
Dual carbon engineering enabling 1T/2H MoS2 with ultrastable potassium ion storage performance†
Rong
Hu
a,
Yanqi
Tong
a,
Jinling
Yin
a,
Junxiong
Wu
*b,
Jing
Zhao
a,
Dianxue
Cao
 a,
Guiling
Wang
*a and
Kai
Zhu
*a
a,
Guiling
Wang
*a and
Kai
Zhu
*a
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China. E-mail: wangguiling@hrbeu.edu.cn; kzhu@hrbeu.edu.cn
bEngineering Research Center of Polymer Green Recycling of Ministry of Education, Fujian Key Laboratory of Pollution Control & Resource Reuse, College of Environmental and Resource Sciences and College of Carbon Neutral Modern Industry, Fujian Normal University, Fuzhou, 350117, Fujian, China. E-mail: jwuba@fjnu.edu.cn
First published on 20th December 2023
Abstract
Potassium-ion batteries (PIBs) as a promising and low-cost battery technology offer the advantage of utilizing abundant and cost-effective K-salt sources. However, the effective adoption of PIBs necessitates the identification of suitable electrode materials. The 1T phase of MoS2 exhibits enhanced electronic conductivity and greater interlayer spacing compared to the 2H phase, leading to a capable potassium ion storage ability. Herein, we fabricated dual carbon engineered 1T/2H MoS2via a secure and straightforward ammonia-assisted hydrothermal method. The 1T/2H MoS2@rGO@C structure demonstrated an expanded interlayer spacing (9.3 Å). Additionally, the sandwich-like structural design not only enhanced material conductivity but also effectively curbed the agglomeration of nanosheets. Remarkably, 1T/2H MoS2@rGO@C exhibited impressive potassium storage ability, delivering capacities of 351.0 mA h g−1 at 100 mA g−1 and 233.8 mA h g−1 at 1000 mA g−1 following 100 and 1000 cycles, respectively. Moreover, the construction of a K-ion full cell was successfully achieved, utilizing perylene tetracarboxylic dianhydride (PTCDA) as the cathode, and manifesting a capacity of 294.3 mA h g−1 at 100 mA g−1 after 160 cycles. This underscores the substantial potential of employing the 1T/2H MoS2@rGO@C electrode material for PIBs.
New conceptsWe have enhanced the K-storage performance of material from a phase perspective. At present, Potassium ion batteries (PIBs) are receiving more attention because potassium is rich in natural resources (2.09 wt%) and has similar physical and chemical properties to lithium. MoS2 is a common anode material in PIBs, which has the advantages of graphite-like layered structure, large interlayer spacing and high theoretical capacity. However, previous studies have focused on the common 2H phase of MoS2, which exhibits smaller capacity and poor cycling stability in PIBs. Here, we introduce the 1T phase (metallic phase) to synthesize the hybrid phase 1T/2H MoS2@rGO@C with double carbon engineering. We found that the content of the 1T phase can be increased by adding graphene and dopamine, which in turn enhances the conductivity of MoS2. The presence of an appropriate amount of 2H phase can also stabilize the 1T phase. During the synthesis process, the addition of NH3·H2O extends the interlayer spacing of MoS2 (9.3 Å). Hence, good performance is obtained even in the K-ion full cell. These results clarify that different phases of the same material have different K-storage properties, and provide valuable information for further understanding the K-storage ability of material. |
Introduction
Lithium-ion batteries (LIBs) have established themselves as enduring energy storage solutions, owing to their extended cycle life, elevated energy density, and remarkable power density. This has rendered them highly suitable for the field of portable electronic devices and electric vehicles over the preceding decades.1 Nevertheless, the disparate geographic distribution and constrained availability (0.0017 wt%) of lithium resources, combined with escalating costs of vital precursor materials, have limited the widespread application of LIBs within stationary energy storage systems and electric vehicles.2,3 Potassium, an elemental resource presenting a higher abundance (2.09 wt%) in the Earth's crust, emerges as a compelling alternative. This substitution capitalizes on its superior affordability and sustainability relative to lithium.4 Furthermore, the notably low redox potential of K/K+ (−2.93 V vs. the standard hydrogen electrode (SHE)) promises improved voltage output and energy density in potassium-ion batteries (PIBs).5,6 Nevertheless, the size of potassium ions (with a radius of 1.40 Å) significantly exceed that of lithium ions (0.78 Å).7 This size difference engenders challenges during the potassiation/depotassiation processes, inducing sluggish diffusion kinetics and enormous volume changes within the electrode material.8 Thus, obtaining high capacity and desirable stability remains a formidable hurdle for PIBs. Consequently, an exhaustive exploration into suitable anode materials becomes imperative for the advancement of PIBs technology.Carbonaceous materials, encompassing hard carbon, soft carbon, as well as pristine and doped graphite, have garnered significant attention as anode materials for potassium-ion batteries (PIBs).9 However, their storage capacities remain confined within the range of 200–300 mA h g−1.10 The difference is that transition metal dichalcogenides (TMDs) have higher theoretical capacity. Moreover, TMDs usually exhibit graphite-like layered structures, such as VS2, MoS2, etc.11,12 Therefore, TMDs are expected to achieve excellent electrochemical performance in PIBs. Among them, MoS2 has been extensively studied, mainly due to two pivotal advantages: (i) Its interlayer spacing is about 6.2 Å, which surpasses that of graphite (3.4 Å),12 facilitating rapid and reversible intercalation/deintercalation of K+ ions.13 (ii) MoS2's abundant availability makes it an appealing contender for large-scale energy storage systems.14 However, the electrochemical performance of pure MoS2 is poor due to challenges such as pronounced agglomeration during cycling,15 substantial volume expansion, and inferior electrical conductivity.5 Addressing these concerns involves two prevalent strategies: the creation of innovative MoS2 microstructures or composites with conductive materials.16,17 For example, Jiao et al.18 synthesized MoS2 porous nanotubes, which are composed of vertically aligned metallic phase MoS2 nanosheets, showing excellent electrochemical performance at high current density even without compounding carbon materials and adding binders. Li et al.19 improved the performance of MoS2 by complexing reduced graphene oxide (rGO), N and P co-doped C with MoS2 nanoparticles. The successfully prepared sheet-like MoS2-C/rGO can still provide a specific capacity of 405.25 mA h g−1 after 100 cycles at 100 mA g−1. Among these methods to improve MoS2 performance, the most common is to composite with carbon materials. However, composite with a single carbon material has been reported more frequently. In fact, these carbon materials have different morphologies and characteristics, and play different roles when compounded with MoS2. This means that appropriate dual carbon (or even multi-carbon) composite may be able to better improve the performance of MoS2. For example, graphene has extreme flexibility, large surface area and excellent electrical conductivity.20 Compared with MoS2, which usually exhibits a nanosheet morphology, graphene can perfectly serve as a carrier for MoS2 and play a role in reducing MoS2 agglomeration and enhancing electrical conductivity. The carbon shell formed by carbon coating on the surface of MoS2 can reduce agglomeration, avoid direct contact between MoS2 and electrolyte, thus buffering the volume change and prolonging the service life.21 Obviously, although graphene and coated carbon are both carbon materials, they play different roles. Combining these two carbon materials appropriately to maximize their strengths, which may be more conducive to solving the inherent problems of MoS2.
It is worth mentioning that MoS2 exhibits three phases (2H, 1T and 3R),22,23 which is a remarkable feature. Because 1T is a metallic phase, 2H and 3R are semiconducting phases. And the electron conductivity of the 1T phase is about 107 times that of the 2H phase.23 Obviously, the 1T phase MoS2 is superior. But in general, the synthetic product is 2H phase MoS2. This is because the thermodynamically metastable 1T MoS2 predisposes it to transition to the more stable 2H MoS2 through interlayer atomic plane sliding.24 Therefore, 1T MoS2 is difficult to exist alone and stably. However, the coexistence of 2H and 1T phase can stabilize the 1T phase.25,26 Unfortunately, the 1T phase of MoS2 is not easy to synthesize and the synthesis process is complicated.27,28 For example, Acerce et al. achieved the exfoliation of bulk 2H MoS2via organolithium chemistry to obtain 1T phase MoS2.29 However, this process involving Li intercalation exfoliation introduces complexity and safety hazards due to the reaction between butyllithium, oxygen and moisture at room temperature.13 Thus, an uncomplicated synthesis method for 1T/2H MoS2 is imperative.
This study introduces an innovative, efficient, and secure approach for synthesizing 1T/2H MoS2. Employing an ammonia-assisted hydrothermal method, we obtained swollen ammoniated multiphasic 1T/2H MoS2. Additionally, a “sandwich” structure of 1T/2H MoS2@rGO@C was devised and synthesized through a simple solvothermal technique. This structure utilizes graphene as a supportive framework, with ultrathin MoS2 nanosheets uniformly grown on its surface, subsequently coated with dopamine-derived thin carbon layers. The incorporation of graphene and carbon coating augments material conductivity, while the “sandwich” design effectively curbs MoS2 nanosheet agglomeration. Furthermore, the addition of dopamine also increases the 1T phase components content, thereby promoting higher conductivity. The resultant 1T/2H MoS2@rGO@C demonstrates exceptional cycling stability, attaining a discharge capacity of 351.0 mA h g−1 at 100 mA g−1 after 100 cycles. Even under the rigorous conditions of 1000 cycles at 1000 mA g−1, the 1T/2H MoS2@rGO@C anode maintains a discharge capacity of 233.8 mA h g−1. Finally, a full cell was successfully assembled using perylenetetracarboxylic dianhydride (PTCDA) as the cathode. The full cell achieves a notably high initial discharge capacity of 345.4 mA h g−1, remarkable cycling stability (294.3 mA h g−1 after 160 cycles) at 100 mA g−1, and exceptional rate performance (210.9 mA h g−1 at 2000 mA g−1).
