
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron transfer between neptunium and sodium chlorite in acidic chloride media†
Brian T.
Arko
 ab,
David
Dan
a,
Sara L.
Adelman
*a,
David B.
Kimball
*a,
Stosh A.
Kozimor
*a and
Jenifer C.
Shafer
*b
ab,
David
Dan
a,
Sara L.
Adelman
*a,
David B.
Kimball
*a,
Stosh A.
Kozimor
*a and
Jenifer C.
Shafer
*b
aLos Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: dkimball@lanl.gov; stosh@lanl.gov; Web: https://mailto:sadelman@lanl.gov
bDepartment of Chemistry, Colorado School of Mines, Golden, CO 90401, USA. E-mail: jshafer@mines.edu
First published on 22nd December 2023
Abstract
Controlling aqueous 5f-element electron transfer chemistry is critical for processing efforts associated with actinide technologies. Often, redox agents are added during actinide processing steps to control actinide redox chemistry and manipulate the actinide oxidation states for the separation. Sodium chlorite, NaClO2(aq), represents one of these useful redox agents. For example, NaClO2(aq) finds widespread application in the processing of plutonium and americium. Surprisingly, however, redox reactivity between NaClO2(aq) and other actinides, like neptunium, has been largely ignored. That knowledge gap is addressed herein. We characterized some redox reactivity between NaClO2(aq) and Np4+(aq) and identified experimental conditions that held neptunium in the +4 oxidation state or converted Np4+(aq) to NpO22+(aq) or NpO21+(aq). This was achieved by carefully adjusting four variables: ingoing concentrations of (1) Np4+(aq), (2) NaClO2(aq), (3) Cl1−(aq), and (4) H1+(aq). We discovered that three neptunium oxidation states (+4, +5, and +6) could be accessed using one ubiquitous redox agent, NaClO2(aq). These results highlight the diverse electron transfer chemistry available to neptunium in aqueous solutions, provide new insight on how neptunium reacts with NaClO2(aq), and are discussed within the context of their importance to plutonium and americium processing.
Introduction
Controlling electron transfer chemistry accessible to the 5f-elements in aqueous media is critical for nearly every actinide-based technological application. These applications span from isotope production, environmental restoration, nuclear fuel rod fabrication, space exploration, and harvesting actinides from weapons program waste streams.1–19 The latter topic was the major motivating factor for this particular research effort. For instance, at Los Alamos National Laboratory (LANL), transuranic actinides – like plutonium (Pu) and americium (Am) – are recovered from aqueous waste streams.20–23 Success associated with this waste stewardship is valuable and responsible. It reduces burden on taxpayers by lowering transuranic waste costs, decreases the amount of transuranic waste generated, and recycles valuable and rare radioisotopes (e.g., 238Pu, 239Pu, 240Pu, 241Pu, 242Pu, and 241Am) for national security and industrial usage.To recover plutonium and americium from aqueous waste streams, LANL implemented the experimental chloride extraction line (EXCEL) and chloride extraction and recovery (CLEAR) aqueous processing methods.15,24–28 In terms of plutonium recovery, waste entering the processing line contains plutonium in a variety of oxidation states. Hence, successful plutonium recovery requires a valence state adjustment that holds plutonium in the +4 oxidation state during subsequent chemical processing steps. This plutonium oxidation-state adjustment can be carried out using sodium chlorite; NaClO2(aq). The robustness associated with using NaClO2(aq) as a plutonium valence adjusting agent has been documented by the successful recovery of plutonium from diverse waste streams using EXCEL for decades.16,29
Accompanying changes to the national nuclear agenda, described in the Nuclear Posture Review, are new chemical challenges that face aqueous recovery and recycling of plutonium and americium.30–32 One obstacle is associated with needs to accommodate increasingly diverse waste feedstocks. Processing these feedstocks is complicated because they contain a wide range of chemical constituents. In addition, their chemical identities and quantities change with time. One relevant example is neptunium-237: 237Np, half-life (t1/2) = 2.144(7) × 106y.33 Concerns regarding 237Np contamination stem from substantial 237Np ingrowth in aged plutonium/americium containing waste. Fig. 1 qualifies these concerns. It shows that the 237Np contaminant is generated via α-decay from its 241Am [t1/2 = 432.6(6) y] parent radionuclide, which in turn is the β− decay product from 241Pu [t1/2 = 14.329(29) y].33 It also highlights how “aged” waste will contain substantial quantities of 237Np.34–38 Unfortunately, it is unknown how 237Np(aq) will respond to the antecedently described plutonium valence adjustment step because redox chemistry between neptunium and NaClO2(aq) has not been studied in depth. Defining electron transfer chemistry between neptunium and NaClO2(aq) in aqueous solutions is relevant to plutonium/americium processing. That information will show how 237Np contaminants move through the EXCEL and CLEAR processing lines and provide insight for maintaining robustness and effectiveness in plutonium/americium waste processing efforts.
 | ||
| Fig. 1 Calculated isotopic decay products for selected transuranic radioisotopes within aged plutonium calculated from aged plutonium isotopic content (WGPu-C, 16–19% 240Pu as defined in the compendium of material composition data for radiation transport modeling).39 | ||
To address the aforementioned 237Np processing concerns, we set out to characterize some redox chemistry between 237Np(aq) and the NaClO2(aq). We discovered that we could force neptunium into three different oxidation states (+4, +5, +6) using the NaClO2(aq) redox agent by manipulating four experimental variables. Those variables were ingoing concentrations of Np4+(aq), NaClO2(aq), Cl1−(aq), and H1+(aq). Although α-particle radiation has been known to influence neptunium oxidation state chemistry, these attributes were not considered.40Scheme 1 and Table 1 showcase that NaClO2(aq) acted as a Np4+(aq) valence holding agent under a wide range of experimental conditions. We also found experimental conditions that converted Np4+(aq) to the NpO21+(aq) mono-cation or the NpO22+(aq) di-cation. Stable solutions of NpO21+(aq) were obtained when the ingoing concentrations of Np4+(aq) (0.81 mM), NaClO2(aq) (3.4 mM), Cl1−(aq) (0.026 M), and H1+(aq) (0.02 M) were all relatively low. Stable solutions of NpO22+(aq) were obtained when the ingoing concentration of Np4+(aq) was high (3.1 mM to 1.5 mM), NaClO2(aq) was high (160 to 32 mM), Cl1−(aq) ranged 0.02 to 9.2 M, and H1+(aq) was between 0.02 and 5.4 M. Overall, these results demonstrated that that reactivity between Np4+(aq) and NaClO2(aq) was diverse, predictable, and therefor controllable. That insight advances understanding about neptunium electron transfer reactions under experimental conditions that are relevant to those described above during EXCEL and CLEAR processing of plutonium and 241Am.
 | ||
| Scheme 1 General Np4+(aq) reaction products under various experimental conditions. The “entry” showed in parentheses references the experimental conditions documented in Table 1. Colors associated with concentrations are depicted as relatively high or low. | ||
| Entry | Dominant species (t = 1 h) | Dominant species (t = 24 h) | Initial Np4+(aq) (mM) | Initial NaClO2(aq) (mM) | Initial H1+(aq) (M) | Ingoing Cl1− totala (M) |
|---|---|---|---|---|---|---|
| (Cl1− from HCl) + (Cl1− from LiCl) + (Cl1− from NpCl4) | ||||||
| a Calculations show ingoing Cl1−(aq) content from HCl(aq), LiCl(aq), and NpCl4(aq) and do not include NaClO2(aq) nor NaClO2(aq) disproportionation products. | ||||||
| 1 | NpO21+(aq) | NpO21+(aq) | 0.81 | 3.4 | 0.02 | 0.023 |
| (0.02) + (0) + 0.003 | ||||||
| 2 | NpO21+(aq) | NpO22+(aq) | 1.5 | 32 | 0.02 | 0.026 |
| (0.02) + (0) + 0.006 | ||||||
| 3 | NpO21+(aq) | NpO22+(aq) | 3.1 | 31 | 0.04 | 0.052 |
| (0.04) + (0) + 0.012 | ||||||
| 4 | NpO21+(aq) | NpO22+(aq) | 1.5 | 160 | 0.02 | 0.026 |
| (0.02) + (0) + 0.006 | ||||||
| 5 | NpO21+(aq) | NpO22+(aq) | 3.1 | 160 | 0.04 | 0.052 |
| (0.04) + (0) + 0.012 | ||||||
| 6 | Np4+(aq) | NpO22+(aq) | 3.1 | 31 | 0.04 | 9.1 |
| (0.04) + (9.0) + 0.012 | ||||||
| 7 | Np4+(aq) | NpO22+(aq) | 1.5 | 32 | 0.02 | 9.2 |
| (0.02) + (9.2) + 0.006 | ||||||
| 8 | Np4+(aq) | NpO22+(aq) | 1.5 | 160 | 0.02 | 9.2 |
| (0.02) + (9.2) + 0.006 | ||||||
| 9 | Np4+(aq) | NpO22+(aq) | 3.1 | 160 | 0.04 | 9.1 |
| (0.04) + (9.0) + 0.012 | ||||||
| 10 | Np4+(aq) | NpO22+(aq) | 2.9 | 160 | 5.4 | 5.4 |
| (5.4) + (0) + 0.012 | ||||||
| 11 | Np4+(aq) | Np4+(aq) | 1.5 | 160 | 6.1 | 6.1 |
| (6.1) + (0) + 0.006 | ||||||
| 12 | Np4+(aq) | Np4+(aq) | 1.5 | 160 | 7.1e | 7.1 |
| (7.1) + (0) + 0.006 | ||||||
| 13 | Np4+(aq) | Np4+(aq) | 2.9 | 160 | 7 | 7.0 |
| (7.0) + (0) + 0.012 | ||||||
| 14 | Np4+(aq) | Np4+(aq) | 2.9 | 32 | 5.4 | 5.4 |
| (5.4) + (0) + 0.012 | ||||||
| 15 | Np4+(aq) | Np4+(aq) | 2.9 | 31 | 6.2 | 6.2 |
| (6.2) + (0) + 0.012 | ||||||
| 16 | Np4+(aq) | Np4+(aq) | 1.5 | 32 | 6.1 | 6.1 |
| (6.1) + (0) + 0.006 | ||||||
| 17 | Np4+(aq) | Np4+(aq) | 2.9 | 31 | 7.0 | 7.0 |
| (7.0) + (0) + 0.012 | ||||||
| 18 | Np4+(aq) | Np4+(aq) | 2.9 | 31 | 5.3 | 5.4 |
| (5.4) + (0) + 0.012 | ||||||
| 19 | Np4+(aq) | Np4+(aq) | 1.5 | 32 | 7.1 | 7.1 |
| (7.1) + (0) + 0.006 | ||||||
Before discussing reactivity between Np4+(aq) and NaClO2(aq), we found it instructive to comment – at a high level – on the aqueous reactivity of NaClO2(aq) in Cl1−(aq) containing solutions. The chemistry of NaClO2(aq) has been the subject of numerous experimental studies described previously.41–47 The NaClO2 salt has been structurally characterized.48–50 The ClO21− anion contains chlorine in the +3 oxidation state, and (based on the standard half-cell potentials) is the strongest oxidizer in the chlorine oxyanion family.45 As testament, many salts containing the ClO21− anion are reported to decompose explosively when exposed to heat and shock, e.g. salts of Ag1+, Hg1+, Tl1+, Pb2+, Cu2+, and NH41+.51 Hence, inorganic ClO21− salts are often regarded as being unstable and highly reactive. In aqueous solutions, the conjugate acid of ClO21− (chlorous acid; HClO2) is a weak acid (pKa ∼ 1.94),52 one of the most reactive (least stable) oxoacids of chlorine,53 and has only been observed in aqueous solution at low concentrations.54 In aqueous chloride solutions, the chemistry of the ClO21−(aq)/HClO2(aq) pair is dominated by disproportionation reactions whose product ratios are heavily influenced by the H1+(aq) and Cl1−(aq) concentrations associated with the aqueous matrix.47,55–57 For example, in aqueous matrixes and in the absence of Cl1−(aq), the ClO21−(aq) anion is reported to disproportionate and form gaseous chlorine dioxide [ClIVO2(g)], chlorate [ClVO31−(aq)], and chloride [Cl1−(aq)] (eqn (1)). In contrast, when Cl1−(aq) concentrations are high (see ref. 58 for details), the disproportionation reaction favors formation of the ClO2(g) and Cl1−(aq) pair (eqn (2)) and the formation of ClO31−(aq) is suppressed. According to Kieffer and Gordon, the rate of ClO21−(aq) disproportionation also varies with Cl1−(aq) content. For example the disproportionation reaction rate increases substantially when moving from low (no ingoing) Cl1−(aq) concentration (t1/2 = 389 ± 3 min without added Cl1−(aq)) to higher (100 mM) Cl1−(aq) concentrations (t1/2 = 6.85 ± 0.05 min).56,57 These disproportionation rates are not exact, and likely represent a combination of multiple chemical events and intertwined reactions.58
ClO21−(aq) disproportionation in low Cl1−(aq) conditions
| 4HClO2 → 2ClO2(g) + ClO31−(aq) + Cl1−(aq) + 2H1+(aq) + H2O | (1) |
ClO21−(aq) disproportionation in high Cl1−(aq) conditions
| 5HClO2 → 4ClO2(g) + Cl1−(aq) + H1+(aq) + 2H2O | (2) |
Results and discussion
Generating oxidation state pure solutions of NpO21+(aq) and NpO22+(aq) with NaClO2(aq)
We discovered experimental conditions that selectively oxidized Np4+(aq) to either NpO21+(aq) or NpO22+(aq) in aqueous acidic solutions of HCl(aq) (0.02 to 5.4 M; entries 1 to 10 in Table 1) without the exclusion of air. To carry out these studies, we generated an oxidation state and chemically pure aqueous stock solution of Np4+(aq) (66 mM) using a previously described method.59 Aliquots from this Np4+(aq) stock solution were combined with the NaClO2(aq) oxidant at fixed Cl1−(aq) concentrations. Transformations that converted Np4+(aq) to either NpO21+(aq) and/or NpO22+(aq) were subsequently monitored using ultraviolet-visible (UV-vis) absorption spectroscopy. Scheme 1 and Table 1 provide a high-level summary of how the matrix compositions impacted product formation.We found that exclusive formation of NpO21+(aq) occurred when ingoing concentrations for the 237Np(aq) analyte (0.81 mM), NaClO2(aq) redox agent (3.4 mM), Cl1−(aq) complexing agent (0.026 M), and H1+(aq) (0.02 M) were all low, relative to the other experimental conditions we examined (entry 1 in Table 1). As documented in Fig. 2, addition of NaClO2(aq) to a solution of Np4+(aq) caused the UV-vis absorption peaks from Np4+(aq) at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1 (960 nm) and 8700 cm−1 (1150 nm) to vanish within 1 day (see Fig. 3). Concomitantly, new peaks from NpO21+(aq) at 10200 cm−1 (980 nm) and 9100 cm−1 (1100 nm) emerged. Fig. 2 and 3 additionally documented that this NpO21+(aq) product was stable during a 5-day monitoring process, as no other neptunium oxidation states were detected by UV-vis spectroscopy. To obtain additional insight into the transformation of Np4+(aq) to NpO21+(aq), we characterized the rate at which absorption peaks from Np4+(aq) decreased in intensity and peaks associated with NpO21+(aq) were prevalent for 5 days (Fig. 3). The reaction rate laws were calculated, and subsequent analyses suggested that oxidation of Np4+(aq) to NpO21+(aq) proceeded by fractional order dependence on both the Np4+(aq) and ClO21−(aq) reagents, 0.300 ± 0.008 and 0.110 ± 0.002, respectively. Fractional order reactions can be indicative of chain reactions propagated by radical species present in solution and many radical reactions pathways could be responsible for the NaClO2(aq) initiated conversion of Np4+(aq) to NpO21+(aq).41,60–63
500 cm−1 (960 nm) and 8700 cm−1 (1150 nm) to vanish within 1 day (see Fig. 3). Concomitantly, new peaks from NpO21+(aq) at 10200 cm−1 (980 nm) and 9100 cm−1 (1100 nm) emerged. Fig. 2 and 3 additionally documented that this NpO21+(aq) product was stable during a 5-day monitoring process, as no other neptunium oxidation states were detected by UV-vis spectroscopy. To obtain additional insight into the transformation of Np4+(aq) to NpO21+(aq), we characterized the rate at which absorption peaks from Np4+(aq) decreased in intensity and peaks associated with NpO21+(aq) were prevalent for 5 days (Fig. 3). The reaction rate laws were calculated, and subsequent analyses suggested that oxidation of Np4+(aq) to NpO21+(aq) proceeded by fractional order dependence on both the Np4+(aq) and ClO21−(aq) reagents, 0.300 ± 0.008 and 0.110 ± 0.002, respectively. Fractional order reactions can be indicative of chain reactions propagated by radical species present in solution and many radical reactions pathways could be responsible for the NaClO2(aq) initiated conversion of Np4+(aq) to NpO21+(aq).41,60–63
 | ||
| Fig. 2 UV-vis absorption spectra documenting oxidation of Np4+(aq) to NpO21+(aq) as a function of time (top, initial; middle, after 60 s; bottom, after 5 days). Entry #1 in Table 1 describe the reaction conditions. Ingoing concentrations were low for Np4+(aq) (0.81 mM), low for NaClO2(aq) (3.4 mM), low for H1+(aq) (0.02 M), and low for Cl1−(aq) (0.026 M). Hence, the ingoing HCl(aq) concentration was 0.02 M and there no added LiCl(aq). | ||
 | ||
| Fig. 3 Peak intensity from UV-vis spectra from Np4+(aq) (teal trace from the absorption peak at 10400 cm−1, 960 nm) and NpO21+(aq) (magenta trace from the absorption peak at 9100 cm−1, 1100 nm) plotted as a function of two different time intervals (left, 0 to 1 min; right, 0.1 to 5 days). Entry #1 in Table 1 describe the reaction conditions. Ingoing concentrations were low for Np4+(aq) (0.81 mM), low for NaClO2(aq) (3.4 mM), low for H1+(aq) (0.02 M), and low for Cl1−(aq) (0.02 M). Hence, the ingoing HCl(aq) concentration was 0.02 M and there was no added LiCl. | ||
Subtle changes in the aqueous matrix identity substantially impacted the Np4+(aq) oxidation process. As a representative example, data displayed in Fig. 4 documented how oxidizing Np4+(aq) to NpO21+(aq) was dependent on the ingoing NaClO2(aq) quantities. To generate these data, we sequentially added aliquots of NaClO2(aq) (0.072 μmol; 10 μL of a 7.2 mM stock solution) to an aqueous solution that was dilute in Np4+(aq) (1.6 mM, 1 μmol, 600 μL) and dilute in HCl(aq) (0.02 M). Analyses by UV-vis spectroscopy showed that the Np4+(aq) reagent reacted immediately with NaClO2(aq). Then, the Np4+(aq) and NpO21+(aq) product-to-reagent ratio stabilized within 60 s. These experiments showed that reacting a small amount [more than two equivalents vs. Np4+(aq)] of NaClO2(aq) (2.2 μmol; 330 μL of a 7.2 mM solution) with one equivalent of Np4+(aq) (1 μmol) generated a one-to-one mixture of Np4+(aq) and NpO21+(aq). Increasing the quantity of NaClO2(aq) increased the amount of NpO21+(aq) generated until approximately 4.5 equivalents of NaClO2(aq) (4.5 μmol; 630 μL of 7.2 mM solution) vs. one equivalent of Np4+(aq) (1 μmol) had been added. In this situation, the Np4+(aq) reagent was completely consumed and NpO21+(aq) formed. The NpO21+(aq) species was stable under ambient conditions during a 5-day monitoring process.
