
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of chiral hexynones for use as precursors to native photosynthetic hydroporphyrins†
Khiem
Chau Nguyen‡
 a,
Duy T. M.
Chung‡
a,
Phattananawee
Nalaoh
b and
Jonathan S.
Lindsey
*a
a,
Duy T. M.
Chung‡
a,
Phattananawee
Nalaoh
b and
Jonathan S.
Lindsey
*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC 27695-8204, USA. E-mail: jlindsey@ncsu.edu
bDepartment of Chemistry, University of Tennessee, Knoxville, TN 37996, USA
First published on 11th January 2024
Abstract
A planned total synthesis of photosynthetic tetrapyrrole macrocycles installs essential stereochemical features in early precursors via established asymmetric methodology. Key building blocks are dihydrodipyrrins that contain a chiral pyrroline unit, for which chiral hex-5-yn-2-ones are valuable precursors. Chiral hex-5-yn-2-ones bearing diverse functional groups at the 1-position were sought that correspond to the chiral pyrroline units (rings B and D) of bacteriochlorophyll a. Three main results are reported herein. First, (2R,3R)-3-ethyl-2-methylpent-4-ynoic acid, prepared via a Schreiber-modified Nicholas reaction, was converted to the analogous chiral Weinreb pentynamide. The latter was treated with tributyltin-one-carbon synthons to create a 1-hydroxyhex-5-yn-2-one scaffold containing the two requisite stereocenters and with the newly introduced hydroxy group in free form or protected as the Me, MOM, THP, MEM, Bn, or SEM derivative. Second, improved routes to intermediates on the path to the pre-B compound, (3R,4R)-1,1-dimethoxy-3-ethyl-4-methylhex-5-yn-2-one, were developed, in part by derivatization of the chiral Weinreb pentynamide. Third, one (semisynthetic) chiral hexynone bearing a native phytyl substituent, a near-universal precursor to ring D of photosynthetic hydroporphyrins, was prepared in 2.5 mmol quantity. The synthesis and manipulation were achieved while maintaining stereochemical integrity in these somewhat densely functionalized chiral hexynones (three functional groups and two stereocenters in a six-carbon scaffold). Altogether, 22 new compounds have been prepared, including 15 chiral hexynones for studies in (bacterio)chlorophyll synthetic methodology. The chiral hexynones were prepared in quantities ranging from 0.11–12 mmol as required for exploratory and/or preparative studies.
Introduction
Chlorophyll a and bacteriochlorophyll a, the chief photoactive molecules of oxygenic and anoxygenic photosynthesis, respectively, are chiral molecules (Chart 1).1–4 The stereocenters are located in the (saturated) pyrroline units, ring E, and in the phytyl chain. Chlorophyll a contains one pyrroline unit (ring D; two stereocenters) whereas bacteriochlorophyll a contains two pyrroline units (rings B and D; four stereocenters). Both macrocycles also contain a common stereocenter in ring E, but the relatively facile epimerization at the β-ketoester causes the stereochemical configuration at this site to be self-setting.5–7 While structural variation occurs across the family of photosynthetic tetrapyrroles,8 the chiral ring D is invariably identical to that of (bacterio)chlorophyll a. The chirality of the macrocycles presents challenges to synthesis. | ||
| Chart 1 Key photosynthetic pigments. | ||
A retrosynthesis9 for bacteriochlorophyll a is shown in Scheme 1: stereochemically defined AD (I) and BC (II)10 halves undergo Knoevenagel condensation followed by double-ring closure, which entails Nazarov cyclization (to form ring E), electrophilic aromatic substitution (SEAr), and elimination of methanol. The incorporation of phytol and magnesium completes the synthesis. The macrocycle-forming strategy has been demonstrated with chemically robust, gem-dimethyl-substituted dihydrodipyrrins (both AD and BC halves)11 and extended to use of a model trans-dialkyl-substituted dihydrodipyrrin AD half,9,12,13 but has not yet been extended to create the native macrocycles. The success of this strategy – where stereochemical features are installed early in the synthesis – requires access to chiral precursors to rings B and D, each of which contains a trans-dialkyl-substituted pyrroline ring. The route also requires a one-carbon functional group at the 1-position of each dihydrodipyrrin; moreover, the two groups must be non-identical for selective joining to give the macrocycle. The two groups employed to date are formyl (attached to ring D) and dimethoxymethyl (attached to ring B). The development of access to hydrodipyrrins bearing distinct one-carbon groups at the 1-position – for reaction and protection – is an essential aspect of methodology studies in this domain.14
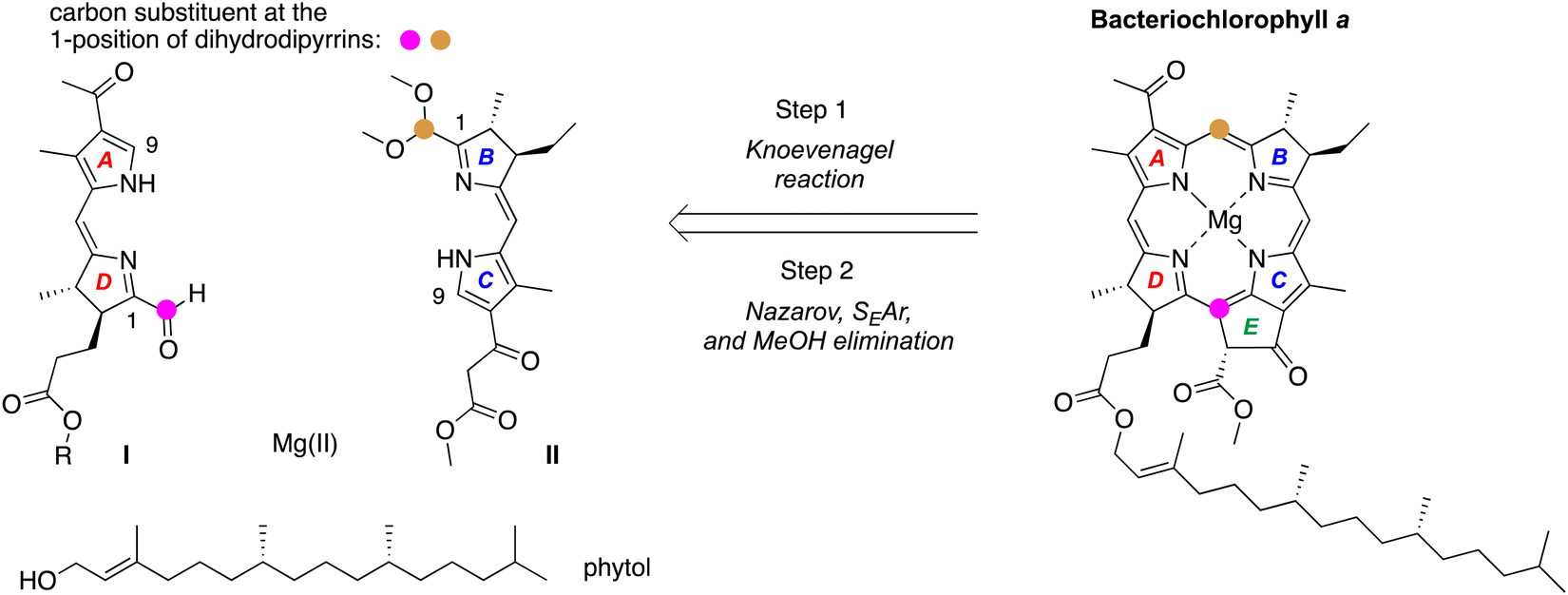 | ||
| Scheme 1 Retrosynthesis of bacteriochlorophyll a. | ||
A route for formation of the pyrroline unit concomitantly with the dihydrodipyrrin was developed through extensive work by Jacobi and coworkers (Scheme 2, top).15–21 The Pd-mediated coupling of a 2-iodopyrrole (III) with a chiral pentynoic acid (IV) is followed by Petasis methenylation22 of lactone V, hydration and Paal–Knorr ring closure23,24 of ene-lactone-pyrrole VI, and Riley oxidation25 of dihydrodipyrrin VII to afford the dihydrodipyrrin-carboxaldehyde AD half (VIII). Two chiral pentynoic acids have been carried through the Jacobi pathway to form dihydrodipyrrins; the structures include R = ethyl (IVa)9,26 and R = 3-(tert-butyldiphenylsilyloxy)propyl (IVb).26 The chiral pentynoic acids (IVa,b) were prepared by use of the Schreiber-modified Nicholas reaction.27–31
 | ||
| Scheme 2 Syntheses of chiral dihydrodipyrrins. | ||
The pentynoic acid route was not viable for the synthesis of the BC half (II) of bacteriochlorophyll a given the presence of the β-ketoester unit. A route that sidesteps such problems was developed very recently (Scheme 2, bottom).10 The route extends the pioneering work of Jacobi with introduction of the following key steps: (1) Sonogashira coupling32,33 of a chiral hex-5-yn-2-one (IX) that bears the requisite 1,1-dimethoxy groups with a 2-iodopyrrole (X); (2) anti-Markovnikov hydration34,35 of the alkyne (XI); and (3) Paal–Knorr ring closure of the diketone (XII). The stereochemical configuration of chiral hexynone IX was also created through use of a Schreiber-modified Nicholas reaction.27–31 The use of a chiral hexynone, which bears a pre-installed one-carbon moiety, thereby bypasses the Petasis methenylation and Riley oxidation required upon use of a chiral pentynoic acid.
To expand the chemistry for preparing native macrocycles and analogues beyond that accessible via the single chiral hexynone (IX) heretofore examined, a family of chiral hex-5-yn-2-ones was prepared and is reported herein. Three objectives were sought at the outset of this work.
(1) For fundamental studies of dihydrodipyrrin coupling reactions, chiral hex-5-yn-2-ones were sought that are equipped with distinct reactive handles at the 1-position (Z = hydroxymethyl and diverse protected hydroxymethyl groups, versus the 1,1-dimethoxymethyl moiety in IX) as is shown in Scheme 3. The 1-position of the chiral hexynone for ring B (or D) gives rise to position 5 (or 15) in the macrocycle. The protected hydroxymethyl group at the hexynone 1-position was highly desired to enable isolation of the intermediate following Nazarov cyclization yet still be able to carry out a subsequent macrocyclization process; such interruption of the double-ring closure process was not possible given the reactivity of the 1,1-dimethoxymethyl group under the conditions for the Nazarov cyclization.
 | ||
| Scheme 3 Rationale for access to diverse chiral hexynones. | ||
(2) A chiral hex-5-yn-2-one was sought that is equipped with the native phytyl propanoate group of ring D (Scheme 3).
(3) Selected precursors were sought at reasonable scale because an objective is to obtain target photosynthetic tetrapyrroles for diverse studies. The desired scale of target photosynthetic hydroporphins is ∼10 μmol, which corresponds to ∼9 mg of bacteriochlorophyll a (911.5 Da). The quantity of A–D constituents is at least 100 μmol, and the quantity of precursors such as chiral hexynones is at least 1–10 mmol to accommodate losses during multistep syntheses and forays in the pursuit of diverse target compounds. The BC half (II) has been prepared at the 0.74 mmol scale (279 mg).10 Here, a chiral hexynone bearing substituents corresponding to ring D was prepared in a quantity of 2.5 mmol, and eight other chiral hexynones corresponding to ring B and bearing diverse 1-substituents were prepared for fundamental studies. In pursuit of the aforementioned objectives, several improved approaches also emerged that facilitate access to key intermediates on the path to chiral hexynone IX, which comprises the pre-B moiety.
Results and discussion
Reconnaissance
Key features of the recent synthesis of compound IX10 are sketched in Scheme 4. Briefly, the Schreiber-Nicholas adduct 1 (from the Schreiber-modified Nicholas reaction27–31) was deprotected with cerium ammonium nitrate (CAN) to give the alkyne 2, a chiral pentynamide. Reduction of the latter with LiAlH4 gave the corresponding chiral pentynol 3. Oxidation with Dess–Martin periodinane (DMP) reagent gave the desired chiral pentynal 4. Addition of 2-lithio-1,3-dithiane gave alcohol 5, which underwent thia-oxa exchange with methanol in the presence of phenyliodine bis(trifluoroacetate) (PIFA) to give 6. Oxidation of the alcohol with DMP followed by removal of the TMS group with AgNO3 gave the target chiral hex-5-yn-2-one IX. The latter constitutes a precursor to ring B of bacteriochlorophyll a. | ||
| Scheme 4 Prior synthesis10 of chiral hexynone IX. | ||
Exploration of the derivatization of Schreiber-Nicholas chiral pentynamide 1
The prior chemistry shown in Scheme 4 afforded chiral hexynone IX, the pre-B compound. Here, means to derivatize the chiral pentynamide 1 were sought to gain more direct access to chiral pentynal 4 and to open the door to the formation of diverse chiral, 1-functionalized hex-5-yn-2-ones. The following paths were pursued (Scheme 5).(1) Direct treatment of Schreiber-Nicholas adduct 1 with diisobutylaluminum hydride (DIBAL-H)36 gave aldehyde 4-Co (75% yield). Subsequent deprotection with CAN to obtain the target pentynal 4 was precarious; at room temperature both the aldehyde 4 and the carboxylic acid 8 were obtained. The same process with strict maintenance of the reaction mixture at 0 °C showed only the aldehyde 4 without the carboxylic acid 8. Aldehyde 4 upon purification was found to spontaneously undergo oxidation to 8 after several hours of exposure to air; hence the compound should be used immediately or stored appropriately under argon at −20 °C at most for a few days (Scheme 5, upper left). In one case, the crude reaction mixture upon treatment with CAN at 0 °C was immediately treated with propane-1,3-dithiol and magnesium bromide etherate37 to give the dithioacetal of the pentynal (9) in 76% yield for the two steps.
 | ||
| Scheme 5 Exploratory derivatization of Nicholas-Schreiber pentynamide 1. | ||
(2) Treatment of 2, which lacks the hexacarbonyldicobalt protection, with DIBAL-H gave selective reduction of the amide exo carbonyl, resulting in the heminal 1′-hydroxyalkyloxazolidin-2-one 10. The chemoselective origin of the DIBAL-H-induced exo carbonyl reduction38,39 of N-acyloxazolidinones is not clear. Single-crystal X-ray diffraction analysis of 10 showed the (R) configuration of the newly generated stereogenic center (Fig. 1). To our knowledge, only one literature report of the stereochemical outcome of such 1′-hydroxyalkyloxazolidin-2-ones has been verified by single-crystal X-ray diffraction, indicating that one single diastereomer was formed in each case.40 Treatment of 10 with K2CO3 in methanol/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio)39 promoted the rapid cleavage of the chiral auxiliary but also brought about epimerization at the Cα position as determined by 1H and 13C{1H} NMR spectroscopy, giving rise to a mixture of 4 and 4-epi in 3:7 ratio (Scheme 5, upper middle). The proton at the Cα position (adjacent to the aldehyde) most likely underwent stereochemical scrambling via enol–keto tautomerization equilibrium in basic media. The 1H NMR spectrum of the mixture of 4 and 4-epi is compared with that of pure 4 in Fig. 1.
:1 ratio)39 promoted the rapid cleavage of the chiral auxiliary but also brought about epimerization at the Cα position as determined by 1H and 13C{1H} NMR spectroscopy, giving rise to a mixture of 4 and 4-epi in 3:7 ratio (Scheme 5, upper middle). The proton at the Cα position (adjacent to the aldehyde) most likely underwent stereochemical scrambling via enol–keto tautomerization equilibrium in basic media. The 1H NMR spectrum of the mixture of 4 and 4-epi is compared with that of pure 4 in Fig. 1.
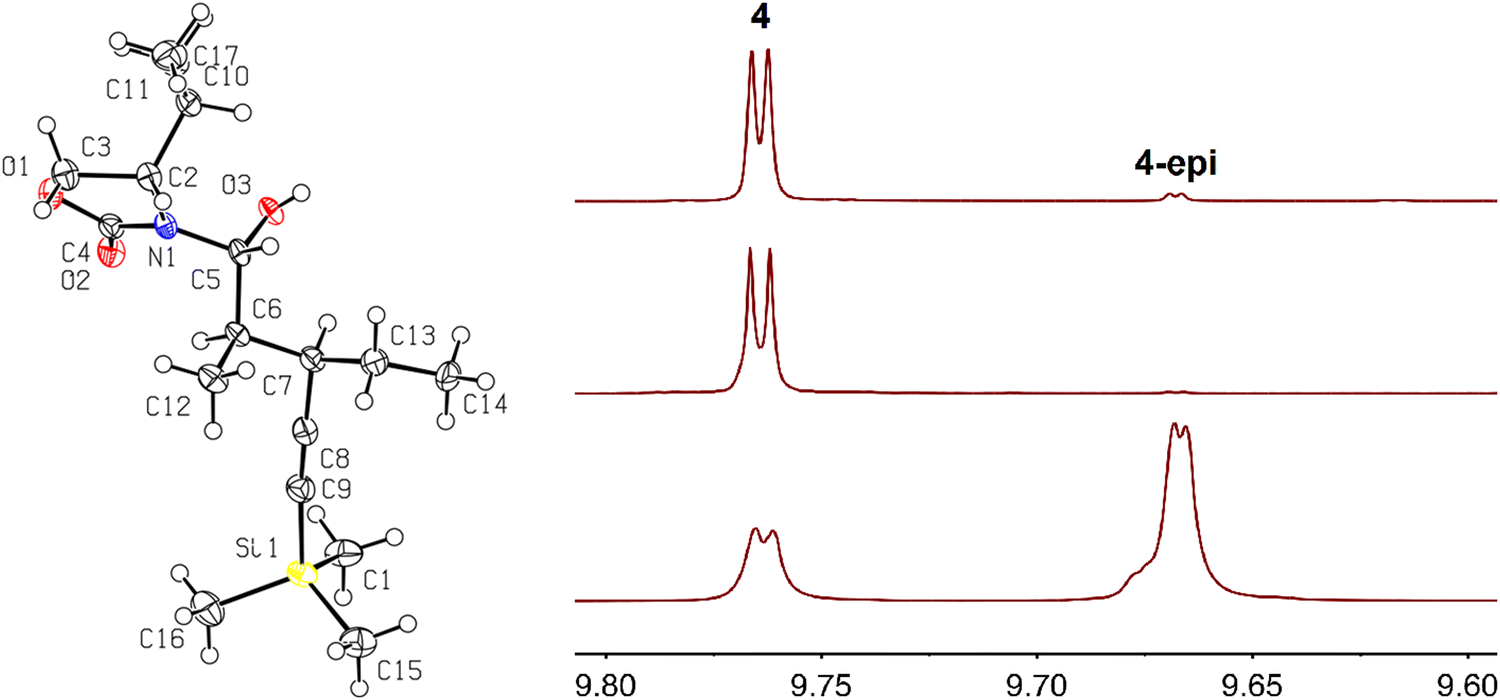 | ||
| Fig. 1 ORTEP diagram with thermal ellipsoids drawn at the 50% probability level of 10 (left) and aldehydic region of the 1H NMR spectrum of 4 (top right, prepared previously),10 product from route 1 (middle right), and products prepared by route 2 (lower right). | ||
Given the oxidative and stereochemical lability of the chiral pentynal 4, a route for the direct conversion of 1 or 2 to a chiral Weinreb pentynamide thereof was sought, given that the latter would be a versatile intermediate. Epimerization at the Cα position to an amide is regarded as far slower than that of an aldehyde.41–43 Hence, routes 3–5 were pursued.
(3) Treatment of Schreiber-Nicholas adduct 1 to aluminum-mediated reductive cleavage in the presence of N,O-dimethylhydroxyamine hydrochloride44–46 caused ring opening to the carbonyl group of the oxazolidinone unit to give undesired carbamate 11 (48%) (Scheme 5, lower left). Such endocyclic nucleophilic cleavage has been observed for hindered substrates44,47 and explained by failure of the Lewis acid to coordinate to the exo carbonyl unit, which is crucial to trigger the exocyclic reduction pathway leading to the Weinreb pentynamide. On the other hand, the formation of such amides via endocyclic nucleophilic cleavage has rarely been reported.48,49 The structure of 11 was confirmed by single-crystal X-ray diffraction (Fig. 2).
 | ||
| Fig. 2 ORTEP diagram of 11 (left) and 12 (right) with thermal ellipsoids drawn at the 50% probability level. | ||
(4) Treatment of 2 to the same reaction conditions gave a mixture of two undesired but separable epimers, 12 (56%, a white solid) and 12-epi (5%, an oil), as established by comparison of the 1H and 13C{1H} NMR spectra as well as HRMS analyses (Scheme 5, lower middle). Examination of the main isomer (12) by single-crystal X-ray diffraction confirmed the nucleophilic ring-opening (Fig. 2). The results show that the trans-dialkyl motif was still sufficiently sterically hindered to suppress the exocyclic nucleophilic cleavage but not sufficiently bulky to impede the epimerization at the Cα site.
(5) In a less direct route, 2 was treated with LiOOH (generated in situ from LiOH and 30% H2O2)47 at 0 °C to afford chiral pentynoic acid 8 in 96% yield (Scheme 5, lower right). The same reaction at room temperature17 exclusively gave (2R,3R)-3-ethyl-2-methylpent-4-ynoic acid, a desilylated derivative of 8 (and isomer of IVa). Compound 8 was then treated50,51 with oxalyl chloride followed by N,O-dimethylhydroxyamine hydrochloride under basic conditions to give desired 13 in 99% yield. The Weinreb pentynamide 13 can be used for sequential transformations without further purification.
In summary, the two approaches (1, 2) with DIBAL-H reduction gave the target pentynal 4 but typically with accompanying side products (carboxylic acid 8, epimeric form 4-epi) derived from oxidation and epimerization, respectively, during the reaction course or work-up procedures. Two direct routes (3, 4) to the Weinreb pentynamide were unsuccessful, whereas hydrolysis to form the carboxylic acid followed by amidation (5) provided access in high yield to the Weinreb pentynamide 13.
Diverse 1-substituted chiral hexynones
The chiral Weinreb pentynamide 13 served as the key scaffold for forming a family of chiral hexynones bearing a protected hydroxy group at the 1-position (Scheme 6). The transformation relied on access to one-carbon stannane synthons, which can undergo lithium-tin transmetalation52,53 at low temperature (≤−78 °C) to afford the corresponding in situ generated α-oxygenated organolithium nucleophiles. Compounds Sn-Me,54Sn-MOM,55Sn-THP,56Sn-MEM,57Sn-Bn,58 and Sn-SEM59 were prepared according to literature procedures from tributyltin hydride via tributyl(iodomethyl)tin or tributyl(hydroxymethyl)tin60 and characterized by 1H and 13C{1H} NMR spectroscopy; the syntheses are listed here for completeness. Chromatographic purification of compound53Sn-(OMe)2 resulted in a significant loss of the product (although the 1H and 13C{1H} NMR spectra still matched the literature data53). To avoid the loss, Sn-(OMe)2 was used in crude form (quantitated by 1H NMR analysis with use of mesitylene as an internal standard) for reactions. | ||
| Scheme 6 Diversification to form chiral 4-hexyn-2-ones (lower panel) with use of tributyltin-one-carbon synthons (top panel). | ||
The use of an organolithium reagent bearing an alkoxy group as nucleophile has been explored in reaction with Weinreb amides to afford compounds equipped with α-hydroxyketone motifs.61,62 Thus, the organolithium reactant derived from Sn-Me, Sn-SEM, Sn-MOM, Sn-Bn, or Sn-MEM (by treatment of 2.0–2.2 equivalent of n-BuLi reagent with an equimolar amount of each (alkoxymethyl)stannane) underwent reaction53 with 13 to give the corresponding adduct in similar yield, including 14-OMe (54%), 14-OSEM (65%), 14-OMOM (61%), 14-OBn (44%), or 14-OMEM (74%). The reaction of 13 with the organolithium reactant derived from Sn-THP afforded two pairs of diastereomers. The first pair (isolated in 70% yield) consisted of two inseparable diastereomers of 14-OTHP due to undefined stereochemistry of the anomeric carbon of the pyran ring. The second pair (24% yield) comprised two diastereomers of 14-OTHP-desTMS due to desilylation of the two parent epimers. The use of Sn-(OMe)2 in the attempted synthesis of 7 (bearing a dimethoxymethyl terminus) was not successful, as starting material 13 was recovered in 82% yield (method C) after chromatographic purification or 93% yield (method A) upon calculation from the crude reaction mixture with use of mesitylene as an internal standard. An alternative approach to convert 13 to 7 is described in the next section.
Altogether, six new chiral hexynones that differ in the nature of the protecting group for the 1-hydroxy substituent (Me, MOM, THP, MEM, Bn, or SEM) have been prepared. Deprotection attempts of selected compounds were also examined, showing that the transformation proceeded smoothly to afford the target compound 14-OH in yields of 72% (from 14-OMEM by treatment with ZnBr2) or 93% (from 14-OTHP by treatment with Amberlyst 15). The integrity of the stereochemical features of the trans-dialkyl moiety remained intact under the deprotection approaches examined. The chiral hexynones with Me, MOM, THP, MEM, Bn, SEM or no protection for the 1-hydroxy group comprise a collection of precursors to dihydrodipyrrins for methodology development in the preparation of (bacterio)chlorophylls.
Improved preparation of intermediates on the path to IX, a pre-B compound
The failure to convert 13 to 7via stannane Sn-(OMe)2 prompted studies using the chiral Weinreb pentynamide 13 as a key scaffold to gain improved access to intermediates on the path to hexynone IX. Thus, one-carbon synthons for nucleophilic addition to the Weinreb amide 13 were explored, with the proviso that the added moieties would be amenable to subsequent transacetalization (Scheme 7). The anticipated advantages of derivatization of pentynamide 13versus pentynal 4 included sidestepping the risk of epimerization as well as avoidance of any oxidation steps following the nucleophilic addition. | ||
| Scheme 7 Refined synthesis of chiral hexynone IX (pre-B) precursors. | ||
First, pentynamide 13 was treated with lithiated 1,3-dithiane at −78 °C63 for 2 h to afford acyldithiane 15 in 80% yield. Treatment of 15 with PIFA in anhydrous methanol10 gave none of the desired product. Such failures of transacetalization of the dithiane group of analogues of 15 are known.63–65 Recourse was achieved by reduction of 15 with NaBH4 to obtain 5-R in nearly quantitative yield. The newly formed stereocenter (carbinol) was assigned by comparing the 1H NMR spectrum of 5-R with the reported spectrum of the same compound.10 Moreover, the highly stereoselective reduction of acyldithiane 15 can be explained via a modified Felkin-Anh model (see inset in Scheme 7).66 In summary, use of 1,3-dithiane as a one-carbon synthon provided access to IX in a slightly better overall yield (73% from 2 here versus 67% from 2 previously10), albeit via a lengthier route, but with the advantage of limited handling of the epimerization-susceptible pentynal 4.
Second, pentynamide 13 was treated with lithiated methoxymethyl(phenyl)sulfane at −78 °C64 to give 16 as a mixture of diastereomers (1:0.3 ratio) in 43% yield. Treatment of 16 with CuCl2/CuO64 or HgCl2/HgO67 in refluxing anhydrous methanol gave a complex mixture. The oxothioacetal 16 upon treatment with 3 equivalents of PIFA in anhydrous methanol gave only ∼50% conversion (on the basis of 1H NMR analysis), and the mixture of 16 and 7 (1:1 ratio) was inseparable by conventional silica gel chromatography. Ultimately, the synergistic effect of PIFA and TFA in anhydrous methanol gave conversion of 16 to 7 but with isolation of the latter in only 39% yield. Thus, both compounds 5-R and 7 can be transformed to chiral hexynone IX (the pre-B compound) via known routes (Scheme 4).10
Phytyl-containing ring D precursor
Chlorophyll a and bacteriochlorophyll a share a common structure of the Southern rim as is shown in Chart 1. Across the photosynthetic tetrapyrroles, a few macrocycles contain a 17-acrylic acid moiety rather than a 17-propionic acid group, and there are variants particularly among bacteriochlorophylls in the nature of the hydrocarbon chain that forms the ester of the propionic acid unit, but otherwise the southern rims are identical. The rim also is similar if not identical across many catabolites of chlorophylls, the phyllobilins.68–72 Both photosynthetic hydroporphyrins73 and phyllobilins74 are largely unexplored with regards to synthesis. A retrosynthesis corresponding to ring D of native photosynthetic hydroporphyrins (and many phyllobilins, although the ring labeling is different) in terms of the chiral hexynone 17 is shown in Scheme 8. | ||
| Scheme 8 Ring D building block equipped with a phytyl tail. | ||
A key objective is to be able to prepare photosynthetic tetrapyrroles in sufficient quantity (e.g., 10 mg each) for studies in the photosciences. As the syntheses are expected to have a modular nature, access to ample quantities of core building blocks is desired. The synthesis of 17 proceeds through intermediates 18–29. The preparation of compounds 18–23 has been reported,26 but was carried out here at larger scale, in streamlined fashion, or in higher yield. The improved synthesis along the series 18–23 is reported first, followed by extension of 23 to form the phytyl target 17.
Treatment of penta-1,5-diol via a four-step synthesis without purification of crude intermediates (method A)26 gave the activated ester 18 in 64% yield. Alternatively, 18 could be approached in 26% yield through a two-step route with use of (diacetoxyiodo)benzene (DIB) and N-hydroxysuccinimide (NHS) as an oxidant and an activating reagent,75 respectively, after monoprotection of pentan-1,5-diol (method B). Compound 18 was obtained as a white solid upon recrystallization in absolute ethanol (method A) but a colorless oil upon column chromatography (method B and previous route26). The conversion of (trimethylsilyl)acetylene to Nicholas substrate 19 (92% yield) was accomplished in one-flask fashion (versus the previous two-flask approach17) followed by short flash column chromatographic purification (Scheme 9). Analogous reactions from methyl 3-trimethylsilyl propargyl ether,30,31 3-(trimethylsilyl)propynal,9 or 4-(trimethylsilyl)but-3-yn-2-ol (two-step synthesis)26 also afforded the same compound 19 (67–85% yield) but in smaller scale. In this manner, 18 and 19 – two key building blocks for ring D synthesis – were obtained in quantities of 75.34 g (166 mmol) and 73.59 g (174 mmol), respectively.
 | ||
| Scheme 9 Streamlined synthesis of precursors. | ||
The formation of the chiral hexynone unit of phytyl target 17 was achieved via the general route described above, which entails the Schreiber-modified Nicholas reaction followed by path 5 in Scheme 5. The reaction scheme beginning with (R)-3-isopropyloxazolidinone via intermediates 20–22 is known26 but was refined here. Thus, reaction of (R)-3-isopropyloxazolidinone with n-butyl lithium followed by 18 gave 20 in 88% yield (Scheme 10). Subsequent reaction with dibutylboron triflate in the presence of N,N-diisopropylethylamine (DIPEA) followed by Nicholas substrate 19 gave the Schreiber-Nicholas product 21 containing two contiguous stereocenters. The higher yield here (90% versus 68% previously26) is attributed to the use of less dibutylboron triflate (1.5 versus 2 equiv.) and shorter reaction time (85 versus 120 minutes). Decomplexation upon treatment with CAN quantitatively gave 22. Previous hydrolysis of 22 with LiOOH at room temperature gave the desilylated product IVb as the dominant product (74% yield) along with that having an intact TMS group (23, 13% yield).26 Here, the same hydrolysis at 0 °C afforded a reversal in the ratio, giving IVb and 23 in 17% and 75%, respectively. A single-crystal X-ray structure of the xylyl amide derivative of IVb was determined to establish the stereochemistry.26
 | ||
| Scheme 10 Synthesis of the chiral phytyl-containing ring D precursor. | ||
Compound 23 was transformed into the corresponding Weinreb pentynamide 24 in 81% yield by treatment51 with oxalyl chloride followed by N,O-dimethylhydroxyamine hydrochloride (Scheme 10). The nucleophilic addition of the organolithium reagent53 derived from Sn-THP to Weinreb pentynamide 24 gave THP-protected hexynone 26 in 64% yield. The same chemistry was carried out on the desilylated compound IVb to give the Weinreb pentynamide 25 in 83% yield. The reaction of 25 with excess Sn-THP and n-BuLi reagents (2.5 to 4.0 equiv.) only afforded the adduct 27 in yields ranging from 14% to 34%, respectively, with 25 recovered in 51–62% yield. Attempts to use even larger amounts (>4 equiv.) of the organotin and n-BuLi reagents required laborious workup to purify the product from unreacted starting material and a large quantity of impurities.
Treatment of 26 with TBAF at 0 °C followed by DIB-TEMPO-mediated oxidation gave a mixture of the aldehyde 28 and the carboxylic acid 29 in 25% and 50% yield, respectively. The aldehyde 28 in its entirety was subjected to Pinnick oxidation76 to quantitatively afford the carboxylic acid 29. The final step for installation of the native phytyl group was achieved by Steglich esterification,77 mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP) in CH2Cl2. Thus, reaction of chiral hexynone 29 and native phytol gave the target 17 as a light-yellow oil in 61% yield (1.43 g). Native phytol was employed, hence the product is semisynthetic. The product 17 contains six stereocenters (two adjacent centers of chirality in the hexynone moiety; two centers of chirality and the E alkene in the phytol unit; and the chiral THP unit) and is a mixture of two diastereomers given the configuration of the THP protecting group.
Outlook
A planned total synthesis of photosynthetic tetrapyrroles installs the requisite stereocenters in the pyrroline rings at a very early stage of the synthesis. An advantage of doing so is the ability to rely on established methodology of asymmetric chemistry; a challenge to doing so is the requirement to carry the stereocenters with fidelity throughout the course of the synthesis. In all of the chiral hexynones and the prior pentynoic acid derivatives, each stereocenter is adjacent to an electron-withdrawing group (ethyne or carbonyl) and hence is vulnerable to epimerization.41–43 The synthetic approaches for installation of the one-carbon handles must be done in a manner compatible with the established stereochemical configuration at the two sites.The Schreiber-modified Nicholas reaction is a time-honored method to install two contiguous stereochemical centers in an alkynyl–amide. Direct reduction of the Schreiber-Nicholas adduct 1 with DIBAL-H and subsequent removal of the hexacarbonyldicobalt protecting group gave pentynal 4, which proved to be quite labile (Scheme 5). Hydrolysis of the amide linking the oxazolidinone chiral auxiliary in 2 released the chiral pentynoic acid (8), which was converted to the chiral Weinreb pentynamide 13 (Scheme 5). The availability of the chiral Weinreb pentynamide 13 opened access to late-stage derivatization with use of a handful of (alkoxymethyl)stannane reagents thereby creating a family of 1-substituted chiral hexynones (Scheme 6). The compound of perhaps greatest immediate interest, 7, the TMS-protected derivative of IX (a pre-B compound), was not accessible via the (1,1-dimethoxymethyl)stannane reagent Sn-(OMe)2 (Scheme 6). Instead, the Weinreb pentynamide (13) was reacted with lithiated 1,3-dithiane or methoxymethyl(phenyl)sulfane to install the 1-carbon of the nascent chiral hex-5-yn-2-one; the former following borohydride treatment afforded dithianyl–alcohol 5-R whereas the latter following PIFA-mediated transacetalization with methanol afforded 7 (Scheme 7). Both 5-R and 7 can funnel into the established route (Scheme 4) to give IX.
The diverse collection of 1-substituted chiral hexynones prepared herein may be of value in development of methodology in the (bacterio)chlorophyll domain. While the immediate target is bacteriochlorophyll a, a broader goal is to be able to create a family of native photosynthetic macrocycles as well as analogues and stereoisomers thereof. Both objectives impose requirements of scale. Here, selected chiral hexynones (17, 26) have been prepared in gram quantities (2.5 mmol, 12 mmol) for use in the total syntheses, which are 250 or 1200 times larger than the quantity of target hydroporphyrin. Known and new routes have been implemented in refined and/or streamlined fashion to enable synthesis at such scale of chiral hexynones, which may be of use in a variety of syntheses aimed at preparing photosynthetic hydroporphyrins.
Experimental section
General information
All chemicals from commercial sources were used as received. Silica for column chromatography was 230–400 mesh (60 Å). THF for use in inert-atmosphere reactions was freshly distilled from sodium/benzophenone ketyl. Other solvents were reagent grade and used as received unless noted otherwise. Phytol was obtained from AmBeed, Inc.Low-temperature reactions
Reaction at temperatures lower than ambient were obtained with ice water (0 °C); ice water/NaCl mixtures (−15 or −10 °C); dry ice/NaCl/water (−20 °C); dry ice/acetone or dry ice/diethyl ether (−78 °C). The dry ice/diethyl ether bath is reported to afford a temperature of −100 °C78 but in our hands was found to be −78 °C by use of a calibrated digital thermometer. The stated temperatures are for the bath, not the contents of a reaction flask immersed therein.Known compounds
Known compounds Sn-(OMe)2,53Sn-Me,54Sn-MOM,55Sn-THP,56Sn-MEM,57Sn-Bn,58 and Sn-SEM59 were prepared as described in the literature. Known compounds 18–23 and IV-b were prepared herein in streamlined syntheses and/or in larger scales than reported previously.26 Known compounds 4,105-R,10 and 710 were prepared by new routes as described herein.Synthesis and characterization
:1)] to afford a dark-brown solid (1.08 g, 75%): 1H NMR (600 MHz, CDCl3) δ 0.34 (s, 9H), 0.92 (t, J = 7.5 Hz, 3H), 1.23 (d, J = 7.2 Hz, 3H), 1.68 (p, J = 7.4 Hz, 2H), 2.64 (q, J = 7.3 Hz, 1H), 3.52 (t, J = 7.2 Hz, 1H), 9.80 (s, 1H); 13C{1H} NMR (150 MHz, CDCl3) δ 1.3, 8.5, 13.6, 27.7, 44.9, 53.2, 80.4, 116.9, 200.4, 203.4; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C17H22Co2O7Si 482.9715; found, 482.9719.
Formation of 4 and 8 by decomplexation of 4-Co
A solution of 4-Co (1.07 g, 2.20 mmol) in acetone (50 mL) at room temperature was treated in portions with CAN (5.00 g in total). Then, the reaction mixture was stirred at room temperature for 10 min, after which water (50 mL) was added and the resulting mixture was concentrated. The residue was extracted with CH2Cl2 (3 × 25 mL). The combined organic extract was dried (Na2SO4) and concentrated to afford a light-yellow oil (366.7 mg) comprised of two components, which were determined to be the aldehyde 4 and the carboxylic acid 8 (3:2 ratio determined by 1H NMR spectroscopy) in isolated yield of 46% and 33%, respectively. The mixture was chromatographed [silica, hexanes/ethyl acetate (20:1) then (1:1)] to afford two fractions (each a colorless oil). The first fraction (92.8 mg) proved to be aldehyde 4 (one spot observed on TLC) but underwent degradation to 8 (upon contact with air in the fume hood) prior to authentication by NMR spectroscopy. The second fraction (208.2 mg) was determined to be 8. The following data are for the crude mixture (366.7 mg) prior to chromatography: 1H NMR (500 MHz, CDCl3) δ 0.14 (s, 9H from 8), 0.15 (s, 9H from 4), 1.01–1.05 (m, 3H for each component), 1.17 (d, J = 7.1 Hz, 3H from 4), 1.23 (d, J = 6.6 Hz, 3H from 8), 1.42–1.54 (m, 2H for each component), 2.39–2.44 (m, 1H from 4), 2.55 (dt, J = 8.8, 5.7 Hz, 1H from 4), 2.63–2.71 (m, 2H from 8), 9.77 (d, J = 2.3 Hz, 1H from 4); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.98, 12.01, 12.1, 13.5, 24.1, 25.6, 36.2, 37.3, 43.4, 49.2, 87.5, 88.5, 106.8, 107.5, 204.7.
:1)] to obtain a viscous oil (14.7 mg, 39%). 1H NMR (500 MHz, CDCl3) δ 0.12 (s, 9H), 1.01 (t, J = 7.3 Hz, 3H), 1.09 (d, J = 7.0 Hz, 3H), 1.39–1.30 (m, 1H), 1.59–1.51 (m, 1H), 2.64–2.60 (m, 1H), 3.13–3.06 (m, 1H), 3.40 (s, 3H), 3.41 (s, 3H), 4.69 (s, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.3, 11.5, 13.6, 23.9, 36.2, 45.0, 54.5, 54.7, 86.9, 103.6, 108.5, 207.1; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C14H27O3Si 271.1724; found, 271.1722.
In a separate flask, a suspension of MgBr2·OEt2 (3.46 g, 13.4 mmol) in anhydrous diethyl ether (31 mL) at room temperature under argon was treated dropwise with propane-1,3-dithiol (1.30 mL, 12.8 mmol) over the course of 15 min.37 Then, the mixture of MgBr2·OEt2 (3.46 g, 13.4 mmol) and propane-1,3-dithiol was treated with a solution of the crude pentynal 4 (assumed 11.1 mmol) in anhydrous diethyl ether (13 mL). The reaction mixture was allowed to stir overnight at room temperature under argon, and then treated with water (150 mL). The mixture was extracted with CH2Cl2 (4 × 100 mL). The combined organic extract was dried (Na2SO4), concentrated under reduced pressure, and chromatographed [silica, hexanes/ethyl acetate (20:1)] to obtain a colorless oil (1.81 g, 76%). 1H NMR (500 MHz, CDCl3) δ 0.17 (s, 9H), 1.01 (t, J = 7.4 Hz, 3H), 1.09 (d, J = 6.9 Hz, 3H), 1.36–1.30 (m, 1H), 1.63–1.57 (m, 1H), 1.96–1.81 (m, 2H), 2.14–2.09 (m, 1H), 2.54 (dt, J = 9.2 and 4.6 Hz, 1H), 2.99–2.81 (m, 4H), 4.57 (d, J = 4.6 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.4, 11.5, 14.0, 23.9, 26.6, 31.0, 31.7, 38.0, 41.8, 54.6, 87.2, 108.9; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C14H27S2Si 287.1318; found, 287.1318.
Formation of 4 and 4-epi by hydrolysis of 10
Following a literature procedure,40 a solution of 10 (931.0 mg, 2.86 mmol) in methanol/water [4:1 (v/v) ratio, 200 mL] was treated with K2CO3 (552.8 mg, 4.00 mmol). The reaction mixture was stirred at room temperature for 15 min followed by concentration and extraction with CH2Cl2 (3 × 30 mL). The combined organic extract was purified through a silica pad [hexanes/ethyl acetate (20:1)] to afford a colorless oil (275.9 mg, 49%) comprised of two epimers of (4 and 4-epi) in 3:7 molar ratio as determined by 1H NMR spectroscopy: 1H NMR (500 MHz, CDCl3) δ 0.13 (s, 9H, major epimer), 0.14 (s, 9H, minor epimer), 0.99–1.05 (m, 3H for each epimer), 1.16–1.18 (m, 3H for each epimer), 1.45–1.54 (m, 2H for each epimer), 2.37–2.45 (m, 1H for each epimer), 2.55 (dt, J = 8.6 and 5.6 Hz, 1H, minor epimer), 2.76 (q, J = 6.9 Hz, 1H, major epimer), 9.67 (d, J = 1.7 Hz, 1H, major epimer), 9.76 (d, J = 2.3 Hz, 1H, minor epimer); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 10.4, 11.97, 12.00, 12.1, 25.6, 26.0, 35.2, 36.2, 49.16, 49.18, 88.3, 88.4, 106.3, 106.8, 204.1, 204.7.
:1)] to afford a dark-red solid (319 mg, 48%): 1H NMR (500 MHz, CDCl3) δ 0.20 (s, 9H), 0.82–0.90 (m, 9H), 1.09 (d, J = 7.1 Hz, 3H), 1.33–1.79 (m, 3H), 2.50 (q, J = 7.3 Hz, 1H), 3.00 (s, 3H), 3.27–3.29 (m, 1H), 3.52 (d, J = 13.6 Hz, 3H), 3.92–4.03 (m, 2H), 4.14–4.21 (m, 1H), 5.67 (t, J = 8.6 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 1.3, 10.7, 13.7, 18.7, 19.3, 27.0, 29.8, 35.6, 47.1, 47.8, 54.0, 61.8, 65.9, 80.6, 117.8, 157.4, 174.3, 200.6; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C25H37Co2N2O10Si 671.0876; found, 671.0872. Compound 11 upon slow evaporation from acetonitrile afforded a crystalline sample that was then examined by single-crystal X-ray diffraction (Bruker D8 Venture instrument using Mo Kα-radiation). The structure was determined and refined using Olex2 and SHELXT programs.
:1)] to afford two products.
12-epi (48 mg, colorless oil): 1H NMR (500 MHz, CDCl3) δ 0.13 (s, 9H), 0.93–1.02 (m, 9H), 1.19 (d, J = 6.6 Hz, 3H), 1.36–1.51 (m, 2H), 1.86 (hept, J = 6.8 Hz, 1H), 2.62–2.68 (m, 2H), 3.08 (s, 3H), 3.66 (s, 3H), 3.85 (ddd, J = 10.5, 8.4, and 5.3 Hz, 1H), 4.08 (dd, J = 11.4 and 4.2 Hz, 1H), 4.25 (dd, J = 11.4 and 6.1 Hz, 1H), 5.82 (d, J = 9.5 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.3, 12.0, 13.7, 18.7, 19.5, 24.1, 29.9, 35.7, 37.4, 43.6, 54.2, 61.5, 64.8, 87.1, 107.8, 160.2, 174.6; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C19H37N2O4Si 385.2517; found, 385.2516.
12 (482 mg, white solid): m.p. 44–46 °C; 1H NMR (500 MHz, CDCl3) δ 0.13 (s, 9H), 0.94–1.00 (m, 9H), 1.20 (d, J = 7.1 Hz, 3H), 1.42 (ddt, J = 17.0, 13.2, and 7.3 Hz, 1H), 1.53 (dtd, J = 14.7, 7.4, and 4.8 Hz, 1H), 1.82 (hept, J = 6.7 Hz, 1H), 2.39 (p, J = 7.0 Hz, 1H), 2.53 (ddd, J = 9.7, 6.8, and 4.7 Hz, 1H), 3.11 (s, 3H), 3.65 (s, 3H), 4.05–4.12 (m, 1H), 4.13–4.20 (m, 2H), 6.07 (d, J = 9.2 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.9, 16.3, 18.7, 19.6, 25.0, 29.7, 35.7, 37.7, 45.3, 53.4, 61.7, 66.2, 88.4, 108.3, 157.2, 174.6; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C19H37N2O4Si 385.2517; found, 385.2519. Compound 12 upon slow evaporation from acetonitrile afforded a crystalline sample that was then examined by single-crystal X-ray diffraction (Bruker D8 Venture instrument using Cu Kα-radiation). The structure was determined and refined using Olex2 and SHELXT programs.
