
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper supported modified magnetic carrageenan as a bio-based catalyst for the synthesis of novel scaffolds bearing the 1,2,3-triazole unit through the click reaction†
Nima
Khaleghi
 a,
Maryam
Esmkhani
b,
Milad
Noori
b,
Navid
Dastyafteh
b,
Minoo Khalili
Ghomi
a,
Mohammad
Mahdavi
a,
Mohammad Hosein
Sayahi
*c and
Shahrzad
Javanshir
*b
a,
Maryam
Esmkhani
b,
Milad
Noori
b,
Navid
Dastyafteh
b,
Minoo Khalili
Ghomi
a,
Mohammad
Mahdavi
a,
Mohammad Hosein
Sayahi
*c and
Shahrzad
Javanshir
*b
aEndocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran
bPharmaceutical and Heterocyclic Compounds Research Laboratory, Department of Chemistry, Iran University of Science and Technology, 16846-13114, Tehran, Iran. E-mail: shjavan@iust.ac.ir
cDepartment of Chemistry, Payame Noor University, Tehran, Iran. E-mail: sayahymh@pnu.ac.ir
First published on 19th March 2024
Abstract
The ongoing work delineates the design of a novel library of 1,2,3-triazole-attached phenylacetamides through molecular hybridization of propargyl and phenylacetamide derivatives. Copper-supported modified magnetic carrageenan serves as a green heterogeneous catalyst, ensuring high yield, efficient reaction times, high atom economy, utilization of an environmentally friendly catalyst from a natural source, and a straightforward workup procedure. The successful synthesis of the catalyst is confirmed and evaluated using various analytical techniques, while the synthetic compounds are characterized through 1H NMR and 13C NMR.
Introduction
The synthesis of new scaffolds and drugs with effective functions for the treatment of diseases is an important topic for researchers. Among these scaffolds with diverse biological activities, N-substituted heterocycles, particularly triazoles, exhibit a broad spectrum of applications in medicinal chemistry.1Triazoles as important compounds with anti-inflammatory,2 antimicrobial,3 antimalarial,4 antiviral,5 and anticancer6 activities can be synthesized through the click reaction between azides and alkynes catalyzed by copper. There have been several reports about copper catalysts used in the click reaction of alkynes and azides over the last few years. However, most of these studies suffer from homogeneous catalyst problems. These problems can include the separation or work-up problems or the production of by-products.
To overcome these, the immobilization of copper on supports such as silica,7 magnetite,8,9 and polymers10,11 can be a good solution. Renewable and biodegradable organic polymers, biopolymers, and especially polysaccharides, have found multiple applications in various industries, such as pharmaceuticals,12 biomedical products,13 cosmetics14,15 and food industries.16,17 Among the mentioned supports, polysaccharides, due to their unique structures and functional groups are the most favorable and abundant supports for metal immobilization.
Incorporating the “green matrix” into our daily lives requires a shift in mindset and behavior. This includes reducing waste, conserving energy, using renewable resources, and minimizing the carbon footprint of our activities. By adopting the “green matrix”, individuals and organizations can help mitigate the negative impact of human activities on the environment.18
The scientific community has been putting a lot of effort into chemically modifying polysaccharides and utilizing them in catalytic systems for environmentally friendly processes and technologies. These polymers have great features such as renewability, biodegradability, non-toxicity, and more, making them widely used in various industries. Due to their unique structures and functional groups, they make for a decent support system for immobilizing metals. These polymers due to their great features such as renewability,19 biodegradability,20 non-toxicity,21etc. are widely used in various industries, but their unique structures and functional groups make them a good support for immobilizing metals.
Carrageenan (CG), a marine polysaccharide with the sulfate group, has mainly been used as a gelling agent in the food industry and a catalyst in chemical processes. It can be extracted from red seaweeds and has three main types based on its sulfate group: iota-carrageenan, kappa-carrageenan and lambda-carrageenan.22 One of the key advantages of CG as a catalyst is its ability to form stable complexes with metal ions, which can enhance the efficiency of catalytic reactions. Additionally, carrageenan is biodegradable and non-toxic, making it an environmentally friendly alternative to traditional catalysts. Its versatility and effectiveness in these reactions make it a valuable tool for researchers and industries looking to develop more sustainable and efficient chemical processes.23–25
Triazoles as the basic building blocks of many organic and inorganic molecules with antitubercular,26 antifungal,27,28 anticancer,29 antibacterial,3 anti-HIV,30 anti-inflammatory,31 and antiallergic32 activities can also be hybridized with other scaffolds to improve their activity. There are several reports on the hybridization of triazoles with different cores such as phthalimides.33–35 Phthalimides, the pharmaceutically active compounds, are also known for their potent biological activities,36 such as anti-HIV,37 anticancer,38 anti-inflammatory,39 hypoglycemic,33 antimalarial,40 histone deacetylase inhibitory,41 cholinesterase inhibitory,42 and COX inhibitory.43 These compounds are also used as starting substances for the synthesis of a wide range of compounds, such as alkaloids, polymers, pesticides, and copolymers.44
In the light of these studies and in continuation of our work on the fabrication of catalysts,45–47 we report the synthesis of a new efficient magnetite-based catalytic system, with its application in the synthesis of new 1,2,3-triazole derivatives. The procedure uses a carrageenan-based magnetic catalyst functionalized with an organic linker. The reaction was performed under mild conditions and the catalyst was removed with an external magnet. The reaction yields were excellent, and the prepared catalyst had good efficiency.
Results and discussion
The synthesis pathway of Fe3O4@CG/CPTMS/OL-Cu(I) is illustrated in Fig. 1. Fe3O4@CG was prepared through a co-precipitation method by dissolving CG, and divalent and trivalent iron salts into distilled water, followed by precipitation with NaOH. Afterward, CPTMS was coated on the surface of Fe3O4@CG nanoparticles to obtain Fe3O4@CG/CPTMS nanoparticles. Subsequently, the organic linker was cross-linked on the surface of Fe3O4@CG/CPTMS. Fe3O4@CG/CPTMS/OL-Cu(I) was obtained by the addition of copper salt to as-prepared magnetic nanoparticles. | ||
| Fig. 1 Schematic presentation of the catalyst preparation. | ||
Characterization of Fe3O4@CG/CPTMS/OL-Cu(I)
The structure, morphology and magnetic properties of the prepared catalyst were characterized by different techniques. The FTIR spectra of Fe3O4@CG/CPTMS/OL-Cu, Fe3O4@CG/CPTMS/OL, and Fe3O4@CG are compared in Fig. 2. The FTIR spectrum of Fe3O4@CG indicates the characteristic band of Fe–O at 574 cm−1. The band at about 1627 cm−1 is assigned to the bending vibration of the hydroxyl group and the band at 1225 cm−1 is attributed to S–O asymmetric sulfate stretching. The absorption band of C–O–S was observed at 845 cm−1. Moreover, the two bands in the range of 1037–1040 cm−1 are assigned to the C–O and C–OH stretching, respectively. The FTIR spectrum of Fe3O4@CG/CPTMS/OL showed the characteristic peaks of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, N–H, and C–N vibrations at 1652, 1631, 1613, and 1521 cm−1 respectively. The peak broadens significantly in Fe3O4@CG/CPTMS/OL-Cu, and extends toward lower stretching frequencies at 3430 cm−1. On the other hand, the slight shift from 1687 cm−1 in Fe3O4@CG/CPTMS/OL to 1652 cm−1 in Fe3O4@CG/CPTMS/OL-Cu can be concluded to be due to the chelation of copper on the surface of the catalyst.
O, CC, N–H, and C–N vibrations at 1652, 1631, 1613, and 1521 cm−1 respectively. The peak broadens significantly in Fe3O4@CG/CPTMS/OL-Cu, and extends toward lower stretching frequencies at 3430 cm−1. On the other hand, the slight shift from 1687 cm−1 in Fe3O4@CG/CPTMS/OL to 1652 cm−1 in Fe3O4@CG/CPTMS/OL-Cu can be concluded to be due to the chelation of copper on the surface of the catalyst.
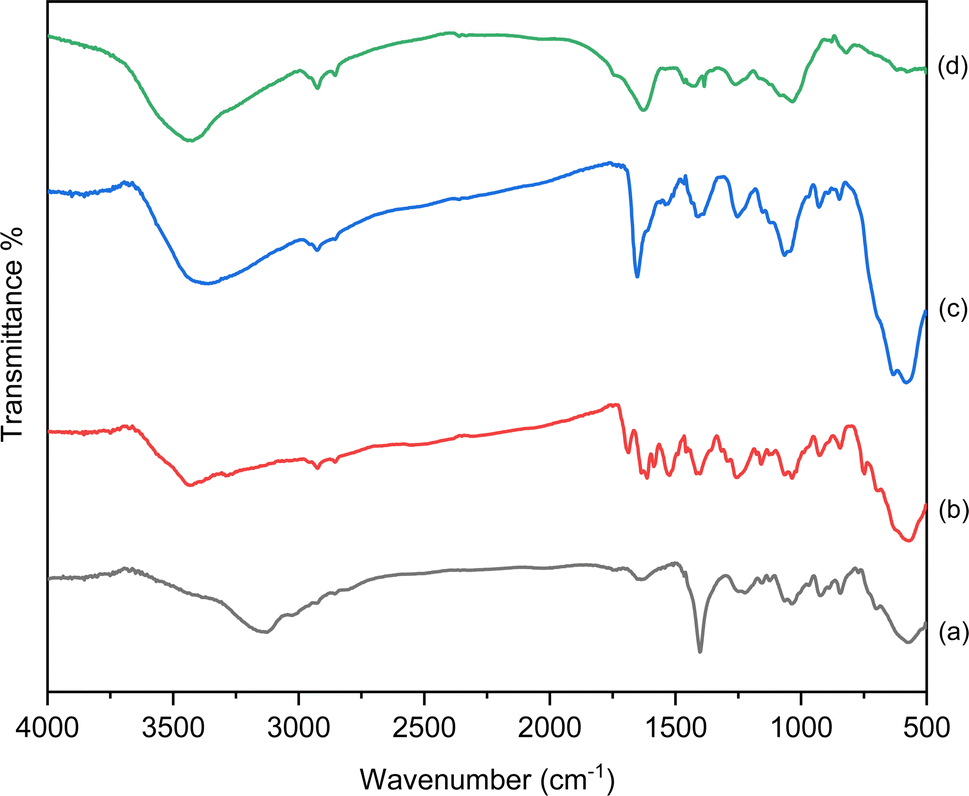 | ||
| Fig. 2 FTIR analysis of (a) Fe3O4@CG, (b) Fe3O4@CG/CPTMS/OL, (c) Fe3O4@CG/CPTMS/OL-Cu(I) and (d) reused Fe3O4@CG/CPTMS/OL-Cu(I). | ||
The morphology and the structure of the prepared catalyst were characterized by SEM analysis (Fig. 3). The almost uniform distribution of the catalyst is clearly observable. Moreover, the good monodispersity and the presence of Fe3O4 cores (the black centers) and SiO2 shells (the brightest areas) are clearly seen in TEM images.
 | ||
| Fig. 3 SEM and TEM images of the prepared catalyst. | ||
Furthermore, the elemental analysis (EDX) revealed the presence of carbon, oxygen, nitrogen, iron, Si and copper elements (ratios of 13.67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44.15:5.62:34.03:1.03 and 0.65 wt%, respectively). It confirms that the incorporation of expected elements into the structure of the prepared catalyst was achieved successfully (Fig. 4).
:44.15:5.62:34.03:1.03 and 0.65 wt%, respectively). It confirms that the incorporation of expected elements into the structure of the prepared catalyst was achieved successfully (Fig. 4).
 | ||
| Fig. 4 The elemental analysis (EDX) of Fe3O4@CG/CPTMS/OL-Cu. | ||
The magnetic feature of Fe3O4@CG/CPTMS/OL-Cu was investigated using VSM analysis and the magnetization cycle is plotted in Fig. 5. As can be noticed, magnetic hysteresis loop measurements indicated that the maximum saturation magnetization value of Fe3O4@CG/CPTMS/OL-Cu is 39 emus g−1.
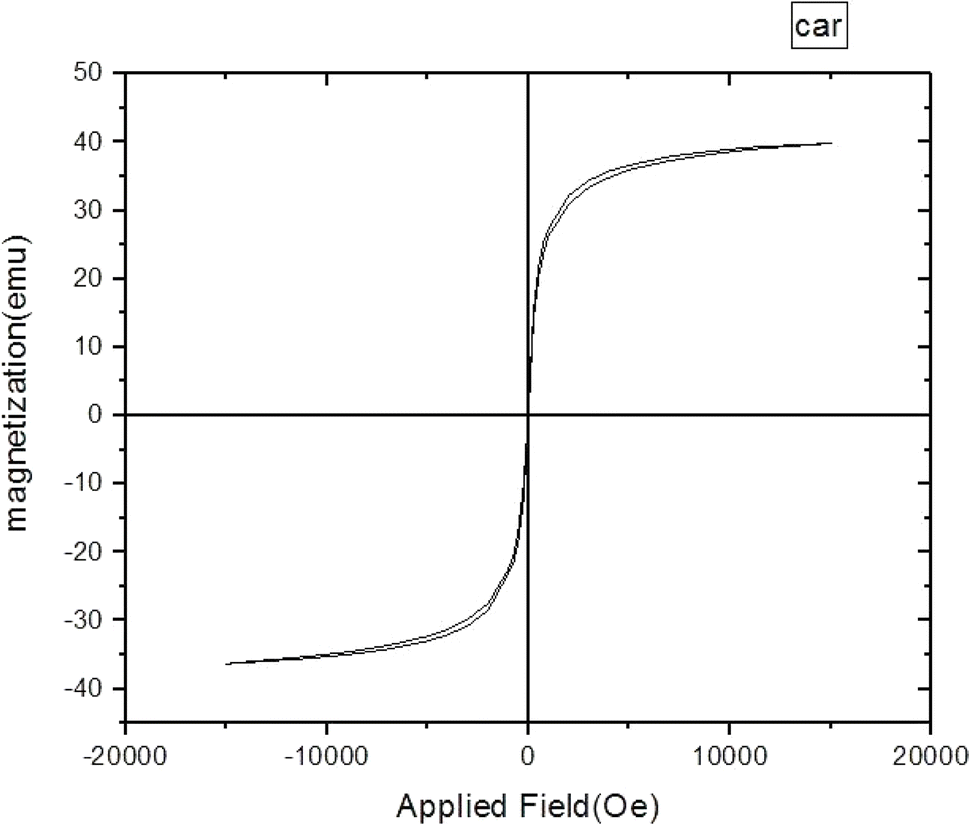 | ||
| Fig. 5 VSM analysis of the prepared Fe3O4@CG/CPTMS/OL-Cu. | ||
In Fig. 6, the XRD pattern of Fe3O4@CG/CPTMS/OL-Cu shows six characteristic diffraction peaks at 2θ = 30.199°, 35.660°, 43.375°, 53.672°, 57.257°, and 62.940° corresponding to the (220), (311), (400), (422), (511), and (440) reflection crystal planes of Fe3O4 respectively (JCPDS card no. 01-075-1609 and 00-002-0459). (The reference card numbers were collected from the X'pert HighScore Plus version 1.0d software developed by the PANalytical B.V.)
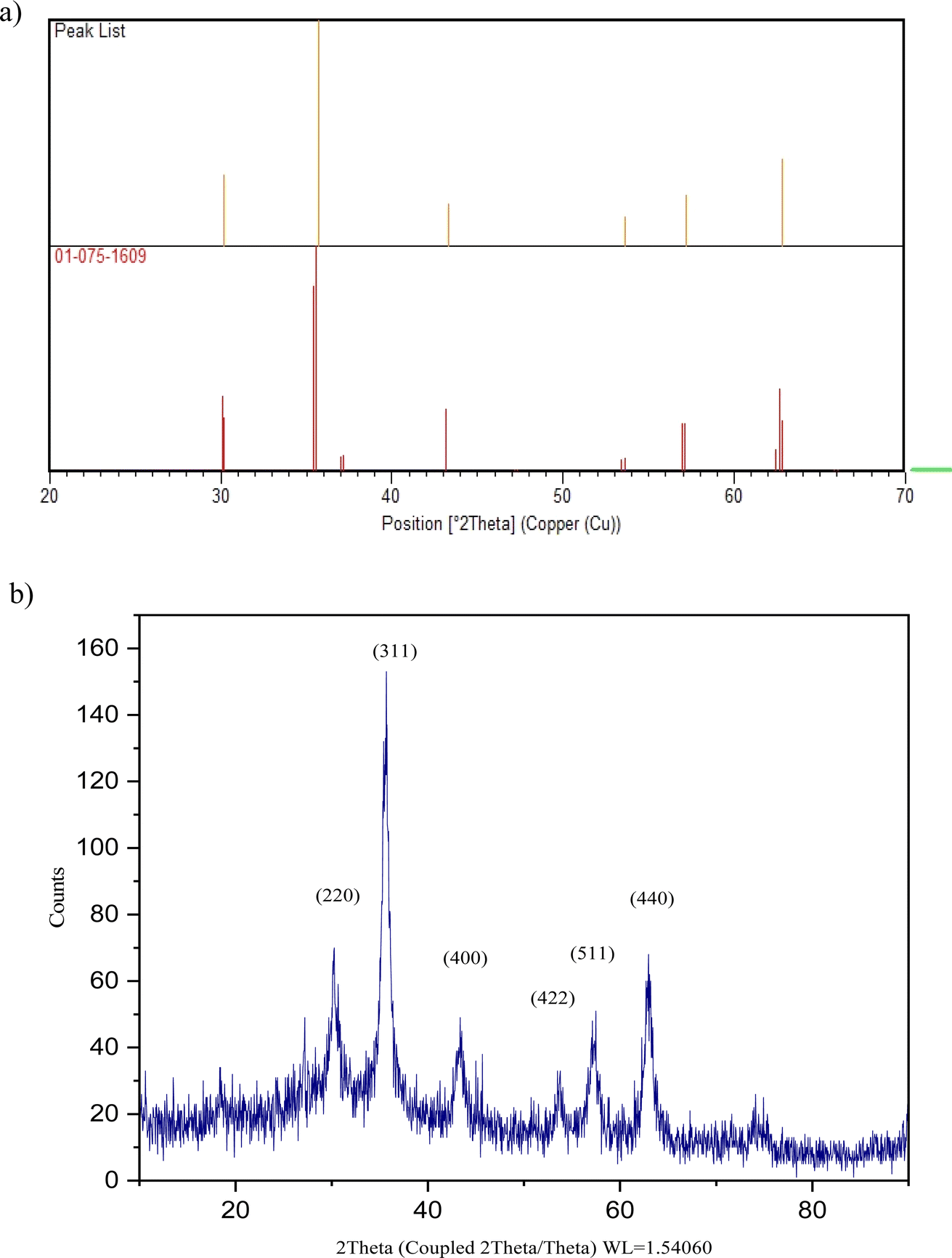 | ||
| Fig. 6 The (a) peak list, reference card number, and (b) XRD pattern of Fe3O4@CG/CPTMS/OL-Cu. | ||
Investigation the catalytic activities of Fe3O4@CG/CPTMS/OL-Cu for the synthesis of triazole derivatives 6a–e, 7a–e and 8a–f
The catalytic behavior of the prepared Fe3O4@CG/CPTMS/OL-Cu was investigated for the synthesis of 1,2,3-triazole derivatives through the reaction between phenylacetamide derivatives and N-propargyl phthalimide under different conditions. To find the optimal reaction conditions, different factors including solvent, catalyst loading, time and reaction temperature were scrutinized in a model reaction including propargyl-based compounds (1), (2) or (3), and 4-nitrophenyl acetamide (4a) to estimate the proper conditions (Fig. 7). Amid different solvents, water led to a shorter reaction time and gave a better yield (Table 1).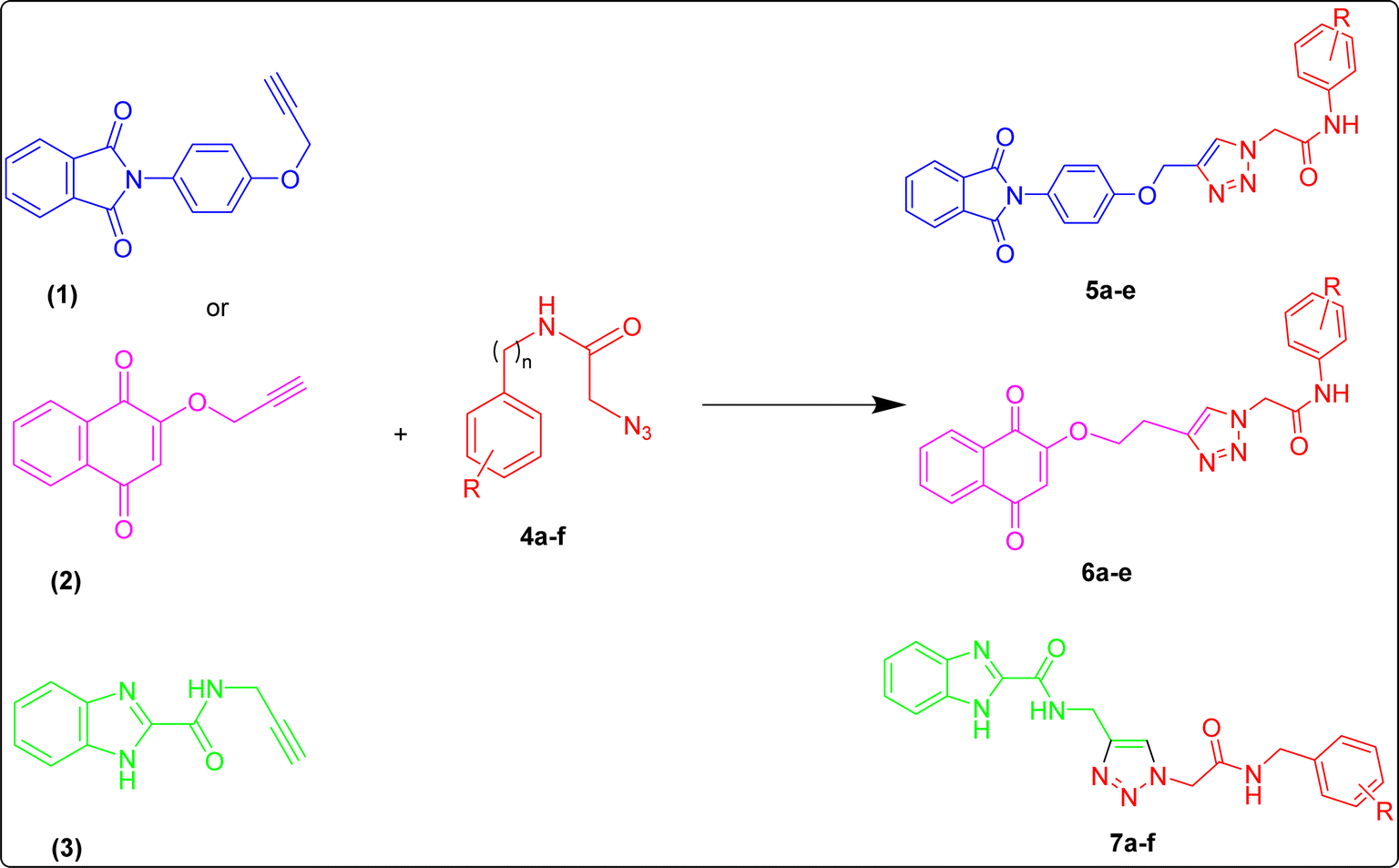 | ||
| Fig. 7 Schematic representation of Fe3O4@CG/CPTMS/OL-Cu and its catalytic activity in the one-pot synthesis of triazole derivatives through the click reaction. | ||
| Entry | Catalyst dosage (g) | Time (minute) | Solvent | Temperature | Yield (%) |
|---|---|---|---|---|---|
| 1 | — | 120 | H2O | r.t. | 0 |
| 2 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | H2O | r.t | 55 |
| 3 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | EtOH | r.t. | 48 |
| 4 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | H2O:EtOH (1:1) |
r.t. | 52 |
| 5 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | Toluene | r.t. | 17 |
| 6 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | CH3CN | r.t. | 41 |
| 7 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | CH3Cl | r.t. | 26 |
| 8 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | H2O:EtOH (1:2) |
r.t. | 50 |
| 9 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | Isopropanol | r.t. | 32 |
| 10 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | PEG-400 | r.t. | Trace |
| 11 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | 1,4-Dioxane | r.t. | 14 |
| 12 | Fe3O4@CG/CPTMS/OL-Cu (0.01 g) | 30 | CH3CN:H2O |
r.t. | 36 |
| 13 | Fe3O4@CG/CPTMS/OL-Cu (0.02 g) | 30 | H2O | r.t. | 59 |
| 14 | Fe3O4@CG/CPTMS/OL-Cu (0.03 g) | 30 | H2O | r.t. | 62 |
| 15 | Fe3O4@CG/CPTMS/OL-Cu (0.04 g) | 30 | H2O | r.t. | 65 |
| 16 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 30 | H2O | r.t. | 67 |
| 17 | Fe3O4@CG/CPTMS/OL-Cu (0.07 g) | 30 | H2O | r.t. | 67 |
| 18 | Fe3O4@CG/CPTMS/OL-Cu (0.1 g) | 30 | H2O | r.t. | 67 |
| 19 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 45 | H2O | r.t. | 71 |
| 20 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 60 | H2O | r.t. | 76 |
| 21 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 75 | H2O | r.t. | 76 |
| 22 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 90 | H2O | r.t. | 76 |
| 23 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 120 | H2O | r.t. | 76 |
| 24 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 30 | H2O | 50 | 74c |
| 25 | Fe 3 O 4 @CG/CPTMS/OL-Cu (0.05 g) | 30 | H 2 O | 75 | 82 d |
| 26 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 30 | H2O | 90 | 82e |
| 27 | Fe3O4@CG/CPTMS/OL-Cu (0.05 g) | 30 | H2O | 100 | 82f |
Moreover, the effects of temperature, type of catalyst, and the catalyst dosage were also evaluated and tabulated in Table 1. The highest yield of 82% was reached for 50 mg catalyst loading at 100 °C. On the other hand, with an amount of catalyst of 50 mg, lowering the temperature leads to a decrease in the reaction yield.
To generalize the optimum conditions, different triazole derivatives were prepared through a one-pot reaction of propargyl derivatives 1, 2 or 3 and phenylacetamide derivatives 4(a–f) in the presence of Fe3O4@CG/CPTMS/OL-Cu (Fig. 7). The results are summarized in Table 2. As expected, the presence of electron-donating groups on phenylacetamide can enhance the rate and yield of the reaction. In contrast, phenylacetamide with an electron-withdrawing group provided the product with a lower yield.
| Comp. |
|
R-N3 | Time (h) | Product structure |
|---|---|---|---|---|
| 5a | 1 |
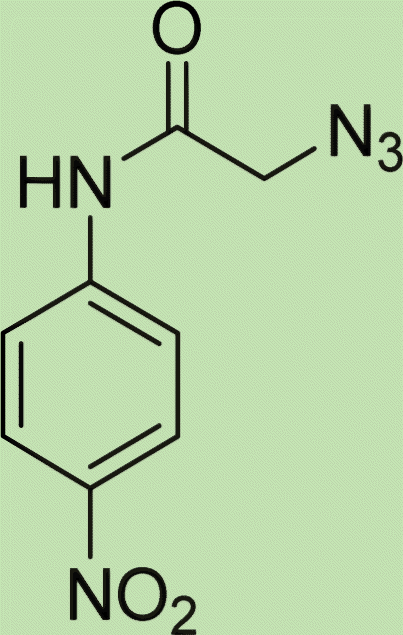
|
6 |

|
| 5b | 1 |

|
4 |
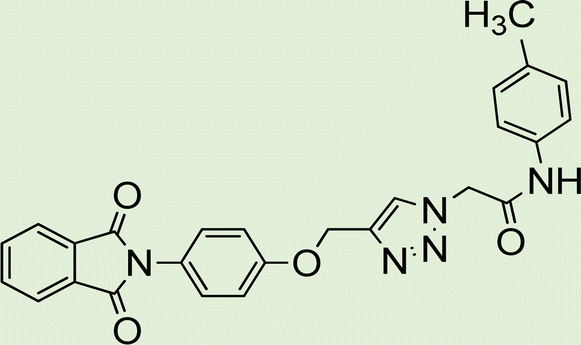
|
| 5c | 1 |
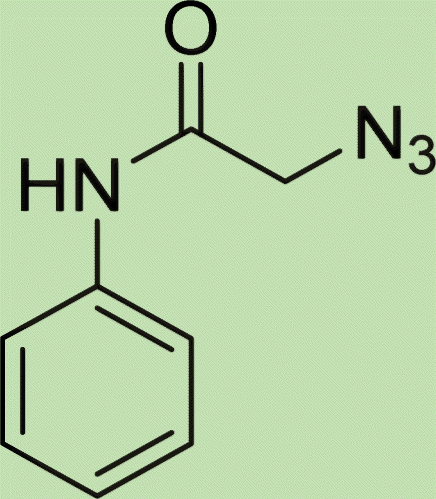
|
5 |
|
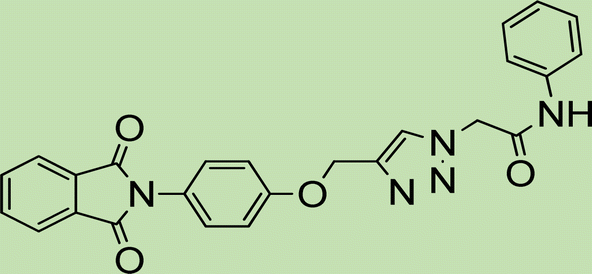
| 5d | 1 |
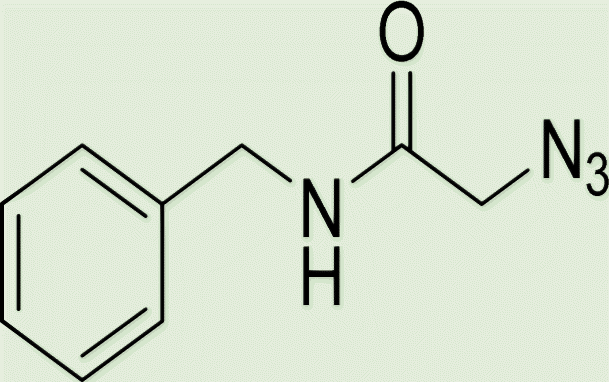
|
4 |
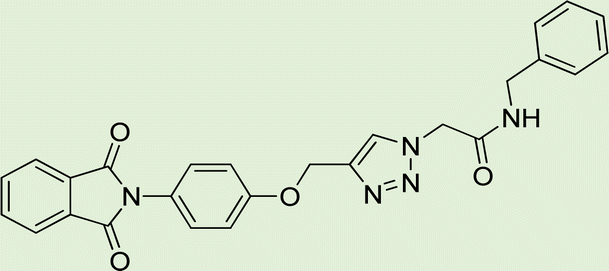
|
| 5e | 1 |
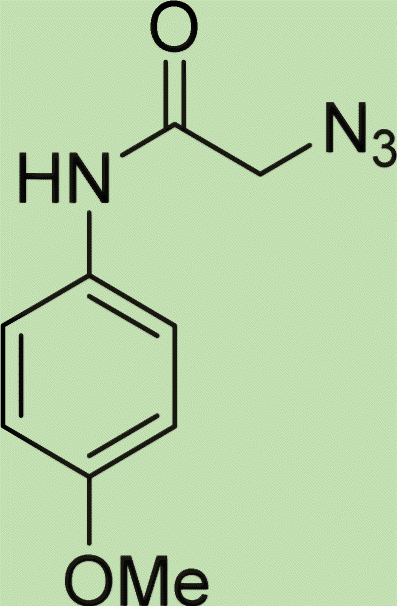
|
3 |
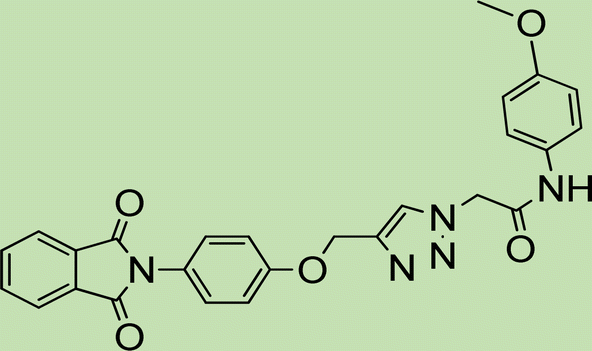
|
| 6a | 2 |
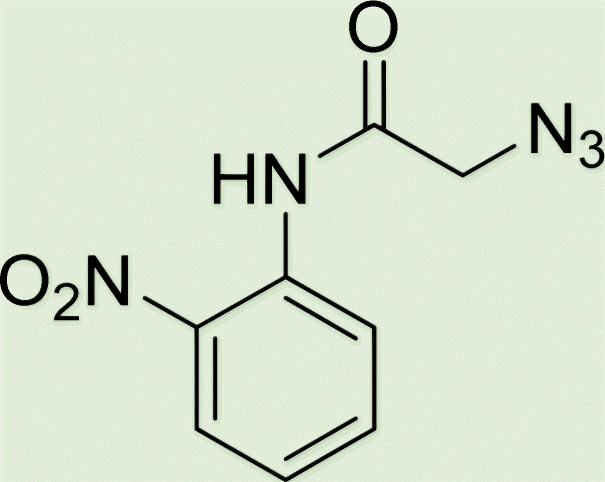
|
7.7 |
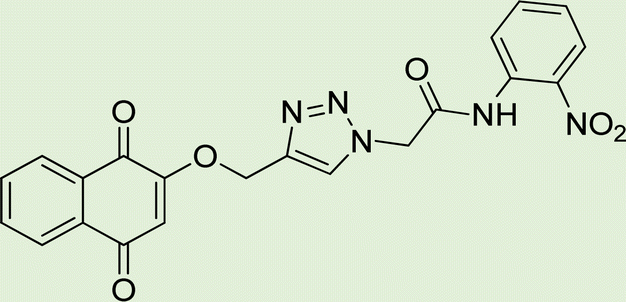
|
| 6b | 2 |
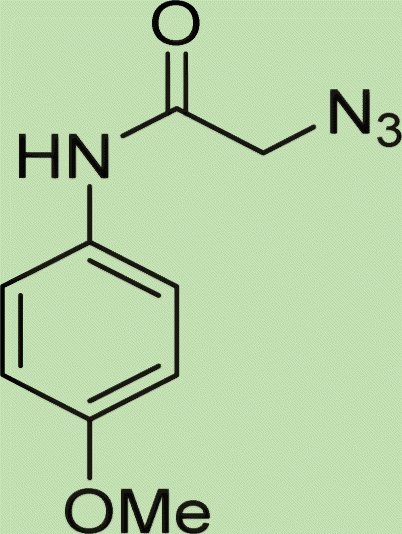
|
7.5 |
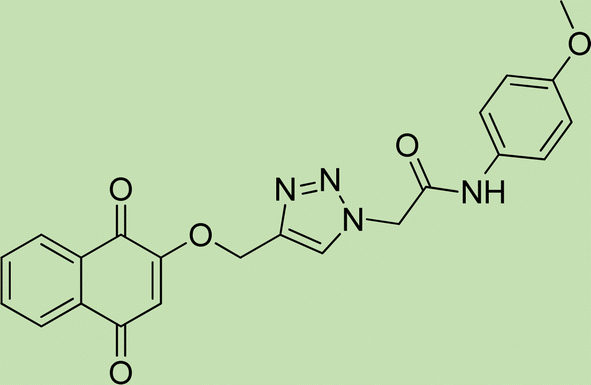
|
| 6c | 2 |

|
5 |

|
| 6d | 2 |

|
3.5 |
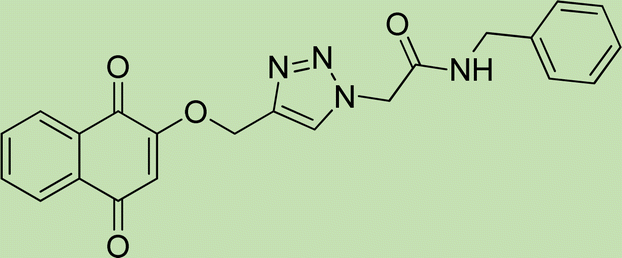
|
| 6e | 2 |

|
4 |
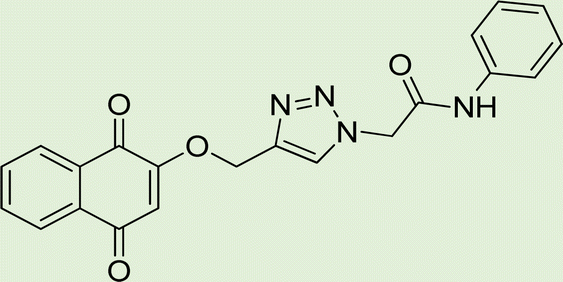
|
| 7a | 3 |
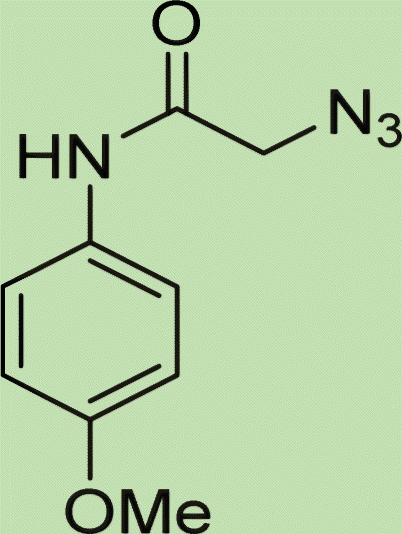
|
5 |

|
| 7b | 3 |

|
6 |
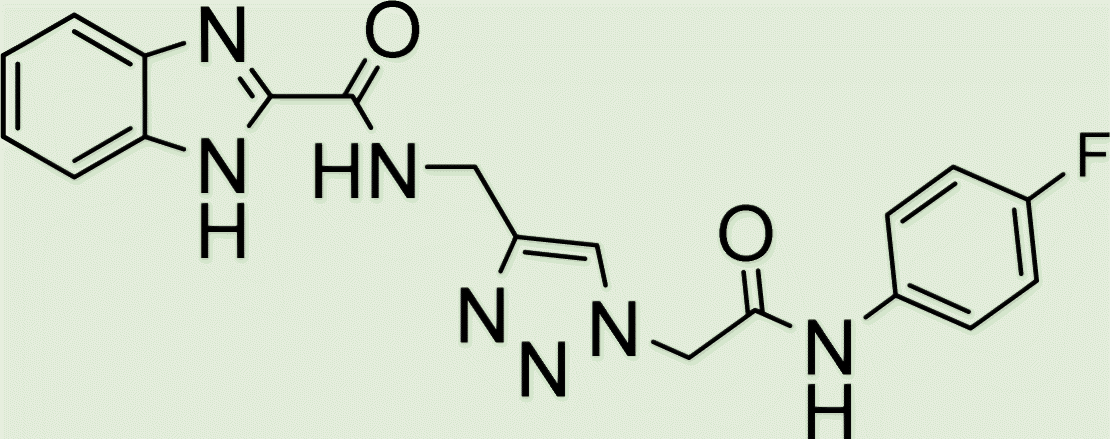
|
| 7c | 3 |
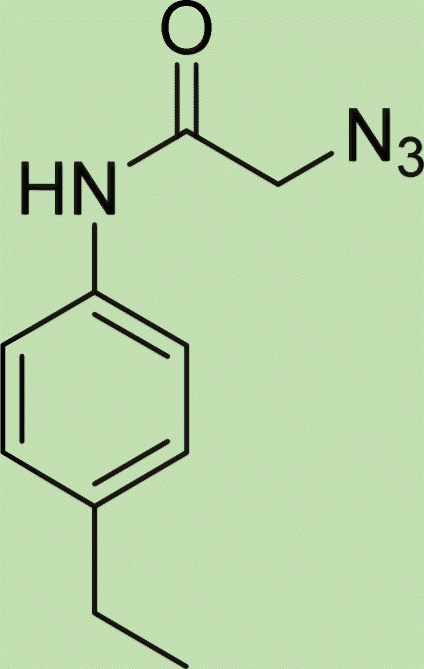
|
6.3 |
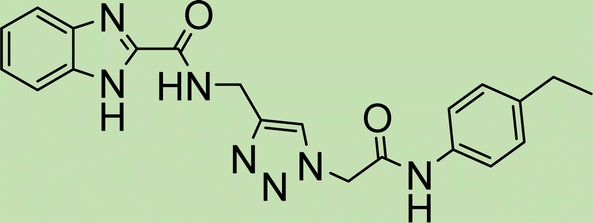
|
| 7d | 3 |
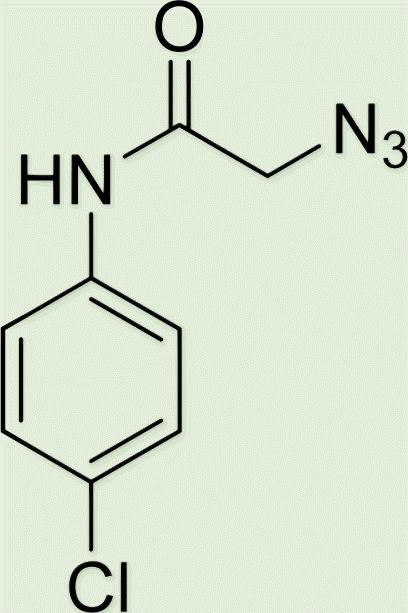
|
7 |

|
| 7e | 3 |
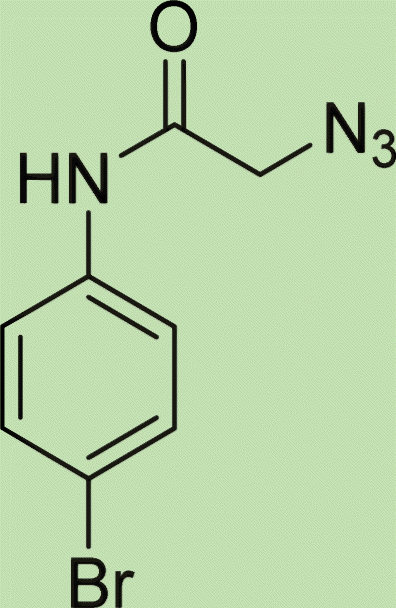
|
7.5 |
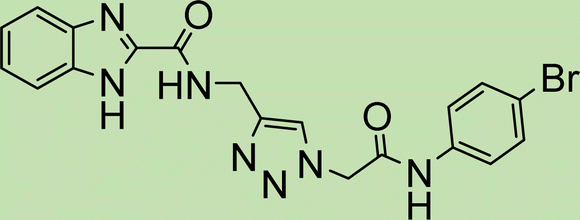
|
| 7f | 3 |
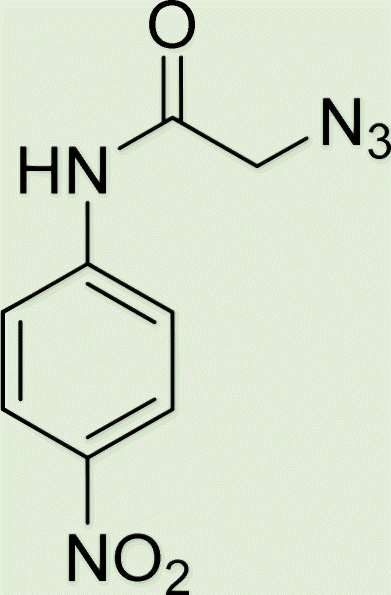
|
7 |
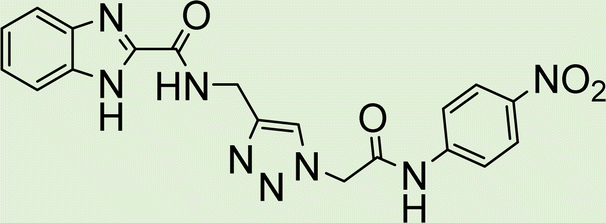
|

Catalyst recyclability
The efficiency of the catalyst's heterogeneity was evaluated through a recyclability test. The nanocatalyst was applied in a model reaction and was collected using an external magnetic field. It was then washed with acetone, ethanol, and water. The dried nanocatalyst was weighed and reused in the model reaction. Fig. 8 shows that the prepared magnetic nanocatalyst can be used up to five times without significant reduction in its catalytic efficiency. This was further confirmed through FTIR analysis shown in Fig. 2d, indicating that the catalyst remained unchanged after several uses.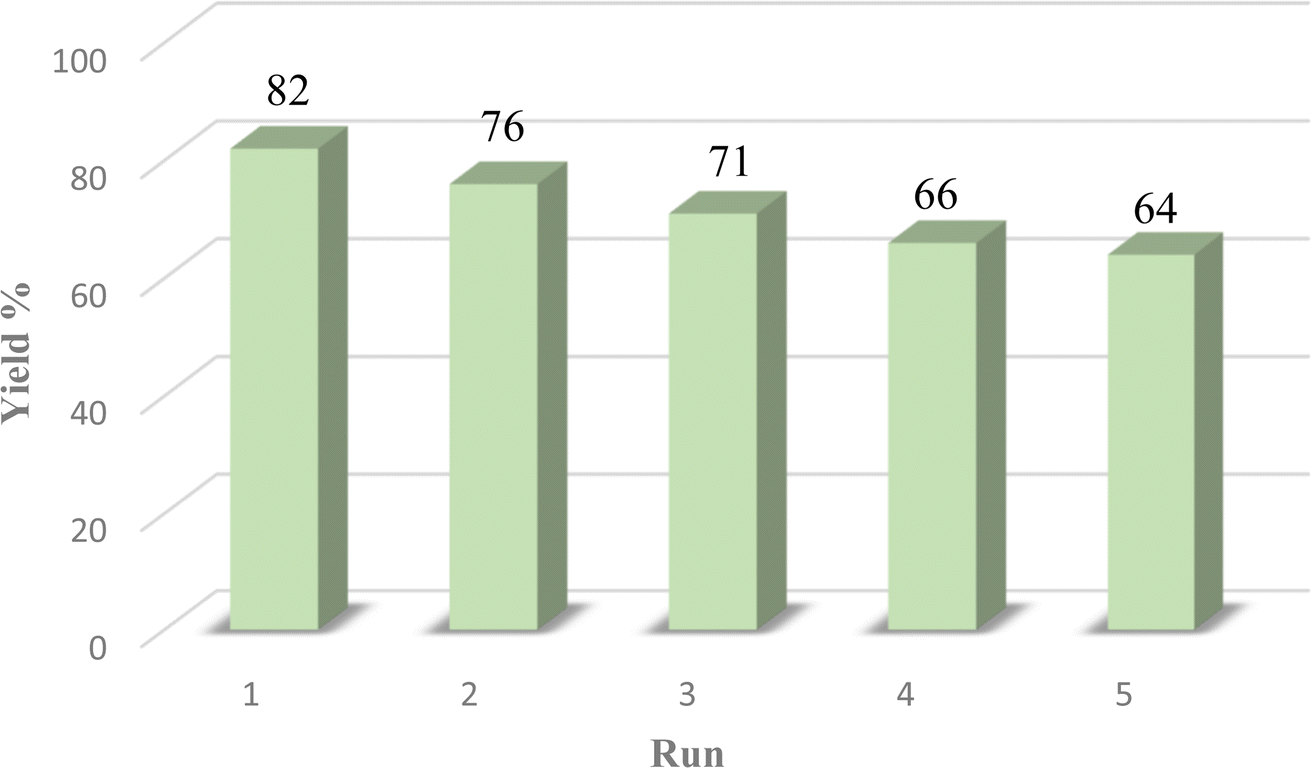 | ||
| Fig. 8 Recyclability of the Fe3O4@CG/CPTMS/OL-Cu catalyst. | ||
To demonstrate the efficacy of the prepared Fe3O4@CG/CPTMS/OL-Cu catalyst, the catalytic activity in the preparation of 1,2,3-triazole derivatives was compared with the previous reports and the results are tabulated in Table 3. Considering the results, the present catalyst has several advantages with respect to reaction time, solvent and temperature over the reported studies.