 | ||
| Fig. 4 A plot showing how oxidation of Np4+(aq) (teal trace from the absorption peak at 10400 cm−1, 960 nm) to NpO21+(aq) (magenta trace from the absorption peak at 10200 cm−1, 980 nm) depended on the amount of added NaClO2(aq). Ingoing concentrations were low for Np4+(aq) (1.6 mM), low for H1+(aq) (0.02 M), and low for Cl1−(aq) (0.026 M). Hence, the ingoing HCl(aq) concentration was 0.02 M and there was no added LiCl. The NaClO2(aq) started at zero and increased with addition of 10 μL aliquots of a NaClO2(aq) (7.2 mM) solution. Concentrations were corrected for dilutions. | ||
The Np4+(aq) cation also oxidized to NpO21+(aq) when the NaClO2(aq) concentration was increased from 3.4 mM to 32 mM; entry 2 in Table 1. However, under these conditions the NpO21+(aq) mono-cation was not stable with time and further oxidation to NpO22+(aq) occurred (Fig. 5). Absorption peaks from NpO21+(aq) at 10400 cm−1 (980 nm) and 8700 cm−1 (1150 nm) were replaced within one hour by features characteristic of NpO22+(aq); 7900 cm−1 (1270 nm). Monitoring this product by UV-vis spectroscopy revealed that NpO22+(aq) was stable for at least 5 days, when monitoring ceased. Comparing this data with that presented above highlighted how formation of NpO22+(aq)vs. NpO21+(aq) could be controlled based on ingoing NaClO2(aq) concentrations. The NpO21+(aq) mono-cation was generated when the ingoing concentration of NaClO2(aq) was low (3.4 mM; entry 1, Table 1). In contrast, the NpO22+(aq) di-cation was generated with the ingoing concentration of NaClO2 was high (32 mM; entry 2, Table 1). There is a disclaimer associated with this conclusion. Controlling oxidation of Np4+(aq) to either NpO21+(aq) or NpO22+(aq) required the ingoing concentrations of Np4+(aq) and NaClO2(aq) as well as ingoing concentrations of Cl1−(aq) and H1+(aq) to be carefully managed; see experiments captured in entries 2 to 19 in Table 1. For example, adding NaClO2(aq) (32 mM or 31 mM to 160 mM entries 2 to 5) to Np4+(aq) (1.5 mM to 3.1 mM) with low Cl1−(aq) and H1+(aq) concentrations (0.02 to 0.04 M) generated NpO21+(aq) at the 1 h timestamp and that NpO21+(aq) converted to NpO22+(aq) within 24 h. Increasing the Cl1−(aq) content changed this outcome. By using the LiCl(aq) to increase Cl1− concentrations to >9 M (entries 6 to 9) and holding the H1+ concentration low (0.02 M to 0.04 M) stabilized the Np4+(aq) regent as the dominant specie present in solution at the 1 h timestamp. Then, the Np4+(aq) cation oxidized to NpO22+(aq) within 24 h. Increasing both H1+(aq) and Cl1− content to >5.4 M shut down the oxidation of Np4+(aq) to NpO22+(aq) and Np4+(aq) was stable throughout the 24 h monitoring duration.
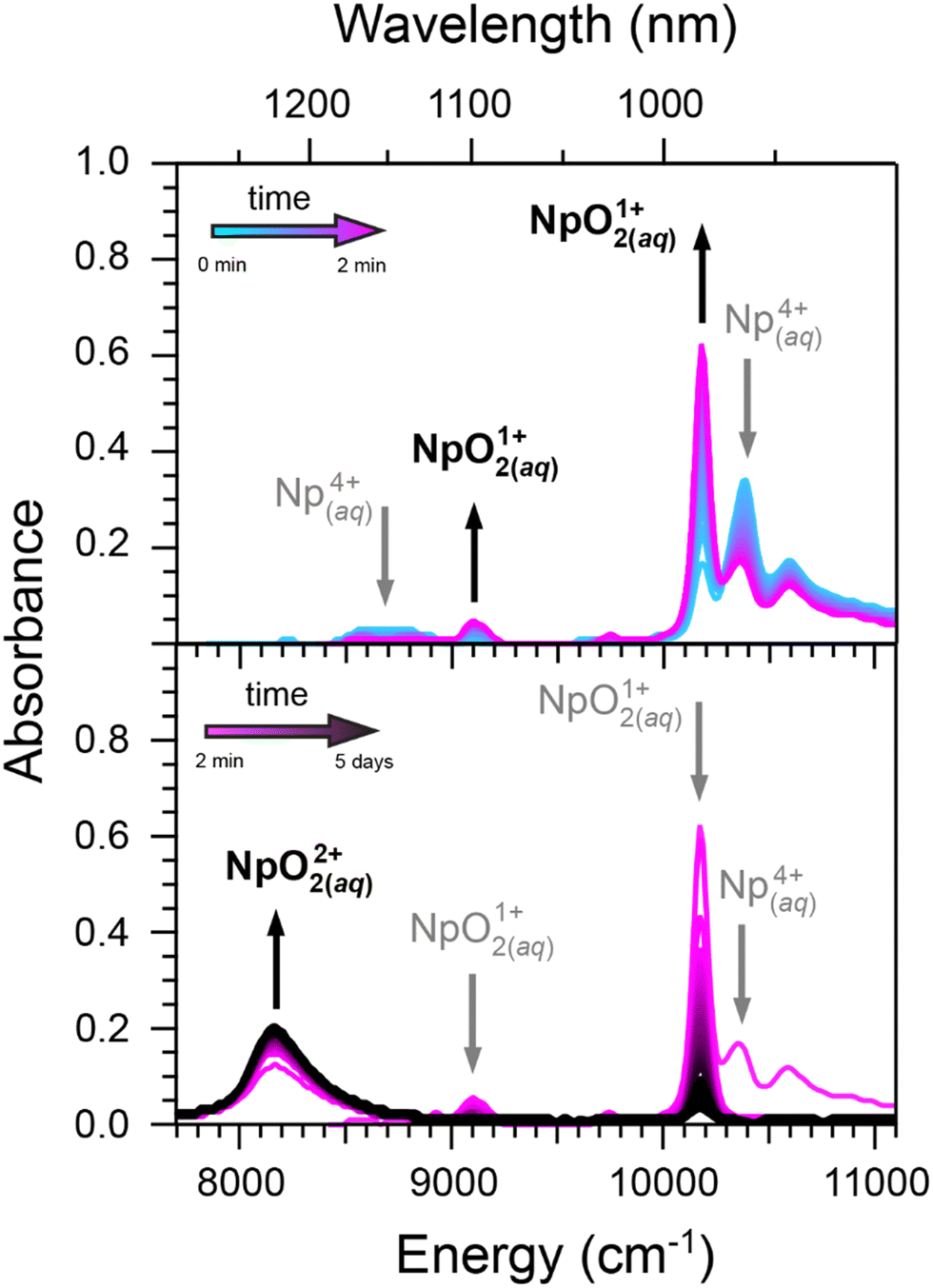 | ||
| Fig. 5 Top: UV-vis absorption spectra documenting oxidation of Np4+(aq) (teal trace) to NpO21+(aq) (magenta trace) as a function of time (2 min). Bottom: Subsequent oxidation of NpO21+(aq) (magenta trace) to NpO22+(aq) (black trace). Entry #2 in Table 1 describe the reaction conditions. Ingoing concentrations were low for Np4+(aq) (1.5 mM), low for NaClO2(aq) (32 mM), low for H1+(aq) (0.02 M), and low for Cl1−(aq) (0.02 M). Hence, the ingoing HCl(aq) concentration was 0.02 M and there was no added LiCl. | ||
We characterized the reaction rates for entry 2 in Table 1 that were associated with the transformation of (1st) Np4+(aq) to NpO21+(aq) and (2nd) NpO21+(aq) to NpO22+(aq) (Fig. 6). The oxidation of Np4+(aq) to NpO21+(aq) proceeded with fractional order dependence on both the Np4+(aq) (0.300 ± 0.008) and ClO21−(aq) (0.110 ± 0.002) reagents. The reaction rate law derived from the oxidation of NpO21+(aq) to NpO22+(aq) was approximately first order in NpO21+(aq) (0.98 ± 0.02) and higher order (1.7 ± 0.01) with regards to NaClO2(aq). No evidence was obtained that suggested the NpO22+(aq) di-cation formed via disproportionation of the NpO21+(aq) mono-cation to Np4+(aq) and NpO22+(aq). For instance, regeneration of Np4+(aq) – or a sustained presence of Np4+(aq) – after initial formation of NpO21+(aq) was not observed. Instead, our data suggested that NpO21+(aq) converted directly to NpO22+(aq). Admittedly, this data does not completely rule out the possibility of a low-concentration Np4+(aq) transient intermediate nor the possibility of disproportionation reaction pathways. However, with the data in hand at this time, it seemed possible that NpO21+(aq) was converted directly NpO22+(aq) by the redox processes accessed with NaClO2(aq) and/or NaClO2(aq) decomposition productions, see eqn (1) and (2).
 | ||
| Fig. 6 Peak intensity from UV-vis spectra from Np4+(aq) (teal trace at 10400 cm−1, 960 nm), NpO21+(aq) (magenta trace at 9100 cm−1, 1100 nm), and NpO22+(aq) (black trace at 8200 cm−1, 1250 nm) plotted as a function of two different time intervals (left, 0 to 2 min; right, approximately 2 min to 10 days). Entry #5 in Table 1 describe the reaction conditions. Ingoing concentrations were high for Np4+(aq) (3.1 mM), high for NaClO2(aq) (160 mM), low for H1+(aq) (0.04 M), and low for Cl1−(aq) (0.052 M). Hence, the ingoing HCl(aq) concentration was 0.04 M and there was no added LiCl. | ||
