd, An Introduction to the Quantum Theory of Atoms in Molecules, 2007 Search PubMed
- G. Saleh, C. Gatti, L. Lo Presti and J. Contreras-García, Chem. – Eur. J., 2012, 18, 15523–15536 CrossRef CAS PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S12 containing ATR-IR, 1H, 13C-NMR, ESI-MS and theoretical calculations, Table S1 presenting the crystallographic parameters of 1, and Tables S2 and S3 presenting the coordinates of the optimized structures. CCDC 2296017. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj04401g |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |