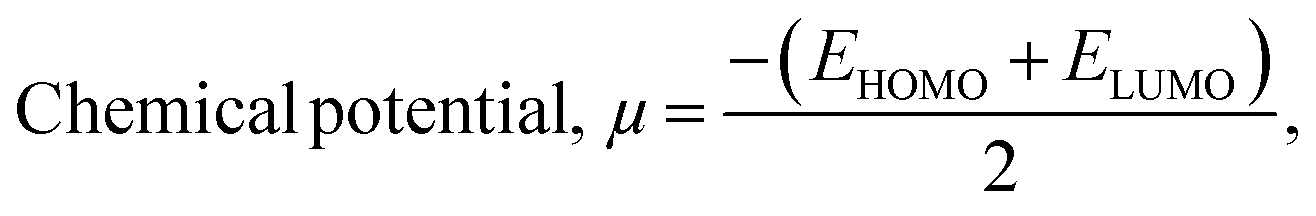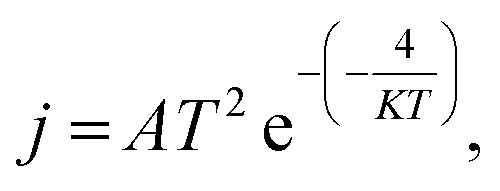DOI:
10.1039/D4NA00472H
(Paper)
Nanoscale Adv., 2024,
6, 5988-6007
Understanding the adsorption performance of hetero-nanocages (C12–B6N6, C12–Al6N6, and B6N6–Al6N6) towards hydroxyurea anticancer drug: a comprehensive study using DFT
Received
7th June 2024
, Accepted 8th September 2024
First published on 3rd October 2024
Abstract
Cancer is a paramount health challenge to global health, which forms tumors that can invade nearby tissues and spread to neighboring cells. Recently, nanotechnology has been used to control the growth of cancer, in which anticancer drugs are delivered to cancerous cells via nanoparticles without damaging healthy tissues. In this study, DFT investigations were carried out to examine the adsorption behavior of C24, B12N12, and Al12N12 nanocages as well as their heterostructures C12–B6N6, C12–Al6N6, and B6N6–Al6N6 towards the hydroxyurea (HU) anticancer drug. In this regard, adsorption energy, interaction distance between the drug and nanocages, charge transfer, energy gap, dipole moment, quantum molecular descriptors, work function, and COSMO surface analysis were analyzed to understand their adsorption performance. Findings demonstrate that the adsorption energies of two hetero-nanocages on their hexagonal (SH) and tetragonal (ST) sites are favorable for the drug delivery process. The computed adsorption energy of B6N6–Al6N6 of the ST/AlN site is 183.59 kJ mol−1, which is higher than that of the C12–Al6N6 nanocage, including minimum adsorption distances. Negative adsorption energy with low adsorption distances implies an attractive interaction between the drug and nanocages. During the interaction, a significant amount of charge is transferred between the drug and nanocages. Furthermore, for both complexes, larger dipole moments were observed in water media compared to gas media. From DOS spectra, prominent peaks were found in the Fermi level after adsorption of HU on the nanocages, implying the reduction of the energy gap. Noticeable overlaps between the PDOS spectra of the nanocages and HU's close contact atom demonstrate the formation of chemical bonds between two specific atoms. Therefore, it can be concluded that among the nanocages, C12–Al6N6 and B6N6–Al6N6 may be suitable carriers for HU drug.
1. Introduction
Uncontrolled growth and abnormal cell proliferation characterize an intricate number of diseases known as cancer. Cancer can develop into benign or harmful tumors, affecting virtually every tissue or organ. Projections allude to its extensive rise over the next two decades, which will cause global health challenges.1 Although chemotherapy and radiotherapy are primary treatments, their impact on both cancerous and normal cells leads to adverse effects. To address this issue, medical researchers worldwide are working diligently to develop new carrier systems. In this system, a nanocarrier carries anticancer drugs to its target cells while causing minimal damage to healthy tissues. Therapy results depend on the cancer type, tumor spot, and progression phase. However, traditional chemotherapy has drawbacks, including harmful side effects, e.g. hair loss (76%), nausea (77%), fatigue (90%), vomiting (75%), and weakness (95%),2,3 and damaging effects on healthy tissues. To address these challenges and enhance traditional methods of cancer treatment, researchers are developing nanotechnology-based drug delivery strategies.4 One of the therapeutic medications generally implemented for the treatment of breast cancer is hydroxyurea (HU), also referred to as hydroxycarbamide. Furthermore, this is an anti-metabolite chemotherapy drug used to treat cervical cancers and leukemia. However, prolonged consumption of HU has been related to inevitable undesirable consequences, such as alopecia (skin), hyperpigmentation (sclera), leukopenia, and leg ulcers. It can, therefore, be utilized in treating cancer diseases under tight supervision and special requirements.5,6 Because HU is one of the most important drugs declared by the World Health Organization, much effort has been invested to develop or conjugate the HU structural components to approach more triumphant treatments.
When compared to other adsorbents, nanomaterials have the following benefits: (i) a larger surface area and smaller particle size for a more stable analyte-nanoparticle interaction that strengthens the capacity of nanomaterials for adsorbing and separating the analytes;7 (ii) enormous surface energy with fast adsorption rates for an immediate adsorption equilibrium state and (iii) ease of functionalization for the removal of specific compounds. Additionally, low-dimensional nanomaterials are highly sensitive sensors.8 Nanotechnology-based drug delivery systems have increased therapeutic efficacy and decreased the side effects of anticancer drugs. Therefore, a suitable nanocarrier design is crucial. However, several types of nanomaterials, such as zero-dimensional (0D), one-dimensional (1D), two-dimensional (2D), and three-dimensional (3D) nanomaterials, are used as nanocarriers in anticancer drug delivery systems. Although these nanomaterials have been evaluated to function as HU drug carriers in previous studies, the results require verification through additional research to move toward a more comprehensive therapy.9 Among all the nanocarriers, such as nanosheets and nanotubes, 0D nanocages are an excellent choice for targeted drug delivery systems due to their spherical form, structural stability, extreme sensitivity to drug molecules, and non-toxicity.10–14 Significantly, in an investigation of the interaction of cathinone anticancer drugs with BN nanotubes, nanocages, and nanosheets, Nejati et al. found that the nanocages were more sensitive and had better adsorption energy than the nanosheets and nanotubes.15
Numerous studies have been conducted on C24 fullerene as an adsorbent because of its remarkable thermodynamic stability.14–17 A stable structure for appropriate drug delivery with a large drug dose is the X12Y12 nanocluster according to previous studies on certain (XY) nanocages (X = Al, B… and Y = P, N).18–21 The Al12N12 and B12N12 nanoclusters are among several types of structures formed by the X12Y12 nanocluster. In drug delivery systems, their use has been thoroughly investigated. Previous studies have demonstrated that the stability of the B12N12 structure is greater than that of BN fullerenes among various geometries.22 In addition, based on the estimated binding energy per AlN unit and disproportionation energy, Wu et al. showed that the Al12N12 nanocarrier had the greatest stability among nitride fullerene-like structures.23 Hosseinian et al.'s study revealed that at first the interactions of C24 fullerene and 5-fluorouracil were poor, but they subsequently became stronger when a boron atom was added in place of one carbon atom.17 This indicates that doping increases the sensitivity and reactivity of the nanocages towards the anticancer drug. Javan et al. extensively investigated the derivatives of C24, such as B12N12, to enhance the interaction of the drug based on DFT and TDFT calculations. It was found that the interaction between 5-FU and the boron substrate affected their electronic characteristics, i.e. the reactivity of the substrate was increased and additional chemical reactions were potentially enabled.24 Following DFT theory, Kaviani et al. reported that an effective biosensor for detecting alprazolam (ALP) drugs is the B12N12 nano-cage.25 A study by Gong et al. showed theoretically that for the delivery of flutamide drugs, the potential nano-vehicle was AlN material.26 A study of the structural and electronic features of nanocage and HU drugs by Saadh et al. evaluated a BN nanocage structure due to adsorb and detected the anticancer medication hydroxyurea to obtain insight into efficient drug delivery systems.9 Because of the special qualities of hetero-nanocage, for instance, their strong interaction with drug molecules, they have been investigated and suggested as ways to deliver drugs nowadays.27–29 Theoretical investigations conducted by Paul et al. and Fan et al. confirmed the energy stability of C12–Al6N6 and C12–B6N6 hetero-nanocages.29,30 Muktadir et al. discussed the adsorption ability, reactivity, and sensitivity of C–BN, C–AlN, and BN–AlN hetero-nanocages concerning the cisplatin (CP) drug. According to their investigation, the B6N6–Al6N6 hetero nanocage was very sensitive to the molecules of CP drug.31
The purpose of this study is to investigate the adsorption capacity of C24 fullerene and its derivatives for the targeted delivery of HU performed using Density Functional Theory (DFT). First, the carbon atoms are replaced by B, Al, and N atoms in equal numbers to form B12N12 and Al12N12. Initially, we investigated the sensitivity of three nanocages, namely C24 to HU and discovered no strong interaction. Therefore, to improve the adsorption properties, 12 carbon atoms were replaced by equal numbers of Al, B, and N atoms in the C24 nanocage. For a better understanding of the biosensing of HU, adsorption simulations using the DFT framework are again performed on the modified C24 nanocage and fullerene-like hetero-nanocages, namely C12–Al6N6, C12–B6N6 and B6N6–Al6N6. To analyze the adsorption behavior of the proposed nanocages towards HU drug adsorption energy, adsorption distance, and charge transfer, the dipole moment, HOMO and LUMO energy, energy gap, work function, density of states (DOS) and projected density of states (PDOS) were investigated to understand the electronic properties of both the gas and water phases. Moreover, we conducted a COSMO surface analysis and quantum molecular descriptors to analyze the reactivity of the suggested nanocages toward HU.
2. Computational details
All the optimizations and calculations were implemented in the Materials Studio software package for both the gas and water phases. Spin-unrestricted density functional theory (DFT) was employed in the Dmol3 module to investigate the adsorption behavior of nanocages towards HU drugs.32,33 In previous studies, it has been observed that the Local Density Approximation (LDA) shows inflated results in bond energy and equilibrium distance. Therefore, instead of LDA, Generalized Gradient Approximations (GGAs) with the Perdew–Burke–Ernzerhof (PBE) functional were applied to describe the exchange–correlation interaction of the nanocages as well as the complexes.34–36 Without regarding symmetry constraints, a Double Numerical basis set plus Polarization (DNP) and DFT Semi-core Pseudopotentials (DSPP) were implemented for core treatments.37 The DNP basis set is more accurate because it can compensate for the impacts of the Basis Set Superposition Error (BSSE) inaccuracy. Grimme's dispersion-corrected Density Functional Theory (DFT-D) was used for all the treatments due to the consideration of van der Waals interactions, which additionally accommodate long-range electron effects.38,39 The global orbital cutoff radius was 5 Å.
To investigate how the HU drug interacts with the nanocages (C24, B12N12, Al12N12, C12–Al6N6, C12–B6N6, and B6N6–Al6N6), the adsorption energy (EAd) is computed using the following formula in both gas and water phases:40,41
| | | Ead = Ecomplex − Edrug − Enanocage | (1) |
where
Edrug and
Enanocage denote the total energy of the drug and nanocage, respectively, and
Ecomplex is the energy of the complexes. The energy of the Highest Occupied Molecular Orbital (
EHOMO) and the Lowest Unoccupied Molecular Orbital (
ELUMO) are used to describe the electronic properties and calculate the energy gap (
Eg) by applying the following formula:
42
The change in energy gap (% ΔEg) is calculated to observe the sensitivity of the nanocages towards the HU drug as follows:43
| |  | (3) |
where
Eg1 and
Eg2 denote the values of the energy gap of the nanocages before and after the adsorption of the HU drug, respectively.
To determine the reactivity and stability of the nanocages, drugs, and complexes, Quantum Mechanical Descriptors such as hardness (η), softness (S), chemical potential (μ), and electrophilicity index (ω) were calculated by applying the following equation:44–47
| |  | (4) |
| |  | (5) |
| |  | (6) |
| |  | (7) |
Mulliken and Hirshfeld charge analysis was used to study the transmission of charges between drugs and nanocages. As a function of space, the electron density is analyzed to determine whether the drug HU functions as an acceptor or donor.48 Because water is the typical transportation medium in the human body, the HU–nanocages interaction in water media was examined using the conductor-like screening model (COSMO) under a dielectric constant of 78.54.49,50 All the investigations (energy calculation and geometry optimization) are conducted again using the solvation model.
3. Results and discussion
3.1. Adsorption behavior of HU on the C24
The adsorption behavior was carried out on two distinct sites of the C24 nanocage to search for the most favorable adsorption configurations of HU on the C24 nanocage. HU drug adsorption occurred on both the hexagonal (SH) and tetragonal (ST) surfaces of the nanocage. According to previous studies, it is understood that the drug's orientation parallel to the nanocage in each instance shows good adsorption behavior.51,52Fig. 1 displays the optimized structure and the interaction distance between the drug and the nanocage after adsorption. Calculations of adsorption energy, adsorption distance, and charge transfer using both Mulliken and Hirshfeld charge analyses were performed for all complexes to fully evaluate the adsorption capacity. The adsorption energy can be positive or negative. If the adsorption energy is positive, then it is called an endothermic reaction, and if it is negative, then it indicates an exothermic reaction.53 From our investigation, for all configurations, we obtain negative adsorption energy, which determines that the interaction between the drug and nanocages is attractive. It is known that a complex with a higher negative adsorption energy value and minimum distance has a stronger adsorption mechanism.54,55 From Table 1, it has been observed that according to the two sites (ST, SH) of the C24 nanocage towards the HU drug, the adsorption energies are −21.13 kJ mol−1 and −21.96 kJ mol−1 at 2.552 Å and 2.531 Å distances for ST and SH sites, respectively. Hence, after observation, the results clearly show that the HU drug in the tetragon site of the C24 nanocage behaves much better than its placement on the hexagon site. This is because these HU/C24 complexes have higher adsorption energies and minimum distances. However, when the adsorption energy is greater than 77.19 kJ mol−1, strong chemisorption and physisorption occur due to the adsorption energy being less than 77.19 kJ mol−1. As the adsorption energies for both complexes of C24 nanocages toward the HU drug are less than 77.19 kJ mol−1, it indicates weak physisorption. An investigation into the net charge transfer between the HU drug and the nanocages was conducted using Mulliken and Hirshfeld charge analysis. When the drug was not adsorbed to the nanocage, the drug molecules had a zero net charge. However, during the adsorption of the HU drug molecule, the drug either receives or transmits a certain amount of charge to the nanocages.56 If the charge transfer has occurred from the nanocage to the drug, then the value of the charge is negative, and if the charge transfer value is positive, then electrons are transferred from the drug to the nanocage.57 During the adsorption of the HU drug on both the ST and SH sites of the C24 nanocage, a small amount of charge 0.075e and 0.063e (in Mulliken) and 0.032e and 0.008e (in Hirshfeld) are transferred from the drug to the nanocage. This indicates that the C24 nanocage acts as a charge acceptor, whereas the HU drug is a donor.
 |
| | Fig. 1 Optimized structures with HOMO and LUMO of (a) C24, (b) HU/C24 (ST), and (c) HU/C24 (SH). | |
Table 1 Calculated adsorption energies (Ead) in kJ mol−1, interaction distances (d) in Å, and charge transfers (Q) in e Mulliken and Hirshfeld for complexes in both gas and water media
| Structure |
Site |
Gas media |
Water media |
|
E
ad
|
d
|
Q
M
|
Q
HS
|
E
ad
|
D
|
Q
M
|
Q
HS
|
| C/HU |
ST |
−21.13 |
2.552 |
0.075 |
0.032 |
−13.99 |
3.023 |
0.069 |
0.055 |
| C/HU |
SH |
−21.96 |
2.531 |
0.063 |
0.008 |
−11.37 |
2.667 |
0.063 |
0.003 |
| BN/HU |
ST |
−105.09 |
1.566 |
0.343 |
0.385 |
−113.88 |
1.524 |
0.45 |
0.53 |
| BN/HU |
SH |
−93.55 |
1.652 |
0.251 |
0.295 |
−127.82 |
1.509 |
0.486 |
0.589 |
| AlN/HU |
ST |
−12.79 |
3.296 |
0.026 |
0.031 |
−115.54 |
1.949 |
0.147 |
0.386 |
| AlN/HU |
SH |
−120.39 |
1.83 |
0.073 |
0.115 |
−197.93 |
1.512 |
0.128 |
0.207 |
| C_BN/HU |
ST |
−123.35 |
1.554 |
0.349 |
0.382 |
−144.05 |
1.51 |
0.463 |
0.521 |
| C_BN/HU |
SH |
−91.19 |
1.622 |
0.265 |
0.329 |
−162.40 |
1.149 |
0.522 |
0.653 |
| C_BN/HU |
SP/T |
−26.61 |
2.118 |
0.079 |
−0.023 |
−18.06 |
2.176 |
0.057 |
−0.042 |
| C_BN/HU |
SP/H |
−19.51 |
2.579 |
0.047 |
−0.032 |
−11.03 |
2.857 |
0.029 |
−0.039 |
| C_AlN/HU |
ST |
−182.31 |
1.706 |
0.124 |
0.215 |
−222.24 |
1.575 |
0.17 |
0.263 |
| C_AlN/HU |
SH |
−137.52 |
1.994 |
0.124 |
0.377 |
−208.54 |
1.9 |
0.308 |
0.743 |
| C_AlN/HU |
SP/T |
−34.62 |
2.467 |
0.04 |
−0.099 |
−9.23 |
3.102 |
0.023 |
−0.036 |
| C_AlN/HU |
SP/H |
−29.43 |
2.312 |
0.031 |
−0.085 |
−23.82 |
1.914 |
−0.012 |
−0.218 |
| BN_AlN/HU |
ST/BN |
−75.18 |
1.623 |
0.241 |
0.275 |
−68.05 |
1.58 |
0.352 |
0.393 |
| BN_AlN/HU |
SH/BN |
−46.79 |
2.098 |
0.019 |
−0.096 |
−25.23 |
1.961 |
−0.02 |
−0.149 |
| BN_AlN/HU |
ST/AlN |
−183.59 |
1.732 |
0.139 |
0.231 |
−189.85 |
1.817 |
0.249 |
0.462 |
| BN_AlN/HU |
SH/AlN |
−162.74 |
1.693 |
0.079 |
0.170 |
−155.34 |
1.897 |
0.187 |
0.480 |
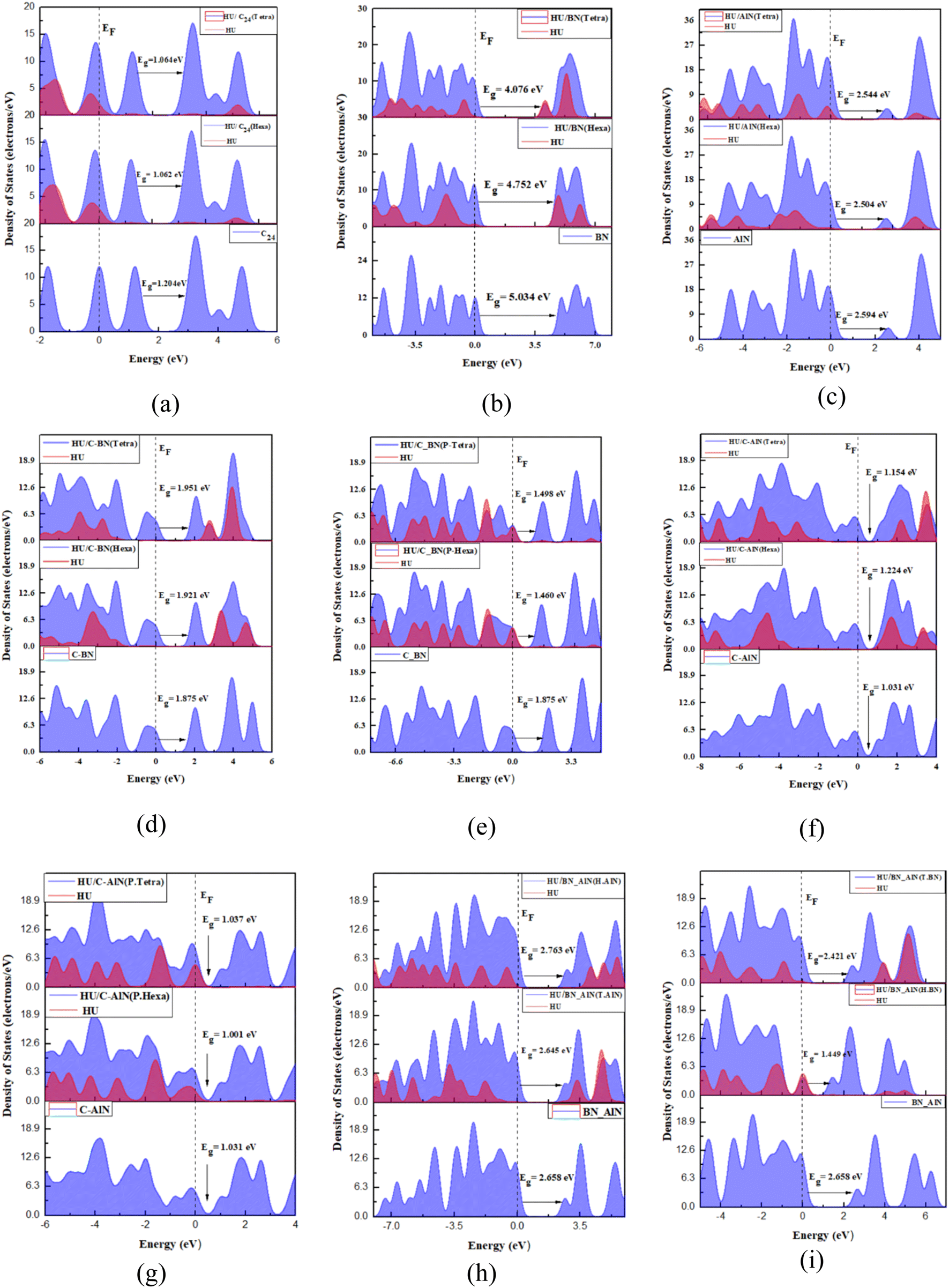
To examine the electronic behavior of the C24 towards the HU anticancer drug, the Frontier Molecular Orbital (FMO) analysis (HOMO and LUMO) and energy gap (Eg) are examined.58 The results are listed in Table 2. The FMO structures of the HU/C24 complex from both tetragon and hexagon site perspectives are illustrated in Fig. 1(b and c). For both sites of the HU/C24 complex, the Highest Occupied Molecular Orbital (HOMO) levels are randomly distributed in both drug and nanocage, and for the Lowest Unoccupied Molecular Orbital (LUMO) levels, the majority are concentrated on the molecules of the nanocage. For the ST and SH sites of C24, the HOMO energies are reduced from −5.812 eV to −5.638 eV and −5.615 eV, and the LUMO energies are decreased from −4.608 eV to −4.551 eV and −4.553 eV, respectively. Consequently, the energy gap of C24 reduces from 1.204 eV to 1.064 eV and 1.062 eV for ST and SH sites, respectively. Thus, 11.63% and 11.79% are the calculated percentage drops in the energy gap (% ΔEg) for the complexes, respectively. Moreover, the Density of States (DOS) is also used to examine the change in the energy gap.59 The DOS spectra of C24 before and after the adsorption of the HU drug are displayed in Fig. 5(a), where it is evident that after drug adsorption, additional states are created at the Fermi level that lessen the energy gap. Minimizing the energy gap increases the conductivity of the electron density exponentially, which is crucial for drug delivery.60 It must be noted that the following relationship exists between the conduction electron density (Nc) and energy gap (Eg):61
| |  | (8) |
where
A (electrons per m
3 per k
3/2) is a constant,
T is the temperature in kelvin, and
K is Boltzmann's constant = 1.38 × 10
−23 m
2 kg s
−2 k
−1. Because the energy gap is greatly minimized for the HU/C
24 complex, the excitation energy is minimal. Consequently, the HU/C
24 complex has a rather large electron density.