There were other experimental conditions that generated a NpO21+(aq) intermediate (within an hour) that in turn oxidized Np4+(aq) to NpO22+(aq) (entries 3 to 10 from Table 1). Each of these scenarios had three commonalities: the ingoing H1+(aq) concentrations ranged 0.02 to 5.4 M, the ingoing Cl1−(aq) concentration ranged from 0.026 to 9.1 M, the ingoing Np4+(aq) concentrations were larger than 0.81 mM (entries 2 through 5 in Table 1), and the ingoing NaClO2(aq) concentrations were high (>31 mM). Modifying these variables changed the reaction outcomes. For example, increasing Cl1−(aq) content to >9 M – using LiCl(aq) – masked observation of a NpO21+(aq) intermediate (entries 6 to 10 in Table 1; Fig. 7 and 8). In these situations, we only observed disappearance of Np4+(aq) and formation of NpO22+(aq) (entries 6 to 9). Monitoring this reaction as a function of time revealed that the loss of Np4+(aq) and formation of NpO22+(aq) proceeded at fourth order (4.11 ± 0.09) dependence with respect to Np4+(aq) and fractional (0.50 ± 0.07) order with respect to NaClO2(aq) (Fig. 8). Although fourth order rate laws have been measured before,64–67 we acknowledge their unusuality and rarity. In our case, we speculated that the measured fourth order rate law resulted from numerous intertwined reactions that were occurring simultaneously. One obvious possibility included NaClO2(aq) decomposition products reacting directly with Np4+(aq) to generate NpO22+(aq) and maybe a transient NpO21+(aq) intermediate. It is also possible that the NaClO2(aq) could react with Np4+(aq) directly. If any NpO21+(aq) formed, that mono-cation could disproportionate to generate NpO22+(aq) and regenerate Np4+(aq). Note, we did not detect evidence for formation of a Np4+(aq) intermediate by UV-vis. Hence, if Np4+(aq) formed, it was generated in relatively small quantities. Regardless, we are intrigued by this reaction and our current efforts are focused on better characterizing how NaClO2(aq) incites Np4+(aq) transformation to NpO22+(aq) in high Cl1−(aq) solutions.
 | ||
| Fig. 7 UV-vis absorption spectra documenting oxidation of Np4+(aq) (teal trace) to NpO22+(aq) (black trace). Entry #7 in Table 1 describe the reaction conditions. Ingoing concentrations were 1.5 mM for Np4+(aq), 32 mM for NaClO2(aq), 0.02 M for H1+(aq), and 9.2 M for Cl1−(aq). Hence the ingoing HCl(aq) concentration was 0.02 M and for LiCl(aq) was 9.2 M. | ||
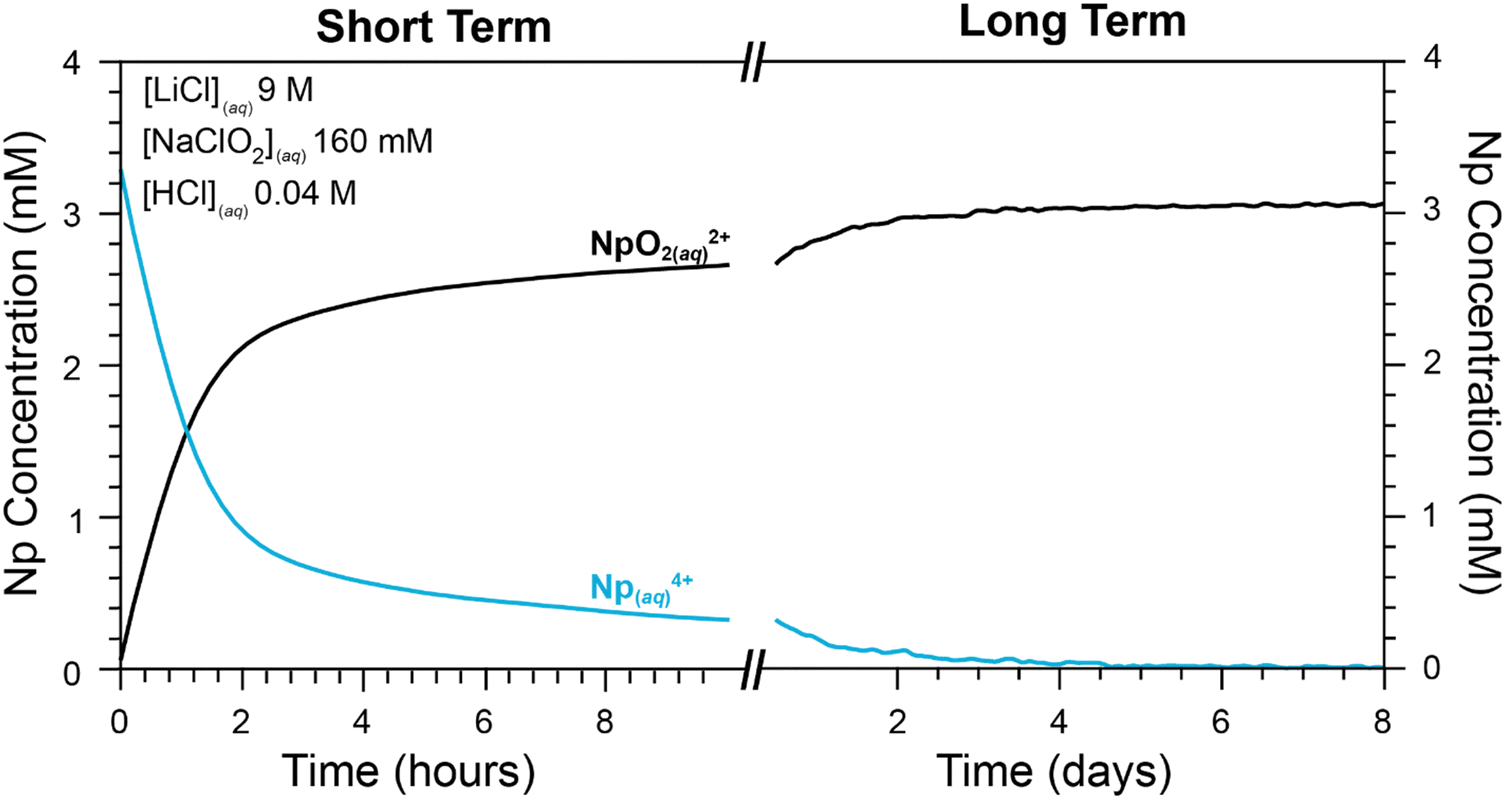 | ||
| Fig. 8 Peak intensity from UV-vis spectra from Np4+(aq) (teal trace at 10400 cm−1, 960 nm) and NpO22+(aq) (black trace at 7900 cm−1, 1270 nm) plotted as a function of two different time intervals (left, 0 to 2 min; right, 2 min to 10 days). Entry #9 in Table 1 describe the reaction conditions. Ingoing concentrations were high for Np4+(aq) (3.1 mM), high for NaClO2(aq) (160 mM), low for H1+(aq) (0.04 M), and high for Cl1−(aq) (9.1 M). Hence, the ingoing HCl(aq) concentration was 0.04 M and for LiCl(aq) was 9.1 M. | ||
As shown in Table 1 (entries 11 to 19), oxidation of Np4+(aq) to NpO22+(aq) was halted when the H1+(aq) and Cl1−(aq) concentrations were ≥5.4 M, when the ingoing Np4+(aq) concentration ranged 1.5 to 2.9 mM, and the NaClO2(aq) concentration was high (31 to 160 mM). It is particularly important to highlight these observations because of their relevance to EXCEL processing methods described in the introduction. Notice, that the NaClO2(aq) redox agent held neptunium in the +4 oxidation state under conditions that mimicked plutonium processing by EXCEL: when the H1+(aq) and Cl1−(aq) concentrations were >5.4 M and the NaClO2(aq) concentrations varied from 31 mM to 160 mM, and Np4+(aq) concentrations varied from 1.5 mM to 2.9 mM (entries 11 to 19 in Table 1). This data suggested that – all things being equal – neptunium should follow plutonium through within the EXCEL processing steps.
Molar extinction coefficients
Table 2 compares molar extinction coefficients (ε) for neptunium absorption peaks calculated for experimental conditions described in Table 1 when Np was in a single oxidation state. These data were important for quantifying Np4+(aq), NpO21+(aq), and NpO22+ abundance in the reactions studied herein and provided insight into the solution phase behavior of Np4+(aq) and NpO22+(aq). In terms of the latter topic, we observed that ε for the Np4+(aq) absorption feature at 10400 cm−1 (960 nm) decreased with increasing Cl1−(aq) concentration. In addition, increasing Cl1−(aq) shifted the Np4+(aq) absorption peaks lower in energy (Fig. 9). These absorption changes are often associated with increased Cl1−(aq) complexation of neptunium.36,37,68 It was interesting to note that ε for the Np4+(aq) peak at 8700 cm−1 (1150 nm) peak was shifted in energy but the ε value was unaffected by changes in Cl1−(aq) content (see Fig. 9). For NpO22+(aq), increasing the Cl1−(aq) caused the absorption feature at 8200 cm−1 (1220 nm) to decrease in energy and shift higher in energy, which is also often attributed to increased Cl1−(aq) complexation.69 Another intriguing observation was that ε for the 10400 cm−1 absorbance peak from Np4+(aq) was more impacted by Cl1−(aq) concentrations than those from NpO22+(aq) at 8200 cm−1. This stronger depended of ε on Cl1−(aq) concentration tracked with the Lewis acidity for the Np4+(aq)vs. NpO22+(aq) cations as well as the variances in Cl1−(aq) complexation constants.37,70
| Oxidation state | Energy (cm−1) | Wavelength (nm) | Table 1 entry # | ε (M−1 cm−1) |
|---|---|---|---|---|
| a In 2 M HClO4(aq) from ref. 37. | ||||
| Np4+(aq) | 10400 |
960 | 1 | 294 |
| Np4+(aq) | 10400 |
960 | 3 | 304 |
| Np4+(aq) | 10400 |
960 | 2 | 341 |
| Np4+(aq) | 10400 |
960 | 7 | 220 |
| Np4+(aq) | 10400 |
960 | 6 | 171 |
| Np4+(aq) | 10400 |
960 | N/A | 162a |
| Np4+(aq) | 8700 | 1150 | 2 | 19 |
| Np4+(aq) | 8700 | 1150 | 3 | 20 |
| Np4+(aq) | 8700 | 1150 | 7 | 26 |
| Np4+(aq) | 8700 | 1150 | 6 | 20 |
| NpO21+(aq) | 10200 |
980 | 1 | 334 |
| NpO21+(aq) | 10200 |