:1 ratio, 440 mL) at 0 °C was treated with a mixture of aqueous 0.5 M LiOH (100.5 mmol, 201 mL) and 30% H2O2 (268 mmol, 31 mL). The resulting solution was stirred at 0 °C for 1 h, then a cold solution of NaHSO3 (prepared from 40 g of solid reagent in 250 mL of cold water) was added. The solution was then acidified by addition of cold concentrated HCl until pH 1. Both tasks were done as quickly as possible (<5 min) while the solution was maintained at 0 °C. The mixture after acidification was quickly extracted with ethyl acetate (3 × 200 mL). The combined organic extract was dried (Na2SO4), concentrated under reduced pressure, and purified by passage through a silica pad [hexanes/ethyl acetate (7:1)] to afford a yellow oil (6.82 g, 96%): 1H NMR (500 MHz, CDCl3) δ 0.14 (s, 9H), 1.02 (t, J = 7.3 Hz, 3H), 1.23 (d, J = 6.6 Hz, 3H), 1.40–1.58 (m, 2H), 2.65–2.71 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 12.1, 13.5, 24.1, 37.3, 43.4, 87.4, 107.5, 180.6; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C11H21O2Si 213.1305; found, 213.1305.
:1)] to give a colorless oil (32 mg, 54%): 1H NMR (500 MHz, CDCl3) δ 0.13 (s, 9H), 1.00 (t, J = 7.3 Hz, 3H), 1.08 (d, J = 6.9 Hz, 3H), 1.34 (ddq, J = 14.5, 9.4, and 7.4 Hz, 1H), 1.57 (dtd, J = 14.8, 7.3, and 4.1 Hz, 1H), 2.54 (td, J = 9.1, 4.0 Hz, 1H), 2.64–2.74 (m, 1H), 3.43 (s, 3H), 4.11–4.25 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.4, 13.8, 24.2, 36.6, 46.0, 59.4, 77.6, 87.6, 108.0, 210.1; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C13H25O2Si 241.1618; found, 241.1615.
:1)] to give a colorless oil (58 mg, 65%): 1H NMR (500 MHz, CDCl3) δ 0.01 (s, 9H), 0.13 (s, 9H), 0.91–0.94 (m, 2H), 1.00 (t, J = 7.3 Hz, 3H), 1.10 (d, J = 7.0 Hz, 3H), 1.33 (ddt, J = 14.5, 9.4, and 7.2 Hz, 1H), 1.55 (dtd, J = 14.6, 7.4, and 4.3 Hz, 1H), 2.55–2.59 (m, 1H), 2.68–2.74 (m, 1H), 3.61–3.69 (m, 2H), 4.28–4.36 (m, 2H), 4.71–4.74 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ −1.3, 0.2, 11.5, 13.7, 18.2, 24.2, 36.5, 46.4, 65.8, 72.3, 87.5, 95.0, 107.9, 209.5; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C18H37O3Si2 357.2276; found, 357.2269.
:1)] to give a colorless oil (41.4 mg, 61%): 1H NMR (500 MHz, CDCl3) δ 0.12 (s, 9H), 1.00 (t, J = 7.4 Hz, 3H), 1.10 (d, J = 7.0 Hz, 3H), 1.30–1.39 (m, 1H), 1.57 (ddh, J = 14.7, 7.4, and 4.0 Hz, 1H), 2.55 (td, J = 9.0 and 4.0 Hz, 1H), 2.67–2.73 (m, 1H), 3.38 (s, 3H), 4.29 (d, J = 17.7 Hz, 1H), 4.35 (d, J = 17.7 Hz, 1H), 4.68 (s, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.4, 13.8, 24.2, 36.5, 46.4, 55.8, 72.1, 87.6, 96.6, 107.8, 209.5; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C14H27O3Si 271.1724; found, 271.1716.
:1)] to give a colorless oil (35 mg, 44%): 1H NMR (600 MHz, CDCl3) δ 0.12 (s, 9H), 1.00 (t, J = 7.4 Hz, 3H), 1.08 (d, J = 6.9 Hz, 3H), 1.34 (ddq, J = 14.2, 9.1, and 7.3 Hz, 1H), 1.56 (dtd, J = 14.7, 7.4, and 4.1 Hz, 1H), 2.57 (td, J = 9.2 and 4.0 Hz, 1H), 2.76 (p, J = 7.2 Hz, 1H), 4.21 (d, J = 17.4 Hz, 1H), 4.26 (d, J = 17.5 Hz, 1H), 4.59 (d, J = 11.8 Hz, 1H), 4.63 (d, J = 11.8 Hz, 1H), 7.28–7.40 (m, 5H); 13C{1H} NMR (150 MHz, CDCl3) δ 0.2, 11.4, 13.7, 24.2, 36.4, 46.1, 73.5, 75.1, 87.4, 108.1, 128.05, 128.08, 128.6, 137.5, 210.2; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C19H29O2Si 317.1931; found, 317.1927.
:1)] to give a colorless oil (1.27 g, 74%): 1H NMR (500 MHz, CDCl3) δ 0.12 (s, 9H), 1.00 (t, J = 7.3 Hz, 3H), 1.10 (d, J = 7.0 Hz, 3H), 1.29–1.38 (m, 1H), 1.55 (dddd, J = 14.8, 11.4, 6.6, and 4.1 Hz, 1H), 2.56 (ddd, J = 9.7, 8.2, and 4.0 Hz, 1H), 2.69 (quint, J = 7.0 Hz, 1H), 3.38 (s, 3H), 3.54 (t, J = 4.7 Hz, 2H), 3.70–3.77 (m, 2H), 4.31–4.38 (m, 2H), 4.76–4.79 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.4, 13.7, 24.2, 36.5, 46.4, 59.1, 67.4, 71.8, 72.3, 87.5, 95.7, 107.9, 209.3; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C16H31O4Si 315.1986; found, 315.1982.
:1)] to give two fractions.
Fraction 1 (1.08 g, 70%) was identified as the title compound comprised of a mixture of two inseparable diastereomers of which the 1H NMR spectra are overlapped: 1H NMR (500 MHz, CDCl3) δ 0.13 (two singlets, 9H), 1.00 (t, J = 7.3 Hz, 3H), 1.10 (two doublets, J = 7.0 Hz for each, 3H), 1.30–1.39 (m, 1H), 1.52–1.63 (m, 4H), 1.70–1.80 (m, 2H), 1.83–1.91 (m, 1H), 2.58 (dddd, J = 9.8, 8.1, 5.9, and 4.0 Hz, 1H), 2.71–2.81 (m, 1H), 3.49–3.53 (m, 1H), 3.81–3.86 (m, 1H), 4.26–4.43 (m, 2H), 4.66 (t, J = 3.5 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.20, 0.22, 11.4, 11.5, 13.6, 13.7, 19.2, 24.1, 24.2, 25.5, 30.4, 36.4, 46.32, 46.33, 62.2, 62.3, 71.8, 71.9, 87.4, 98.7, 108.0, 108.1, 210.0, 210.1; HRMS (ESI-FTMS) m/z: [M + Na]+ calcd for C17H30O3SiNa 333.1856; found, 333.1849.
Fraction 2 (289 mg, 24%) was identified as the desilylated derivative of the desired product (14-OTHP-desTMS), comprised of a mixture of two inseparable diastereomers of which the 1H NMR spectra are overlapped: 1H NMR (500 MHz, CDCl3) δ 1.03 (t, J = 7.3 Hz, 3H), 1.12 (two doublets, J = 7.0 Hz for each, 3H), 1.34–1.43 (m, 1H), 1.52–1.64 (m, 4H), 1.70–1.81 (m, 2H), 1.83–1.90 (m, 1H), 2.09–2.10 (m, 1H), 2.56–2.62 (m, 1H), 2.75–2.85 (m, 1H), 3.48–3.53 (m, 1H), 3.81–3.87 (m, 1H), 4.25–4.42 (m, 2H), 4.66–4.68 (m, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 11.4, 11.5, 13.75, 13.77, 19.15, 19.19, 24.1, 25.45, 25.47, 30.3, 30.4, 35.2, 35.3, 45.9, 46.0, 62.27, 62.32, 70.98, 71.04, 71.7, 71.8, 85.4, 85.5, 98.6, 98.7, 210.07, 210.09; HRMS (ESI-FTMS) m/z: [M + Na]+ calcd for C14H22O3Na 261.1461; found, 261.1459.
From 14-OMEM: a solution of 14-OMEM (393.4 mg, 1.25 mmol) in CH2Cl2 (20 mL) was treated with anhydrous ZnBr2 (638.0 mg, 2.50 mmol). The reaction mixture was stirred at room temperature for 4 h and then concentrated under reduced pressure. The crude material was purified by chromatography (silica, CH2Cl2) to afford a colorless oil (203 mg, 72%): 1H NMR (500 MHz, CDCl3) δ 0.13 (s, 9H), 1.01 (t, J = 7.4 Hz, 3H), 1.14 (d, J = 6.9 Hz, 3H), 1.32–1.41 (m, 1H), 1.53–1.61 (m, 1H), 2.52–2.56 (m, 1H), 2.61–2.67 (m, 1H), 3.12 (t, J = 4.7 Hz, 1H), 4.30–4.41 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.1, 11.4, 13.9, 24.5, 37.0, 46.4, 68.4, 88.2, 107.2, 212.3; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C12H23O2Si 227.1462; found, 227.1459.
:1)] to obtain a colorless oil (251.0 mg, 80%). 1H NMR (500 MHz, CDCl3) δ 0.14 (s, 9H), 1.01 (t, J = 7.4 Hz, 3H), 1.17 (d, J = 6.7 Hz, 3H), 1.45–1.30 (m, 1H), 1.63 (dt, J = 13.4, 7.4, and 3.7 Hz, 1H), 2.06–1.95 (m, 1H), 2.11 (dddd, J = 11.1, 5.2, 4.0, and 2.7 Hz, 1H), 2.65–2.42 (m, 3H), 3.00–2.89 (m, 2H), 3.39 (ddd, J = 14.2, 12.0, and 2.7 Hz, 1H), 4.45 (s, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 11.2, 15.3, 24.4, 25.4, 25.9, 26.0, 37.6, 47.3, 47.7, 88.2, 107.6, 204.4; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C15H27OS2Si 315.1267; found, 315.1265.
:1)] to obtain a light-yellow oil (148.2 mg, 43%).1H NMR (500 MHz, CDCl3) δ 0.89–0.85 (m, 9H for each epimer), 1.25–1.15 (m, 1H for each epimer), 1.38–1.31 (m, 1H, minor epimer), 1.52–1.44 (m, 1H, major epimer), 2.29–2.25 (m, 1H, major epimer), 2.52–2.48 (m, 1H, minor epimer), 2.97–2.87 (m, 1H for each epimer), 3.45 (s, 3H, minor epimer), 3.49 (s, 3H, major epimer), 5.04 (s, 1H, minor epimer), 5.23 (s, 1H, major epimer), 7.18–7.13 (m, 3H for each epimer), 7.32–7.30 (m, 2H, major epimer), 7.44–7.40 (m, 2H, minor epimer); 13C NMR (125 MHz, CDCl3, for the major epimer only) δ 0.0, 10.8, 13.7, 24.1, 37.5, 45.1, 56.2, 92.1, 107.5, 128.0, 128.9, 131.7, 132.5, 133.4, 202.6; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C19H29O2Si 349.1652; found, 349.1647.