Table 2 Calculated HOMO energies (EHOMO), LUMO energies (ELUMO), HOMO–LUMO energy gap (Eg) in eV, change in energy gap (% ΔEg), and dipole moment (D. M.) in Debye (D) of the studied complexes in both gas and water media
| Structure |
Site |
Gas media |
Water media |
|
E
HOMO
|
E
LUMO
|
E
g
|
% Eg |
D. M. |
E
HOMO
|
E
LUMO
|
E
g
|
% Eg |
D. M. |
| C24 |
|
−5.812 |
−4.608 |
1.204 |
0 |
0 |
−5.76 |
−4.57 |
1.203 |
0 |
0 |
| C/HU |
ST |
−5.638 |
−4.551 |
1.064 |
−11.63 |
2.88 |
−5.60 |
−4.48 |
1.117 |
−7.15 |
5.55 |
| C/HU |
SH |
−5.615 |
−4.553 |
1.062 |
−11.79 |
3.37 |
−5.64 |
−4.54 |
1.107 |
−7.98 |
5.46 |
| B12N12 |
|
−7.158 |
−2.124 |
5.034 |
0 |
0 |
−7.17 |
−2.03 |
5.136 |
0 |
0 |
| BN/HU |
ST |
−6.242 |
−2.166 |
4.076 |
−19.03 |
8.70 |
−6.28 |
−1.58 |
4.705 |
−8.39 |
13.80 |
| BN/HU |
SH |
−6.494 |
−1.742 |
4.752 |
−5.60 |
7.61 |
−6.43 |
−1.55 |
4.877 |
−5.04 |
15.72 |
| Al12N12 |
|
−5.632 |
−3.038 |
2.594 |
0 |
0 |
−5.87 |
−3.19 |
2.679 |
0 |
0 |
| AlN/HU |
ST |
−5.545 |
−3.001 |
2.544 |
−1.93 |
2.94 |
−5.43 |
−2.88 |
2.553 |
−4.70 |
12.47 |
| AlN/HU |
SH |
−5.493 |
−2.989 |
2.504 |
−3.47 |
2.24 |
−5.48 |
−2.94 |
2.534 |
−5.41 |
10.63 |
| C12B6N6 |
|
−5.770 |
−3.895 |
1.875 |
0 |
2.12 |
−5.77 |
−3.84 |
1.93 |
0 |
3.70 |
| C_BN/HU |
ST |
−4.956 |
−3.005 |
1.951 |
4.05 |
11.49 |
−5.39 |
−3.37 |
2.013 |
4.30 |
17.55 |
| C_BN/HU |
SH |
−5.183 |
−3.262 |
1.921 |
2.45 |
9.34 |
−5.25 |
−3.16 |
2.086 |
8.08 |
21.50 |
| C_BN/HU |
SP/T |
−5.493 |
−3.995 |
1.498 |
−20.11 |
5.24 |
−5.61 |
−3.85 |
1.765 |
−8.55 |
8.28 |
| C_BN/HU |
SP/H |
−5.493 |
−4.033 |
1.460 |
−22.13 |
5.74 |
−5.74 |
−3.85 |
1.889 |
−2.12 |
8.96 |
| C12Al6N6 |
|
−4.526 |
−3.495 |
1.031 |
0 |
8.83 |
−4.90 |
−3.51 |
1.39 |
0 |
13.92 |
| C_AlN/HU |
ST |
−4.220 |
−3.066 |
1.154 |
11.93 |
12.13 |
−4.73 |
−3.22 |
1.516 |
9.06 |
21.97 |
| C_AlN/HU |
SH |
−4.088 |
−2.864 |
1.224 |
18.72 |
13.76 |
−4.41 |
−2.82 |
1.587 |
14.17 |
31.99 |
| C_AlN/HU |
SP/T |
−4.830 |
−3.793 |
1.037 |
0.58 |
14.15 |
−4.86 |
−3.49 |
1.377 |
−0.94 |
11.97 |
| C_AlN/HU |
SP/H |
−4.681 |
−3.680 |
1.001 |
−2.91 |
11.51 |
−5.02 |
−3.63 |
1.397 |
0.50 |
20.28 |
| B6N6_Al6N6 |
|
−5.896 |
−3.238 |
2.658 |
0 |
7.53 |
−6.29 |
−3.30 |
2.985 |
0 |
11.35 |
| BN_AlN/HU |
ST/BN |
−5.234 |
−2.813 |
2.421 |
−8.92 |
4.24 |
−5.67 |
−3.04 |
2.634 |
−11.76 |
7.37 |
| BN_AlN/HU |
SH/BN |
−4.903 |
−3.454 |
1.449 |
−45.49 |
10.76 |
−5.56 |
−3.37 |
2.187 |
−26.73 |
15.04 |
| BN_AlN/HU |
ST/AlN |
−5.469 |
−2.824 |
2.645 |
−0.49 |
13.23 |
−5.88 |
−2.86 |
3.014 |
0.97 |
23.67 |
| BN_AlN/HU |
SH/AlN |
−5.755 |
−2.992 |
2.763 |
3.95 |
6.99 |
−5.89 |
−2.90 |
2.987 |
0.07 |
22.30 |
In the water phase, the HU drug interacts attractively with C24, similar to the gas phase. Placing the drug parallel to the C24 nanocage results in adsorption energies of −21.13 kJ mol−1 and −21.96 kJ mol−1 for the tetragon (ST) and hexagon (SH) sites of the HU/C24 complex, respectively (Table 1). As presented in Table 1, the distances between the HU drug and C24 nanocage are 3.023 Å and 2.667 Å for the ST and SH sites, respectively. For both sites, the transmitted electrons between the drug and nanocage are 0.069e and 0.055e in the Mulliken charge analysis and 0.063e and 0.003e in the Hirshfeld charge analysis, respectively. The electrical properties (e.g., Eg, EHOMO, and ELUMO energies) change after being HU adsorbed on C24 nanocage surfaces. The Eg of C24 was reduced by about 7.15% for ST and by 7.98% for the SH state after adsorption. In water media, adsorption energy reduces, while the interaction distance increases slightly compared to the gas phase. The drug molecules are shielded from interacting directly with C24 by molecules of water, which results in a reduction in adsorption energy. Compared to the gas phase, the electrical conductivity decreased as the energy difference (Eg) increases. Therefore, we may deduce that the adsorption behavior of C24 in water media decreases after drug molecule adsorption.
3.2. Adsorption behavior of HU on the B12N12
The discourse mentioned above makes it clear that C24's sensitivity to the HU drug molecules is insufficient. Therefore, we modified 50% of the C24 nanocage by boron atoms, and the other 50% was replaced by nitrogen atoms to form B12N12. HU drug molecules were adsorbed on the B12N12 nanocage at various adsorption regions after completing its geometry optimization. Two distinct configurations were assessed. The two sites are the ST and SH sites of the B12N12 nanocage [Fig. 2(b–c)]. The optimized structure of B12N12 and the relaxed complexes with minimal adsorption distances are illustrated in Fig. 2. Drug absorption occurred on the ST and SH sites of the nanocage, and it was kept at least 1.566 Å and 1.652 Å apart, respectively, with adsorption energies of −105.09 kJ mol−1 and −93.55 kJ mol−1 for the HU/B12N12 complex. The adsorption energy values are relatively large. Electrons are donated by drugs through charge transfer, while electrons are accepted by nanocages. Charges shifted to B12N12 from the HU drug include 0.343e and 0.385e in the Mulliken charge analysis and 0.251e and 0.295e in the Hirshfeld charge analysis for both ST and SH sites. Thus, compared to the SH site, the complex with the ST site has a larger adsorption energy and minimal distance. Even though complexes with SH sites transport charge more than those with ST sites, the variance is trivial. Consequently, it can be said that the ST site is a more suitable configuration for HU adsorption on B12N12. The definition of adsorption energy clarifies that chemisorption occurs on both sites. Chemisorption is significantly better in the long run when it comes to drug delivery based on previous studies. This is because in the drug delivery system, strong covalent bonds are created during chemisorption, which improves stability and enables more regulated and prolonged drug release.62
 |
| | Fig. 2 Optimized structure with HOMO and LUMO of (a) B12N12, (b) HU/B12N12 (ST), and (c) HU/B12N12 (SH). | |
After the drug is adsorbed, it has been observed that B12N12 nanocages deform close to the adsorption region. The FMO analysis includes data on the HOMO and LUMO energies, energy gap (Eg), and change in energy gap (Table 2). FMO analysis is employed to analyze the electronic properties of the molecules, such as chemical stability, reactivity, and biological activity.63 HOMO levels are observed on the nanocage for both the SH and ST sites of the HU/B12N12 complex. Additionally, the LUMO level appears on the drug for the ST site, whereas for the SH site, the LUMO level appears on the nanocage and drug molecules. After HU drug adsorption on B12N12, there is a considerable variation in the HOMO and LUMO energies, which results in a decrease in the energy gaps from 5.034 eV to 4.076 eV (ST) and 4.752 eV (SH), respectively. There is a 19.03% and 5.60% reduction in the energy gap between the drug and nanocage. When Eg decreases, B12N12 nanocages become more reactive and sensitive to the HU drug.64Fig. 5(b) shows the Fermi level and energy gap in the DOS spectra before and after the drug HU is adsorbed on B12N12. It is confirmed that the drug may significantly boost the conductivity of B12N12 by reducing the energy gap, which improves the electrical characteristics.