| Entry | Catalyst | Conditions | Yield% | Ref. |
|---|---|---|---|---|
| 1 | SBA-15-tris(triphenylphosphine)ruthenium(II) dichloride | 12 h, H2O, 90 °C | 88 | 48 |
| 2 | (E)-6-(2-((2-Hydroxynaphthalen-1-yl)methylene)hydrazinyl)nicotinohydrazide-Cu | 240 min, H2O, RT | 94 | 49 |
| 3 | Bis[(tetrabutylammonium)di-m-iodo-diiododicuprate(I)] complex | 240 min, H2O, 60 °C | 97 | 50 |
| 4 | Fe3O4@LDH@cysteine-Cu(I) | 25 min, choline azide/70 °C | 90 | 51 |
| 5 | Fe3O4@CG/CPTMS/OL-Cu | 180 min, DMF, RT | 82 | This work |
Conclusions
In summary, Fe3O4@CG/CPTMS/OL-Cu, a novel hybrid magnetic material was found to be a highly efficient nano-biocatalyst in the synthesis of triazole derivatives via the click reaction. The catalyst was fully characterized through various techniques including FTIR, XRD, SEM, EDS, and VSM. The prepared Fe3O4@CG/CPTMS/OL-Cu catalyst offers several advantages, such as non-usage of toxic solvents, high yields, good reaction time, no waste production, simple work-up, and facile separation through a magnetic field. The remarkable efficiency of this catalyst holds promising potential for its application in various chemical reactions beyond the scope of the present study. These findings not only contribute to the development of efficient and environmentally friendly catalytic systems but also underscore the versatility of Fe3O4@CG/CPTMS/OL-Cu for broader applications in diverse chemical transformations.Experimental section
Reagents and apparatus
All reagents and materials were analytical grade and purchased from commercial sources. All of them were used without purification. The known products were identified by comparison of their melting points. Melting points were determined in open capillaries using an Electrothermal 9100 instrument. Infrared (IR) spectra were acquired on a Shimadzu FT-IR-8400S spectrometer with spectroscopic grade KBr. The 1H NMR (500 MHz) was performed on a Bruker Avance DPX-300 instrument. The spectra were obtained in DMSO-d6 relative to TMS as an internal standard. Scanning electron microscopy (SEM) was performed on a VEG2/TESCAN 30 kV with gold coating, and energy dispersive X-ray spectroscopy (EDX) was performed on a VEG//TESCAN-XMU.General procedure for the preparation of Fe3O4@CG@CPTMS
The superparamagnetic iron oxide nanoparticles (SPIONs) were synthesized using a co-precipitation method described before.24,52 Typically, FeCl3·6H2O (1.215 g) and FeCl2·4H2O (0.637 g) (Fe2+/Fe3+ = 1:2) were dissolved in deionized water (20 ml) under an inert atmosphere to get a homogenous solution. Then CG powder was added into the solution and chemical precipitation was carried out by the slow addition of NaOH solution (25%), stirring vigorously until the pH = 11 was attained. The obtained magnetic particles were separated by an external magnetic field, washed several times with water and ethanol (25 ml), and dried in a 65 °C oven for 24 h.
After that 1 g of the prepared Fe3O4@CG was ultrasonically dispersed in toluene (50 ml). Then CPTMS was slowly added and the mixture was refluxed for 2 h under an inert atmosphere. The prepared Fe3O4@CG@CPTMS was magnetically separated, washed sequentially with absolute ethanol and dried in a vacuum oven at 50 °C.
Synthesis of the organic linker (OL)
To prepare the organic linker, 4-(amino methyl) benzoic acid (10 mmol, 1.511 g) was poured into a round bottom flask containing 20 ml water and stirred for 30 min at 40 °C. Then, isatoic anhydride (10 mmol, 1.613 g) was slowly added and the mixture was stirred for 3 h. The resulting precipitate was then filtered, washed with water to remove unreacted reagents, and dried for the next step.General procedure for the preparation of Fe3O4@CG/CPTMS/OL-Cu(I)
Initially, 1.5 g of the prepared OL and dry toluene were poured into a flask and triethylamine (0.07 g) was added. Then 1 g of the dried magnetic NPs was added and the mixture was refluxed for 24 h. Finally, the obtained precipitate was magnetically separated, washed with water and ethanol and dried at 50 °C.In the last step, for copper immobilization, the obtained NPs were refluxed with CuCl in EtOH for 24 h and the prepared catalyst was washed with EtOH and water and oven dried.
Synthesis of propargyl-based compounds 1, 2, and 3
A mixture of phthalic anhydride (1 mmol, 0.148 g) and 4-aminophenol (1 mmol, 0.109 g) was refluxed in acetic acid for 6 h. After completion of the reaction as indicated by TLC, the mixture was filtered, washed with MeOH and dried. In the next step, the as-obtained powder was stirred for 3 h with 3-bromopropylene in the presence of K2CO3 and DMF to obtain compound 1.The procedure for the preparation of compound 2 is the same as 1 with the substitution of 2-hydroxy-1,4-naphthoquinone with phthalic anhydride.
For the preparation of compound 3, 1H-benzo[d]imidazole-2-carboxylic acid was prepared according to the literature.53 Then, 3 mmol of the prepared acid, TBTU (3.6 mmol, 1.155 g) and DIEA (9 mmol, 1.567 ml) were added to 4 ml DMF and stirred at room temperature for 20 minutes. Next, propargyl amine (4.5 mmol, 0.247 g) was added to the reaction mixture and stirred at room temperature for 24 hours. After the completion of the reaction, indicated by TLC, the reaction mixture was quenched with water and a light brown precipitate was filtered and dried.
General procedure for the preparation of N-phenyl-2-chloroacetamide
To a solution of 1 mmol aniline/benzylamine derivatives in 4 ml DMF, 1 mmol chloroacetylchloride was added at 0 °C. The mixture was stirred at room temperature for 5 h and poured into water and then filtered to get phenylacetamide derivatives. The obtained solids were then filtered, dried, and recrystallized from ethanol.General experimental procedure for the click reaction
In a round bottom flask, phenylacetamide derivatives were mixed with sodium azide in DMF and triethylamine was added. The mixture was stirred for an hour at room temperature to prepare compound 4(a–f).Lastly, the as-prepared compounds were mixed in water and in the presence of the prepared catalyst for an appropriate period of time to obtain triazoles.
Spectral data for the synthetic compounds
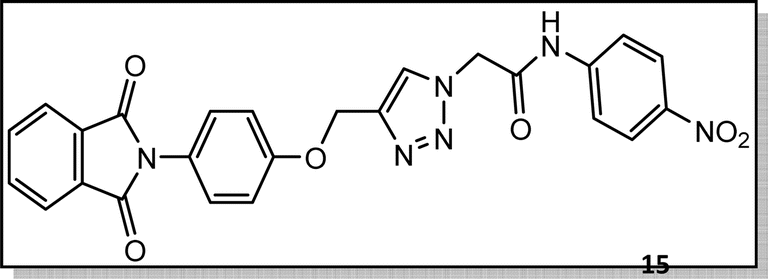 Light brown crystal, isolated yield: 66% (328 mg), m.p. 260–262 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.34 (s, 1H), 8.29–8.18 (m, 2H), 8.00–7.93 (m, 2H), 7.91 (td, J = 5.6, 5.1, 2.0 Hz, 2H), 7.87–7.77 (m, 2H), 7.45–7.31 (m, 2H), 7.27–6.94 (m, 2H), 5.48 (s, 2H), 5.27 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.82, 158.10, 144.97, 143.05, 142.88, 135.08, 132.06, 129.27, 126.90, 125.60, 125.18, 123.80, 119.50, 115.34, 61.68, 52.82 ppm.
Light brown crystal, isolated yield: 66% (328 mg), m.p. 260–262 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.34 (s, 1H), 8.29–8.18 (m, 2H), 8.00–7.93 (m, 2H), 7.91 (td, J = 5.6, 5.1, 2.0 Hz, 2H), 7.87–7.77 (m, 2H), 7.45–7.31 (m, 2H), 7.27–6.94 (m, 2H), 5.48 (s, 2H), 5.27 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.82, 158.10, 144.97, 143.05, 142.88, 135.08, 132.06, 129.27, 126.90, 125.60, 125.18, 123.80, 119.50, 115.34, 61.68, 52.82 ppm.
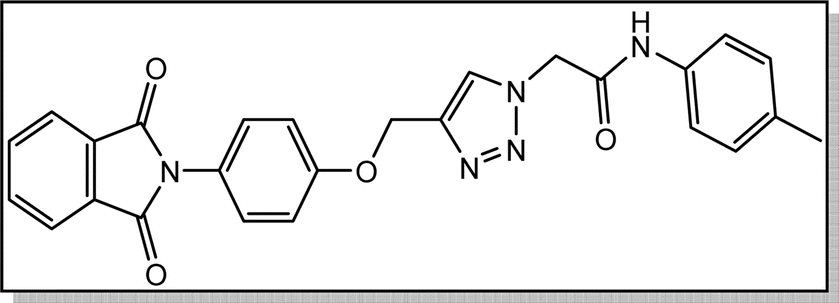 White crystal, isolated yield: 81% (378 mg), m.p. 230–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.33 (s, 1H), 8.02–7.73 (m, 4H), 7.49 (d, J = 8.0 Hz, 2H), 7.41–7.32 (m, 2H), 7.27–7.17 (m, 2H), 7.14 (d, J = 8.0 Hz, 2H), 5.37 (s, 2H), 5.26 (s, 2H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.38, 158.12, 142.82, 136.37, 135.07, 133.22, 132.04, 129.76, 129.25, 126.89, 125.17, 123.80, 119.68, 115.34, 61.71, 52.70, 20.92 ppm.
White crystal, isolated yield: 81% (378 mg), m.p. 230–232 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.33 (s, 1H), 8.02–7.73 (m, 4H), 7.49 (d, J = 8.0 Hz, 2H), 7.41–7.32 (m, 2H), 7.27–7.17 (m, 2H), 7.14 (d, J = 8.0 Hz, 2H), 5.37 (s, 2H), 5.26 (s, 2H), 2.26 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.38, 158.12, 142.82, 136.37, 135.07, 133.22, 132.04, 129.76, 129.25, 126.89, 125.17, 123.80, 119.68, 115.34, 61.71, 52.70, 20.92 ppm.
 White crystal, isolated yield: 77%, m.p. 218 – 220 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.34 (s, 1H), 7.92 (ddt, J = 20.0, 5.5, 3.2 Hz, 4H), 7.62 (dd, J = 8.8, 4.9 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.33–7.10 (m, 4H), 5.38 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.59, 158.11, 143.06, 135.25, 135.06, 132.03, 129.24, 126.96, 125.17, 123.79, 121.54, 121.46, 115.87, 115.34, 61.71, 52.66 ppm.
White crystal, isolated yield: 77%, m.p. 218 – 220 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.59 (s, 1H), 8.34 (s, 1H), 7.92 (ddt, J = 20.0, 5.5, 3.2 Hz, 4H), 7.62 (dd, J = 8.8, 4.9 Hz, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.33–7.10 (m, 4H), 5.38 (s, 2H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.59, 158.11, 143.06, 135.25, 135.06, 132.03, 129.24, 126.96, 125.17, 123.79, 121.54, 121.46, 115.87, 115.34, 61.71, 52.66 ppm.
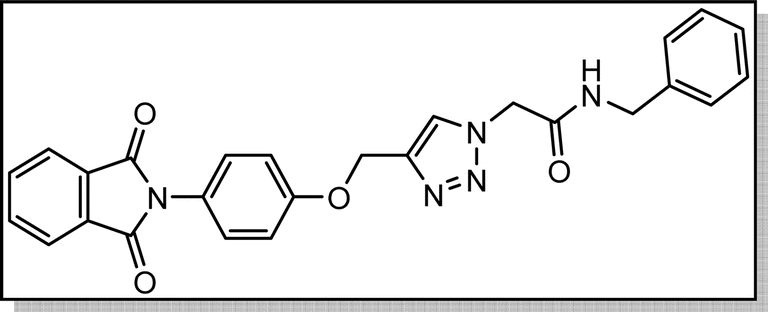 White crystal, isolated yield: 80%, m.p. 201 – 203 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.8 Hz, 1H), 8.28 (s, 1H), 7.93 (ddd, J = 24.4, 5.6, 3.1 Hz, 4H), 7.49–6.82 (m, 9H), 5.24 (s, 2H), 5.23 (s, 2H), 4.35 (d, J = 5.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.90, 158.13, 142.78, 139.17, 135.08, 132.04, 129.25, 128.84, 127.87, 127.48, 126.80, 125.16, 123.80, 115.34, 61.71, 52.12, 42.87 ppm.
White crystal, isolated yield: 80%, m.p. 201 – 203 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.8 Hz, 1H), 8.28 (s, 1H), 7.93 (ddd, J = 24.4, 5.6, 3.1 Hz, 4H), 7.49–6.82 (m, 9H), 5.24 (s, 2H), 5.23 (s, 2H), 4.35 (d, J = 5.7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 165.90, 158.13, 142.78, 139.17, 135.08, 132.04, 129.25, 128.84, 127.87, 127.48, 126.80, 125.16, 123.80, 115.34, 61.71, 52.12, 42.87 ppm.
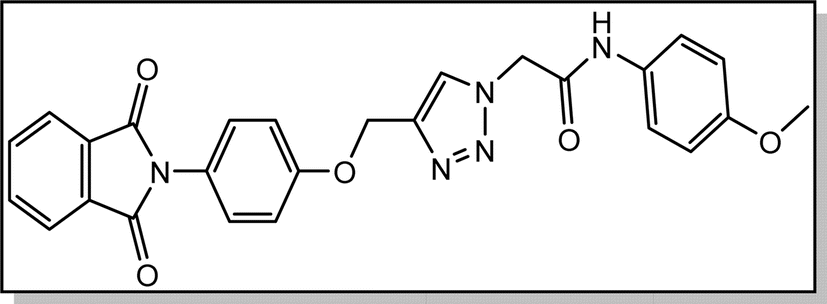 Yield: 90%. White crystal. M.p. 208–210 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.33 (s, 1H), 7.92 (ddt, J = 20.6, 5.5, 3.2 Hz, 4H), 7.52 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 5.36 (s, 2H), 5.26 (s, 2H), 3.73 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.11, 158.12, 156.00, 142.83, 135.06, 132.03, 131.97, 129.24, 126.88, 125.17, 123.79, 121.24, 115.34, 114.47, 61.71, 55.61, 52.66 ppm.
Yield: 90%. White crystal. M.p. 208–210 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.40 (s, 1H), 8.33 (s, 1H), 7.92 (ddt, J = 20.6, 5.5, 3.2 Hz, 4H), 7.52 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.9 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 5.36 (s, 2H), 5.26 (s, 2H), 3.73 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.72, 164.11, 158.12, 156.00, 142.83, 135.06, 132.03, 131.97, 129.24, 126.88, 125.17, 123.79, 121.24, 115.34, 114.47, 61.71, 55.61, 52.66 ppm.
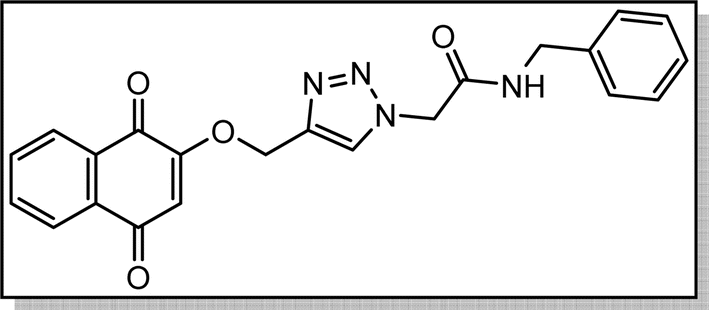
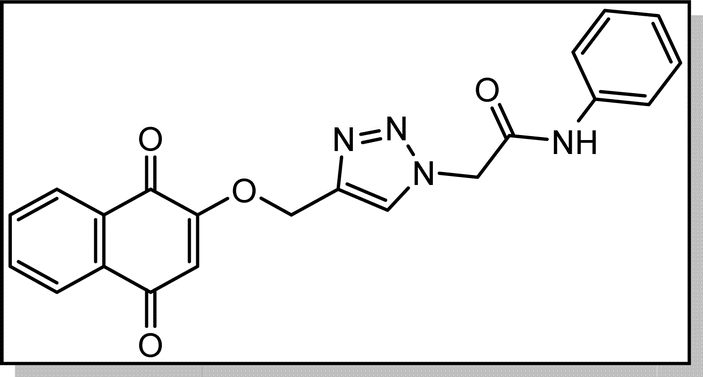 Cream solid; isolated yield: 70%, m.p. 200–202 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52, 8.37 (s, 1H), 8.01 (td, 2H), 7.90–7.82 (m, 2H), 7.59 (d, 2H), 7.34 (t, 2H), 7.10 (t, 1H), 6.68 (s, 1H), 5.40 (s, 2H), 5.29 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 185.05, 179, 164.61, 159.46, 141.06, 138., 135.02, 134.16, 131.96, 131.31, 129.41, 127.72, 126.61, 126.05, 124.26, 119.66, 111.38, 62.83, 52.72 ppm.
Cream solid; isolated yield: 70%, m.p. 200–202 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.52, 8.37 (s, 1H), 8.01 (td, 2H), 7.90–7.82 (m, 2H), 7.59 (d, 2H), 7.34 (t, 2H), 7.10 (t, 1H), 6.68 (s, 1H), 5.40 (s, 2H), 5.29 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 185.05, 179, 164.61, 159.46, 141.06, 138., 135.02, 134.16, 131.96, 131.31, 129.41, 127.72, 126.61, 126.05, 124.26, 119.66, 111.38, 62.83, 52.72 ppm.
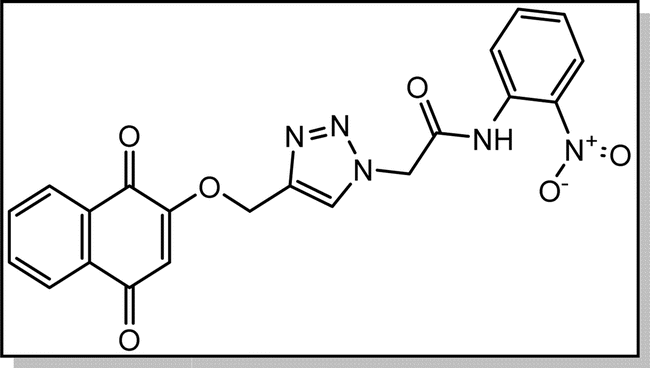 Yellow solid; isolated yield: 83%, m.p. 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.34 (s, 1H), 8.05–7.95 (m, 3H), 7.91–7.80 (m, 2H), 7.81–7.64 (m, 2H), 7.43 (t, 1H), 6.67 (s, 1H), 5.49 (s, 2H), 5.29 (s, 2H).13C NMR (100 MHz, DMSO-d6) δ 185.05, 179.97, 165.54, 165.39, 159.44, 142.93, 134.66, 134.15, 131.95, 131.30, 130.79, 130.74, 126.60, 126.42, 126.36, 126.05, 125.96, 125.57, 111.38, 62.77, 52.44 ppm.
Yellow solid; isolated yield: 83%, m.p. 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.80 (s, 1H), 8.34 (s, 1H), 8.05–7.95 (m, 3H), 7.91–7.80 (m, 2H), 7.81–7.64 (m, 2H), 7.43 (t, 1H), 6.67 (s, 1H), 5.49 (s, 2H), 5.29 (s, 2H).13C NMR (100 MHz, DMSO-d6) δ 185.05, 179.97, 165.54, 165.39, 159.44, 142.93, 134.66, 134.15, 131.95, 131.30, 130.79, 130.74, 126.60, 126.42, 126.36, 126.05, 125.96, 125.57, 111.38, 62.77, 52.44 ppm.
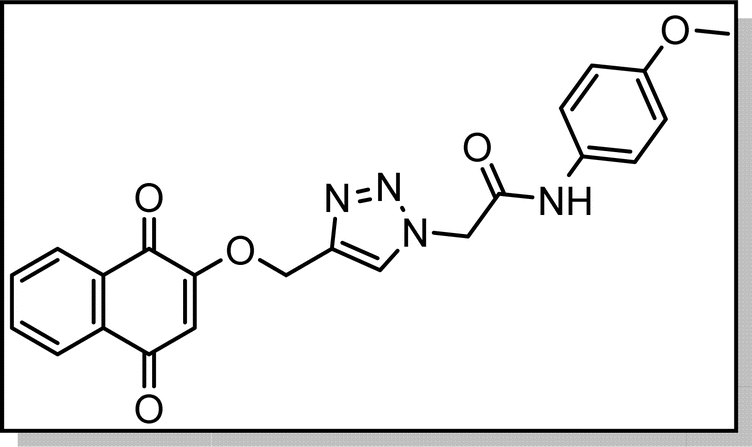 Cream solid; isolated yield: 62%, m.p. 235–237 °C.1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.37 (s, 1H), 8.00 (td, J = 7.4, 1.6 Hz, 2H), 7.85 (dtd, J = 16.6, 7.4, 1.5 Hz, 2H), 7.50 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 6.67 (s, 1H), 5.36 (s, 2H), 5.29 (s, 2H), 3.72 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 185.04, 179.97, 164.06, 159.44, 155.99, 141.06, 135.00, 134.15, 131.97, 131.94, 131.29, 127.71, 126.59, 126.04, 121.21, 114.48, 111.37, 62.83, 55.61, 52.64 ppm.
Cream solid; isolated yield: 62%, m.p. 235–237 °C.1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.37 (s, 1H), 8.00 (td, J = 7.4, 1.6 Hz, 2H), 7.85 (dtd, J = 16.6, 7.4, 1.5 Hz, 2H), 7.50 (d, J = 9.0 Hz, 2H), 6.91 (d, J = 9.0 Hz, 2H), 6.67 (s, 1H), 5.36 (s, 2H), 5.29 (s, 2H), 3.72 (s, 3H).13C NMR (100 MHz, DMSO-d6) δ 185.04, 179.97, 164.06, 159.44, 155.99, 141.06, 135.00, 134.15, 131.97, 131.94, 131.29, 127.71, 126.59, 126.04, 121.21, 114.48, 111.37, 62.83, 55.61, 52.64 ppm.
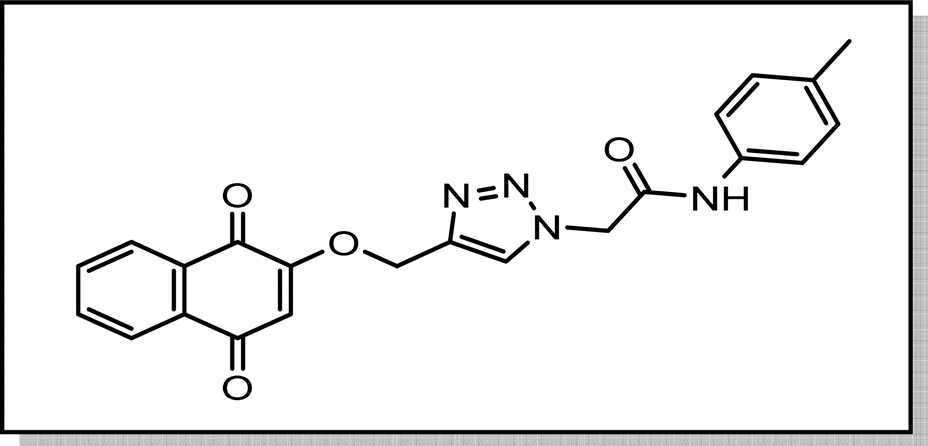 Cream solid; isolated yield: 68%, m.p. 244–246 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.37 (s, 1H, C–H), 8.00 (tdd, J = 7.6, 1.6 Hz, 2H), 7.91–7.77 (m, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 6.67 (s, 1H), 5.38 (s, 2H), 5.29 (s, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.05, 179.99, 164.33, 159.46, 141.05, 136.36, 135.01, 134.16, 133.23, 131.95, 131.30, 129.78, 127.71, 126.60, 126.05, 119.66, 111.37, 62.83, 52.69, 20.93 ppm.
Cream solid; isolated yield: 68%, m.p. 244–246 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.43 (s, 1H), 8.37 (s, 1H, C–H), 8.00 (tdd, J = 7.6, 1.6 Hz, 2H), 7.91–7.77 (m, 2H), 7.47 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.1 Hz, 2H), 6.67 (s, 1H), 5.38 (s, 2H), 5.29 (s, 2H), 2.26 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 185.05, 179.99, 164.33, 159.46, 141.05, 136.36, 135.01, 134.16, 133.23, 131.95, 131.30, 129.78, 127.71, 126.60, 126.05, 119.66, 111.37, 62.83, 52.69, 20.93 ppm.
 Cream solid; isolated yield: 67%, m.p. 218–220 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.9 Hz, 1H), 8.32 (s, 1H, C–H), 8.08–7.96 (m, 2H), 7.94–7.73 (m, 2H), 7.53–7.09 (m, 5H), 6.67 (s, 1H), 5.27 (s, 2H), 5.23 (s, 2H), 4.34 (d, J = 5.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 185.05, 179.99, 165.84, 159.46, 140.98, 139.16, 135.01, 134.16, 131.95, 131.31, 128.85, 127.89, 127.59, 127.50, 126.60, 126.05, 111.37, 62.81, 52.10, 42.85 ppm.
Cream solid; isolated yield: 67%, m.p. 218–220 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.89 (t, J = 5.9 Hz, 1H), 8.32 (s, 1H, C–H), 8.08–7.96 (m, 2H), 7.94–7.73 (m, 2H), 7.53–7.09 (m, 5H), 6.67 (s, 1H), 5.27 (s, 2H), 5.23 (s, 2H), 4.34 (d, J = 5.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 185.05, 179.99, 165.84, 159.46, 140.98, 139.16, 135.01, 134.16, 131.95, 131.31, 128.85, 127.89, 127.59, 127.50, 126.60, 126.05, 111.37, 62.81, 52.10, 42.85 ppm.
 White solid, isolated yield: 89%, m.p. 235–237 °C. IR (KBr, ν): 3356, 3287, 1650, 1331, 1189 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.28 (s, 1H), 10.28 (s, 1H), 9.42 (t, J = 6.2 Hz, 1H), 8.01 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.7 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 5.25 (s, 2H), 4.59 (d, J = 6.0 Hz, 2H), 3.70 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.17, 159.27, 156.00, 145.95, 145.02, 142.99, 134.95, 131.98, 125.09, 124.61, 123.04, 121.24, 120.37, 114.47, 113.03, 55.62, 52.57, 35.03. Anal. Calcd. for C20H19N7O3: C 59.25; H 4.72; N 24.18; Found: C 59.04; H 4.54; N 24.28.
White solid, isolated yield: 89%, m.p. 235–237 °C. IR (KBr, ν): 3356, 3287, 1650, 1331, 1189 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.28 (s, 1H), 10.28 (s, 1H), 9.42 (t, J = 6.2 Hz, 1H), 8.01 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 8.1 Hz, 1H), 7.47 (d, J = 8.5 Hz, 2H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.7 Hz, 1H), 6.88 (d, J = 8.5 Hz, 2H), 5.25 (s, 2H), 4.59 (d, J = 6.0 Hz, 2H), 3.70 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.17, 159.27, 156.00, 145.95, 145.02, 142.99, 134.95, 131.98, 125.09, 124.61, 123.04, 121.24, 120.37, 114.47, 113.03, 55.62, 52.57, 35.03. Anal. Calcd. for C20H19N7O3: C 59.25; H 4.72; N 24.18; Found: C 59.04; H 4.54; N 24.28.
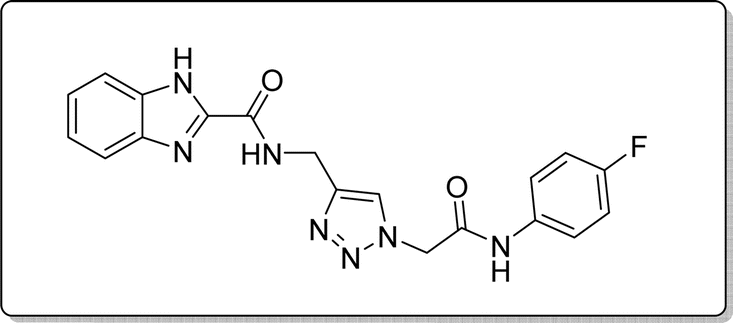 White solid, isolated yield: 57%, m.p. 245–247 °C. IR (KBr, ν): 3347, 3297, 1672, 1324, 1009 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.27 (s, 1H), 10.48 (s, 1H), 9.42 (t, J = 6.2 Hz, 1H), 8.01 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.57 (ddt, J = 8.9, 4.9 Hz, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.15 (t, J = 8.7 Hz, 2H), 5.28 (s, 2H), 4.58 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 164.64, 161.56 (1JC–F = 240 Hz), 159.24, 145.92, 145.04, 142.96, 135.25 (4JC–F = 1.25 Hz), 134.92, 125.09, 124.60, 123.03, 121.52 (3JC–F = 8.75 Hz), 121.45, 120.35, 116.05 (2JCVF = 22.5 Hz), 113.02, 52.54, 35.01. Anal. Calcd. for C19H16FN7O2: C 58.01; H 4.10; N 24.92; Found: C 57.88; H 4.26; N 24.75.
White solid, isolated yield: 57%, m.p. 245–247 °C. IR (KBr, ν): 3347, 3297, 1672, 1324, 1009 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.27 (s, 1H), 10.48 (s, 1H), 9.42 (t, J = 6.2 Hz, 1H), 8.01 (s, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.57 (ddt, J = 8.9, 4.9 Hz, 2H), 7.53 (d, J = 8.0 Hz, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.15 (t, J = 8.7 Hz, 2H), 5.28 (s, 2H), 4.58 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 164.64, 161.56 (1JC–F = 240 Hz), 159.24, 145.92, 145.04, 142.96, 135.25 (4JC–F = 1.25 Hz), 134.92, 125.09, 124.60, 123.03, 121.52 (3JC–F = 8.75 Hz), 121.45, 120.35, 116.05 (2JCVF = 22.5 Hz), 113.02, 52.54, 35.01. Anal. Calcd. for C19H16FN7O2: C 58.01; H 4.10; N 24.92; Found: C 57.88; H 4.26; N 24.75.
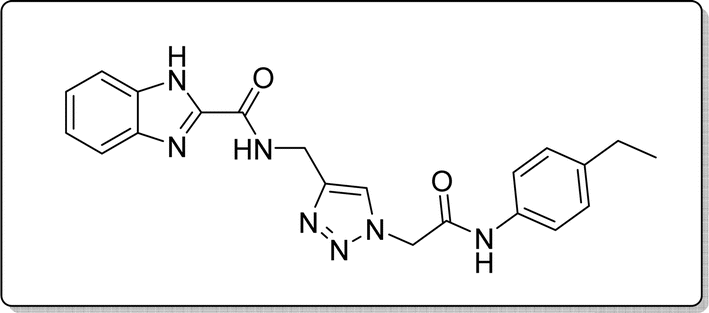 White solid, isolated yield: 76%, m.p. 213–215 °C. IR (KBr, ν): 3344, 3281, 1648, 1357, 1279 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13. 27 (s, 1H), 10.34 (s, 1H), 9.41 (t, J = 6.1 Hz, 1H), 8.00 (s, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 8.1 Hz, 2H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 8.1 Hz, 2H), 5.26 (s, 2H), 4.58 (d, J = 6.1 Hz, 2H), 2.53 (q, J = 7.7 Hz, 2H), 1.13 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.42, 159.23, 145.93, 145.00, 142.96, 139.61, 136.55, 134.93, 128.53, 125.08, 124.59, 123.02, 120.35, 119.74, 113.01, 52.59, 35.01, 28.03, 16.08. Anal. Calcd. for C21H21N7O2: C 62.52; H 5.25; N 24.30; Found: C 62.38; H 5.49; N 24.14.
White solid, isolated yield: 76%, m.p. 213–215 °C. IR (KBr, ν): 3344, 3281, 1648, 1357, 1279 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13. 27 (s, 1H), 10.34 (s, 1H), 9.41 (t, J = 6.1 Hz, 1H), 8.00 (s, 1H), 7.71 (d, J = 8.1 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.46 (d, J = 8.1 Hz, 2H), 7.31 (t, J = 7.5 Hz, 1H), 7.26 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 8.1 Hz, 2H), 5.26 (s, 2H), 4.58 (d, J = 6.1 Hz, 2H), 2.53 (q, J = 7.7 Hz, 2H), 1.13 (t, J = 7.5 Hz, 3H). 13C NMR (125 MHz, DMSO-d6) δ 164.42, 159.23, 145.93, 145.00, 142.96, 139.61, 136.55, 134.93, 128.53, 125.08, 124.59, 123.02, 120.35, 119.74, 113.01, 52.59, 35.01, 28.03, 16.08. Anal. Calcd. for C21H21N7O2: C 62.52; H 5.25; N 24.30; Found: C 62.38; H 5.49; N 24.14.
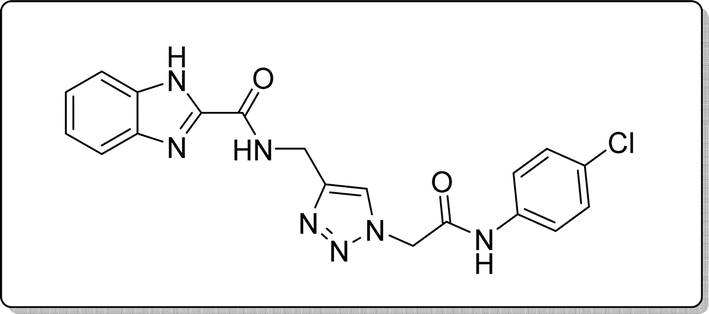 White solid, isolated yield: 69%, m.p. 236–238 °C. IR (KBr, ν): 3349, 3290, 1655, 1369, 767 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 10.71 (s, 1H), 9.43 (t, J = 6.2 Hz, 1H), 8.02 (s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.34–7.25 (m, 2H), 5.32 (s, 2H), and 4.59 (d, J = 6.1 Hz, 2H).13C NMR (125 MHz, DMSO-d6) δ 164.66, 160.03, 145.39, 145.04, 141.70, 137.88, 135.30, 129.26, 127.75, 125.10, 124.60, 123.01, 121.22, 120.10, 113.01, 52.61, 35.00. Anal. Calcd. for C19H16ClN7O2: C 55.68; H 3.94; N 23.92; Found: C 55.39; H 4.16; N 24.11.
White solid, isolated yield: 69%, m.p. 236–238 °C. IR (KBr, ν): 3349, 3290, 1655, 1369, 767 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 10.71 (s, 1H), 9.43 (t, J = 6.2 Hz, 1H), 8.02 (s, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.61 (d, J = 8.4 Hz, 2H), 7.54 (d, J = 8.0 Hz, 1H), 7.38 (d, J = 8.4 Hz, 2H), 7.34–7.25 (m, 2H), 5.32 (s, 2H), and 4.59 (d, J = 6.1 Hz, 2H).13C NMR (125 MHz, DMSO-d6) δ 164.66, 160.03, 145.39, 145.04, 141.70, 137.88, 135.30, 129.26, 127.75, 125.10, 124.60, 123.01, 121.22, 120.10, 113.01, 52.61, 35.00. Anal. Calcd. for C19H16ClN7O2: C 55.68; H 3.94; N 23.92; Found: C 55.39; H 4.16; N 24.11.
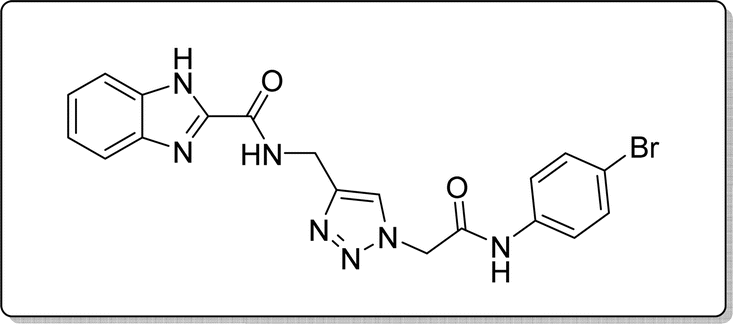 White solid, isolated yield: 73%, m.p. 227–229 °C. IR (KBr, ν): 3353, 3285, 1649, 1342, 620 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 10.58 (s, 1H), 9.43 (t, J = 6.3 Hz, 1H), 8.02 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.65–7.38 (m, 5H), 7.30 (dt, J = 17.2, 7.4 Hz, 2H), 5.31 (s, 2H), 4.60 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 164.93, 159.24, 145.93, 145.06, 142.95, 138.24, 132.19, 125.10, 124.59, 123.03, 122.98, 121.61, 120.36, 115.83, 113.02, 52.62, 35.02. Anal. Calcd. for C19H16BrN7O2: C 50.23; H 3.55; N 21.58; Found: C 50.02; H 3.81; N 21.33.
White solid, isolated yield: 73%, m.p. 227–229 °C. IR (KBr, ν): 3353, 3285, 1649, 1342, 620 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 10.58 (s, 1H), 9.43 (t, J = 6.3 Hz, 1H), 8.02 (s, 1H), 7.73 (d, J = 8.0 Hz, 1H), 7.65–7.38 (m, 5H), 7.30 (dt, J = 17.2, 7.4 Hz, 2H), 5.31 (s, 2H), 4.60 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 164.93, 159.24, 145.93, 145.06, 142.95, 138.24, 132.19, 125.10, 124.59, 123.03, 122.98, 121.61, 120.36, 115.83, 113.02, 52.62, 35.02. Anal. Calcd. for C19H16BrN7O2: C 50.23; H 3.55; N 21.58; Found: C 50.02; H 3.81; N 21.33.
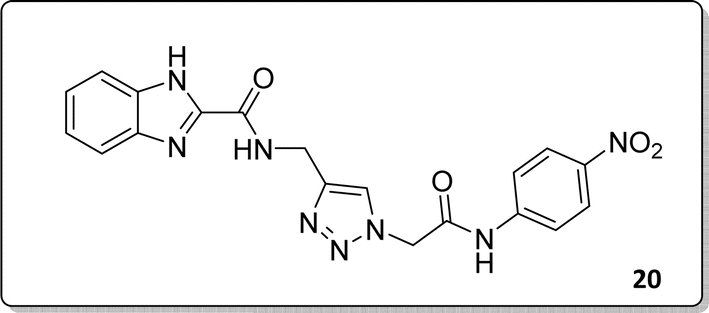 White solid, isolated yield: 81%, m.p. 259–261 °C. IR (KBr, ν): 3341, 3276, 1684, 1551, 1352, 1223 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 11.05 (s, 1H), 9.41 (t, J = 6.3 Hz, 1H), 8.23 (d, J = 8.6 Hz, 2H), 8.05 (s, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 7.4 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 5.40 (s, 2H), 4.61 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 165.85, 159.25, 145.92, 145.14, 144.96, 143.03, 142.97, 134.87, 125.55, 125.15, 124.59, 123.01, 120.33, 119.48, 113.01, 52.73, 35.02. Anal. Calcd. for C19H16N8O4: C 54.29; H 3.84; N 26.66; Found: C 54.09; H 4.03; N 26.40.
White solid, isolated yield: 81%, m.p. 259–261 °C. IR (KBr, ν): 3341, 3276, 1684, 1551, 1352, 1223 cm−1. 1H NMR (500 MHz, DMSO-d6) δ 13.29 (s, 1H), 11.05 (s, 1H), 9.41 (t, J = 6.3 Hz, 1H), 8.23 (d, J = 8.6 Hz, 2H), 8.05 (s, 1H), 7.82 (d, J = 8.7 Hz, 2H), 7.73 (d, J = 8.0 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 7.4 Hz, 1H), 7.28 (d, J = 7.5 Hz, 1H), 5.40 (s, 2H), 4.61 (d, J = 6.1 Hz, 2H). 13C NMR (125 MHz, DMSO-d6) δ 165.85, 159.25, 145.92, 145.14, 144.96, 143.03, 142.97, 134.87, 125.55, 125.15, 124.59, 123.01, 120.33, 119.48, 113.01, 52.73, 35.02. Anal. Calcd. for C19H16N8O4: C 54.29; H 3.84; N 26.66; Found: C 54.09; H 4.03; N 26.40.
Conflicts of interest
There are no conflicts to declare.References
- M. Henary, C. Kananda, L. Rotolo, B. Savino, E. A. Owens and G. Cravotto, RSC Adv., 2020, 10, 14170–14197 RSC.
- T.-Y. Zhang, C.-S. Li, L.-T. Cao, X.-Q. Bai, D.-H. Zhao and S.-M. Sun, Mol. Diversity, 2022, 26, 1129–1139 CrossRef CAS PubMed.
- M. Strzelecka and P. Świątek, Pharmaceuticals, 2021, 14, 224 CrossRef CAS PubMed.
- F. Eya’ane Meva, T. J. Prior, D. J. Evans, S. Shah, C. F. Tamngwa, H. G. L. Belengue, R. E. Mang, J. Munro, T. Qahash and M. Llinás, Res. Chem. Intermed., 2022, 48, 885–898 CrossRef.
- K. Ji, G.-N. Zhang, J.-Y. Zhao, M. Zhu, M.-H. Wang, J.-X. Wang, S. Cen, Y.-C. Wang and W.-Y. Li, Bioorg. Med. Chem. Lett., 2022, 64, 128681 CrossRef CAS PubMed.
- M. M. Alam, Arch. Pharm., 2022, 355, 2100158 CrossRef CAS PubMed.
- P. Shiri and J. Aboonajmi, Beilstein J. Org. Chem., 2020, 16, 551–586 CrossRef CAS PubMed.
- S. Sabaqian, F. Nemati, M. M. Heravi and H. T. Nahzomi, Appl. Organomet. Chem., 2017, 31, e3660 CrossRef.
- A. Salamatmanesh, M. Kazemi Miraki, E. Yazdani and A. Heydari, Catal. Lett., 2018, 148, 3257–3268 CrossRef CAS.
- M. Gholinejad and N. Jeddi, ACS Sustain. Chem. Eng., 2014, 2, 2658–2665 CrossRef CAS.
- L. Bahsis, H. Ben El Ayouchia, H. Anane, A. Pascual-Álvarez, G. De Munno, M. Julve and S. E. Stiriba, Appl. Organomet. Chem., 2019, 33, e4669 CrossRef.
- R. Gheorghita, L. Anchidin-Norocel, R. Filip, M. Dimian and M. Covasa, Polymers, 2021, 13, 2729 CrossRef CAS PubMed.
- S. Deshmukh, M. Kathiresan and M. A. Kulandainathan, Biomater. Sci., 2022, 10, 4424–4442 RSC.
- A. Madni, A. Khalid, F. Wahid, H. Ayub, R. Khan and R. Kousar, Curr. Nanosci., 2021, 17, 365–379 CrossRef CAS.
- S. Mitura, A. Sionkowska and A. Jaiswal, J. Mater. Sci.: Mater. Med., 2020, 31, 1–14 CrossRef PubMed.
- T. Saha, M. E. Hoque and T. Mahbub, in Advanced Processing, Properties, and Applications of Starch and Other Bio-Based Polymers, Elsevier, 2020, pp. 197–214 Search PubMed.
- Y. Liu, S. Ahmed, D. E. Sameen, Y. Wang, R. Lu, J. Dai, S. Li and W. Qin, Trends Food Sci. Technol., 2021, 112, 532–546 CrossRef CAS.
- S. Ullah, F. U. Khan and N. Ahmad, Environ. Sci. Pollut. Res., 2022, 1–21 Search PubMed.
- H. B. Ahmed and H. E. Emam, in Polysaccharide Nanoparticles, Elsevier, 2022, pp. 375–413 Search PubMed.
- A. S. Volokhova, K. J. Edgar and J. B. Matson, Mater. Chem. Front., 2020, 4, 99–112 RSC.
- J. Kurczewska, Polymers, 2022, 14, 4189 CrossRef CAS PubMed.
- A. Mirzaei, M. Esmkhani, M. Zallaghi, Z. Nezafat and S. Javanshir, J. Polym. Environ., 2022, 1–27 CAS.
- S. Darvishi, S. Sadjadi, E. Monflier and M. M. Heravi, J. Mol. Struct., 2024, 1296, 136827 CrossRef CAS.
- Z. Dolatkhah, S. Javanshir, A. Bazgir and B. Hemmati, Appl. Organomet. Chem., 2019, 33, e4859 CrossRef.
- S. Amirnejat, A. Nosrati, R. Peymanfar and S. Javanshir, Res. Chem. Intermed., 2020, 46, 3683–3701 CrossRef CAS.
- R. Reddyrajula and U. Dalimba, Bioorg. Med. Chem. Lett., 2020, 30, 126846 CrossRef CAS PubMed.
- N. Poonia, A. Kumar, V. Kumar, M. Yadav and K. Lal, Curr. Top. Med. Chem., 2021, 21, 2109–2133 CrossRef CAS PubMed.
- Z. Salehi, N. Fatahi, M. Taran, A. Izadi, H. Badali, S. Hashemi, S. Rezaie, R. D. Ghazvini, M. Ghaffari and F. Aala, J. Mycol. Med., 2020, 30, 100935 CrossRef CAS PubMed.
- İ. Şahin, F. B. Özgeriş, M. Köse, E. Bakan and F. Tümer, J. Mol. Struct., 2021, 1232, 130042 CrossRef.
- A. R. Nesaragi, R. R. Kamble, P. K. Bayannavar, S. K. J. Shaikh, S. R. Hoolageri, B. Kodasi, S. D. Joshi and V. M. Kumbar, Bioorg. Med. Chem. Lett., 2021, 41, 127984 CrossRef CAS PubMed.
- D. Dixit, P. K. Verma and R. K. Marwaha, J. Iran. Chem. Soc., 2021, 18, 2535–2565 CrossRef CAS.
- F. Laffafchi, M. Tajbakhsh, Y. Sarrafi, B. Maleki and M. Ghani, Polycyclic Aromat. Compd., 2022, 1–17 Search PubMed.
- A. Tan, J. Mol. Struct., 2022, 1261, 132915 CrossRef CAS PubMed.
- S. P. D. O. Assis, M. Silva, F. Silva, M. P. Sant'Anna, C. D. A. Tenório, C. Santos, C. Fonseca, G. Seabra, V. L. Lima and R. D. Oliveira, Chem. Pharm. Bull., 2019, 67, 96–105 CrossRef PubMed.
- S. K. Ihmaid, S. Y. Alraqa, M. R. Aouad, A. Aljuhani, H. M. Elbadawy, S. A. Salama, N. Rezki and H. E. Ahmed, Bioorg. Chem., 2021, 111, 104835 CrossRef CAS PubMed.
- H. Jelali, L. Mansour, E. Deniau, M. Sauthier and N. Hamdi, Polycyclic Aromat. Compd., 2022, 42, 1806–1813 CrossRef CAS.
- S. Das, S. K. Parida, T. Mandal, L. Sing, S. De Sarkar and S. Murarka, Chem.–An Asian J., 2020, 15, 568–572 CrossRef CAS PubMed.
- A. R. Oliveira, F. A. Dos Santos, L. P. de Lima Ferreira, M. G. da Rocha Pitta, M. V. de Oliveira Silva, M. V. de Oliveira Cardoso, A. F. Pinto, P. Marchand, M. J. B. de Melo Rêgo and A. C. L. Leite, Chem.-Biol. Interact., 2021, 347, 109597 CrossRef CAS PubMed.
- I. A. Khan, S. Ahmad, F. Ullah, F. Niaz, S. Iqbal, E. A. Khan, M. Khan and S. W. Khan, Pak. J. Med. Health Sci., 2022, 16, 810 Search PubMed.
- V. Singh, R. S. Hada, R. Jain, M. Vashistha, G. Kumari, S. Singh, N. Sharma, M. Bansal, M. Zoltner and C. R. Caffrey, Eur. J. Med. Chem., 2022, 239, 114534 CrossRef CAS PubMed.
- B. W. Matore, P. Banjare, T. Guria, P. P. Roy and J. Singh, Eur. J. Med. Chem. Rep., 2022, 100058 CAS.
- M. Asadi, M. Ebrahimi, M. Mohammadi-Khanaposhtani, H. Azizian, S. Sepehri, H. Nadri, M. Biglar, M. Amanlou, B. Larijani and R. Mirzazadeh, Chem. Biodiversity, 2019, 16, e1900370 CrossRef CAS PubMed.
- S. Mor, M. Khatri, R. Punia, S. Nagoria and S. Sindhu, Mini-Rev. Org. Chem., 2022, 19, 717–778 CrossRef CAS.
- M. Ribeiro da Silva and C. P. Santos, J. Therm. Anal. Calorim., 2007, 87, 21–25 CrossRef CAS.
- S. G. Azarnier, M. Esmkhani, Z. Dolatkhah and S. Javanshir, Sci. Rep., 2022, 12, 1–11 CrossRef PubMed.
- B. K. Heragh, S. Javanshir, G. R. Mahdavinia and M. R. N. Jamal, Int. J. Biol. Macromol., 2021, 190, 351–359 CrossRef CAS PubMed.
- F. Jafarzadeh, Z. Dolatkhah, S. Molaei and S. Javanshir, Arabian J. Chem., 2022, 15, 103838 CrossRef CAS.
- P. Sharma, J. Rathod, A. Singh, P. Kumar and Y. Sasson, Catal. Sci. Technol., 2018, 8, 3246–3259 RSC.
- M. Emami, R. Bikas, N. Noshiranzadeh, A. Kozakiewicz and T. Lis, ACS Omega, 2020, 5, 13344–13357 CrossRef CAS PubMed.
- S. Nekkanti, K. Veeramani, S. S. Kumari, R. Tokala and N. Shankaraiah, RSC Adv., 2016, 6, 103556–103566 RSC.
- F. Pazoki, A. Salamatmanesh, S. Bagheri and A. Heydari, Catal. Lett., 2020, 150, 1186–1195 CrossRef CAS.
- S. Rostamnia, B. Zeynizadeh, E. Doustkhah, A. Baghban and K. O. Aghbash, Catal. Commun., 2015, 68, 77–83 CrossRef CAS.
- Y. Shang, Q. Hao, K. Jiang, M. He and J. Wang, Bioorg. Med. Chem. Lett., 2020, 30, 127118 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4na00022f |
| This journal is © The Royal Society of Chemistry 2024 |