:1)] to afford a light-yellow oil (1.43 g, 61%): 1H NMR (700 MHz, CDCl3) δ 0.84–0.87 (m, 12H), 1.02–1.10 (m, 4H), 1.11–1.46 (m, 18H), 1.49–1.62 (m, 6H), 1.68 (s, 3H), 1.71–1.79 (m, 2H), 1.84–1.89 (m, 1H), 1.91–2.03 (m, 4H), 2.11 (t, J = 2.7 Hz, 1H), 2.22 (dtd, J = 16.0, 7.9, and 5.2 Hz, 1H), 2.31 (dddd, J = 16.1, 8.7, 6.0, and 3.8 Hz, 1H), 2.71–2.83 (m, 2H), 3.48–3.52 (m, 1H), 3.83 (tdd, J = 11.3, 9.0, and 3.0 Hz, 1H), 4.19–4.42 (m, 2H), 4.56–4.61 (m, 1H), 4.67 (dt, J = 9.2, 3.5 Hz, 1H), 5.30–5.32 (m, 1H); 13C{1H} NMR (175 MHz, CDCl3) δ 16.5, 17.9, 18.0, 19.16, 19.19, 19.86, 19.90, 22.8, 22.9, 23.7, 23.9, 24.6, 24.9, 25.2, 25.4, 25.5, 27.4, 27.5, 28.1, 30.3, 31.88, 31.90, 32.8, 33.0, 36.8, 37.4, 37.5, 37.6, 39.5, 40.0, 51.46, 51.53, 61.7, 62.27, 62.30, 70.4, 70.5, 73.0, 73.1, 86.1, 86.2, 98.6, 98.8, 118.01, 118.04, 143.06, 143.11, 172.90, 172.93, 209.6, 209.7; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C35H61O5 561.4514; found, 561.4521.
Method A. A literature route26 was modified to derivatize pentan-1,5-diol in a streamlined four-step synthesis at a larger scale.
In the first step, a sample of NaH (60% dispersion in mineral oil, 10.00 g, 250 mmol) after being washed with hexanes (3 × 50 mL) was suspended in freshly distilled THF (500 mL). The resulting suspension under argon with stirring was treated dropwise with pentan-1,5-diol (26.04 g, 250 mmol) through an addition funnel. The reaction mixture was stirred at room temperature for 45 min, then tert-butyldiphenylsilyl chloride (68.72 g, 250 mmol) was added dropwise through an addition funnel. The resulting mixture was further stirred at room temperature for 1 h, treated with water (500 mL), and extracted with Et2O (3 × 250 mL). The combined organic extract was dried (Na2SO4) and concentrated under reduced pressure to afford the crude 5-((tert-butyldiphenylsilyl)oxy)pentan-1-ol as a yellow oil (85.14 g).
In the second step, the entire crude sample from the first step was dissolved in CH2Cl2 (1.2 L) followed by treatment with DMP (116.64 g, 275 mmol) in one batch. The reaction mixture was stirred at room temperature for 1 h, then 10% aqueous Na2S2O3 solution (800 mL) was added. The organic phase was collected, washed with saturated aqueous NaHCO3 solution (800 mL), dried (Na2SO4), and concentrated under reduced pressure to give the crude 5-((tert-butyldiphenylsilyl)oxy)pentanal as a yellow oil (90.10 g).
In the third step, the entire crude sample from the second step was dissolved in tert-butanol (600 mL) and water (200 mL) followed by addition of 2-methyl-2-butene (264.8 mL, 2.5 mol), NaH2PO4·H2O (103.49 g, 750 mmol), and NaClO2 (84.79 g, 750 mmol). The reaction mixture was stirred at room temperature for 30 min, diluted with brine solution (500 mL), acidified by adding 2 M aqueous HCl until pH 1, and extracted with ethyl acetate (3 × 250 mL). The combined organic extract was dried (Na2SO4) and concentrated under reduced pressure to give a residue, which upon dilution with CH2Cl2 gave rise to insoluble materials. Filtration through an F-fritted funnel followed by concentration under reduced pressure afforded the crude 5-((tert-butyldiphenylsilyl)oxy)pentanoic acid as a yellow oil (98.12 g).
In the final step, the entire crude sample from the third step was dissolved in CH2Cl2 (1 L) followed by addition of N-hydroxysuccinimide (34.53 g, 300 mmol). The solution was cooled at 0 °C, treated with DCC (61.90 g, 300 mmol), and stirred at 0 °C for 1 h and then at room temperature for 29 h, whereupon a white suspension was present. The reaction mixture was filtered. The filtered cake was washed with cold CH2Cl2. The filtrates were combined and concentrated under reduced pressure to give a thick yellow oil, which upon recrystallization in absolute ethanol (400 mL) afforded a white solid (75.34 g, 66% from 1,5-pentadiol): mp 57–60 °C; 1H NMR (500 MHz, CDCl3) δ 1.06 (s, 9H), 1.64–1.71 (m, 2H), 1.87 (quint, J = 7.5 Hz, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.82 (br, 4H), 3.69 (t, J = 6.1 Hz, 2H), 7.36–7.44 (m, 6H), 7.67 (d, J = 6.4 Hz, 4H); 13C{1H} NMR (125 MHz, CDCl3) δ 19.3, 21.3, 25.7, 27.0, 30.7, 31.6, 61.3, 127.8, 129.7, 133.9, 135.7, 168.7, 169.3; HRMS (ESI-FTMS) m/z: [M – H]− calcd for C25H30NO5Si 452.1899; found, 452.1916.
Method B. Following reported procedures26,75 with modification, a mixture of pentan-1,5-diol (5.20 g, 49.9 mmol) and imidazole (4.90 g, 72.0 mmol) in anhydrous CH2Cl2 (250 mL) under argon at 0 °C was treated dropwise with a solution of tert-butyldiphenylsilyl chloride (16.55 g, 60.2 mmol) in anhydrous CH2Cl2 (75 mL) over 1 h. Then, the reaction mixture was allowed to warm overnight to room temperature. The mixture was treated with saturated aqueous NH4Cl solution (150 mL). The aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic extract was dried (Na2SO4) and concentrated under reduced pressure to obtain a crude yellow oil. The crude oil was used without any further purification. The oil was dissolved in anhydrous acetonitrile (100 mL) under argon at 0 °C. The solution was then treated with DIB (17.71 g, 55.0 mmol) and NHS (6.33 g, 55.0 mmol). The resulting reaction mixture was stirred for 0.5 h at 0 °C under argon. The mixture was filtered through a Celite pad. The filter cake was washed with ethyl acetate (3 × 100 mL). The filtrate was dried (Na2SO4), concentrated under reduced pressure, and chromatographed [silica, hexanes/ethyl acetate (3
:1)] to afford a colorless oil (5.90 g, 26%): 1H NMR (CDCl3, 500 MHz) δ 1.05 (s, 9H), 1.64–1.69 (m, 2H), 1.84–1.90 (m, 2H), 2.61 (t, J = 7.5 Hz, 2H), 2.82–2.83 (m, 4H), 3.69 (t, J = 5 Hz, 2H), 7.37–7.43 (m, 6H), 7.65–7.67 (m, 4H); 13C{1H} NMR (CDCl3, 125 MHz) δ 19.3, 21.3, 25.7, 27.0, 30.8, 31.6, 63.2, 127.8, 129.7, 133.9, 135.7, 168.7, 169.3.
:1)] to afford a dark brown solid (73.59 g, 92%): 1H NMR (500 MHz, CDCl3) δ 0.31 (s, 9H), 1.47 (d, J = 6.2 Hz, 3H), 3.48 (s, 3H), 4.47 (q, J = 6.2 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.1, 23.0, 57.3, 78.0, 113.5, 200.5.
:1)] to furnish a colorless oil (32.82 g, 88%): 1H NMR (600 MHz, CDCl3) δ 0.87 (d, J = 6.9 Hz, 3H), 0.91 (d, J = 7.0 Hz, 3H), 1.04 (s, 9H), 1.61–1.65 (m, 2H), 1.72–1.79 (m, 2H), 2.33–2.41 (m, 1H), 2.87 (ddd, J = 16.9, 8.1, and 6.8 Hz, 1H), 2.99 (ddd, J = 16.9, 8.2, and 6.6 Hz, 1H), 3.69 (t, J = 6.3 Hz, 2H), 4.20 (dd, J = 9.1 and 3.0 Hz, 1H), 4.25 (dd, J = 9.1 and 8.3 Hz, 1H), 4.43 (ddd, J = 8.3, 3.9, and 3.0 Hz, 1H), 7.36–7.43 (m, 6H), 7.66 (dd, J = 8.0 and 1.5 Hz, 4H); 13C{1H} NMR (150 Hz, CDCl3) δ 14.8, 18.1, 19.4, 21.0, 27.0, 28.5, 32.1, 35.4, 58.5, 63.5, 63.7, 127.7, 129.7, 134.1, 135.7, 154.2, 173.3; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C27H38NO4Si 468.2565; found, 468.2568.
:1)] to afford a dark-red paste (55.59 g, 90%): 1H NMR (600 MHz, CDCl3) δ 0.29 (s, 9H), 0.87 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 7.0 Hz, 3H), 1.03 (s, 9H), 1.21 (d, J = 7.1 Hz, 3H), 1.47–1.56 (m, 3H), 2.01–2.07 (m, 1H), 2.29–2.34 (m, 1H), 3.40 (qd, J = 7.1 Hz and 2.3 Hz, 1H), 3.63 (t, J = 6.5 Hz, 2H), 4.09 (dt, J = 11.9 and 2.1 Hz, 1H), 4.19 (dd, J = 9.1 and 3.2 Hz, 1H), 4.25 (t, J = 8.8 Hz, 1H), 4.50 (dt, J = 8.4 and 3.4 Hz, 1H), 7.34–7.43 (m, 6H), 7.63–7.66 (m, 4H); 13C{1H} NMR (150 MHz, CDCl3) δ 1.3, 14.7, 18.1, 18.4, 19.3, 20.9, 27.0, 28.7, 31.2, 41.5, 49.3, 58.4, 63.3, 64.3, 79.7, 115.2, 127.73, 127.74, 129.6, 134.0, 134.1, 135.7, 153.4, 173.9; HRMS (ESI-FTMS) m/z: [M − H]− calcd for C40H48Co2NO10Si2 876.1486; found, 876.1483.
:1) with 2% (v/v) AcOH] to afford two fractions.
Fraction 1 (colorless oil) was characterized as 23 (11.34 g, 75%): 1H NMR (500 MHz, CDCl3) δ 0.12 (s, 9H), 1.04 (s, 9H), 1.20 (d, J = 7.0 Hz, 3H), 1.53–1.68 (m, 2H), 1.69–1.78 (m, 2H), 2.47 (ddd, J = 9.2, 6.6, and 5.1 Hz, 1H), 2.80 (quint, J = 7.0 Hz, 1H), 3.67 (tq, J = 6.9 and 3.9 Hz, 2H), 7.36–7.43 (m, 6H), 7.66 (dd, J = 8.0 and 1.6 Hz, 4H); 13C{1H} NMR (125 MHz, CDCl3) δ 0.2, 18.0, 19.3, 25.5, 27.0, 29.3, 30.6, 50.5, 63.7, 86.3, 108.6, 127.8, 127.9, 129.7, 129.8, 134.01, 134.03, 134.9, 135.7; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C28H41O3Si2 481.2589; found, 481.2587.
Fraction 2 (yellow oil) was characterized as IVb (2.22 g, 17%): 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 9H), 1.23 (d, J = 7.2 Hz, 3H), 1.53–1.69 (m, 2H), 1.71–1.83 (m, 2H), 2.10 (d, J = 2.5 Hz, 1H), 2.48 (ddd, J = 9.8, 6.6, and 4.6 Hz, 1H), 2.81 (quintd, J = 7.0 and 2.5 Hz, 1H), 3.68 (qd, J = 10.2 and 6.0 Hz, 2H), 7.36–7.43 (m, 6H), 7.66 (dd, J = 8.0 and 1.6 Hz, 4H); 13C{1H} NMR (125 MHz, CDCl3) δ 18.0, 19.3, 25.5, 27.0, 28.1, 30.4, 50.0, 63.5, 70.1, 86.0, 127.8, 129.7, 134.0, 135.7, 179.1; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C25H33O3Si2 409.2194; found, 409.2202.