In the water medium, the adsorption energy for both sites increases. For this phase, the complex with SH configuration shows higher adsorption energy (−127.82 kJ mol−1) than ST (−113.88 kJ mol−1) with minimum distance (Table 1), which is vice versa to gas medium. The HU drug molecule was observed to be adsorbed slightly perpendicularly with B12N12 at a distance of 1.509 Å from the central top B atom. The HU drug molecules lose 0.53e (in Mulliken) and 0.589e (in Hirshfeld) charges, belonging to the SH configuration. After adsorption in the water phase, the Eg is minimal at the ST site, which is similar to the gas phase. After adsorbing the HU, the energy gap was reduced by approximately 8.39% (ST site). The variations in the mentioned parameters indicate that HU and B12N12 interact strongly in both the gas and aqueous phases.
3.3. Adsorption behavior of HU on the Al12N12
The Al12N12 nanocage was generated by replacing equal numbers of Al and N atoms in place of the C atoms, which improved the adsorption behavior of the C24 nanocage. After Al12N12 optimization, the HU drug was adsorbed on the surface of Al12N12 nanocages; specific reactive sites were targeted, and then two of these sites were chosen (Fig. 3), just as we had done with the two preceding nanocages. The calculated adsorption energies for the ST and SH sites are −12.79 kJ mol−1 and −120.39 kJ mol−1, respectively. According to the definition of adsorption energy, the SH configuration exhibits chemisorption, while the ST configuration exhibits physisorption. For both locations, adsorption distances were found at 3.296 Å and 1.830 Å. Therefore, the SH site exhibits the least adsorption distance with the highest adsorption energy, which indicates that this site shows better adsorption behavior than the ST site. According to the Mulliken and Hirshfeld charge analyses, the HU drug molecule transfers 0.073e and 0.115e charges to the Al12N12 nanocage, respectively, during the interaction with the nanocage's hexagonal site. The FMO maps of the SH site of the HU/Al12N12 complex are illustrated in Fig. 3(c). Here, the HOMO levels are localized maximum on the nanocage and a few are on the drug molecules, and the LUMO levels are located only on the nanocage, which means a significant amount of charge has been transferred between the HU and the nanocage. The energy gap Eg, reduces from 2.594 eV to 2.544 eV and 2.504 eV for the ST and SH sites, respectively. Additionally, Eg is shown through the DOS spectra (Fig. 5(c)).
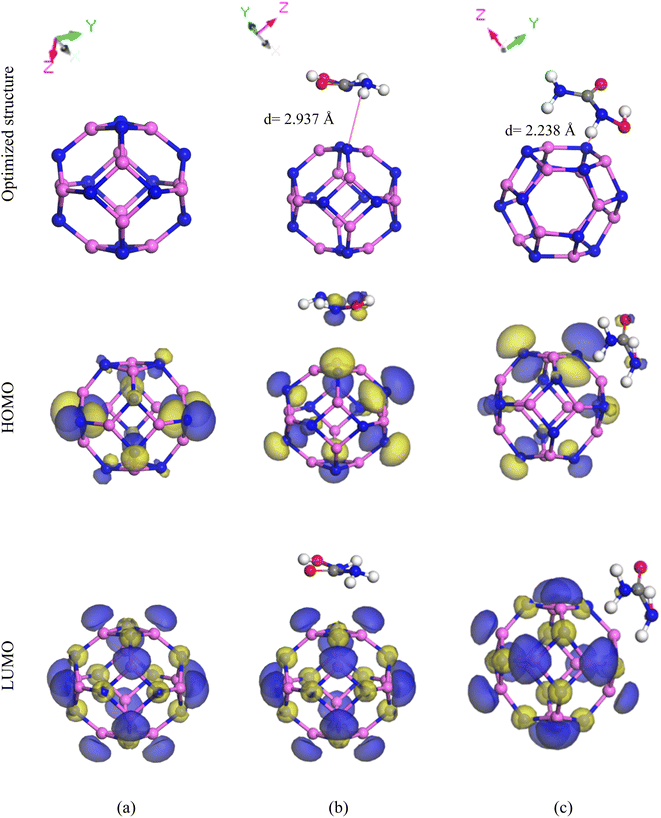 |
| | Fig. 3 Optimized structure with HOMO and LUMO of (a) Al12N12, (b) HU/Al12N12 (ST), and (c) HU/Al12N12 (SH). | |
Similar to gas media, Al12N12's SH site has a higher adsorption energy in the water phase, measuring around −197.93 kJ mol−1 at the 1.512 Å distance. However, there was also further charge transfer at the ST state, which was around 0.147e in Mulliken and 0.386e in Hirshfeld. Considerable changes in the electronic properties were also noted after the HU adsorption on the Al12N12 nanocage. Therefore, in both gas and water media, Al12N12's adsorption behaviors, particularly at the hexagon site, are desirable.
3.4. Adsorption behavior of HU on the C12–B6N6
An enhancement in adsorption behavior was noted upon doping the fullerene nanocage with Al, B, and N atoms. This is why we customized 50% of the C24 nanocage by adding B and N atoms and keeping the other 50% as C atoms to form the C12–B6N6 hetero-nanocage. The B6N6 site can be called the doped site, and the C12 site can be called the pristine site. Four different sites on the complexes are analyzed in our hetero-nanocage analysis: (i) the tetragonal site of C12 (SP/T), (ii) the hexagonal site of C12 (SP/H), (iii) the tetragonal site of B6N6 (ST), and (iv) the hexagonal site of B6N6 (SH) of the C12–B6N6 hetero-nanocage. These HU/C12–B6N6 complexes are illustrated in Fig. 4(b–e). Notably, as shown in Table 1, the pristine sites show physisorption properties, whereas the doped sites exhibit chemisorption in both the gas and water phases. The adsorption energies in the gas phase are −123.35 kJ mol−1, −91.19 kJ mol−1, −26.61 kJ mol−1, and −19.51 kJ mol−1, with corresponding adsorption distances of 1.554 Å, 1.622 Å, 2.118 Å, and 2.579 Å for the ST, SH, SP/T, and SP/H sites, respectively. It is noticeable that the ST configuration indicates greater adsorption energy (Ead) in the gas phase. According to the Mulliken charge analysis, charges for the ST, SH, SP/T, and SP/H sites were transferred from the drug to the nanocage in the amounts of 0.349e, 0.265e, 0.079e, and 0.047e, respectively. Furthermore, according to the Hirshfeld charge analysis, charges for the ST and SH configurations transferred from the drug to the nanocage at a rate of 0.382e and 0.329e, respectively, but for the SP/T and SP/H sites, the minimum amounts of charges transferred from the C12–B6N6 nanocage to the HU drug molecule at rates of 0.023e and 0.032e, respectively. Based on FMO studies, the doped sites ST show better HOMO–LUMO values and a smaller energy gap than SH. Fig. 9 illustrates these favorable results, which indicate that the C12–B6N6 nanocage contains the majority of the HOMO and LUMO levels. As shown in Table 2, at the ST and SH sites, the energy gaps decrease in the gas phase by around 4.05% and 2.45%, respectively. The full and partial DOS spectra for both the doped and pristine sites of the complex are displayed in Fig. 5(d and e), where the energy gap of the nanocage before and after drug adsorption is illustrated. Based on the adsorption behavior and electronic properties, it can be decided that the ST is the most stable configuration in the C12–B6N6 complex.
 |
| | Fig. 4 Optimized structure of (a) the C12–B6N6, HU/C12–B6N6 complex on (b) the tetragonal site of B6N6, (c) hexagonal site of B6N6, (d) tetragonal site of C12, and (e) hexagonal site of C12. | |
 |
| | Fig. 5 DOS spectra of (a) HU/C24, (b) HU/B12N12, (c) HU/Al12N12, (d and e) HU/C12–B6N6, (f and g) HU/C12–Al6N6, (h and i) HU/B6N6–Al6N6 complexes on tetragon and hexagon sites. | |
On the contrary, within the water phase, the adsorption energies are calculated as −144.05 kJ mol−1, −162.40 kJ mol−1, −18.06 kJ mol−1, and −11.03 kJ mol−1, with interaction distances found at 1.51 Å, 1.149 Å, 2.176 Å, and 2.857 Å for the ST, SH, SP/T, and SP/H sites, respectively. The results of the investigation of the water media indicate that the SH site has a much greater adsorption energy. Conversely, in gas media, the ST site showcases a higher adsorption energy, which indicates a reversal behavior between the two phases. This means that the complex may react differently inside the human body, as there is a solvent effect in water media. The drug molecules function as donors at the SH site, where a transfer of 0.522e is also shown by Mulliken charge analysis. However, in the Hirshfeld charge analysis, the doped sites show better charge transfer than the pristine sites, especially the SH site with a value of 0.653e. These results highlight the effectiveness of doping, as demonstrated by the increased adsorption behavior for the gas and water phases, thereby signifying the achievement of the doping target.
3.5. Adsorption behavior of HU on the C12–Al6N6
The formation of the C12–Al6N6 hetero-nanocage involves substituting 6 Al and 6 N atoms in place of 12C atoms. To enhance the HU drug molecule's targeted delivery, we examined its adsorption on the tetragonal and hexagonal surfaces of the doped (ST, SH) and pristine sites (SP/T, and SP/H) of the C12–Al6N6 hetero-nanocage [Fig. 7]. In this case, the Al6N6 portion is referred to as the doped site, while the C12 portion corresponds to the pristine site. The measured charge transfer, interaction distances, and adsorption energies for the four specified configurations of HU/C12–Al6N6 are shown in Table 1. Adsorption of the HU drug molecule on the surface of C12–Al6N6 has been demonstrated by analysis, with adsorption energies of −182.31 kJ mol−1, −137.52 kJ mol−1, −34.62 kJ mol−1, and −29.43 kJ mol−1 for the gas phase for the ST, SH, SP/T, and SP/H configurations, respectively. In accordance with this, the interaction distances for each configuration are measured at 1.706 Å, 1.994 Å, 2.467 Å, and 2.312 Å, respectively. Particularly, ST, which is positioned near the doping site, is the configuration that shows lower close contacts and greater adsorption energy. Furthermore, as Table 1 shows, the pristine sites exhibit greater adsorption behavior in comparison to the fullerene nanocage after doping. Therefore, it can be said that adsorption behavior improves after doping.
In the water phase, the ST site exhibits a higher adsorption energy of −222.24 kJ mol−1, accompanied by a minimum interaction distance of 1.575 Å. Furthermore, both Mulliken and Hirshfeld's charge investigations show that the charge transfer for this site in water is similar to that in the gas phase but slightly greater. More precisely, in Hirshfeld charge analysis, a transfer of 0.263e (ST) and 0.743e (SH) charges occurs from the drug to the nanocage in doped sites, whereas in pristine sites, the nanocage operates as the electron donor. The HOMO–LUMO levels and energy gap are displayed in Table 2, which shows a significant variation in the energy gap of 9.06% (ST) and 14.17% (SH) in the water phase.
Fig. 9 and 5(f–g) depict the HOMO–LUMO structure of the doped (SH) site and DOS spectra of the hetero-nanocage, respectively. More importantly, the results of the FMO study show that the energy gap of the doped sites in the water phase is narrower than in the gas phase, indicating a favorable outcome. The Projected Density of State (PDOS) graph shows how the drug molecules interact with the nanocage at the atomic level. We can understand the bonding or interaction by observing the overlapping areas in the drug's and nanocage's PDOS. For example, when a drug exhibits a large PDOS contribution at a given energy level, it suggests a considerable interaction with the nanocage, which may impact the drug's release mechanism.65,66 Changes in the PDOS peaks may signify modifications in the electronic distribution of the drug, which could impact its solubility, reactivity, and overall effectiveness.67,68 Strong bonding contacts are suggested by the large overlaps, which might indicate that the drug is safely retained inside the nanocage until it reaches the target region. Furthermore, if there is minimal overlap between the drug and nanocage close to the Fermi level, this might mean that the electronic interaction is poor,69,70 and that the drug is either released too soon or is not sufficiently safeguarded during delivery. Thus, as illustrated in Fig. 6(a), the PDOS of the close contact atoms of the drug and nanocage was taken. It can be observed that the large PDOS overlaps occur between the C atom of the nanocage and H/O atoms of the drug at the Fermi level of the HU/C12–Al6N6 complex.
 |
| | Fig. 6 PDOS spectra of the close contact atoms of (a) HU/C12–Al6N6 and (b) HU/B6N6–Al6N6 complexes on tetragonal and hexagonal sites. | |
 |
| | Fig. 7 Optimized structure of (a) the C12–Al6N6, HU/C12–Al6N6 complex on (b) the tetragonal site of Al6N6, (c) hexagonal site of Al6N6, (d) tetragonal site of C12, and (e) hexagonal site of C12. | |
3.6. Adsorption behavior of HU on the B6N6–Al6N6
Many studies have convincingly revealed that doped nanocarriers or heterostructures are suitable nanocarriers.71,72 By doping pristine nanocages, we witness effective adsorption behavior as observed in prior cases. In previous hetero-nanocages, the doped sites exhibited better adsorption behavior than the pristine sites. Therefore, by combining the two doped sites (B6N6 and Al6N6), we improve these features. Fig. 8 displays the optimized structure of our preferred hetero-nanocage. In the gas phase, the Ead values of the tetragon site of the AlN portion (ST/AlN) and the hexagon site of the AlN portion (SH/AlN) are −183.59 kJ mol−1 and −162.74 kJ mol−1, respectively, while their interaction distances are 1.732 Å and 1.693 Å, respectively. Of all the four configurations, these two sites show higher adsorption energies. Among the configurations in the gas phase, B6N6–Al6N6 is remarkable for having optimum adsorption distances and significantly greater negative Ead values presented in Table 1. The ST/AlN configuration appears to be the most stable due to its better adsorption energy. The significant amount of charge transition from the HU drug to the tetragonal site of the AlN portion of the nanocage is around 0.231e according to the results of the Hirshfeld charge analysis. The energy gaps for the ST/AlN and SH/AlN sites gradually decreased from 2.658 eV to 2.645 eV and 2.763 eV, respectively, according to the FMO analysis displayed in Table 2. Consequently, the ST/AlN configuration demonstrates a smaller energy gap, which indicates higher conductivity.
 |
| | Fig. 8 Optimized structure of (a) the B6N6–Al6N6, HU/B6N6–Al6N6 complex on (b) the tetragonal site of Al6N6, (c) hexagonal site of Al6N6, (d) tetragonal site of B6N6, and (e) hexagonal site of B6N6. | |
In the water phase, compared to the other selected sites, we can observe that the Ead of the ST/AlN site has an impressive adsorption energy of around 189.85 kJ mol−1 with an optimal distance of 1.817 Å, as listed in Table 1. This site transferred the greatest quantity of charge, around 1.817e and 0.249e in Mulliken and Hirshfeld charge analyses, respectively, where the drug is the electron donor, as compared to other adsorption sites of the HU drug on the nanocage. The energy gap of the ST/AlN site is less than that of the SH/AlN site, as listed in Table 2, based on the FMO research. Fig. 9 illustrates the positioning of both HOMO and LUMO levels, which are only on the nanocage molecules exclusively for the ST/AlN site. Fig. 5(h) shows the full and partial DOS spectra of both states, confirming the ST/AlN configuration's energy gap minimization phenomenon. Therefore, in comparison to other previously investigated nanocages, the B6N6–Al6N6 hetero-nanocage exhibits a more efficient adsorption behavior for the HU drug. From the PDOS spectra of Fig. 6(b), it is observed that the close contact atoms form a strong chemical bond between the HU drug and B6N6–Al6N6 nanocage due to strong PDOS overlapping at or near the Fermi level in HU/B6N6–Al6N6 complex. Therefore, the drug may not be released before approaching the target cell.
 |
| | Fig. 9 HOMO–LUMO of (a) the HU/C12–B6N6 complex on the tetragonal site, (b) HU/C12–Al6N6 complex on the hexagonal site, and (c) HU/B6N6–Al6N6 complex on the tetragonal site of the Al6N6 portion of the nanocages. | |
3.7. Dipole moment and quantum molecular descriptors
The dipole moment is a crucial parameter for investigating the charge transfer and reactivity between the drug and the nanocage. The solubility of a complex may be gained by studying its dipole moment in the water phase. The strong dipole moment indicates good charge distribution and high reactivity between drugs and nanocages.73,74
Using dipole moment analysis, the asymmetry in the charge allocation of our researched complexes was evaluated. It was found that the dipole moments for C24, B12N12, and Al12N12 were all equal to 0 D. However, as depicted in Table 2, there was a noticeable increase in the dipole moment values for every selected site after adsorption. This post-adsorption rise in dipole moment guarantees the solubility of the complexes in the human body. The ST, SH, and ST sites of the HU/C24, HU/B12N12, and HU/Al12N12 complexes in water media showed an enormous increase in dipole moments after adsorption, by around 5.55 D, 15.72 D, and 12.47 D, respectively. The C12–B6N6, C12–Al6N6, and B6N6–Al6N6 nanocages differ from the C24, B12N12, or Al12N12 nanocages, with preliminary non-zero dipole moments of 2.12 D, 8.83 D, and 7.53 D in the gas phase, respectively. These dipole moments increase to 3.70 D, 13.92 D, and 11.53 D, respectively, when shifting to water media from the gas phase. Specifically, the dipole moments show higher magnitudes in the gas phase than in the water phase. After adsorption, for SH, SH, and ST/AlN sites, the dipole moments of the C12–B6N6, C12–Al6N6, and B6N6–Al6N6 nanocages further increase in the water media to 21.50 D, 31.99 D, and 23.67 D, respectively. The maximum dipole moment is observed at the tetragonal site of the AlN portion of the B6N6–Al6N6 nanocage, which indicates a higher amount of charge transportation and high reactivity between the drug and the nanocage.
Quantum molecular descriptors, such as chemical potential (μ), global hardness (η), global softness (S), and electrophilicity index (ω), were extensively studied to elucidate the reaction mechanisms and explore the stability of the complexes under investigation (Table 3). It is desirable in drug delivery systems to enhance chemical potential, global softness, and electrophilicity after drug adsorption and, conversely, a decrease in global hardness.47 The resistance of a system to deformation in the existence of an external electric field is represented by its global hardness.75 A lower value of η indicates high chemical reactivity. After adsorption, the computed hardness was reduced from 0.60 eV to 0.532 eV and 0.531 eV for C24, 2.52 eV to 2.04 eV and 2.38 eV for B12N12, and 1.29 eV to 1.27 eV and 1.25 eV for Al12N12, at the ST and SH sites, respectively. In summary, reactivity increases, while chemical stability decreases in the complexes of HU/C24, HU/B12N12, and HU/Al12N12. Table 3 shows that the calculated hardness of C12–B6N6 and C12–Al6N6 is increased in doped sites (ST and SH) and reduced in pristine sites (SP/T and SP/H). However, for B6N6–Al6N6 hetero-nanocage, η decreases from 1.33 eV to 1.21 eV at ST/BN, 0.72 eV at SH/BN, 1.32 eV at ST/AlN, and increases to 1.38 eV at SH/AlN. The chemical potential increased within all the configurations, with a particular emphasis on the B6N6–Al6N6 structure. This improvement is most prominent at two specific sites in the structure, in particular at the tetragon sites of the AlN and BN portions in both the gas and water phases, as shown in Table 3. Further evidence of increased reactivity was provided by an apparent upward change in the calculated softness and electrophilicity index values of the five nanocages. The rise in global softness for HU/C24, HU/B12N12, and HU/Al12N12 complexes in both the gas and water phases is shown in Table 3. Specifically, the softness at the ST/BN, SH/BN, and ST/AlN sites of the HU/B6N6–Al6N6 complex enhanced from 0.376 to 0.413 eV−1, 0.690 eV−1, and 0.378 eV−1, respectively. However, the SH/AlN site showed a decrease. The electrophilicity index of C12–B6N6 and C12–Al6N6 nanocages showed a noticeable downward trend, except for the SP/T and SP/H sites. However, only the B6N6–Al6N6 nanocage at the SH/BN site showed an increment in the electrophilicity index from 7.847 eV to 12.04 eV in the gas phase and from 7.701 eV to 9.902 eV in the water phase.
Table 3 Computed chemical potential (μ) in eV, global hardness (η) in eV, electrophilicity index (ω) in eV, and global softness (S) in eV−1 of the studied complexes in gas and water phases
| Structure |
Site |
Gas phase |
Water phase |
|
μ
|
η
|
ω
|
S
|
μ
|
η
|
ω
|
S
|
| C24 |
|
−5.21 |
0.60 |
22.544 |
0.831 |
−5.17 |
0.60 |
22.197 |
0.8312 |
| C/HU |
ST |
−5.09 |
0.53 |
24.392 |
0.940 |
−5.04 |
0.56 |
22.763 |
0.895 |
| C/HU |
SH |
−5.08 |
0.53 |
24.338 |
0.942 |
−5.09 |
0.55 |
23.390 |
0.903 |
| B12N12 |
|
−4.64 |
2.52 |
4.278 |
0.199 |
−4.60 |
2.57 |
4.124 |
0.195 |
| BN/HU |
ST |
−4.20 |
2.04 |
4.336 |
0.245 |
−3.93 |
2.35 |
3.282 |
0.213 |
| BN/HU |
SH |
−4.12 |
2.38 |
3.568 |
0.210 |
−3.99 |
2.44 |
3.264 |
0.205 |
| Al12N12 |
|
−4.34 |
1.29 |
7.244 |
0.386 |
−4.53 |
1.34 |
7.672 |
0.373 |
| AlN/HU |
ST |
−4.27 |
1.27 |
7.177 |
0.393 |
−4.15 |
1.28 |
6.751 |
0.392 |
| AlN/HU |
SH |
−4.24 |
1.25 |
7.182 |
0.399 |
−4.21 |
1.27 |
6.988 |
0.395 |
| C12B6N6 |
|
−4.83 |
0.94 |
12.455 |
0.533 |
−4.81 |
0.97 |
11.972 |
0.518 |
| C_BN/HU |
ST |
−3.98 |
0.98 |
8.121 |
0.513 |
−4.38 |
1.01 |
9.524 |
0.497 |
| C_BN/HU |
SH |
−4.22 |
0.96 |
9.281 |
0.521 |
−4.21 |
1.04 |
8.477 |
0.479 |
| C_BN/HU |
SP/T |
−4.74 |
0.75 |
15.023 |
0.668 |
−4.73 |
0.88 |
12.668 |
0.567 |
| C_BN/HU |
SP/H |
−4.76 |
0.73 |
15.538 |
0.685 |
−4.79 |
0.94 |
12.159 |
0.529 |
| C12Al6N6 |
|
−4.01 |
0.52 |
15.600 |
0.969 |
−4.21 |
0.70 |
12.745 |
0.719 |
| C_AlN/HU |
ST |
−3.64 |
0.58 |
11.500 |
0.867 |
−3.97 |
0.76 |
10.417 |
0.660 |
| C_AlN/HU |
SH |
−3.48 |
0.61 |
9.871 |
0.817 |
−3.62 |
0.79 |
8.237 |
0.630 |
| C_AlN/HU |
SP/T |
−4.31 |
0.52 |
17.925 |
0.964 |
−4.17 |
0.69 |
12.649 |
0.726 |
| C_AlN/HU |
SP/H |
−4.18 |
0.50 |
17.459 |
0.999 |
−4.33 |
0.70 |
13.392 |
0.716 |
| B6N6_Al6N6 |
|
−4.57 |
1.33 |
7.847 |
0.376 |
−4.79 |
1.49 |
7.701 |
0.335 |
| BN_AlN/HU |
ST/BN |
−4.02 |
1.21 |
6.687 |
0.413 |
−4.35 |
1.32 |
7.197 |
0.380 |
| BN_AlN/HU |
SH/BN |
−4.18 |
0.72 |
12.04 |
0.690 |
−4.46 |
1.09 |
9.102 |
0.457 |
| BN_AlN/HU |
ST/AlN |
−4.15 |
1.32 |
6.500 |
0.378 |
−4.37 |
1.51 |
6.338 |
0.332 |
| BN_AlN/HU |
SH/AlN |
−4.37 |
1.38 |
6.923 |
0.362 |
−4.39 |
1.49 |
6.465 |
0.335 |
3.8. Work function
The work function is the minimal energy required to remove a single electron from the Fermi level. The purpose of this study is to determine if the HU impacts the work function of the nanocages, and it is computed using the following equation:76where EF, φ, and Vel(+∞) denote the Fermi level energy, work function, and electrostatic potential of an electron far away from the surface, respectively. However, Vel(+∞) is considered to be 0. Eqn (9) may be expressed as φ = −EF. Changes in the work function can influence the field emission properties of nanocages, which cause the alteration of gate voltage. The Richardson–Dushman equation can be used to characterize this connection:77| |  | (10) |
where j stands for the electron current density released from the material's surface, A is the Richardson constant (A m−2), T is the temperature, and K is the Boltzmann constant. Now, the following formula is used to calculate the change in work function after adsorption:78| |  | (11) |
The calculated values of Fermi level (EF), work function (φ), and change in work function (Δφ) are tabulated in Table 4. In our investigation, the both HU/C24 and HU/B12N12 complexes show a rise in the work function change at the hexagonal site (SH) in the gas phase, whereas Δφ enhances at the tetragonal site (ST) in the water phase. However, for the Al12N12 configuration, in both the gas and water phases, Δφ increases at the tetragonal site. In the water phase, the change in work function at the hexagonal site increases by around 12.08% and 13.84% for HU/C12–B6N6 and HU/C12–Al6N6 complexes, respectively. However, the HU/C12B6N6 complex exhibits a fluctuation of 17.42% in Δφ during the gas phase, with an increase at the SH site and a decrease at the ST site. The measured values of the work function at two distinct sites of the HU/B6N6–Al6N6 complex are 0.151 eV (ST/AlN) and 0.147 eV (ST/BN). In the gas phase, the corresponding changes in work function for these sites are 11.43% and 8.88%, respectively while in the water phase, the corresponding changes are 8.60% and 8.58%, respectively. Consequently, there are greater Δφ values at the tetragonal areas of the nanocages. Comparing these results to other nanocages, the estimated values show that B6N6, C12–B6N6, C12–Al6N6, and B6N6–Al6N6 nanocages are more effective φ-type sensors for HU. Importantly, the lowest Δφ values are observed at the pristine sites of the hetero-nanocages. Thus, this indicates that Al6N6 and C24 are unsuitable for use as φ-type sensors for HU drugs.
Table 4 Fermi level energies (EF) in eV, work function (φ) in eV, and change in work function (% Δφ) for the nanocages and complexes in gas and water phases
| Structure |
Site |
Gas phase |
Water phase |
|
E
F
|
φ
|
% Δφ |
E
F
|
φ
|
% Δφ |
| C24 |
|
−0.191 |
0.191 |
0 |
−0.190 |
0.190 |
0 |
| C/HU |
ST |
−0.188 |
0.188 |
−1.990 |
−0.186 |
0.186 |
−2.236 |
| C/HU |
SH |
−0.187 |
0.187 |
−2.084 |
−0.187 |
0.187 |
−1.291 |
| B12N12 |
|
−0.171 |
0.171 |
0 |
−0.169 |
0.169 |
0 |
| BN/HU |
ST |
−0.153 |
0.153 |
−10.021 |
−0.146 |
0.146 |
−13.739 |
| BN/HU |
SH |
−0.151 |
0.151 |
−11.512 |
−0.147 |
0.147 |
−13.035 |
| Al12N12 |
|
−0.156 |
0.156 |
0 |
−0.164 |
0.164 |
0 |
| AlN/HU |
ST |
−0.154 |
0.154 |
−1.268 |
−0.151 |
0.151 |
−7.727 |
| AlN/HU |
SH |
−0.154 |
0.154 |
−1.169 |
−0.153 |
0.153 |
−6.252 |
| C12B6N6 |
|
−0.178 |
0.178 |
0 |
−0.178 |
0.178 |
0 |
| C_BN/HU |
ST |
−0.147 |
0.147 |
−17.424 |
−0.162 |
0.162 |
−8.697 |
| C_BN/HU |
SH |
−0.156 |
0.156 |
−12.501 |
−0.156 |
0.156 |
−12.076 |
| C_BN/HU |
SP/T |
−0.175 |
0.175 |
−1.926 |
−0.174 |
0.174 |
−1.748 |
| C_BN/HU |
SP/H |
−0.176 |
0.176 |
−1.467 |
−0.176 |
0.176 |
−0.962 |
| C12Al6N6 |
|
−0.147 |
0.147 |
0 |
−0.155 |
0.155 |
0 |
| C_AlN/HU |
ST |
−0.134 |
0.134 |
−9.091 |
−0.146 |
0.146 |
−5.459 |
| C_AlN/HU |
SH |
−0.128 |
0.128 |
−13.075 |
−0.133 |
0.133 |
−13.846 |
| C_AlN/HU |
SP/T |
−0.157 |
0.157 |
6.949 |
−0.153 |
0.153 |
−0.853 |
| C_AlN/HU |
SP/H |
−0.153 |
0.153 |
4.076 |
−0.159 |
0.159 |
2.778 |
| B6N6_Al6N6 |
|
−0.166 |
0.166 |
0 |
−0.174 |
0.174 |
0 |
| BN_AlN/HU |
ST/BN |
−0.147 |
0.147 |
−11.435 |
−0.159 |
0.159 |
−8.603 |
| BN_AlN/HU |
SH/BN |
−0.154 |
0.154 |
−7.523 |
−0.164 |
0.164 |
−5.744 |
| BN_AlN/HU |
ST/AlN |
−0.151 |
0.151 |
−8.884 |
−0.159 |
0.159 |
−8.584 |
| BN_AlN/HU |
SH/AlN |
−0.159 |
0.159 |
−4.071 |
−0.160 |
0.160 |
−8.064 |
3.9. COSMO surface analysis
Previous studies have examined the impact of complexes on solvation by calculating the dipole moment, electrical properties, and adsorption energy in the water phase. In this work, conductor-like screening model (COSMO) surface analysis was carried out to provide an understanding of the biological environment within the human body. Significantly, we performed a COSMO surface analysis to obtain a thorough understanding of the polarity of the complexes. The COSMO surfaces of the nanocages and their complexes with the most stable configurations are depicted in Fig. 10.
 |
| | Fig. 10 COSMO surfaces for the nanocages and their complexes with the most stable configurations: (a) C24, (b) B12N12, (c) Al12N12, (d) HU/C24 (ST), (e) HU/B12N12 (SH), (f) HU/Al12N12 (ST), (g) C12–B6N6, (h) C12–Al6N6, (i) B6N6–Al6N6, (j) HU/C12–B6N6 (SH), (k) HU/C12–Al6N6 (ST), and (l) HU/B6N6–Al6N6 (ST/AlN). | |
The green sections in the molecular structures are non-polar neutral areas. There is no discernible distribution of charges in these regions. Highly positive sections are shown by the blue segments, which act as hydrogen bond donors (HBD). However, the red segments act as hydrogen bond acceptors (HBA) and indicate highly negative regions. The COSMO surface of C24 is primarily green in Fig. 10(a), suggesting its overall neutral polarity before adsorption. Conversely, the blue segments observed in the B12N12 and Al12N12 nanocages indicate the HBD areas. These sections are mostly situated above the nitrogen atoms in the respective nanocages. As shown in Fig. 10(b), the HBA regions are located on the B atoms in the B12N12 nanocage. In the C12–B6N6 and C12–Al6N6 hetero-nanocages, the HBA regions are observed in the B6N6 and Al6N6 segments, respectively, while the HBD regions are located in the C12 segment of the nanocages. Similarly, in the B6N6–Al6N6 nanocage, the HBA sections are mostly found in the B6N6 part, the HBD segments are primarily found on the nitrogen atoms of the Al6N6 portion, and the HBA regions are found in the aluminum atoms [see Fig. 10(i)]. In all the complexes, the largest parts of the HU drug molecule show a negative polarity area, whereas the maximum part of the nanocages represents a positive polarity region. To be more precise, two oxygen atoms in the HU/B6N6–Al6N6 complex have the HBD segment, while every other drug molecule, including a portion of the tetragon component of the AlN section of the nanocage, contains the HBA segment (Fig. 10(l)). This study indicates that because drug molecules are negatively polar, their adsorption increases the polarity of the complexes in solvent circumstances, which is consistent with our dipole moment analysis.
4. Conclusions
Initially, the adsorption behavior of the drug towards several nanocages, namely C24, B12N12, Al12N12, C12–B6N6, C12–Al6N6, and B6N6–Al6N6, was examined to determine an appropriate nanocarrier for the HU drug. The doped sites of C12–Al6N6 and the tetragonal site of the AlN portion of B6N6–Al6N6 where HU was adsorbed showed advantageous adsorption energies for drug delivery systems in both gas and water media. The ST/AlN site of B6N6–Al6N6 was determined to have a higher adsorption energy than the adsorption energies for the ST and SH sites of C12–Al6N6 with minimum adsorption distances. Negative adsorption energy and low adsorption distances represent favorable interactions between the drug and nanocages. An FMO analysis was carried out to provide a deeper understanding of the sensitivity and reactivity of the nanocages. The results showed that the adsorption of HU on the C12–Al6N6 and B6N6–Al6N6 nanocages caused the Eg to move to the upper and downward values, respectively. The B6N6–Al6N6 nanocage's energy gap significantly reduced in the gas phase from 2.658 eV to 2.645 eV. In contrast, there is no preferable energy gap in the case of the C12–Al6N6 nanocage (from 1.031 eV to 1.154 eV and 1.1224 eV) after the adsorption of HU. Minimum charge transfer between HU and C12–Al6N6 was observed at the tetragonal site by the Hirshfeld charge analysis; values for the ST and SH configurations are 0.215e and 0.263e in the gas phase and 0.377e and 0.743e in the water phase, respectively. The B6N6–Al6N6 structure, however, exhibits a notable charge transfer, with charges of 0.231e and 0.462e in the gas and water phases, respectively. The tetragonal and hexagonal sites of the HU/C12–Al6N6 combination have lower global softness and higher global hardness, which are unfavorable. In the instance of B6N6–Al6N6, the global softness rises and the global hardness drops according to the quantum molecular descriptors of the hetero-nanocages. The tetragonal position of B6N6–Al6N6 is a promising option for HU drug delivery, as supported by dipole moment, work function, and COSMO surface analysis. Consequently, adsorption energy, Mulliken and Hirshfeld charge analysis, electronic properties, and quantum molecular descriptors reveal that B6N6–Al6N6 could be a potential nanocarrier for targeted HU delivery.
Data availability
The data supporting this article have been included in the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thankfully acknowledge the Research Cell of Mawlana Bhashani Science and Technology University funded by UGC of Bangladesh (Grant No. 3631108) under the Ministry of Science and Technology (MOST) and the Bangladesh Research and Education Network (BdREN) for their computational access.
References
- Y. Xi and P. Xu, Global colorectal cancer burden in 2020 and projections to 2040, Transl. Oncol., 2021, 14(10), 101174 CrossRef PubMed.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra and D. K. Pandey,
et al., Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics, Genes Dis., 2022, 1367–1401 Search PubMed.
- M. S. Aslam, S. Naveed, A. Ahmed, Z. Abbas, I. Gull and M. A. Athar, Side effects of chemotherapy in cancer patients and evaluation of patients opinion about starvation based differential chemotherapy, J. Cancer Ther., 2014, 5, 817–822 CrossRef.
- P. Parhi, C. Mohanty and S. K. Sahoo, Nanotechnology-based combinational drug delivery: an emerging approach for cancer therapy, Drug Discovery Today, 2012, 17(17–18), 1044–1052 CrossRef CAS PubMed.
- S. Kumaran, V. Vetrivelan, S. Muthu and A. A. Al-Saadi, Computational analysis of anti-cancer drug hydroxyurea adsorption on nanocages of gold, silver and copper: SERS and DFT assessment, Heliyon, 2024, 10(5), e24475 CrossRef CAS PubMed.
- M. M. Salem-Bekhit, Z. S. Al, N. A. Alhabib, R. R. Maaliw III, M. Da'i and M. Mirzaei, Metal-doped fullerenes as promising drug carriers of hydroxycarbamide anticancer: insights from density functional theory, Chem. Phys. Impact, 2023, 7, 100347 CrossRef.
- H. Karimi-Maleh, B. G. Kumar, S. Rajendran, J. Qin, S. Vadivel and D. Durgalakshmi,
et al., Tuning of metal oxides photocatalytic performance using Ag nanoparticles integration, J. Mol. Liq., 2020, 314, 113588 CrossRef CAS.
- A. Abbasi and J. J. Sardroodi, Adsorption of O3, SO2 and SO3 gas molecules on MoS2 monolayers: a computational investigation, Appl. Surf. Sci., 2019, 469, 781–791 CrossRef CAS.
- M. J. Saadh, M. Mirzaei, B. S. Abdullaeva, R. R. Maaliw III, M. Da'i and M. M. Salem-Bekhit,
et al., Explorations of structural and electronic features of an enhanced iron-doped boron nitride nanocage for adsorbing/sensing functions of the hydroxyurea anticancer drug delivery under density functional theory calculations, Phys. B, 2023, 671, 415445 CrossRef CAS.
- A. Al-Jumaili, S. Alancherry, K. Bazaka and M. V. Jacob, Review on the antimicrobial properties of carbon nanostructures, Materials, 2017, 10(9), 1066 CrossRef PubMed.
- Q. Sun, R. Zhang, J. Qiu, R. Liu and W. Xu, On-Surface Synthesis of Carbon Nanostructures, Adv. Mater., 2018, 30(17), 1705630 CrossRef PubMed.
- B. T. Tomić, C. S. Abraham, S. Pelemiš, S. J. Armaković and S. Armaković, Fullerene C 24 as a potential carrier of ephedrine drug–a computational study of interactions and influence of temperature, Phys. Chem. Chem. Phys., 2019, 21(42), 23329–23337 RSC.
- M. H. Miah, M. R. Hossain, M. S. Islam, T. Ferdous and F. Ahmed, A theoretical study of allopurinol drug sensing by carbon and boron nitride nanostructures: DFT, QTAIM, RDG, NBO and PCM insights, RSC Adv., 2021, 11(61), 38457–38472 RSC.
- P. R. C. Kent, M. D. Towler, R. J. Needs and G. Rajagopal, Carbon clusters near the crossover to fullerene stability, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62(23), 15394 CrossRef CAS.
- K. Nejati, A. Hosseinian, E. Vessally, A. Bekhradnia and L. Edjlali, A comparative DFT study on the interaction of cathinone drug with BN nanotubes, nanocages, and nanosheets, Appl. Surf. Sci., 2017, 422, 763–768 CrossRef CAS.
- B. T. Tomić, C. S. Abraham, S. Pelemiš, S. J. Armaković and S. Armaković, Fullerene C 24 as a potential carrier of ephedrine drug–a computational study of interactions and influence of temperature, Phys. Chem. Chem. Phys., 2019, 21(42), 23329–23337 RSC.
- A. Hosseinian, E. Vessally, S. Yahyaei, L. Edjlali and A. Bekhradnia, A Density Functional Theory Study on the Interaction Between 5-Fluorouracil Drug and C 24 Fullerene, J. Cluster Sci., 2017, 28, 2681–2692 CrossRef CAS.
- N. Saikia, M. Seel and R. Pandey, Stability and electronic properties of 2D nanomaterials conjugated with pyrazinamide chemotherapeutic: a first-principles cluster study, J. Phys. Chem. C, 2016, 120(36), 20323–20332 CrossRef CAS.
- H. Jahangirian, E. G. Lemraski, T. J. Webster, R. Rafiee-Moghaddam and Y. Abdollahi, A review of drug delivery systems based on nanotechnology and green chemistry: green nanomedicine, Int. J. Nanomed., 2017, 2957–2978 CrossRef CAS PubMed.
-
J. Müssig and N. Graupner, Test methods for fibre/matrix adhesion in cellulose fibre-reinforced thermoplastic composite materials: A critical review, Progress in Adhesion and Adhesives, 2021, vol. 6, pp. 69–130 Search PubMed.
- H. R. A. El-Mageed, F. M. Mustafa and M. K. Abdel-Latif, Boron nitride nanoclusters, nanoparticles and nanotubes as a drug carrier for isoniazid anti-tuberculosis drug, computational chemistry approaches, J. Biomol. Struct. Dyn., 2022, 40(1), 226–235 CrossRef CAS PubMed.
- M. R. Hossain, M. M. Hasan, S. U. D. Shamim, T. Ferdous, M. A. Hossain and F. Ahmed, First-principles study of the adsorption of chlormethine anticancer drug on C24, B12N12 and B12C6N6 nanocages, Comput. Theor. Chem., 2021, 1197, 113156 CrossRef.
- H. S. Wu, F. Q. Zhang, X. H. Xu, C. J. Zhang and H. Jiao, Geometric and energetic aspects of aluminum nitride cages, J. Phys. Chem. A, 2003, 107(1), 204–209 CrossRef CAS.
- M. B. Javan, A. Soltani, Z. Azmoodeh, N. Abdolahi and N. Gholami, DFT study on the interaction between 5-fluorouracil and B12N12 nanocluster, RSC Adv., 2016, 6(106), 104513–104521 RSC.
- S. Kaviani, S. Shahab and M. Sheikhi, Adsorption of alprazolam drug on the B12N12 and Al12N12 nano-cages for biological applications: A DFT study, Phys. E, 2021, 126, 114473 CrossRef CAS.
- X. Gong, L. Guo and R. Zhou, Exploring the potential use of aluminium nitride nanoparticles in the delivery of flutamide anticancer drug, Inorg. Chem. Commun., 2023, 158, 111478 CrossRef CAS.
- A. Hosseinian, A. Bekhradnia, E. Vessally, L. Edjlali and M. D. Esrafili, A theoretical study on the C30X15Y15 (X= B, and Al; Y= N, and P) heterofullerenes, Comput. Theor. Chem., 2017, 1115, 114–118 CrossRef CAS.
- M. K. Hazrati, Z. Bagheri and A. Bodaghi, Application of C30B15N15 heterofullerene in the isoniazid drug delivery: DFT studies, Phys. E, 2017, 89, 72–76 CrossRef CAS.
- X. F. Fan, Z. Zhu, Z. X. Shen and J. L. Kuo, On the use of bond-counting rules in predicting the stability of C12B6N6 fullerene, J. Phys. Chem. C, 2008, 112(40), 15691–15696 CrossRef CAS.
- D. Paul, J. Deb, B. Bhattacharya and U. Sarkar, Electronic and optical properties of C 24, C 12 X 6 Y 6, and X 12 Y 12 (X= B, Al and Y= N, P), J. Mol. Model., 2018, 24, 1–13 CrossRef CAS PubMed.
- M. G. Muktadir, A. Alam and A. A. Piya, Shamim SUD. Exploring the adsorption ability with sensitivity and reactivity of C 12–B 6 N 6, C 12–Al 6 N 6, and B 6 N 6–Al 6 N 6 heteronanocages towards the cisplatin drug: a DFT, AIM, and COSMO analysis, RSC Adv., 2022, 12(45), 29569–29584 RSC.
- M. Fasnacht, K. Butenhof, A. Goupil-Lamy, F. Hernandez-Guzman, H. Huang and L. Yan, Automated antibody structure prediction using Accelrys tools: results and best practices, Proteins: Struct., Funct., Bioinf., 2014, 82(8), 1583–1598 CrossRef CAS PubMed.
- Y. Kolokoltsev, O. Amelines-Sarria, T. Y. Gromovoy and V. A. Basiuk, Interaction of meso-tetraphenylporphines with C60 fullerene: comparison of several density functional theory functionals implemented in DMol3 module, J. Comput. Theor. Nanosci., 2010, 7(6), 1095–1103 CrossRef CAS.
- T. Ahmed, M. A. Rahman, R. Islam, A. A. Piya and S. U. D. Shamim, Unravelling the adsorption performance of BN, AlN, GaN and InN 2D nanosheets towards the
ciclopirox, 5-fluorouracil and nitrosourea for anticancer drug delivery motive: A DFT-D with QTAIM, PCM and COSMO investigations, Comput. Theor. Chem., 2022, 1214, 113797 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865 CrossRef CAS PubMed.
- J. Paier, R. Hirschl, M. Marsman and G. Kresse, The Perdew–Burke–Ernzerhof exchange-correlation functional applied to the G2-1 test set using a plane-wave basis set, J. Chem. Phys., 2005, 122(23), 234102 CrossRef PubMed.
- V. A. Basiuk, Interaction of porphine with closed-end zigzag (6, 0) single-walled carbon nanotube: the effect of parameters in DMol3 DFT calculations, J. Comput. Theor. Nanosci., 2008, 5(11), 2114–2118 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27(15), 1787–1799 CrossRef CAS PubMed.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92(1), 508–517 CrossRef CAS.