Results and discussion
The synthesis process for 1T/2H MoS2@rGO@C is illustrated in Fig. 1. The preparation of the 1T/2H MoS2@rGO@C composite involves two consecutive steps. Initially, a typical solvothermal reaction was used to synthesize 1T/2H MoS2@rGO, in which the Mo and S precursors in the graphene oxide (GO) solution were Na2MoO4·2H2O and thiourea, respectively. Subsequently, magnetic stirring facilitated the coating of dopamine onto MoS2@rGO, which underwent carbonization at a high temperature of 600 °C under an Ar atmosphere, culminating in the 1T/2H MoS2@rGO@C sample. Notably, the ultra-thin MoS2 nanosheets are uniformly covered on the translucent graphene sheet in the MoS2@rGO sample (Fig. S1a and b, ESI†). Whereas pure MoS2 nanosheets (Fig. S1c and d, ESI†) agglomerated into bulks. This phenomenon underscores the role of graphene in furnishing essential nucleation sites for the uniform growth of MoS2, facilitating enhanced contact between MoS2 and the electrolyte and bolstering conductivity.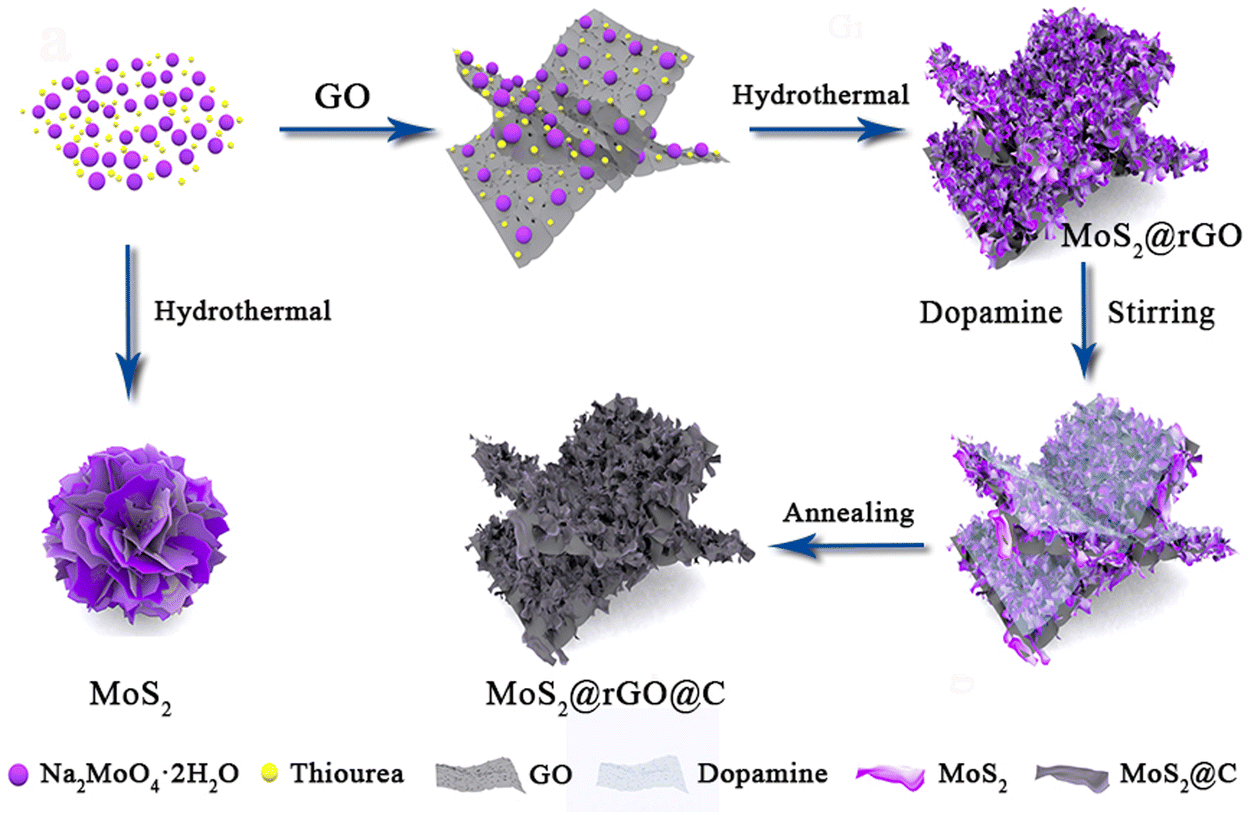 | ||
| Fig. 1 Schematic diagram for the synthesis process of 1T/2H MoS2@rGO@C. | ||
Observing the morphology of 1T/2H MoS2@rGO@C, the thin GO stretches out as a supporting skeleton, on which ultrathin MoS2 nanosheets of about 100 nm in diameter are supported (Fig. 2a and b). At room temperature, dopamine introduced through magnetic stirring uniformly envelops the MoS2 nanosheets and GO, and form a uniform carbon shell after calcination, as depicted in the TEM image of MoS2@rGO@C (Fig. 2c). In Fig. S2 (ESI†), the above-described morphological characteristics can be observed more intuitively and clearly. A large flake of thin rGO is spread out as a supporting skeleton, on which are scattered MoS2 nanosheets. The carbon shell coated on the MoS2 surface can also be clearly observed. It is obvious that in the 1T/2H MoS2@rGO@C composite, two kinds of carbon materials are introduced, namely rGO and coated carbon shell. This is because different carbon materials can play different roles. First, The crinkled graphene provides abundant active sites where MoS2 can grow, thereby inhibiting the massive agglomeration of MoS2 and favoring the direct contact between more MoS2 and electrolyte. Secondly, the entirely coated carbon shells effectively anchor MoS2 nanosheets onto the graphene and isolate them from each other. And interstices among the MoS2 nanosheets proficiently thwart material pulverization. The coated carbon shell is formed by the calcination of dopamine, and subsequent XPS analysis showed that the added dopamine could effectively increase the content of 1T phase component. In addition, both the carbon shell and rGO can further enhance the conductivity of MoS2.30 Therefore, as observed in SEM and TEM images, 1T/2H MoS2@rGO@C forms a unique sandwich-like structure under the synergistic effect of rGO and carbon shell. In the HRTEM image of 1T/2H MoS2@rGO@C (Fig. 2d), rGO is more clearly observed. And there are clear lattice fringes that can correspond to the (002) plane of MoS2.31 In particular, in the 1T/2H MoS2@rGO@C composite, the interlayer spacing of (002) plane is 0.930 nm, which is significantly larger than that of conventional MoS2 (0.616 nm).32 The unique synthesis method of 1T/2H MoS2@rGO@C composites is the main reason for the expanded interlayer spacing. The HRTEM image of other positions in the sample (Fig. 2e) also show the same lattice fringes. Meanwhile, it can be seen from the EDS mapping that Mo, S and C are uniformly distributed in the 1T/2H MoS2@rGO@C composites (Fig. 2f).
 | ||
| Fig. 2 (a and b) SEM, (c) TEM, (d) and (e) HRTEM and (f) EDS mapping images of 1T/2H MoS2@rGO@C. | ||
Notably, rGO is all mentioned in the above description of the morphology of 1T/2H MoS2@rGO@C. This is because most of the added GO is reduced to rGO during the synthesis process. In order to prove that 1T/2H MoS2@rGO@C does contain rGO, XPS analysis of GO and 1T/2H MoS2@rGO@C was performed. The EDS images of 1T/2H MoS2@rGO and 1T/2H MoS2@rGO@C are also given. In the XPS survey scan spectra of GO and 1T/2H MoS2@rGO@C (Fig. S3a, ESI†), the C 1s (284.5 eV) and O 1s (531.5 eV) peaks can be clearly observed. Among them, the intensity of the O 1s peak represents the number of oxygen-containing functional groups on the surface of the sample. Obviously, compared with GO, the intensity of O 1s Peak in 1T/2H MoS2@rGO@C is significantly lower than that in GO, which means that a large number of oxygen-containing functional groups on the surface of GO are removed during the synthesis process, proving that most of GO is reduced to rGO. The conclusion can be further confirmed by the O 1s spectra of GO and 1T/2H MoS2@rGO@C. The O 1s spectra of GO (Fig. S3b, ESI†) can be divided into five peaks, namely C–OH (533.4 eV), C–O (532.7 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (532.2 eV), OC–OH (531.4 eV), and Quinone (530.5 eV).33,34 Similarly, the O 1s spectra of 1T/2H MoS2@rGO@C (Fig. S3c, ESI†) can be fitted into these five peaks, with only a slight shift in peak position. The biggest difference between the two samples is the proportion of different oxygen-containing functional groups (Fig. S3d, ESI†). The content of C–O (37.1%) and CO (29.5%) is the highest in GO. When GO is reduced to rGO, the content of C–O (7.4%) and CO (13.7%) is significantly reduced, suggesting a significant reduction of oxygen-containing functional groups on the surface of rGO. However, oxygen-containing functional groups still exist on the rGO surface, such as OC–OH (46.0%) and Quinone (21.4%) with significantly increased contents. The presence of these two oxygen-containing functional groups can, on the contrary, enhance the wettability and pseudocapacitive property of material, which helps to improve the K-storage capacity of 1T/2H MoS2@rGO@C.35 In addition, the EDS mapping images of 1T/2H MoS2@rGO@C, 1T/2H MoS2@rGO and GO can also visually reflect the difference of O element content on the surface of three materials (Fig. S4, ESI†). Obviously, the content of O element in 1T/2H MoS2@rGO@C is the least, while the content of O element in GO is the most.
O (532.2 eV), OC–OH (531.4 eV), and Quinone (530.5 eV).33,34 Similarly, the O 1s spectra of 1T/2H MoS2@rGO@C (Fig. S3c, ESI†) can be fitted into these five peaks, with only a slight shift in peak position. The biggest difference between the two samples is the proportion of different oxygen-containing functional groups (Fig. S3d, ESI†). The content of C–O (37.1%) and CO (29.5%) is the highest in GO. When GO is reduced to rGO, the content of C–O (7.4%) and CO (13.7%) is significantly reduced, suggesting a significant reduction of oxygen-containing functional groups on the surface of rGO. However, oxygen-containing functional groups still exist on the rGO surface, such as OC–OH (46.0%) and Quinone (21.4%) with significantly increased contents. The presence of these two oxygen-containing functional groups can, on the contrary, enhance the wettability and pseudocapacitive property of material, which helps to improve the K-storage capacity of 1T/2H MoS2@rGO@C.35 In addition, the EDS mapping images of 1T/2H MoS2@rGO@C, 1T/2H MoS2@rGO and GO can also visually reflect the difference of O element content on the surface of three materials (Fig. S4, ESI†). Obviously, the content of O element in 1T/2H MoS2@rGO@C is the least, while the content of O element in GO is the most.
The carbon content of 1T/2H MoS2@rGO and 1T/2H MoS2@rGO@C can be determined by TGA (Fig. S5, ESI†). The TGA curve of 1T/2H MoS2 shows significant weight loss in the ranges of 25–250 °C, 250–310 °C and 310–430 °C. Weight loss in the range of 25–250 °C and 250–310 °C is attributed to loosely adsorbed water on the surface of the material and tightly bound water between MoS2 layers, respectively.36,37 Weight loss in the range of 310–430 °C means that MoS2 is oxidized to MoO3,38 which can be verified by the XRD pattern of the final residue after the TGA test (Fig. S6, ESI†). According to the TGA curve, the carbon content in 1T/2H MoS2@rGO and 1T/2H MoS2@rGO@C is about 2.9 wt% and 13.3 wt%, respectively. It is worth noting that the TGA curve of 1T/2H MoS2@rGO@C lags behind the other two samples, indicating that 1T/2H MoS2@rGO@C has better thermal stability. This advantage benefit from two factors. One is the carbon coating design of 1T/2H MoS2@rGO@C. 1T/2H MoS2@rGO is completely coated by a carbon layer, which acts as a barrier during heating, retarding the dispersion and emission of small gas molecules.39 The other is the calcination process (600 °C, 3 h, Ar) during the synthesis of 1T/2H MoS2@rGO@C. This calcination step is capable of removing water, which is present in the material by chemisorption and physisorption, in large quantities. Based on the above two points, 1T/2H MoS2@rGO@C shows better thermal stability.
Fig. 3a illustrates the XRD patterns of the three specimens. Notably, the (002) peaks exhibit distinct shifts: 8.9° for 1T/2H MoS2, 8.9° for 1T/2H MoS2@rGO, and 9.49° for 1T/2H MoS2@rGO@C, in comparison to the (002) peak of standard MoS2 (JCPDS No. 37–1492) at 14.4°.40 This shift signifies the expansion of the (002) interplanar spacing, computed via the Scherer equation, from 0.616 nm to 0.996 nm, 0.996 nm, and 0.930 nm, respectively. This expanded interlayer spacing could be attributed to NH4+ intercalation,26 where its diameter (0.350 nm) is close to the expanded interlayer dimensions (0.380 nm and 0.314 nm). Moreover, 1T/2H MoS2@rGO@C exhibits a notably diminished (002) peak intensity relative to the other samples, indicative of successful carbon coating introduction.41
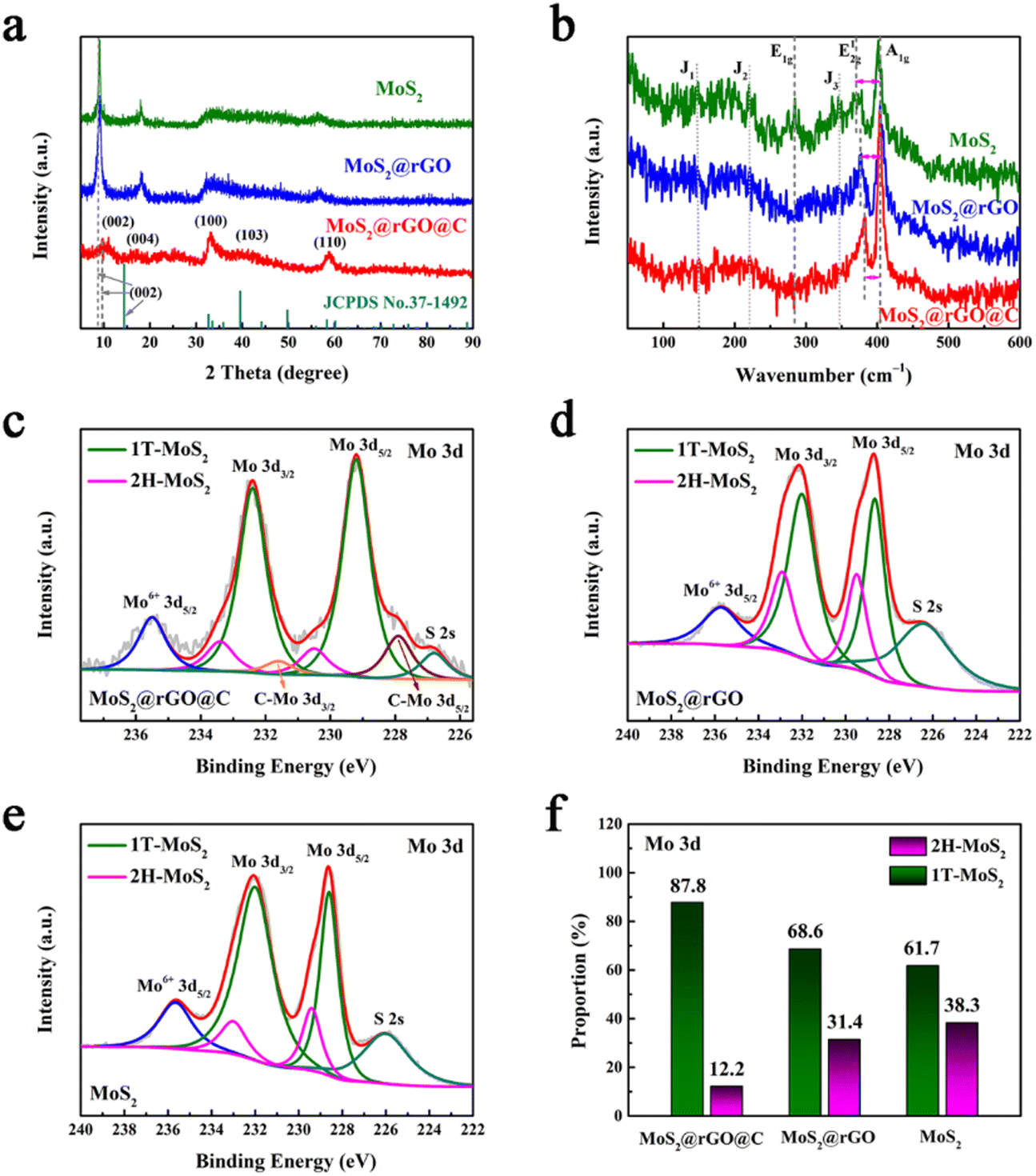 | ||
| Fig. 3 (a) XRD and (b) Raman of 1T/2H MoS2@rGO@C, 1T/2H MoS2@rGO and 1T/2H MoS2. XPS spectra from Mo 3d of (c) 1T/2H MoS2@rGO@C, (d) 1T/2H MoS2@rGO and (e) 1T/2H MoS2. (f) The proportion of 2H-MoS2 and 1T-MoS2 containing components in the three samples, respectively. | ||
Fig. 3b illustrates the Raman spectra of 1T/2H MoS2, 1T/2H MoS2@rGO, and 1T/2H MoS2@rGO@C. The Raman spectra of MoS2 exhibit characteristic E1g (283.9 cm−1), E12g (370.5 cm−1), and A1g (404.0 cm−1) modes, signifying the presence of the semiconductor 2H MoS2 phase.42 Notably, discernible J1 (147.0 cm−1), J2 (220.8 cm−1), and J3 (346.9 cm−1) peaks indicate the metallic 1T MoS2 phase.26 Evidently, the synthesized MoS2 sample contains both 2H and 1T phases. Remarkably, in the Raman spectra of 1T/2H MoS2@rGO and 1T/2H MoS2@rGO@C, the intensity of J1, J2, J3, and E1g peaks is notably reduced or even nearly indiscernible, possibly due to the poor crystallinity of MoS2.26 Concurrently, the frequency separation between E12g and A1g peaks correlates with the layer numbers of MoS2 nanosheets.40 A smaller frequency separation indicates fewer MoS2 layers. Compared to 1T/2H MoS2 (33.6 cm−1) and 1T/2H MoS2@rGO (26.7 cm−1), the frequency separation in 1T/2H MoS2@rGO@C is merely 20.7 cm−1, implying that the number of layers of MoS2 in 1T/2H MoS2@rGO@C is less.