:1)] to afford a colorless oil (10.01 g, 81%). 1H NMR (700 MHz, CDCl3) δ 0.11 (s, 9H), 1.04 (s, 9H), 1.20 (d, J = 7.0 Hz, 3H), 1.48–1.60 (m, 3H), 1.76 (dtd, J = 12.9, 6.3, and 2.9 Hz, 1H), 2.76 (dq, J = 9.0 and 7.0 Hz, 1H), 2.91 (br, 1H), 3.19 (s, 3H), 3.60–3.67 (m, 2H), 3.69 (s, 3H), 7.36–7.38 (m, 4H), 7.40–7.42 (m, 2H), 7.64–7.66 (m, 4H); 13C{1H} NMR (150 MHz, CDCl3) δ 18.1, 19.4, 25.9, 27.0, 29.3, 30.4, 32.1, 45.9, 61.5, 63.8, 85.0, 127.7, 129.7, 134.1, 134.2, 135.7, 175.8; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C30H46NO3Si2 524.3011; found, 524.3019.
:1)] to afford a colorless oil (638.8 mg, 86%): 1H NMR (600 MHz, CDCl3) δ 1.05 (s, 9H), 1.22 (d, J = 7.0 Hz, 3H), 1.48–1.57 (m, 2H), 1.62–1.68 (m, 1H), 1.77–1.83 (m, 1H), 2.06 (d, J = 1.2 Hz, 1H), 2.75–2.81 (m, 1H), 2.94 (br s, 1H), 3.20 (s, 3H), 3.61–3.72 (m, 5H), 7.36–7.43 (m, 6H), 7.66 (d, J = 6.5 Hz, 4H); 13C NMR (150 MHz, CDCl3) δ 18.0, 19.4, 25.8, 27.0, 28.0, 30.3, 32.1, 45.5, 61.5, 63.7, 69.2, 87.6, 127.7, 129.7, 134.0, 134.1, 135.7, 175.5; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C27H38NO3Si 452.2616; found, 452.2627.
:8)] to give a colorless thick oil (7.03 g, 64%) consisting of two inseparable diastereomers (due to the presence of the THP unit): 1H NMR (700 MHz, CDCl3) δ 0.13 (s, 9H), 1.04 (s, 9H), 1.16 (d, J = 6.9 Hz, 3H), 1.43–1.62 (m, 5H), 1.66–1.71 (m, 2H), 1.72–1.78 (m, 2H), 1.83–1.91 (m, 1H), 2.59–2.71 (m, 2H), 3.47–3.51 (m, 1H), 3.59–3.65 (m, 2H), 3.80–3.84 (m, 1H), 4.21–4.41 (m, 2H), 4.65 (dt, J = 9.2 and 3.5 Hz, 1H), 7.38 (t, J = 7.4 Hz, 4H), 7.42 (t, J = 7.2 Hz, 2H), 7.65 (t, J = 6.9 Hz, 4H); 13C{1H} NMR (175 MHz, CDCl3) δ 18.31, 18.33, 19.1, 19.2, 19.3, 25.49, 25.51, 25.53, 27.0, 28.67, 28.70, 30.30, 30.35, 30.4, 53.2, 53.6, 62.1, 62.2, 63.7, 72.8, 73.1, 86.26, 86.30, 98.5, 98.7, 109.2, 109.3, 127.8, 129.7, 133.97, 134.01, 135.7, 209.9, 210.1; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C34H51O4Si2 579.3320; found, 579.3316.
:1)] to give a colorless oil (33.7 mg, 34%): 1H NMR (500 MHz, CDCl3) δ 1.05 (s, 9H), 1.19 (d, J = 6.6 Hz, 3H), 1.41–1.93 (m, 10H), 2.09–2.10 (m, 1H), 2.63–2.75 (m, 2H), 3.46–3.51 (m, 1H), 3.59–3.67 (m, 2H), 3.79–3.86 (m, 1H), 4.18–4.42 (m, 2H), 4.65–4.67 (m, 1H), 7.36–7.44 (m, 6H), 7.60–7.71 (m, 4H); 13C NMR (125 MHz, CDCl3) δ 18.1, 18.2, 19.10, 19.14, 19.3, 25.3, 25.36, 25.43, 25.5, 27.0, 27.4, 27.5, 30.2, 30.3, 52.5, 52.6, 62.1, 62.2, 63.6, 70.1, 70.2, 72.8, 73.0, 86.5, 86.6, 98.46, 98.50, 127.8, 129.7, 133.90, 133.94, 135.7, 209.96, 210.03; HRMS (ESI-FTMS) m/z: [M + Na]+ calcd for C31H42O4SiNa 529.2745; found, 529.2741.
:1, 26 mL) at 0 °C was treated with DIB (8.60 g, 26.71 mmol) and TEMPO (189.1 mg, 1.21 mmol). The reaction mixture was stirred at 0 °C for 2 days followed by addition of 5% aqueous Na2S2O3 solution (130 mL) and extraction with ethyl acetate (3 × 50 mL). The combined organic extract was dried (Na2SO4), concentrated under reduced pressure, and chromatographed to afford the partially oxidized product, aldehyde 28, as a colorless oil (798 mg, 25%) followed by the title compound as a yellowish oil (1.70 g, 50%).
29, as a mixture of two diastereomers: 1H NMR (700 MHz, CDCl3) δ 1.18–1.20 (m, 3H), 1.50–1.62 (m, 3H), 1.68–1.79 (m, 2H), 1.83–1.88 (m, 1H), 1.90–2.02 (m, 2H), 2.12–2.13 (m, 1H), 2.25–2.30 (m, 1H), 2.34–2.39 (m, 1H), 2.73–2.88 (m, 2H), 3.48–3.53 (m, 1H), 3.83 (dddd, J = 14.3, 11.7, 9.0, and 3.0 Hz, 1H), 4.17–4.43 (m, 2H), 4.67 (dt, J = 9.7 and 3.5 Hz, 1H); 13C{1H} NMR (175 MHz, CDCl3) δ 17.8, 17.9, 19.1, 19.2, 23.3, 23.4, 25.38, 25.41, 27.4, 27.5, 30.26, 30.28, 31.51, 31.54, 51.31, 51.33, 62.3, 62.4, 70.5, 70.6, 73.0, 73.1, 86.0, 86.1, 98.6, 98.9, 177.91, 177.93, 209.6, 209.7; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C15H23O5 283.1540; found, 283.1536.
28, as a mixture of two diastereomers: 1H NMR (700 MHz, CDCl3) δ 1.19 (dd, J = 7.0 and 5.8 Hz, 3H), 1.49–1.62 (m, 3H), 1.67–1.73 (m, 1H), 1.74–1.80 (m, 1H), 1.85 (qq, J = 12.0 and 3.8 Hz, 1H), 1.95 (quint, J = 7.2 Hz, 2H), 2.12 (dd, J = 3.2 and 2.3 Hz, 1H), 2.35–2.51 (m, 2H), 2.73–2.89 (m, 2H), 3.46–3.55 (m, 1H), 3.82 (dddd, J = 13.8, 11.5, 8.9, and 3.1 Hz, 1H), 4.14–4.39 (m, 2H), 4.65 (dt, J = 7.0 and 3.6 Hz, 1H), 9.73 (dd, J = 2.2 and 1.2 Hz, 1H); 13C{1H} NMR (175 MHz, CDCl3) δ 17.7, 17.8, 19.2, 19.3, 20.5, 20.7, 25.38, 25.41, 27.4, 27.5, 30.29, 30.33, 41.3, 51.3, 51.4, 62.4, 62.5, 70.5, 70.6, 72.96, 72.98, 86.05, 86.12, 98.7, 99.0, 201.2, 201.3, 209.67, 209.70; HRMS (ESI-FTMS) m/z: [M + H]+ calcd for C15H23O4 267.1591; found, 267.1589.
Formation of 29 from 28 could be achieved through Pinnick oxidation as follows. The entire sample of 28 (798 mg, 3.00 mmol) in tert-butanol (11.50 mL) and water (3.83 mL) at room temperature was sequentially treated with 2-methyl-2-butene (3.18 mL, 30.0 mmol), NaH2PO4·H2O (1.24 g, 9.00 mmol), and NaClO2 (80% purity, 1.02 g, 9.00 mmol). The reaction mixture was stirred at room temperature for 20 min followed by dilution in brine (30 mL) and extraction with ethyl acetate (3 × 50 mL). The combined organic extract was dried (Na2SO4), concentrated under reduced pressure, and dried overnight under vacuum to afford 29 as a yellowish oil (853 mg, quant). Characterization by 1H and 13C{1H} NMR spectroscopy indicated that the sample was identical to that obtained from the above DIB-TEMPO-mediated oxidation.
:1)] to yield a colorless oil (32.84 g, 79%): 1H NMR (500 MHz, CDCl3) δ 0.85–0.99 (m, 15H), 1.27–1.35 (m, 6H), 1.44–1.57 (m, 6H), 4.02 (d, J = 4.4 Hz, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 8.7, 13.9, 27.5, 29.3, 53.8.
:1)] to give a colorless oil (16.89 g, 84%): 1H NMR (500 MHz, CDCl3) δ 0.84–0.98 (m, 15H), 1.26–1.34 (m, 6H), 1.43–1.56 (m, 6H), 3.29 (s, 3H), 3.69–3.72 (m, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 9.1, 13.9, 27.5, 29.3, 63.4, 64.6.
:1)] to afford a colorless oil (7.24 g, 89%): 1H NMR (500 MHz, CDCl3) δ 0.84–0.97 (m, 15H), 1.27–1.34 (m, 6H), 1.43–1.60 (m, 10H), 1.64–1.69 (m, 1H), 1.74–1.82 (m, 1H), 3.47–3.52 (m, 1H), 3.59 (d, J = 10.6 Hz, 1H); 3.80 (ddd, J = 11.4, 8.9, and 2.7 Hz, 1H), 3.99 (d, J = 10.6 Hz, 1H), 4.39 (t, J = 3.4 Hz, 1H); 13C{1H} NMR (125 MHz, CDCl3) δ 9.2, 13.9, 19.4, 25.8, 27.5, 29.3, 30.8, 57.7, 61.8, 101.6.
:1)] to afford a colorless oil (4.48 g, 66%): 1H NMR (500 MHz, CDCl3) δ 0.84–0.97 (m, 15H), 1.30 (sext, J = 7.3 Hz, 6H), 1.41–1.57 (m, 6H), 3.40 (s, 3H), 3.57 (dd, J = 5.9 and 3.6 Hz, 2H), 3.65 (dd, J = 6.0 and 3.5 Hz, 2H), 3.75 (s, 2H), 4.61 (s, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ 9.0, 13.9, 27.5, 29.3, 57.9, 59.2, 66.6, 72.0, 98.7.
:1)] to afford a colorless oil (647.8 mg, 79%): 1H NMR (500 MHz, CDCl3) δ 0.85–0.98 (m, 15H), 1.26–1.34 (m, 6H), 1.44–1.58 (m, 6H), 3.75 (s, 2H), 4.42 (s, 2H), 7.24–7.35 (m, 5H); 13C{1H} NMR (125 MHz, CDCl3) δ 9.1, 13.9, 27.5, 29.3, 61.6, 77.4, 127.4, 127.7, 128.3, 139.1.
:1)] to give a colorless oil (8.38 g, 93%): 1H NMR (500 MHz, CDCl3) δ 0.02 (s, 9H), 0.84–0.97 (m, 17H), 1.27–1.34 (m, 6H), 1.43–1.56 (m, 6H), 3.55–3.59 (m, 2H), 3.73–3.76 (m, 2H), 4.56 (s, 2H); 13C{1H} NMR (125 MHz, CDCl3) δ −1.3, 9.0, 13.9, 18.3, 27.5, 29.3, 57.7, 64.9, 98.0.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by a grant from the NSF (CHE-2054497). NMR and mass spectrometry measurements were carried out in the Molecular Education, Technology, and Research Innovation Center (METRIC) at NC State University. XRD analyses were performed at University of Tennessee at Knoxville.References
- M. Taniguchi and J. S. Lindsey, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Publishing Co. Pte. Ltd., Singapore, 2012, pp. 1–80 Search PubMed.