980 | 3 | 660 |
| NpO21+(aq) | 10200 |
980 | 4 | 790 |
| NpO21+(aq) | 10200 |
980 | 5 | 446 |
| NpO21+(aq) | 10200 |
980 | N/A | 375a |
| NpO21+(aq) | 9100 | 1100 | 1 | 41 |
| NpO21+(aq) | 9100 | 1100 | 3 | 53 |
| NpO21+(aq) | 9100 | 1100 | 4 | 60 |
| NpO21+(aq) | 9100 | 1100 | 5 | 50 |
| NpO22+(aq) | 8200 | 1220 | 4 | 127 |
| NpO22+(aq) | 8200 | 1220 | 5 | 115 |
| NpO22+(aq) | 8200 | 1220 | N/A | 43a |
| NpO22+(aq) | 7900 | 1270 | 8 | 190 |
| NpO22+(aq) | 7900 | 1270 | 7 | 190 |
| NpO22+(aq) | 7900 | 1270 | 9 | 174 |
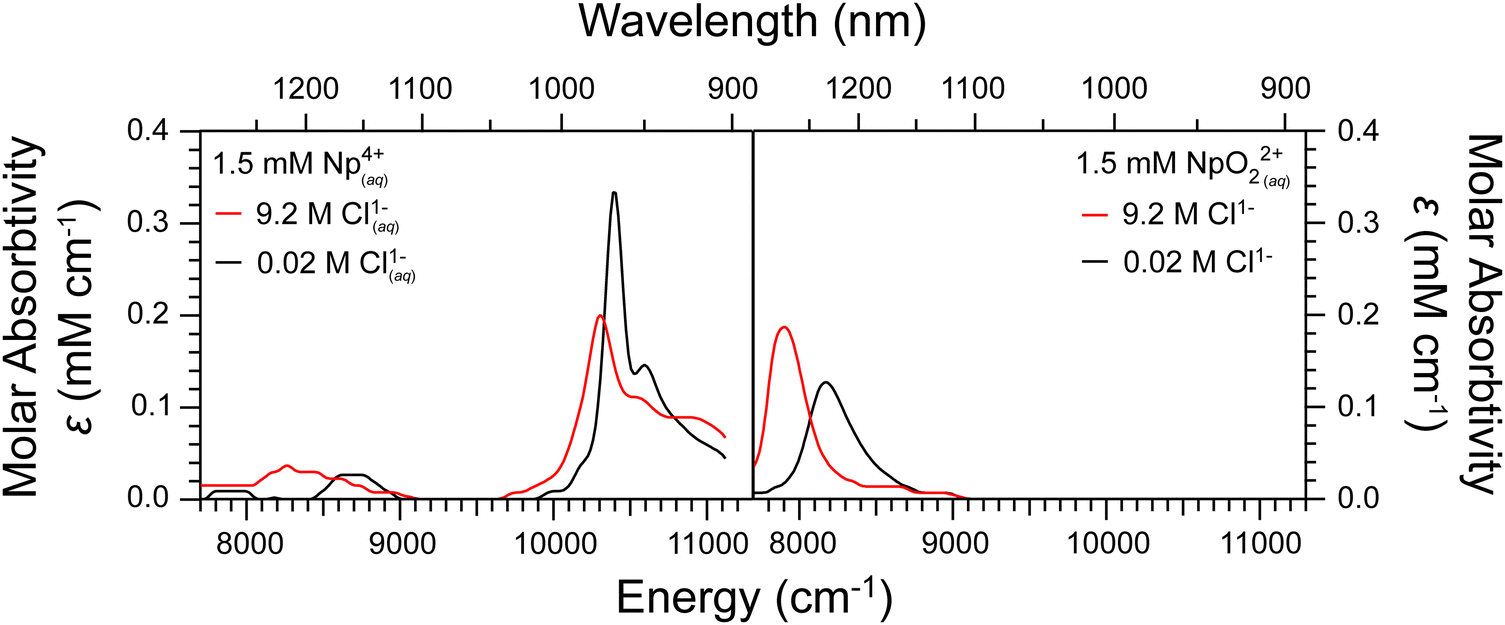 | ||
| Fig. 9 UV-vis absorption spectra documenting oxidation of the Np4+(aq) reagent (left) to the NpO21+(aq) product (right) from entries #4 (black trace) and #8 (red trace). This figure highlights how moving from low concentration Cl1− (0.026 M; entry #4, black trace) to high concentration Cl1− (9.2 M, entry #8, red trace) impacts the absorption features. Note, other than Cl1− concentration, the other experimental variables were held constant between these two experiments. Ingoing concentrations associated with the entry #4 (black trace) experiment were 1.5 mM for Np4+(aq), 160 mM for NaClO2(aq), 0.02 M for H1+(aq), and 0.026 M for Cl1−(aq). In this case, the ingoing HCl(aq) concentration was 0.02 M and there was no LiCl added. Ingoing concentrations associated with the entry #8 (red trace) experiment were 1.5 mM for Np4+(aq), 160 mM for NaClO2(aq), 0.02 M for H1+(aq), and 9.2 M for Cl1−(aq). | ||
Outlook
Herein, we observed that NaClO2(aq) enabled Np4+(aq) to access many redox events in acidic aqueous solutions (Scheme 1). Moreover, oxidation product identities could be controlled as a function of time by manipulating four variables; ingoing concentrations of (1) Np4+(aq), (2) NaClO2(aq), (3) Cl1−(aq), and (4) H1+(aq). For example, NpO21+(aq) exclusively formed when Np4+(aq), NaClO2(aq), Cl1−(aq), and H1+(aq) concentrations were all relatively low. Increasing the Cl1−(aq) concentration and/or increasing the NaClO2(aq) concentration generated NpO22+(aq), provided that the H1+(aq) concentration was also low. Finally, Np4+(aq) was stabilized when the H1+(aq) and Cl1−(aq) concentrations were high (>5.4 M), regardless of the ingoing NaClO2(aq) and Np4+(aq) concentrations.Regarding applications for plutonium processing methods described in the introduction, we make the following prediction. Our data suggested that the neptunium oxidation state during plutonium processing will be either maintained at +4 or a mixture of +4 and +6 if plutonium separations are carried out at high NaClO2(aq), high Cl1−(aq), high H1+(aq), and low Np4+(aq) concentrations. If we are correct, the 237Np(aq) contaminant will follow Pu4+(aq) through processing lines. However, we acknowledge that there are substantial differences between the fundamental studies carried out here vs. real-world processing environments (e.g. radiation effects, scaling effects from other impurities, etc.). To improve relevancy, future work will center on understanding how other constituents present in various plutonium and americium waste streams impact redox chemistry between 237Np(aq) and NaClO2(aq).
The data in Table 1 and Scheme 1 also demonstrated that the neptunium, NaClO2(aq), H1+(aq), and Cl1−(aq) concentration variables were intertwined and must be delicately managed to control the Np4+(aq) oxidation to NpO21+(aq)vs. NpO22+(aq) by way of NaClO2(aq). These studies provided a more sophisticated understanding of aqueous actinide electron transfer chemistry and offer opportunity to better predict and control actinide redox processes. Future efforts will center on developing a better understanding on what reaction pathways are responsible for the oxidation of Np4+(aq) by NaClO2(aq). On an applied level, these types of studies – alongside the data reported herein – will impact waste stewardship programs, production of pure isotopes, and touch on many processing campaigns associated with actinide reliant technologies.
Methods
General considerations
¡CAUTION! Neptunium-237 [237Np, t1/2 = 2.144(6) × 106y]33 and its daughter products constitute serious health threats because of radioactive decay. Hence, all experiments that involved manipulation of these radionuclides were conducted in a radiological buffer area that contained HEPA filtered hoods, continuous air monitors, negative pressure gloveboxes, and monitoring equipment appropriate for α-, β-, and γ-particle detection. Entrance into the laboratory space was controlled with a hand and foot monitoring instrument for α-, β-, and γ-emitting isotopes and a full body personal contamination monitoring station.Aqueous hydrochloric acid [HCl(aq), Fisher Scientific, Optima® grade] and sodium chlorite (NaClO2, 80%, Sigma Aldrich) was obtained commercially and used as received. Water (H2O) was deionized, passed through a Barnstead water purification system to achieve resistivity of 18 MΩ cm, and purified further via distillation using a Teflon distillation apparatus. Chemically pure and oxidation state pure stock solutions of Np4+(aq) were obtained from 237Np samples that had been recovered from previous experimental campaigns, as described previously.59 In general, these 237Np samples were combined and processed using a series of precipitations, valence adjustments, and ion exchange chromatography. The end result was a chemically pure, emerald green stock solution that contained Np4+(aq) (66 mM) in HCl(aq) (1 M).59
UV-Vis spectroscopy
