
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid cellular uptake of citrate-coated iron oxide nanoparticles unaffected by cell-surface glycosaminoglycans†
Lena
Kampen
 *abc,
Amani
Remmo
d,
Shailey Gale
Twamley
ce,
Andrea
Weller
ab,
Anke
Stach
ab,
Paul
Turko
f,
Norbert
Löwa
d,
Frank
Wiekhorst
d and
Antje
Ludwig
*abc
*abc,
Amani
Remmo
d,
Shailey Gale
Twamley
ce,
Andrea
Weller
ab,
Anke
Stach
ab,
Paul
Turko
f,
Norbert
Löwa
d,
Frank
Wiekhorst
d and
Antje
Ludwig
*abc
aDeutsches Herzzentrum der Charité, Department of Cardiology, Angiology and Intensive Care Medicine, Charitéplatz 1, 10117 Berlin, Germany. E-mail: lena.kampen@dhzc-charite.de; antje.ludwig@dhzc-charite.de
bCharité – Universitätsmedizin Berlin, Corporate member of Freie Universität Berlin and Humboldt-Universität zu Berlin, Department of Cardiology, Angiology and Intensive Care Medicine, Charitéplatz 1, 10117 Berlin, Germany
cDZHK (German Centre for Cardiovascular Research), Partner Site Berlin, Germany
dPhysikalisch-Technische Bundesanstalt, Working Group 8.23 Metrology for Magnetic Nanoparticles, Abbestraße 2-12, 10587 Berlin, Germany
eCharité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Institute of Functional Anatomy, Charitéplatz 1, 10117 Berlin, Germany
fCharité-Universitätsmedizin Berlin, Corporate Member of Freie Universität Berlin and Humboldt Universität zu Berlin, Institute of Integrative Neuroanatomy, Charitéplatz 1, 10117 Berlin, Germany
First published on 13th June 2024
Abstract
Citrate-coated iron oxide nanoparticles, specifically Synomag®-COOH (SynC), are promising tracers in magnetic particle imaging (MPI) due to their high magnetic moments and rapid cellular uptake. The mechanisms driving efficient SynC uptake remain unclear. Previous observations suggest a role of the extracellular glycocalyx during nanoparticle uptake. Here, we ascertain whether the cell-surface glycosaminoglycans (GAGs) regulate the uptake of SynC. Using transmission electron microscopy (TEM), we visualized SynC uptake by THP-1 cells, a human acute monocytic leukemia cell line. We investigated the interaction of SynC with GAGs in living cells using click-chemistry-based labeling. Upon treating THP-1 cells with chondroitinase or hyaluronidase and with a xylosyltransferase-deficient cell line, we quantified SynC uptake and measured interactions of SynC with cells in real time using magnetic particle spectroscopy (MPS). The THP-1 cell membrane engulfed or formed extensions around SynC, indicating uptake through pinocytosis and phagocytosis. We measured an increased MPS signal of SynC within seconds of cell contact, suggesting an interaction with extracellular components like the glycocalyx. Upon adding SynC to THP-1 cells, we could not observe disruption of fluorescently labeled GAGs or an enhanced intracellular fluorescence, implying that SynC does not accelerate the turnover of GAGs by binding. Lack of chondroitin sulfate, heparan sulfate, and hyaluronic acid did not affect the rapid magnetic behavior increase of SynC upon cell contact. Accordingly, we measured no significant differences in SynC uptake between wild type cells and our GAG-deficient models. These findings suggest that GAGs act as a permeable bandpass for SynC nanoparticles with a minor negative surface charge of −13.8 mV. This finding has significant implications for MPI-based cell tracking because it facilitates efficient tracking of cell types that lack a strong repulsion by cell-surface GAGs. It will be crucial to investigate whether the rapid uptake of SynC is cell-type specific and influenced by different extracellular matrix compositions.
1. Introduction
The unique magnetic properties of magnetic nanoparticles make them promising tools for disease therapy and diagnosis. Their biomedical applications range from facilitating magnetically-guided drug delivery,1,2 destruction of tumors via magnetic hyperthermia,3 serving as contrast agents in magnetic resonance imaging (MRI),4 or acting as magnetic tracers in magnetic particle imaging (MPI).5,6 MPI is a powerful tool for tracking magnetic nanoparticle-containing cells as it offers high temporal resolution predicted to be <0.1 s, a spatial resolution <1 mm, and negligible background.7–9 To take full advantage of MPI, magnetic nanoparticle tracers must be identified and characterized appropriately. Suitable MPI tracers must exhibit a high magnetic moment to achieve a high MPI signal intensity.10 This requirement is fulfilled by Synomag®-COOH (SynC), a citrate-coated iron oxide nanoparticle. SynC has a mean hydrodynamic diameter of 30 nm and a negative surface charge of −13.8 mV.11 These nanoparticles show a high signal intensity in MPI due to their nanoflower-shaped multicore, which exhibits high magnetic moments caused by ferromagnetic-like interactions between the cores.10 The potential of SynC was previously demonstrated during MPI-based imaging, cellular uptake quantification, and the use of SynC as an MPI tracer for THP-1 monocytes.7,12 MPI-based cell tracking requires high cellular uptake of magnetic nanoparticles.11,13 Monocytes effectively take up SynC within 10 min, unlike differently coated iron oxide nanoparticles. The strong MPI signal of SynC, coupled with their biocompatibility and rapid cellular uptake, renders these nanoparticles ideal MPI tracers.11How the efficient and rapid uptake of SynC is facilitated is still unclear. Prior research considered the connection between cellular uptake and nanoparticle properties, such as shape, size, and charge.14–16 The highest uptake rates were reported for nanoparticles with a diameter of 50 nm compared to larger and smaller nanoparticles.16–18 Moreover, higher uptake was determined for charged nanoparticles compared to their neutral counterparts.19,20 While this research identified trends regarding the connection between nanoparticle uptake and physiochemical properties, it does not fully explain the complex processes determining whether a specific nanoparticle will be taken up by cells.21 Cellular nanoparticle uptake is influenced by multiple biological and experimental factors, including nanoparticle aggregation,22 diffusion,23 interactions with proteins,24 and the often neglected, cell-surrounding glycocalyx. Nanoparticles must pass the glycocalyx before cellular uptake.25–27 The cell surrounding glycocalyx consists of lipids, proteins, and glycoconjugates in the form of glycolipids, glycoproteins, proteoglycans and glycosaminoglycans (GAG).28,29 The long GAG chains contain repeating disaccharides with a type of uronic acid bound to an amino sugar, either N-acetylgalactosamine or N-acetylglucosaminamine. By differentially combining these saccharides, five distinct GAG classes form: chondroitin sulfate (CS), dermatan sulfate (DS), heparan sulfate (HS), keratan sulfate (KS), and hyaluronic acid (HA). CS, DS, HS, and KS are covalently linked to core proteins forming proteoglycans.30 It is well recognized that the glycocalyx regulates cellular uptake through unspecific electrostatic interactions and by specifically binding molecules.31,32 Recently, the glycocalyx has emerged as a potential binding partner or barrier for nanoparticle uptake.33,34 Lieleg et al. proposed that the glycocalyx might act as a charge-based bandpass, allowing particle diffusion within a charge range of −30 mV to +10 mV.25 Olivieri et al. observed that binding to chondroitin and heparan sulfate facilitated the uptake of positively charged nanoparticles while acting as a barrier for strongly negative particles.27
Whether and how the glycocalyx plays a role during the rapid uptake of slightly negatively charged SynC remains to be elucidated. Here, we investigate the role of glycosaminoglycans during the uptake of SynC. We observe the uptake processes of SynC with transmission electron microscopy (TEM) in THP-1 cells. With magnetic particle spectroscopy (MPS), we measure the interaction of SynC with GAGs, THP-1 cells, or Chinese hamster ovary cells (CHO) in real time. Upon click-chemistry-based labeling of THP-1 amino sugars, we investigate whether SynC binds or disrupts the labeled glycocalyx structures, facilitating their uptake. Subsequently, we investigated how removing GAGs from the cell surface affects SynC interaction and uptake efficiency.
2. Results and discussion
2.1. Uptake of SynC by THP-1 cells through endocytosis
Transmission electron microscopy (TEM) has become state-of-the-art for studying nanoparticle uptake and their interaction with cellular structures. Iron-oxide nanoparticles can be clearly distinguished from cells with TEM due to their high electron density (Fig. S1†).35 We imaged cells incubated with SynC for 10 min to visualize processes at the cell membrane and nanoparticle uptake. We could observe uptake processes with high recurrence using a high SynC concentration of 6.5 mmol L−1. Initially, SynC nanoparticles co-localized with the plasma membrane individually or as clusters. The latter came in different organizational forms, such as long chains extending from the membrane (Fig. 1A1), or larger clusters mainly associated with the membrane (Fig. 1B1). Generally, the cell membrane is negatively charged, suggesting a repulsion of SynC with a negative surface charge of −13.8 mV. However, the membrane contains cationic plasma membrane sites, which could explain large SynC aggregates forming at certain sides of the cell membrane.36–38 We observed the formation of membrane protrusions around large nanoparticle clusters (Fig. 1A2, B1 and B2). The extension of the plasma membrane occurs in phagocytosis and macropinocytosis due to actin reorganization. These two processes mediate the unspecific uptake of relatively large particles.39,40 The majority of SynC nanoparticles were surrounded by membrane extensions, indicating that unspecific uptake through micropinocytosis and phagocytosis drives the efficient uptake of SynC nanoparticles. To a lesser extent, we detected membrane engulfment around smaller nanoparticle clusters (Fig. 1A2 and B2). The formation of small membrane pockets is characteristic of clathrin- and caveolin-mediated pinocytosis, which results in small endocytic vesicles with a size of 60–120 nm. In contracts, endosomes forming upon macropinocytosis range from 200 nm to 5 μm.40 We observed either a few or a large number of SynC nanoparticles in endosomes (Fig. 1A3 and B3). It is well established that nanoparticles are initially located in early endosomes, which fuse and mature into multivesicular endosomes.41 Our findings suggest that a substantial amount of the cell-associated iron quantified in previous studies was internalized after 10 minutes rather than merely bound to the cell surface.7,11 In summary, we observed aggregation of SynC at the cell surface followed by efficient uptake through several endocytic processes within 10 min. | ||
| Fig. 1 Endocytic uptake of SynC by THP-1 monocytes. Transmission electron microscopy of THP-1 cells that were preserved with HPF/FS after incubation with cFe = 6.5 mM SynC for 10 min. Iron oxide SynC nanoparticles are visible as electron-dense structures. (A and B) Overview of THP-1 cells. (A1 and B1) SynC aggregates and individual nanoparticles concentrated at the cell surface pointed out by arrows. (A2, B2) Arrows demonstrate membrane engulfment and extension around SynC particles. (A3 and B3) Arrows point at the intracellular localization of SynC particles inside endosomes. | ||
2.2. Rapid alteration of the magnetic signal upon the interaction of SynC with cells
Next, we focused on the initial processes at the cell surface facilitating SynC uptake. To monitor the initial processes occurring during the interaction of SynC with cells, we used magnetic particle spectroscopy (MPS). MPS is proven to be a sensitive method for depicting effects on magnetic nanoparticles with high temporal resolution.42 Magnetic nanoparticles undergo physicochemical and magnetic changes upon interacting with cells. The magnetic change can be expressed by the ratio of the third and fifth harmonic (A5/A3), providing a measure independent of the nanoparticle concentration.43 We previously determined that the A5/A3 ratio of SynC remains stable in PBS, in contrast to FCS-containing medium.11 Consequently, we normalized our measurements to SynC nanoparticles in PBS, eliminating a potential effect of media components on the nanoparticles. We started the measurement immediately after adding SynC to monitor the initial cell–particle interaction. The measurement ran for 667 s, with additional measurement points during the first 100 s. To ensure cell viability, we used a concentration of cFe = 0.5 mmol L−1 in all following uptake and interaction experiments.11 When adding the nanoparticles to THP-1 cells in PBS, the A5/A3 signal of SynC changed within seconds. During the first 11 s of cell contact, the A5/A3 ratio increased steeply by Δ14.54% (SD: ±1.73%) compared to the baseline in PBS (Fig. 2). The rapid increase shows that cell contact immediately affected the magnetic response of SynC in MPS. In the following minutes, the A5/A3 ratio approached a plateau at Δ16.25% (SD: ±0.25%). These observations suggest that initial cell contact had a more substantial effect on the magnetic behavior of SynC compared to the process of cellular uptake, which steadily increases. The rapidity of the effect within just 10 s implies that extracellular structures, either secreted or bound to the cell surface, caused the alteration due to their immediate availability for interaction. To determine whether secreted or cell-bound structures caused the rapid A5/A3 increase, we repeated the measurement with conditioned PBS, containing only molecules secreted from the same number of cells within 10 min. The A5/A3 ratio only elevated by Δ2.72% (SD: ±3.32%) after 11 s and increased to Δ3.77% (SD: ±4.43%) after 667 s in the presence of conditioned PBS. We concluded that the steep A5/A3 increase in the presence of cells was mainly caused by contact with cell-bound structures and only marginally through interaction with molecules secreted during the 10 minutes measurement. The underlying processes causing the A5/A3 ratio to increase could be a cooperative character of induced dipolar interactions between the moments of aggregated nanoparticles at the cell surface.42,44–48 Taken together, we observed a rapid alteration of SynC's magnetic behavior upon contact with cellular structures. We hypothesized that the binding of SynC to the extracellular glycocalyx caused the rapid A5/A3 increase. | ||
| Fig. 2 Magnetic particle spectroscopy of SynC in PBS, conditioned PBS, or THP-1 cells in PBS. The A5/A3 ratio investigates external effects on cFe = 0.5 mmol L−1 SynC. Measurements were performed at bexcit = 12 mT and boff = 0.05 mT at 37 °C. N = 3, mean with error bars showing SD. Measurements normalized to the stable A5/A3 ratio of SynC in PBS. In the presence of THP-1 cells, the A5/A3 ratio increased rapidly by Δ14.54% (SD: ±1.73%) within 11 s. Upon incubation with conditioned PBS, the A5/A3 ratio of SynC increased by Δ2.72% (SD: ±3.32%) within the initial 11 s. After 667 s, the A5/A3 ratio increased by Δ16.25% (SD: ±0.25%) in the presence of THP-1 cells and by Δ3.77% (SD: ±4.43%) in conditioned PBS. | ||
2.3. Fluorescent glycosaminoglycan labeling revealed no binding of SynC to GAGs
To test the hypothesis that interactions with molecules of the glycocalyx trigger the magnetic behavior change of SynC, we aspired to visualize potential interactions between SynC and the glycocalyx in living cells. To label glycosaminoglycans under physiological conditions, we used copper-free click chemistry. This technique provides a powerful tool for labeling the glycocalyx in real time by attaching a fluorescent probe to a previously incorporated sugar mimic.49–52 We introduced modified N-acetylglucosamine (GlcNAz), a component of heparan sulfate, keratan sulfate, hyaluronic acid, and N-glycans into the cells. Alternatively, we incorporated N-acetylgalactosamine (GalNAz), a component of chondroitin sulfate, dermatan sulfate, and O-glycans (Fig. 3A).30,53 Upon incorporation, we performed click-chemistry-based labeling of the modified sugars.50,54 When omitting the addition of modified sugars, background staining was located intracellularly (Fig. 3B). After staining cells incubated with modified GlcNAz, we only observed enhanced intracellular fluorescence and no extracellular fluorescent, indicating that the glycocalyx of THP-1 cells contains minor levels of GlcNAz-containing structures (Fig. 3C). In agreement with our observation, prior HPLC measurements suggested that THP-1 cells contain negligible levels of GlcNAz-containing heparan sulfate.55,56 Upon fluorescently labeling GalNAz-containing cells, we observed an enhanced intracellular fluorescence, a bright membrane labeling, and fluorescent structures expanding from the membrane, implying that GalNAz-containing molecules are abundant in the THP-1 glycocalyx (Fig. 3D). Labeling of GalNAz-containing structures in the glycocalyx verifies previously conducted HPLC analyses, suggesting that Gal-NAz-containing chondroitin sulfate is the predominant glycosaminoglycan in THP-1 cells.55–57 | ||
| Fig. 3 Fluorescent labeling of GalNAz or GlcNAz in THP-1 cells using copper-free click-chemistry. (A) Scheme depicting the incorporation of azide-modified GalNAz or GlcNAz into the glycocalyx of THP-1 cells. Chondroitin (CS) and dermatan sulfate (DS) contain GalNAz. Heparan sulfate (HS), keratan sulfate (KS), and hyaluronic acid (HA) contain GlcNAz. After azide-modified sugars were incorporated, an alkyne-coupled fluorescent molecule was added for 1 h at 37 °C. The fluorescently coupled alkyne group reacts with the modified sugars' azido group, thereby fluorescently labeling GalNAz and GlcNAz. (B) Intracellular background fluorescence of THP-1 cells previously not incubated with modified sugars. (C) THP-1 cells labeled with 50 μM GlcNAz show enhanced intracellular fluorescence compared to control THP-1 cells (D). Upon incubation with 50 μM GalNAz, fluorescent staining is present intracellularly and in extracellular structures attached to the cell membrane. Labeling of extracellular structures indicates that GalNAz-containing molecules, such as CS and DS, are present in the glycocalyx of THP-1 cells. Representative images are shown. | ||
Having identified significant expression of GalNAz-containing molecules in THP-1 cells, we next aimed to exploit this method to investigate interactions between GalNAz-containing glycocalyx structures and SynC (Fig. 4A). First, we confirmed the stability of fluorescently labeled GAGs in PBS. We imaged labeled cells 5 or 20 min after adding SynC and included a control for each time point to control for fluorescent reduction due to photobleaching or turnover of labeled glycosaminoglycans. As shown in Fig. 4B, we could not observe the disruption of fluorescently labeled glycocalyx components upon incubation with SynC for up to 20 min, indicating that glycocalyx disruption is not associated with SynC uptake. It was previously proposed that nanoparticles bind to glycosaminoglycans, facilitating their uptake.27,58,59 To test this hypothesis, we quantified the intracellular fluorescence upon adding SynC. If SynC binding to labeled GalNAz structures accelerates the turnover rate of GalNAz-containing molecules, we would expect to see an increase in fluorescently labeled endosomes (Fig. 4A). We could not observe a significant enhancement of the intracellular fluorescence per cytosol area upon adding SynC (two-tailed unpaired Student's t-test; pctrl short vs. SynC 5 min = 0.14, pctrl long vs. SynC 20 min = 0.35) (Fig. 4C). We determined a cytosolic fluorescent intensity/cytosol area of 56.49 (a.u.) for short control cells and 62.89 (a.u.) for THP-1 cells incubated with SynC for 5 min. For longer control cells, we measured a cytosolic fluorescent intensity/cytosol area of 56.75 (a.u.) and 52.70 (a.u.) for cells incubated with SynC for 20 min. These observations suggest that SynC nanoparticles do not bind to GalNAz-containing structures before their uptake. Alternatively, cells could take up GAG-bound nanoparticles at the same rate as membrane-bound GAGs. A further possibility is that SynC nanoparticles do not bind to fluorescently labeled GAGs prior to their uptake. The interaction of SynC with GalNAz-containing molecules could be hindered by incorporating biorthogonal GalNAz and attaching a fluorescent label. In summary, upon labeling GalNAz-containing structures, we could show no disruption of fluorescently labeled glycocalyx components upon adding SynC. We could not detect an accelerated uptake of fluorescently labeled glycocalyx components in the presence of SynC nanoparticles. With further methods, we aimed to distinguish whether SynC nanoparticles do not bind to GalNAz-containing glycocalyx structures before cellular uptake or whether SynC binds to GalNAz-containing structures but is taken up at the regular turnover rate of GalNAz structures.
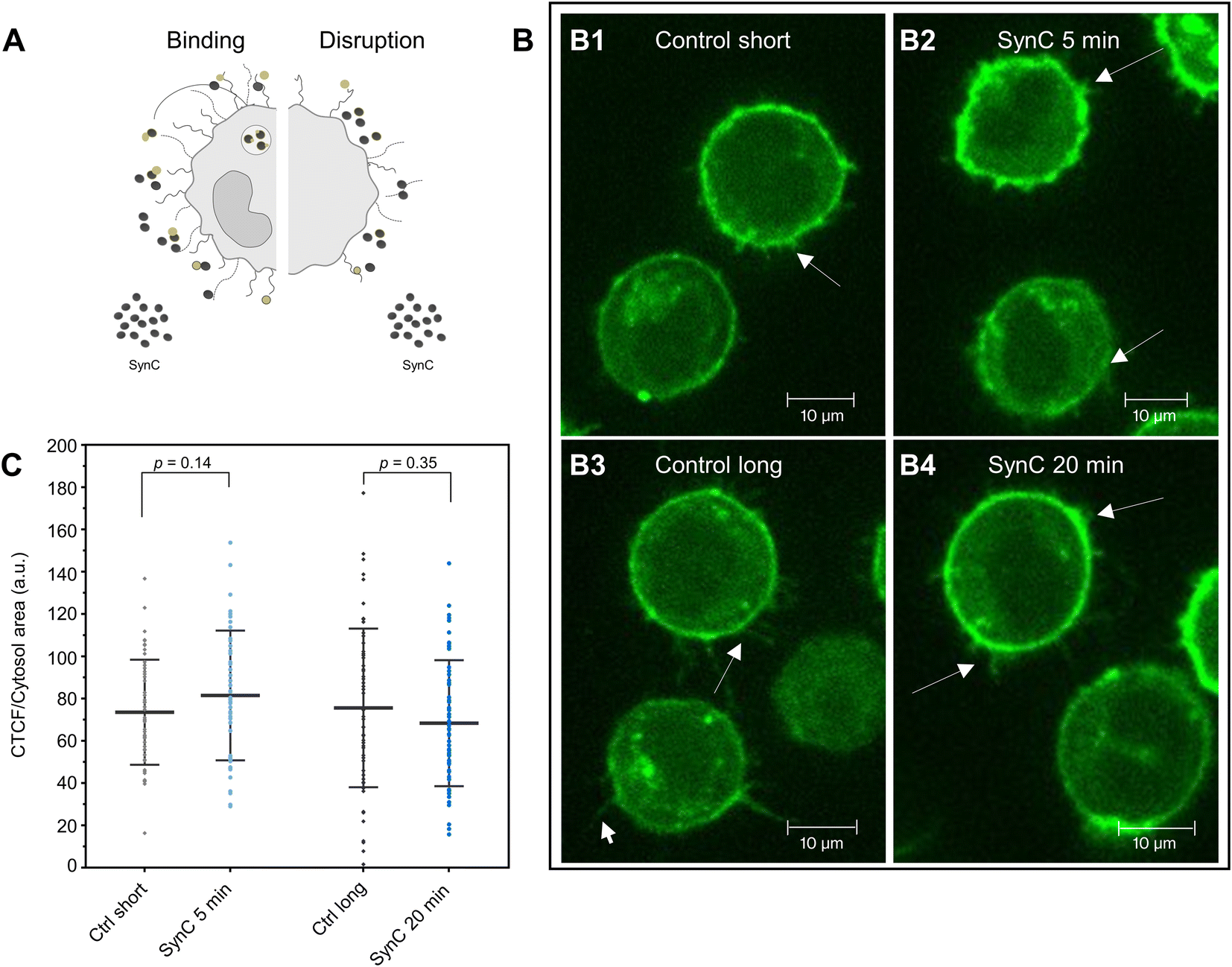 | ||
| Fig. 4 Interaction of SynC with THP-1 cells upon fluorescent labeling of GalNAz. (A) Scheme showing potential hypotheses of SynC interacting with the glycocalyx of THP-1 monocytes either by binding or disrupting the glycocalyx. (B) THP-1 cells labeled with 50 μM GalNAz for three days and stained with 3 μM DIBO488 for 1 h. (B1 and B3) GalNAz labeled THP-1 cells without the addition of SynC imaged (B1) directly after washing (n = 72 cells) or (B3) or upon incubation for 20 min at 37 °C (n = 67 cells). (B2 and B4) GalNAz labeled THP-1 cells incubated with cFe = 0.5 mM SynC for (B2) 5 min (n = 83 cells), (B4) or 20 min (n = 62 cells) in PBS at 37 °C. SynC nanoparticles do not disrupt fluorescently labeled GalNAz structures (arrows). Representative images are shown. (C) Quantification of the cytosolic fluorescent intensity as corrected total cytoplasmic fluorescent (CTCF)/cytosol area (a.u). Control short: 56.49, SynC 5 min: 62.89, control long: 56.75, SynC 20 min: 52.70. We determined no significant difference in the cytosolic fluorescent intensity upon the addition of SynC (two-tailed unpaired Student's t-test; pctrl short vs. SynC 5 min = 0.14, pctrl long vs. SynC 20 min = 0.35). We conclude that SynC does not cause enhanced uptake of GalNAz-containing structures upon SynC addition. | ||
2.4. Rapid SynC uptake by THP-1 cells independent of cell-surface GAGs
To determine whether SynC uptake is facilitated by binding to chondroitin sulfate, the main glycosaminoglycan in THP-1 cells, we treated cells with chondroitinase ABC (ChABC). The enzyme breaks down chondroitin sulfate and dermatan sulfate. We initially verified that a concentration of 10 mU ChABC for 1 h was sufficient to break down 5 μg of purified CS (Fig. 5B) and CS in the glycocalyx of THP-1 cells using stains-all for differentially glycosaminoglycans staining (Fig. S4†). Upon treatment with ChABC, we performed real-time MPS measurements to test whether chondroitin sulfate triggers the A5/A3 increase of SynC upon cell contact (Fig. 5C). We could not measure any significant differences in the A5/A3 ratio change between cells pretreated with ChABC and untreated THP-1 cells. ChABC-treated cells caused an increase of Δ10.40% (SD: ±0.39%) compared to an increase of Δ10.19% (SD: ±0.30%) triggered by untreated THP-1 cells within 11 s of cell contact (two-tailed unpaired Student's t-test; p11 s = 0.41). These results imply that interaction with chondroitin sulfate on the cell surface does not solely cause the rapid magnetic behavior change of SynC. To test whether chondroitin sulfate alone affects the A5/A3 ratio of SynC, we repeated the measurement with purified chondroitin sulfate dissolved in dH2O (Fig. S2A†). While purified CS does not resemble the cellular chondroitin sulfate organization, the general composition remains. During the incubating of SynC with purified chondroitin sulfate, the A5/A3 ratio remained consistent with the baseline in dH2O, with an A5/A3 difference of +0.02% (SD: ±0.11%). Based on that, we concluded that interaction with chondroitin sulfate does not trigger the rapid magnetic behavior increase of SynC we observed upon cell contact. We tested whether the MPS results also translate into similar SynC uptake efficiency upon chondroitin sulfate removal. We incubated ChABC-treated and untreated cells with cFe = 0.5 mmol L−1 SynC for 10 min and quantified intracellular iron per cell using Prussian blue iron staining (Fig. 5D and E). As shown in Fig. 5F, we did not measure any significant differences in the uptake of SynC between untreated and ChABC-treated THP-1 cells (two-tailed unpaired Student's t-test; p = 0.88). Untreated THP-1 cells had a mean Prussian blue stained area per cell of 9.14% (SD: ±8.36%) compared to 8.30% (SD: ±6.06%) in ChABC-treated THP-1 cells. In conclusion, digesting chondroitin sulfate did not influence the uptake efficiency of SynC, indicating that chondroitin sulfate does not act as an essential binding partner facilitating SynC uptake nor provides a significant barrier for SynC.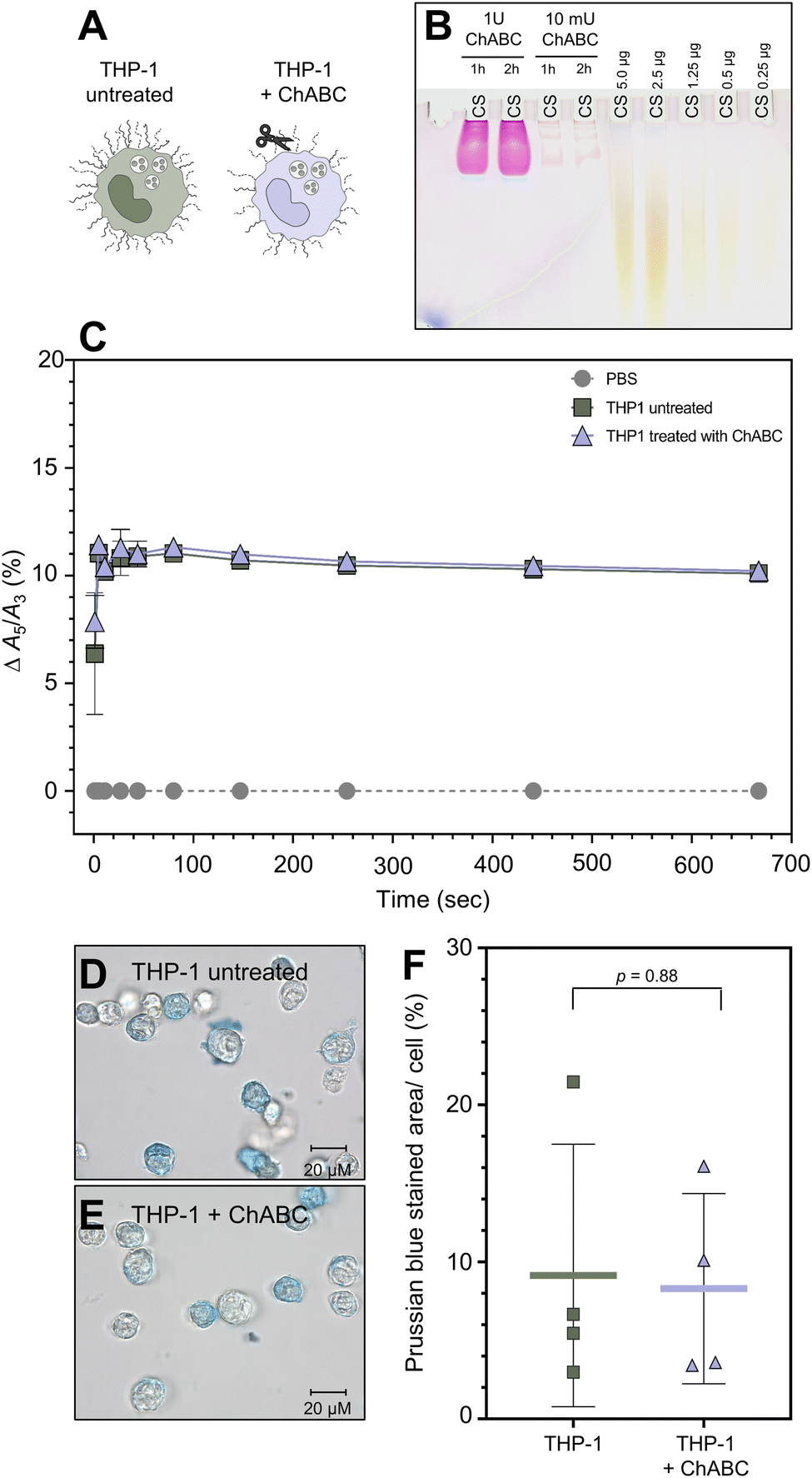 | ||
| Fig. 5 Interaction of SynC with ChABC-treated THP-1 cells. (A) Scheme showing the experimental setup. (B) Stains-all gel showing 5 μg chondroitin sulfate (CS) digested with 1 U or 10 mU chondroitinase ABC for 2 h or 1 h at 37 °C. 10 mU ChABC for 1 h was sufficient to digest 5 μg CS. Standard range of 5–0.25 μg CS. (C) Magnetic particle spectroscopy of SynC in the presence of untreated THP-1 cells or THP-1 cells pretreated with 10 mU ChABC for 1 h. Measurements at 12 mT, boff = 0.05 mT at 37 °C. N = 4 mean with error bars showing SD. The measurements were normalized to the A5/A3 baseline in PBS. ChABC-treated THP-1 cells caused an A5/A3 increase within 11 s by Δ10.40% ± 0.39% compared to an increase of Δ10.19% ± 0.30% in the presence of untreated THP-1 cells (two-tailed unpaired Student's t-test; p11 s = 0.41). (D and E) Prussian blue iron staining after incubation with cFe = 0.5 mM SynC for 10 min of (D) control THP-1 cells or (E) THP1 cell pretreated with 10 mU ChABC for 1 h. Representative images shown. (F) Uptake quantification of SynC with Prussian blue iron staining by untreated and THP-1 cells treated with 10 mU ChABC for 1 h. An average of 91 cells per condition were quantified. No significant differences in the uptake of SynC were measured between untreated THP-1 (9.14% ± 8.36%) and ChABC-treated THP-1 cells (8.30% ± 6.06%) (two-tailed unpaired Student's t-test; p = 0.88). Mean ± SD (n = 4). | ||
Previously, Zhang et al. proposed that citrate-coated gold nanoparticles bind to purified hyaluronic acid.60 We could not conclusively eliminate the potential presence of hyaluronic acid at low levels in the THP-1 glycocalyx with differential labeling of glycosaminoglycans (Fig. S3†). Therefore, we decided to investigate the role of hyaluronic acid during the uptake of SynC (Fig. 6A). We initially verified that 2 U hyaluronidase (HyA) was sufficient to break down 10 μg of hyaluronic acid with differential glycosaminoglycan labeling (Fig. 6B). To guarantee a breakdown of smaller HA fragments, we increased the concentration of HyA to 10 U. With real-time MPS, we measured no significant differences between untreated and HyA-treated cells regarding their ability to affect the A5/A3 ratio of SynC during the initial seconds of cell contact (two-tailed unpaired Student's t-test; p11 s = 0.83). Untreated THP-1 cells enhanced the A5/A3 ratio by Δ9.45% (SD: ±0.62%) during the first 11 s, compared to an increase of Δ9.26% (SD: ±1.6%) triggered by HyA-treated cells (Fig. 6C). We concluded that the rapid A5/A3 increase of SynC upon cell contact was not exclusively caused by hyaluronic acid. We repeated the measurement with purified high and low-molecular-weight hyaluronic acid. In the presence of low-molecular-weight HA, the A5/A3 increased by Δ0.11% (SD: ±0.10%) compared to SynC in dH2O. High molecular weight HA caused a decrease in the A5/A3 ratio by Δ−0.09% (SD: ±0.34%) (Fig. S2C and D†). Based on these measurements, we propose that hyaluronic acid does not cause the steep increase of the A5/A3 ratio of SynC upon cell contact. Next, we quantified SynC uptake by THP-1 cells upon HyA treatment using Prussian blue iron staining (Fig. 6D–F). Our quantification revealed no significant differences between HyA-treated and untreated cells (two-tailed unpaired Student's t-test; p = 0.60). Untreated THP-1 cells showed a mean stained area per cell of 3.75% (SD: ±1.00%) compared to 3.26% (SD: ±1.49%) in THP-1 cells pretreated with HyA (Fig. 6C). We concluded that hyaluronic acid is not essential for SynC uptake by THP-1 cells.
 | ||
| Fig. 6 Interaction of SynC with HyA-treated THP-1 cells. (A) Scheme showing the experimental setup. (B) Stains-all gel showing 10 μg of high molecular weight hyaluronic acid (HA) (control) or digested with 0.3 U, 1 U, 2 U, or 4 U HyA at 37 °C for 1 h. 2 U HyA were sufficient to break down 10 μg of high weight hyaluronic acid. (C) Magnetic particle spectroscopy of SynC in the presence of untreated THP-1 cells or pretreated with 10 U HyA for 1 h at 37 °C. Measurements were performed at 12 mT, boff = 0.05 mT at 37 °C. N = 4, mean ± SD. Measurements normalized to stable A5/A3 baseline of SynC in PBS. Contact with untreated THP-1 cells for 11 s caused an A5/A3 increase of Δ9.45% (SD: ±0.62%) compared to Δ9.26% (SD: ±1.6%) caused by HyA-treated cells (two-tailed unpaired Student's t-test; p11 s = 0.83). (D and E) Representative images showing Prussian blue iron staining after incubation with cFe = 0.5 mM SynC for 10 min of (D) control THP-1 cells or (E) THP1 cell pretreated with 10 U HyA for 1 h. (F) Uptake quantification with Prussian blue iron staining of untreated THP-1 cells compared to THP-1 cells pretreated with 10 U HyA for 1 h at 37 °C. On average 90 cells were quantified per condition. Untreated THP-1 cells had a mean Prussian blue stained area per cell of 3.75% ± 1.00% compared to a mean of 3.26% ± 1.49% in HyA-treated cells (two-tailed unpaired Student's t-test; p = 0.60. Mean ± SD (n = 4)). | ||
In summary, removing chondroitin sulfate and hyaluronic acid did not enhance or reduce the uptake of SynC, suggesting that CS and HA do not form a significant barrier for SynC nor demonstrate an essential binding partner for SynC uptake. The involvement of other glycosaminoglycans during the uptake of SynC by THP-1 cells is unlikely due to minor levels of Gal-NAz containing heparan sulfate and keratan sulfate (Fig. 3C). Note that THP-1 cells could have resynthesized chondroitin sulfate or hyaluronic acid during the incubation with SynC due to absence of the digesting enzymes.
2.5. Xylosyltransferase-deficient CHO mutants efficiently take up SynC
To exclude GAG resynthesize as a confounding factor, we investigated SynC uptake of an established GAG-deficient cell line. We compared two Chinese hamster ovarian carcinoma cell lines (CHO): wild type CHO-K1 cells and psgA-745 mutants, lacking xylosyltransferase (Fig. 7A).61 The enzyme facilitates the rate-limiting step of attaching xylose to the proteoglycan core protein during glycosaminoglycan synthesis. Thus, xylosyltransferase-deficient mutants lack chondroitin and heparan sulfate.27,62,63 Using the CHO cell models, Olivieri et al. observed a significantly reduced uptake of positively charged polystyrene nanoparticles (+59 mV) in psgA-745 mutants, indicating that CS and HS binding facilitates their uptake. The uptake of negative polystyrene nanoparticles (−58.9 mV) increased in psgA-745 mutants compared to wild type cells, indicating that CS and HS repelled these nanoparticles.27 We aimed to investigate whether CS and HS also regulate the uptake of SynC in CHO cells by incubating adherently grown cells with SynC nanoparticles for 3 h. We quantified the intracellular uptake of SynC with Prussian blue iron staining (Fig. 7B). Mutant psgA-745 cells showed a trend towards a slightly increased uptake of SynC compared to wildtype CHO-K1 cells (mean stained area 47.60% (SD: ±16.90%) vs. 31.05% (SD: ±9.58%), two-tailed unpaired Student's t-test; p = 0.06). These results indicate that CS and HS are not essential for SynC uptake, as shown by efficient SynC uptake in GAG-deficient psgA-745 cells. Additionally, HS and CS did not form a substantial barrier for SynC uptake in adherently grown CHO cells. | ||
| Fig. 7 SynC uptake and cell-surface interaction with xylosyltransferase-deficient CHO cells. (A) Scheme of wild type CHO-K1 cells and xylosyltransferase-deficient psgA-745 cells. (B) Prussian blue iron staining of wild type and mutant CHO cells grown adherent upon incubation with SynC for 3 h (n = 6). On average 36 cells were analyzed per condition for each experiment. CHO-K1 had a mean Prussian blue stained area per cell of 31.05% ± 9.58%. PsgA-745 cells had a stained area of 47.60% ± 16.90% per cell (two-tailed unpaired Student's t-test; p = 0.06). (C) Magnetic particle spectroscopy of SynC in the presence of CHO-K1 or psgA-745 cells grown in suspension. Measurements were performed at 12 mT and Boff = 0.05 mT at 37 °C for 667 s. N = 4 mean with SD. Measurements normalized to the A5/A3 baseline of SynC in PBS. Wild type CHO-K1 cells caused an A5/A3 increase of Δ12.50% ± 1.06% after 11 s of cell contact. PsgA-745 cells caused an A5/A3 increase of Δ11.92% ± 0.98% within 11 s (two-tailed unpaired Student's t-test; p11 s = 0.43). (D) Prussian blue iron staining of CHO cells after growth in suspension. Incubation with cFe = 0.5 mM SynC for 10 min or 60 min. We quantified on average 38 cells per condition for each experiment. No significant differences were determined in SynC uptake by CHO-K1 and psgA-745 cells after growth in suspension (two-tailed unpaired Student's t-test; p10 min = 0.76; p60 min = 0.93) (n = 4). CHO-K1 cells had a mean Prussian blue stained area per cell of 20.45% ± 8.84% after 10 min and 39.62% ± 14.18% after 60 min. Compared to psgA-745 cells 18.30% ± 10.34% (10 min) and 38.55% ± 17.21% (60 min). | ||
Due to our prior focus on suspension cells and the restriction of the MPS setup requiring cells in solution, we investigated CHO cells grown in suspension for at least 24 h. Initially, we measured the effect of xylosyltransferase deficiency on the A5/A3 ratio of SynC upon cell contact. With real-time MPS measurements, we could not measure significant differences in the ability to induce a steep A5/A3 increase of SynC upon cell contact (two-tailed unpaired Student's t-test; p11 s = 0.43) (Fig. 7C). CHO-K1 cells caused an increase of Δ12.50% (SD: ±1.06%) compared to Δ11.92% (SD: ±0.98%) in psgA-745 cells within 11 s. Subsequently, the A5/A3 ratio approached a plateau of Δ15.32% (SD: ±0.64%) in the presence of wild type CHO-K1 cells and Δ16.08% (SD: ±0.48%) upon incubation with psgA-745 cells. We concluded that lack of chondroitin and heparan sulfate does not alter the A5/A3 ratio of SynC upon cell contact. Accordingly, the A5/A3 ratio remained stable during the incubation with purified chondroitin sulfate and heparin, which caused A5/A3 ratio changes of Δ0.02% (SD: ±0.11%) or a decrease by Δ−0.11% (SD: ±0.07%) after 667 s, compared to SynC in dH2O (Fig. S2A and B†). Subsequently, we quantified the uptake of SynC by wild type and mutant CHO cells after growth in suspension. Here, we could investigate nanoparticle uptake after SynC incubation for 10- and 60 min. Quantification of SynC uptake by CHO-K1 and mutant psgA-745 cells with Prussian blue staining showed no significant differences between the two cell lines after growth in suspension (Fig. 7D). CHO-K1 cells showed a mean stained area per cell of 20.45% (SD: ±8.84%) after 10 min compared to 18.30% (SD: ±10.34%) in psgA-745 cells (two-tailed unpaired Student's t-test; p10 min = 0.76). Upon a 60 minutes incubation with SynC CHO-K1, cells showed a mean Prussian blue stained area of 39.62% (SD: ±14.18%), and psgA-745 cells had a mean stained area of 38.55% (SD: ±17.21%) (two-tailed unpaired Student's t-test; p60 min = 0.93). In summary, our results show no significant differences in the uptake of SynC by CHO-K1 and chondroitin and heparan sulfate deficient psgA-745 cells. With CHO cells grown in suspension, we could not verify the trend towards slightly increased uptake of SynC as observed in adherently grown psgA-745 cells compared to CHO-K1 cells. This further supports the conclusions that CS and HS are not essential for efficient SynC uptake and do not form a substantial barrier for SynC in CHO cells. SynC uptake could be facilitated by binding to other components in the glycocalyx, such as glycoproteins associated with N-glycans or O-glycans, glycolipids, or proteins. In wild type and mutant CHO cells, we could not determine the presence of hyaluronic acid upon differential glycosaminoglycan staining with stains-all (Fig. S3†).
Upon reaching the cell membrane, SynC nanoparticles undergo endocytosis. The mechanism by which nanoparticles initiate endocytosis remains to be fully elucidated. Rees et al. suggested that nanoparticles arrive into currently forming endosomes by chance and are internalized as additional cargo.21 Variability in the cellular endocytic capacity might influence the uptake efficiency of nanoparticles. Wild type and mutant CHO cells exhibited different endocytic capacities for dextran, potentially affecting the uptake efficiency of SynC quantified here.64 However, our prior digestion experiments quantified SynC uptake in THP-1 cells with identical endocytic baseline capacities.
3. Conclusion
Magnetic nanoparticles need to pass the extracellular glycocalyx to undergo cellular uptake. Our study focused on the role of glycosaminoglycans during the uptake of citrate-coated iron oxide nanoparticles (SynC) with a surface charge of −13.82 mV. Herein, we show that THP-1 monocytes and CHO cells internalize SynC independent of chondroitin sulfate, heparan sulfate, and hyaluronic acid on the cell surface, indicating that glycosaminoglycans are not essential binding partners that facilitate SynC uptake nor present a significant barrier for SynC uptake. We propose that SynC nanoparticles reach the cell membrane without experiencing significant repulsion by extracellular glycosaminoglycans due to their minor negative surface charge. The absence of SynC repulsion by cell surface GAGs could explain the efficient uptake of SynC within 10 minutes. Prior research showed that the uptake efficiency of strongly negatively or positively charged nanoparticles is affected by cell surface glycosaminoglycans.27 Based on our results, we conclude that glycosaminoglycans do not have the same effect on weakly-negative particles such as SynC. Our concluding model summarizes the role of cell-surface glycosaminoglycans as a barrier, binding partner, and permeable bandpass dependent on the charge of the nanoparticles in Fig. 8.25,65 | ||
| Fig. 8 Proposed model for the glycocalyx as a charged-based bandpass for magnetic nanoparticles. (A) Repulsion of strongly negative nanoparticles with a surface charge below −30 mV. (green). Binding of positive nanoparticles to negatively charged glycosaminoglycans (orange). (B) Permeable bandpass for nanoparticles within a charge range between −30 mV and +10 mV. | ||
By understanding the relationship between the glycocalyx and nanoparticle uptake, we may better use citrate-coated iron oxide nanoparticles as tools in diagnosis and therapy. The here-reported efficient uptake of SynC unaffected by cell-surface glycosaminoglycans in THP-1 monocytes further underscores the potential of SynC as MPI tracers. Moving forward, it will be crucial to investigate whether the rapid uptake of SynC is cell-type specific and influenced by different extracellular matrix compositions. Such cell-type specific uptake, for instance, by immune cells or cancer cells, could be exploited in MPI-based cell tracking of distinct cell types that lack significant SynC repulsion by cell-surface glycosaminoglycans.
4. Methods
4.1. Cell culture and treatments
We used the human acute monocytic THP-1 cell line (ACC 16, DSMZ) cultivated in suspension at 37 °C with 5% CO2 in 75 cm2 flasks (Sigma-Aldrich) in RPMI 1640 media (Gibco) supplemented with 10% FCS (Biochrom), 100 U mL−1 penicillin (Invitrogen) and 100 mg mL−1 streptomycin (cRPMI) (Invitrogen). The cell number was determined with a Fuchs-Rosenthal counting chamber. Conditioned medium was prepared from 1 × 106 THP-1 cells incubated in PBS for 10 min at 37 °C in a 5% CO2 incubator. The supernatant was collected upon centrifugation for 2 min at 800g. For GAG digestion experiments, 1 × 106 THP-1 cells were centrifuged at 1000g for 5 min. The pellet was resuspended in a total volume of 100 μL Dulbecco's phosphate-buffered saline (PBS) (Gibco) supplemented with 10 mU chondroitinase ABC (Sigma-Aldrich) or 10 U hyaluronidase (Sigma-Aldrich). Digestion was performed in PBS for 1 h in a 5% CO2 incubator at 37 °C. Additionally, we used two Chinese hamster epithelial-like ovary cell lines (CHO): wild type CHO-K1 cells (CCL-61, ATCC) and psgA-745 cells, a xylosyltransferase I/II deficient mutant (CRL2242, ATCC). CHO cells were cultivated in T-75 cm2 flasks (Sigma-Aldrich) with F-12K medium (Gibco), 10% FCS (Biochrom), 100 U mL−1 penicillin (Invitrogen) and 100 mg mL−1 streptomycin (Invitrogen) in a 5% CO2 incubator at 37 °C. Upon trypsin treatment (Gibco), cells grew at least 24 h, either adherent or in suspension, in 50 mL Falcon tubes (Thermo Fisher Scientific).4.2. Magnetic nanoparticles
We used citrate-coated multicore iron oxide nanoparticles called Synomag®-COOH (SynC) (article no. 103-02-301m, Micromod), with an iron concentration of 130.9 mmol L−1. The incubation of cells with SynC was performed in PBS at 37 °C with 5% CO2. We used a concentration of cFe = 0.5 mmol L−1 SynC unless stated otherwise. The SynC particles used had a zeta potential of −13.8 ±2.1 mV at pH 7.0 and a hydrodynamic diameter of 23.4 ±6.2 nm, as previously determined.114.3. Transmission electron microscopy
1 × 106 THP-1 cells were centrifuged for 3 min at 1000g and washed with 1 mL PBS. The pellet was resuspended in 20 μL PBS or 19 μL PBS supplemented with 1 μL SynC (final conc. 6.5 mmol L−1). The particle incubation was performed for 10 min. 7 μL of the cell suspension was placed in the 200 μM indented side of a type A aluminum carrier (Ø 0.3 × 0.5 mm type A, Nr. 16770141, Leica). Cells were enclosed between the carriers by placing the flat side of a type B aluminum carrier on top (Ø 3.0 × 0.5 mm, Nr. 16770142, Leica). The cells underwent high-pressure freezing (HPF) (EM HPM 100 High Pressure Freezer, Leica) at 2100 bar in liquid nitrogen. Afterwards, the samples were freeze-substituted (FS) (EM AFS2, Leica) in a cocktail containing acetone with 1% methanol (Carl Roth), 0.5% osmium tetroxide (Electron Microscopy Science), and 5% dH2O. Freeze substitution began at −90 °C, followed by a temperature increase of 5 °C per hour until reaching 0 °C. In between, the samples were kept at −50 °C for 2 h. (T1: −90 °C for 2 h, T2: −50 °C for 2 h, T3: 0 °C). The samples were washed with acetone (J. T. Baker) four times for 5 min, followed by two 10 minutes washing steps. To avoid cellular disruption, we waited for sedimentation instead of using centrifugation in between washing steps. The samples were embedded in Epon (Serva), which solidified at 60 °C for 48 h. Ultra-thin sections (60 nm) were cut with an ultramicrotome (Ultracut E, Reicht-Jung) equipped with a diamond knife (Diatome) and placed on copper grids (3.05 mm with a slot of 2 mm × 1 mm, Plano). We used a Zeiss Leo 906 electron microscope at 80 kV acceleration voltage equipped with a Proscan slow scan 2K CCD camera (Carl Zeiss) and the ImageSP software for imaging.4.4. Differential staining of glycosaminoglycans
We used stains-all (1-ethyl-2-[3-(1-ethylnaphtho[1,2-d]thiazolin-2-ylidene)-2-methylpropenyl]naphtho[1,2-d]thiazolium bromide) (Sigma-Aldrich), a cationic carbocyanine dye to differentially stain glycosaminoglycans.59 As glycosaminoglycan standards, we used 5 μg chondroitin sulfate (BioSYNTH), heparin (Sigma-Aldrich), hyaluronic acid with a high molecular weight of 750–1000 kD (Sigma-Aldrich), or a low molecular weight between 130 kD and 150 kD (Sigma-Aldrich). The GAG profile of our cell models was visualized upon glycosaminoglycan isolation from 15 × 106 cells. We visualized the GAG profile of equally treated duplicates to verify the successful digestion of chondroitin sulfate in the following uptake experiments. We pooled THP-1 cells from four independent experiments with 1 × 106 cells per condition. We initially performed protein digestion with 2 U papain (Sigma-Aldrich) in PBS supplemented with 24 mg mL−1L-cysteine (Sigma-Aldrich) for 41 h at 65 °C. The enzymes were inactivated the next day by increasing the temperature to 95 °C for 15 min. The samples were centrifuged for 5 min at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g. Upon transferring the supernatant into a new tube, we added 100 μL 1 M NaBH4 (Thermo Fisher Scientific) in 0.5 M NaOH (Merck) and incubated the samples for 2 h at 37 °C. Subsequently, the samples were placed on ice, and 16 μL 25% HCl (Merck) was added in 2 μL steps. We added 15% TCA (Carl Roth) and stored the samples at 4 °C for 20 h. The samples were centrifuged for 10 min at 19000g the next day. The supernatant was kept and underwent precipitation at −20 °C overnight using 5× the volume of 100% ethanol (Carl Roth) supplemented with 5% potassium acetate (Roth). The samples were centrifuged for 30 min at 21000g. The pellets were washed twice with 2 mL 80% ethanol and centrifuged for 30 min at 21000g between washing steps. We resuspended the pellet in dH2O, added the suspension to a 30 kDa Amicon® Ultra Centrifugal Filter (Millipore), and centrifuged for 20 min at 5000g. Afterward, the samples were washed 10 times using 500 μL dH2O and centrifugation at 5000g for 15 min. In the last washing step centrifugation was performed for 16 min. Glycosaminoglycans were eluted at a speed of 1000 g for 2 min. 12 μL of the sample were evaporated at 65 °C. We used stains-all to verify the digestion of chondroitin sulfate and hyaluronic acid. We digested 5 μg of chondroitin sulfate in 10 μL PBS for 1 h or 2 h with 10 mU or 1 U ChABC under the previously described conditions. We digested 10 μg of high molecular weight hyaluronic acid as an additional control with 0.3 U, 1 U, 2 U, or 4 U hyaluronidase for 1 h under the previously described conditions.
000g. Upon transferring the supernatant into a new tube, we added 100 μL 1 M NaBH4 (Thermo Fisher Scientific) in 0.5 M NaOH (Merck) and incubated the samples for 2 h at 37 °C. Subsequently, the samples were placed on ice, and 16 μL 25% HCl (Merck) was added in 2 μL steps. We added 15% TCA (Carl Roth) and stored the samples at 4 °C for 20 h. The samples were centrifuged for 10 min at 19000g the next day. The supernatant was kept and underwent precipitation at −20 °C overnight using 5× the volume of 100% ethanol (Carl Roth) supplemented with 5% potassium acetate (Roth). The samples were centrifuged for 30 min at 21000g. The pellets were washed twice with 2 mL 80% ethanol and centrifuged for 30 min at 21000g between washing steps. We resuspended the pellet in dH2O, added the suspension to a 30 kDa Amicon® Ultra Centrifugal Filter (Millipore), and centrifuged for 20 min at 5000g. Afterward, the samples were washed 10 times using 500 μL dH2O and centrifugation at 5000g for 15 min. In the last washing step centrifugation was performed for 16 min. Glycosaminoglycans were eluted at a speed of 1000 g for 2 min. 12 μL of the sample were evaporated at 65 °C. We used stains-all to verify the digestion of chondroitin sulfate and hyaluronic acid. We digested 5 μg of chondroitin sulfate in 10 μL PBS for 1 h or 2 h with 10 mU or 1 U ChABC under the previously described conditions. We digested 10 μg of high molecular weight hyaluronic acid as an additional control with 0.3 U, 1 U, 2 U, or 4 U hyaluronidase for 1 h under the previously described conditions.
Glycosaminoglycans isolated from cells and control samples were analyzed with a 10% polyacrylamide gel (Rotiphorese® Gel 40 (29:1), Carl Roth) that underwent a pre-run for 30 min at 100 V. We used 50% glycerol in 60 mmol L−1 tris (pH 8.7) as loading buffer. Upon sample application, the gel was run for 1 h at 100 V using 60 mmol L−1 tris (pH 8.7) as a buffer. The gel was stained with 3 mg stains-all dissolved in 3 mL chloroform (Merck) in 60 mL 50% ethanol. The staining was performed for 15 min at RT. To remove background staining, the gel was rinsed twice with dH2O, washed twice for 15 min with dH2O, and kept overnight in a dH2O bath.
4.5. Continuous magnetic particle spectroscopy
With magnetic particle spectroscopy (MPS), the interaction of magnetic nanoparticles with cells was measured. MPS is a fast and sensitive measurement technique applying an oscillatory magnetic field to magnetic nanoparticles at a standard frequency of 25 kHz and amplitudes up to 25 mT. MPS detects the non-linear magnetic response of the nanoparticle's magnetic moment. For the measurement, 1 × 106 cells in suspension were used. Before the measurement, the cells were centrifuged at 800g for 2 min. The pellet was washed once with 1 mL PBS and resuspended in 100 μL PBS. The interaction with the previously described purified glycosaminoglycans (GAGs) was investigated using 100 μL of GAGs (1 mg mL−1) dissolved in dH2O. For each measurement, 100 μL of sample was transferred into an NMR glass tube (Bruker 180 NMS PC). MPS measurements were performed at 37 °C with an excitation field amplitude of 12 mT (MPS-3, Bruker). We measured at 12 mT to achieve a balance between a strong magnetic response and a low potential magnetic influence to prevent chain formation of nanoparticles. Therefore, the field was reduced to 0.05 mT between the measurements. Immediately after starting the measurement, 100 μL of 1 mmol L−1 SynC in PBS or dH2O was added to the sample (final iron concentration cFe = 0.5 mmol L−1). The MPS spectrum was measured for 667 s. We measured continuously after 1 s, 5 s, 11 s, 27 s, 44 s, 80 s, 147 s, 254 s, 441 s, and 667 s. The data were analyzed and visualized using Microsoft Excel (Microsoft) and Prism version 8 (GraphPad).4.6. Click-chemistry-based labeling of THP-1 cells
Based on copper-free click chemistry, we stained THP-1 cells fluorescently.50 We added 50 μM azido-modified sugars to 1 × 106 THP-1 cells for 72 h under the previously described cell culture conditions. We used Click-iT™ GalNAz (N-azidoacetylgalactosamine tetraacetylated) (Thermo Fisher Scientific) or Click-iT™ GlcNAz (N-azidoacetylglucosamine tetraacetylated) (Thermo Fisher Scientific). After three days, the cells were centrifuged at 800g for 2 min. The pellet was washed twice with cRPMI medium. Fluorescent staining was performed with 3 μM Click-iT™ sDIBO-Alexa Fluor 488 (Thermo Fisher Scientific) for 1 h at 37 °C with 5% CO2. The cells were washed four times with cRPMI medium and centrifuged for 3 min at 800g between washing steps. For imaging, the cells were resuspended in PBS and transferred into a μ-slide 4-well ibi-Treat (ibidi). Microscopy (BZX-810, Keyence) was performed at 60× magnification. To investigate the interaction of cells with cFe = 0.5 mmol L−1 SynC, we added the nanoparticles and repeated imaging 5 or 20 min after addition. We imaged a control for the short and long incubation with SynC to control for photobleaching and turnover of fluorescently labeled structures. Imaging was performed with a confocal laser scanning microscope (Spinning Disk Confocal CSU-X, Nikon) with a 60× water immersion objective (Plan Apo IR 60× WI DIC N2, numerical aperture of 1.27, Nikon). The intracellular fluorescence was determined with ImageJ according to Ansari et al.66 We set a threshold to the green channel and measured the cytosolic area and integrated density of the cytosol for each cell. We calculated the corrected total cytoplasmic fluorescence (CTCF) for each cell using the formula: CTCF = integrated density cytoplasm − (area cytoplasm × mean fluorescence of background readings). We measured three background measurements for each image. The calculated CTCF was divided by the cytosolic area. The data is shown as mean ± SD. With Prism version 8 (GraphPad) we performed a two-tailed unpaired Student's t-test to compare the control cells with cells incubated with SynC. Statistical significance was defined as p < 0.05.4.7. Quantification of SynC uptake
We quantified the cellular uptake of SynC with Prussian blue iron staining. We used 1 × 106 suspension cells. Adherent cells were seeded at a density of 1 × 104 per well for 24 h in μ-slides with multi-cell μ-pattern (ibidi). Medium changes were performed by tilting over the slide. Prior to SynC incubation, cells were washed once with PBS. SynC incubation was performed for 10 min, 60 min, or 3 h in PBS. Afterwards, suspension cells were washed once with PBS, and adherent cells three times. We fixed the cells with 0.05% glutaraldehyde (Merck) for 45 min at RT. Suspension cells were transferred into an uncoated μ-slide 4-well (ibidi). Subsequently, 2% potassium ferricyanide (Sigma-Aldrich) and 1% HCl (Merck) were added for 30 min (suspension) or 20 min (adherent) at RT. Imaging was performed in dH2O with a light microscope (BZX-810 microscope, Keyence) at 100× magnification. To minimize the influence of the time related increase in staining intensity we imaged the cells quickly and alternated between the two groups during image acquisition. SynC uptake was quantified as Prussian blue stained area per cell upon background reduction and threshold setting with ImageJ. We excluded dead cells from subsequent analysis. Cells with a destroyed membrane did not show the characteristic distribution pattern of endosomal iron accumulation but rather a homogeneous strong staining. The data is shown as mean ± SD. Prism version 8 (GraphPad) was used for statistical analysis and visualization. We performed a two-tailed unpaired Student's t-test to determine the differences in SynC uptake in treated vs. untreated THP-1 cells and between CHO-K1 cells and psgA-745 cells. Statistical significance was defined as p < 0.05.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Study conception and design, AL, LK; methodology, AL, NL, FW, LK, AR, ST, AW, PT; data collection, LK, AR, AW, ST, AS; formal analysis, LK, AR; interpretation of results, AL, LK; writing—original draft preparation, LK; writing—review and editing, LK, AL, NL, FW, ST, AR, PT; visualization, LK; supervision AL; funding acquisition, AL, FW, NL. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Dr Sara Timm and Petra Schrade at the Core Facility for Electron Microscopy of the Charité for support in image acquisition. We thank Katja Dörfel and John Horn for freeze-substitution, embedding, and ultrathin sections. We thank the Advanced Medical BIOimaging Core Facility (AMBIO) at the Charité Universitätsmedizin Berlin for their help in confocal image acquisition. This work was funded by the German Research Foundation in the scope of the collaborative research center “Matrix in Vision” (SFB 1340/2, project number 372486779).References
- E. Kianfar, J. Supercond. Novel Magn., 2021, 34, 1709–1735 CrossRef CAS.
- A. Senyei, K. Widder and G. Czerlinski, J. Appl. Phys., 1978, 49, 3578–3583 CrossRef CAS.
- J. Wells, S. Twamley, A. Sekar, A. Ludwig, H. Paysen, O. Kosch and F. Wiekhorst, Nanoscale, 2020, 12, 18342–18355 RSC.
- S. Caspani, R. Magalhães, J. P. Araújo and C. T. Sousa, Materials, 2020, 13, 2586 CrossRef CAS PubMed.
- X. Han, Y. Li, W. Liu, X. Chen, Z. Song, X. Wang, Y. Deng, X. Tang and Z. Jiang, Diagnostics, 2020, 10, 800 CrossRef PubMed.
- N. Shalaby, J. J. Kelly, O. C. Sehl, J. J. Gevaert, M. S. Fox, Q. Qi, P. J. Foster, J. D. Thiessen, J. W. Hicks and T. J. Scholl, Nanoscale, 2023, 15, 3408–3418 RSC.
- H. Paysen, N. Loewa, A. Stach, J. Wells, O. Kosch, S. Twamley, M. R. Makowski, T. Schaeffter, A. Ludwig and F. Wiekhorst, Sci. Rep., 2020, 10, 1922 CrossRef CAS PubMed.
- B. Gleich and J. Weizenecker, Nature, 2005, 435, 1214–1217 CrossRef CAS PubMed.
- Y. Du, P. T. Lai, C. H. Leung and P. W. Pong, Int. J. Mol. Sci., 2013, 14, 18682–18710 CrossRef CAS PubMed.
- H. Kratz, M. Taupitz, A. Ariza de Schellenberger, O. Kosch, D. Eberbeck, S. Wagner, L. Trahms, B. Hamm and J. Schnorr, PLoS One, 2018, 13, e0190214 CrossRef PubMed.
- A. Remmo, N. Löwa, O. Kosch, D. Eberbeck, A. Ludwig, L. Kampen, C. Grüttner and F. Wiekhorst, Cells, 2022, 11, 2892 CrossRef CAS PubMed.
- A. Remmo, O. Kosch, L. Kampen, A. Ludwig, F. Wiekhorst and N. Löwa, Sci. Rep., 2024, 14, 4253 CrossRef CAS PubMed.
- D. L. Thorek, A. K. Chen, J. Czupryna and A. Tsourkas, Ann. Biomed. Eng., 2006, 34, 23–38 CrossRef PubMed.
- S. Haque, C. W. Pouton, M. P. McIntosh, D. B. Ascher, D. W. Keizer, M. R. Whittaker and L. M. Kaminskas, Nanomed. Nanotechnol. Biol. Med., 2020, 30, 102291 CrossRef CAS PubMed.
- P. Sabourian, G. Yazdani, S. S. Ashraf, M. Frounchi, S. Mashayekhan, S. Kiani and A. Kakkar, Int. J. Mol. Sci., 2020, 21, 8019 CrossRef CAS PubMed.
- J. Yue, T. J. Feliciano, W. Li, A. Lee and T. W. Odom, Bioconjugate Chem., 2017, 28, 1791–1800 CrossRef CAS PubMed.
- B. D. Chithrani, A. A. Ghazani and W. C. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- F. Lu, S. H. Wu, Y. Hung and C. Y. Mou, Small, 2009, 5, 1408–1413 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- A. Villanueva, M. Canete, A. G. Roca, M. Calero, S. Veintemillas-Verdaguer, C. J. Serna, M. del Puerto Morales and R. Miranda, Nanotechnology, 2009, 20, 115103 CrossRef PubMed.
- P. Rees, J. W. Wills, M. R. Brown, C. M. Barnes and H. D. Summers, Nat. Commun., 2019, 10, 2341 CrossRef PubMed.
- J. W. Wills, H. D. Summers, N. Hondow, A. Sooresh, K. E. Meissner, P. A. White, P. Rees, A. Brown and S. H. Doak, ACS Nano, 2017, 11, 11986–12000 CrossRef CAS PubMed.
- Y. Song, M. Luo and L. L. Dai, Langmuir, 2010, 26, 5–9 CrossRef CAS PubMed.
- Y. Portilla, S. Mellid, A. Paradela, A. Ramos-Fernandez, N. Daviu, L. Sanz-Ortega, S. Perez-Yague, M. P. Morales and D. F. Barber, ACS Appl. Mater. Interfaces, 2021, 13, 7924–7944 CrossRef CAS PubMed.
- O. Lieleg, R. M. Baumgärtel and A. R. Bausch, Biophys. J., 2009, 97, 1569–1577 CrossRef CAS PubMed.
- M. J. Cheng, R. Kumar, S. Sridhar, T. J. Webster and E. E. Ebong, Int. J. Nanomed., 2016, 3305–3315 CAS.
- P. H. Olivieri, M. B. Jesus, H. B. Nader, G. Z. Justo and A. A. Sousa, Nanoscale, 2022, 14, 7350–7363 RSC.
- Q. Li, Y. Xie, M. Wong, M. Barboza and C. B. Lebrilla, Nat. Protoc., 2020, 1–37 Search PubMed.
- J. Tarbell and L. Cancel, J. Intern. Med., 2016, 280, 97–113 CrossRef CAS PubMed.
- A. Zappe, R. L. Miller, W. B. Struwe and K. Pagel, Mass Spectrom. Rev., 2022, 41, 1040–1071 CrossRef CAS PubMed.
- J. Fernández-Sarmiento, L. M. Salazar-Peláez and J. A. Carcillo, Pediatr. Crit. Care Med., 2020, 21, e291 CrossRef PubMed.
- A. Ueda, M. Shimomura, M. Ikeda, R. Yamaguchi and K. Tanishita, Am. J. Physiol. Heart Circ. Physiol., 2004, 287, H2287–H2294 CrossRef CAS PubMed.
- B. Peter, N. Kanyo, K. D. Kovacs, V. Kovács, I. Szekacs, B. Pécz, K. Molnár, H. Nakanishi, I. Lagzi and R. Horvath, ACS Appl. Bio Mater., 2022, 6, 64–73 CrossRef PubMed.
- L. Möckl, S. Hirn, A. A. Torrano, B. Uhl, C. Bräuchle and F. Krombach, Nanomedicine, 2017, 12, 207–217 CrossRef PubMed.
- M. Malatesta, Int. J. Mol. Sci., 2021, 22, 12789 CrossRef CAS PubMed.
- C. Wilhelm, C. Billotey, J. Roger, J. Pons, J.-C. Bacri and F. Gazeau, Biomaterials, 2003, 24, 1001–1011 CrossRef CAS PubMed.
- E. C. Cho, J. Xie, P. A. Wurm and Y. Xia, Nano Lett., 2009, 9, 1080–1084 CrossRef CAS PubMed.
- N. Ghinea and N. Simionescu, J. Cell Biol., 1985, 100, 606–612 CrossRef CAS PubMed.
- R. R. Kay, Cells Dev., 2021, 168, 203713 CrossRef CAS PubMed.
- Y.-X. Li and H.-B. Pang, J. Controlled Release, 2021, 329, 1222–1230 CrossRef CAS PubMed.
- S. Patel, J. Kim, M. Herrera, A. Mukherjee, A. V. Kabanov and G. Sahay, Adv. Drug Delivery Rev., 2019, 144, 90–111 CrossRef CAS PubMed.
- W. C. Poller, N. Löwa, M. Schleicher, A. Münster-Wandowski, M. Taupitz, V. Stangl, A. Ludwig and F. Wiekhorst, Sci. Rep., 2020, 10, 3591 CrossRef CAS PubMed.
- K. Wu, D. Su, R. Saha, J. Liu, V. K. Chugh and J.-P. Wang, ACS Appl. Nano Mater., 2020, 3, 4972–4989 CrossRef CAS.
- A. M. Rauwerdink and J. B. Weaver, Med. Phys., 2010, 37, 2587–2592 CrossRef PubMed.
- D. Berkov and N. Gorn, J. Phys.: Condens. Matter, 2001, 13, 9369 CrossRef CAS.
- G. Barrera, P. Allia and P. Tiberto, Nanoscale, 2021, 13, 4103–4121 RSC.
- L. Lartigue, P. Hugounenq, D. Alloyeau, S. P. Clarke, M. Lévy, J.-C. Bacri, R. Bazzi, D. F. Brougham, C. Wilhelm and F. Gazeau, ACS Nano, 2012, 6, 10935–10949 CrossRef CAS PubMed.
- J. G. Ovejero, D. Cabrera, J. Carrey, T. Valdivielso, G. Salas and F. J. Teran, Phys. Chem. Chem. Phys., 2016, 18, 10954–10963 RSC.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851 RSC.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS PubMed.
- N. Al-Nakouzi, C. K. Wang, H. Z. Oo, I. Nelepcu, N. Lallous, C. B. Spliid, N. Khazamipour, J. Lo, S. Truong and C. Collins, Nat. Commun., 2022, 13, 4760 CrossRef CAS PubMed.
- H. C. Hang, C. Yu, D. L. Kato and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14846–14851 CrossRef CAS PubMed.
- J. C. Brazil and C. A. Parkos, Mucosal Immunol., 2022, 15, 211–222 CrossRef CAS PubMed.
- C. M. McLeod and R. L. Mauck, Sci. Rep., 2016, 6, 38852 CrossRef CAS PubMed.
- M. Ambrosius, K. Kleesiek and C. Götting, Cell Biol. Int., 2009, 33, 848–852 CrossRef CAS PubMed.
- E. Makatsori, N. Karamanos, N. Papadogiannakis, A. Hjerpe, E. Anastassiou and T. Tsegenidis, Biomed. Chromatogr., 2001, 15, 413–417 CrossRef CAS PubMed.
- E. Makatsori, F. N. Lamari, A. D. Theocharis, S. Anagnostides, A. Hjerpe, T. Tsegenidis and N. K. Karamanos, Anticancer Res., 2003, 23, 3303–3309 CAS.
- A. Ludwig, W. C. Poller, K. Westphal, S. Minkwitz, G. Lättig-Tünnemann, S. Metzkow, K. Stangl, G. Baumann, M. Taupitz and S. Wagner, Basic Res. Cardiol., 2013, 108, 1–13 CrossRef PubMed.
- D. Berndt, J. M. Millward, J. Schnorr, M. Taupitz, V. Stangl, F. Paul, S. Wagner, J. T. Wuerfel, I. Sack and A. Ludwig, Nanomed. Nanotechnol. Biol. Med., 2017, 13, 1411–1421 CrossRef CAS PubMed.
- S. Zhang, Y. Moustafa and Q. Huo, ACS Appl. Mater. Interfaces, 2014, 6, 21184–21192 CrossRef CAS PubMed.
- J. D. Esko, T. E. Stewart and W. H. Taylor, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3197–3201 CrossRef CAS PubMed.
- M. B. Piva, E. R. Suarez, C. M. Melo, R. P. Cavalheiro, H. B. Nader and M. A. Pinhal, Glycobiology, 2015, 25, 976–983 CrossRef CAS PubMed.
- D. C. Briggs and E. Hohenester, Structure, 2018, 26, 801–809 CrossRef CAS PubMed.
- E. Wesén, A. Gallud, A. Paul, D. J. Lindberg, P. Malmberg and E. K. Esbjörner, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 2204–2214 CrossRef.
- J. D. Clogston and A. K. Patri, Characterization of Nanoparticles Intended for Drug Delivery, 2011, pp. 63–70 Search PubMed.
- N. Ansari, S. Müller, E. H. Stelzer and F. Pampaloni, in Methods in Cell Biology, Elsevier, 2013, vol. 113, pp. 295–309 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00277f |
| This journal is © The Royal Society of Chemistry 2024 |