- M. O. Senge, A. A. Ryan, K. A. Letchford, S. A. MacGowan and T. Mielke, Symmetry, 2014, 6, 781–843 CrossRef.
- T. Oba and H. Tamiaki, J. Porphyrins Phthalocyanines, 2014, 18, 919–932 CrossRef CAS.
- M. Taniguchi and J. S. Lindsey, Chem. Rev., 2017, 117, 344–535 CrossRef CAS PubMed.
- P. H. Hynninen, in Chlorophylls, ed. H. Scheer, CRC Press, Inc., Boca Raton, Florida, 1991, pp. 145–209 Search PubMed.
- H. Mazaki, T. Watanabe, T. Takahashi, A. Struck and H. Scheer, Bull. Chem. Soc. Jpn., 1992, 65, 3080–3087 CrossRef CAS.
- Y. Saga and S. Nakagawa, J. Porphyrins Phthalocyanines, 2020, 24, 499–504 CrossRef CAS.
- H. Scheer, in Chlorophylls and Bacteriochlorophylls. Biochemistry, Biophysics, Functions and Applications, ed. B. Grimm, R. J. Porra, W. Rüdiger and H. Scheer, Springer, Dordrecht, The Netherlands, 2006, pp. 1–26 Search PubMed.
- K. Chau Nguyen, P. Wang, R. D. Sommer and J. S. Lindsey, J. Org. Chem., 2020, 85, 6605–6619 CrossRef PubMed.
- K. Chau Nguyen and J. S. Lindsey, J. Org. Chem., 2023, 88, 11205–11216 CrossRef PubMed.
- S. Zhang and J. S. Lindsey, J. Org. Chem., 2017, 82, 2489–2504 CrossRef CAS PubMed.
- K. Chau Nguyen, P. Wang and J. S. Lindsey, New J. Chem., 2021, 45, 569–581 RSC.
- D. T. M. Chung, P. V. Tran, K. Chau Nguyen, P. Wang and J. S. Lindsey, New J. Chem., 2021, 45, 13302–13316 RSC.
- H.-J. Kim, D. K. Dogutan, M. Ptaszek and J. S. Lindsey, Tetrahedron, 2007, 63, 37–55 CrossRef CAS PubMed.
- P. A. Jacobi and S. Rajeswari, Tetrahedron Lett., 1992, 33, 6235–6238 CrossRef CAS.
- P. A. Jacobi and W. Zheng, Tetrahedron Lett., 1993, 34, 2581–2584 CrossRef CAS.
- P. A. Jacobi, S. Murphree, F. Rupprecht and W. Zheng, J. Org. Chem., 1996, 61, 2413–2427 CrossRef CAS.
- W. G. O’Neal, W. P. Roberts, I. Ghosh and P. A. Jacobi, J. Org. Chem., 2005, 70, 7243–7251 CrossRef PubMed.
- W. G. O’Neal, W. P. Roberts, I. Ghosh, H. Wang and P. A. Jacobi, J. Org. Chem., 2006, 71, 3472–3480 CrossRef PubMed.
- W. G. O’Neal and P. A. Jacobi, J. Am. Chem. Soc., 2008, 130, 1102–1108 CrossRef PubMed.
- P. A. Jacobi, H. L. Brielmann, M. Chiu, I. Ghosh, S. I. Hauck, S. Lanz, S. Leung, Y. Li, H. Liu, F. Löwer, W. G. O’Neal, D. Pippin, E. Pollina, B. A. Pratt, F. Robert, W. P. Roberts, C. Tassa and H. Wang, Heterocycles, 2011, 82, 1029–1081 CrossRef CAS PubMed.
- N. A. Petasis and E. I. Bzowej, J. Am. Chem. Soc., 1990, 112, 6392–6394 CrossRef CAS.
- G. P. Bean, in Pyrroles. Part One. The Synthesis and the Physical and Chemical Aspects of the Pyrrole Ring, ed. R. A. Jones, John Wiley & Sons, Inc., New York, 1990, pp. 105–303 Search PubMed.
- S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95–146 CrossRef CAS.
- P. Wang and J. S. Lindsey, Molecules, 2020, 25, 1858 CrossRef CAS PubMed.
- P. Wang and J. S. Lindsey, J. Org. Chem., 2021, 86, 11794–11811 CrossRef CAS PubMed.
- R. F. Lockwood and K. M. Nicholas, Tetrahedron Lett., 1977, 18, 4163–4166 CrossRef.
- K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207–214 CrossRef CAS.
- K. M. Nicholas, J. Org. Chem., 2015, 80, 6943–6950 CrossRef CAS PubMed.
- S. L. Schreiber, T. Sammakia and W. E. Crowe, J. Am. Chem. Soc., 1986, 108, 3128–3130 CrossRef CAS.
- S. L. Schreiber, M. T. Klimas and T. Sammakia, J. Am. Chem. Soc., 1987, 109, 5749–5759 CrossRef CAS.
- K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467–4470 CrossRef.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- K. Imi, K. Imai and K. Utimoto, Tetrahedron Lett., 1987, 28, 3127–3130 CrossRef CAS.
- Y. Fukuda, H. Shiragami, K. Utimoto and H. Nozaki, J. Org. Chem., 1991, 56, 5816–5819 CrossRef CAS.
- Y.-J. Kim, Y. Yoon and H.-Y. Lee, Tetrahedron, 2013, 69, 7810–7816 CrossRef CAS.
- V. A. Keller, J. R. Martinelli, E. R. Streiter and S. D. Burke, Org. Lett., 2002, 4, 467–470 CrossRef CAS PubMed.
- J. Bach, S. D. Bull, S. G. Davies, R. L. Nicholson, H. J. Sanganee and A. D. Smith, Tetrahedron Lett., 1999, 40, 6677–6680 CrossRef CAS.
- J. Bach, C. Blachère, S. D. Bull, S. G. Davies, R. L. Nicholson, P. D. Price, H. J. Sanganee and A. D. Smith, Org. Biomol. Chem., 2003, 1, 2001–2010 RSC.
- S. G. Davies, I. A. Hunter, R. L. Nicholson, P. M. Roberts, E. D. Savory and A. D. Smith, Tetrahedron, 2004, 60, 7553–7577 CrossRef CAS.
- S. Forsén and M. Nilsson, in The Chemistry of the Carbonyl Group, ed. J. Zabicky, Interscience Publishers, London, 1970, pp. 157–240 Search PubMed.
- J. Toullec, Adv. Phys. Org. Chem., 1982, 18, 1–77 CrossRef CAS.
- J. Wirz, Adv. Phys. Org. Chem., 2010, 44, 325–356 CrossRef CAS.
- D. A. Evans and S. L. Bender, Tetrahedron Lett., 1986, 27, 799–802 CrossRef CAS.
- G. C. Micalizio, A. N. Pinchuk and W. R. Roush, J. Org. Chem., 2000, 65, 8730–8736 CrossRef CAS PubMed.
- T. Kobayashi, K. Tanaka, M. Ishida, M. Yamakita, H. Abe and H. Ito, Chem. Commun., 2018, 54, 10316–10319 RSC.
- D. A. Evans, T. C. Britton and J. A. Ellman, Tetrahedron Lett., 1987, 28, 6141–6144 CrossRef CAS.
- B. Chen, R. Y. Y. Ko, M. S. M. Yuen, K.-F. Cheng and P. Chiu, J. Org. Chem., 2003, 68, 4195–4205 CrossRef CAS PubMed.
- G. J. Kramp, M. Kim, H.-J. Gais and C. Vermeeren, J. Am. Chem. Soc., 2005, 127, 17910–17920 CrossRef CAS PubMed.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815–3818 CrossRef CAS.
- A. Innitzer, L. Brecker and J. Mulzer, Org. Lett., 2007, 9, 4431–4434 CrossRef CAS PubMed.
- D. Seyferth and M. A. Weiner, J. Am. Chem. Soc., 1961, 83, 3583–3586 CrossRef CAS.
- C. S. Shiner, T. Tsunoda, B. A. Goodman, S. Ingham, S.-H. Lee and P. E. Vorndam, J. Am. Chem. Soc., 1989, 111, 1381–1392 CrossRef CAS.
- J. W. Labadie and J. K. Stille, J. Am. Chem. Soc., 1983, 105, 6129–6137 CrossRef CAS.
- R. L. Danheiser, S. K. Gee and J. J. Perez, J. Am. Chem. Soc., 1986, 108, 806–810 CrossRef CAS.
- D. C. Kapeller and F. Hammerschmidt, Tetrahedron, 2010, 66, 591–598 CrossRef CAS.
- G. C. M. Lee, US 5376676, 1994.
- W. C. Still, J. Am. Chem. Soc., 1978, 100, 1481–1487 CrossRef CAS.
- K. H. Lee and H. W. Moore, J. Korean Chem. Soc., 2003, 47, 229–236 CrossRef CAS.
- J. Åhman and P. Somfai, Synth. Commun., 1994, 24, 1117–1120 CrossRef.
- S. R. Gilbertson and W. D. Wulff, Synlett, 1989, 47–49 CAS.
- E. Fernández-Megía and S. V. Ley, Synlett, 2000, 455–458 Search PubMed.
- F. D. Wael, G. G. Muccioli, D. M. Lambert, T. Sergent, Y.-J. Schneider, J.-F. Rees and J. Marchand-Brynaert, Eur. J. Med. Chem., 2010, 45, 3564–3574 CrossRef PubMed.
- C.-M. Chou, Y.-W. Tung, M.-I. Ling, D. Chan, W. Phakhodee and M. Isobe, Heterocycles, 2012, 86, 1323–1339 CrossRef CAS PubMed.
- A. Flores-Parra and F. Khuong-Huu, Tetrahedron, 1986, 42, 5925–5930 CrossRef CAS.
- N. T. Anh, Top. Curr. Chem., 1980, 88, 145–166 CrossRef.
- D. Seebach and A. K. Beck, Org. Synth., 1971, 51, 39–43 CrossRef CAS.
- S. Hörtensteiner and B. Kräutler, Biochim. Biophys. Acta, 2011, 1807, 977–988 CrossRef PubMed.
- B. Kräutler, Chem. Soc. Rev., 2014, 43, 6227–6238 RSC.
- S. Hörtensteiner, M. Hauenstein and B. Kräutler, Adv. Bot. Res., 2019, 90, 213–271 Search PubMed.
- P. Wang, C. A. Karg, N. Frey, J. Frädrich, A. M. Vollmar and S. Moser, Arch. Pharm., 2021, 354, e2100061 CrossRef PubMed.
- C. A. Karg, M. Taniguchi, J. S. Lindsey and S. Moser, Planta Med., 2023, 89, 637–662 CrossRef CAS PubMed.
- Y. Liu, S. Zhang and J. S. Lindsey, Nat. Prod. Rep., 2018, 35, 879–901 RSC.
- A. T. Nguyen Tran, Z. Wu, D. T. M. Chung, P. Nalaoh and J. S. Lindsey, New J. Chem., 2023, 47, 13626–13637 RSC.
- N. Wang, R. Liu, Q. Xu and X. Liang, Chem. Lett., 2006, 35, 566–567 CrossRef CAS.
- B. O. Lindgren and T. Nilsson, Acta Chem. Scand., 1973, 27, 888–890 CrossRef CAS.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- A. J. Gordon and R. A. Ford, The Chemist's Companion, Wiley-Interscience, New York, 1972, p. 451 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C{1H} NMR spectra for all new compounds; 1H NMR and 13C{1H} NMR spectra for known compounds 4, 5-R, 7, 18–23, Sn-Me, Sn-MOM, Sn-THP, Sn-MEM, Sn-Bn, Sn-SEM, and Sn-(OMe)2; single-crystal X-ray diffraction data. CCDC 2287122 (10), 2287123 (11) and 2287124 (12). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj03900e |
| ‡ Equal contributions by both coauthors. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |