
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of comb length on the in vitro and in vivo properties of self-assembled poly(oligoethylene glycol methacrylate)-based block copolymer nanoparticles†
Andrew
Singh
a,
Andrew
Lofts
a,
Ramya
Krishnan
b,
Matthew
Campea
a,
Lan
Chen
b,
Yonghong
Wan
b and
Todd
Hoare
 *a
*a
aDepartment of Chemical Engineering, McMaster University, 1280 Main St. W., Hamilton, Ontario L8S 4L7, Canada. E-mail: hoaretr@mcmaster.ca
bDepartment of Pathology and Molecular Medicine, McMaster University, 1280 Main St. W., Hamilton, Ontario L8S 4L7, Canada
First published on 15th April 2024
Abstract
Comb copolymer analogues of poly(lactic acid)-polyethylene glycol block copolymers (PLA-b-PEG) offer potential to overcome the inherent chemistry and stability limitations of their linear block copolymer counterparts. Herein, we examine the differences between P(L)LA10K-b-PEG10K and linear-comb copolymer analogues thereof in which the linear PEG block is replaced by poly(oligo(ethylene glycol) methacrylate) (POEGMA) blocks with different side chain (comb) lengths but the same overall molecular weight. P(L)LA10K-b-POEGMA47510K and P(L)LA10K-b-POEGMA200010K block copolymers were synthesized via activators regenerated by electron transfer atom transfer radical polymerization (ARGET ATRP) and fabricated into self-assembled nanoparticles using flash nanoprecipitation via confined impinging jet mixing. Linear-comb copolymer analogues based on PLA-b-POEGMA yielded smaller but still well-controlled nanoparticle sizes (88 ± 2 nm and 114 ± 1 nm respectively compared to 159 ± 2 nm for P(L)LA10K-b-PEG10K nanoparticles) that exhibited improved colloidal stability relative to linear copolymer-based nanoparticles over a 15 day incubation period while maintaining comparably high cytocompatibility, although the comb copolymer analogues had somewhat lower loading capacity for doxorubicin hydrochloride. Cell spheroid studies showed that the linear-comb copolymers promoted enhanced tumor transport and thus cell killing compared to conventional linear block copolymers. In vivo studies showed all NP types could passively accumulate within implanted CT26 tumors but with different accumulation profiles, with P(L)LA10K-b-POEGMA200010K NPs showing continuous accumulation throughout the full 24 h monitoring period whereas tumor accumulation of P(L)LA10K-b-POEGMA47510K NPs was significant only between 8 h and 24 h. Overall, the linear-comb copolymer analogues exhibited superior stability, biodistribution, spheroid penetration, and inherent tunability over linear NP counterparts.
Introduction
Polymeric nanoparticles (NPs) have been used in a range of biomedical applications including ocular and nutraceutical drug delivery as well as oncologic diagnostics and treatments.1–3 While many types of NPs can be fabricated, self-assembled NPs based on amphiphilic block copolymers consisting of one hydrophobic and one hydrophilic polymer block have attracted particular interest due to their facile fabrication, tunable surface chemistry and/or particle size, potential for stimulus-responsive disassembly/degradation, and capacity to deliver poorly soluble drugs within their hydrophobic core.4–9Among the many amphiphilic block copolymers assessed for their potential for drug delivery, copolymers consisting of the water soluble polymer poly(ethylene glycol) (PEG) and a water-insoluble degradable polyester (e.g. poly(lactide) – PLA, poly(lactide-co-glycolic acid) – PLGA, or poly(caprolactone) – PCL) are the most widely investigated.4,10,11 The hydrophobic polyester enables self-assembly of the block copolymers into NPs in water, while the choice of polyester governs the hydrolytic half life and thus the degradation time/stability of the nanoparticle.12 PLA is particularly widely used in biomedical applications due to it being relatively inexpensive, produced from renewable resources, possessing low immunogenicity, and being biodegradable under physiologically relevant conditions into a common metabolic by-product (lactic acid).13 Complementarily, the PEG hydrophilic block creates a hydrophilic interface for the NPs, reducing their detection by the immune system, typically increasing circulation times, as well as imparting favorable dispersibility in physiological media.14–16 These benefits have led PEGylated NPs to be of interest in applications including biosensing, bioimaging, chemotherapy, and tissue engineering.17 Specific to drug delivery, both PEG and PLA copolymers are approved for in vivo use in multiple jurisdictions, with block copolymers thereof explored for the delivery of proteins, genes, photosensitizers, small molecules, and other therapeutics for ophthalmic, vaginal, oral, chemotherapeutic, and other drug delivery applications.4,18–21 Chemotherapeutic applications of such NPs have attracted particular interest given that the size range and surface chemistry achievable with self-assembled NPs can overcome many of the multiple physiological barriers toward drug transport to tumors by exploiting their potential to evade immune detection14 and penetrate through trans-endothelial pathways and ultimately into cancer cells.22 However, PEG-based block copolymers also pose several limitations for the rational engineering of drug delivery NPs in terms of their chain-end only functionalization, limiting the degree to which a PEG-based interface can be altered to customize delivery pathways. In addition, reports of the presence of PEG antibodies (estimated to be present in as many as 72% of the population23,24) may offer an additional limitation to the wide-spread use of PEG-based materials in drug formulation, although the existence and ultimate importance of PEG antibodies remains a point of debate in the literature.
As an alternative to PEG, amphiphilic brush or comb block copolymers have recently been explored that can retain many of the beneficial properties of linear PEG while providing a significant additional level of chemical and interfacial tunability. Poly(oligoethylene glycol methacrylate) (POEGMA) is one such comb PEG derivative that has a suite of attractive properties including: (1) facile modification of interfacial properties by the length of the oligo(ethylene glycol) side chains, enabling responses ranging from thermoresponsive behavior at short side chain lengths to highly protein-repellent behavior at longer side chain lengths;25–27 (2) tunable PEG densities depending on the mixture of OEGMA side chain length comonomers used;27 and (3) facile polymerization via free radical or controlled free radical techniques, enabling a broad range of potential functionalization within the combs themselves (instead of only at the chain ends) via judicious selection of functional comonomers that are also amenable to free radical polymerization. In particular, controlled radical polymerization techniques such as reversible addition–fragmentation chain-transfer (RAFT) and atom transfer radical polymerization (ATRP) have both been noted to enable POEGMA polymerization, with ATRP in particular leveraged to create well-defined POEGMA-based copolymers with tunable molecular weights and side chain lengths/functionalization.27 Such controlled/living radical polymerization techniques bypass many of the challenges associated with fabricating comb copolymers (e.g. steric bulk at graft from/to sites that results in irregular comb densities or inconsistent molecular weight growth) that can result in low degrees of polymerization and high polydispersities in other types of comb polymers,28 thus enabling a systematic study of how the architecture of the combs influences drug delivery characteristics (akin to previous studies on graft PEGylated polymers29,30) using a more scalable and direct polymerization strategy. The commercial availability of OEGMA monomers with different side chain lengths also facilitates the fabrication of comb polymers compared to other reported techniques in which a secondary grafting step typically must be performed after the initial polymerization reaction, offering a key translational advantage relative to other reported comb polymer-based nanoparticle vehicles. Furthermore, although the mechanism remains unclear, POEGMA has been reported to avoid PEG-related immunogenicity,31 potentially related to the steric crowding around each oligo(ethylene glycol) side chain that may inhibit immune cell recognition. As such, POEGMA is a viable alternative to PEG in multiple applications but specifically of interest in biological applications in which circulation time is vital.
Herein, we directly compare the physical properties and in vivo tumor model biodistribution properties of NPs self-assembled from block copolymers with the same molecular weight and same relative hydrophobic/hydrophilic block lengths fabricated using PLA as the hydrophobic block and one of three hydrophilic blocks: linear PEG (P(L)LA10K-b-PEG10K), POEGMA with n = 8–9 ethylene oxide repeat units in the side chains (P(L)LA10K-b-POEGMA47510K) and POEGMA with n = 40 ethylene oxide repeat units in the side chains (P(L)LA10K-b-POEGMA200010K). The equivalent molecular weight of each of the three block copolymers synthesized, both within each block as well as overall, enables a direct assessment of the effect of the comb architecture (i.e. the number of side-chains versus side-chain length) of linear-comb copolymers on properties relevant to nanoparticle-based drug delivery including particle size, particle stability, anti-cancer drug encapsulation efficiency, cytocompatibility, and biodistribution in a mouse tumor model. We demonstrate that the comb copolymers offer the potential for lower sizes, improved stability, prolonged circulation times, enhanced spheroid penetration and thus killing, and improved tumour localization without compromising cytocompatibility, albeit at the cost of drug loading capacity.
Experimental
Materials
All reagents unless otherwise stated were purchased from Sigma Aldrich and all solvents were purchased from Caledon Laboratories Ltd. Amine-terminated P(L)LA10K-b-PEG10K and Cy5-P(L)LA10K-b-PEG10K were purchased from Ruixibiotech (Xi’an, China). Cyanine5 NHS ester was purchased from Lumiprobe. All monomers were purified using a basic alumina column to remove inhibitors prior to use. NIH 3T3 cells were purchased from ATCC: Cedarlane Laboratories.Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, PDI ≤ 1.1), 1.0 g (105 eq.) of OEGMA475, 5 mL of anisole, 1 mg (0.22 eq.) of CuBr2, and 4.0 μL (7.5 × 10−2 eq.) Me6TREN were mixed in a 25 mL round bottom flask, sealed, and degassed with N2 for 30 minutes. Following, 16.0 μL (2.47 eq.) of Sn(Oct)2 was injected into the flask and the reaction mixture was heated to 70 °C and stirred for 35 minutes. The reaction mixture was then exposed to oxygen, filtered through a basic alumina column, and purified via dialysis (SnakeSkin™ Dialysis Tubing, 10 K MWCO, 22 mm) in a methanol bath (6 × 6 hours). The purified product was dried using a rotary evaporator followed by drying under high vacuum for 8 hours or until dry.
000, PDI ≤ 1.1), 2.0 g of OEGMA2000 (50 eq.), 5 mL of anisole, 1 mg (0.22 eq.) of CuBr2, 4.0 μL (7.5 × 10−2 eq.) Me6TREN were mixed in a 25 mL round bottom flask, sealed, and degassed with N2 for 30 minutes. 16.0 μL (2.47 eq.) of Sn(Oct)2 was then injected into the round bottom flask, and the reaction mixture was heated to 70 °C and stirred for 10 minutes. The reaction mixture was then exposed to oxygen, filtered through a basic alumina column, and purified via dialysis (SnakeSkin™ Dialysis Tubing, 10 K MWCO, 22 mm) in a methanol bath (6 × 6 hours). The purified product was dried using a rotary evaporator followed by drying under high vacuum for 8 hours or until dry.
000 cells per well in 150 μL of DMEM containing 10% FBS and 1% penicillin/streptomycin and incubated (37 °C, 5% CO2) for 24 hours. Polymer samples were sterilized via filtration through a non-pyrogenic Acrodisc® syringe filter (0.20 μm Supor® membrane) and then added to the cell wells at concentrations between 0.1–1 mg mL−1. After 24 h of incubation, the DMEM solution was replaced with 150 μL of a DMEM/resazurin (10 μg mL−1) solution and incubated for 4 hours. Converted resorufin was measured using a BioTek Synergy HTX Multi-Mode Reader plate reader (λexcitation = 575 ± 10 nm, λemission = 600 ± 10 nm). The results are presented as relative viabilities compared to non-treated controls (i.e. cells plated at the same density but not exposed to NPs), with the error bars representing the standard deviations of the percent viability measurements (n = 6).
000, PDI ≤ 1.1), 1.0 g (105 eq.) of OEGMA475, 5 mL of anisole, 1 mg (0.22 eq.) of CuBr2, and 4.0 μL (7.5 × 10−2 eq.) Me6TREN were mixed in a 25 mL round bottom flask, sealed, and degassed with N2 for 30 minutes. Following, 16.0 μL (2.47 eq.) of Sn(Oct)2 was injected into the flask and the reaction mixture was heated to 70 °C and stirred for 35 minutes. The reaction mixture was then exposed to oxygen, filtered through a basic alumina column, and purified via dialysis (SnakeSkin™ Dialysis Tubing, 10 K MWCO, 22 mm) in a methanol bath (6 × 6 hours). The purified product was dried using a rotary evaporator followed by drying under high vacuum for 8 hours or until dry.
000, PDI ≤ 1.1), 2.0 g of OEGMA2000 (50 eq.), 5 mL of anisole, 1 mg (0.22 eq.) of CuBr2, 4.0 μL (7.5 × 10−2 eq.) Me6TREN were mixed in a 25 mL round bottom flask, sealed, and degassed with N2 for 30 minutes. 16.0 μL (2.47 eq.) of Sn(Oct)2 was then injected into the round bottom flask, and the reaction mixture was heated to 70 °C and stirred for 10 minutes. The reaction mixture was then exposed to oxygen, filtered through a basic alumina column, and purified via dialysis (SnakeSkin™ Dialysis Tubing, 10 K MWCO, 22 mm) in a methanol bath (6 × 6 hours). The purified product was dried using a rotary evaporator followed by drying under high vacuum for 8 hours or until dry.
000 cells per well in 150 μL of DMEM containing 10% FBS and 1% penicillin/streptomycin and incubated (37 °C, 5% CO2) for 24 hours. Polymer samples were sterilized via filtration through a non-pyrogenic Acrodisc® syringe filter (0.20 μm Supor® membrane) and then added to the cell wells at concentrations between 0.1–1 mg mL−1. After 24 h of incubation, the DMEM solution was replaced with 150 μL of a DMEM/resazurin (10 μg mL−1) solution and incubated for 4 hours. Converted resorufin was measured using a BioTek Synergy HTX Multi-Mode Reader plate reader (λexcitation = 575 ± 10 nm, λemission = 600 ± 10 nm). The results are presented as relative viabilities compared to non-treated controls (i.e. cells plated at the same density but not exposed to NPs), with the error bars representing the standard deviations of the percent viability measurements (n = 6).
Transport of P(L)LA-PEG-based nanomaterials in 3D in vitro tumor cell cultures
Toxicity of PLA-PEG nanomaterials in spheroids
The toxicity of DOX-loaded P(L)LA10K-b-PEG10K, P(L)LA10K-b-POEGMA47510K, and P(L)LA10K-b-POEGMA200010K NPs was tested in CT26 spheroids cultured as described above. DOX was loaded as described in the Fabrication of loaded and blank nps section, after which 1 μL of the 7.5 mg mL−1 drug-loaded nanoparticle suspension was added to spheroids in triplicate and incubated for 72 h. Following, Invitrogen™ LIVE/DEAD™ Cell Imaging Kit (488/570) (10 μL of the mixed stock solution) was added directly to each treated spheroid as well as untreated spheroids (control) and incubated in the dark at room temperature for 15 minutes. Z-stacks of the spheroids were then collected using a confocal microscope at 10 μm depth intervals using both the FITC (live cells; brightness = 400, gain = 2, offset = −1; TD brightness = 105, gain = 1.25, offset = 2) and Texas Red (dead cells; brightness = 350, gain = 2, offset = −1; TD brightness = 105, gain = 1.25, offset = 2) filters. All z-stack images in each channel were then projected onto a single composite image using ImageJ and the average intensity was projected. A circle was manually drawn to encompass the area of each spheroid, with the area and IntDen (representing the pixel density within the circle) calculated separately in each channel for all spheroids measured. The IntDen/area ratio was calculated in each or the red and green channels, after which the ratio of greenIntDen/area:redIntDen/area was calculated to assess the ratio of live/dead cells within each spheroid. The average ± greenIntDen/area:redIntDen/area was plotted for each treatment group. One-way ANOVA was performed with Dunnett's post-hoc test to compare the DOX-loaded nanoparticle results to control (untreated) spheroids.
Biodistribution studies
BALB/c mice (n = 5) were shaved and implanted intradermally on the left flank with 1 × 106 CT26 tumor cells using a 30 μL injection volume in PBS and allowed to grow into tumors for 10 days or until palpable (∼200 mm3). Following, to perform biodistribution studies, mice were injected via the tail vein with 200 μL of a 7.5 mg mL−1 Cy5-labelled nanoparticle suspension and sacrificed at 2 h, 8 h, and 24 h post-injection. The mass concentration of Cy5-labeled polymer was conserved between all NPs; however, while both PLA-POEGMA polymers were labeled with the same 5 wt% Cy5 content and prepared from the same polymer batches, the Cy5-labeled P(L)LA10K-b-PEG10K is a commercial product with a higher per gram fluorescence value. As such, comparisons are restricted to relative fluorescence distributions within single treatment groups rather than total fluorescence values between the different treatment groups. The lungs, heart, spleen, liver, tumor, and kidneys were excised and imaged using an ex vivo imaging system (PerkinElmer IVIS Spectrum CT, λexcitation = 640, λemission = 680 at a height of 0.1 cm). Following, heart, spleen, lung, kidney, and tumor tissues were collected into 2 mL Eppendorf tubes and liver tissue was collected into a 15 mL Falcon tube, covered in foil, and stored in a −80 °C freezer for further analysis.Organ homogenization
Organs were briefly thawed at −4 °C before the experiment. Each full organ (or a < 150 mg portion of the liver) was weighed and placed in a 15 mL Falcon tube with 1× PBS to achieve known tissue concentrations within the concentration range of 10–40 mg mL−1 (heart: 30 mg mL−1, liver: 20 mg mL−1, spleen: 30 mg mL−1, lung: 40 mg mL−1, kidney: 40 mg mL−1, and tumor: 10 mg mL−1); these concentrations were chosen in light of the total mass of each tissue available to ensure all samples had >1.5 mL total volume of PBS (maximum 6 mL). The tissues were then homogenized (Kinematica Polytron, Blade: PT 10/35 Model: PCU-2-110, Brinkmann Instruments, New York) for 1 min, followed immediately by running the homogenizer probe in 70% ethanol 30% MQW for 30 seconds for cleaning. The organ mixture was vortexed and immediately pipetted into black edged, clear bottom 96 well plates at a volume of 10-40 μL (proportionate to the above concentrations listed for each organ), after which PBS was used to dilute each well to a 200 μL total volume containing 2 mg mL−1 tissue in each well. A PBS control was used in addition to the control animal organs to ensure there was no autofluorescence of any of the organ homogenates in the excitation/emission range of Cy5. An excitation wavelength of 640 nm was used for spectrum scanning the liver (chosen given that it gave the highest signal from the IVIS experiments) to determine the optimal excitation wavelength (680 nm) and the optimal gain (150) to be used on the remaining plates. Results correspond to tissue fluorescence across n = 5 animals per group, with each plate scanned 3 times for technical replication.Results
P(L)LA-b-POEGMA block copolymers were prepared via ARGET ATRP using a 2-bromoisobutyryl-terminated poly(L-lactide) macroinitiator (Scheme 1). POEGMA polymers were prepared using OEGMA monomers with either n = 8–9 repeat unit (475 Da molecular weight) or n = 40 repeat unit (2000 Da molecular weight) side chains, both of which are sufficiently long to promote high protein repellency rather than thermoresponsive behavior;34 indeed, that polymers fabricated with n = 2–3 repeat unit side chains were not colloidally stable at physiological temperature due to the LCST behavior of short side chain POEGMA polymers. | ||
| Scheme 1 General synthesis of P(L)LA-b-POEGMA (top) and Cy5-labelled P(L)LA-b-POEGMA (bottom) via ARGET ATRP. | ||
Table 1 summarizes the molecular weight properties of the block copolymers produced as well as corresponding information for the commercial P(L)LA-PEG polymer used as the linear polymer control (see ESI, Fig. S4–S6† for full traces). Both POEGMA-based blocks had number average molecular weights (Mn) of ∼10 kDa as measured via gel permeation chromatography, comparable to the 10 kDa macroinitiator used and the 10 kDa PEG chain present on the P(L)LA-PEG commercial polymer. As such, all three polymers tested have an equivalent hydrodynamic radius, a parameter we anticipate to be most predictive of nanoparticle assembly (given its direct correlation with polymer geometry) and thus chosen for use as the basis for direct comparisons between the samples. 1H NMR analysis was also conducted to calculate the Mn by comparing the relative amounts of both lactic acid and ethylene oxide repeat units incorporated into the polymer (see ESI Fig. S5 and S6 and Table S1†). Based on the known 10 kDa molecular weight of the PLA macroinitiator, the Mn values of the POEGMA blocks for P(L)LA10K-b-POEGMA47510K and P(L)LA10K-b-POEGMA200010K were both calculated to be ∼45 kDa, significantly higher than the GPC result and consistent with the compact comb structure of the OEGMA-based blocks resulting in lower effective hydrodynamic diameters at equivalent molecular weights. Of note, the equivalent measured hydrodynamic diameters and molecular weights of the two P(L)LA-b-POEGMA polymers facilitate highly relevant direct comparisons between those samples. Đ values of the synthesized polymers were also found to be consistently low (<1.1), indicative of tight control over the polymerization process using the ARGET ATRP protocol. Based on the equivalent hydrodynamic diameters of both the P(L)LA and PEG-based blocks among all three copolymers, the effects of comb length can be assessed independent of the effects of block length in subsequent experiments.
| Polymer | M n (GPC) | M n (1H NMR) | M w (GPC) | Đ (GPC) |
|---|---|---|---|---|
| 2-Bromoisobutyryl terminated poly(L-lactide) macroinitiator | 10000 |
7600 | — | <1.10 |
| P(L)LA10K-b-PEG10K | 19500 |
19300 |
23000 |
1.18 |
| P(L)LA10K-b-POEGMA47510K | 19000 |
53300 |
20200 |
1.06 |
| P(L)LA10K-b-POEGMA200010K | 19900 |
53500 |
21500 |
1.08 |
NPs were then fabricated via flash nanoprecipitation based on each of the linear and comb copolymers fabricated using a CIJ-M device. Each of the linear-comb copolymers maintains a critical micelle concentration (CMC) < 40 mg L−1 to ensure facile nanoassembly (ESI Fig. S1†), although the longer comb (and thus sterically bulkier) P(L)LA10K-b-POEGMA200010K polymer CMC is significantly higher than that of the shorter comb P(L)LA10K-b-POEGMA47510K polymer or the linear–linear P(L)LA10K-b-PEG10K polymer. Flash nanoprecipitation facilitates the production of NPs within or very close to the targeted circulation size range of ∼100–150 nm for each of the three block copolymers (Table 2), with TEM images showing that all NPs showed spherical morphologies (Fig. 1A–C). However, NPs fabricated from the linear-comb analogues were both initially smaller (particularly those prepared with the shorter n = 8–9 side chains), exhibit lower polydispersity, and better maintained their size over extended incubation at 37 °C in a PBS solution to emulate physiological conditions compared to the linear–linear P(L)LA10K-b-PEG10K copolymer (Fig. 1D and E). Changing the solvent from THF to DMF significantly reduced NP size for the linear-comb copolymer NPs by a factor of up to ∼2 for the P(L)LA10K-b-POEGMA47510K NPs but had little impact on the size of the linear–linear copolymer NPs; the very high solubility of the POEGMA475 block in DMF can be correlated directly to this result, with the longer PEG linear chain or the longer comb length POEGMA2000 exhibiting still sufficient but somewhat lower solubility. Correspondingly, the P(L)LA10K-b-POEGMA47510K NPs fabricated in DMF retained high colloidal stability over at least two weeks (Fig. 1E) without significant alterations in the observed particle size distribution (ESI Fig. S7†) while NPs prepared with both the POEGMA2000 and PEG hydrophilic blocks using DMF showed reduced stability relative to their THF-fabricated counterparts consistent with the potential for trace amounts of DMF to drive block copolymer rearrangements/disassembly in suspension. The very low particle size achievable with the shorter comb P(L)LA10K-b-POEGMA47510K copolymers in DMF is notable for a rapid nanoparticle fabrication method such as flash nanoprecipitation.35
| Solvent | Polymer | Hydrodynamic diameter (nm) | Polydispersity | Zeta potential (mV) | Drug loading capacity (%) | Encapsulation efficiency (%) |
|---|---|---|---|---|---|---|
| THF | P(L)LA10K-b-PEG10K | 159 ± 2 | 0.34 | −4 ± 1 | — | — |
| P(L)LA10K-b-POEGMA47510K | 88 ± 2 | 0.27 | −15 ± 2 | — | — | |
| P(L)LA10K-b-POEGMA200010K | 114 ± 1 | 0.21 | −11 ± 1 | — | — | |
| DMF | P(L)LA10K-b-PEG10K | 151 ± 1 | 0.29 | −1 ± 1 | — | — |
| P(L)LA10K-b-POEGMA47510K | 43 ± 1 | 0.28 | −1 ± 1 | — | — | |
| P(L)LA10K-b-POEGMA200010K | 81 ± 3 | 0.39 | −1 ± 1 | — | — | |
| DMF + 1 mg mL−1 Dox·HCl) | P(L)LA10K-b-PEG10K | 343 ± 3 | 0.25 | 1 ± 2 | 3.3 ± 0.1 | 33 ± 1 |
| P(L)LA10K-b-POEGMA47510K | 97 ± 3 | 0.33 | 1 ± 4 | 2.5 ± 0.1 | 25 ± 1 | |
| P(L)LA10K-b-POEGMA200010K | 102 ± 5 | 0.37 | 1 ± 1 | 1.4 ± 0.2 | 14 ± 2 |
 | ||
| Fig. 1 Particle size, morphology, and stability of linear-comb PLA-POEGMA copolymer nanoparticles compared to linear–linear PLA-PEG nanoparticles: (A–C) transmission electron microscopy images of nanoparticles fabricated using (A) P(L)LA10K-b-PEG10K; (B) P(L)LA10K-b-POEGMA47510K; and (C) P(L)LA10K-b-POEGMA200010K (scale bars = 200 nm); (D and E) DLS stability data for linear and comb block copolymer-based nanoparticles fabricated using (D) THF or (E) DMF as a solvent via flash nanoprecipitation over a 15 day incubation time in PBS at 37 °C (n = 3, data points represent the mean values, error bars represent the standard deviation). Beyond day 3 in DMF (panel E, grey arrow) P(L)LA10k-b-POEGMA200010k exhibited large sizes and polydispersity values indicative of nanoparticle aggregation. | ||
To assess the effect of the PEG block morphology on drug loading, DMF was selected to as the solvent phase during flash nanoprecipitation to facilitate the solubilization of doxorubicin hydrochloride in the precursor organic solution. Both encapsulation efficiency (EE) and drug loading capacity (DLC) are reduced by the introduction of a comb morphology in the PEG block (Table 2), with only minimal reductions observed for the shorter POEGMA475 hydrophilic block (n = 8–9 side chains, ∼15% decrease) but significantly higher reductions observed with the longer POEGMA2000 hydrophilic block (n = 40 side chains, >50% decrease). As such, while the introduction of a comb morphology in the PEG block reduces the particle size and improves the colloidal stability of the NPs, the concurrent reduction in drug loading capacity must also be considered depending on the required dose of therapeutic to be delivered. Of note, doxorubicin loading significantly increased the particle size of the linear–linear P(L)LA10K-b-PEG10K copolymer above the particle size range typically cited to enable long-term circulation (20–250 nm) while both comb copolymers exhibit only small increases in particle size that keep the loaded particle size of the NPs within the efficient circulation range (∼50–100 nm), suggesting another benefit of the comb copolymer building blocks.36–38
The cytotoxicity of each NP type was assessed by conducting a resazurin assay on 3T3 NIH mouse fibroblasts at concentration of up to 1 mg mL−1 for 24 hours. None of the NP formulations displayed significant cytotoxicity in vitro at any tested concentrations, using 80% cell viability as the threshold for toxicity (Fig. 2). As such, NPs based on both the linear and comb PEG block copolymers are non-cytotoxic to cells.
 | ||
| Fig. 2 Relative cell viability of NIH 3T3 fibroblasts as measured via a resazurin assay over a 24 hour exposure time to various concentrations (n = 6, data points represent the mean values, error bars represent the standard deviation) of P(L)LA10K-b-PEG10K, P(L)LA10K-b-POEGMA47510K, and P(L)LA10K-b-POEGMA200010K NPs. | ||
To assess the effect of the comb structure of the hydrophilic blocks on tumor transport, P(L)LA10K-b-POEGMA10K NPs were fluorescently labeled with Cy5 and incubated with CT26 colon carcinoma spheroids. Confocal laser scanning microscopy taken after 2 hours of incubation (Fig. 3) show that NPs with both comb lengths can penetrate effectively into the spheroids, with significant fluorescence observable at least 60 μm into the spheroids. The effectiveness of DOX-loaded P(L)LA10K-b-POEGMA10K NPs in killing these spheroids was then assessed via LIVE/DEAD staining, comparing to non-drug loaded spheroid controls (Fig. 4A). Even in control (untreated) spheroids, a significant amount of dead cells is observed consistent with the necrotic cores of in vivo tumors; however, treatment with any DOX-loaded nanoparticle shows increased red fluorescence compared to control spheroids. Comparing the live:dead ratio (calculated by dividing green fluorescence by red fluorescence in each treatment group in triplicate, Fig. 4B), control spheroids had ∼1:1 live/dead cells, while higher ratios of dead cells were seen in P(L)LA10K-b-PEG10K (0.72 ± 0.06), P(L)LA10K-b-POEGMA47510K (0.58 ± 0.05), and P(L)LA10K-b-POEGMA200010K (0.59 ± 0.08). Interestingly, while the DOX-loaded linear–linear copolymer P(L)LA10K-b-PEG10K did not induce a significantly lower live:dead ratio compared to the control, both P(L)LA10K-b-POEGMA10K formulations did; we attribute this result to the high penetration capacity of the P(L)LA10K-b-POEGMA10K formulations. Fig. 4A also shows increased dead cells at deeper regions of the spheroid (60 μm) for both P(L)LA10K-b-POEGMA47510K and P(L)LA10K-b-POEGMA200010K compared to P(L)LA10K-b-PEG10K as well as a thin veil of dead cells at all penetration depths, likely indicative of dead cancer cells breaking from the surface of the spheroid. As such, the linear-comb P(L)LA10K-b-POEGMA47510K and P(L)LA10K-b-POEGMA200010K NPs can both penetrate deep into tumor cells and efficiently release DOX to enhance spheroid toxicity compared to that observed with the linear–linear P(L)LA10K-b-PEG10K NPs.
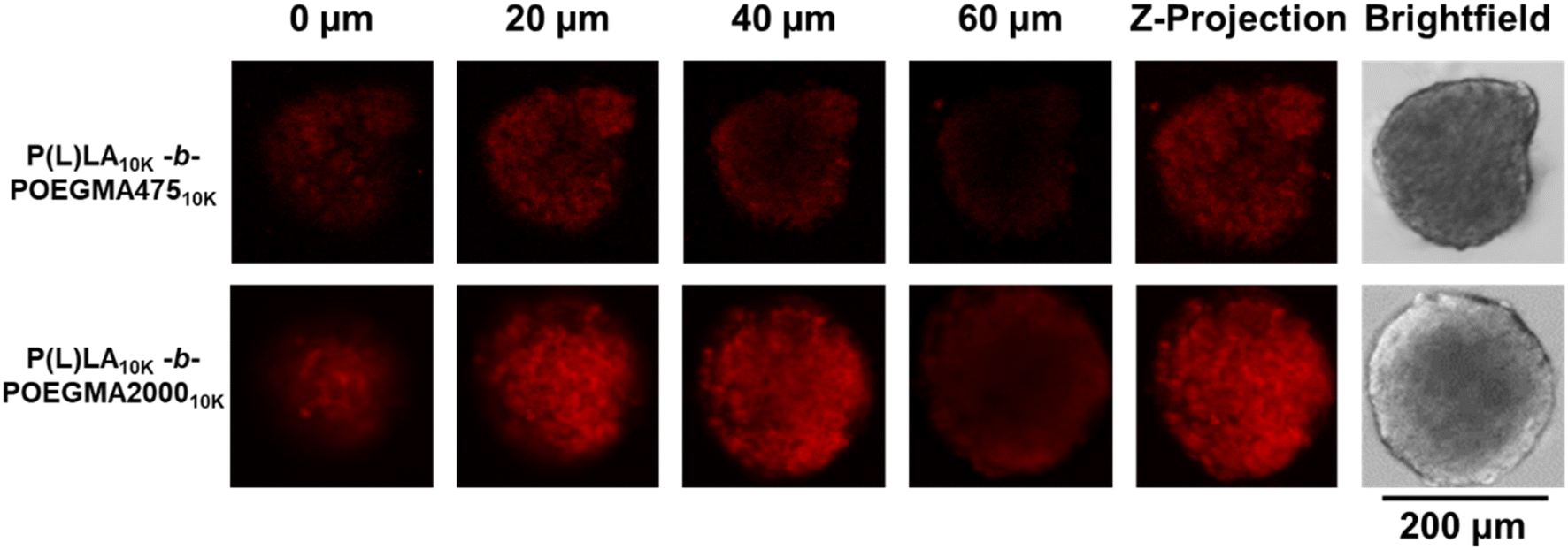 | ||
| Fig. 3 Penetration of Cy5-labeled P(L)LA10k-b-POEGMA47510k and P(L)LA10k-b-POEGMA200010k into CT26 tumor spheroids. | ||
 | ||
| Fig. 4 Spheroid toxicity at varying penetration depths using a LIVE/DEAD® assay and imaging live (green) and dead (red) cells using a confocal laser scanning microscope. (A) Confocal images after incubating spheroids with either no DOX (control), or DOX-loaded P(L)LA10K-b-PEG10K, P(L)LA10K-b-POEGMA47510K, or P(L)LA10K-b-POEGMA200010K for 72 h and staining with LIVE/DEAD®. (B) Ratio of green:red fluorescence (live:dead ratio) within the z-projections for each spheroid. n = 3 per formulation tested, ns not significant, *p < 0.05. | ||
To assess the differences in biodistribution achieved according to the different sizes and colloidal stabilities of NPs formed from linear–linear versus linear-comb block copolymers, Cy5-labeled NPs were injected in the tail vein of mice and the biodistribution of the fluorescently labeled NPs was tracked using both ex vivo imaging as well as fluorescence quantification of homogenized tissue samples. Cy5 was incorporated in sufficient quantities to maximize signal but to minimize any interfacial changes in the nanoparticles that may impact cell–nanoparticle interactions (<5 mol% total monomer residues functionalized), consistent with or lower than approaches used in other studies.39,40Fig. 5 shows the ex vivo IVIS images of each organ excised after 2 h, 8 h, and 24 h post-administration (refer to ESI, Fig. S8† for the corresponding measured irradiance values). P(L)LA10K-b-PEG10K showed high fluorescence in all organs and the tumor (right-most column) even at the very short 2 hour time point that does not significantly change over the subsequent 22 hours, indicating relatively rapid deposition of the linear–linear NPs in organs; note that the Cy5 density is higher on the commercial linear–linear block copolymer than on the internally-synthesized linear-comb copolymers, such that absolute fluorescence values cannot be compared between the different samples but can be compared within single samples at different timepoints. Conversely, a relatively lower fluorescence signal was noted for both linear-comb copolymers at 2 h but continuing accumulation of the NPs (particularly in the tumour) was observed over the full 24 hour period of the circulation time experiment.
 | ||
| Fig. 5 Representative images of the biodistribution of fluorescently-labelled NPs (experimental groups n = 6, PBS group n = 5) 2 h, 8 h, and 24 h after intravenous injection the heart, liver, spleen, lungs, kidneys, and implanted tumors (left to right). A PBS control is displayed at the bottom of P(L)LA10K-b-PEG10K 2 h, P(L)LA10K-b-POEGMA200010K 8 h, and all P(L)LA10K-b-PEG10K 24 h timepoints to confirm the absence of tissue auto-fluorescence at this wavelength. | ||
Homogenization of the organs to facilitate improved quantification of the fluorescence single yielded a similar trend (Fig. 6), with the linear–linear P(L)LA10K-b-PEG10K copolymer showing significant tumor accumulation at short times but little if any additional accumulation over the subsequent 22 hours (Fig. 6A) while significant increases in tumor fluorescence (both in absolute terms as well as in relative terms to the off-target organs) were observed for both comb copolymer-based NPs (Fig. 6B and C) over the full 24 hour period of the circulation time test. Furthermore, at 24 h, a significantly larger percentage of the total fluorescent signal (Fig. S9†) was observed from the tumor tissue with P(L)LA10K-b-POEGMA47510K (34.2 ± 0.5%) and P(L)LA10K-b-POEGMA200010K (28.5 ± 2.8%) relative to the linear P(L)LA10K-b-PEG10K control (19.0 ± 2.1%) (p < 0.05 in both pair-wise comparison), suggesting improved overall accumulation of the NPs in the tumor with the comb copolymers.
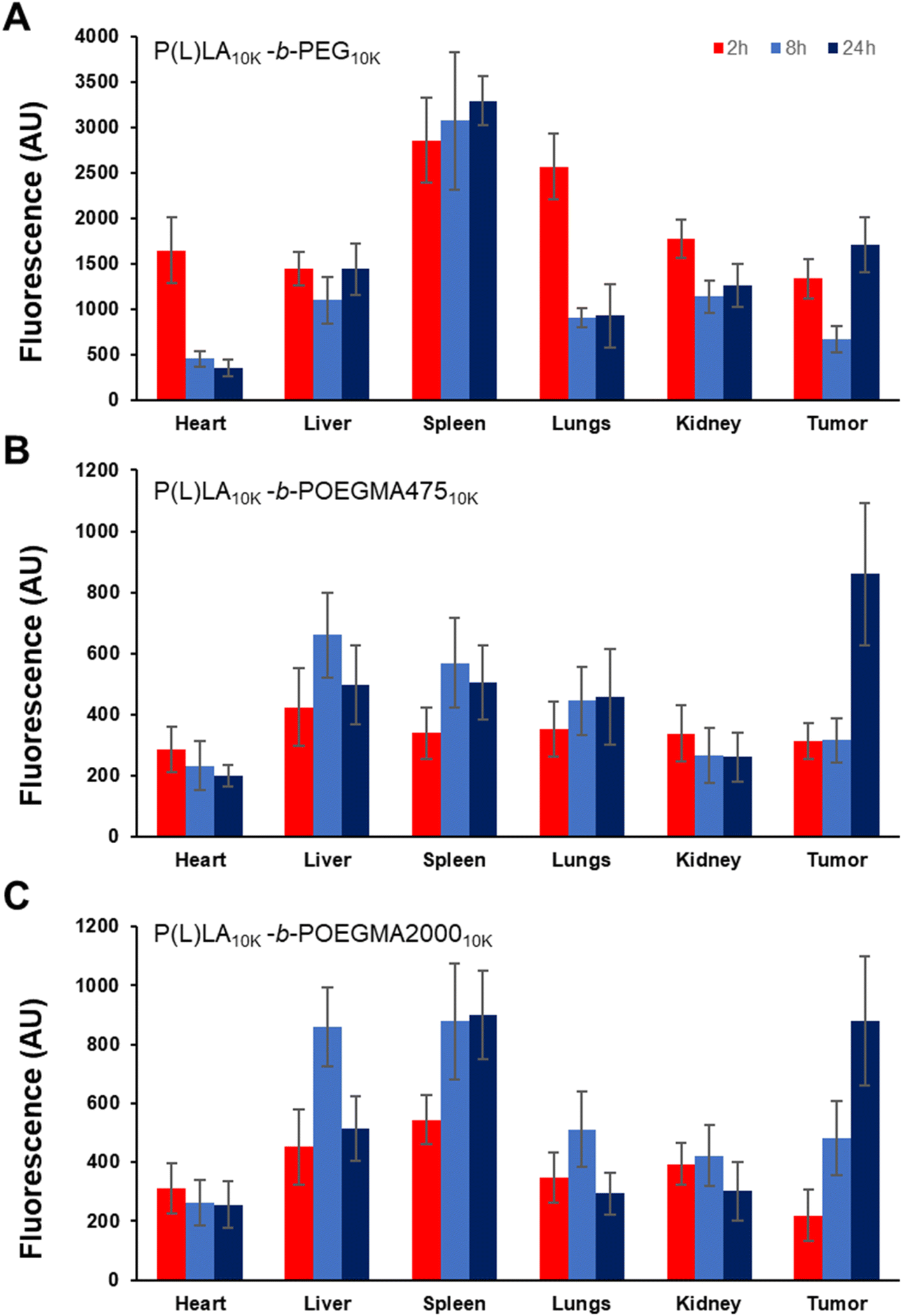 | ||
| Fig. 6 Biodistribution of Cy5-labeled polymer NPs in ground organs of BALB/c mice implanted with CT26 tumors as a function of time post-injection: (A) P(L)LA10K-b-PEG10K; (B) P(L)LA10K-b-POEGMA47510K; and (C) P(L)LA10K-b-POEGMA200010K (n = 5, data points represent the mean values, error bars represent the standard deviation). | ||
Discussion
Linear-comb block copolymers can be synthesized via ARGET-ATRP with low Đ values, allowing for full matching of both the total and individual block molecular weights between a commercial PLA-PEG linear polymer and the comb analogue copolymers synthesized herein. Unlike conventional PLA-PEG linear polymers that are challenging to functionalize aside from the chain ends (and even then often requiring expensive initiators), using ARGET-ATRP allows for facile incorporation of a comonomer in the hydrophilic POEGMA comb block, as demonstrated herein using (2-Boc-amino)ethyl methacrylate comonomers to generate functional primary amine residues in the POEGMA block following hydrolysis. Any polymerizable protected monomer compatible with ARGET-ATRP at any desired comonomer ratio could be incorporated using the same strategy, allowing for a plethora of functional groups to be added in a manner that makes the linear-comb block copolymers significantly more chemically tunable than conventional linear–linear block copolymers.NPs fabricated from linear-comb copolymers of comparable length were significantly smaller than those fabricated using their linear–linear counterparts. PEG comb copolymers have been reported to present a more sterically stable interface due to the high density of PEG repeat units covalently tethered in close proximity within the comb structure, enabling the stabilization of more surface area (i.e. smaller particle sizes).41 When prepared in THF, PEG10K-b-P(L)LA10K demonstrated a significant change in size over a 15 day period of incubation at 37 °C whereas neither P(L)LA10K-b-POEGMA47510K nor P(L)LA10K-b-POEGMA200010K showed significant changes in particle size over the same time period; similarly, when the NPs were prepared in DMF, the POEGMA475 linear-comb copolymer-based NPs exhibited good particle size stability while the linear–linear copolymer-based NPs showed very high size increases. These observations both also support the higher degree of steric stabilization facilitated by the comb structure in the hydrophilic block.41 However, the linear-comb block copolymers facilitated lower drug loading than the linear–linear block copolymers, with the longer chain P(L)LA10K-b-POEGMA200010K block copolymer showing a particularly reduced drug loading. In this context, we hypothesize that the same high comb density that enables improved colloidal stability also hinders the tight packing of the P(L)LA domains to reduce the size and/or packing density of the P(L)LA domains within the self-assembled NPs, leading to a reduced affinity and/or volume within the nanoparticle for hydrophobic drug loading. As such, depending on the dose required of a specific drug, the lower sizes and higher colloidal stability of the linear-comb block copolymers may be most relevant to apply with more potent drugs that require lower functional doses to be effective, with the shorter side chain POEGMA475-based NPs offering an appealing balance of both high colloidal stability and still relatively high drug loading. However, both POEGMA-based linear-comb block copolymers tested can enable both highly effective penetration into spheroids (attributed to the dense hydrophilic interface, higher nanoparticle stability, and smaller nanoparticle sizes enabled by the comb POEGMA polymer) as well as more efficient tumor killing, even compared to the PEG-based linear copolymer analogue that enables enhanced DOX loading. Indeed, the observation that the POEGMA-based NPs facilitate improved anti-tumor activity despite having lower drug loading capacities shows the importance of the PEG architecture in facilitating tumor transport, with shorter main chains and longer/denser PEG comb lengths enabling improved transport into tumors and thus improved tumor killing.
In vivo biodistribution experiments demonstrated significant accumulation of all NPs in tumors (Fig. 5 and 6); however, comb NPs displayed increased accumulation over a 24 hour time period and continuous localization of the NPs in the tumor over the full 24 hour test window. Specifically, P(L)LA10K-b-POEGMA47510K displayed a spike in NP concentration between the 8 and 24 hour timepoints whereas P(L)LA10K-b-POEGMA200010K displayed showed continuous increases over all timepoints, in contrast to P(L)LA10K-b-PEG10K which did not show any significant enhancement in tumor localization over the time period of the test. Furthermore, the spleen was the only other organ to show significant NP accumulation over time, specifically in the case of P(L)LA10K-b-POEGMA47510K NPs (although the accumulation is relatively low over the tested time window). Taken together, we interpret these results as the linear-comb block copolymers facilitating prolonged circulation times and thus continual accumulation in the tumor relative to the linear–linear block copolymer. While the precise mechanism leading to this result would require further study (i.e. if PEG antibody interactions are indeed suppressed by using POEGMA or if the brush structure otherwise alters the interfacial hydration layer to suppress more general opsonization), these results suggest potential advantages of the linear-comb copolymers for passive tumor drug targeting that can reduce the risk of off-target effects that are strongly detrimental to the safety and efficacy of anti-cancer therapies.
Conclusion
The polymer architecture of the hydrophilic block of self-assembled NPs can be engineered to regulate key nanoparticle properties, specifically by replacing linear PEG with comb POEGMA. ARGET-ATRP is shown to be a reproducible method to synthesize such linear-comb block copolymers with relatively low Đ values while providing a facile method to incorporate comonomers in the hydrophilic block to allow for covalent fluorescent labeling or affinity-based tuning of the nanoparticle interface. Using POEGMA-based hydrophilic blocks enables the fabrication of particles with significantly lower particle sizes and higher stabilities as a function of time while maintaining high cytocompatibility as well as enhanced tumor spheroid penetration and killing capacity, although such benefits must be balanced with the reduced drug loading capacities observed particularly with longer comb length copolymers. The linear-comb copolymers also accumulate in tumors following tail vein injections in mice to a higher degree over significantly longer time periods than observed with the conventional linear–linear polymers at the same molecular weight, suggesting a potentially improved capacity for passive tumor-targeted drug delivery. Overall, the linear-comb block copolymer platform described herein provides numerous options for tuning the size, chemistry, surface properties, colloidal stability, and thus circulation properties of block copolymer-based self-assembled NPs, opening new possibilities for designing nanoparticle-based delivery vehicles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Paul Gatt for the construction of the CIJ-M. Financial support from the Canadian Cancer Society Research Institute (CCSRI – grant # 702142), the Natural Sciences and Engineering Research Council of Canada (NSERC – grant # RGPIN-06455-17), and the Canada Research Chairs program (grant # 950-230943) is gratefully acknowledged.References
- B. Begines, T. Ortiz, M. Perez-Aranda, G. Martinez, M. Merinero, F. Arguelles-Arias and A. Alcudia, Nanomaterials, 2020, 10, 1403 CrossRef CAS PubMed.