- S. S. U. Daula, M. K. Hossain, S. M. Hasan, A. Hossain and F. Ahmed, Ab initio study of N-doped graphene oxide (NDGO) as a promising anode material for Li-ion rechargeable battery, Mol. Simul., 2020, 46(14), 1135–1145 CrossRef.
- S. U. D. Shamim, M. K. Hossain, S. M. Hasan, A. A. Piya, M. S. Rahman and M. A. Hossain,
et al., Understanding Na-ion adsorption in nitrogen doped graphene oxide anode for rechargeable sodium ion batteries, Appl. Surf. Sci., 2022, 579, 152147 CrossRef CAS.
- O. Prasad, Investigations on Molecular Structure, Electronic Properties, NLO Properties, HOMO–LUMO Analysis and Comparison of Drug-likeness of Triazolothiadiazole Derivatives by Quantum methods and QSAR Analysis, Rev. Theor. Sci., 2015, 3, 1–10 CrossRef.
- B. Mohammadi, S. Musavi Raziabadi and E. Vessally, DFT Study on the Adsorption of Propylthiouracil on Si-and Ga-doped Carbon Nanoflake and Aluminum Nitride Nanosheets, Silicon, 2023, 15(6), 2693–2702 CrossRef CAS.
- S. U. D. Shamim, M. H. Miah, M. R. Hossain, M. M. Hasan, M. K. Hossain and M. A. Hossain,
et al., Theoretical investigation of emodin conjugated doped B12N12 nanocage by means of DFT, QTAIM and PCM analysis, Phys. E, 2022, 136, 115027 CrossRef CAS.
- M. M. Hasan, A. C. Das, M. R. Hossain, M. K. Hossain, M. A. Hossain and B. Neher,
et al., The computational quantum mechanical investigation of the functionalized boron nitride nanocage as the smart carriers for favipiravir drug delivery: a DFT and QTAIM analysis, J. Biomol. Struct. Dyn., 2022, 40(23), 13190–13206 CrossRef CAS PubMed.
- M. R. Hossain, M. M. Hasan, H. Rahman, M. S. Rahman, F. Ahmed and T. Ferdous,
et al., Adsorption behaviour of metronidazole drug molecule on the surface of hydrogenated graphene, boron nitride and boron carbide nanosheets in gaseous and aqueous medium: A comparative DFT and QTAIM insight, Phys. E, 2021, 126, 114483 CrossRef CAS.
- A. A. Piya, T. Ahmed, M. A. Khaleque, K. Ahmed and S. U. D. Shamim, Trivalent and pentavalent atoms doped boron nitride nanosheets as Favipiravir drug carriers for the treatment of COVID-19 using computational approaches, Comput. Theor. Chem., 2022, 1217, 113902 CrossRef PubMed.
- M. Mohammadzaheri, S. Jamehbozorgi, M. D. Ganji, M. Rezvani and Z. Javanshir, Toward functionalization of ZnO nanotubes and monolayers with 5-aminolevulinic acid drugs as possible nanocarriers for drug delivery: a DFT based molecular dynamic simulation, Phys. Chem. Chem. Phys., 2023, 25(32), 21492–21508 RSC.
- Z. M. Shabavi, E. Shakerzadeh, T. Yadav, E. Tahmasebi, S. Kaviani and E. C. Anota, A DFT study on the potential application of metal-encapsulated B12N12 nanocage for efficient removal of gemifloxacin in aqueous medium, Comput. Theor. Chem., 2024, 114535 CrossRef CAS.
- D. Farmanzadeh and M. Keyhanian, Computational assessment on the interaction of amantadine drug with B 12 N 12 and Zn 12 O 12 nanocages and improvement in adsorption behaviors by impurity Al doping, Theor. Chem. Acc., 2019, 138, 1–10 Search PubMed.
- E. Vessally, M. D. Esrafili, R. Nurazar, P. Nematollahi and A. Bekhradnia, A DFT study on electronic and optical properties of aspirin-functionalized B 12 N 12 fullerene-like nanocluster, Struct. Chem., 2017, 28, 735–748 CrossRef CAS.
- N. Wazzan, K. A. Soliman and W. S. A. Halim, Theoretical study of gallium nitride nanocage as a carrier for 5-fluorouracil anticancer drug, J. Mol. Model., 2019, 25, 1–19 CrossRef CAS PubMed.
- P. Saha and S. Chowdhury, Insight into adsorption thermodynamics, Thermodynamics, 2011, 16, 349–364 Search PubMed.
- S. U. D. Shamim, D. Roy, S. Alam, A. A. Piya, M. S. Rahman and M. K. Hossain,
et al., Doubly doped graphene as gas sensing materials for oxygen-containing gas molecules: A first-principles investigation, Appl. Surf. Sci., 2022, 596, 153603 CrossRef CAS.
- M. N. Madadi, F. Mostaghni and H. Shafiekhani, A density functional theory study on the adsorption of Mercaptopurine anti-cancer drug and Cu/Zn-doped boron nitride nanocages as a drug delivery, J. Biomol. Struct. Dyn., 2023, 1–8 Search PubMed.
- X. Chen, L. Liu and C. Jiang, Charge-reversal nanoparticles: novel targeted drug delivery carriers, Acta Pharm. Sin. B, 2016, 6(4), 261–267 CrossRef PubMed.
- S. N. Ema, M. A. Khaleque, A. Ghosh, A. A. Piya, U. Habiba and S. U. D. Shamim, Surface adsorption of nitrosourea on pristine and doped (Al, Ga and In) boron nitride nanosheets as anticancer drug carriers: The DFT and COSMO insights, RSC Adv., 2021, 11(58), 36866–36883 RSC.
- E. U. Ejiofor, J. E. Ishebe, I. Benjamin, G. A. Okon, T. E. Gber and H. Louis, Exploring the potential of single-metals (Cu, Ni, Zn) decorated Al12N12 nanostructures as sensors for flutamide anticancer drug, Heliyon, 2023, 9(10), e20682 CrossRef CAS PubMed.
-
E. Canadell, M. L. Doublet and C. Iung, Orbital Approach to the Electronic Structure of Solids, Oxford University Press, 2012 Search PubMed.
- R. O. Saleh, D. O. Bokov, M. N. Fenjan, W. K. Abdelbasset, U. S. Altimari and A. T. Jalil,
et al., Application of aluminum nitride nanotubes as a promising nanocarriers for anticancer drug 5-aminosalicylic acid in drug delivery system, J. Mol. Liq., 2022, 352, 118676 CrossRef.
- P. A. Ahmadi, N. L. Hadipour and Z. Bagheri, Effects of Al doping and double-antisite defect on the adsorption of HCN on a BC2N nanotube: density functional theory studies, J. Phys. Chem. C, 2013, 117(5), 2427–2432 CrossRef.
- A. A. Walsh, Chemisorption of iodine-125 to gold nanoparticles allows for real-time quantitation and potential use in nanomedicine, J. Nanopart. Res., 2017, 19, 1–14 CrossRef CAS PubMed.
- G. Serdaroğlu, Harmine derivatives: a comprehensive quantum chemical investigation of the structural, electronic (FMO, NBO, and MEP), and spectroscopic (FT-IR and UV–Vis) properties, Res. Chem. Intermed., 2020, 46, 961–982 CrossRef.
- A. Mortazavifar, H. Raissi and M. Shahabi, Comparative prediction of binding affinity of Hydroxyurea anti-cancer to boron nitride and carbon nanotubes as smart targeted drug delivery vehicles, J. Biomol. Struct. Dyn., 2019, 37(18), 4852–4862 CrossRef CAS PubMed.
- R. M. Del Castillo, E. Ramos and A. Martínez, Interaction of graphene with antipsychotic drugs: Is there any charge transfer process?, J. Comput. Chem., 2021, 42(1), 60–65 CrossRef CAS PubMed.
- N. F. Andriambelaza, R. E. Mapasha and N. Chetty, Band gap engineering of a MoS2 monolayer through oxygen alloying: an ab initio study, Nanotechnology, 2018, 29(50), 505701 CrossRef CAS PubMed.
- A. Abbasi and J. J. Sardroodi, The adsorption of sulfur trioxide and ozone molecules on stanene nanosheets investigated by DFT: Applications to gas sensor devices, Phys. E, 2019, 108, 382–390 CrossRef CAS.
- Z. Hasanzade and H. Raissi, Solvent/co-solvent effects on the electronic properties and adsorption mechanism of anticancer drug Thioguanine on Graphene oxide surface as a nanocarrier: Density functional theory investigation and a molecular dynamics, Appl. Surf. Sci., 2017, 422, 1030–1041 CrossRef CAS.
- A. Abbasi and J. J. Sardroodi, Investigation of the adsorption of ozone molecules on TiO2/WSe2 nanocomposites by DFT computations: Applications to gas sensor devices, Appl. Surf. Sci., 2018, 436, 27–41 CrossRef CAS.
- A. Abbasi and J. J. Sardroodi, Electronic structure tuning of stanene monolayers from DFT calculations: Effects of substitutional
elemental doping, Appl. Surf. Sci., 2018, 456, 290–301 CrossRef CAS.
- M. H. Fekri, R. Bazvand, M. Soleymani and M. Razavi Mehr, Adsorption of Metronidazole drug on the surface of nano fullerene C60 doped with Si, B and Al: A DFT study, Int. J. Nano Dimens., 2020, 11(4), 346–354 CAS.
- M. Vatanparast and Z. Shariatinia, Computational studies on the doped graphene quantum dots as potential carriers in drug delivery systems for isoniazid drug, Struct. Chem., 2018, 29, 1427–1448 CrossRef CAS.
- Z. Khodadadi and L. Torkian, Studying metal-doped graphene nanosheet as a drug carrier for anticancer drug β-lapachone using density functional theory (DFT), Mater. Res. Express, 2019, 6(6), 065058 CrossRef CAS.
- E. E. Havinga and P. Van Pelt, Intramolecular charge transfer, studied by electrochromism of organic molecules in polymer matrices, Mol. Cryst. Liq. Cryst., 1979, 52(1), 145–155 CrossRef.
- R. Kar, K. R. S. Chandrakumar and S. Pal, The influence of electric field on the global and local reactivity descriptors: reactivity and stability of weakly bonded complexes, J. Phys. Chem. A, 2007, 111(2), 375–383 CrossRef CAS PubMed.
- N. Yuksel and M. F. Fellah, Host–guest complex properties of calix [4] arene derivatives: a DFT study of adsorption and sensing of an anticancer drug, 5-fluorouracil, Monatsh. fur Chem., 2021, 152, 217–228 CrossRef CAS.
- C. Wang, L. Shen and L. Wu, Adsorption and sensing of an anticancer drug on the boron nitride nanocones; a computational inspection, Comput. Methods Biomech. Biomed. Eng., 2021, 24(2), 151–160 CrossRef PubMed.
- S. N. Ema, M. A. Khaleque, A. Ghosh, A. A. Piya, U. Habiba and S. U. D. Shamim, Surface adsorption of nitrosourea on pristine and doped (Al, Ga and In) boron nitride nanosheets as anticancer drug carriers: The DFT and COSMO insights, RSC Adv., 2021, 11(58), 36866–36883 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Umme
Habiba
and
Siraj Ud Daula
Shamim
,
Umme
Habiba
and
Siraj Ud Daula
Shamim