Although Raman analysis confirms the presence of both 1T and 2H phases in MoS2, the relatively weak characteristic peaks, especially for the 1T phase, hinder a convincing phase analysis of MoS2 and quantitative assessment of the 1T and 2H phase components. To address this, XPS analysis was conducted on the three samples. The Mo 3d spectra distinctly indicate the coexistence of 2H and 1T phases in all three materials (Fig. 3c–e). For instance, in the 1T/2H MoS2@rGO@C spectrum (Fig. 3c), two prominent peaks at 229.2 and 232.4 eV signify the 1T phase,28 while the two relatively weaker peaks at 230.5 and 233.4 eV indicate 2H-MoS2.43 Notably, detailed analysis of the Mo 3d region in the 1T/2H MoS2@rGO@C spectrum (Fig. 3f) reveals a significantly higher composition proportion of the 1T phase (87.8%) compared to the 2H phase (12.2%). Moreover, for the 1T phase components in the three samples, 1T/2H MoS2@rGO@C (87.8%) exhibits the highest 1T phase composition, surpassing 1T/2H MoS2@rGO (68.6%) and 1T/2H MoS2 (61.7%) (Fig. 3f). And, for the 1T/2H MoS2@rGO@C synthesized by the scheme, the 1T phase content is also better than that of the traditional MoS2 exfoliation method reported in the literature.26,42 The content of 1T phase component in 1T/2H MoS2@rGO@C is the largest, which is mainly attributed to two aspects. One is that graphene provides more active sites for MoS2 growth, thus avoiding MoS2 stacking to obtain more monolayer 1T-MoS2. The second is that dopamine may promptly exfoliate 2H-MoS2 and coat it during the carbon coating process, thus retaining more 1T phase. Furthermore, for 1T/2H MoS2@rGO@C, a peak at 226.8 eV in the Mo 3d spectra corresponds to S 2s.44 While a peak at 235.5 eV, corresponding to Mo6+, indicates the minor presence of MoO3 oxide species in the sample.44 Analogous characteristic peaks of S 2s and Mo6+ are found in the other two samples (1T/2H MoS2@rGO and 1T/2H MoS2). Based on these results, the successful synthesis of MoS2 is confirmed in all three samples. In the Mo 3d spectra of 1T/2H MoS2@rGO@C, a doublet peak at 227.9 and 231.6 eV implies the C–Mo bond (Fig. 3c),45 indicating a close adhesion and contact between MoS2@rGO and the carbon coating.15
In addition to the Mo 3d spectra, the S 2p spectra of the three samples can also be used to confirm the content of the 2H and 1T phase components. Notably, in the S 2p spectra of 1T/2H MoS2@rGO@C, two primary peaks at 162.1 and 163.9 eV correspond to the 1T phase (Fig. S7a, ESI†),44 while the other two peaks at 163.4 and 165.2 eV indicate the 2H phase.46 This same conclusion holds for 1T/2H MoS2@rGO and 1T/2H MoS2 in their respective S 2p spectra (Fig. S7b and c, ESI†). As with the Mo 3d spectra, quantitative analysis of the S 2p spectra of all three samples (Fig. S7d, ESI†) reveals similar results regarding the 2H and 1T phase components. Specifically, for 1T/2H MoS2@rGO@C, the 1T phase content is 80.6%, whereas the 2H phase content stands at 19.4%. This proportion of the 1T phase (80.6%) also outperforms 1T/2H MoS2@rGO (67.4%) and 1T/2H MoS2 (58.9%) (Fig. S7d, ESI†). In the S 2p spectrum of 1T/2H MoS2@rGO@C, the S–O peak at 168.2 eV suggests partial oxidation of the sulfur element, implying slight MoS2 oxidation.44 Notably, in the S 2p spectrum of 1T/2H MoS2@rGO, the S–O peak splits into two peaks, S–O 2p1/2 (169.4 eV) and S–O 2p3/2 (168.3 eV).47 Similarly, in the S 2p spectrum of MoS2, the S–O peak is also deconvoluted into S–O 2p1/2 (169.3 eV) and S–O 2p3/2 (168.2 eV). Furthermore, a obvious observation emerges from the C 1s spectrum analysis: 1T/2H MoS2@rGO lacks the C–Mo (283.9 eV) peak, but 1T/2H MoS2@rGO@C exhibits this peak (Fig. S8, ESI†).15 This observation underscores a tight bridge between MoS2@rGO and the carbon coating, aligning with the analysis of Mo 3d spectra in 1T/2H MoS2@rGO@C. In addition, the C 1s spectrum of 1T/2H MoS2@rGO@C contains the following five peaks: C–C/CC at 284.5 eV, C–O at 285.1 eV, C–O–C/S at 285.8 eV, C–O–Mo at 286.5 eV, and CO at 287.5 eV.30,45–48 And the C 1s spectrum of 1T/2H MoS2@rGO contains only these five peaks. Among them, the C–O–Mo bond demonstrates a tight bridge between rGO and MoS2.
To delve into the intricate connection between the electrochemical performance of 1T/2H MoS2@rGO@C and the 1T phase content, CV tests were performed on three materials with a voltage range of 0.2 to 2.8 V. In the case of 1T/2H MoS2@rGO@C, the peak within the 0.73 to 1.16 V range corresponds to the intercalation of K ions into MoS2, forming KxMoS2 during the initial cathodic scan (Fig. S9a, ESI†).49 At 0.50 V, the peak signifies the transformation from KxMoS2 to Mo and K2S4,32 along with the irreversible formation of SEI.50 The 1.61 V peak indicates the conversion of Mo to MoS2 and the deintercalation of K+ during the first anodic scan.51 Remarkably, this identical cathodic reaction process persists across the subsequent two cycles: K ion insertion generates KxMoS2 (1.40 V), followed by conversion to Mo and K2S4 (1.11–0.53 V).52 Yet, for 1T/2H MoS2@rGO and 1T/2H MoS2, only one cathodic peak representing the conversion reaction is retained in the second and third cycles (Fig. S9b and c, ESI†). The vanishing intercalation peak might stem from structural impairment due to phase conversion.53 Put differently, a heightened 1T phase content aids in preserving the crystal structure's integrity and stability, ensuring complete intercalation and conversion reactions within each cycle. This reaction mechanism underpins the impressive potassium storage capacity exhibited by 1T/2H MoS2@rGO@C composites.
Simultaneously, galvanostatic charge–discharge profiles at 100 mA g−1 were analyzed for 1T/2H MoS2@rGO@C, 1T/2H MoS2@rGO, and 1T/2H MoS2. 1T/2H MoS2@rGO@C exhibited a first discharge capacity of 900.4 mA h g−1, a charge capacity of 409.1 mA h g−1, and an initial Coulombic efficiency of 45.4% (Fig. S9d, ESI†). The lower initial Coulombic efficiency typically stems from SEI film formation and electrolyte decomposition. Conversely, in the absence of carbon coating (1T/2H MoS2@rGO), the first discharge capacity reached 1143.9 mA h g−1, while the first charge capacity is 460.7 mA h g−1, resulting in an initial Coulombic efficiency of 40.3% (Fig. S9e, ESI†). In contrast, pure 1T/2H MoS2 demonstrated a first discharge capacity of 798.6 mA h g−1, a charge capacity of 262.9 mA h g−1, and an initial Coulomb efficiency of a mere 32.9% (Fig. S9f, ESI†). This clearly underscores that a higher content of 1T phase components enhances the material's initial Coulombic efficiency.
Electrochemical measurements were conducted on 1T/2H MoS2, 1T/2H MoS2@rGO, and 1T/2H MoS2@rGO@C materials to assess their potassium storage capacity. Initially, the cycling performance of the three materials was compared at 100 mA g−1 (Fig. 4a). Evidently, the 1T/2H MoS2@rGO@C material demonstrates remarkable cycle stability from the 15th cycle onward, maintaining a capacity of 388.8 mA h g−1. Impressively, even after 500 cycles, the specific capacity of the 1T/2H MoS2@rGO@C material remains at 351.0 mA h g−1, indicating an outstanding capacity retention rate of 90.3%. During the initial 15 cycles, 1T/2H MoS2@rGO@C experiences a slight capacity decline, attributed to SEI film formation, minor irreversible potassium storage, and electrolyte-related side reactions.54 In contrast, the cycling profiles of 1T/2H MoS2@rGO and 1T/2H MoS2 exhibit a pronounced descending trend. Particularly, the specific capacity of the 1T/2H MoS2@rGO material sharply diminishes before the 60th cycle, plummeting to a mere 86.3 mA h g−1 by the 300th cycle. This observation further corroborates the heightened potassium storage capacity resulting from carbon coating and a substantial content of 1T phase components in MoS2 materials.
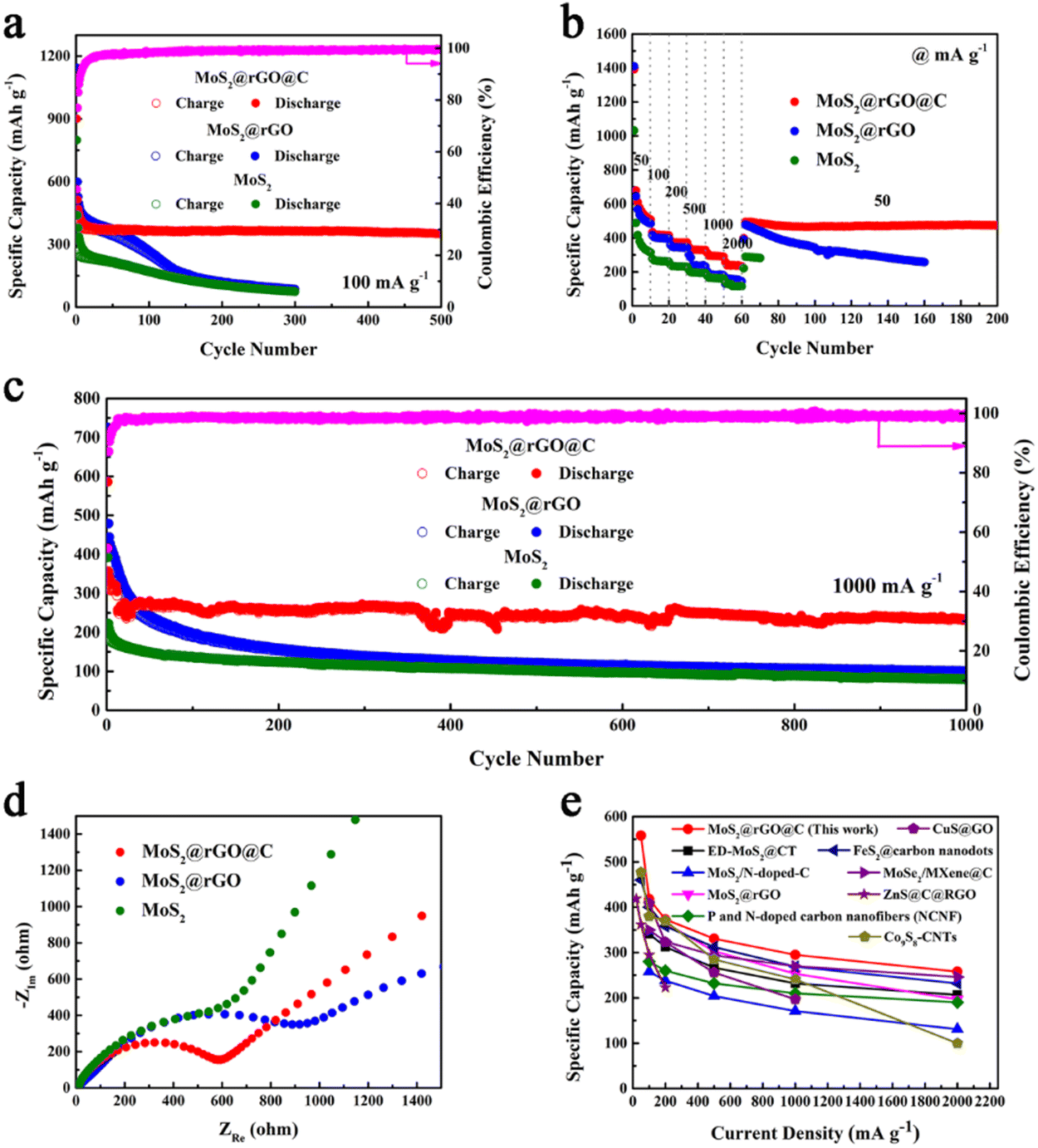 | ||
| Fig. 4 Electrochemical performance of 1T/2H MoS2@rGO@C, 1T/2H MoS2@rGO and 1T/2H MoS2: (a) cycling performance at 100 mA g−1, (b) rate performance from 50 to 2000 mA g−1, (c) long-term cycling stability at 1000 mA g−1, (d) Nyquist plots. (e) Comparison of the specific capacity with the reported anode materials of PIBs. | ||
The rate characteristics of 1T/2H MoS2, 1T/2H MoS2@rGO, and 1T/2H MoS2@rGO@C were also assessed (Fig. 4b). Notably, 1T/2H MoS2@rGO@C showcases robust specific capacities across various current densities: 508.1, 414.7, 372.7, 359.1, 292.7, and 238.2 mA h g−1 within the range of 50–2000 mA g−1. Impressively, even with a recovery to 50 mA g−1, the specific capacity swiftly returns to 493.3 mA h g−1. Furthermore, it maintains a commendable 473.8 mA h g−1 after 200 cycles. Similarly, 1T/2H MoS2@rGO attains specific capacities of 488.3, 396.6, and 344.8 mA h g−1 at 50, 100, and 200 mA g−1. However, the specific capacity experiences precipitous decline under high current densities (500, 1000, and 2000 mA g−1), reaching only 235.9, 181.7, and 144.1 mA h g−1. Notably, after reverting to a current density of 50 mA g−1, the specific capacity still gradually decreased with cycling, reaching 257.3 mA h g−1 after 160 cycles. Comparatively, pure 1T/2H MoS2 manifests worst rate performance. In summary, the augmented 1T phase content and carbon coating contribute to enhanced structural stability and improved electronic conductivity. These attributes bestow the 1T/2H MoS2@rGO@C composite with exceptional potassium storage capabilities.
Furthermore, it is crucial to investigate the long-term cycling stability of 1T/2H MoS2@rGO@C (Fig. 4c). Initially, at a high current density of 1000 mA g−1, 1T/2H MoS2@rGO@C exhibits an impressive initial discharge specific capacity of 586.1 mA h g−1, followed by a capacity decline prior to the 30th cycle (266.5 mA h g−1). Subsequently, the specific capacity stabilizes, maintaining 233.8 mA h g−1 after 1000 cycles. Conversely, for 1T/2H MoS2@rGO, the initial discharge specific capacity is 725.1 mA h g−1, followed by severe capacity decay, resulting in a mere 100.7 mA h g−1 after 1000 cycles. While the initial discharge specific capacity of 1T/2H MoS2 is only 392.4 mA h g−1, and even decreased to 79.7 mA h g−1 after 1000 cycles. As a result, 1T/2H MoS2@rGO@C exhibits remarkable long-term cycling stability.
Notably, in the long-term cycling stability test of 1000 mA g−1, the specific capacity of 1T/2H MoS2@rGO is greater than that of the other two materials in the first 50 cycles. This phenomenon is also observed in the cyclic performance at 100 mA g−1 (Fig. 4a). First, in the first 50 cycles, the specific capacity of 1T/2H MoS2@rGO is higher than that of 1T/2H MoS2, because rGO itself shows better potassium storage capacity in PIBs. For example, as early as 2015, Jian et al. have demonstrated that graphite can achieve a specific capacity of 273 mA h g−1 in PIBs.55 Therefore, compared to 1T/2H MoS2, the specific capacity of 1T/2H MoS2@rGO compounded with rGO will be higher. Secondly, in the first 50 cycles, the specific capacity of 1T/2H MoS2@rGO is also higher than that of 1T/2H MoS2@rGO@C. This is mainly attributed to the structural design of the carbon coating in 1T/2H MoS2@rGO@C: the fully coated carbon layer on the surface of 1T/2H MoS2@rGO acts as a barrier to slow down the penetration of the electrolyte. This allows the K+ first contact the carbon shell, and then slowly contact the MoS2. This means that the carbon shell stores potassium first, followed by MoS2. In contrast, in 1T/2H MoS2@rGO, the electrolyte is in direct contact with MoS2. As we all know, the theoretical specific capacity of MoS2 is higher than that of carbon materials, but its stability is lower than that of carbon materials. As a result, in the first 50 cycles, even if 1T/2H MoS2 is combined with two carbon materials (rGO and carbon coating layer), the specific capacity of 1T/2H MoS2@rGO@C is still lower than that of 1T/2H MoS2@rGO. And this comes with the benefit that the cycling stability of 1T/2H MoS2@rGO@C is superior to that of 1T/2H MoS2@rGO.
Moreover, to delve deeper into the long-term cycling stability of 1T/2H MoS2@rGO@C, SEM images of these three materials after 1000 cycles were analyzed (Fig. S10, ESI†). Impressively, following 1000 cycles, the multilayer 3D structure composed of graphene, MoS2 nanosheets, and carbon coating in 1T/2H MoS2@rGO@C remains intact (Fig. S10a and b, ESI†). In contrast, in 1T/2H MoS2@rGO material, while the graphene structure remains complete, the MoS2 nanosheets layered atop it are absent, potentially due to exfoliation or pulverization (Fig. S10c and d, ESI†). As for 1T/2H MoS2, its MoS2 nanosheets exhibit denser aggregation, but the bulk structure formed by these aggregated nanosheets is no longer evident (Fig. S10e and f, ESI†). Collectively, these findings validate that the structural blueprint of 1T/2H MoS2@rGO@C ensures greater overall framework stability, safeguarding against MoS2 nanosheet exfoliation and pulverization during cycling. Furthermore, the remarkable long-term cycling stability of 1T/2H MoS2@rGO@C holds true even at a higher current density of 2000 mA g−1 (Fig. S11, ESI†): The initial discharge specific capacity is 312.7 mA h g−1 and exhibits rapid decline before the 10th cycle (198.1 mA h g−1), but the specific capacity remained stable at 163.3 mA h g−1 after 1500 cycles.
Furthermore, the exceptional electrochemical performance of 1T/2H MoS2@rGO@C is further demonstrated using EIS measurements. Typically, the Nyquist plots (Fig. 4d) elucidate this, with the semicircle signifying the charge transfer resistance (Rct), and the inclined line representing the Warburg resistance (W).56 Notably, in Fig. 4d, the semicircle diameter of 1T/2H MoS2@rGO@C is significantly smaller than that of 1T/2H MoS2@rGO and 1T/2H MoS2. This suggests that 1T/2H MoS2@rGO@C possesses the least charge transfer resistance. Evidently, synergizing high-content 1T phase components with carbon-coated structures improves electrical conductivity, thereby augmenting the potassium storage capacity of the 1T/2H MoS2@rGO@C composite. In conclusion, 1T/2H MoS2@rGO@C exhibits an exceptional potassium storage capacity. Furthermore, it is worth noting that the 1T/2H MoS2@rGO@C composite stands as a competitive counterpart to recently reported anode materials for PIBs (Fig. 4e), such as ED-MoS2@CT,31 MoS2/N-doped-C,51 MoS2@rGO,40 porous and nitrogen-doped carbon nanofibers (NCNF),57 FeS2@carbon nanodots,58 MoSe2/MXene@C,59 CuS@GO,60 ZnS@C@RGO,61 and Co9S8-CNTs.62
The potassium storage capability of 1T/2H MoS2@rGO@C is further understood through a detailed kinetics analysis (Fig. 5). Circular voltammetry (CV) profiles at varying scan rates (0.2–1.2 mV s−1) were acquired (Fig. 5a). The connection between peak current (i) and scan rate (v) is mathematically represented as:63
| i = avb | (1) |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) i = blogv + loga i = blogv + loga | (2) |
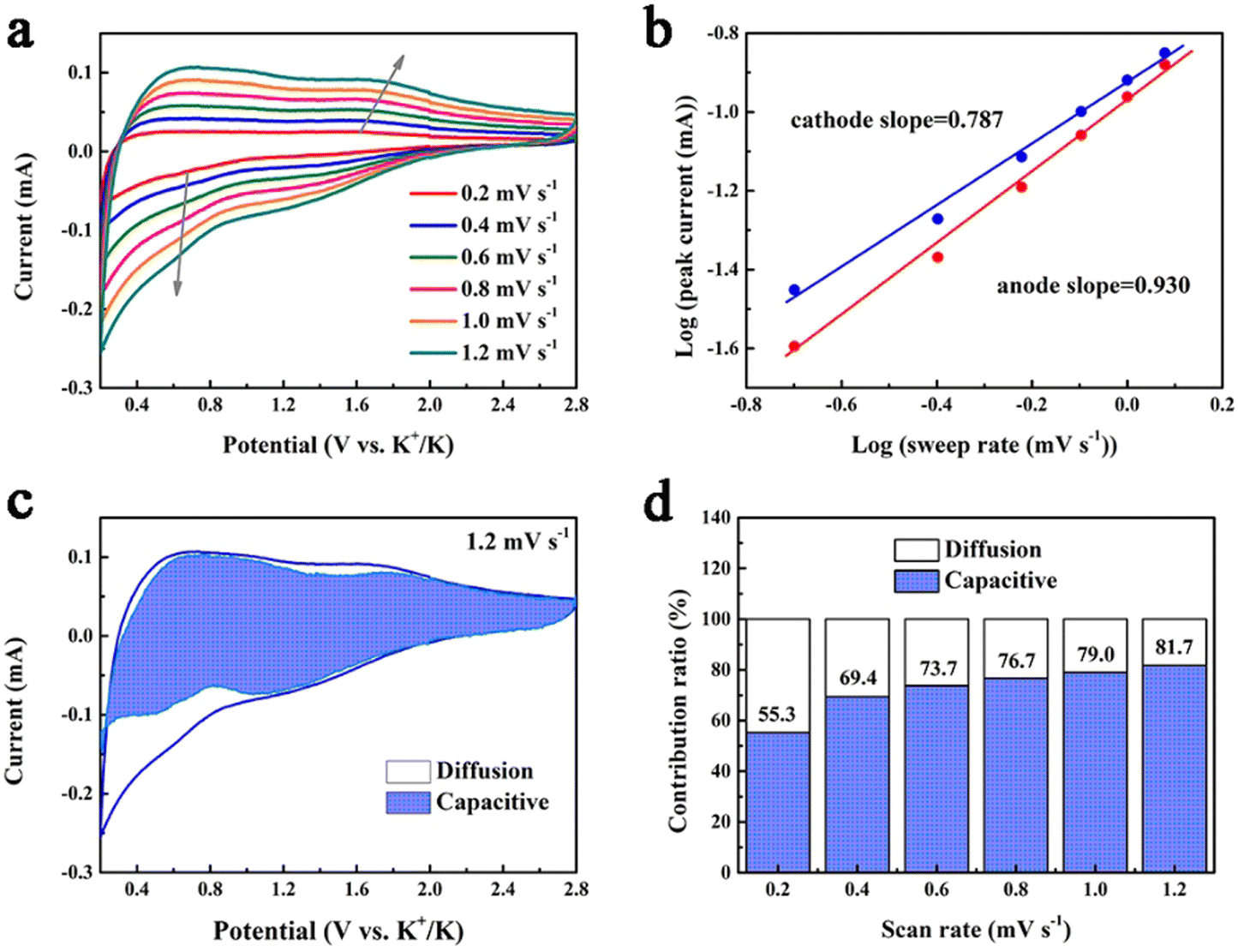 | ||
| Fig. 5 (a) CV curves of 1T/2H MoS2@rGO@C at various scan rates. (b) A linear relationship between peak currents (i) and scan rates (v) for the two peaks labeled in (a). (c) Capacitive contribution at a scan rate of 1.2 mV s−1. (d) The percentages of capacitive and diffusion contributions at various scan rates. | ||
The constants a and b hold significance in this context. Specifically, when the b value approximates 0.5, diffusion-driven behavior predominates in the electrochemical storage process; conversely, the b value nearing 1 signifies dominance of pseudocapacitive behavior.64 For the 1T/2H MoS2@rGO@C electrode, the application of eqn (2) enables the calculation of the b-values for the cathodic (0.787) and anodic peaks (0.930), implying that the pseudocapacitive behavior is dominant. The pseudocapacitive behavior is related to interfacial ion storage,65 which also reveals the reason why the 1T/2H MoS2@rGO@C electrode exhibits excellent rate capability.66 The relative contribution of diffusion-controlled (k1v1/2) and pseudocapacitance-driven (k2v) behaviors at various scan rates is quantitatively determinable via the ensuing equation:67
| i = k1v1/2 + k2v | (3) |
Typically, at a scan rate of 1.2 mV s−1, the pseudocapacitance contribution ratio in 1T/2H MoS2@rGO@C is 81.7% (Fig. 5c). Furthermore, this contribution ratio progressively increases (from 55.3% to 81.7%) as the scan rate rises from 0.2 to 1.2 mV s−1 (Fig. 5d). This underscores that a high proportion of pseudocapacitance-controlled behavior augments the potassium storage capacity of electrode materials.
In-depth understanding of the potassium storage mechanism within 1T/2H MoS2@rGO@C composites was pursued through ex situ XRD and HRTEM analysis (Fig. 6). The XRD results for 1T/2H MoS2@rGO@C are depicted in Fig. 6a. At completely discharge to 0.2 V, two peaks emerge at 30.9° and 31.5°, corresponding to K2S4 (JCPDS No. 30-0992) of the (032) and (212) planes.68 The amorphous phase of Mo nanograins potentially accounts for the absence of discernible Mo diffraction peaks.69 Meanwhile, the absence of conspicuous MoS2 diffraction peak at 0.2 V discharge indicates the final conversion of 1T/2H MoS2@rGO@C into Mo and K2S4, aligning with the prior analysis of electrochemical reaction mechanism in Fig. S9 (ESI†). Notably, the strong peaks at 43.3° and 50.4° are attributed to (111) and (200) planes of Cu, potentially stemming from the test sample scraped from a Cu foil-based electrode. When the electrode is fully charged to 2.8 V, the K2S4 diffraction peak vanishes, and a faint peak at 32.3°, indexed to (100) plane of MoS2 (JCPDS No. 37-1492), arises. Compared to pristine MoS2 (32.7°), the peak corresponding to the (100) plane is shifted, result from the intercalation and extraction of K+. Reappearance of MoS2 signals reversible conversion and deintercalation reactions during charging. Fig. 6a reveals relatively weak intensities for both K2S4 and MoS2 peaks, likely due to the incomplete conversion reaction between MoS2 and K+, as well as interference from residual electrode components such as activated carbon and binder. In summation, the electrochemical reaction mechanism can be delineated as follows:
| MoS2 + xK+ + xe− ↔ KxMoS2 | (4) |
| KxMoS2 + (2 − x)K+ + (2 − x)e− ↔ Mo + K2S4 | (5) |
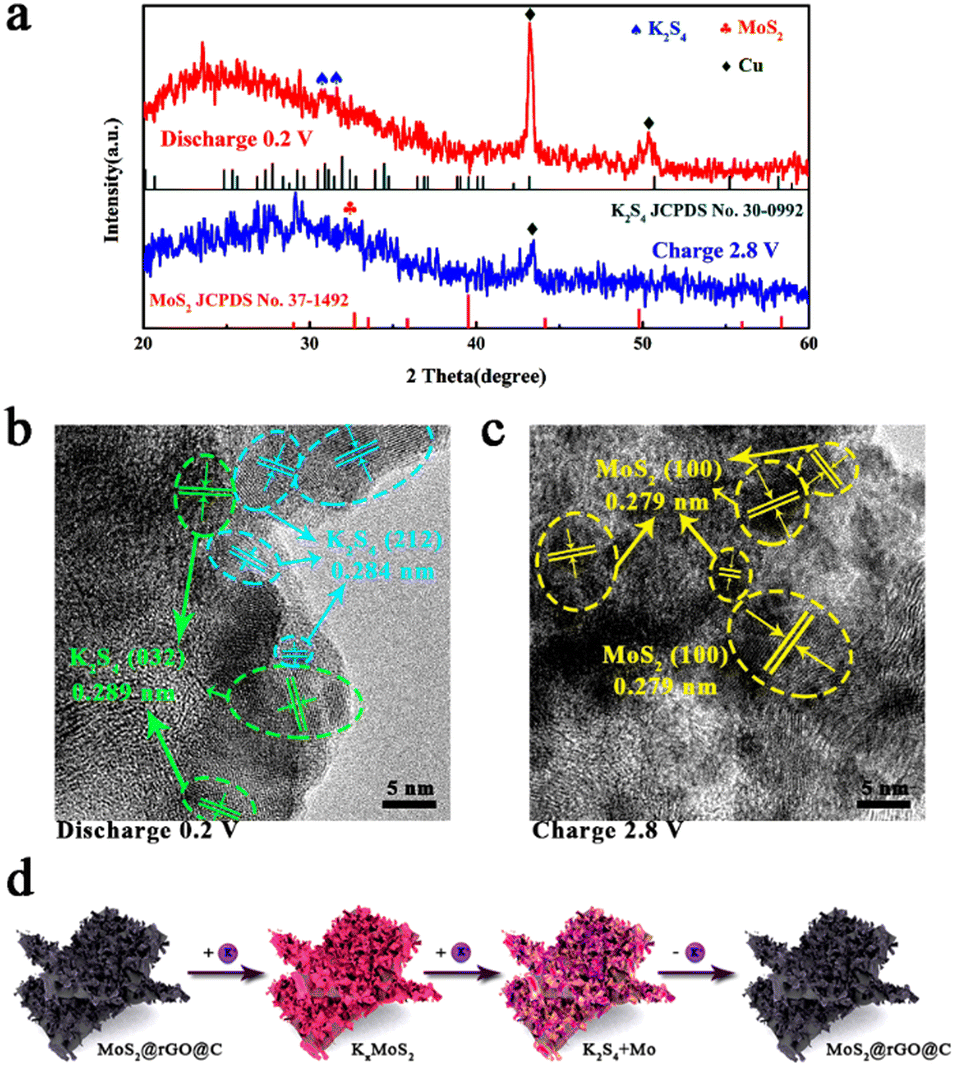 | ||
| Fig. 6 Potassium storage mechanism analysis of 1T/2H MoS2@rGO@C electrode at different charge–discharge states: (a) ex situ XRD patterns and (b and c) ex situ HRTEM analysis in the initial charge–discharge processes. (d) Schematic illustration of K+ ion storage mechanism in the 1T/2H MoS2@rGO@C. | ||
Additionally, ex situ HRTEM analysis in Fig. 6b and c elucidates the structural changes and phase transformation within 1T/2H MoS2@rGO@C during electrochemical reactions. Distinct lattice spacings of 0.289 nm and 0.284 nm emerge when fully discharge to 0.2 V (Fig. 6b), corresponding to (032) and (212) planes of K2S4, respectively. When fully charge to 2.8 V, lattice fringes of MoS2 (0.279 nm) are observed, identified as (100) planes of MoS2. Notably, this lattice spacing (0.279 nm) is larger than that of the (100) plane of pristine MoS2, potentially attributed to K+ intercalation. For the 1T/2H MoS2@rGO@C composite, these HRTEM results align with ex situ XRD findings, corroborating the plausibility of the potassium storage mechanism analysis. The electrochemical mechanism is succinctly depicted in Fig. 6d.
To assess the practical viability of 1T/2H MoS2@rGO@C, we constructed a potassium ion full cell employing 1T/2H MoS2@rGO@C as the anode and PTCDA as the cathode (Fig. 7a). Fig. 7b presents the charge–discharge curves of the 1T/2H MoS2@rGO@C//PTCDA full cell, operating within a voltage window of 1.0–4.2 V, at 100 mA g−1. The initial charge–discharge capacities are 476.4 mA h g−1 and 345.4 mA h g−1, respectively, with an initial Coulombic efficiency of 72.5%. The lower initial Coulombic efficiency could be attributed to electrolyte decomposition and SEI layer formation. Simultaneously, the 1T/2H MoS2@rGO@C//PTCDA full cell demonstrates exceptional cycling stability at 100 mA g−1 (Fig. 7c), maintaining a discharge capacity of 294.3 mA h g−1 and 74.2% capacity retention after 160 cycles. Notably, the initial 30 cycles show a rapid decline in charge–discharge capacity, potentially stemming from an unoptimized cathode/anode mass ratio.70 Additionally, impressive rate performance is exhibited by the 1T/2H MoS2@rGO@C//PTCDA full cell (Fig. 7d), with discharge capacities of 362.6, 310.0, 274.9, 244.2, and 210.9 mA h g−1 at 100, 200, 500, 1000, and 2000 mA g−1, respectively. Upon reverting to a current density of 100 mA g−1, the discharge capacity is maintained at 342.1 mA h g−1. These results underscore the prospective practical applications of the 1T/2H MoS2@rGO@C composite in full cells.
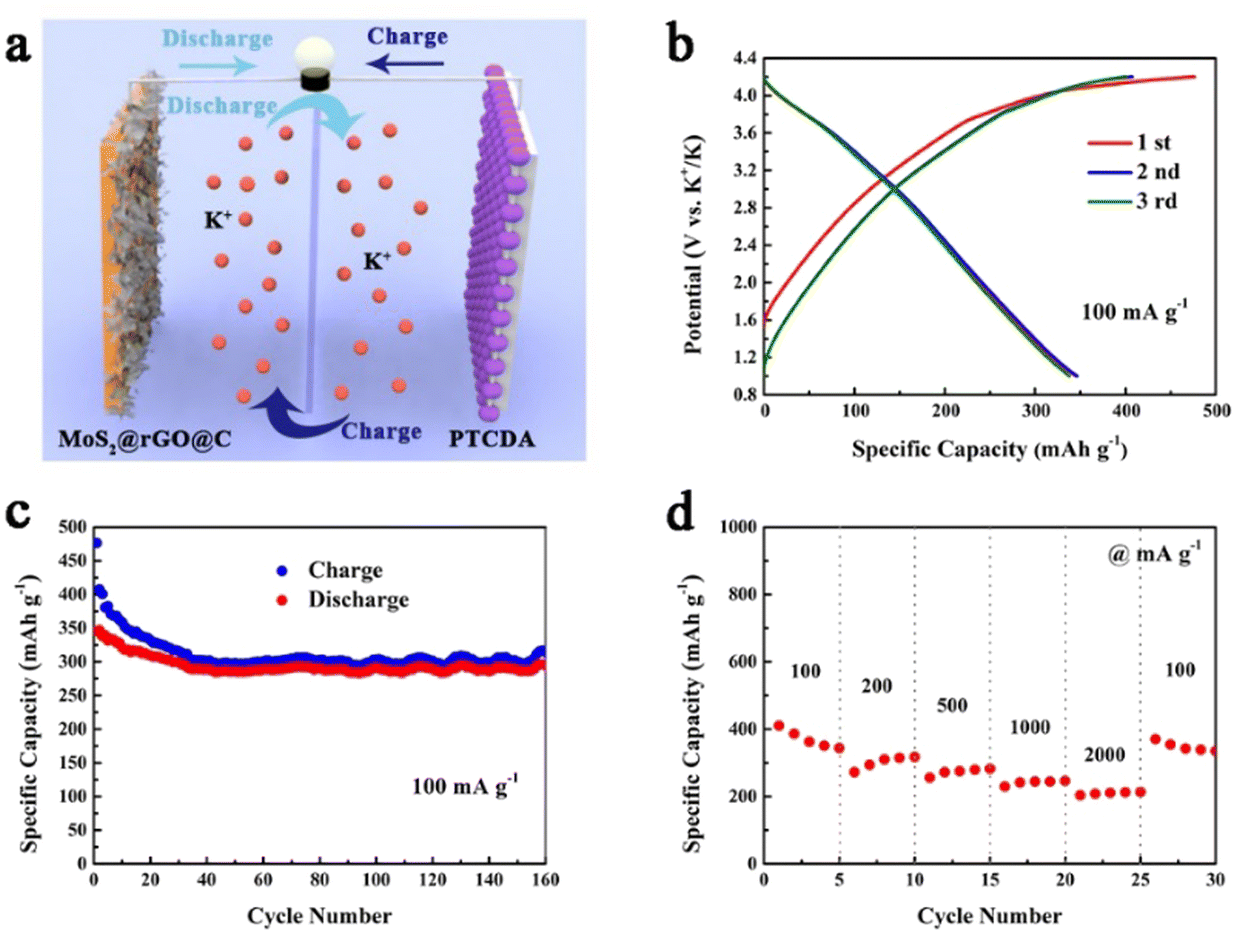 | ||
| Fig. 7 Electrochemical performance of the 1T/2H MoS2@rGO@C//PTCDA full cell: (a) Schematic illustration. (b) Initial three charge–discharge curves at 100 mA g−1. (c) The cycling performance at 100 mA g−1. (d) Rate performance. | ||
Conclusions
In conclusion, we propose an innovative synthesis method for fabricating 1T/2H MoS2@rGO@C composites with a distinctive “sandwich” structure. This synthesis endows MoS2 with extended (002) crystal planes. The uniform dispersion of ultrathin MoS2 nanosheets on rGO effectively mitigates volume changes during the long-term cycling. Furthermore, carbon coating significantly enhances material conductivity and reversible capacity. In particular, the introduction of dopamine in 1T/2H MoS2@rGO@C composites elevates the 1T phase component content, culminating in remarkable potassium storage capacity. At a current density of 100 mA g−1, 1T/2H MoS2@rGO@C displays satisfactory cycling stability as a PIBs anode, retaining a discharge capacity of 351.0 mA h g−1 after 500 cycles. Concurrently, it demonstrates long cycling stability over 1000 cycles (233.8 mA h g−1) at 1000 mA g−1, along with favorable rate performance (238.2 mA h g−1 at 2000 mA g−1). Furthermore, a 1T/2H MoS2@rGO@C//PTCDA full cell is constructed, achieving a substantial initial discharge capacity (345.4 mA h g−1 at 100 mA g−1), exceptional cycling stability over 160 cycles (294.3 mA h g−1 at 100 mA g−1), and notable rate performance (210.9 mA h g−1 at 2000 mA g−1). This work introduces a pioneering concept for designing high-performance PIBs electrode materials.Author contributions
R. Hu conceived, designed and wrote the original manuscript. Y. Tong optimised the language of the manuscript and provided comments, support, and formal analysis. J. Yin was involved in the investigation, formal analysis, conceptualization, and project administration. J. Wu directed, supported, and supervised the completion of the manuscript. J. Zhao participated in the formal analysis, conceptualization, and investigation of the experiments and manuscripts. D. Cao provided financial support for the experiment, conducted project administration and formal analysis. Both G. Wang and K. Zhu directed and supervised the completion of the experiment and manuscript, provided financial support, and conducted project administration. All authors edited, commented, and reviewed on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51702063, 51672056), Natural Science Foundation of Heilongjiang (LC2018004), China Postdoctoral Science Foundation (2018M630340, 2019T120254) and the Fundamental Research Funds for the Central University.References
- C. D. Quilty, D. Wu, W. Li, D. C. Bock, L. Wang, L. M. Housel, A. Abraham, K. J. Takeuchi, A. C. Marschilok and E. S. Takeuchi, Chem. Rev., 2023, 123(4), 1327–1363 CrossRef CAS PubMed.
- Z. Gong, Z. Li, P. Wang, K. Jiang, Z. Bai, K. Zhu, J. Yan, K. Ye, D. Cao and G. Chen, Energy Mater. Adv., 2023, 4, 0035 CrossRef CAS.
- J. Wu, M. Ihsan-Ul-Haq, F. Ciucci, B. Huang and J.-K. Kim, Energy Storage Mater., 2021, 34, 582–628 CrossRef.
- K. Wu, X. Cao, M. Li, B. Lei, J. Zhan and M. Wu, Small, 2020, 16(43), 2004178 CrossRef CAS PubMed.
- H. Lei, J. Li, X. Zhang, L. Ma, Z. Ji, Z. Wang, L. Pan, S. Tan and W. Mai, InfoMat, 2022, 4(2), e12272 CrossRef CAS.
- X. Sun, Z. Li, Z. Liu, X. Lv, K. Shi, R. Chen, F. Wu and L. Li, Adv. Funct. Mater., 2023, 33(22), 2300125 CrossRef CAS.
- M. Kim, L. Ma, Z. Li, W. Mai, N. Amiralian, A. E. Rowan, Y. Yamauchi, A. Qin, R. A. Afzal, D. Martin, A. K. Nanjundan and J. Li, J. Mater. Chem. A, 2023, 11, 16626–16635 RSC.
- W. Feng, H. Wang, Y. Jiang, H. Zhang, W. Luo, W. Chen, C. Shen, C. Wang, J. Wu and L. Mai, Adv. Energy Mater., 2022, 12, 2103343 CrossRef CAS.
- T. Wu, W. Zhang, J. Yang, Q. Lu, J. Peng, M. Zheng, F. Xu, Y. Liu and Y. Liang, Carbon Energy, 2021, 3(4), 554–581 CrossRef CAS.
- Y. Feng, S. Chen, J. Wang and B. Lu, J. Energy Chem., 2020, 43, 129–138 CrossRef.
- J. Zhou, Y. Shen, F. Lv, W. Zhang, F. Lin, W. Zhang, K. Wang, H. Luo, Q. Wang, H. Yang and S. Guo, Adv. Funct. Mater., 2022, 32(34), 2204495 CrossRef CAS.
- J. Li, F. Hu, H. Wei, J. Hei, Y. Yin, G. Liu, N. Wang and H. Wei, Composites, Part B, 2023, 250, 110424 CrossRef CAS.
- Q. Zang, Q. Zhang, Z. Luo, L. Liao, X. Ouyang and S. Xie, J. Power Sources, 2022, 543, 231800 CrossRef CAS.
- X. Ren, Q. Zhao, W. D. McCulloch and Y. Wu, Nano Res., 2017, 10, 1313–1321 CrossRef CAS.
- Q. Pan, Z. Tong, Y. Su, Y. Zheng, L. Shang and Y. Tang, Adv. Mater., 2022, 34(39), 2203485 CrossRef CAS PubMed.
- J. Wu, J. Liu, J. Cui, S. Yao, M. Ihsan-Ul-Haq, N. Mubarak, E. Quattrocchi, F. Ciucci and J.-K. Kim, J. Mater. Chem. A, 2020, 8, 2114–2122 RSC.
- T. Xiong, X. Yao, D. Adekoya, H. Yang and M.-S. Balogun, J. Mater. Sci. Technol., 2023, 145, 14–24 CrossRef CAS.
- Y. Jiao, A. Mukhopadhyay, Y. Ma, L. Yang, A. M. Hafez and H. Zhu, Adv. Energy Mater., 2018, 8, 1702779 CrossRef.
- J. Li, F. Hu, H. Wei, J. Hei, Y. Yin, G. Liu, N. Wang and H. Wei, Composites, Part B, 2023, 250, 110424 CrossRef CAS.
- L. Ma, J. Ye, W. Chen, D. Chen and J. Y. Lee, Nano Energy, 2014, 10, 144–152 CrossRef CAS.
- H.-H. Lu, C.-S. Shi, N.-Q. Zhao, E.-Z. Liu, C.-N. He and F. He, Rare Met., 2018, 37, 107–117 CrossRef CAS.
- G. Ye, Y. Gong, J. Lin, B. Li, Y. He, S. T. Pantelides, W. Zhou, R. Vajtai and P. M. Ajayan, Nano Lett., 2016, 16(2), 1097–1103 CrossRef CAS PubMed.
- Y. Xiao, M. Zhou, J. Liu, J. Xu and L. Fu, Sci. China Mater., 2019, 62, 759–775 CrossRef CAS.
- T. Zhu, C. Liu, X. Tan, B. Huang, G.-Q. Bian, Q. Shao, S. Bai, Y. Qian, Y. Li and X. Huang, ACS Nano, 2019, 13(10), 11303–11309 CrossRef CAS PubMed.
- C. K. Chua, A. H. Loo and M. Pumera, Chem. – Eur. J., 2016, 22, 14336 CrossRef CAS PubMed.
- D. Wang, X. Zhang, S. Bao, Z. Zhang, H. Fei and Z. Wu, J. Mater. Chem. A, 2017, 5, 2681–2688 RSC.
- X. Fan, P. Xu, D. Zhou, Y. Sun, Y. C. Li, M. A. T. Nguyen, M. Terrones and T. E. Mallouk, Nano Lett., 2015, 15(9), 5956–5960 CrossRef CAS PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135(28), 10274–10277 CrossRef CAS PubMed.
- M. Acerce, D. Voiry and M. Chhowalla, Nat. Nanotechnol., 2015, 10, 313–318 CrossRef CAS PubMed.
- Q. Sun, D. Li, L. Dai, Z. Liang and L. Ci, Small, 2020, 16, 2005023 CrossRef CAS PubMed.
- Y. Cui, W. Liu, W. Feng, Y. Zhang, Y. Du, S. Liu, H. Wang, M. Chen and J. Zhou, Adv. Funct. Mater., 2020, 30, 1908755 CrossRef CAS.
- B. Luo, P. Wu, J. Zhang, L. Cao, C. Wang, B. Lu, B. Zhang and X. Ou, Nano Res., 2021, 14, 3854–3863 CrossRef CAS.
- Y. J. Oh, J. J. Yoo, Y. Il Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- Z. Xiong, C. Liaoa and X. Wang, J. Mater. Chem. A, 2014, 2, 19141–19144 RSC.
- Y. Zhang, G. Wen, P. Gao, S. Bi, X. Tang and D. Wang, Electrochim. Acta, 2016, 221, 167–176 CrossRef CAS.
- Y. Liang, H. D. Yoo, Y. Li, J. Shuai, H. A. Calderon, F. C. R. Hernandez, L. C. Grabow and Y. Yao, Nano Lett., 2015, 15(3), 2194–2202 CrossRef CAS PubMed.
- T. Xiang, Q. Fang, H. Xie, C. Wu, C. Wang, Y. Zhou, D. Liu, S. Chen, A. Khalil, S. Tao, Q. Liu and L. Song, Nanoscale, 2017, 9, 6975–6983 RSC.
- Y. Liu, L. Zhang, Y. Zhao, T. Shen, X. Yan, C. Yu, H. Wang and H. Zeng, J. Alloys Compd., 2019, 787, 996–1003 CrossRef CAS.
- P. Bhawal, S. Ganguly, T. K. Chakia and N. C. Das, RSC Adv., 2016, 6, 20781–20790 RSC.
- S. Chong, L. Sun, C. Shu, S. Guo, Y. Liu, W. A. Wang and H. K. Liu, Nano Energy, 2019, 63, 103868 CrossRef CAS.
- L. Li, W. Zhang, X. Wang, S. Zhang, Y. Liu, M. Li, G. Zhu, Y. Zheng, Q. Zhang, T. Zhou, W. K. Pang, W. Luo, Z. Guo and J. Yang, ACS Nano, 2019, 13(7), 7939–7948 CrossRef CAS PubMed.
- D. Wang, B. Su, Y. Jiang, L. Li, B. K. Ng, Z. Wu and F. Liu, Chem. Eng. J., 2017, 330, 102–108 CrossRef CAS.
- Y. Qi, Q. Xu, Y. Wang, B. Yan, Y. Ren and Z. Chen, ACS Nano, 2016, 10(2), 2903–2909 CrossRef CAS PubMed.
- Y. You, Y. Ye, M. Wei, W. Sun, Q. Tang, J. Zhang, X. Chen, H. Li and J. Xu, Chem. Eng. J., 2019, 355, 671–678 CrossRef CAS.
- R. Hu, Y. Fang, K. Zhu, X. Yang, J. Yin, K. Ye, J. Yan, D. Cao and G. Wang, Appl. Surf. Sci., 2021, 564, 150387 CrossRef CAS.
- B. Gao, X. Du, Y. Ma, Y. Li, Y. Li, S. Ding, Z. Song and C. Xiao, Appl. Catal., B, 2020, 263, 117750 CrossRef CAS.
- M. Song, H. Tan, X. Li, A. I. Y. Tok, P. Liang, D. Chao and H. J. Fan, Small Methods, 2020, 4(6), 1900274 CrossRef CAS.
- B. Chen, H. Lu, J. Zhou, C. Ye, C. Shi, N. Zhao and S.-Z. Qiao, Adv. Energy Mater., 2018, 8(15), 1702909 CrossRef.
- G. Ma, Y. Zhou, Y. Wang, Z. Feng and J. Yang, Nano Res., 2021, 14, 3523–3530 CrossRef CAS.
- J. Hu, Y. Xie, X. Zhou and Z. Zhang, ACS Appl. Mater. Interfaces, 2020, 12(1), 1232–1240 CrossRef CAS PubMed.
- B. Jia, Q. Yu, Y. Zhao, M. Qin, W. Wang, Z. Liu, C.-Y. Lao, Y. Liu, H. Wu, Z. Zhang and X. Qu, Adv. Funct. Mater., 2018, 28(40), 1803409 CrossRef.
- J. M. Ge, L. Fan, J. Wang, Q. Zhang, Z. Liu, E. Zhang, Q. Liu, X. Yu and B. Lu, Adv. Energy Mater., 2018, 8(29), 1801477 CrossRef.
- J. Sheng, T. Wang, J. Tan, W. Lv, L. Qiu, Q. Zhang, G. Zhou and H.-M. Cheng, ACS Nano, 2020, 14(10), 14026–14035 CrossRef CAS PubMed.
- W. Miao, Y. Zhang, H. Li, Z. Zhang, L. Li, Z. Yu and W. Zhang, J. Mater. Chem. A, 2019, 7, 5504–5512 RSC.
- Z. Jian, W. Luo and X. Ji, J. Am. Chem. Soc., 2015, 137(36), 11566–11569 CrossRef CAS PubMed.
- N. Cheng, W. Zhou, J. Liu, Z. Liu and B. Lu, Nano-Micro Lett., 2022, 14, 146 CrossRef CAS PubMed.
- F. Liu, J. Meng, F. Xia, Z. Liu, H. Peng, C. Sun, L. Xu, G. V. Tendeloo, L. Mai and J. Wu, J. Mater. Chem. A, 2020, 8, 18079–18086 RSC.
- K. Han, W. Zhao, Q. Yu, Z. Liu, P. Li, W. A. Wang, L. Song, F. An, P. Cao and X. Qu, J. Power Sources, 2020, 469, 228429 CrossRef CAS.
- H. Huang, J. Cui, G. Liu, R. Bi and L. Zhang, ACS Nano, 2019, 13(3), 3448–3456 CrossRef CAS PubMed.
- X. Jia, E. Zhang, X. Yu and B. Lu, Energy Technol., 2020, 8(1), 1900987 CrossRef CAS.
- J. Chu, W. A. Wang, J. Feng, C.-Y. Lao, K. Xi, L. Xing, K. Han, Q. Li, L. Song, P. Li, X. Li and Y. Bao, ACS Nano, 2019, 13(6), 6906–6916 CrossRef CAS PubMed.
- J. Zhou, H. Zhao, Q. Zhang, T. Li, Y. Li, N. Lin and Y. Qian, Chem. Commun., 2019, 55, 1406–1409 RSC.
- R. Hu, K. Zhu, K. Ye, J. Yan, Q. Wang, D. Cao and G. Wang, Appl. Surf. Sci., 2021, 536, 147832 CrossRef CAS.
- M. Han, Y. Mu, J. Guo, L. Wei, L. Zeng and T. Zhao, Nano-Micro Lett., 2023, 15, 80 CrossRef CAS PubMed.
- X. Wei, C.-C. Lin, C. Wu, N. Qaiser, Y. Cai, A.-Y. Lu, K. Qi, J.-H. Fu, Y.-H. Chiang, Z. Yang, L. Ding, O. S. Ali, W. Xu, W. Zhang, M. B. Hassine, J. Kong, H.-Y. Chen and V. Tung, Nat. Commun., 2022, 13, 6006 CrossRef CAS PubMed.
- X. Zhu, F. Xia, D. Liu, X. Xiang, J. Wu, J. Lei, J. Li, D. Qu and J. Liu, Adv. Funct. Mater., 2023, 33(2), 2207548 CrossRef CAS.
- M. Xie, Z. Lv, W. Zhao, Y. Fang, J. Huang and F. Huang, Chem. Eng. J., 2023, 470, 144282 CrossRef CAS.
- J. Xie, Y. Zhu, N. Zhuang, H. Lei, W. Zhu, Y. Fu, M. S. Javed, J. Li and W. Mai, Nanoscale, 2018, 10(36), 17092–17098 RSC.
- J. Zhang, P. Cui, Y. Gu, D. Wu, S. Tao, B. Qian, W. Chu and L. Song, Adv. Mater. Interfaces, 2019, 6(22), 1901066 CrossRef CAS.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26(23), 3854–3859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00404j |
| This journal is © The Royal Society of Chemistry 2024 |