- E. B. Souto, J. Dias-Ferreira, A. Lopez-Machado, M. Ettcheto, A. Cano, A. Camins Espuny, M. Espina, M. L. Garcia and E. Sanchez-Lopez, Pharmaceutics, 2019, 11, 460 CrossRef CAS PubMed.
- S. Sur, A. Rathore, V. Dave, K. R. Reddy, R. S. Chouhan and V. Sadhu, Nano-Struct. Nano-Objects, 2019, 20, 100397 CrossRef CAS.
- A. M. Bodratti and P. Alexandridis, Expert Opin. Drug Delivery, 2018, 15, 1085–1104 CrossRef CAS PubMed.
- S. Puntawee, M. Theerasilp, S. Reabroi, R. Saeeng, P. Piyachaturawat, A. Chairoungdua and N. Nasongkla, Pharm. Dev. Technol., 2016, 21, 437–444 CAS.
- Y. Tian and S. Mao, Expert Opin. Drug Delivery, 2012, 9, 687–700 CrossRef CAS PubMed.
- S. Yorulmaz Avsar, M. Kyropoulou, S. Di Leone, C. A. Schoenenberger, W. P. Meier and C. G. Palivan, Front. Chem., 2018, 6, 645 CrossRef PubMed.
- J. Han, Z. Zhu, H. Qian, A. R. Wohl, C. J. Beaman, T. R. Hoye and C. W. Macosko, J. Pharm. Sci., 2012, 101, 4018–4023 CrossRef CAS PubMed.
- C. Vauthier and G. Ponchel, Polymer Nanoparticles for Nanomedicines, Springer, 2017 Search PubMed.
- J. Wang, S. Li, Y. Han, J. Guan, S. Chung, C. Wang and D. Li, Front. Pharmacol, 2018, 9, 202 CrossRef PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- Y. Yi, G. Lin, S. Chen, J. Liu, H. Zhang and P. Mi, Mater. Sci. Eng., C, 2018, 83, 218–232 CrossRef CAS PubMed.
- R. P. Pawar, S. U. Tekale, S. U. Shisodia, J. T. Totre and A. J. Domb, Recent Pat. Regener. Med., 2014, 4, 40–51 CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- G. Chedid and A. Yassin, Nanomaterials, 2018, 8, 916 CrossRef PubMed.
- L. Bai, S. Z. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC.
- F. Zhao, H. Liu, S. D. R. Mathe, A. Dong and J. Zhang, Nanomaterials, 2017, 8, 15 CrossRef PubMed.
- S. K. Lai, Y. Y. Wang and J. Hanes, Adv. Drug Delivery Rev., 2009, 61, 158–171 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- L. M. Ensign, R. Cone and J. Hanes, J. Controlled Release, 2014, 190, 500–514 CrossRef CAS PubMed.
- L. M. Ensign, R. Cone and J. Hanes, Adv. Drug Delivery Rev., 2012, 64, 557–570 CrossRef CAS PubMed.
- S. Sindhwani, A. M. Syed, J. Ngai, B. R. Kingston, L. Maiorino, J. Rothschild, P. MacMillan, Y. Zhang, N. U. Rajesh, T. Hoang, J. L. Y. Wu, S. Wilhelm, A. Zilman, S. Gadde, A. Sulaiman, B. Ouyang, Z. Lin, L. Wang, M. Egeblad and W. C. W. Chan, Nat. Mater., 2020, 19, 566–575 CrossRef CAS PubMed.
- Q. Yang and S. K. Lai, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 655–677 CAS.
- Q. Yang, T. M. Jacobs, J. D. McCallen, D. T. Moore, J. T. Huckaby, J. N. Edelstein and S. K. Lai, Anal. Chem., 2016, 88, 11804–11812 CrossRef CAS PubMed.
- J.-F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- J.-F. Lutz, H. G. Borner and K. Weichenhan, Macromolecules, 2006, 39, 6376–6383 CrossRef CAS.
- J.-F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- H. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- J.-M. Rabanel, J. Faivre, G. D. Paka, C. Ramassamy, P. Hildgen and X. Banquy, Eur. J. Pharm. Biopharm., 2015, 96, 409–420 CrossRef CAS PubMed.
- J.-L. Wang, X.-J. Du, J.-X. Yang, S. Shen, H.-J. Li, Y.-L. Luo, S. Iqbal, C.-F. Xu, X.-D. Ye and J. Cao, Biomaterials, 2018, 182, 104–113 CrossRef CAS PubMed.
- I. Ozer, G. Kelly, R. Gu, X. Li, N. Zakharov, P. Sirohi, S. K. Nair, J. H. Collier, M. S. Hershfield, A. M. Hucknall and A. Chilkoti, Adv. Sci., 2022, 9, e2103672 CrossRef PubMed.
- T. Ren, A. Wang, W. Yuan, L. Li and Y. Feng, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2303–2313 CrossRef CAS.
- N. E. Timmins and L. K. Nielsen, Tissue Eng., 2007, 141–151 CAS.
- Z. Jin, W. Feng, K. Beisser, S. Zhu, H. Sheardown and J. L. Brash, Colloids Surf., B, 2009, 70, 53–59 CrossRef CAS PubMed.
- W. N. Sharratt, V. E. Lee, R. D. Priestley and J. T. Cabral, ACS Appl. Polym. Mater., 2021, 3, 4746–4768 CrossRef CAS.
- X. Duan and Y. Li, Small, 2013, 9, 1521–1532 CrossRef CAS PubMed.
- I. V. Zelepukin, A. V. Yaremenko, M. V. Yuryev, A. B. Mirkasymov, I. L. Sokolov, S. M. Deyev, P. I. Nikitin and M. P. Nikitin, J. Controlled Release, 2020, 326, 181–191 CrossRef CAS PubMed.
- E. P. Frank Alexis, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505–515 CrossRef PubMed.
- R. Tong, V. J. Coyle, L. Tang, A. M. Barger, T. M. Fan and J. Cheng, Microsc. Res. Tech., 2010, 73, 901–909 CrossRef CAS PubMed.
- X. Shi, X. Ma, M. Hou, Y.-E. Gao, S. Bai, B. Xiao, P. Xue, Y. Kang, Z. Xu and C. M. Li, J. Mater. Chem. B, 2017, 5, 6847–6859 RSC.
- B. A. Aguilar-Castillo, J. L. Santos, H. Luo, Y. E. Aguirre-Chagala, T. Palacios-Hernandez and M. Herrera-Alonso, Soft Matter, 2015, 11, 7296–7307 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na01156a |
| This journal is © The Royal Society of Chemistry 2024 |