All UV-vis spectroscopy measurements were conducted using a Stellar Net EXtended Range NIR Spectrometer and/or Ocean Insight NIR Quest Spectrometer. To mitigate hazards associated with making optical measurements on radioactive samples, the cuvette holder was housed within a HEPA filtered chemical fume hood. This holder was connected to the UV-vis spectrometer using fiber optics and the neptunium samples were contained within the fume hood in screw top quartz cuvettes (Starna Scientific). Neptunium concentrations were determined by monitoring the following absorption peaks: 10400 cm−1 (960 nm) and/or 8700 cm−1 (1150 nm) for Np4+(aq), 10200 cm−1 (980 nm) and/or 9100 cm−1 (1100 nm) for NpO21+(aq), 8200 cm−1 (1220 nm) and/or 7900 cm−1 (1270 nm) for NpO22+(aq).
Data acquisition and analysis
Before handling the Np stock, a reference UV-vis spectra was initially obtained from pure HCl(aq) (0, 6, 7, or 8 M) or LiCl(aq) (10 M) solutions prior to assaying Np4+(aq) and NaClO2(aq) containing solutions. Data from Np4+(aq) and NaClO2(aq) reactions were background subtracted by setting the intensity at 9520 cm−1 (1050 nm) to zero. Reaction rate constants were determined using the systematic numerical procedure by which the concentration of one variable was varied (e.g. ingoing Np4+(aq)) whilst the other three variables were held constant [in this scenario, ingoing NaClO2(aq), H1+(aq), and Cl1−(aq)].71,72 The linear initial reaction rate was taken to be the rate by which Np4+(aq) reacted (after mixing) by using linear regression. Errors for the analysis of reactions rates were calculated through propagation of error in measurements of ingoing NaClO2(aq) mass, pipetting, and volumetric flasks. An example calculation may be found in the ESI.† Molar extinction coefficients were calculated using the Beer–Lambert Law.73Using dilute aqueous sodium chlorite solutions, NaClO2(aq), to generate oxidation state pure solutions of neptunyl(V), NpO21+(aq)
In a fume hood and with no attempt to exclude air and moisture, an aliquot (15 μL; 0.2 mg, 1 μmol of Np4+) of the aforementioned Np4+(aq) stock solution (66 mM Np4+) in HCl(aq) (1 M) was added to a screw-top quartz cuvette charged with H2O (600 μL). The Np4+(aq) concentration for this new 615 μL solution was now 1.6 mM. Meanwhile, in a separate beaker, a dilute solution of NaClO2(aq) (6.9 mM) was prepared by dissolving NaClO2(s) (0.0389 g, 80%, 0.344 mmol) in H2O (50 mL). An aliquot (600 μL, 4 μmol) from this NaClO2(aq) solution was added all at once to the Np4+(aq) containing cuvette. ¡CAUTION! The combination of HCl(aq) and NaClO2(aq) is vigorous, can bubble, and generates toxic gases (like ClO2). To mitigate these hazards, we implemented the following engineering and administrative controls. First, the aliquot of the NaClO2(aq) was added slowly (over the course of 5 seconds to the Np4+(aq) containing cuvette). Second, the manipulation was carried out in a chemical fume hood, behind glass shielding, to guard the worker from evolved gases. Third, secondary containment was used to avoid spreading 237Np-contamination, which can result from potential splattering. Finally, the screw top cuvette was only loosely capped, to avoid pressurization. The reagents were mixed by pumping the solution (from bottom to top) with a transfer pipette at least 4 times. This mixing action marked zero time for the reaction. It is relevant to note that the concentrations at the zero time for this solution (1.2 mL) were 0.81 mM for Np(aq), 3.4 mM for NaClO2(aq), and 0.01 M for HCl(aq). The cuvette was inserted into the UV-vis holder, the apparatus covered (to exclude ambient light), and the spectra were collected repeatedly over the course of a 5-day monitoring process.Using concentrated aqueous sodium chlorite solutions, NaClO2(aq), to generate oxidation state pure solutions of neptunyl(VI), NpO22+(aq)
In a fume hood and with no attempt to exclude air and moisture, oxidation state pure solutions of NpO22+(aq) were prepared using an adaptation of the procedure described in the preceding section titled, “Using Dilute Aqueous Sodium Chlorite Solutions, NaClO2(aq), to Generate Oxidation State Pure Solutions of Neptunyl(V).” The major difference between these procedures was associated with the amount of added NaClO2(aq) used to oxidize Np4+(aq). Toward this end, a more concentrated solution of NaClO2(aq) (2.6 M) was prepared by dissolving NaClO2(s) (298 mg, 2.6 mmol) in H2O (1 mL). Then, the NaClO2(aq) (40 μL, 2.6 M, 104 μmol) and Np4+(aq) (615 μL, 1.6 mM, 1 μmol) solutions were combined in a cuvette. CAUTION!See precautions described above that were implemented to mitigate this hazardous combination activity. The reagents were mixed by pumping the solution (from bottom to top) with a transfer pipette at least 4 times. This mixing action marked zero time for the reaction. The reagent concentrations at the zero time for this solution (655 μL) were 1.5 mM for Np4+(aq), 160 mM for NaClO2(aq) (approximate), and 0.02 M for HCl(aq). The cuvette was inserted into the UV-vis holder, the apparatus covered (to exclude ambient light), and the spectra were collected repeatedly over the course of a 5 day monitoring period.Evaluating how four variables impacted oxidation of neptunium by sodium chlorite; Np4+(aq) + NaClO2(aq)
In a fume hood and with no attempt to exclude air and moisture, oxidation of Np4+(aq) by NaClO2(aq) was investigated in HCl as a function of the following four variables, (1) Np4+(aq) reagent concentration, (2) NaClO2(aq) oxidant concentration, (3) Cl1−(aq) complexant concentration, and (4) H1+(aq) concentration. This was achieved by modifying the experimental conditions described above in the antecedent section titled, “Using Dilute Aqueous Sodium Chlorite Solutions, NaClO2(aq), to Generate Oxidation State Pure Solutions of Neptunyl(V).” See Table 1 for a summary of those tested parameters and the primary neptunium oxidation states that formed within 1 h and within 24 h. These solutions were prepared by modifying the concentration of the ingoing reagents, oxidant, complexing agents, and acid. The ingoing Np4+(aq) concentration was varied (1) by combining 15 μL of the Np4+(aq) (66 mM) stock solution with the other reagents in a cuvette to generate a final solution that was dilute in Np4+(aq) (1.5 mM) or (2) by combining 30 μL of the Np4+(aq) (66 mM) stock solution with the other regents in a cuvette to generate a final solution that was highly concentrated in Np4+(aq) (3.1 mM). The ingoing NaClO2(aq) concentration was varied (1) by combining 40 μL of a NaClO2(aq) (520 mM) stock solution with the other reagents in a cuvette to generate a final solution that was intermediately concentrated in NaClO2(aq) (31 or 32 mM) or (2) by combining 40 μL of a NaClO2(aq) (2.6 M) stock solution with the other reagents in a cuvette to generate a final solution that was highly concentrated in NaClO2(aq) (160 mM). The ingoing HCl(aq) concentration was varied (1) by combining either 600 μL of an HCl(aq) (6 to 8 M) stock solution with the other reagents in a cuvette to generate a final solution that was more concentrated in HCl(aq) (5.3 to 7.1 M) or (2) by adding an aliquot (15 or 30 μL, described above) from the neptunium stock solution [1 M HCl(aq)] to a cuvette containing Teflon distilled H2O (600 μL). This gave final volumes and concentrations of either 615 μL at 0.02 M HCl(aq) or 630 μL at 0.05 M HCl(aq). Then, NaClO2(aq) was added. These dilute HCl(aq) solutions also served as low ionic strength solutions, relatively speaking. High ionic strength solutions were prepared by combining 600 μL of a LiCl(aq) (10 M) stock solution with the other reagents in a cuvette to generate a final solution that was 9.0 to 9.2 M in LiCl(aq).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was primarly supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry Programs (number 2022LANLE3M1 for Los Alamos and DE-SC0020189 for Colorado School of Mines). Additional support was provided by LANL's Laboratory Directed Research and Development program (LDRD-DR, 20220054DR), the US Department of Energy, National Nuclear Security Association (NNSA), Plutonium Modernization Program (NA-191), and the by the U.S. Department of Energy Isotope Program, managed by the Office of Science for Isotope R&D and Production. Los Alamos National Laboratory (LANL) is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (contract no. 89233218CNA000001).References
- P. Paviet-Hartmann, C. Riddle, K. Campbell and E. Mausolf, Overview of Reductants Utilized in Nuclear Fuel Reprocessing/Recycling, Report INL/CON-12-28006, Idaho National Labs, Idaho Falls, Idaho, 2013 Search PubMed.
- B. J. Mincher, L. R. Martin and N. C. Schmitt, Inorg. Chem., 2008, 47, 6984–6989 CrossRef CAS PubMed.
- Z. Karoutas, J. Brown, A. Atwood, L. Hallstadius, E. Lahoda, S. Ray and J. Bradfute, Prog. Nucl. Energy, 2018, 102, 68–78 CrossRef CAS.
- T. Allen, J. Busby, M. Meyer and D. Petti, Mater. Today, 2010, 13, 14–23 CrossRef CAS.
- W. H. E. Schwarz, N. M. Shavaleev, S. N. Kalmykov, A. Y. Romanchuk and I. E. Vlasova, Front. Chem., 2020, 1, 630 Search PubMed.
- B. J. Mincher, G. Modolo and S. P. Mezyk, Solvent Extr. Ion Exch., 2009, 27, 1–25 CrossRef CAS.
- P. Baron, S. M. Cornet, E. D. Collins, G. DeAngelis, G. Del Cul, Y. Fedorov, J. P. Glatz, V. Ignatiev, T. Inoue, A. Khaperskaya, I. T. Kim, M. Kormilitsyn, T. Koyama, J. D. Law, H. S. Lee, K. Minato, Y. Morita, J. Uhlíř, D. Warin and R. J. Taylor, Prog. Nucl. Energy, 2019, 117 Search PubMed.
- J. E. Birkett, M. J. Carrott, O. Danny, C. J. Jones, C. J. Maher, C. V. Roube, R. J. Taylor and D. A. Woodhead, J. Nucl. Sci. Technol., 2007, 44, 337–343 CrossRef CAS.
- R. C. Thompson, Radiat. Res., 1982, 90, 1–32 CrossRef CAS PubMed.
- D. L. Clark, G. D. Jarvinen, C. Kowalczyk, J. Rubin and M. A. Stroud, Actinide Research Quarterly: Plutonium Processing at Los Alamos, LALP-08-004, Los Alamos National Labs, Los Alamos, New Mexico, 2008 Search PubMed.
- B. C. Reed, The history and science of the Manhattan Project, Springer, New York, 2014 Search PubMed.
- T. Feder, Phys. Today, 2015, 68, 22–24 Search PubMed.
- S. K. Cary, K. S. Boland, J. N. Cross, S. A. Kozimor and B. L. Scott, Polyhedron, 2017, 126, 220–226 CrossRef CAS.
- A. E. Waltar, Encyclopedia of Nuclear Energy, Elsevier, 2021, vol. 4, pp. 451–464 Search PubMed.
- C. Adam, N. Olga and L. Duane, Final Radiological Assessment of External Exposure for CLEAR-Line Americium Recovery Operations, LA-UR-13-28160, Los Alamos National Labs, Los Alamos, NM, 2014 Search PubMed.
- G. F. Vandegrift, Sep. Sci. Technol., 2006, 23, 1409–1421 Search PubMed.
- E. P. Horwitz, H. Diamond and K. A. Martin, Solvent Extr. Ion Exch., 2007, 6299, 447–470 Search PubMed.
- R. E. Isaacson and B. F. Judson, Neptunium Recovery and Purification at Hanford, HW-SA-3283, Hanford Site, Hanford, Washington, 1963 Search PubMed.
- H. E. Henry, D. G. Karraker and C. S. Schlea, Neptunium Behavior in Solvent Extraction of Uranium at Savannah River Plant, DP-638, Savanah River National Labs, Aiken, South Carolina, 1961 Search PubMed.
- M. Barr, L. Schulte, G. Jarvinen, J. Espinoza, T. Ricketts, Y. Valdez, K. Abney and R. Bartsch, J. Radioanal. Nucl. Chem., 2001, 248, 457–465 CrossRef CAS.
- R. A. Barr, M. E. Jarvinen, G. D. Schulte, L. D. Stark, P. C. Chamberlin, R. M. Abney, K. D. Ricketts, T. E. Valdez and Y. E. Bartsch, Americium Separations from High Salt Solutions, LA-13676-MS, Los Alamos National Labs, Los Alamos, New Mexico, 2000 Search PubMed.
- A. C. 74 and M. E. Killion, Proceedings of the 55th Conference: American Nuclear Society winter meeting, Los Angeles, CA, USA, 1987.
- J. A. Schramke, E. F. U. Santillan and R. T. Peake, Appl. Geochem., 2020, 116, 104561 CrossRef CAS PubMed.
- B. T. Arko, D. Dan, S. L. Adelman, D. L. Huber, D. B. Kimball, S. A. Kozimor, M. N. Lam, V. Mocko, J. C. Shafer, B. W. Stein and S. L. Thiemann, Ind. Eng. Chem. Res., 2021, 60, 14282–14296 CrossRef CAS.
- B. T. Arko, D. Dan, S. Adelman, D. B. Kimball, S. A. Kozimor, M. M. Martinez, T. Mastren, D. L. Huber, V. Mocko, J. Rim, J. C. Shafer and W. Stein, Mater. Adv., 2023, 265–283 RSC.
- L. D. Schulte, J. R. FitzPatrick, R. R. Salazar, B. S. Schake and B. T. Martinez, Sep. Sci. Technol., 1995, 30, 1833–1847 CrossRef CAS.
- D. Christensen and P. Cunningham, Los Alamos National Laboratory Qualifications For Lead Laboratory in Plutonium Pit Technology, LA-UR-92-1729, Los Alamos National Labs, Los Alamos, New Mexico, 1992 Search PubMed.
- K. S. Gardner, D. B. Kimball and B. E. Skidmore, Aqueous Chloride Operations Overview: Plutonium and Americium Purification/Recovery, LA-UR-16-27346, Los Alamos National Labs, Los Alamos, NM, 2016 Search PubMed.
- W. H. Smith, Evaluation of Chloride-Ion-Specific Electrodes as in situ Chemical Sensors for Monitoring Total Chloride Concentration in Aqueous Solutions Generated During the Recovery of Plutonium from Molten Salts Used in Plutonium Electrorefining Operations, LA—122461992, Los Alamos National Labs, Los Alamos, New Mexico, 1992 Search PubMed.
- Nuclear Posture Review Report, US Department of Defense, Washington, DC, 2010.
- B. Roberts, Wash Q, 2021, 44, 123–142 CrossRef.
- K. Reif, Arms Control Today, 2018, 48, 29–30 Search PubMed.
- National Nuclear Data Center, https://www.nndc.bnl.gov/nudat2/, (accessed 21 April 2021).
- L. R. Morss, N. M. Edelstein and J. Fuger, The Chemistry of the Actinide and Transactinide Elements, Springer, Dordrecht, The Netherlands, 3rd edn, 2006 Search PubMed.
- T. Hindman, J. C. Magnusson and L. B. LaChapelle, J. Am. Chem. Soc., 1949, 71, 687–693 CrossRef.
- J. R. Yoshida, Z. Johnson, S. G. Kimura and T. Krsul, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein, and J. Fuger, Springer, Dordrecht, The Netherlands, 3rd edn, 2008, vol. 2 ch. 6, pp. 699–812 Search PubMed.
- G. A. Burney and R. M. Harbour, Radiochemistry of Neptunium, NAS-NS-3060, Savanah River National Labs, Aiken, South Carolina, 1974 Search PubMed.
- R. C. Thompson, Radiat. Res., 1982, 90, 1–32 CrossRef CAS.
- R. McConn Jr, C. Gesh, R. Pagh, R. Rucker and R. G. Williams III, Compendium of Material Composition Data for Radiation Transport Modeling, PIET-43741-TM-963 PNNL-15870 Rev. 1, Pacific Northwest National Lab, Richland, Washington, 2011 Search PubMed.
- T. Fukasawa, C. Lierse and J. I. Kim, J. Nucl. Sci. Technol., 1996, 33, 486–491 CrossRef CAS.
- B. R. Deshwal, H. D. Jo and H. K. Lee, Can. J. Chem. Eng., 2004, 82, 619–623 CrossRef CAS.
- G. Gordon in Progress in Inorganic Chemistry, ed. S. J. Lippard, Wiley, New York, 1972, vol. 15 ch. 3, pp. 201–286 Search PubMed.
- G. Holst, Ind. Eng. Chem., 1950, 42, 2359–2371 CrossRef CAS.
- M. C. Taylor, J. F. Whitte, G. P. Vincent and G. I. Cunnigham, Ind. Eng. Chem., 1940, 32, 899–903 CrossRef CAS.
- N. F. Gray, in Microbiology of Waterborne Diseases, ed. S. L. Percival, M. V. Yates, D. W. Williams, R. M. Chalmers and N. F. Gray, Academic Press, London, 2nd edn, 2014, pp. 591–598 Search PubMed.
- E. M. Aieta and P. V. Roberts, Environ. Sci. Technol., 1986, 20, 50–55 CrossRef CAS PubMed.
- A. K. Horva, Inorg. Chem., 2008, 47, 7914–7920 CrossRef PubMed.
- A. Earnshaw and N. N. Greenwood, Chemistry of the Elements, Elsiver, Burlington, MA, 2nd edn, 1997, vol. 60 Search PubMed.
- C. Tarimci, R. D. Rosenstein and E. Schempp, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 610–612 CrossRef.
- V. Tazzoli, V. Riganti, G. Giuseppetti and A. Coda, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 1032–1037 CrossRef.
- N. Langerman, ACS Chem. Health Saf., 2021, 28, 402–409 CrossRef CAS.
- CRC handbook of chemistry and physics, CRC Press, Boca Raton, 2004, vol. 85 Search PubMed.
- M. A. Busch, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2018 Search PubMed.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced inorganic chemistry, John Wiley & Sons, Chichester, 5th edn, 1988 Search PubMed.
- E. M. Aieta and P. V. Roberts, Environ. Sci. Technol., 1986, 20, 50–55 CrossRef CAS PubMed.
- R. G. Kieffer and G. Gordon, Inorg. Chem., 1968, 7, 235–239 CrossRef CAS.
- R. G. Kieffer and G. Gordon, Inorg. Chem., 1968, 7, 239–244 CrossRef CAS.
- C. C. Hong and W. H. Rapson, Can. J. Chem., 1967, 1, 2053–2060 Search PubMed.
- S. K. Cary, M. Livshits, J. N. Cross, M. G. Ferrier, V. Mocko, B. W. Stein, S. A. Kozimor, B. L. Scott and J. Rack, Inorg. Chem., 2018, 57, 3782–3797 CrossRef CAS PubMed.
- V. Bondet, W. Brand-Williams and C. Berset, LWT – Food Sci. Technol., 1997, 30, 609–615 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- C. Liang and C. J. Bruell, Ind. Eng. Chem. Res., 2008, 47, 2912–2918 CrossRef CAS.
- S. Ašperger, Chemical Kinetics and Inorganic Reaction Mechanisms, Plenum Publishers, New York, 2nd edn, 2003 Search PubMed.
- X. Yang, Z. Zheng, J. Hu, J. Qu, D. Ma, J. Li, C. Guo and C. M. Li, iScience, 2021, 24, 103500 CrossRef CAS PubMed.
- H. A. Young and W. C. Bray, J. Am. Chem. Soc., 1932, 54, 4284–4296 CrossRef CAS.
- G. B. Kolski and D. W. Margerum, Inorg. Chem., 1969, 8, 1125–1131 CrossRef CAS.
- W. C. Bray and H. A. Liebhafsky, J. Am. Chem. Soc., 1935, 57, 51–56 CrossRef CAS.
- S. K. Patil, V. V. Ramakrishna and M. V. Ramaniah, Coord. Chem. Rev., 1978, 25, 133–171 CrossRef CAS.
- D. Cohen and B. Taylor, J. Inorg. Nucl. Chem., 1961, 22, 151–153 CrossRef CAS.
- P. R. Danesi, R. Chiarizia, G. Scibona and G. D’Alessandro, J. Inorg. Nucl. Chem., 1971, 33, 3503–3510 CrossRef CAS.
- M. R. Wright, An Introduction to Chemical Kinetics, John Wiley & Sons, Ltd, Hoboken, 2004, vol. 8 Search PubMed.
- G. B. Skinner, in Introduction to Chemical Kinetics, ed. G. B. Skinner, Academic Press, Dayton, 1974, pp. 108–140 Search PubMed.
- D. F. Swinehart, J. Chem. Educ., 1962, 39, 333 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj03730d |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2024 |